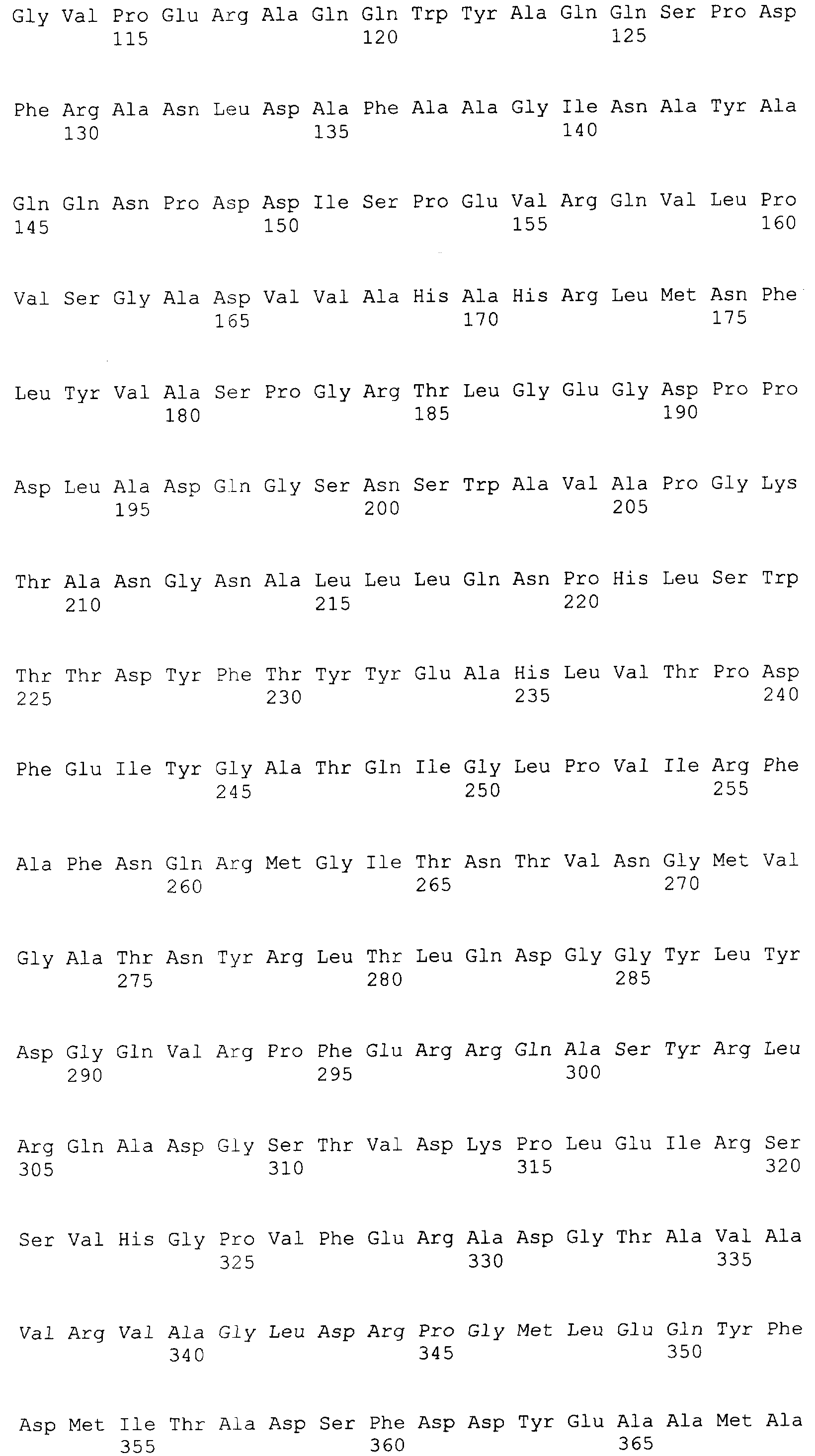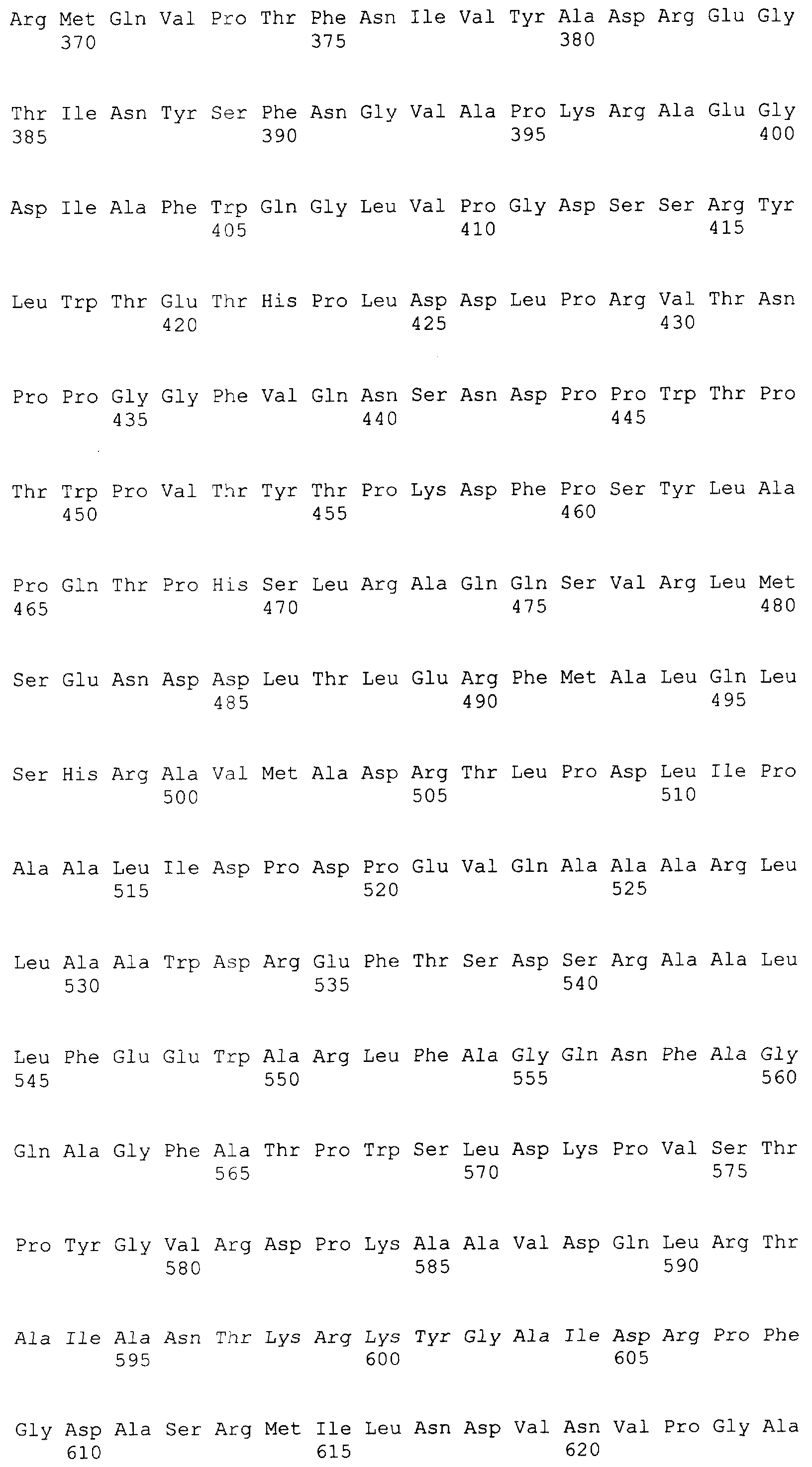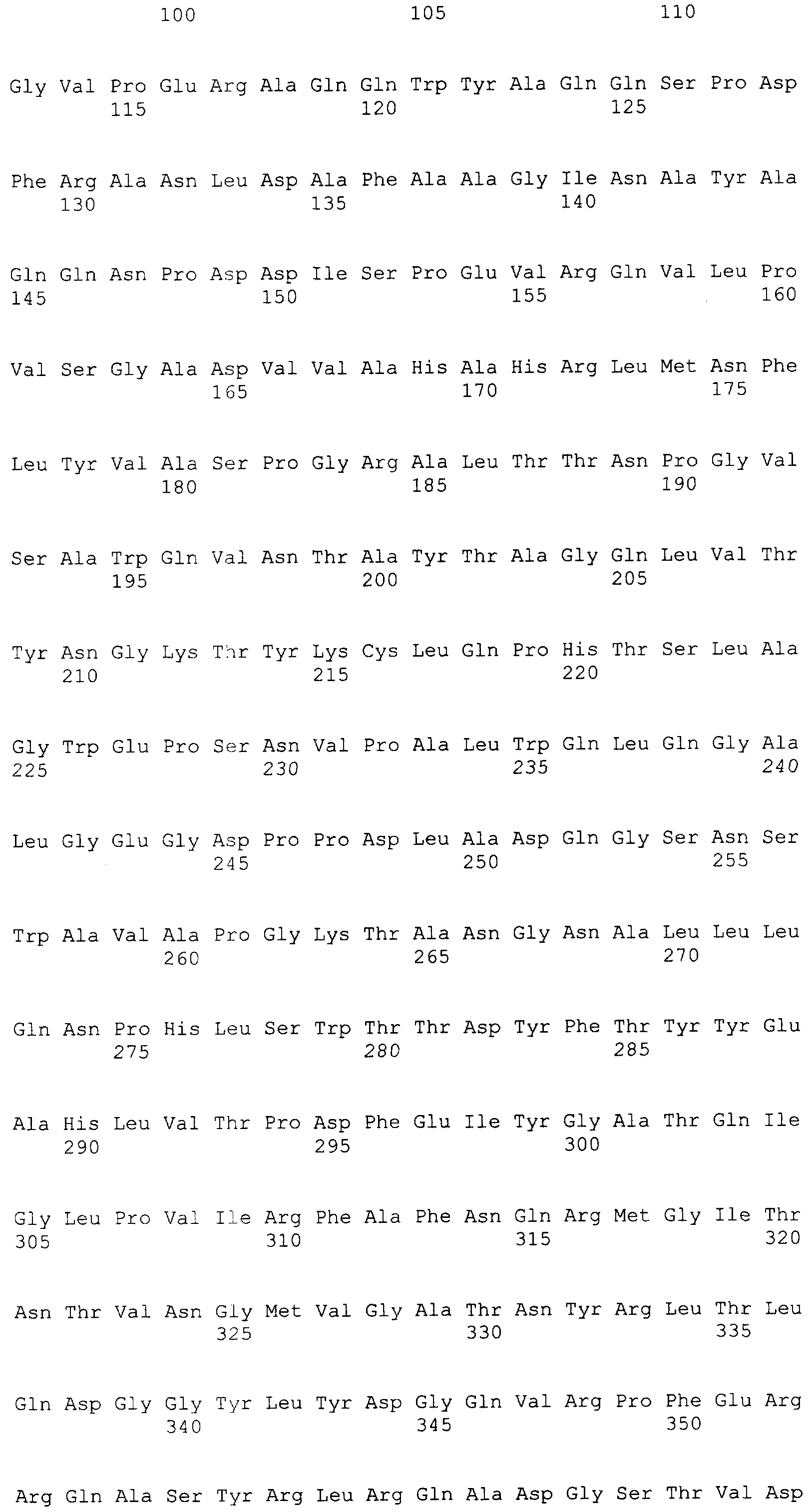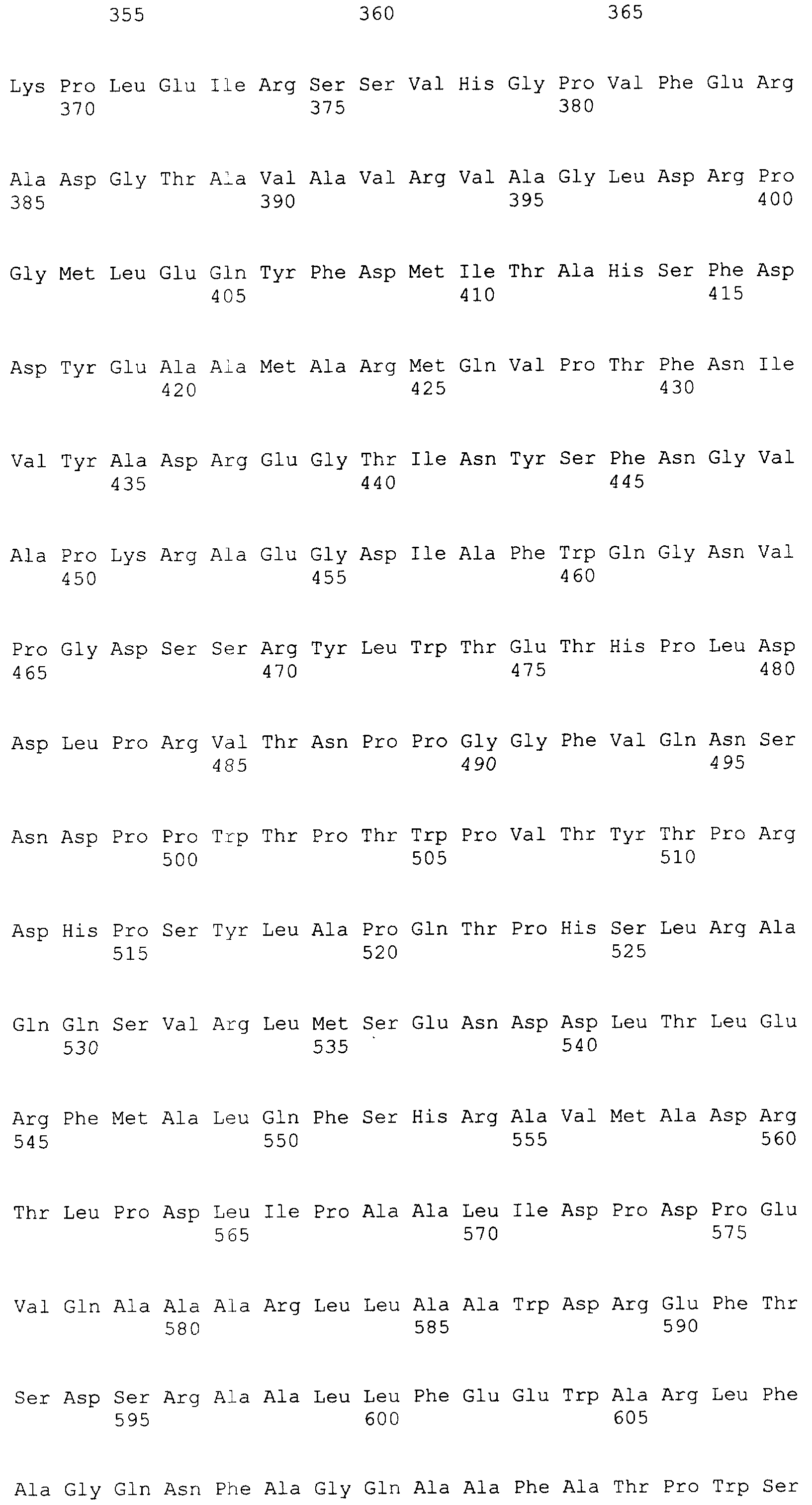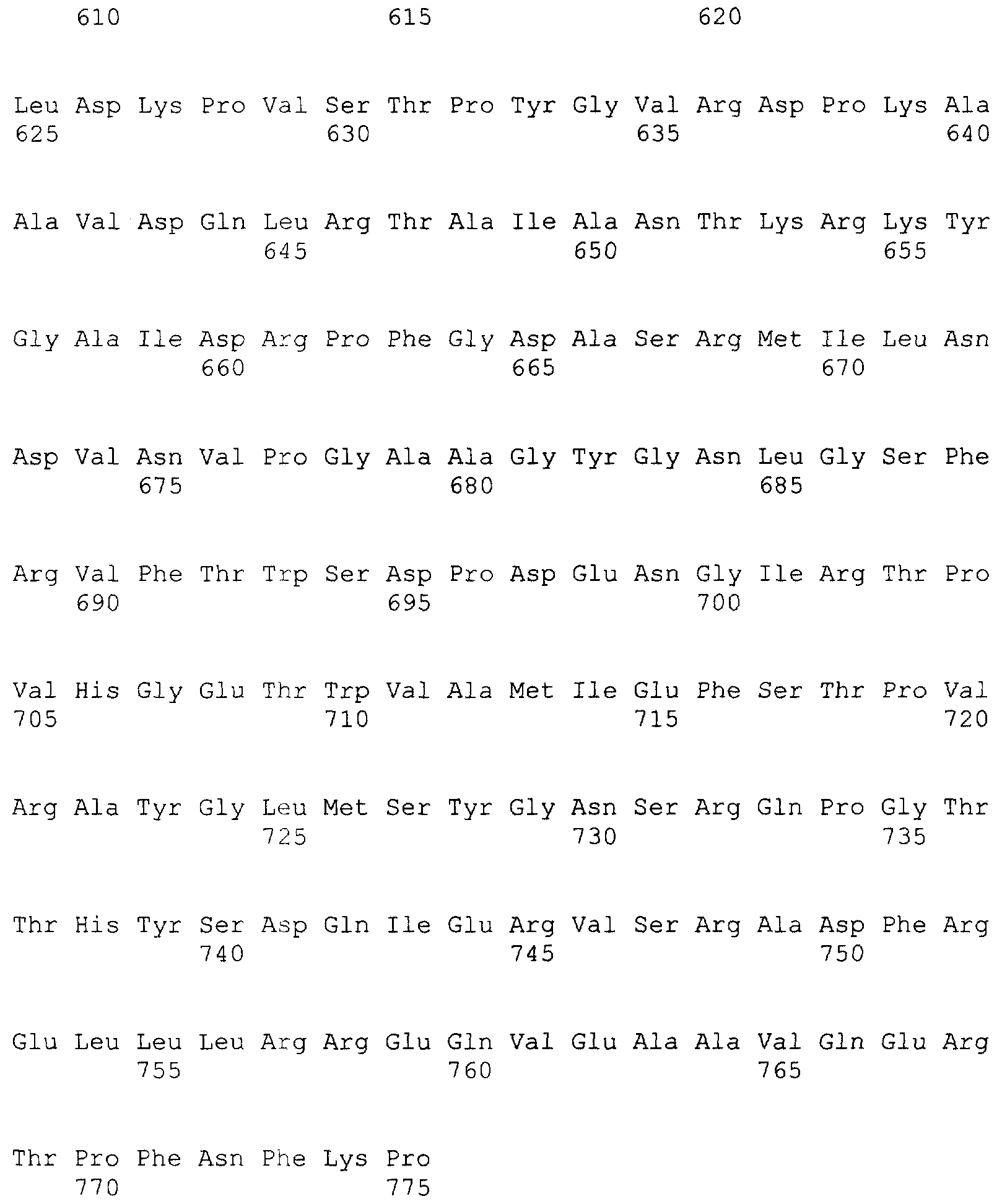Патент на изобретение №2388826
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) РЕКОМБИНАНТНАЯ ДНК, КОДИРУЮЩАЯ ФУНКЦИОНАЛЬНО АКТИВНЫЙ ГИБРИДНЫЙ БЕЛОК G17ACA-АЦИЛАЗЫ С ХИТИН-СВЯЗЫВАЮЩИМ ДОМЕНОМ (BrdG17ACA-cbd), РЕКОМБИНАНТНАЯ ПЛАЗМИДА pSVH0108, ОБЕСПЕЧИВАЮЩАЯ ЕГО СИНТЕЗ В КЛЕТКАХ Escherichia coli, И РЕКОМБИНАНТНЫЙ ШТАММ Escherichia coli BL21(DE3)/pSVH0108-ПРОДУЦЕНТ BrdG17ACA-cbd
(57) Реферат:
Настоящее изобретение относится к области биотехнологии, в частности к генетической инженерии, и может быть использовано в микробиологической промышленности при получении полусинтетических цефалоспориновых антибиотиков нового поколения. Получена рекомбинантная ДНК, которая кодирует функционально активный гибридный белок (BrdG17ACA-cbd), состоящий из аминокислотной последовательности ацилазы глутарил-7-аминоцефалоспориновой кислоты штамма Brevundimonas diminuta BKM В-1297 и хитин-связывающего домена хитиназы Al Bacillus circulans. Сконструирована рекомбинантная плазмида pSVH0108 для экспрессии BrdG17ACA-cbd в клетках E.coli, содержащая кодирующую гибридный белок последовательность рекомбинантной ДНК под контролем промотора и терминатора РНК-полимеразы фага Т7. В результате трансформации штамма E.coli данной рекомбинантной плазмидой и селекции трансформированных клонов получен новый штамм E.coli BL21(DE3)/pSVH0108 cbd – продуцент гибридного белка BrdG17ACA-cbd, обеспечивающий высокий выход рекомбинантного фермента.3 н.п.ф-лы, 4 ил., 2 табл.
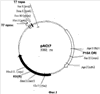
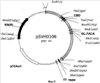
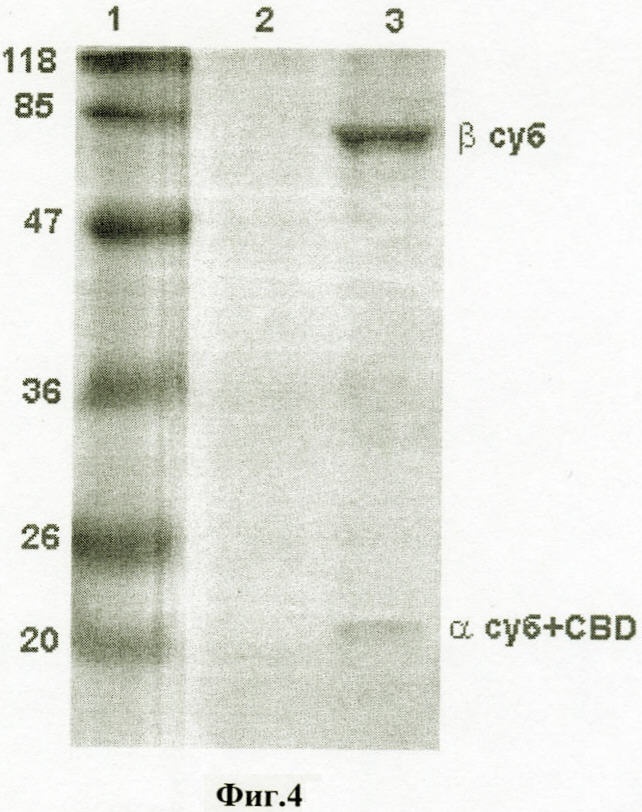
Настоящее изобретение относится к области биотехнологии, в частности к генетической инженерии, и может быть использовано в микробиологической промышленности при получении полусинтетических цефалоспориновых антибиотиков нового поколения. Предлагаются: – рекомбинантная ДНК, кодирующая функционально активный гибридный белок BrdGl7ACA-cbd, который состоит из ацилазы глутарил-7-аминоцефалоспориновой кислоты штамма Brevundimonas diminuta ВКМ B-1297 и хитин-связывающего домена хитиназы А1 Bacillus circulans; – рекомбинантная плазмида pSVH0108, содержащая фрагмент ДНК, который кодирует полную аминокислотную последовательность BrdGl7ACA-cbd, и обеспечивающая высокий уровень экспрессии названного гибридного белка в клетках Escherichia coli; – рекомбинантный штамм E.coli BL21(DE3)/pSVH0108 – продуцент гибридного белка BrdGl7ACA-cbd. Уровень техники Gl7ACA-ацилазы [1] относятся к обширному семейству пенициллин-амидаз (EC 3.5.1.11), способных превращать пенициллины и цефалоспорины в ценные промежуточные соединения, используемые при производстве полусинтетических бета-лактамных антибиотиков нового поколения [2, 3]. В связи с потребностью в наращивании объема производства этой широко применяемой ныне группы антибиотиков [4-6] усовершенствование известных методов получения необходимых для их синтеза ферментов, в том числе с помощью технологий рекомбинантных ДНК, представляет несомненный практический интерес. К настоящему времени известны первичные структуры генов бактериальных Gl7ACA-ацилаз из различных штаммов Pseudomonas; описаны рекомбинантные плазмидные ДНК, обеспечивающие синтез этих ферментов в клетках E.coli и Bacillus subtilis, а также способы получения рекомбинантных форм ферментов с их использованием [1, 7-12]. Общими недостатками известных методов гетерологичной экспрессии Gl7ACA-ацилаз являются невысокий уровень синтеза рекомбинантного продукта [7], сложности процессов масштабирования ферментации штаммов [8-10] и освобождения полученных препаратов Gl7ACA-ацилаз от примесей, в частности от неспецифических бета-лактамаз [5]. Устранение указанных недостатков может быть достигнуто на пути разработки новых систем экспрессии Gl7ACA-ацилазы, обеспечивающих повышение выхода активного фермента и упрощение методов его очистки. Поскольку одним из наиболее эффективных способов очистки белка является аффинная хроматография [13], в последние годы получили широкое распространение системы экспрессии для получения рекомбинантных белковых продуктов в форме гибридных белков, содержащих вспомогательный полипептид, специфически связывающийся с соединением, которое применяется в качестве лиганда при последующей очистке белка аффинной хроматографией. Получение гибридных белков, хотя и является достаточно простым и эффективным методом, сопряжено с некоторыми сложностями, одной из которых является возможность частичной или полной утраты активности вследствие модификации структуры молекулы белка после связывания его с вспомогательным полипептидом. В подобных ситуациях может быть предусмотрено последующее расщепление гибридного белка (дополнительная стадия) на составляющие под действием химических или энзиматических агентов, однако введение процедуры расщепления экспрессированного гибридного белка требует проведения последующей дополнительной очистки. С учетом указанной особенности получения ферментов в виде гибридных белков принципиальным моментом является поиск комбинации целевого и вспомогательного белка, в которой присутствие дополнительного компонента в препарате фермента не будет негативно сказываться на свойствах целевого продукта, а объединение предназначенного для очистки полипептида с интересующим белком, в свою очередь, не приведет к уменьшению его специфической связывающей активности. Конструирование гибридного белка с такими свойствами упрощает очистку целевого продукта и при этом гарантирует полное сохранение его активности в составе слитого белка. При удачном выборе дополнительного компонента рекомбинантные ферменты в гибридной конструкции оказываются полностью подготовленными к использованию в качестве иммобилизованного препарата, если это предусмотрено технологическим процессом. Что касается Gl7ACA-ацилазы, то попытки осуществить ее экспрессию в форме слитого белка по существу ограничиваются получением производного, несущего 6 дополнительных N-концевых гистидиновых остатков [14], которые традиционно используются для последующей хроматографической очистки многих рекомбинантных белков. Однако сорбенты для His-tag форм, как правило, являются достаточно дорогостоящими и мало пригодными для последующего прямого использования в составе биокатализаторов, что существенно ограничивает область их практического применения. В настоящем изобретении была поставлена задача экспрессии гибридного белка, включающего Gl7ACA-ацилазу, в котором бы полностью сохранялась функциональная активность обеих его составляющих, а его иммобилизованная форма при этом могла бы непосредственно применяться в процессе биотрансформации, а также разработки векторов и систем, необходимых для его получения. Раскрытие изобретения Решение задачи получения любого функционально-активного гибридного белка включает следующие этапы: а) дизайн конструкции гена гибридного белка, предполагающий оптимальную с точки зрения сохранения функциональной активности комбинацию целевого и вспомогательного белка; б) получение и объединение с помощью методов рекомбинантных ДНК нуклеотидных последовательностей, кодирующих компоненты гибридного белка; в) выбор клеток-хозяев и создание векторных конструкций для экспрессии гибридного белка в избранной гетерологичной системе; г) оптимизацию условий продукции и очистки гибридного белка. Поставленная цель получить функционально-активный гибридный белок на основе Gl7ACA-ацилазы, пригодный для последующего применения в качестве иммобилизованного препарата, достигнута за счет того, что 1) предложена рекомбинантная ДНК, кодирующая гибридный белок BrdGl7ACA-cbd, в котором в С-концевую область последовательности альфа-субъединицы ацилазы глутарил-7-аминоцефалоспориновой кислоты штамма Brevundimonas diminuta ВКМ B-1297 встроен хитин-связывающий домен хитиназы А1 Bacillus circulans; 2) сконструирована рекомбинантная плазмида pSVH0108, содержащая рекомбинантную последовательность ДНК, которая кодирует гибридный белок BrdGl7ACA-cbd, и обеспечивающая его синтез в клетках кишечной палочки с высоким выходом; 3) в результате трансформации экспрессирующей плазмидой pSVH0108 клеток штамма E.coli BL21(DE3) получен рекомбинантный штамм Escherichia coli BL21(DE3)/pSVH0108, характеризующийся высоким уровнем индуцируемого синтеза и стабильной продукцией гибридного белка BrdGl7ACA-cbd, в котором активность сохраняют оба входящих в его состав белковых домена. Выбор хитин-связывающего домена хитиназы А1 Bacillus circulans (CBD) в качестве вспомогательного полипептида был обусловлен тем, что хитин (хитиновый носитель), с которым он взаимодействует, имеет природное происхождение, является более дешевым по сравнению со многими другими носителями для аффинной хроматографии, а связь, которую образует включающий СВD гибридный белок с носителем, отличается физической и химической стабильностью, что позволяет использовать его многократно. Кроме того, в известных случаях использования хитин-связывающих доменов в комбинации с активными белками они, как правило, не оказывали неблагоприятного воздействия на функцию активной составляющей. Gl7ACA-ацилазы представляют собой гетеротетрамеры, состоящие из 2-х Нуклеотидную последовательность, кодирующую хитин-связывающий домен хитиназы А1 Bacillus circulans (SEQ ID Положение CBD в структуре гибридного белка было определено на основании данных о пространственной структуре Gl7ACA-ацилазы Brevundimoinas diminuta, комплексов этого фермента с субстратом и продуктом реакции и свойствах двухсубъединичных периплазматических ферментов, подвергающихся автокаталитическому процессингу [9, 15, 16]. Проведенный дизайн модификаций концевых участков полипептидной цепи субъединиц показал, что предпочтительным участком для связывания CBD является С-концевая область альфа- субъединицы Gl7ACA-ацилазы, поскольку она находится на достаточно удаленном расстоянии от активного центра фермента и не принимает участия в катализе. Хотя в настоящее время нет однозначного представления о механизме удаления спейсерного пептида [15, 17], есть основания полагать, что для правильного расщепления альфа-субъединицы и спейсерного пептида важна сохранность С-концевых аминокислот альфа-субъединицы, поэтому с целью снижения вероятности отщепления СBD вместе со спейсерным участком представлялось целесообразным включить последовательность хитин-связывающего домена так, чтобы после него оставалось несколько аминокислот С-конца альфа-субъединицы. Соответственно, положение CBD в структуре гибридного белка было определено между аминокислотами R184 и T185, что предполагало встраивание последовательности, кодирующей хитин-связывающий домен, между нуклеотидами 551 и 554 гена BrdGl7ACA-ацилазы (SEQ ID Объединение последовательности, кодирующей CBD (SEQ ID Для создания вектора экспрессии, направляющего в клетках E.coli синтез гибридного белка, на основе плазмиды pSVH1811 был сконструирован промежуточный вектор pSVH0107, полученный путем введения в нее дополнительного сайта рестрикции Vne1 в положении 551-554 SEQ ID Сравнение аминокислотных последовательностей BrdGl7ACA-cbd, BrdGl7ACA-Vne с последовательностью BrdGl7ACA-ацилазы приведено на фиг.3. Путем трансформации клеток штамма Escherichia coli BL21(DE3) [21] сконструированной плазмидой pSVH0108, отбора и культивирования клонов трансформантов с высоким уровнем синтеза функционально-активного гибридного белка получен рекомбинантный штамм Escherichia coli BL21(DE3)/ pSVH0108 – продуцент гибридного белка, состоящего из BrdGl7ACA-ацилазы, содержащей в С-концевой области последовательности альфа-субъединицы хитин-связывающий домен хитиназы А1 Bacillus circulans. Синтез BrdGl7ACA-cbd в полученном рекомбинантном штамме осуществляется при культивировании на обычных селективных средах с добавлением индуктора изопропил-D-тиогалактозида (ИПТГ) или лактозы. Таким образом, настоящее изобретение включает три объекта: Первый объект – рекомбинантная ДНК, которая кодирует гибридный белок BrdGl7ACA-cbd, состоящий из аминокислотной последовательности ацилазы глутарил-7- аминоцефалоспориновой кислоты штамма Brevundimonas diminuta ВКМ B-1297 и хитин-связывающего домена хитиназы А1 Bacillus circulans, и характеризуется нуклеотидной последовательностью SEQ ID Второй объект – рекомбинантная плазмида pSVH0108, обеспечивающая синтез гибридного белка BrdGl7ACA-cbd в клетках Escherichia coli, которая образована вектором рАСТ7, состоящим из фрагмента модифицированной в области полилинкера плазмиды рЕТ21d, содержащего промотор и терминатор РНК-полимеразы фага Т7, разделенные участком полилинкера, и фрагмента плазмиды рACYC177, содержащего репликон р15А и ген устойчивости к канамицину, и последовательностью рекомбинантной ДНК по п.1, встроенной в полилинкерную область указанного вектора. Третий объект – рекомбинантный штамм Escherichia coli BL21 (DE3)/pSVH0108 – продуцент гибридного белка BrdGl7ACA-cbd. Краткое описание чертежей Фиг.1 – физическая и генетическая карты вектора pACT7. Обозначены положения индикаторных сайтов рестрикции, участки промотора и терминатора РНК-полимеразы фага Т7 (Т7 пром и Т7 терм), область начала репликации (p15Aori), ген устойчивости к канамицину (обозначен KN(R), заполнение черным). Фиг.2 – физическая и генетическая карты плазмиды pSVH0108. Обозначено положение гена BrdGl7ACA (заполнение серым), положение хитин, связывающего домена (заполнение штрихом), остальные обозначения – как на Фиг.1. Фиг.3 – сравнение аминокислотных последовательностей BrdGl7ACA-cbd, BrdGl7ACA-Vne с последовательностью BrdGl7ACA-ацилазы. Черным выделены последовательности, соответствующие С-концу альфа-субъединицы, подчеркнуты последовательности спейсерного пептида, курсив – CBD-домен, в рамке – rjlbhe.ofz последовательности N-конца бета-субъединицы. Фиг.4 – электрофореграмма разделения в 12% ДСН-ПААГ фракций, полученных при очистке рекомбинантного белка BrdGl7ACA-cbd. Дорожки: 1 – маркер мол. веса, 2 – фракция после промывки колонки с хитининовыми гранулами после иммобилизации белка BrdGl7ACA-cbd, линия 3 – очищенный препарат BrdGl7ACA-cbd. Осуществление изобретения При осуществлении изобретения помимо методов, подробно раскрытых в нижеследующих примерах, использовали хорошо известные специалистам методики, описанные в руководствах по молекулярной биологии и генетической инженерии [19, 20]. Пример 1. Выделение гена Gl7ACA-ацилазы Первичные структуры генов Gl7ACA-ацилаз, выделенных из различных штаммов Pseudomonas и Brevundimonas известны (см. номера доступа в GenBank AAC34685, AAN39264, AAP68796). Эти последовательности отличаются высокой степенью консервативности, что значительно упрощает задачу выделения гена Gl7ACA-ацилазы методом ПЦР-амплификации. В качестве матрицы для получения гена Gl7ACA-ацилазы используют хромосомную ДНК штамма Brevundiminas diminuta ВКМ B-1297, а в качестве праймеров – олигонуклеотиды 7ACA_F (SEQ ID 1 мкг геномной ДНК Brevundiminas diminuta ВКМ B-1297, выделенной традиционными методами, денатурируют путем нагревания при 100° в течение 5 минут, помещают в лед и подвергают 30 циклам ПЦР с использованием набора Expand Long Template PCR system («Roche Diagnostics GmbH Смесь для ПЦР (50 мкл): 5 мкл 10-кратного ПЦР-буфера («Roche Diagnostics GmbH 5 мкл геномной ДНК (200 нг/мкл); 5 мкл 3 мкМ праймера 7ACA_F; 5 мкл 3 мкМ праймера 7ACA_R; 5 мкл 2,5 мМ dNTP каждого вида; 25 мкл деионизованной воды; 1 мкл ДНК полимеразы («Roche Diagnostics GmbH Условия проведения ПЦР: 94°, 5′ (денатурация), 94°, 30”; 50°, 30”; 72°,2′ (амплификация). После амплификации 5 мкл ПЦР смеси анализируют электрофорезом в 1% агарозном геле и выявляют гомогенный фрагмент размером около 2,2 тпн. Фрагмент выделяют из геля с помощью набора Wizard PCR Preps Kit (Promega, США) в соответствии с инструкцией производителя и подвергают автоматическому секвенированию на приборе ABIPrizm 3100 DNA Sequencer с использованием праймеров BRD1 (SEQ ID Сравнение полученной нуклеотидной последовательности с базой данных GenBank с помощью программы BLAST показало, что она обладает высокой степенью гомологии с соответствующими кодирующими последовательностями других бактериальных Gl7ACА-ацилаз. На этом основании был сделан вывод о том, что полученный фрагмент соответствует гену Gl7ACA-ацилазы штамма Brevundimonas diminuta ВКМ B-1297. Установленная нуклеотидная последовательность гена BrdGl7ACA-ацилазы приведена в перечне под номером SEQ ID Пример 2. Конструирование вектора-носителя pACT7 Векторы на основе промотора фага Т7 обеспечивают высокую экспрессию гетерологичных генов в штаммах E.coli, синтезирующих T7 полимеразу, и коммерчески доступны [21]. В то же время препараты Gl7ACA-ацилаз, используемые для биотрансформации цефалоспориновых антибиотиков, не должны содержать примесей бета-лактамаз, гены которых представлены в большинстве коммерческих векторных конструкций этого типа. Поэтому для экспрессии BrdGl7ACA-ацилазы и других ферментов биотрансформации антибиотиков, свободных от примесей бета-лактамаз, целесообразно использовать векторы, несущие маркеры плазмидной устойчивости, отличные от AmpR. Такие векторы к тому же обладают более высокой сегрегационной стабильностью [21], которая может быть дополнительно повышена за счет использования репликона p15A вместо репликона ColE1 [22]. Конструирование вектора pACT7, удовлетворяющего указанным условиям, проводили в несколько стадий. А) Конструирование промежуточной плазмиды pET21dRI. C использованием метода «инвертированной ПЦР» и праймеров pETR1_F (SEQ ID Б) Конструирование плазмиды pACYCpET. Для получения производного вектора pET21dRI с репликоном p15A ДНК плазмиды pACYC177 [22] гидролизовали совместно рестриктазами BamHI+PstI и выделяли из агарозного геля фрагмент размером 3000 п.н., включающий область репликона р15А, ген устойчивости к канамицину и С-концевую кодирующую часть гена бета-лактамазы (устойчивости к ампициллину). Плазмиду pET21dR1 гидролизовали совместно BglII+PstI и выделяли из агарозного геля фрагмент размером 1,5 т.п.н., включающий участок полилинкера, промотор и терминатор Т7-фага и N-концевую кодирующую часть гена бета-лактамазы. Два фрагмента лигировали, полученной лигазной смесью трансформировали штамм E. Coli Xl1Blue и отбирали трансформанты, устойчивые одновременно к ампициллину и канамицину. Выделенные из культур полученных клонов плазмидные ДНК анализировали путем рестрикции эндонуклеазой SmaI и отбирали клоны, образующие фрагменты размером 3,2 и 1,2 т.п.н. Один из отобранных клонов обозначили как pACYCpET. В) Конструирование вектора pACT7 Для удаления гена бета-лактамазы из вектора pACYCpET препарат плазмидной ДНК гидролизуют совместно рестриктазами NaeI/FspI, выделяют из агарозного геля фрагмент размером 3,4 т.п.н., лигируют «сам на себя» по «тупым концам» и лигазной смесью трансформируют компетентные клетки штамма E.сoli JM109. Из отобранных канамицин-устойчивых и ампициллин-чувствительных трансформантов выделяют плазмидную ДНК и идентифицируют нужный клон по данным рестриктного анализа (наличие фрагментов размером 1,2 и 2,2 т.п.н. в SmaI-гидролизатах препаратов плазмидной ДНК). Пример 3. Конструирование плазмиды pSVH1811. Для создания плазмиды, способной направлять синтез BrdGL7ACA-ацилазы в клетках E.coli, проводили встраивание выделенного гена BrdGL7ACA-ацилазы под контроль промотора фага Т7 вектора pACT7. Полученный методом ПЦР фрагмент геномной ДНК штамма Brevundiminas diminuta ВКМB-1297 содержит полную кодирующую последовательность BrdGl7ACA-ацилазы и фланкирован уникальными сайтами рестрикции EcoRI и SacI, отсутствующими в кодирующей последовательности гена для последующего направленного его встраивания под контроль промотора Т7 созданных экспрессионных векторов. Выделенный из агарозного геля фрагмент гидролизуют рестриктазами EcoRI/SacI и клонируют в EcoRI/SacI вектор pACT7. Отбирают канамицин-устойчивые трансформанты штамма Escherichia coli JM109 по критерию образования фрагментов размером 2,2 и 3,4 т.п.н. в EcoRI/SacI гидролизатах полученных препаратов плазмидной ДНК. Для селекции клона, несущего вставку с интактным геном BrdGL7ACA-ацилазы без возможных мутаций, привнесенных за счет ПЦР-амплификации, проверяют способность отобранных плазмид направлять синтез функционально-активной BrdGL7ACA-ацилазы при их переносе в штамм E.coli BL21(DE3). Для этого индивидуальные трансформанты штамма E.coli BL21(DE3), несущие полученные плазмиды, выращивают на LB-среде, содержащей 30 мкг/мл канамицина, в объеме 5 мл до А600 1,0 ОЕ. После этого в культуры вносят ИПТГ до конечной концентрации 1 мкМ и продолжают культивирование в течение 18 часов при температуре 25-30°С. Клетки собирают центрифугированием и проводят определение активности Gl7ACA-ацилазы, для чего используют колориметрический метод, аналогичный методу, предложенному для определения 6-аминопенициллиновой кислоты [24]. 100 мкл 65 мM раствора глутарил-7-АСА в 0.1M фосфатном буфере (рН 7,0) добавляют к 900 мкл суспензии клеток, приготовленной на том же (0,1M фосфатном) буфере, содержащем 3 мг/мл клавулината калия, и инкубируют смесь при 37°С от 30 мин до 4 часов. Через определенные промежутки времени смесь центрифугируют, отбирают 500 мкл супернатанта и смешивают его с 3 мл смеси (2:1) 20% уксусной кислоты и 0,5 М раствора NaOH для остановки реакции. К пробе добавляют 0,5 мл p-диметиламинобензальдегида (PDAB) и после развития окраски измеряют поглощение при 415 нм. За одну единицу активности фермента принимается его количество, необходимое для превращения 1 мкмоль субстрата в указанных условиях в течение 1 мин инкубации. Уровень определенной таким образом ацилазной активности в проанализированных штаммах колебался от 1,0 ед/мл до примерно 1,4 ед/мл культуры. Отобранные «положительные клоны» секвенируют и идентифицируют клон, несущий вставку с интактным геном BrdGl7ACA-ацилазы, кодирующая последовательность которого идентична последовательности, приведенной в перечне последовательностей под номером SEQ ID Пример 4. Конструирование вектора экспрессии гибридного белка BrdGl7ACA-cbd А) Получение промежуточной плазмиды pSVH0107. Для получения вектора, позволяющего осуществлять экспрессию целевого фермента в виде гибрида с хитин-связывающим доменом, сначала проводили олигонуклеотид-направленный мутагенез с использованием набора QuiсkChange® Site-Directed Mutagenesis Kit (Stratagene, США) и праймеров Apa_up (SEQ ID После проведения реакции смесь очищали от примеси праймеров, используя «Wizard PCR preps Purification System» (Promega, США) и гидролизовали 5 единицами рестриктазы Dpn1 ((Fermentas). Полученной смесью трансформировали компетентные клетки штамма Escherichia coli XL1-Blue recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac [F’proAB lacIqZ Б) Выделение последовательности, кодирующей CBD домен хитиназы A1 B.circulans. В качестве источника гена CBD домена хитиназы A1 B.circulans использовали плазмиду pTYB4 (New England Biolabs). C помощью праймеров СBD_F (SEQ ID Фрагмент выделяли из геля с помощью набора «Wizard PCR Preps Kit» (Promega, США) в соответствии с инструкцией фирмы-производителя и подвергали автоматическому секвенированию на приборе ABIPrizm 3100 DNA Sequencer с использованием праймеров СBD_F и CBD_R и набора Applera «fluorescent Big dye Cycle sequencing kit». Полученная последовательность (SEQ ID В) Получение плазмиды pSVH0108. Последовательность, кодирующую гибридный белок BrdGl7ACA-cbd, получали путем объединения содержащегося в векторе pSVH0107 фрагмента, кодирующего BrdGl7ACA и включающего дополнительный сайт Vne1, и фрагмента, кодирующего СВD-домен (см. выше). Для этого 2 мкг плазмидной ДНК pSVH0107 гидролизовали рестриктазой Vne1, дефосфорилировали с помощью фосфатазы кишки теленка для предотвращения замыкания вектора «сам на себя» и выделяли из агарозного геля фрагмент размером 5,6 т.п.н. 5 мкл смеси использовали для трансформации компетентных клеток штамма E.сoli JM109 [19]. ПЦР фрагмент, кодирующий CBD-домен, также гидролизовали рестриктазой Vne1. Использовали следующие условия лигирования: 1 мкл вектора (20 нг/мкл), 1 мкл фрагмента (50 нг/мкл), 2 мкл 5-кратного лигазного буфера (Gibco-BRL), 5 мкл воды и 1 мкл Т4 ДНК лигазы (1 ед/мкл, Сибэнзим). Смесь инкубировали в течение 14 часов при 12°С, затем прогревали 15 мин при 65°С, охлаждали во льду. Полученной лигазной смесью трансформировали компетентные клетки штамма Escherichia coli XL1-Blue. Из полученных канамицин-устойчивых трансформантов выделяли плазмидную ДНК и отбирали «положительные» клоны методом ПЦР-скрининга с праймерами BRD2 и CBD_F по образованию ПЦР-продукта размером 1000 п.н. Несколько «положительных» клонов дополнительно проверяли сиквенированием и отбирали клон с инсерцией CBD-кодирующего фрагмента, не содержащий неспецифических мутаций. Полученную из него плазмиду обозначили как pSVH0108 (фиг.2). Данная плазмида содержала последовательность, кодирующую полный гибридный белок BrdGl7ACA-cbd (SEQ ID Пример 5. Получение рекомбинантного штамма-продуцента BrdGl7ACA-cbd. Полученной рекомбинантной плазмидой pSVH0108 трансформировали штамм E.coli BL21 (DE3) [21] [F-, ompT, hsdSB (rB-, mB-), dcm, gal (DE3)]. Поскольку первичные трансформанты DE3 – реципиентов могут заметно отличаться по уровню экспрессии целевого белка [25], для селекции наиболее активного продуцента белка BrdGl7ACA-cbd отдельные клоны трансформантов выращивали в предварительно определенных, оптимальных для продукции Gl7ACA-ацилазы условиях: клетки вносили в 50 мл среды LB, культивировали при температуре 25°С до 1,0 ОЕ при А600, после чего добавляли индуктор ИПТГ до конечной концентрации 0,2 мМ и продолжали инкубацию в течение 22 часов. Периодически отбирали аликвоты суспензии клеток выросших индивидуальных трансформантов и использовали их для определения параметров роста культуры и активности фермента. Для получения грубого экстракта осадок клеток разрушали ультразвуком с периодическим охлаждением во льду (4×30 сек с интервалом 1 мин). Клеточный дебрис удаляли центрифугированием. Для определения активности фермента, которое проводили, как описано в примере 3, использовали 50 мкл полученного осветленного лизата. Удельную активность выражали в единицах активности фермента (количество мкмолей субстрата в мин при 25°С) на мг общего белка осветленного экстракта. Содержание общего белка определялось методом Брэдфорда [19]. На основании полученных данных был отобран трансформант
Пример 6. Частичная очистка гибридного белка BrdGl7ACA-cbd и его иммобилизация на хитиновом сорбенте. Достаточно высокий уровень экспрессии вариантов глутарил-ацилазы в полученном штамме в определенной степени упростил задачу разработки методики частичной очистки гибридного белка, которую осуществляли, как описано ниже. Полученную биомассу рекомбинантного штамма (5 г) суспендировали в 20 мл буфера А (100 мМ фосфат натрия, рН 8,0, 5 мМ 2-меркаптоэтанол, 10 мМ ЕДТА, 0,3% цетилтриметил аммоний бромид (СТАВ), 2% глицерин) и разрушали ультразвуковой дезинтеграцией. Супернатант собирали, дебрис после промывания тем же буфером отбрасывали. Балластные белки из супернатанта осаждали сульфатом аммония при 35% насыщении, рекомбинантную BrdGl7ACA-cbd получали в виде осадка при насыщении сульфатом аммония 60%. Осадок растворяли в 10 мл буфера Б (10 мМ пирофосфат натрия, рН 8,0, 5 мМ 2-меркаптоэтанол, 2 мМ ЕДТА, 10% глицерин, 300 мМ NaCl) и наносили на колонку с хитиновыми гранулами (2×5 см). Колонку промывали буфером Б до исчезновения поглощения при А280, отбирали аликвоту сорбента и использовали для определения активности ацилазы и общего белка. Этапы очистки и иммобилизации отражены в Таблице 2. Методом электрофореза мы установили достаточно высокую чистоту рекомбинатной ацилазы, состоящей из двух субъединиц: бета – 54 кДа и альфа+CBD – 20 кДа. Полученные данные позволяют сделать вывод, что в составе созданной конструкции химерного белка полностью сохраняются свойства обеих полипептидных составляющих.
Пример 7. Характеристика рекомбинантного штамма E.coli BL21(DE3)/ pSVH0108. Клетки полученного рекомбинантного штамма Escherichia coli BL21(DE3)/ pSVH0108 характеризуется следующими признаками. Морфологические признаки Клетки имеют продолговатую палочковидную форму, при делении не почкуются. Культуральные признаки Клетки хорошо растут на обычно используемых питательных средах. Время генерации около 30 мин в жидкой LB-среде. На 2-2,5% питательном агаре “Difco” образуются круглые, гладкие, желтоватые колонии с ровными краями. При выращивании на жидких LB- и YT-средах образуется интенсивная ровная мутность. Физиолого-биохимические признаки Оптимальная температура культивирования – от 25 до 30°C, оптимум рН – 7,6. Источником азота служат органические соединения (в виде триптона, дрожжевого экстракта). Уровень синтеза BrdGl7ACA-cbd в сконструированном штамме составляет около 100 мг/л при титре культуры 1×109 кл/мл, что следует из данных определения GL7ACA-ацилазы в образцах биомассы штамма-продуцента с помощью специфической ферментативной реакции. Величина удельной активности гибридного белка BrdGl7ACA-cbd составляет около 10000 мЕ/мг белка (Таблица 2) и близка к показателю удельной активности очищенных препаратов рекомбинантных «немодифицированных» Gl7ACA-ацилаз, описаных ранее [9-12, 15]. Cписок цитированных источников 1. Matsuda, A. & Komatsu, K.I. Molecular cloning and structure of the gene for 7 beta-(4-carboxybutanamido)cephalosporanic acid acylase from a Pseudomonas strain. J. Bacteriol. 163, 1222-1228 (1985). 2. Arroyo, M., de, 1.M., I, Acebal, C., & Castillon, M.P. Biotechnological applications of penicillin acylases: state-of-the-art. Appl. Microbiol. Biotechnol. 60, 507-514 (2003). 3. Valle, F., Balbas, P., Merino, E., & Bolivar, F. The role of penicillin amidases in nature and in industry. Trends Biochem. Sci. 16, 36-40 (1991). 4. Barber, M.S., Giesecke, U., Reichert, A., & Minas, W. Industrial enzymatic production of cephalosporin-based beta-lactams. Adv. Biochem. Eng Biotechnol. 88, 179-215 (2004). 5. Kumar, K.K., Sudhakaran, V., Deshpande, B.S., Ambedkar, S.S., & Shewale, J.G. Cephalosporin acylases: enzyme production, structure and application in the production of 7-ACA. Hindustan Antibiot. Bull.35, 111-125 (1993). 6. Parmar, A., Kumar, H., Marwaha, S.S., & Kennedy, J.F. Recent Trends in Enzymatic Conversion of Cephalosporin C to 7-Aminocephalosporanic Acid (7-ACA). Critical Reviews in Biotechnology 18, 1-12 (1998). 7. Aramori, I., Fukagawa, M., Tsumura, M., Iwami, M., Ono, H., Kojo, H., Kohsaka, M., Ueda, Y., & Imanaka, H. Cloning and nucleotide sequencing of a novel 7 beta-(4-carboxybutanamido)cephalosporanic acid acylase gene of Bacillus laterosporus and its expression in Escherichia coli and Bacillus subtilis. J. Bacteriol. 173, 7848-7855 (1991). 8. Battistel, E., Bianchi, D., Bortolo, R., & Bonoldi, L. Purification and stability of glutaryl-7-ACA acylase from Pseudomonas sp.Appl. Biochem. Biotechnol. 69, 53-67 (1998). 9. Lee, Y.S. & Park, S.S. Two-step autocatalytic processing of the glutaryl 7-aminocephalosporanic acid acylase from Pseudomonas sp. strain GK16. J. Bacteriol. 180, 4576-4582 (1998). 10. Li, Y., Jiang, W., Yang, Y., Zhao, G., & Wang, E. Overproduction and purification of glutaryl 7-amino cephalosporanic acid acylase. Protein Expr. Purif. 12, 233-238 (1998). 11. Ishiye, M. & Niwa, M. Nucleotide sequence and expression in Escherichia coli of the cephalosporin acylase gene of a Pseudomonas strain. Biochim. Biophys. Acta 1132, 233-239 (1992). 12. Wang, E.D., Zheng, Y.G., Li, Y., Jiang, W.H., & Yang, Y.L. Expression of gene encoding GL-7ACA acylase in Escherichia coli. Sheng Wu Hua Xue. Yu Sheng Wu Wu Li Xue. Bao. (Shanghai) 34, 526-531 (2002). 13. Sassenfeld HM. Engineering proteins for purification. Trends Biotechnol., 8, 88-93 (1990). 14. Garcia Lopez et all. Process for modifying the enzyme 7 beta-(4-carboxybutanamido) cephalosporinacylase and purifying said enzyme in single chromatographic step. European patent EP 0839914, 1997-10-30. 15. Kim, J.K., Yang, I.S., Rhee, S., Dauter, Z., Lee, Y.S., Park, S.S., & Kim, K.H. Crystal structures of glutaryl 7-aminocephalosporanic acid acylase: insight into autoproteolytic activation. Biochemistry 42, 4084-4093 (2003). 16. Lee, Y.S., Kim, H.W., Lee, K.B., & Park, S.S. Involvement of arginine and tryptophan residues in catalytic activity of glutaryl 7-aminocephalosporanic acid acylase from Pseudomonas sp.strain GK16. Biochim. Biophys. Acta 1523, 123-127 (2000). 17. Kim, Y. & Hol, W.G. Structure of cephalosporin acylase in complex with glutaryl-7-aminocephalosporanic acid and glutarate: insight into the basis of its substrate specificity. Chem.Biol. 8, 1253-1264 (2001). 18. Studier FW, Moffatt BA Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol 189, 113-30 (1986). 19. Sambrook, J., Fritsch, E.F., and Maniatis, T. Molecular Cloning; A Laboratory Manual, 2nd ed. Cold Spring Harbor Laboratory Press, New York [1989]. 20. Ausubel, F.M., Brent, R., Kingston, R. E., Moore, D.D., Seidman, J.G., Smith, J.A., & Struhl, K. Current Protocols in Molecular Biology, John Wiley and Sons, New York (1997). 21. pET System Manual, 10th Edition Rev. B 0403, Novagen Inc. (2003) 22. Rose, R.E. The nucleotide sequence of pACYC177. Nucleic Acids Res. 16, 356 (1988). 23. Reece, K.S. & Phillips, G.J. New plasmids carrying antibiotic-resistance cassettes. Gene 165, 141-142(1995). 24. Balasingham, K., Warburton, D., Dunnill, P., & Lilly, M.D. The isolation and kinetics of penicillin amidase from Escherichia coli. Biochim. Biophys. Acta 276, 250-256 (1972). 25. Vethanayagam J., Flower A. Decreased gene expression from T7 promoters may be due to impaired production of active T7 RNA polymerase. Microbial Cell Factories 4, 3-10 (2005).
Формула изобретения
1. Рекомбинантная ДНК, которая кодирует функционально активный гибридный белок (BrdG17ACA-cbd), состоящий из аминокислотной последовательности ацилазы глутарил-7-аминоцефалоспориновой кислоты штамма Brevundimonas diminuta BKM В-1297 и хитин-связывающего домена хитиназы Al Bacillus circulans, и характеризуется нуклеотидной последовательностью SEQ ID 2. Рекомбинантная плазмида pSVH0108, обеспечивающая синтез гибридного белка BrdG17ACA-cbd в клетках E.coli, которая образована вектором рАСТ7, состоящим из фрагмента модифицированной в области полилинкера плазмиды pET21d, содержащего промотор и терминатор РНК-полимеразы фага Т7; разделенные участком полилинкера, и фрагмента плазмиды pACYC177, содержащего репликон р15А и ген устойчивости к канамицину, объединенных между собой как показано на фиг.2, и последовательностью рекомбинантной ДНК по п.1, встроенной в полилинкерную область указанного вектора. 3. Рекомбинантный штамм E.coli BL21(DE3)/pSVH0108 cbd – продуцент гибридного белка BrdG17ACA-cbd.
РИСУНКИ
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
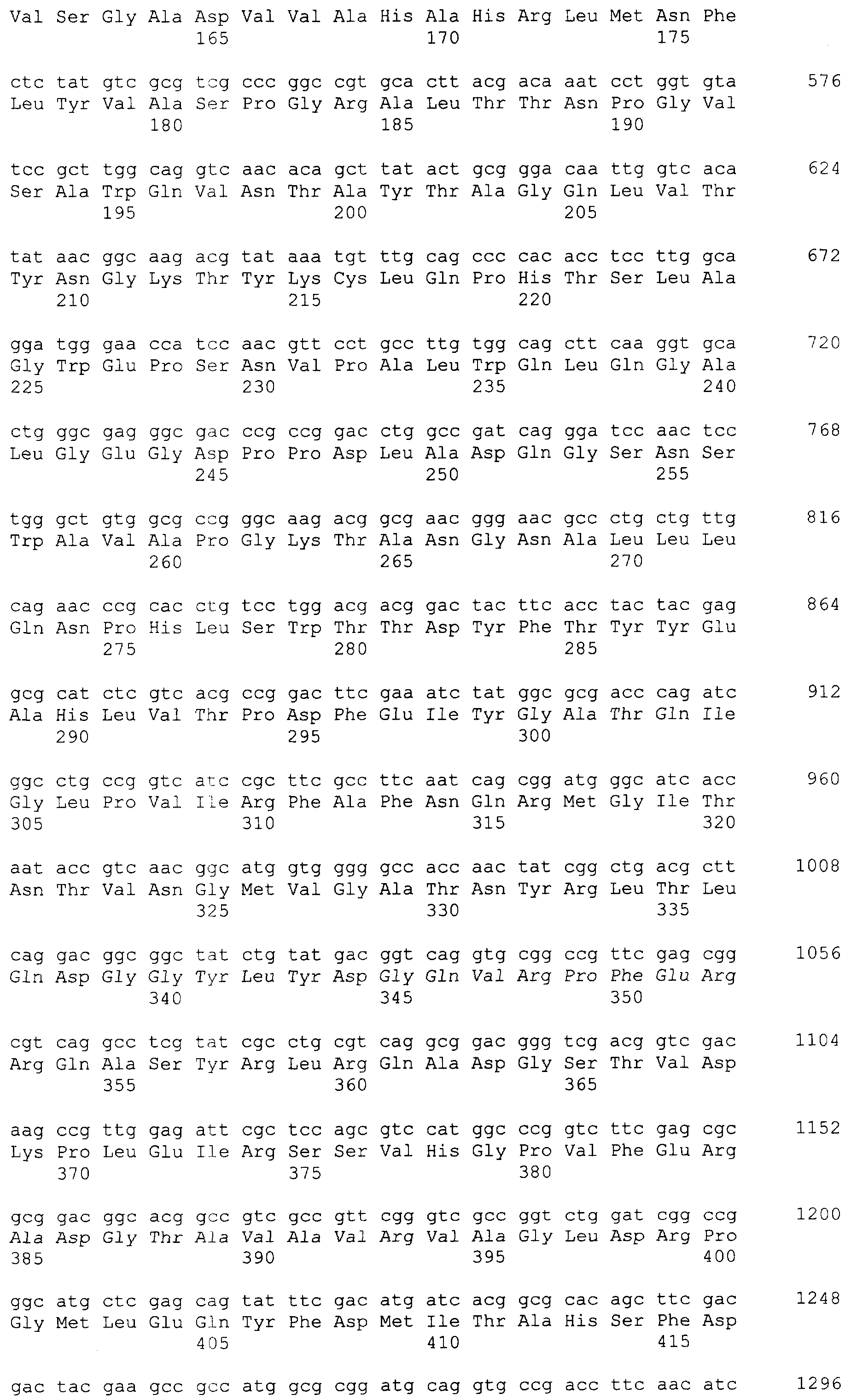
 и 2-х
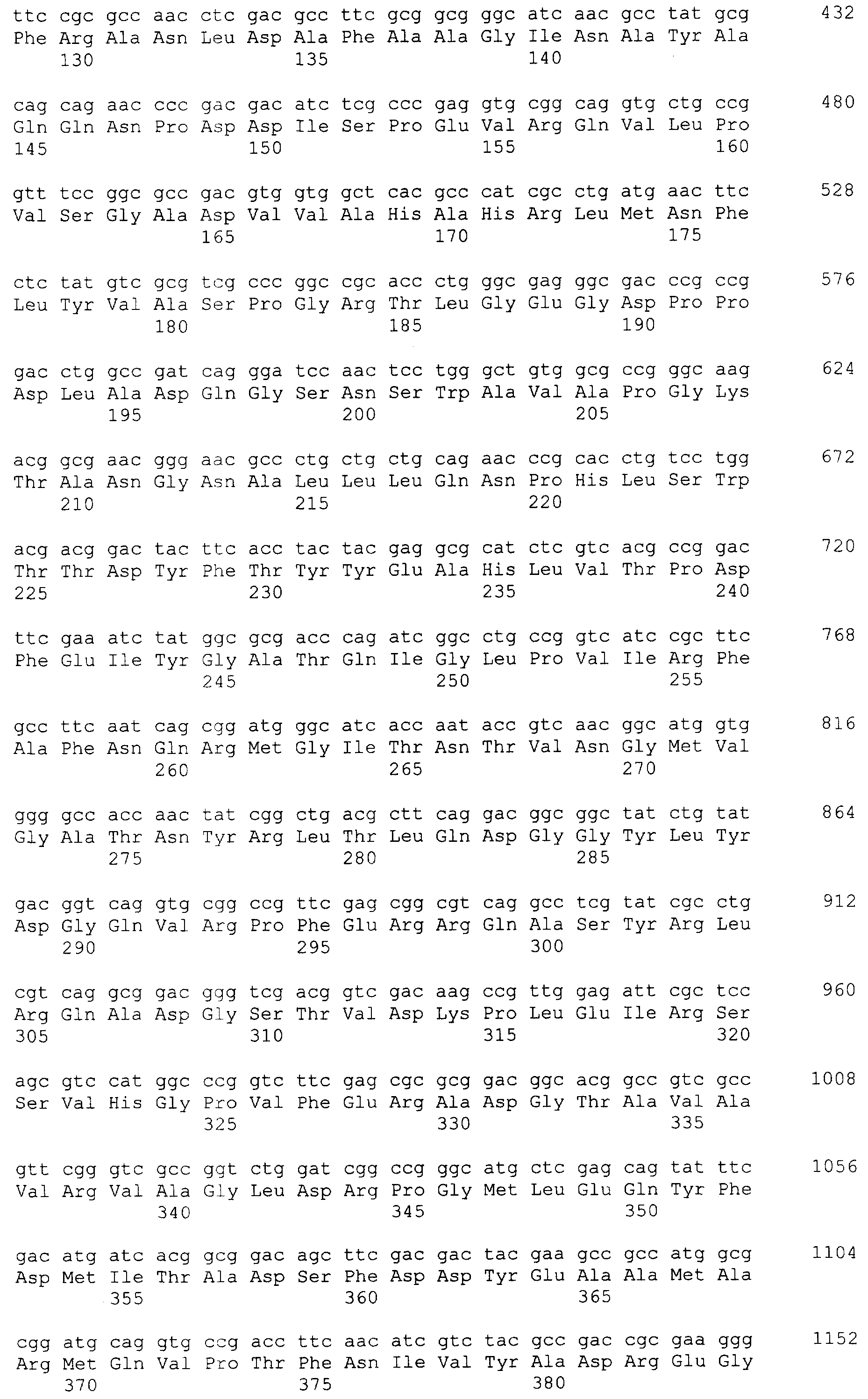
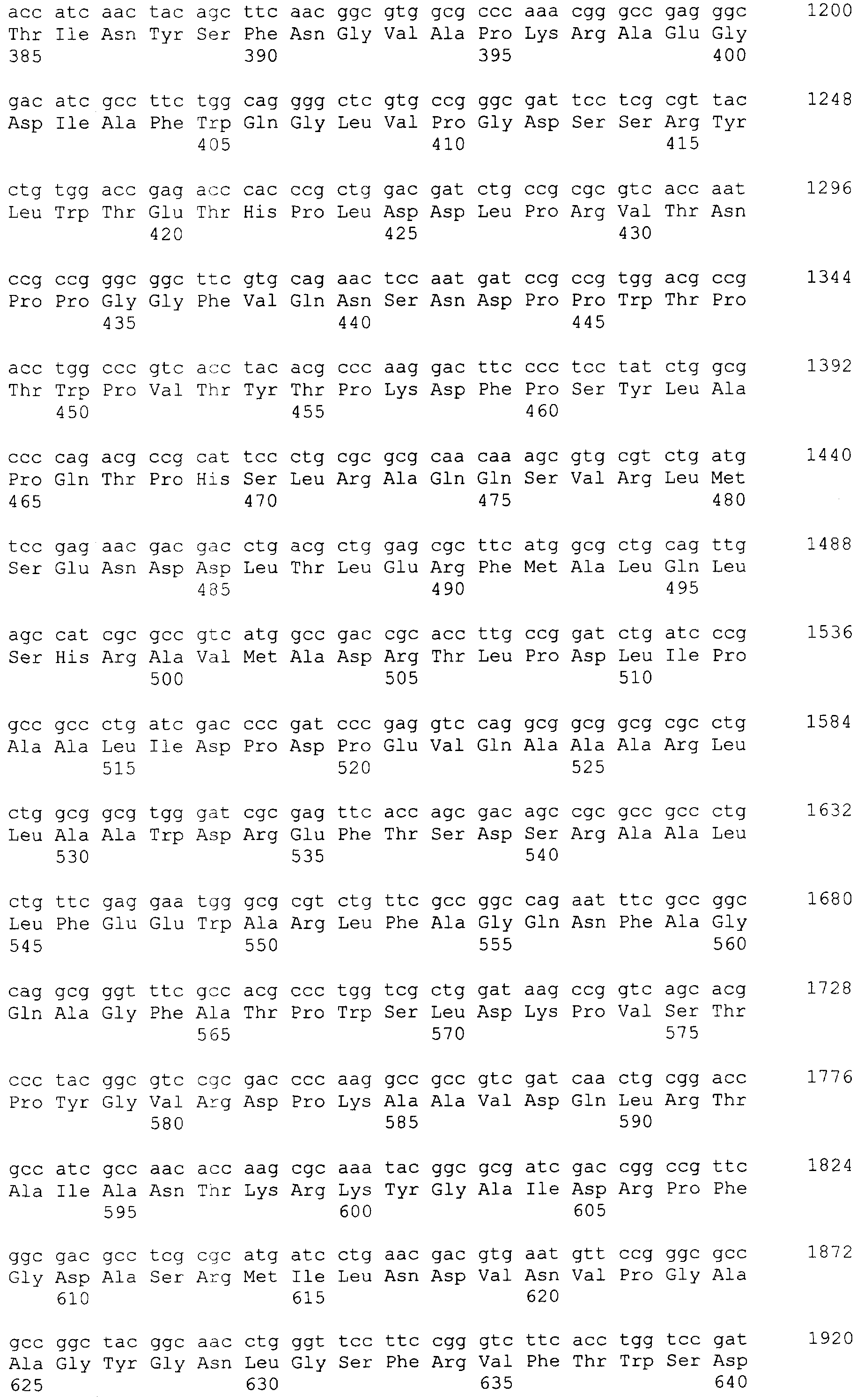
и 2-х  субъединиц [15], которые формируются из одноцепочечного полипептидного предшественника. Полная кодирующая последовательность гена BrdGl7ACA-ацилазы (SEQ ID
субъединиц [15], которые формируются из одноцепочечного полипептидного предшественника. Полная кодирующая последовательность гена BrdGl7ACA-ацилазы (SEQ ID  5) получена методом полимеразной цепной реакции (ПЦР) с использованием в качестве матрицы хромосомной ДНК, выделенной из штамма Brevundimonas diminuta ВКМ В-1297, а в качестве праймеров – синтетических олигонуклеотидов с нуклеотидными последовательностями SEQ ID
5) получена методом полимеразной цепной реакции (ПЦР) с использованием в качестве матрицы хромосомной ДНК, выделенной из штамма Brevundimonas diminuta ВКМ В-1297, а в качестве праймеров – синтетических олигонуклеотидов с нуклеотидными последовательностями SEQ ID  , Mannheim, Германия) в соответствии с инструкцией изготовителя.
, Mannheim, Германия) в соответствии с инструкцией изготовителя. M15 Tn10 (Tetr)] (Stratagene, США), затем из полученных ампициллин-устойчивых трансформантов выделяют препараты плазмидной ДНК с использованием набора Wizard MiniPreps Kit (Promega, США). Полученные образцы ДНК анализируют совместным гидролизом рестриктазами EcoRI/PstI и BamHI/PstI и отбирают «положительные» клоны, содержащие EcoRI/PstI фрагменты размером 1300 и 4100 п.н. При этом за счет делеции сайта BamHI в BamHI/PstI гидролизате ДНК плазмидных клонов присутствует уникальный фрагмент размером 5400 п.н., в то время как в препарате плазмидной ДНК pET21d, гидролизованной BamHI/PstI, обнаруживаются фрагменты размером 1300 и 4100 п.н. Несколько «положительных» клонов проверяют секвенированием с использованием праймеров Т7prom (SEQ ID
M15 Tn10 (Tetr)] (Stratagene, США), затем из полученных ампициллин-устойчивых трансформантов выделяют препараты плазмидной ДНК с использованием набора Wizard MiniPreps Kit (Promega, США). Полученные образцы ДНК анализируют совместным гидролизом рестриктазами EcoRI/PstI и BamHI/PstI и отбирают «положительные» клоны, содержащие EcoRI/PstI фрагменты размером 1300 и 4100 п.н. При этом за счет делеции сайта BamHI в BamHI/PstI гидролизате ДНК плазмидных клонов присутствует уникальный фрагмент размером 5400 п.н., в то время как в препарате плазмидной ДНК pET21d, гидролизованной BamHI/PstI, обнаруживаются фрагменты размером 1300 и 4100 п.н. Несколько «положительных» клонов проверяют секвенированием с использованием праймеров Т7prom (SEQ ID