Патент на изобретение №2386629
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) НОВЫЕ ГИДАНТОИНОВЫЕ ПРОИЗВОДНЫЕ ДЛЯ ЛЕЧЕНИЯ ОБСТРУКТИВНЫХ ЗАБОЛЕВАНИЙ ДЫХАТЕЛЬНЫХ ПУТЕЙ
(57) Реферат:
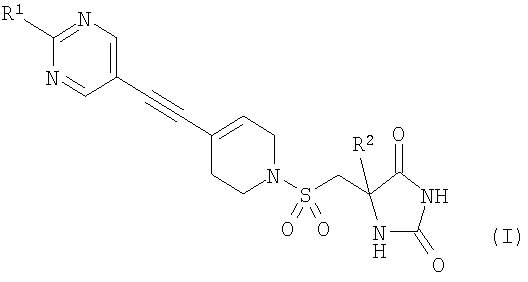
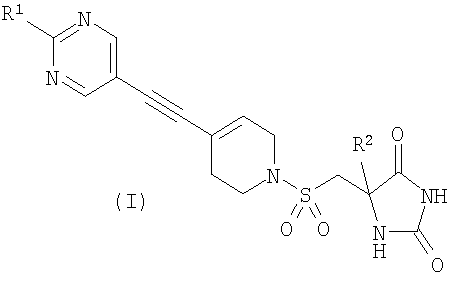
Изобретение относится к новым соединениям формулы (I) в виде (S)-стереоизомеров или к их фармацевтически приемлемым солям, которые обладают свойствами ингибитора ММР12 или ММР9. В формуле (I)
R1 представляет собой С1-2алкил, циклопропил, ОСН3 или SCH3, причем указанная алкильная группа возможно дополнительно замещена одним или более чем одним атомом фтора, и R2 представляет собой С1-2алкил. Изобретение также относится к способам получения соединений формулы (I), фармацевтической композиции и способу ее изготовления, а также к применению соединений формулы (I). 8 н. и 9 з.п. ф-лы, 1 табл.
Изобретение относится к новым гидантоиновым производным, способам их получения, содержащим их фармацевтическим композициям и их применению в терапии. Металлопротеиназы представляют собой суперсемейство протеиназ (ферментов), количество которых в последние годы значительно возросло. Исходя из их структуры и функции, эти ферменты были классифицированы в семейства и подсемейства, как описано у N.M.Hooper (1994) FEBS Letters 354:1-6. Примеры металлопротеиназ включают матриксные металлопротеиназы (ММР), такие как коллагеназы (ММР1, ММР8, ММР13), желатиназы (ММР2, ММР9), стромелизины (ММР3, ММР10, ММР11), матрилизин (ММР7), металлоэластазу (ММР12), энамелизин (ММР19), металлопротеиназы мембранного типа (МТ-ММР) (ММР14, ММР15, ММР16, ММР17); репролизин, или адамализин, или семейство MDC, включающее секретазы и шеддазы, такие как TNF (фактор некроза опухоли) конвертирующие ферменты (ADAM10 и ТАСЕ); семейство астацинов, включающее такие ферменты, как протеиназа, процессирующая проколлаген (РСР); и другие металлопротеиназы, такие как аггреканаза, семейство эндотелин-конвертирующих ферментов и семейство ангиотензин-конверирующих ферментов. Полагают, что металлопротеиназы играют важную роль во множестве физиологических болезненных процессов, включающих реконструкцию ткани, такую как эмбриональное развитие, образование кости и реконструкция матки при менструации. Это основано на способности металлопротеиназ расщеплять широкий спектр матриксных субстратов, таких как коллаген, протеогликан и фибронектин. Также полагают, что металлопротеиназы важны при процессинге, или секреции, биологически значимых клеточных медиаторов, таких как фактор некроза опухоли (TNF), и при посттрансляционном протеолитическом процессинге, или шеддинге, биологически значимых мембранных белков, таких как низкоаффинный рецептор IgE CD23 (более полный перечень смотри в N.М.Hooper et at., (1997) Biochem. J. 321:265-279). Металлопротеиназы были ассоциированы со множеством заболеваний или состояний. Ингибирование активности одной или более чем одной металлопротеиназы может быть благоприятно при таких заболеваниях или состояниях, как, например, различные воспалительные и аллергические заболевания, такие как воспаления суставов (в частности, ревматоидный артрит, остеоартрит и подагра), воспаление желудочно-кишечного тракта (в частности, воспалительное заболевание кишечника, неспецифический язвенный колит и гастрит), воспаление кожи (в частности, псориаз, экзема, дерматит); при опухолевых метастазах или инвазии; при заболевании, ассоциированном с неконтролируемой деградацией внеклеточного матрикса, таком как остеоартрит; при резорбции кости (такой как остеопороз и болезнь Педжета); при заболеваниях, ассоциированных с аномальным ангиогенезом; при усиленной реконструкции коллагена, ассоциированной с диабетом, периодонтальным заболеванием (таким как гингивит), изъязвлением роговицы, изъязвлением кожи, послеоперационными состояниями (такими как анастомоз прямой кишки) и заживлением ран кожи; при демиелинизирующих заболеваниях центральной и периферической нервной системы (таких как рассеянный склероз); при болезни Альцгеймера; при реконструкции внеклеточного матрикса, наблюдаемой при сердечно-сосудистых заболеваниях, таких как рестеноз и атеросклероз; при астме; при рините и хроническом обструктивном заболевании легких (ХОЗЛ). ММР 12, также известную как эластаза макрофагов или металлоэластаза, Shapiro et al [1992, Journal of Biological Chemistry 267: 4664] изначально клонировали в мыши, а затем та же группа в 1995 году клонировала ее в человеке. ММР12 предпочтительно экспрессируется в активированных макрофагах, и показано, что она секретируется из альвеолярных макрофагов у курильщиков [Shapiro et al, 1993, Journal of Biological Chemistry, 268: 23824], a также в пенистых клетках при атеросклеротических поражениях [Matsumoto et al, 1998, Am. J. Pathol. 153: 109]. Мышиная модель ХОЗЛ основана на воздействии на мышей сигаретным дымом в течение шести месяцев, по две сигареты в день в течение шести дней в неделю. После такой обработки у мышей дикого типа развивалась легочная эмфизема. Когда в этой модели тестировали мышей, нокаутированных по ММР 12, у них значительная эмфизема не развивалась, что убедительно свидетельствовало о том, что ММР 12 представляет собой ключевой фермент в патогенезе ХОЗЛ. Роль ММР, таких как ММР 12, в ХОЗЛ (эмфизема и бронхит) обсуждается в Anderson and Shinagawa, 1999, Current Opinion in Anti-inflammatory and Immunomodulatory Investigational Drugs 1(1): 29-38. Недавно обнаружили, что курение увеличивает инфильтрацию макрофагов и экспрессию макрофагальной ММР-12 в бляшках Кангавари (Kangavari) сонной артерии человека [Matetzky S, Fishbein MC et al., Circulation 102:(18). 36-39 Suppl. S, Oct 31, 2000]. MMP9 (желатиназа В; коллагеназа IV типа массой 92 кДа; желатиназа 92 кДа) представляет собой секретируемый белок, который впервые был очищен, затем клонирован и секвенирован в 1989 году [S.M.Wilhelm et al (1989) J. Biol. Chem. 264 (29): 17213-17221; опечатки опубликованы в J. Biol. Chem. (1990) 265 (36): 22570]. Недавний обзор MMP9 представляет собой превосходный источник подробной информации и ссылок, касающихся этой протеазы: Т.Н.Vu & Z.Werb (1998) (в: Matrix Metalloproteinases, 1998, edited by W.C.Parks & R.P.Mecham, pp.115-148, Academic Press. ISBN 0-12-545090-7). Следующая информация взята из этого обзора Т.Н.Vu & Z.Werb (1998). Экспрессия MMP9 обычно ограничена несколькими типами клеток, включающими трофобласты, остеокласты, нейтрофилы и макрофаги. Однако экспрессия может быть индуцирована несколькими медиаторами в тех же самых клетках и в клетках других типов, включая воздействие на клетки факторов роста или цитокинов. Они представляют собой те медиаторы, которые часто вовлечены в инициацию воспалительного ответа. Как и другие секретируемые ММР, MMP9 высвобождается в качестве неактивного профермента (pro-фермент), который затем расщепляется с образованием ферментативно активного фермента. Протеазы, которые требуются для этой активации in vivo, не известны. Баланс активной MMP9 и неактивного фермента дополнительно регулируется in vivo путем взаимодействия с природным белком TIMP-1 (тканевый ингибитор металлопротеиназ-1). TIMP-1 связывается с С-концевой областью MMP9, приводя к ингибированию каталитического домена MMP9. Баланс индуцированной экспрессии ProMMP9, расщепления Pro- в активную MMP9 и присутствия TIMP-1 комбинируют для определения количества каталитически активной MMP9, присутствующей в локальном сайте. Протеолитически активная ММР9 атакует субстраты, которые включают желатин, эластин и нативные коллагены IV и V типов; она не обладает активностью в отношении нативного коллагена I типа, протеогликанов или ламининов. Имеется все растущий объем данных, указывающих на роль ММР9 в различных физиологических и патологических процессах. Физиологическая роль включает инвазию эмбриональных трофобластов через эпителий матки на ранних стадиях имплантации эмбриона; некоторое участие в росте и формировании костей и миграцию воспалительных клеток из сосудистой сети в ткани. Высвобождение ММР9, измеренное с использованием иммуноферментного анализа, значительно усилено в жидкостях и в супернатантах AM, полученных от не подвергавшихся лечению астматиков, по сравнению с высвобождением ММР9 в других группах [Am. J. Resp. Cell & Mol. Biol., Nov 1997, 17 (5):583-591]. Кроме того, усиленную экспрессию ММР9 наблюдают при некоторых других патологических состояниях, тем самым указывая на вовлеченность ММР9 в развитие таких заболеваний, как ХОЗЛ, артрит, опухолевое метастазирование, болезнь Альцгеймера, рассеянный склероз и отрыв бляшек при атеросклерозе, приводящий к острым коронарным состояниям, таким как инфаркт миокарда. Известно множество ингибиторов металлопротеиназ (смотри, например, обзор ингибиторов ММР в Beckett R.P. and Whittaker M., 1998, Exp. Opin. Ther. Patents, 8(3):259-282, и Whittaker et al, 1999, Chemical Reviews 99(9); 2735-2776). В WO 02/074767 раскрыты гидантоиновые производные формулы
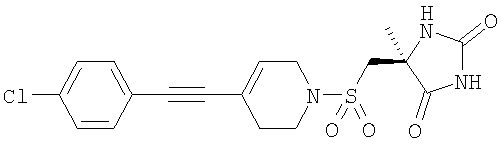
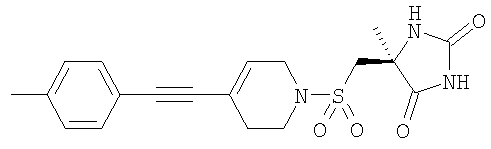
которые полезны в качестве ингибиторов ММР, в частности в качестве сильных ингибиторов ММР12. Следующие три соединения конкретно раскрыты в WO 02/074767
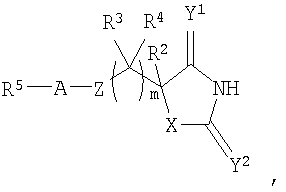
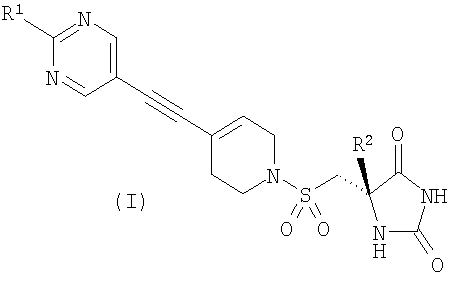
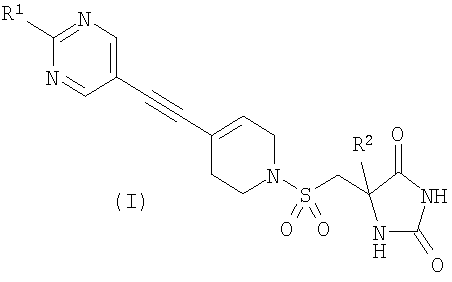
Авторы изобретения обнаружили группу соединений, являющихся ингибиторами металлопротеиназ и представляющих особенный интерес в плане ингибирования таких ММР, как ММР12 и ММР9. Соединения по настоящему изобретению обладают благоприятной активностью, избирательностью и/или фармакокинетическими свойствами. Соединения по настоящему изобретению попадают в общий объем WO 02/074767, но относятся к типу, конкретно не раскрытому в этом документе. В соответствии с настоящим изобретением предложено соединение формулы (I)
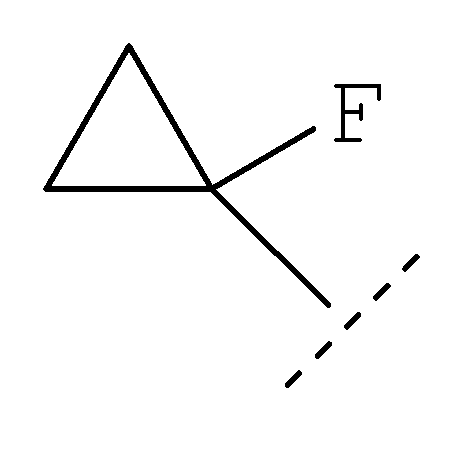
где R1 представляет собой С1-2алкил, циклопропил, ОСН3, SCH3 или OCF3, причем указанная алкильная или циклопропильная группа возможно дополнительно замещена одним или более чем одним атомом фтора; и R2 представляет собой C1-3алкил; и его фармацевтически приемлемые соли. Соединения формулы (I) могут существовать в энантиомерных формах. Понятно, что все энантиомеры, диастереоизомеры, рацематы и их смеси включены в объем изобретения. Соединения формулы (I) также могут существовать в различных таутомерных формах. Все возможные таутомерные формы и их смеси включены в объем изобретения. В одном из воплощений R1 представляет собой С1-2алкил или циклопропил, причем указанная алкильная или циклопропильная группа возможно дополнительно замещена одним или более чем одним атомом фтора. В еще одном воплощении R1 представляет собой С1-2алкил, возможно дополнительно замещенный одним или более чем одним атомом фтора. В одном из воплощений R1 представляет собой циклопропил, возможно дополнительно замещенный одним или более чем одним атомом фтора. В одном из воплощений R1 представляет собой циклопропил. В одном из воплощений R1 представляет собой трифторметил. В одном из воплощений R1 представляет собой ОСН3 или SCH3. В одном из воплощений R2 представляет собой метил или этил. В одном из воплощений R2 представляет собой метил. В одном из воплощений R1 представляет собой С1-2алкил или циклопропил, причем указанная алкильная или циклопропильная группа возможно дополнительно замещена одним или более чем одним атомом фтора, и R2 представляет собой метил или этил. В одном из воплощений R1 представляет собой С1-2алкил или циклопропил, причем указанная алкильная или циклопропильная группа дополнительно замещена одним или более чем одним атомом фтора, и R2 обозначает метил. В одном из воплощений R1 представляет собой С1-2алкил, возможно дополнительно замещенный одним или более чем одним атомом фтора, и R2 обозначает метил или этил. В одном из воплощений R1 представляет собой CF3 и R2 представляет собой метил или этил. В одном из воплощений R1 представляет собой циклопропил и R2 представляет собой метил или этил. Если не указано иначе, используемый здесь термин “C1-3алкил” означает алкильную группу с прямой или разветвленной цепью, имеющую от 1 до 3 атомов углерода. Примеры таких групп включают метил, этил, н-пропил и изо-пропил. Термин “С1-2алкил” означает метил или этил. Примеры С1-2алкила, возможно дополнительно замещенного одним или более чем одним атомом фтора, включают CF3, CH2F, CH2CF3, CF2CH3 и CF2CF3. Примеры циклопропильного кольца, возможно дополнительно замещенного одним или более чем одним атомом фтора, включают 1-фтор-1-циклопропил, 2,2-дифтор-1-циклопропил и 2,3-дифтор-1-циклопропил:
Примеры соединений по изобретению включают: (5S)-5-({[4-[(2-циклопропилпиримидин-5-ил)этинил]-3,6-дигидропиридин-1(2H)ил]сульфонил}метил)-5-метилимидазолидин-2,4-дион; (5S)-5-метил-5-({[4-{[2-(метилтио)пиримидин-5-ил]этинил}-3,6-дигидропиридин-1(2Н)ил]сульфонил}метил)имидазолидин-2,4-дион; (5S)-5-метил-5-({[4-{[2-(трифторметил)пиримидин-5-ил]этинил}-3,6-дигидропиридин-1(2H)ил]сульфонил}метил)имидазолидин-2,4-дион; (5S)-5-метил-5-({[4-[(2-метилпиримидин-5-ил)этинил]-3,6-дигидропиридин-1(2H)ил]сульфонил}метил)имидазолидин-2,4-дион; (5S)-5-({[4-[(2-этилпиримидин-5-ил)этинил]-3,6-дигидропиридин-1(2H)ил]сульфонил}метил)-5-метилимидазолидин-2,4-дион; (5S)-5-({[4-[(2-метоксипиримидин-5-ил)этинил]-3,6-дигидропиридин-1(2H)-ил]сульфонил}метил)-5-метилимидазолидин-2,4-дион; и их фармацевтически приемлемые соли. Каждое приведенное в качестве примера соединение представляет собой конкретный и независимый аспект изобретения. Соединения формулы (I) могут существовать в энантиомерных формах. Таким образом, все энантиомеры, диастереоизомеры, рацематы и их смеси включены в объем изобретения. Различные оптические изомеры могут быть выделены путем разделения рацемической смеси соединений с использованием обычных способов, например фракционной кристаллизации или высокоэффективной жидкостной хроматографии (ВЭЖХ). Альтернативно, оптические изомеры могут быть получены путем асимметричного синтеза или путем синтеза из оптически активных исходных веществ. Когда у соединений по изобретению существуют оптические изомеры, авторы изобретения раскрывают все индивидуальные оптически активные формы и их комбинации в качестве индивидуальных специфических воплощений изобретения, так же как их соответствующие рацематы. Предпочтительно соединения формулы (I) обладают (5S)-стереохимией в соответствии с изображенным ниже:
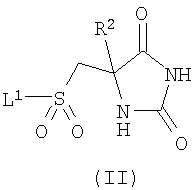
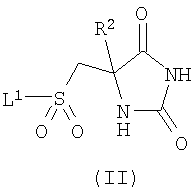
Когда у соединений по изобретению существуют таутомеры, авторы изобретения раскрывают все индивидуальные таутомерные формы и их комбинации в качестве индивидуальных специфических воплощений изобретения. Настоящее изобретение включает соединения формулы (I) в форме солей. Подходящие соли включают соли, образованные с органическими или неорганическими кислотами, либо с органическими или неорганическими основаниями. Такие соли обычно представляют собой фармацевтически приемлемые соли, хотя фармацевтически неприемлемые соли могут быть полезны при получении и очистке конкретных соединений. Такие соли включают соли присоединения кислот, такие как соль гидрохлорид, гидробромид, цитрат, тозилат и малеат, и соли, образованные с фосфорной кислотой или серной кислотой. В еще одном аспекте подходящие соли представляют собой соли с основаниями, такие как соль щелочного металла, например натрия или калия, соль щелочно-земельного металла, например кальция или магния, или соль органического амина, например триэтиламина. Соли соединений формулы (I) могут быть получены путем взаимодействия свободного основания или другой его соли с одним или более чем одним эквивалентом соответствующей кислоты или основания. Соединения формулы (I) полезны, поскольку обладают фармакологической активностью у животных и поэтому потенциально полезны в качестве фармацевтических средств. В частности, соединения по изобретению представляют собой ингибиторы металлопротеиназ и поэтому могут быть использованы в лечении заболеваний или состояний, опосредованных ММР12 и/или ММР9, таких как астма, ринит, хроническое обструктивное заболевание легких (ХОЗЛ), артрит (такой как ревматоидный артрит и остеоартрит), атеросклероз и рестеноз, рак, инвазия и метастазирование, заболевания, в которые вовлечено разрушение ткани, ослабление реплантатов тазобедренного сустава, периодонтальное заболевание, фиброз, инфаркт и сердечное заболевание, печеночный и почечный фиброз, эндометриоз, заболевания, связанные с ослаблением внеклеточного матрикса, сердечная недостаточность, аневризмы аорты, заболевания, связанные с центральной нервной системой (ЦНС), такие как болезнь Альцгеймера и рассеянный склероз (PC), и гематологические расстройства. Как правило, соединения по настоящему изобретению представляют собой сильные ингибиторы ММР9 и ММР12. Соединения по настоящему изобретению также демонстрируют хорошую избирательность в плане относительного отсутствия ингибирования различных других ММР, таких как ММР8, ММР14 и ММР19. Кроме того, соединения по настоящему изобретению также, в общем, обладают улучшенными значениями log D, в частности имеют значения log D, находящиеся в диапазоне 0,5 Соответственно, в настоящем изобретении предложено соединение формулы (I) или его фармацевтически приемлемая соль, как они определены ранее, для применения в терапии. В еще одном аспекте изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как они определены ранее, в изготовлении лекарственного средства для использования в терапии. В еще одном аспекте изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как они определены ранее, в изготовлении лекарственного средства для использования в лечении заболеваний или состояний, при которых благоприятным является ингибирование ММР12 и/или ММР9. В еще одном аспекте изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как они определены ранее, в изготовлении лекарственного средства для использования в лечении воспалительного заболевания. В еще одном аспекте изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как они определены ранее, в изготовлении лекарственного средства для использования в лечении обструктивного заболевания дыхательных путей, такого как астма или ХОЗЛ. В контексте настоящего описания термин “лечение”, если конкретно не указано иного, также включает “профилактику”. Термины “терапевтический” и “терапевтически” должны быть истолкованы соответственно. Предполагают, что профилактика особенно существенна для лечения лиц, у которых уже имел место случай рассматриваемого заболевания или состояния или которые по иным причинам рассматриваются как имеющие повышенный риск рассматриваемого заболевания или состояния. Лица с риском развития конкретного заболевания или состояния, как правило, включают лиц, у которых имеется семейная история заболевания или состояния, или лиц, которые были идентифицированы путем генетического тестирования или скрининга как особенно подверженные развитию такого заболевания или состояния. В изобретении дополнительно предложен способ лечения заболевания или состояния, при котором благоприятным является ингибирование ММР12 и/или ММР9, включающий введение пациенту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как они определены ранее. В изобретении также предложен способ лечения обструктивного заболевания дыхательных путей, например астмы или ХОЗЛ, включающий введение пациенту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как они определены ранее. Для упомянутых выше терапевтических применений вводимая доза, безусловно, будет зависеть от используемого соединения, способа введения, желаемого лечения и расстройства, которое лечат. Суточная доза соединения формулы (I)/соли (активный ингредиент) может находиться в диапазоне от 0,001 мг/кг до 75 мг/кг, в частности от 0,5 мг/кг до 30 мг/кг. Эта суточная доза при необходимости может быть введена в виде разделенных доз. Как правило, стандартные лекарственные формы содержат приблизительно от 1 мг до 500 мг соединения по изобретению. Соединения формулы (I) и их фармацевтически приемлемые соли могут быть использованы сами по себе, но, как правило, их вводят в форме фармацевтической композиции, в которой соединение формулы (I) / соль (активный ингредиент) находится в сочетании с фармацевтически приемлемым адъювантом, разбавителем или носителем. В зависимости от способа введения фармацевтическая композиция предпочтительно содержит от 0,05 до 99 мас.% (процент по массе), более предпочтительно от 0,10 до 70 мас.% активного ингредиента, и от 1 до 99,95 мас.%, более предпочтительно от 30 до 99,90 мас.% фармацевтически приемлемого адъюванта, разбавителя или носителя, все проценты по массе основаны на массе всей композиции. Обычные способы выбора и приготовления подходящих фармацевтических препаратов описаны, например, в “Pharmaceuticals – The Science of Dosage Form Designs”, M.E.Aulton, Churchill Livingstone, 1988. Таким образом, в настоящем изобретении также предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, как они определены ранее, в сочетании с фармацевтически приемлемым адъювантом, разбавителем или носителем. В изобретении также предложен способ приготовления фармацевтической композиции по изобретению, включающий смешивание соединения формулы (I) или его фармацевтически приемлемой соли, как они определены ранее, с фармацевтически приемлемым адъювантом, разбавителем или носителем. Фармацевтические композиции по изобретению могут быть введены стандартным образом при заболевании или состоянии, которое желательно лечить, например, путем перорального, местного, парентерального, трансбуккального, назального, вагинального или ректального введения или путем ингаляции. Для этих целей соединения по изобретению могут быть приготовлены в виде препаратов с помощью способов, известных в данной области техники, например, в форме таблеток, капсул, водных или масляных растворов, суспензий, эмульсий, кремов, мазей, гелей, назальных спреев, суппозиториев, тонкоизмельченных порошков или аэрозолей для ингаляции, а для парентерального применения (включающего внутривенное, внутримышечное введение или инфузию) в виде стерильных водных или масляных растворов, или суспензий, или стерильных эмульсий. Дополнительно к соединениям по настоящему изобретению фармацевтическая композиция по изобретению также может содержать один или более чем один фармакологический агент, имеющий ценность при лечении одного или более чем одного заболевания или состояния, упомянутого ранее, такой как продукт “Symbicort” (товарный знак). В настоящем изобретении дополнительно предложен способ получения соединения формулы (I) или его фармацевтически приемлемой соли, как они определены выше, включающий: а) взаимодействие соединения формулы (II)
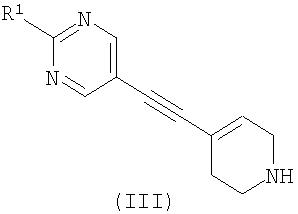
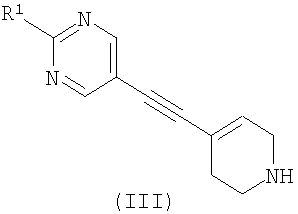
где R2 является таким, как определено в формуле (I), и L1 представляет собой уходящую группу, с соединением формулы (III) (или его солью)
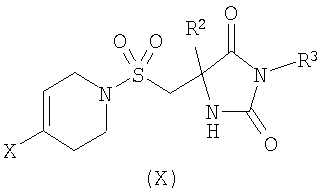
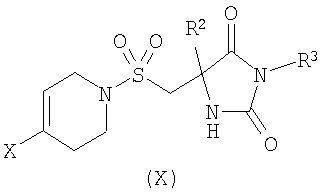
где R1 является таким, как определено в формуле (I); или б) взаимодействие соединения формулы (X)
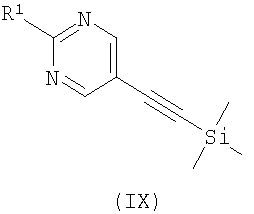
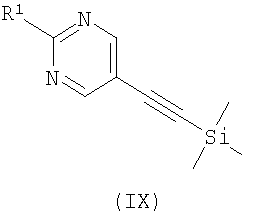
где R2 является таким, как определено в формуле (I), R3 представляет собой Н или подходящую защитную группу, и Х представляет собой уходящую группу, такую как галогенид или трифлат, с ацетиленовым соединением формулы (IX)
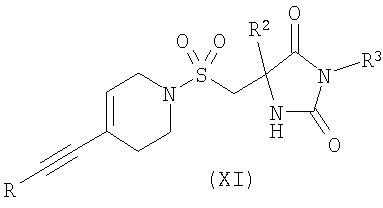
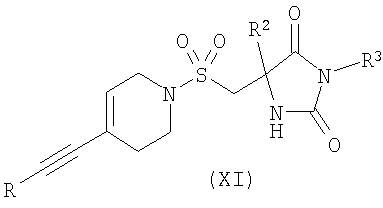
где R1 является таким, как определено в формуле (I); или в) взаимодействие соединения формулы (XI)
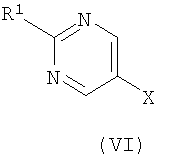
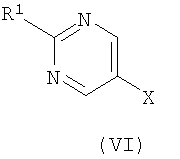
где R представляет собой Н или триметилсилил, R2 является таким, как определено в формуле (I), и R3 представляет собой Н или подходящую защитную группу, с арилгалогенидом или трифлатом формулы (VI)
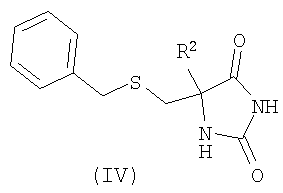
где R1 является таким, как определено в формуле (I), и Х представляет собой галогенид или трифлат; и возможно после этого образование его фармацевтически приемлемой соли. В вышеописанном способе (а) подходящие уходящие группы L1 включают галогено, в частности хлоро. Взаимодействие предпочтительно осуществляют в подходящем растворителе, возможно в присутствии добавленного основания в течение подходящего периода времени, как правило от 0,5 до 24 ч, при температуре от температуры окружающей среды до температуры дефлегмации. Обычно используют такие растворители, как пиридин, диметилформамид, тетрагидрофуран, ацетонитрил или дихлорметан. Когда используют добавленное основание, оно может представлять собой органическое основание, такое как триэтиламин, диизопропилэтиламин, N-метилморфолин или пиридин, либо неорганическое основание, такое как карбонат щелочного металла. Взаимодействие обычно осуществляют при температуре окружающей среды в течение 0,5-16 ч либо до достижения завершения реакции, что определяют при помощи хроматографических или спектроскопических способов. Реакции взаимодействия сульфонилгалогенидов с различными первичными и вторичными аминами хорошо известны в литературе, и вариации условий очевидны для специалистов в данной области техники. Сульфонилхлориды формулы (II) (где L1 представляет собой хлор) удобным образом получают путем окислительного хлорирования соединений формулы (IV)
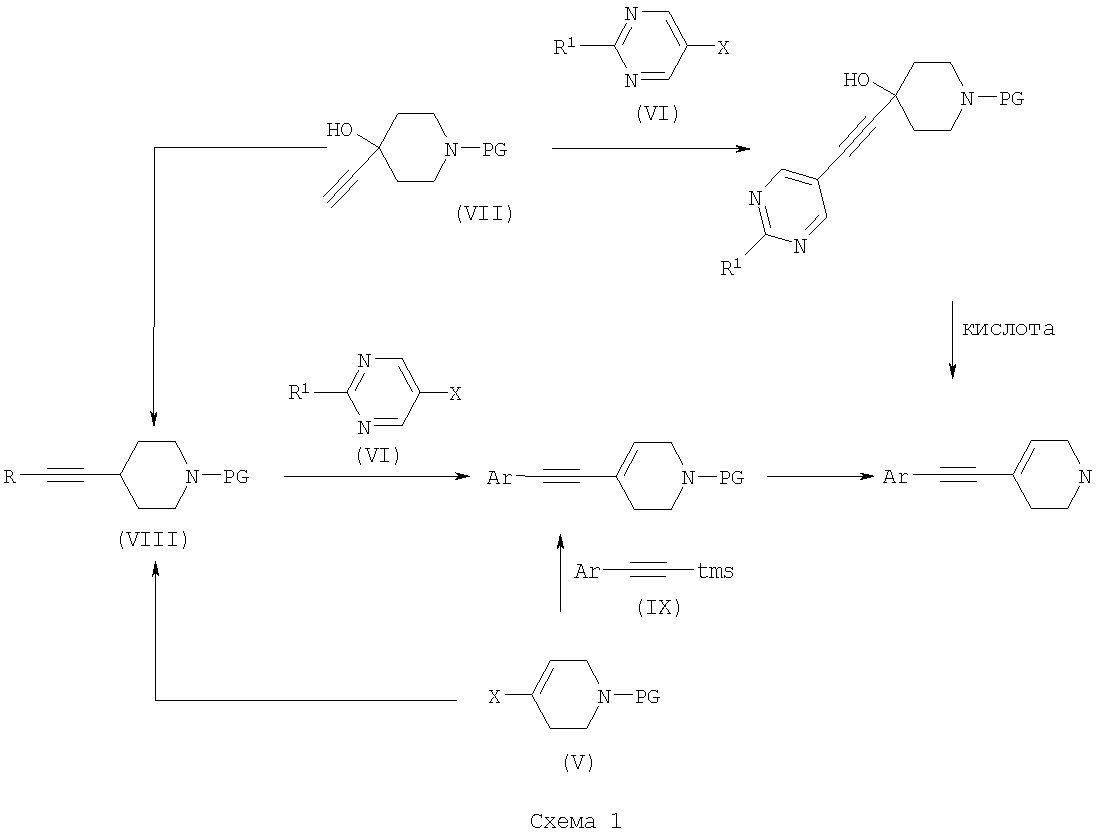
с использованием способов, которые очевидны для специалистов в данной области техники (Mosher, J., J. Org. Chem. 1958. 23, 1257; Griffith, O., J. Biol. Chem. 1983. 258, (3), 1591; WO 02/074767). Соединения формулы (III) могут быть получены с использованием различных способов, описанных в литературе, или их вариантов, очевидных для специалистов в области органической химии. Подходящие способы включают способы, описанные ниже и изображенные на Схеме 1, но не ограничиваются ими:
На Схеме 1 PG представляет собой подходящую защитную группу, такую как t-Boc (трет-бутоксикарбонил); Х представляет собой уходящую группу, такую как галогенид или трифлат; R представляет собой водород или триметилсилил; tms представляет собой триметилсилил; Ar представляет собой 5-пиримидинильное кольцо, замещенное по положению 2 радикалом R1; и R1 является таким, как определено в формуле (I). Взаимодействие между арильным или винильным производным [(V) или (VI)] и ацетиленом [(VII), (VIII) или (IX)] может быть осуществлено возможно в подходящем растворителе с использованием катализатора, такого как подходящая соль палладия, например PdCl2(PPh3)2, с добавлением соли меди или без нее и с аминным основанием, таким как пиперидин, триэтиламин, диизопропиламин или диизопропилэтиламин. При использовании добавленного растворителя он может представлять собой, например, тетрагидрофуран, ацетонитрил или N,N-диметилформамид. Взаимодействие осуществляют при температуре от температуры окружающей среды до температуры дефлегмации в течение промежутка времени от 20 минут до нескольких часов до тех пор, пока хроматографические или спектроскопические методы не укажут на завершение реакции. Реакции, катализируемые палладием, включающие ацетиленовые соединения, хорошо известны в литературе, и варианты условий очевидны для специалистов в данной области техники. Общая методология этого типа описана, например, в Brandsma, L., Synthesis of Acetylenes, Atlenes and Cumulenes: Methods and Techniques, 2004, Elsiever Academic Press, chapter 16, pages 293-317; Transition Metals-Catalysed Couplings of Acetylenes with sp2-halides, Sonogashira, K., J. Organomet. Chem., 2002, 653, 46-49; Tykwinski, R.R., Angew. Chem. Int. Ed., 2003, 42, 1566-1568. Винилтрифлат (V), где Х представляет собой O-трифлат и PG представляет собой t-Boc, может быть получен в соответствии с описанным в литературе (Wustrow, D.J., Synthesis, 1991, 993-995). Подходящие замещенные пиримидинилгалогениды или трифлаты формулы (VI) могут быть получены с использованием различных способов, описанных в литературе, например Budesinsky, Z. et al., Coll. Czech. Chem. Commun., 1949, 14, 223-235; Takahashi et al., Chem. Pharm. Bull, 1958, 6, 334-337; US 4558039. Ацетиленовое соединение (VIII) может быть получено из трифлата (V) посредством катализируемой палладием реакции сочетания с триметилсилилацетиленом, при необходимости с последующим удалением защитной триметилсилильной группы с использованием, например, фторида калия в подходящем растворителе. Альтернативно, получение соединения (VIII), где R представляет собой Н и PG представляет собой t-Boc, может быть осуществлено путем дегидратации соединения формулы (VII), например путем мезилирования с последующей обработкой подходящим основанием, например диизопропилэтиламином. Ацетиленовые гетероарильные соединения формулы (IX) могут быть получены с использованием различных способов, описанных в литературе. В способе (б) реакции осуществляют с использованием способов, сходных с описанными выше для получения соединений формулы (VIII). При необходимости один из атомов азота в гидантоиновом кольце соединений формулы (X) может быть защищен с использованием 2-(триметилсилил)этоксиметилхлорида SEMCI (R3=SEM) перед осуществлением взаимодействия, катализируемого палладием. Соединения формулы (X) могут быть получены путем катализируемого кислотой удаления защиты с соединений формулы (V) (PG=t-Boc) с последующим взаимодействием с соединением формулы (II) по аналогии с описанным выше для получения соединений формулы (I). В способе (в) реакции осуществляют по аналогии с описанным выше для получения соединений формулы (VIII). При необходимости один из атомов азота в гидантоиновом кольце соединений формулы (XI) может быть защищен с использованием SEMCI (R3=SEM) перед осуществлением взаимодействия, катализируемого палладием. Соединение (XI) обычно получают из соединения (VIII), где R представляет собой триметилсилил, a PG представляет собой t-Вос, путем катализируемого кислотой удаления группы t-Boc (например, с использованием ацетилхлорида в метаноле) с последующим взаимодействием с соединением формулы (II) в соответствии с описанным выше для взаимодействия между соединениями формул (II) и (III). Специалисту в данной области понятно, что в способах по настоящему изобретению может оказаться необходимым защитить подходящими защитными группами некоторые потенциально реакционно-способные функциональные группы, такие как гидроксильная или аминогруппа, в исходных реагентах или промежуточных соединениях. Таким образом, получение соединений по изобретению на различных стадиях может включать введение и удаление одной или более чем одной защитной группы. Подходящие защитные группы и информация о способах введения и удаления таких групп описаны в “Protective Groups in Organic Chemistry”, edited by J.W.F. McOmie, Plenum Press (1973)” и “Protective Groups in Organic Synthesis”, 3rd edition, T.W.Greene and P.G.M.Wuts, Wiley-Interscience (1999). Соединения по изобретению и их промежуточные соединения могут быть выделены из их реакционных смесей и при необходимости дополнительно очищены с использованием стандартных способов. Настоящее изобретение дополнительно раскрыто путем ссылки на следующие иллюстративные примеры. Общие способы Спектры ядерно-магнитного резонанса 1H ЯМР и 13С ЯМР регистрировали на приборе Varian Inova 400 МГц или Varian Mercury-VX 300 МГц. Центральные пики хлороформа-d ( Следующий способ использовали для анализа путем жидкостной хроматографии/масс-спектрометрии (ЖХ/МС): Прибор Agilent 1100; колонка Waters Symmetry 2,1×30 мм; масс-спектрометрия с химической ионизацией при атмосферном давлении (ХИАД); скорость потока 0,7 мл/мин; длина волны 254 или 220 нм; растворитель А: вода + 0,1% ТФУ; растворитель Б: ацетон + 0,1% ТФУ; Градиент 15-95%/Б 2,7 мин, 95% Б 0,3 мин. Следующий способ использовали для анализа путем ЖХ: Способ А. Прибор Agilent 1100; колонка: Kromasil C18 100×3 мм, размер частиц 5 мкм, растворитель А: 0,1% ТФУ/вода, растворитель Б: 0,08% ТФУ/ацетонитрил. Скорость потока 1 мл/мин, градиент 10-100%/Б 20 мин, 100% Б 1 мин. Адсорбцию измеряли при 220, 254 и 280 нм. Способ Б. Прибор Agilent 1100; колонка: XTerra C8, 100×3 мм, размер частиц 5 мкм, растворитель А: 15 мМ NH3/вода, растворитель Б: ацетонитрил скорость потока 1 мл/мин, градиент 10-100%/Б 20 мин, 100% Б 1 мин. Адсорбцию измеряли при 220, 254 и 280 нм. Сокращения:
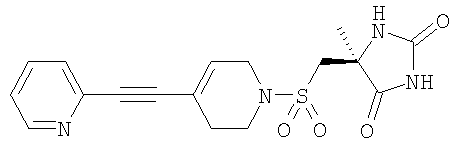
Пример 1 (5S)-5-({[4-[(2-Циклопропилпиримидин-5-ил)этинил]-3,6-дигидропиридин-1(2Н)ил]сульфонил}метил)-5-метилимидазолидин-2,4-дион Указанное в заголовке соединение получали в соответствии с общим способом Yamanaka et al, Synth. Commun., 1983, 312-314. К 5-бром-2-циклопропилпиримидину (110 мг, 0,55 ммоль) и (55)-5-{[(4-этинил-3,6-дигидропиридин-1(2Н)ил)сульфонил]метил}-5-метилимидазолидин-2,4-диону (180 мг, 0,61 ммоль) в ТГФ (3 мл) при 35°С добавляли Et3N (1 мл) и ДМФ (1 мл). После образования раствора добавляли CuI (4 моль.%) и PdCl2(PPh3)2 (2 моль.%) и смесь нагревали при 72°С в течение 6 часов. Смесь распределяли между EtOAc (15 мл) и водой (10 мл), и водный слой трижды экстрагировали EtOAc. Объединенные органические слои сушили и концентрировали с получением неочищенного продукта в виде желтого масла. Указанное в заголовке соединение (65 мг) получали путем очистки с использованием препаративной ВЭЖХ. 1H-ЯМР (ДМСО-d6): ХИАД-МС m/z: 416 [МН+]. a) 5-Бром-2-циклопропилпиримидин 5-Бром-2-циклопропилпиримидин получали с использованием способа Budesinsky, Z., Coll. Czech. Chem. Commun., 1949, 14, 223-235. Гидрохлорид циклопропанкарбоксимидамида (2,5 г, 20,7 ммоль) растворяли в EtOH (4 мл), добавляли свежеприготовленный 4,1М NaOEt в EtOH (4,8 мл), а затем мукобромную кислоту (2,7 г; 10,3 ммоль). Смесь нагревали до 56°С в течение 30 минут, добавляли дополнительное количество NaOEt в EtOH (4,1М, 3,2 мл) и реакционную смесь перемешивали при 56°С в течение еще 15 минут и затем при комнатной температуре в течение ночи. Растворитель выпаривали, добавляли водную HCl (2M, 10 мл) и коричневое твердое вещество отфильтровывали. Водный слой трижды экстрагировали дихлорметаном. Объединенные органические слои сушили и концентрировали с получением коричневого масла, которое вместе с твердым веществом давало неочищенное промежуточное соединение 5-бром-2-циклопропилпиримидин-4-карбоновой кислоты (1,6 г). Неочищенное промежуточное соединение нагревали при 140°С в течение 8 минут с получением коричневого вязкого масла, которое затем частично растворяли в дихлорметане. Раствор декантировали из смеси и концентрировали с получением указанного в подзаголовке соединения в виде масла (673 мг). 1H-ЯМР (CDCl3): ХИАД-МС m/z: 199/201 1:1 [MH+]. б) (5S)-5-{[(4-Этинил-3,6-дигидропиридин-1(2Н)-ил)сульфонил]метил}-5-метилимидазолидин-2,4-дион (5S)-5-Метил-5-({[4-[(триметилсилил)этинил]-3,6-дигидропиридин-1(2Н)-ил]сульфонил}метил)имидазолидин-2,4-дион (2,27 г, 6,0 ммоль) и фторид калия (1,07 г, 18,4 ммоль) перемешивали в течение ночи при комнатной температуре в метаноле (50 мл). Растворитель выпаривали, остаток растворяли в EtOAc, промывали водой, затем рассолом, сушили (сульфат натрия) и упаривали. Остаток очищали путем колоночной хроматографии, элюируя смесью изогексан/EtOAc 1:1 с получением твердого продукта (1,81 г). 1H ЯМР (CDCl3) ХИАД-МС m/z: 298 [MH+]. в) (5S)-5-Метил-5-({[4-[(триметилсилил)этинил]-3,6-дигидропиридин-1(2H)-ил]сульфонил}метил)имидазолидин-2,4-дион Гидрохлорид 4-[(триметилсилил)этинил]-1,2,3,6-тетрагидропиридина (3,43 г, 15,9 ммоль) перемешивали в ТГФ (100 мл) с [(4S)-4-метил-2,5-диоксоимидазолидин-4-ил]метансульфонилхлоридом (3,39 г, 15 ммоль) и охлаждали в ледяной солевой бане (температура приблизительно -10°С). По каплям в течение 2 часов добавляли N-этилдиизопропиламин (5,13 мл, 30 ммоль) в ТГФ (100 мл) и смесь перемешивали в течение еще 2 часов. Реакционную смесь промывали водой, водный слой экстрагировали в EtOAc (х2), органические фазы объединяли, промывали 2М HCl (х2), насыщенным раствором бикарбоната (х2), затем рассолом, сушили (сульфат натрия) и выпаривали с получением неочищенного продукта (5,06 г). Его использовали без дополнительной очистки. 1H ЯМР (ДМСО-d6) ХИАД-МС m/z: 370 [MH+]. г) Гидрохлорид 4-[(триметилсилил)этинил]-1,2,3,6-тетрагидропиридина трет-Бутил-4-[(триметилсилил)этинил]-3,6-дигидропиридин-1(2Н)-карбоксилат (2,75 г, 9,8 ммоль) перемешивали в метаноле (10 мл) и по каплям добавляли ацетилхлорид (2,1 мл, 29,2 ммоль). Во время добавления температура увеличивалась с 18°С до 30°С и смесь поддерживали при 40°С до тех пор, пока исходное вещество не перестали обнаруживать путем тонкослойной хроматографии (ТСХ). Смесь охлаждали до комнатной температуры, добавляли EtOAc (15 мл) и твердое вещество отфильтровывали с получением не совсем белого твердого вещества (1,6 г). 1H ЯМР (ДМСО-d6) ХИАД-МС m/z: 180 [MH+]. д) трет-Бутил-4-[(триметилсилил)этинил]-3,6-дигидропиридин-1(2Н)-карбоксилат Получали из N-Вос-пиперидин-4-она, как в WO 96/05200. 1Н ЯМР (CDCl3) Газовая хроматография/масс-спектрометрия/масс-спектрометрия (ГХ/МС-МС) m/z: 223 [М-55]. е) [(4S)-4-Метил-2,5-диоксоимидазолидин-4-ил]метансульфонилхлорид Получали в соответствии со способами, описанными в следующих публикациях: Mosher, J., J. Org. Chem. 1958. 23, 1257; Griffith, O., J. Biol. Chem. 1983. 258, (3), 1591; и WO 02/074767. Пример 2 (5S)-5-Метил-5-({[4-{[2-(метилтио)пиридин-5-ил]этинил}-3,6-дигидропиридин-1(2Н)-ил]сульфонил}метил)имидазолидин-2,4-дион Указанное в заголовке соединение получали с использованием общего способа, описанного Nishihara et al., J. Org. Chem., 2000, 65, 1780-1787. К раствору 2-(метилтио)-5-[(триметилсилил)этинил]пиримидина (0,55 г, 2,47 ммоль) и трифторметансульфоната 1-{[(4-метил-2,5-диоксоимидазолидин-4-ил)метил]сульфонил}-1,2,3,6-тетрагидропиридин-4-ила (0,94 г, 2,22 ммоль) в ДМФ (5 мл) добавляли CuI (10 моль.%) и PdCl2(PPh3)2 (5 моль.%) и смесь нагревали при 85°С в течение 6 часов. Смесь распределяли между EtOAc (20 мл) и водой (10 мл) и водный слой трижды экстрагировали EtOAc. Объединенные органические слои промывали рассолом, водой и концентрировали до получения коричневого масла (1,6 г). Указанное в заголовке соединение получали в виде твердого вещества (10 мг) после очистки путем препаративной ВЭЖХ (с использованием колонки Xterra-Prep-MC-C18 (50×19) с 12-минутным градиентом 5-35% ацетонитрила в воде с 0,06% NH3). 1H-ЯМР (ДМСО-d6): ХИАД-МС m/z: 422 [MH+]. а) 5-Бром-2-(метилтио)пиримидин Указанное в подзаголовке соединение получали в соответствии со способом Takahashi et al., Chem. Pharm. Bull, 1958, 6, 334-337. К раствору 5-бром-2-хлорпиримидина (1,0 г, 5,2 ммоль) в ЕtOН при комнатной температуре добавляли метантиолят натрия (0,36 г, 5,2 ммоль) и реакционную смесь перемешивали в течение ночи. Смесь распределяли между EtOAc (15 мл) и водой (10 мл). Водный слой дважды экстрагировали EtOAc и промывали рассолом. Объединенные органические слои сушили и концентрировали с получением указанного в подзаголовке соединения в виде белого твердого вещества (1,1 г). 1H-ЯМР (CD3OD): ХИАД-МС m/z: 204/206 1:1 [MH+]. б) 2-(Метилтио)-5-[(триметилсилил)этинил]пиримидин Указанное в подзаголовке соединение получали в соответствии со способом Yamanaka et al, Synth. Commun., 1983, 312-314. К 5-бром-2-(метилтио)пиримидину (0,60 г, 2,9 ммоль) в Et3N (3 мл) добавляли ДМФ (0,5 мл), CuI (5 моль.%) и PdCl2(PPh3)2 (3 моль.%). Смесь нагревали при 95°С в течение 12 часов в запаянной пробирке и затем распределяли между Et2O (30 мл) и водой (10 мл). Водный слой дважды экстрагировали Et2O и объединенные органические слои промывали водой, сушили и концентрировали с получением неочищенного продукта в виде коричневого масла. Соединение очищали путем флэш-хроматографии с использованием градиента 10-60% EtOAc в гептане, что позволило получить указанное в подзаголовке соединение в виде бесцветного масла (0,55 г). 1H ЯМР (CDCl3): ХИАД-МС m/z: 223 [MH+]. в) Трифторметансульфонат 1-({[(4S)-4-метил-2,5-диоксоимидазолидин-4-ил]метил}сульфонил)-1,2,3,6-тетрагидропиридин-4-ила Хлорид 4-{[(трифторметил)сульфонил]окси}-1,2,3,6-тетрагидропиридиния приводили во взаимодействие с [(4S)-4-метил-2,5-диоксоимидазолидин-4-ил]метансульфонилхлоридом (Пример 1е) тем же самым путем, как для Примера 1в. 1H ЯМР (ДМСО-d6) ХИАД-МС m/z: 422 [MH+]. г) Хлорид 4-{[(трифторметил)сульфонил]окси}-1,2,3,6-тетрагидропиридиния трет-Бутил-4-{[(трифторметил)сульфонил]окси}-3,6-дигидропиридин-1(2Н)карбоксилат (3,77 г, 11,4 ммоль) смешивали с ТГФ (15 мл) и концентрированной соляной кислотой (15 мл). Через 1 час смесь упаривали и сушили путем азеотропного упаривания с толуолом и метанолом с получением бежевого твердого вещества (88%), которое может быть использовано без дополнительной очистки. 1H ЯМР (CDCl3) ХИАД-МС m/z: 232 [MH+]. д) трет-Бутил-4-{[(трифторметил)сульфонил]окси}-3,6-дигидропиридин-1(2Н)-карбоксилат Раствор N-boc-пиперидин-4-она (10,14 г, 50 ммоль) в ТГФ (80 мл) добавляли к охлажденному раствору (-78°С) 2М LDA в ТГФ (30 мл, 60 ммоль, 1,2 эквив.) и ТГФ (80 мл) в течение приблизительно 30 минут. После перемешивания в течение еще 10 минут добавляли раствор 1,1,1-трифтор-N-фенил-N-[(трифторметил)сульфонил]метансульфонамида (20 г, 56 ммоль, 1,1 эквив.) в ТГФ (80 мл) и смеси давали возможность нагреться до комнатной температуры. Раствор промывали водой, водный слой промывали EtOAc (х2), органические фазы объединяли и промывали насыщенным раствором хлорида аммония, рассолом, сушили (сульфат натрия) и упаривали. Остаток фильтровали через нейтральный оксид алюминия (200 г), элюируя н-гептаном, а затем смесью н-гептан/EtOAc 9:1. После упаривания 1H-ЯМР демонстрировал присутствие некоторого количества трифлатирующего агента, но неочищенный продукт использовали без дополнительной очистки. Выход (13,17 г, 79,5%) (Wustrow, D.J., Synthesis, 1991, 993-995). 1H ЯМР (CDCl3) ГХМС-МС m/z: 274 [M-57]. Пример 3 (5S)-5-Метил-5-({[4-{[2-(трифторметил)пиримидин-5-ил]этинил}-3,6-дигидропиридин-1(2Н)-ил]сульфонил}метил)имидазолидин-2,4-дион Указанное в заголовке соединение получали с выходом 48% из 2-(трифторметил)-5-[(триметилсилил)этинил]пиримидина с использованием того же самого способа, как описанный для Примера 2. Белое твердое вещество из 95% EtOH, разлож. 240-245°С. 1H ЯМР (ДМСО-d6) ХИАД-МС m/z: 444 [MH+]. а) 2-(Трифторметил)-5-[(триметилсилил)этинил]пиримидин Трифторметансульфонат 2-(трифторметил)пиримидин-5-ила (0,45 г, 1,5 ммоль) и безводный триэтиламин (1,0 мл) смешивали в виале с завинчивающейся пробкой. Раствор в течение 10 минут продували безводным аргоном. Добавляли триметилсилилацетилен (0,43 мл, 3,0 ммоль), тонкоизмельченный CuI (0,010 г, 0,05 ммоль) и PdCl2(PPh3)2 (0,020 г, 0,030 ммоль). Виалу закрывали и нагревали в алюминиевом блоке при 80°С. После перемешивания в течение 5 часов летучие вещества выпаривали при комнатной температуре (продукт возгоняется при 35-40°С/1000 Па (10 мбар)). Черный остаток переносили в EtOAc (20 мл) и досуха концентрировали с использованием диоксида кремния (приблизительно 5-10 г). Флэш-хроматография на диоксиде кремния с EtOAc/гептан (1:30) позволила получить 2-(трифторметил)-5-[(триметилсилил)этинил]пиримидин в виде белого твердого вещества (0,35 г, 95%), т.пл. 75,5-76,0°С. 1H ЯМР (CDCl3) ХИАД-МС m/z: 245 [MH+]. б) Трифторметансульфонат 2-(трифторметил)пиримидин-5-ила Трифликовый ангидрид (1,01 мл, 6,0 ммоль) по каплям добавляли к перемешиваемой смеси 2-(трифторметил)пиримидин-5-ола (полученного в соответствии с патентом США 1Н ЯМР (CDCl3) Пример 4 (5S)-5-Метил-5-({[4-[(2-метилпиримидин-5-ил)этинил]-3,6-дигидропиридин-1(2H)-ил]сульфонил}метил)имидазолидин-2,4-дион трет-Бутил-4-[(2-метилпиримидин-5-ил)этинил]-3,6-дигидропиридин-1(2H)карбоксилат обрабатывали ТФУ в EtOH и после завершения реакции растворитель упаривали и смесь сушили вымораживанием. Остаток суспендировали в ДМФ (1,5 мл) и смесь охлаждали до 4°С. Добавляли N-этилдиизопропиламин (2,2 эквив.) и смесь перемешивали в течение 20 минут перед добавлением [(4S)-4-метил-2,5-диоксоимидазолидин-4-ил]метансульфонилхлорида (Пример 1е) (1,1 эквив.) в ДМФ (1 мл). Смесь перемешивали в течение 10 мин при 4°С и затем перемешивали в течение 2 ч при комнатной температуре перед упариванием растворителя. Продукт очищали путем препаративной ВЭЖХ с получением указанного в заголовке соединения (0,022 г, 30%). 1H ЯМР (ДМСО-d6); 10.75 (1Н, s); 8.80 (2Н, s); 8.02 (1Н, s); 7.80 (1Н, m); 7.32 (1Н, d, J=8.1 Гц); 6.24 (1Н, s); 3.81 (2Н, d, J=3.2 Гц); 3.34-3.21 (2Н, m); 3.30 (3H, s); 2.75 (2Н, q, J=20.8 Гц); 2.34 (2Н, m); 1.29 (3H, s); 1.19 (3H, t, J=7.6 Гц). ХИАД-МС m/z: 390 [MH+]. а) 2-Метил-5-[(триметилсилил)этинил]пиримидин 5-Бром-2-метил-пиримидин (полученный в соответствии с заявкой на патент Великобритании ХИАД-МС m/z: 191 [МН+]. б) трет-Бутил-4-[(2-метилпиримидин-5-ил)этинил]-3,6-дигидропиридин-1(2H)карбоксилат К раствору CuCl (1 мг, 0,01 ммоль) и PdCl2(PPh3)2 (0,003 г, 0,004 ммоль) в ДМФ (2 мл) при комнатной температуре добавляли 2-метил-5-[(триметилсилил)этинил]пиримидин (0,088 г, 0,462 ммоль) и трет-бутил-4-{[(трифторметил)сульфонил]окси}-3,6-дигидропиридин-1(2Н)карбоксилат (Пример 2д) (0,183 г, 0,555 ммоль). Реакционную смесь перемешивали в течение 8 ч при 80°С. После охлаждения смесь гасили 1 н. HCl и экстрагировали простым эфиром (3х). Объединенные органические слои промывали насыщенным водным раствором NaHCO3, рассолом и сушили. Фильтрация и упаривание позволили получить коричневое масло, которое очищали с использованием ВЭЖХ с получением указанного в подзаголовке соединения (0,062 г, 45%). ХИАД-МС m/z: 300 [MH+]. Пример 5 Трифторацетат(5S)-5-({[4-[(2-этилпиримидин-5-ил)этинил]-3,6-дигидропиридин-1(2H)-ил]сульфонил}метил)-5-метилимидазолидин-2,4-диона Указанное в заголовке соединение получали тем же самым путем, как описано для Примера 4. 5-Бром-2-этилпиримидиновое исходное вещество получали с использованием способа из GB 2157288. 1H ЯМР (ДМСО-d6); 10.75 (1Н, s); 8.82 (2Н, s); 8.05 (1Н, s); 6.24 (1H, s); 3.81 (2Н, d, J=3.2 Гц); 3.34-3.21 (2Н, m); 3.30 (3H, s); 2.92 (2Н, q, J=17.8 Гц); 2.75 (2Н, q, J=20.8 Гц); 2.34 (2Н, m); 1.26 (3H, t, J=12.8 Гц); 1.19 (1Н, s). ХИАД-МС m/z: 404 [MH+]. Пример 6 (5S)-5-({[4-[(2-Метоксипиримидин-5-ил)этинил]-3,6-дигидропиридин-1(2H)-ил]сульфонил)метил)-5-метилимидазолидин-2,4-дион Указанное в заголовке соединение получали из 5-бром-2-метоксипиримидина и (5S)-5-{[(4-этинил-3,6-дигидропиридин-1(2H)-ил)сульфонил]метил}-5-метилимидазолидин-2,4-диона тем же самым путем, как описано в Примере 1. 1H ЯМР (ДМСО-d6): ХИАД-МС m/z: 406 [MH+]. Фармакологический пример Анализы выделенных ферментов ММР12 Каталитический домен рекомбинантной человеческой ММР12 может быть экспрессирован и очищен в соответствии с описанным Parkar A.A. et at, (2000), Protein Expression and Purification, 20, 152. Очищенный фермент может быть использован для мониторинга ингибиторов активности следующим образом: ММР12 (конечная концентрация 50 нг/мл) инкубируют в течение 60 минут при комнатной температуре с синтетическим субстратом Mca-Pro-Cha-Gly-Nva-His-Ala-Dpa-NH2 (10 мкМ) в буфере для анализа (0,1М буфер “Tris-HCl” (товарный знак) рН 7,3, содержащий 0,1М NaCl, 20 мМ CaCl2, 0,020 мМ ZnCl и 0,05% (мас./об.) детергента “Brij 35” (товарный знак)) в присутствии (10 концентраций) или в отсутствие ингибиторов. Активность определяют путем измерения флуоресценции при % ингибирования равен отношению [Флуоресценцияплюс ингибитор – Флуоресценцияфон] к [Флуоресценцияминус ингибитор – Флуоресценцияфон]. ММР8 Очищенный pro-MMPS приобретен в Calbiochem. Фермент (при 10 мкг/мл) в течение 2,5 ч активируют ацетатом пара-аминофенилртути (АРМА) в концентрации 1 мМ, 35°С. Активированный фермент может быть использован для мониторинга ингибиторов активности следующим образом: ММР8 (конечная концентрация 200 нг/мл) инкубируют в течение 90 минут при 35°С (80% Н2О) с синтетическим субстратом Mca-Pro-Cha-Gly-Nva-His-Ala-Dpa-NH2 (12,5 мкМ) в буфере для анализа (0,1М буфер “Tris-HCl” (товарный знак) рН 7,5, содержащий 0,1М NaCl, 30 мМ CaCl2, 0,040 мМ ZnCl и 0,05% (мас./об.) детергента “Brij 35” (товарный знак)) в присутствии (10 концентраций) или в отсутствие ингибиторов. Активность определяют путем измерения флуоресценции при % ингибирования равен отношению [Флуоресценцияплюс ингибитор – Флуоресценцияфон] к [Флуоресценцияминус ингибитор – Флуоресценцияфон]. ММР9 Каталитический домен рекомбинантной человеческой ММР9 экспрессировали и затем очищали путем колоночной хроматографии с хелатом Zn с последующей аффинной колоночной хроматографией с гидроксаматом. Фермент может быть использован для мониторинга ингибиторов активности в соответствии со следующим: ММР9 (конечная концентрация 5 нг/мл) инкубируют в течение 30 минут при комнатной температуре (KT) с синтетическим субстратом Mca-Pro-Cha-Gly-Nva-His-Ala-Dpa-NH2 (5 мкМ) в буфере для анализа (0,1М буфер “Tris-HCl” (товарный знак) рН 7,3, содержащий 0,1М NaCl, 20 мМ CaCl2, 0,020 мМ ZnCl и 0,05% (мас./об.) детергента “Brij 35” (товарный знак)) в присутствии (10 концентраций) или в отсутствие ингибиторов. Активность определяют путем измерения флуоресценции при % ингибирования равен отношению [Флуоресценцияплюс ингибитор – Флуоресценцияфон] к [Флуоресценцияминус ингибитор – Флуоресценцияфон]. ММР14 Каталитический домен рекомбинантной человеческой ММР14 может быть экспрессирован и очищен в соответствии с описанным Parkar A.A. et at, (2000), Protein Expression and Purification, 20, 152. Очищенный фермент может быть использован для мониторинга ингибиторов активности в соответствии со следующим: ММР14 (конечная концентрация 10 нг/мл) инкубируют в течение 60 минут при комнатной температуре с синтетическим субстратом Mca-Pro-Cha-Gly-Nva-His-Ala-Dpa-NH2 (10 мкМ) в буфере для анализа (0,1М буфер “Tris-HCl” (товарный знак) рН 7,5, содержащий 0,1 М NaCl, 20 мМ CaCl2, 0,020 мМ ZnCl и 0,05% (мас./об.) детергента “Brij 35” (товарный знак)) в присутствии (5 концентраций) или в отсутствие ингибиторов. Активность определяют путем измерения флуоресценции при % ингибирования равен отношению [Флуоресценцияплюс ингибитор – Флуоресценцияфон] к [Флуоресценцияминус ингибитор – Флуоресценцияфон]. Протокол тестирования в отношении других матриксных металлопротеиназ, включая ММР9, с использованием экспрессированной и очищенной pro ММР описан, например, в С.Graham Knight et al, (1992) FEBS Lett, 296(3), 263-266. MMP19 Каталитический домен рекомбинантной человеческой MMP19 может быть экспрессирован и очищен в соответствии с описанным Parkar А.А. et al, (2000), Protein Expression and Purification, 20, 152. Очищенный фермент может быть использован для мониторинга ингибиторов активности в соответствии со следующим: MMP19 (конечная концентрация 40 нг/мл) инкубируют в течение 120 минут при 35°С с синтетическим субстратом Mca-Pro-Leu-Ala-Nva-Dpa-Ala-Arg-NH2 (5 мкМ) в буфере для анализа (0,1М буфер “Tris-HCl” (товарный знак) рН 7,3, содержащий 0,1М NaCl, 20 мМ CaCl2, 0,020 мМ ZnCl и 0,05% (мас./об.) детергента “Brij 35” (товарный знак)) в присутствии (5 концентраций) или в отсутствие ингибиторов. Активность определяют путем измерения флуоресценции при % ингибирования равен отношению [Флуоресценцияплюс ингибитор – Флуоресценцияфон] к [Флуоресценцияминус ингибитор – Флуоресценцияфон]. Связывание с белками Связывание с белками плазмы определяли путем ультрафильтрации в автоматизированном 96-луночном формате анализа. В каждом тесте параллельно контролировали связывание с белками плазмы референсного соединения (будесонида). Тестируемые соединения (10 мМ, растворенные в ДМСО) добавляли к плазме до конечной концентрации 10 мкМ и уравновешивали при комнатной температуре в течение 10 минут. 350 мкл плазмы переносили в ячейку для ультрафильтрации Microcon-96 (номинально отсекаемая молекулярная масса 10 кДа, Millipore). Ячейку для ультрафильтрации центрифугировали при 3000G в течение 70 минут при комнатной температуре. После центрифугирования при помощи ЖХ-МС/МС с использованием 3-точечного калибровочного графика определяли концентрацию соединений в полученной жидкости плазмы крови (несвязавшаяся фракция) и сравнивали с концентрацией в исходной спайкированной плазме. Анализы осуществляли с использованием градиентной хроматографической системы с подвижными фазами уксусная кислота/ацетонитрил. Обнаружение осуществляли с использованием квадрупольного масс-спектрометра API3000 или API4000 производства Applied Biosystems с электрораспылительным интерфейсом. Протокол определения растворимости Растворимость тестируемых соединений в 0,1М фосфатном буфере, рН 7,4, определяли следующим образом. Тестируемое соединение (1 мг) взвешивали в стеклянную виалу объемом 2 мл с завинчивающейся крышкой и добавляли 0,1М фосфатный буфер рН 7,4 (1,00 мл). Виалу с образцом затем подвергали ультразвуковому воздействию в течение приблизительно 10 минут и затем затем помещали на панель аппарата для встряхивания на ночь при 20°С. Содержимые виал с образцом затем фильтровали через фильтр Millipore Millex-LH 0,45 мкм в новую стеклянную виалу объемом 2 мл с получением прозрачного раствора. Прозрачный раствор (40 мкл) переносили в новую стеклянную виалу объемом 2 мл и разбавляли 0,1М фосфатным буфером, рН 7,4 (960 мкл). Стандартную калибровочную кривую для каждого конкретного тестируемого соединения получали с использованием растворов с известной концентрацией. Эти растворы с известными концентрациями обычно выбирали таким образом, чтобы концентрации составляли приблизительно 10 мкг/мл и 50 мкг/мл. Их готовили путем растворения известной массы соединения в 99,5% этаноле (500 мкл) и затем при необходимости обрабатывали ультразвуком в течение одной минуты. Если соединение все еще не было растворено полностью, добавляли ДМСО (500 мкл) и смесь обрабатывали ультразвуком в течение еще одной минуты. Получающийся в результате раствор затем разбавляли до соответствующего объема смесью ацетонитрил/100 мМ ацетат аммония, рН 5,5 20-50/80-50. При необходимости дополнительный более разведенный стандартный раствор получали путем разбавления. Растворы тестируемого соединения и стандартные растворы затем анализировали при помощи ВЭЖХ с ультрафиолетовым (УФ) обнаружением с использованием следующих параметров и таким образом определяли растворимость тестируемого соединения в 0,1М фосфатном буфере:
Система обработки данных хроматографии: ATLAS/Xchrome Протокол определения Log D Значения Log D при рН 7,4 определяли с использованием способа со встряхиванием колбы. Соответствующее небольшое количество тестируемого соединения помещали в стеклянную виалу объемом 2 мл с завинчивающейся крышкой при комнатной температуре и добавляли 600 мкл 1-октанола (насыщенного 10 мМ фосфатным буфером с рН 7,4). Виалу затем обрабатывали ультразвуком в течение одной минуты для полного растворения соединения. Затем добавляли 600 мкл 10 мМ фосфатного буфера с рН 7,4 (насыщенного 1-октанолом) и виалу встряхивали в течение 4 минут для смешивания двух фаз. Две фазы затем разделяли путем центрифугирования образца при 1000 g в течение 10 минут при комнатной температуре. Наконец разделенные водную и органическую фазы анализировали в двух параллелях путем ВЭЖХ с использованием следующих условий:
Система обработки данных хроматографии: ATLAS/Xchrome Значение log DpH 7.4 рассчитывали автоматически (смотр уравнение ниже) при помощи Excel после ручного ввода ответов в виде площадей пиков для соединений, полученных в результате использования системы обработки данных хроматографии ATLAS. Расчет log DpH 7,4 в соответствии с уравнением:
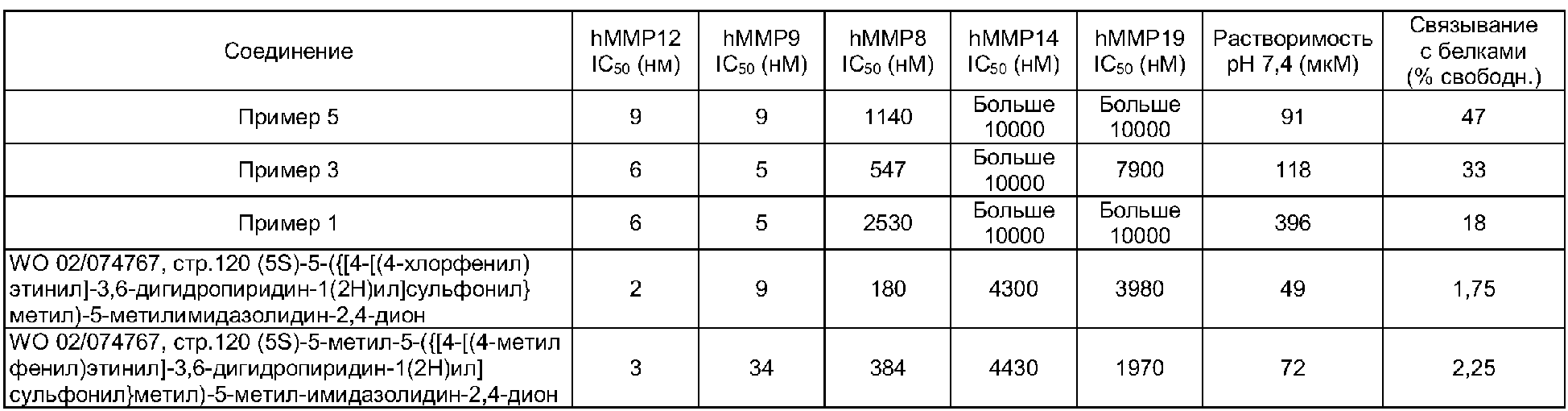
В следующей таблице представлены данные для типичного выбора соединения по настоящему изобретению и для избранных соединений из WO 02/074767.
Формула изобретения
1. Соединение формулы (I) в виде (S)-стереоизомера или его фармацевтически приемлемая соль 2. Соединение по п.1, где R1 представляет собой С1-2алкил или циклопропил, причем указанная алкильная группа возможно дополнительно замещена одним или более чем одним атомом фтора. 3. Соединение по п.2, где R1 представляет собой С1-2алкил, возможно дополнительно замещенный одним или более чем одним атомом фтора. 4. Соединение по п.3, где R1 представляет собой CF3. 5. Соединение по п.2, где R1 представляет собой циклопропил. 6. Соединение по п.1, где R2 представляет собой метил или этил. 7. Соединение по п.6, где R2 представляет собой метил. 8. Соединение по п.1, выбранное из группы, состоящей из 9. Соединение (5S)-5-({[4-[(2-циклопропилпиримидин-5-ил)этинил]-3,6-дигидропиридин-1(2Н)-ил]сульфонил}метил)-5-метилимидазолидин-2,4-дион и его фармацевтически приемлемые соли. 10. Способ получения соединения формулы (I), как оно определено в п.1, или его фармацевтически приемлемой соли, включающий 11. Способ получения соединения формулы (I), как оно определено в п.1, или его фармацевтически приемлемой соли, включающий 12. Способ получения соединения формулы (I), как оно определено в п.1, или его фармацевтически приемлемой соли, включающий 13. Фармацевтическая композиция, обладающая свойствами ингибитора ММР12 или ММР9, содержащая соединение формулы (I) или его фармацевтически приемлемую соль по любому из пп.1-9 в эффективном количестве в сочетании с фармацевтически приемлемым адъювантом, разбавителем или носителем. 14. Способ изготовления фармацевтической композиции по п.13, включающий смешивание соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп.1-9 с фармацевтически приемлемым адъювантом, разбавителем или носителем. 15. Соединение формулы (I) или его фармацевтически приемлемая соль по любому из пп.1-9 для изготовления лекарственного средства для лечения обструктивного заболевания дыхательных путей, опосредованного ММР12 и/или ММР9. 16. Применение соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп.1-9 в изготовлении лекарственного средства для использования в лечении обструктивного заболевания дыхательных путей, опосредованного ММР12 и/или ММР9. 17. Применение по п.16, где обструктивное заболевание дыхательных путей представляет собой астму или хроническое обструктивное заболевание легких.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
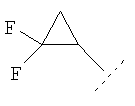
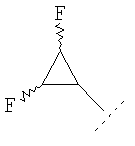
 ,
, ,
,
 ,
, ,
, ,
,
 H 7,27 млн-1), диметилсульфоксида-d6 (
H 7,27 млн-1), диметилсульфоксида-d6 ( 4558039) (0,82 г, 5,0 ммоль), толуола (10 мл) и водного трикалия фосфата (30% по массе, 10 мл) при температуре ледяной бани (Frantz et al., Organic Letters, 2002, 4(26), 4717-4718). После завершения добавления ледяную баню убирали и раствор перемешивали при температуре окружающей среды в течение 30 минут. Прозрачные фазы отделяли и органический слой промывали водой, затем рассолом. Сушка органической фазы над безводным сульфатом натрия, фильтрация и концентрирование путем роторного упаривания при комнатной температуре позволило получить трифторметансульфонат 2-(трифторметил)пиримидин-5-ила в виде бесцветного масла (1,38 г, 93%). Т.пл. 75-77°С (1000 Па (10 мбар)).
4558039) (0,82 г, 5,0 ммоль), толуола (10 мл) и водного трикалия фосфата (30% по массе, 10 мл) при температуре ледяной бани (Frantz et al., Organic Letters, 2002, 4(26), 4717-4718). После завершения добавления ледяную баню убирали и раствор перемешивали при температуре окружающей среды в течение 30 минут. Прозрачные фазы отделяли и органический слой промывали водой, затем рассолом. Сушка органической фазы над безводным сульфатом натрия, фильтрация и концентрирование путем роторного упаривания при комнатной температуре позволило получить трифторметансульфонат 2-(трифторметил)пиримидин-5-ила в виде бесцветного масла (1,38 г, 93%). Т.пл. 75-77°С (1000 Па (10 мбар)). ех 320 нм и
ех 320 нм и 
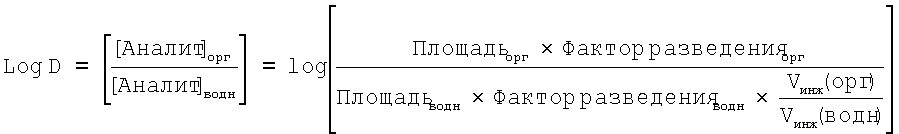
 ,
, ,
,