Патент на изобретение №2386623
|
||||||||||||||||||||||||||
(54) НОВЫЕ МОДУЛЯТОРЫ ДОФАМИНОВОЙ НЕЙРОТРАНСМИССИИ
(57) Реферат:
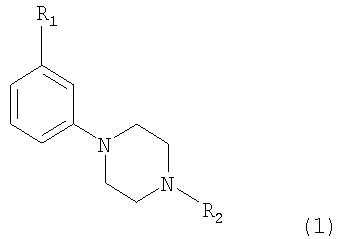
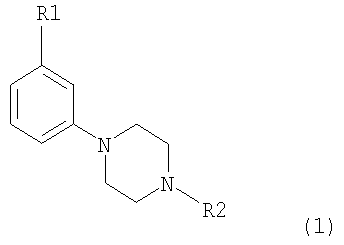
Изобретение относится к новым соединениям 3-замещенного 4-(фенил-N-алкил)пиперазина формулы (1)
R1 представляет собой OSO2CF3, OSO2CH3, SO2R3, COCF3 и СОСН2СН3, R2 представляет собой C1-C4 алкил или аллил, R3 представляет собой C1-С3 алкил или CF3, а также к их фармацевтически приемлемым солям. Соединения по настоящему изобретению обладают способностью модулировать нейротрансмиссию допамина. Также в настоящем изобретении раскрываются фармацевтические композиции вышеназванных соединений, где вышеназванные соединения используются для лечения расстройств центральной нервной системы. 2 н. и 2 з.п. ф-лы.
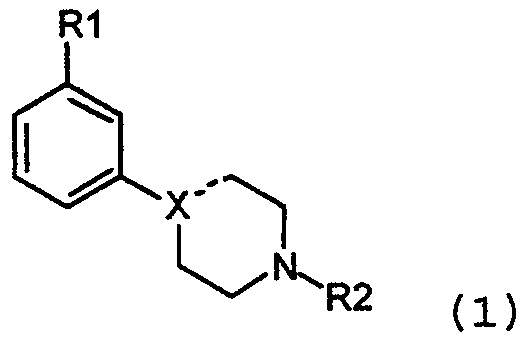
Настоящее изобретение относится к новым модуляторам дофаминовой нейротрансмиссии, более конкретно к новым замещенным 4-(фенил-N-алкил)пиперазинам и 4-(фенил-N-алкил)пиперидинам, и к их использованию. Дофамин является нейротрансмиттером в головном мозге. С момента его открытия, происшедшего в 1950 году, функция дофамина в мозге широко использовалась. К настоящему времени точно установлено, что дофамин играет существенную роль в различных аспектах функции мозга, включая моторную, познавательную, чувствительную, эмоциональную и вегетативные функции (например, регуляция аппетита, температуры тела, сна). Так, модулирование функции дофамина может быть полезно при лечении широкого спектра заболеваний, затрагивающих функцию мозга. В действительности, как неврологические, так и психиатрические заболевания лечатся медикаментозными средствами, основанными на взаимодействии дофаминовых систем и дофаминовых рецепторов в мозге. Лекарственные средства, которые непосредственно или опосредовано действуют на центральные дофаминовые рецепторы, обычно используют в лечении неврологических и психиатрических заболеваний, например болезни Паркинсона и шизофрении. В настоящее время доступные дофаминэргические фармацевтические средства обладают серьезными побочными эффектами, такими как экстрапирамидальные побочные эффекты и отдаленная дискинезия при использовании дофаминэргических антагонистов в качестве антипсихотических агентов, и дискинезии и психозов при использовании дофаминэргических антагонистов в качестве агентов, направленных на лечение болезни Паркинсона. Терапевтические действия неудовлетворительны во многих отношениях. Продолжаются поиски улучшения эффективности и снижения побочных эффектов лигандов дофаминэргических фармацевтических средств, новых лигандов дофаминовых рецепторов с избирательностью по отношению к специфическим подтипам дофаминовых рецепторов или региональной избирательностью. В этом контексте для достижения оптимального уровня стимулирования дофаминовых рецепторов также разрабатываются частичные антагонисты дофаминовых рецепторов, то есть лиганды дофаминовых рецепторов с некоторой, но не полной присущей им активностью относительно дофаминовых рецепторов, что позволяет избежать блокады дофаминового рецептора или его избыточного стимулирования. Ранее были сообщения и о соединениях, принадлежащих к классу замещенного 4-(фенил-N-алкил)пиперазина и замещенных 4-(фенил-N-алкил)пиперидинов. Среди этих соединений некоторые являются неактивными в ЦНС, некоторые показывают серотонэргический или смешанный серотонэргический/дофаминэргический фармакологический профиль, в то время, как некоторые являются частичными или полными агонистами или антагонистами дофаминового рецептора с высоким сродством относительно дофаминовых рецепторов. Ряд производных 4-фенилпиперазинов и 4-фенилпиперидина известен и описан, например, Costall et al. European J. Pharm. 31, 94, (1975), Newshaw et al. Bioorg. Med. Chem. Lett., 8, 295, (1998). Описанные соединения являются замещенными 4-фенилпиперазинами, причем большинство из них является 2-, 3- или 4-ОН фенилзамещенными и проявляющими свойства агонистов дофаминовых (DA) ауторецепторов. Fuller R.W. et al., J. Pharmacol. Exp. Therapeut. 218, 636, (1981) раскрывает замещенные пиперазины (например, 1-(м-трифторметилфенил)пиперазин), которые, как сообщают, действуют как агонисты серотонина и ингибируют поглощение серотонина. Fuller R.W. et al., Res. Commun. Chem. Pathol. Pharmacol. 17, 551, (1977) раскрывают сравнительное воздействие на 3,4-дигидроксифенилуксусную кислоту, и Res. Commun. Chem. Pathol. Pharmacol. 29, 201, (1980) раскрывает сравнительное воздействие на концентрацию 5-гидроксииндолуксусной кислоты в мозге крысы посредством 1-(парахлорфенол)пиперазина. Boissier J. еt al., Chem Abstr. 61:10691c, раскрывает двузамещенные пиперазины. Соединения представлены как адренолитические, антигипертензивные, потенциаторы барбитуратов и депрессанты центральной нервной системы. Имеются публикации о ряде различных замещенных пиперазинов, таких как лиганды 5-HT1А рецепторов, например, Glennon R.A. et al., J. Med. Chem., 31, 1968, (1988), van Steen B.J., J. Med. Chem., 36, 2751, (1993), Mokrosz, J. et al., Arch. Pharm. (Weinheim) 328, 143-148 (1995), and Dukat M.-L., J. Med. Chem., 39, 4017, (1996). Glennon R.A. в международных патентных заявках WO 93/00313 и WO 91/09594 раскрывает различные амины, и среди них замещенные пиперазины, как лиганды сигма рецепторов. Клинические работы, исследующие свойства лигандов сигма рецепторов у больных шизофренией, не делают очевидным антипсихотическую активность или активность по отношению к каким-либо другим заболеваниям ЦНС. С обоими из наиболее изученных селективных антагонистов сигма рецепторов, BW234U (римказол) и BMY14802, проводились клинические исследования на больных шизофренией (Borison et al., 1991, Psychopharmacol Bull 27(2): 103-106; Gewirtz et al., 1994, Neuropsychopharmacology 10:37-40). Кроме того, WO 93/04684 и GB 2027703 также описывают конкретные замещенные пиперазины, пригодные для лечения заболеваний ЦНС. Изложение сущности изобретения Задачей настоящего изобретения является создание новых фармакологически активных соединений, особенно пригодных в лечении заболеваний центральной нервной системы, которые не обладают недостатками вышеобсужденных веществ. В результате разработки данного изобретения обнаружено, что желательно создание веществ со специфическими фармакологическими свойствами, а именно веществ, которые обладают модулирующим действием на нейротрансмиссию дофамина. Подобные свойства ранее не обсуждались, и их невозможно получить с ранее известными соединениями. Соединения в соответствии с настоящим изобретением обладают удивительным и интересным дуалистическим профилем допаминэргической активности с антагонист-подобными эффектами на нейрохимию головного мозга и умеренными агонист-подобными эффектами на нормальное поведение, но они вызывают подавление поведения при состояниях гиперактивности. Таким образом настоящее изобретение относится к новым 3-замещенным 4-(фенил-N-алкил)пиперазинам и 3-замещенным 4-(фенил-N-алкил)пиперидинам в форме свободного основания или к их фармацевтически приемлемой соли, фармацевтическим композициям, содержащим названные соединения и использованию названных соединений в терапии. Одним объектом изобретения является создание новых соединений для использования в терапии, и более точно, соединения для модулирования допаминэргических систем в головном мозге млекопитающего, включая мозг человека. Другим объектом настоящего изобретения является создание соединений, обладающих терапевтическим действием после орального введения. Более точно, настоящее изобретение относится к соединениям 3-замещенного 4-(фенил-N-алкил)пиперазина и 4-(фенил-N-алкил)пиперидина формулы 1:
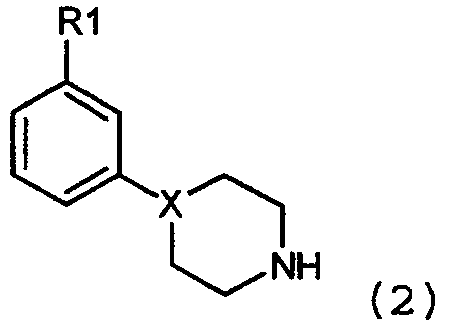
и к их фармацевтически приемлемым солям, где Х выбран из группы, состоящей из N, СН и С, однако, Х может представлять собой только С, если соединение содержит двойную связь на участке, обозначенном пунктирной линией; R1 выбран из группы, состоящей из OSO2CF3, OSO2CH3, SOR3, SO2R3, COR3, NO2 и CONНR3, где R3 такой, как определено выше, и когда Х представляет собой СН или С, R1 может также быть выбран из группы, состоящей из CF3, CN, F, Cl, Br и I; R2 выбран из группы, состоящей из C1-C4 алкилов, аллилов, CH2SCH3, CН2CH2OCH3, CH2CH2CH2F, CH2CF3, 3,3,3-трифторпропила, 4,4,4-трифторбутила и -(CH2)-R4, где R4 такой, как определено ниже; R3 выбран из группы, состоящей из С1-С3 алкилов, CF3 и N(R2)2, где R2 такой, как определено выше; R4 выбран из группы, состоящей из С3-С6 циклоалкилов, 2-тетрагидрофурана и 3-тетрагидрофурана. Соединения по настоящему изобретению обладают дофамин-модулирующими свойствами и пригодны в лечении ряда заболеваний центральной нервной системы, включая как психиатрическую, так и неврологическую симптоматику. Заболевания, при которых могут быть полезны соединения с модулирующим действием на дофаминэргические системы, представляют собой заболевания, относящиеся к старению, для предупреждения брадикинезии и депрессии и для улучшения психических функций. Они также могут быть использованы для улучшения познавательных функций и связанных с ними эмоциональных расстройств при нейродегенеративных заболеваниях и нарушениях развития, а также после повреждения мозга. Соединения по настоящему изобретению также могут быть использованы для улучшения всех симптомов психозов, включая шизофрению и шизофреноподобные заболевания, а также психотические нарушения, вызванные лекарственными средствами. Соединения по настоящему изобретению также могут использоваться при нарушениях поведения, обычно впервые диагносцирующиеся в младенчестве, детстве или юности, а также при нарушениях импульсного контроля. Также могут быть улучшены нарушения речи, такие как заикание. Они также могут быть использованы для лечения токсикомании, а также заболеваний, характеризующихся неправильным употреблением пищи. Нарушения настроения и тревожные состояния, изменение личности и конверсивная истерия могут также лечиться с помощью соединений настоящего изобретения. Неврологические показания включают лечение болезни Хантингтона и других расстройств движения, а также расстройств движения, вызванных лекарственными средствами. «Restless legs» и родственные заболевания, а также нарколепсия также могут лечиться соединениями, по настоящему изобретению. Они также могут улучшать психические и моторные функции при болезни Паркинсона, и при родственной паркинсоноподобной симптоматике. Они также могут использоваться для улучшения тремора различного происхождения. Они могут использоваться в лечении головных болей и для улучшения функции мозга после сосудистого или травматического повреждения мозга. Кроме того, они могут использоваться для ослабления боли при состояниях, характеризующихся повышением мышечного тонуса. Неожиданно было обнаружено, что соединения по настоящему изобретению специфически воздействуют на дофаминэргические системы мозга. Они обладают воздействием на биохимические импульсы в мозге с характерными чертами избранных антагонистов дофамина, например, вызывая повышение концентрации метаболитов дофамина. Кроме того, антагонисты рецепторов дофамина специфически подавляют поведенческую активность и вызывают каталепсию, в то время как соединения по изобретению не показывают или только ограничивают ингибиторные эффекты на спонтанную локомоторную активность. Напротив, они могут вызывать легкую поведенческую активизацию с сопутствующим усилением мелкомасштабных движений, например, остановки в центре области регистрации поведения, подобные тем, которые вызывают дофаминэргические антагонисты. Поведенческая активация является ограниченной, не достигая такого же полного повышения активности, какое вызывают прямые или непрямые допаминэргические антагонисты. С другой стороны, предпочтительные вещества снижают повышение активности, вызванное прямыми или непрямыми допаминэргическими агонистами, то есть д-амфетамином и веществами аналогичного действия. Таким образом, соединения по настоящему изобретению неожиданно показали интересный дуалистический профиль дофаминэргической активности с антагонист-подобным действием на нейрохимию мозга и умеренным агонист-подобным действием на нормальное поведение, но подавлением поведения в состоянии гиперактивности. Профиль активности предлагает модуляторное действие на дофаминэргические функции, явно отличающиеся от известных соединений, принадлежащих к данным химическим классам или эффектам, ожидаемым от типичных антагонистов или агонистов дофаминовых рецепторов этих или других химических классов. Учитывая участие дофамина в большом разнообразии функций ЦНС и клинические недостатки в настоящее время доступных фармацевтических средств, воздействующих на дофаминовые системы, можно доказать превосходство новых классов дофаминэргических модуляторов, присутствующих в данном изобретении, над известными в настоящее время дофаминэргическими соединениями в лечении некоторых заболеваний, относящихся к дисфункциям ЦНС, с точки зрения эффективности, а также побочных эффектов. Обнаружено, что некоторые соединения по настоящему изобретению обладают удивительно хорошими фармакокинетическими свойствами, включая высокую степень биодоступности при оральном применении. Таким образом, они являются пригодными для получения фармацевтических средств для орального применения. В предыдущем уровне техники не существует руководства для получения соединений с такими эффектами на дофаминовые системы в мозге. Подробное описание изобретения Фармакология Очевидно, что при психиатрических и неврологических заболеваниях нарушается нейротрансмиссия в ЦНС. Во многих случаях, например, при шизофрении или болезни Паркинсона, является применимой, но не оптимальной, фармакотерапия, основанная на антагонизме или агонизме дофаминовых рецепторов. В недавние годы много усилий было положено на открытие новых и селективных лигандов для субстратов дофаминового рецептора (D1, D2, D3, D4, D5) с целью улучшения эффективности и снижения побочных эффектов. Настоящее изобретение предлагает другие принципы для новых терапевтических средств, основанных на взаимодействии с дофаминовыми системами. Соединения по настоящему изобретению обладают воздействием на нейрохимию мозга, подобным воздействию антагонистов дофаминовых D2 рецепторов. В противоположность используемым в настоящее время антагонистам дофаминового рецептора, соединения настоящего изобретения показывают отсутствие или ограничение воздействия на спонтанную двигательную активность. Они могут вызывать поведенческую активацию с одновременным повышением мелкомасштабных движений, например, остановки в центре области регистрации поведения, подобно остановкам, вызванным антагонистами дофамина. Поведенческая активация является ограниченной, не достигая такого же полного усиления активности, какое вызывают прямые или непрямые антагонисты дофамина. Удивительно, что предпочтительные вещества могут действительно снижать повышение активности, вызванное прямыми или непрямыми дофаминэргическими агонистами, то есть д-амфетапином и веществами аналогичного действия. Предпочтительные структуры являются замещенными в мета-положении на ароматическом кольце. Примером такого соединения является 3-(1-пропилпиперидин-4-ил)фениловый эфир метансульфоновой кислоты, который представлен ниже в примере 14. У крыс это соединение повышает 3,4-дигидроксифенилуксусную кислоту в полосатом теле с 1265±74 (контроли) до 3208±236 нг/г ткани при 50 мкмоль/кг подкожно в сочетании с легким повышением поведенческой активности; 1485±328 см/30 мин (контроли) до 2126±240 см/30 мин при 50 мкмоль/кг подкожно, n=4. Другой предпочтительный пример соединения по изобретению представляет собой 4-(3-метансульфонилфенил)-1-пропилпиперидин, далее иллюстрированный в примере 6. У крыс это соединение повышает 3,4-дигидроксифенилуксусную кислоту в полосатом теле с 914±19 (контроли) до 1703±19 нг/г ткани при 50 мкмоль/кг подкожно. За подобным повышением оборота допамина следует тенденция в направлении повышения моторной активности с 2030±299 см/60 мин до 2879±398 см/60 мин р=0,14. У животных, приученных к коробке измерения подвижности, соединение в примере 6, 4-(3-метансульфонилфенил)-1-пропилпиперидин, повышает поведенческую активность с 476±279 см/60 мин (контроли) до 1243±72 см/60 мин, р<0,05, n=4, и 4-дигидроксифенилуксусную кислоту в полосатом теле с 975±23 (контроли) до 2074±144 нг/г ткани при 50 мкмоль/кг подкожно р<0,05, n=4. Кроме того, соединение, описанное в примере 6, 4-(3-метансульфонилфенил)-1-пропилпиперидин, имеет предпочтительную способность снижать поведенческую активность, вызванную как д-амфетамином (1,5 мг/кг подкожно), так и дизолципином (Mk-801, 0,7 мг/кг интраперитонеально). д-Амфетаминовая гиперактивность снижается с 10694±2165 см/60 мин до 1839±344 см/60 мин, р<0,05 n=4, при 50 мкмоль/кг подкожно соединения, описанного в примере 6, и поведенческая активность, вызванная дизолципином (Mk-801) снижается с 32580±4303 см/60 мин до 18197±1389 см/60 мин р<0,05 при 50 мкмоль/кг подкожно. К удивлению соединение, описанное в примере 6, доступно при оральном введении (F) 85% у крыс. В отличие от схожих соединений, описанных в WO 91/09594, у соединения из примера 6, 4-(3-метансульфонилфенил)-1-пропилпиперидин, отсутствует сродство к сигма-рецептору, <50% подавления [3H]-DTG связывания (в соответствии со способом измерения сигма-связывания, описанного Shirayama Y. еt al., 1993, Eur. J. Pharmacol. 237, p.117) при 10 мкмоль/л в мембранах головного мозга крыс. С целью демонстрации удивительного действия соединений в соответствии с изобретением, некоторые из соединений сравнивают с подобными соединениями известного уровня техники. Таким образом, соединения, пригодные для сравнения с соединениями по изобретению в сравнительных примерах, не входят в объем настоящего изобретения, так как они не проявляют желательных свойств. Сравнительный пример 1:4-(4-метансульфонилфенил)-1-пропилпиперидин иллюстрирует, что замещение в пара-положении дает неактивные соединения. 4-(4-метансульфонилфенил)-1-пропилпиперидин не оказывает эффекта на 3,4-дигидроксифенилуксусную кислоту в полосатом теле, как показано в нейрохимическом эксперименте; 988±70 (контроли) нг/г ткани и 928±51 нг/г ткани при 50 мкмоль/кг подкожно. 4-(4-метансульфонилфенил)-1-пропилпиперидин не обладает желательными свойствами. Сравнительный пример 2: Для дальнейшей иллюстрации важности замещения ароматического кольца для получения желательных свойств, 4-фенил-1-пропилпиперидин демонстрирует отсутствие активности при исследовании поведенческой активности у крыс, не подверженных предварительной обработке, с 3661±494 см/60 мин, контроли, до 2553±471 см/60 мин, р>0,05, n=4, при 33 мкмоль/кг и отсутствие воздействия на 3,4-дигидроксифенилуксусную кислоту в полосатом теле, как показано в нейрохимическом эксперименте; 1027±31 (контроли) нг/г ткани и 1190±70 нг/г ткани при 33 мкмоль/кг подкожно, р>0,05, 4-фенил-1-пропилпиперидин также не оказывает желательного подавления поведенческой активности при стимулировании д-амфетамином (с 17295±4738 см/60 мин, д-амфетамин, до 13764±2919 см/60 мин, n=4, р>>0,05 при 33 мкмоль/кг. Сравнительный пример 3: Кроме того обнаружено, что 1-фенил-4-пропилпиперазин, описанный в WO 91/09594, как соединение связывающее сигма-рецептор, снижает поведенческую активность у предварительно не обработанных животных с 3370±227, контроли, до 1923±204 см/60 мин, n=4, р<0,05 при 33 мкмоль/кг подкожно, таким образом обеспечивая желаемые свойства. Сравнительный пример 4: Замещение в орто-положении, как проиллюстрировано на примере 1-(2-метоксифенил)-4-пропилпиперазина, дает соединение, которое повышает содержание 3,4-дигидроксифенилуксусной кислоты в полосатом теле с 1028±9 (контроли) нг/г ткани до 3836±65 нг/г ткани при 50 мкмоль/кг подкожно, n=4, р<0,05. Это сопровождается ингибированием поведенческой активности, нежелательной в настоящем изобретении; с 1651±300 см/60 мин (контроли) до 67±34 см/60 мин при 50 мкмоль/кг подкожно, р<0,05, n=4. Сравнительный пример 5: Очень важны свойства заместителей в мета-положении. 1-пропил-4-(3-трифторметилфенил)пиперазин повышает содержание 3,4-дигидроксифенилуксусной кислоты в полосатом теле с 1066±46 (контроли) нг/г ткани до 3358±162 нг/г ткани при 50 мкмоль/кг подкожно, р<0,05, n=4, однако, затем следует подавление поведения с 1244±341 см/60 мин (контроли) до 271±137 при 50 мкмоль/кг подкожно, р<0,05, n=4, таким образом не обеспечивая желательные свойства. Сравнительный пример 6: Далее, соединение 3-(4-пропилпиперазин-1-ил)бензонитрила повышает содержание 3,4-дигидроксиуксусной кислоты в полосатом теле с 1432±57 (контроли) нг/г ткани до 4498±234 нг/г ткани при 100 мкмоль подкожно р<0,05, n=4, и снижает содержание 5-гидроксииндолуксусной кислоты с 630±16 (контроли) нг/г ткани до 484±26 нг/г ткани при 100 мкмоль/кг, р<0,05, n=4. За этими эффектами следует подавление поведенческой активности с 3959±688 см/60 мин (контроли) до 634±266 при 100 мкмоль/кг подкожно, р<0,05, n=4, таким образом не обеспечивая свойства, желательные в настоящем изобретении. 3-(4-Пропилпиперазин-1-ил)бензонитрил обладает следующими свойствами: т.пл. 159°С (фумарат). MС m/z (относительная интенсивность 70 эВ) 229 (М+, 28), 200 (bp), 157 (27), 129 (22), 70 (25). Сравнительный пример 7: Другой пример значения заместителя представляет собой препаративный пример 14, который также не оказывает воздействия на 3,4-дигидроксифенилуксусную кислоту в полосатом теле; с 1121±36 (контроли) нг/г ткани до 1169±42 нг/г ткани при 50 мкмоль/кг подкожно. Сравнительный пример 8: Физико-химические свойства, оказываемые заместителем на основном азоте, также важны для получения желаемого профиля. Невозможно использовать любой заместитель, например, 1-фенетил-4-(3-трифторметилфенил)пиперазин, описанный как лиганд-сигма рецептора в WO 91/09594 и WO 93/00313, который имеет некоторое воздействие на 3,4-дигидроксифенилуксусную кислоту в полосатом теле; с 852±33 (контроли) до 1406±77 нг/г ткани при 50 мкмоль/кг подкожно, р<0,05, n=4, а также снижает как количество 5-гидроксииндолуксусной кислоты в полосатом теле с 358±20 (контроли) до 289±16 нг/г ткани при 50 мкмоль/кг подкожно р<0,05, n=4, так и серотонин (5-НТ) с 379±10 (контроли) до 282±6 нг/г ткани при 50 мкмоль/кг подкожно, р<0,05, n=4, что является нежелательным свойством в соответствии с данным изобретением, но как сообщается IC50 20,3 при 5-НТ1А рецепторе (WO 93/00313). Сравнительный пример 9: Кроме того, 1-бензил-4-(3-метансульфонилфенил)пиперидин, и 3-(1-бензилпиперидин-4-ил)фенол, соединения с бензильным замещением на основном азоте, оба имеют нежелательные свойства для связывания с серотониновыми системами мозга. 1-Бензил-4-(3-метансульфонилфенил)пиперидин повышает уровень 5-гидроксииндолуксусной кислоты в полосатом теле с 428±20 (контроли) до 487±7 нг/г ткани при 50 мкмоль/кг подкожно, р<0,05, n=4, и снижает серотонин (5-НТ) с 442±15 (контроли) до 345±18 нг/г ткани при 50 мкмоль/кг подкожно, р<0,05, n=4, и вызывает синдром поведенческой активности обусловленный серотонином (синдром поведенческой активности, обусловлен серотонином, например, Tricklebank et al., 1985, Eur. J. Pharmacol, 106, pp.271-282). 3-(1-Бензилпиперидин-4-ил)фенол обладает нежелательной способностью повышать уровень 5-гидроксииндолуксусной кислоты в полосатом теле с 404±10 нг/г ткани до 492±26 нг/г ткани при 50 мкмоль подкожно, р<0,05, n=4, и снижает серотонин в лимбической области (5-НТ) с 734±8 (контроли) до 677±20 нг/г ткани при 50 мкмоль/кг подкожно, р<0,05, n=4. Сравнительный пример 10: Замещение на основном азоте в соответствии с 2-[4-(3-метансульфонилфенил)пиперазин-1-ил]-этанолом, (описанным в GB 20277030), дает соединения, которые неактивны в тесте поведенческой активности; с 3238±1089 см/60 мин (контроли) до 3782±962 см/60 мин при 33 мкмоль/кг, подкожно р<0,05, n=4, а также в нейрохимическом тесте; воздействует на 3,4-дигидроксифенилуксусную кислоту в полосатом теле; с 1158±126 (контроли) до 1239±162 нг/г ткани при 33 мкмоль/кг подкожно, р<0,05, n=4. Соединения по изобретению особенно пригодны для лечения заболеваний центральной нервной системы и в особенности для лечения заболеваний, опосредованных дофамином. Они могут, например, использоваться для снижения симптомов при нарушении настроения, при ожирении, в качестве аноректического агента и при других нарушениях питания, для улучшения когнитивных функций и связанных с ним эмоциональных расстройств, для улучшения когнитивной и моторной дисфункции, связанной с нарушениями развития, для исправления всех симптомов шизофрении и шизофреноподобных заболеваний, а также и других психозов, для улучшения имеющихся симптомов, а также для предупреждения проявления новых психических проявлений, для регулирования патологических состояний, связанных с употреблением пищи, кофе, чая, алкоголя, лекарственных средств, вызывающих привыкание, и т.д. Таким образом, соединения по изобретению могут использоваться для лечения симптомов при, например: – шизофрении и других психотических заболеваниях, таких как кататоническая, гебефреническая, параноидная, резидуальная или дифференциальная шизофрения; шизофреноподобные заболевания; шизоаффективные заболевания; бредовые состояния; краткие психотические расстройства, психотическое расстройство в результате общего болезненного состояния с бредом и/или галлюцинациями; – расстройства настроения, такие как, например, дистимические расстройства и большие депрессивные расстройства; биполярные расстройства, например, биполярное I расстройство, биполярное II расстройство и циклотимическое расстройство; расстройство настроения в результате общего болезненного состояния с депрессивными и/или маниакальными чертами; и расстройство настроения вызванное определенными веществами; – тревожные расстройства, такие как острое стрессовое расстройство, агорафобия без панического расстройства в эпикризе; тревожное расстройство в результате общего болезненного состояния, генерализованное тревожное расстройство, обсессивно-компульсивное расстройство, паническое расстройство с агарофобией, паническое расстройство без агарофобии, посттравматическое стрессовое расстройство, специфическая фобия, социальная фобия и тревожное расстройство, вызванное определенными веществами; – расстройства питания, такие как нервная анорексия, нервная булимия и ожирение; – расстройства сна, такие как диссомния, например, расстройство сна, связанное с дыханием, расстройство сна с нарушением циркадного ритма, гиперсомния, бессонница, нарколепсия и расстройство нормального циркадного ритма; – не классифицированные расстройства импульсивного контроля, такие как расстройство импульсивного контроля, характеризующееся несколькими отдельными эпизодами потери контроля за агрессивными побуждениями, клептомания, патологическая азартность, пиромания и трихокриптомания; – личностные расстройства, такие как параноидное, шизоидное или шизотипическое расстройство, антисоциальное, пограничное, лицемерие и нарциссизм; и замкнутость, зависимость, обсессивно-компульсивное расстройство; – вызванные медикаментозными средствами расстройства движения, такие как нейролептический индуцированный паркинсонизм, нейролептический злокачественный синдром, нейролептически индуцированная и отдаленная дистония, нейролептически индуцированная акатизия, нейролептически индуцированная отдаленная дискинезия, медикаментозно-индуцированный тремор и медикаментозно-индуцированные дискинезии; – вызванные веществами расстройства, такие как злоупотребление, зависимость, тревожные расстройства, интоксикация, токсический делирий, психотические расстройства, психотические расстройства с бредом, расстройства настроения, стойкое амнестическое расстройство, стойкая деменция, стойкое расстройство восприятия, половые дисфункции, расстройство сна, синдром отмены, абстинентный делирий вследствие злоупотребления алкоголем, амфетамином (или амфетамин-подобными веществами), кофеином, гашишем, кокаином, галлюциногенами, летучими препаратами, никотином, препаратами опия, фенциклидином (или фенциклидин-подобными веществами), седативными веществами, гипнотическими веществами, и/или транквилизаторами; – расстройства, впервые диагносцируемые в младенчестве, детстве или юности, такие как олигофрения, расстройство обучения, расстройство моторных навыков, например, расстройство развития координации; коммуникационные расстройства, например, расстройство выразительности языка, фонологическое расстройство, расстройство восприятия-выражения языка и заикание; распространяющееся расстройство развития, например, болезнь Аспергера, аутизм, детская дезинтеграция, болезнь Ретта, дефицит внимания и разрушительное поведение, например, дефицит внимания/гиперактивность, расстройство поведения и оппозиционное неповиновение; расстройства питания и кормления младенцев или детей раннего возраста, например, расстройство кормления младенцев или детей раннего возраста, пикацизм, расстройство жевания; тики, например, хронический моторный или голосовой тик и расстройство Туретта; другие расстройства младенческого, детского или юношеского возраста, например, избирательный мутизм и стереотипическое расстройство движения; – делирий, деменция, амнестические и другие родственные заболевания, такие как болезнь Альцгеймера, Крейтцфельдта-Якоба, dead травма, болезнь Гентингтона, ВИЧ, болезнь Пика, и диффузная деменция Леви; – конверсивная истерия; – состояния, связанные с нормальным старением, такие как расстройство двигательных функций и ментальных функций; – болезнь Паркинсона и родственные заболевания, такие как множественная системная атрофия, например, стриальная дегенерация, оливопонтоцеребеллярная атрофия, синдром Шая-Джейджера; прогрессивный супрануклеарный паралич; кортикобазальная дегенерация; и сосудистый паркинсонизм; – треморы, такие как существенный, ортостатический, тремор в покое, церебеллярный и вторичный тремор; – головные боли, такие как мигрень, гемикрания, головная боль напряжения и параксизмальная головная боль; – расстройства движения, такие как дискинезии, например, при общих медицинских состояниях, вторичные после травматического или сосудистого инсульта, гемибаллизм, атетозная, хорея Сиденгама и пароксизмальная дискинезия; синдром Экбома (restless legs), болезнь Вильсона, болезнь Галлервордена-Шпатца; – реабилитационное лечение, например, реабилитация после сосудистого или травматического повреждения мозга; – боль при состояниях, характеризующихся повышением мышечного тонуса, таких как фибромиалгия, мышечно-лицевой синдром, дистония и паркинсонизм; а также – состояния, родственные вышеперечисленным, которые попадают в более широкий класс заболеваний, но не соответствуют критериям какого-либо конкретного заболевания в границах этих классов. Синтез Синтез настоящих соединений проводится способами, обычными для синтеза родственных им известных соединений. Синтез соединений формулы 1, в целом, включает взаимодействие промежуточных веществ, содержащих алкильную группу, с промежуточными пиперидином или пиперазином, содержащих амино группу формулы 2:
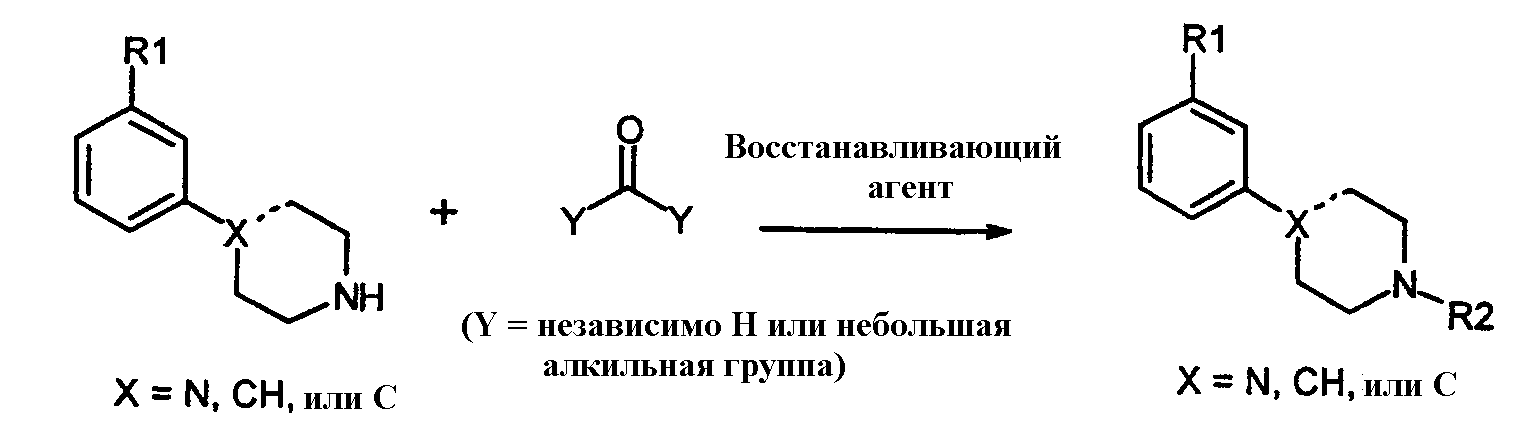
Обычный способ синтеза настоящих соединений состоит в использовании алкилйодида (например, 1-пропил-йодида). Альтернативно, на алкильной группе, конечно, могут использоваться другие уходящие группы кроме йодида, такие как сульфонаты, особенно метансульфонат или толуолсульфонат, группа брома и им подобные. Алкильные промежуточные соединения взаимодействуют с соответствующим амином в присутствии любого подходящего уловителя кислоты. Подходящими уловителями кислоты являются обычные основания, такие как карбонаты, бикарбонаты и гидроксиды щелочных или щелочноземельных металлов, как и некоторые органические основания, такие как триалкиламины и триалканоламины. Реакционной средой для подобных реакций может быть любой подходящий органический растворитель, инертный в щелочных условиях; могут использоваться ацетонитрил, сложные эфиры, такие как этилацетат и ему подобные, и галогенированные алкановые растворители. Обычно реакции проводятся при повышенной температуре реакционной смеси, например, от температуры окружающей среды до температуры кипения с обратным холодильником, в особенности от 50°С до около 100°С. Другой удобный способ синтеза настоящих соединений включает восстановительное аминирование амином формулы 2:
с альдегидом или кетоном, или в присутствии восстанавливающего агента, такого как цианоборгидрид натрия или триацетоксиборгидрид натрия, или с последующим восстановлением, например, используя каталитическое гидрирование, получая соответствующее соединение формулы 1. Соединение формулы 3
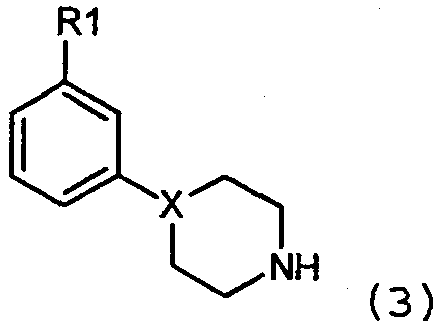
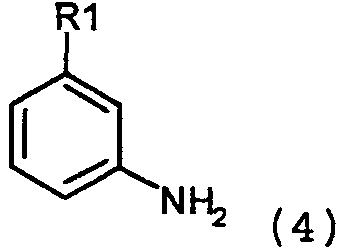
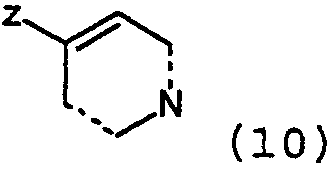
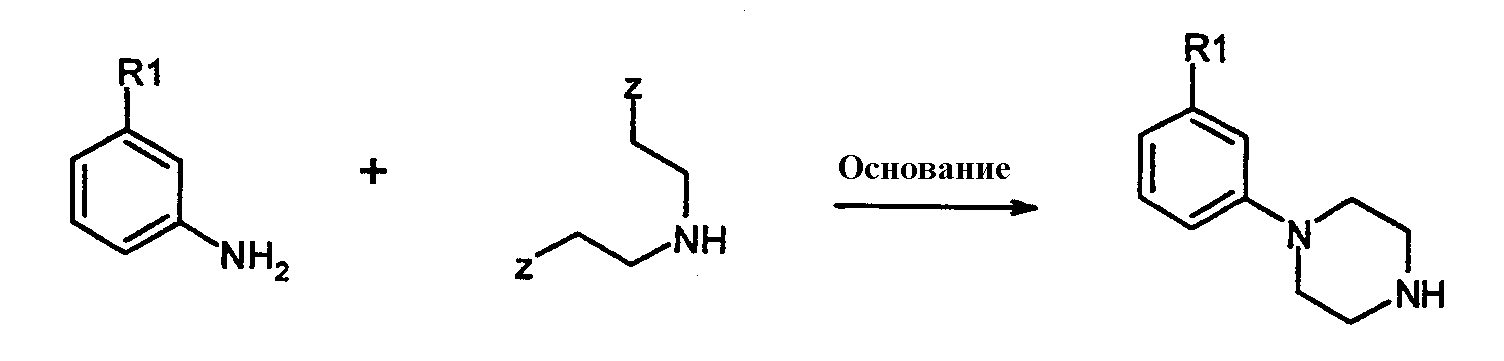
где Х=N, получают взаимодействием соединений формулы 4:
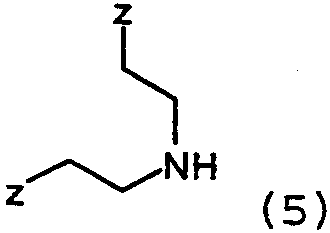
с соединениями формулы 5:
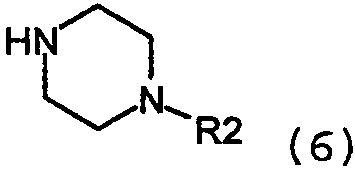
где Z представляет собой уходящую группу, подобную йодиду. Конечно, на алкильной группе могут использоваться и другие уходящие группы, кроме йодида, такие как сульфонаты, в особенности метансульфонат, или толуолсульфонат, группа брома и им подобные. Алкильное промежуточное вещество взаимодействует с соответствующим амином в присутствии любого подходящего уловителя кислоты. Подходящими уловителями кислоты являются обычные основания, такие как карбонаты, бикарбонаты и гидроксиды щелочных или щелочноземельных металлов, как и органические основания, такие как триалкиламины и триалканоламины. Реакцию проводят в подходящем растворителе, таком как н-бутанол, с нагреванием при около 50-150°С. Соединения формулы 1, где Х=N также получают взаимодействием соединений формулы 6:
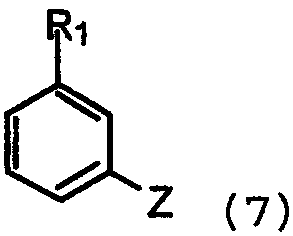
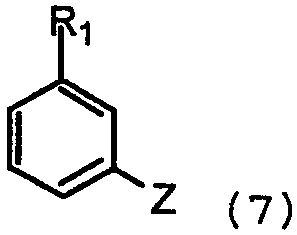
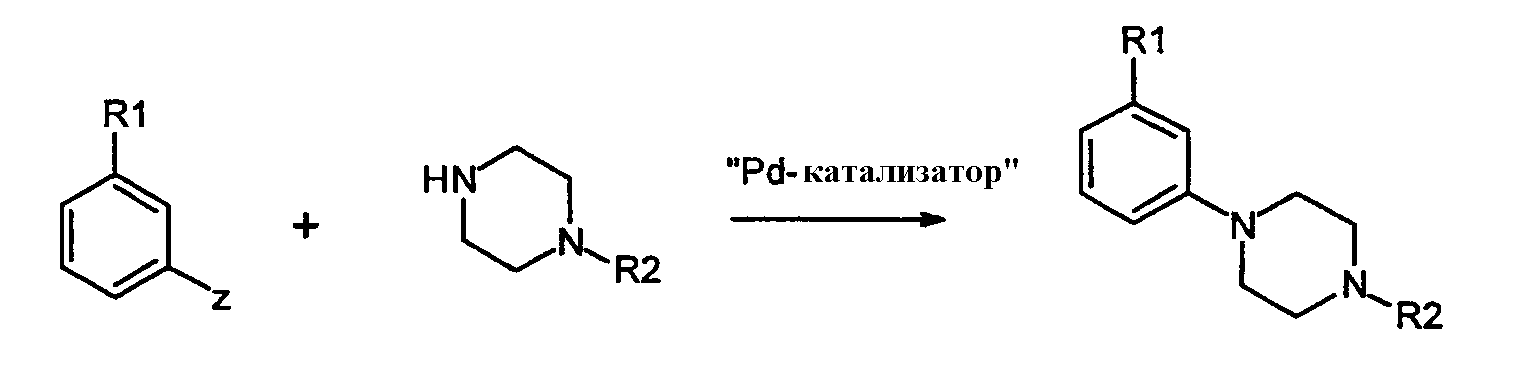
с арилом, замещенным уходящей группой формулы 7:
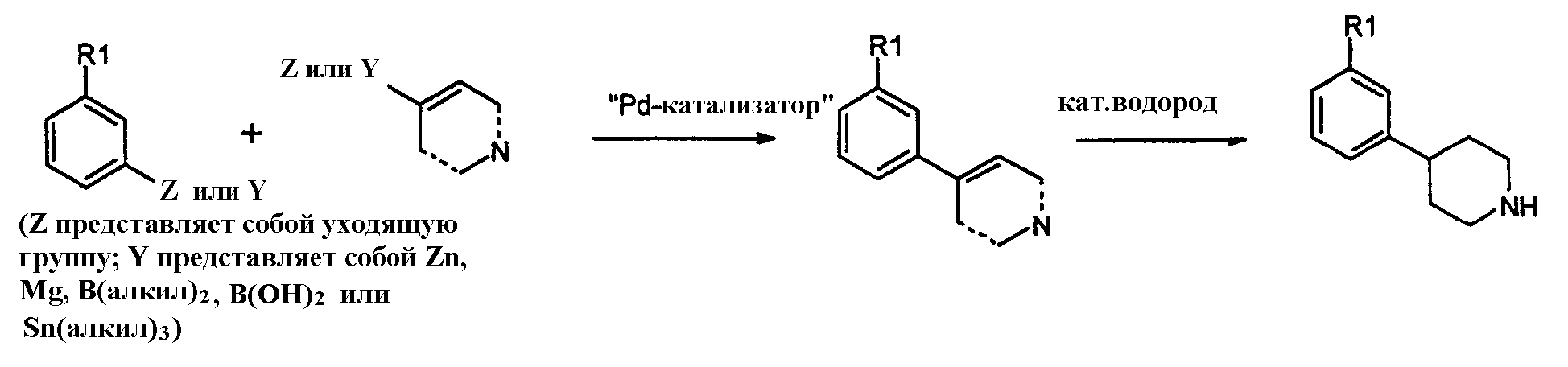
где Z представляет собой галогенид, например хлор, бром, йод, или сульфонат, например, -OSO2CF3 или -OSO2F в присутствии основания и катализатора в виде переходного металла с нулевой валентностью, такого как Pb или Ni, в соответствии с известным способом (Tetrahedron Letters, vol.37, 1996, 4463-4466, J. Org. Chem., vol.61, 1996, 1133-1135). Катализатор, предпочтительно Pd, обладает способностью образовывать лигандный комплекс и подвергаться окислительному присоединению. Типичный Pd катализатор представляет собой Pd2(dba)3, где dba относится к дибeнзилиденацетону), Pd(PPh3)4, Pd(OAc)2 или PdCl2[P(o-tol)3]2, и типичные фосфиновые лиганды представляют собой BINAP, P(o-tol)3, dppf и им подобные. Подходящими уловителями кислоты являются обычные основания, такие как карбонаты, бикарбонаты и алкилоксиды щелочных или щелочноземельных металлов, как и некоторые органические основания, такие как триалкиламины и триалканоламины. Реакционной средой для подобных реакций может быть любой общепринятый органический растворитель, инертный в щелочных условиях; подходящими растворителями являются ацетонитрил, толуол, диоксан, НMП (N-метил-2-пирролидон), ДМЕ (диметоксиэтан), ДМФ (N,N-диметилформамид), ДМСО (диметилсульфоксид) и ТГФ (тетрагидрофуран). Обычно реакции проводят при повышенной температуре реакционной смеси, например, от температуры окружающей среды до температуры кипения с обратным холодильником, особенно от 50°С до около 120°С. Соединения формулы 1, где Х=N, также получают взаимодействием соединений формулы 6 с арилом, замещенным уходящей группой (например, F или Cl), посредством реакции нуклеофильного ароматического замещения в присутствии основания, как объяснялось выше. Соединения формулы 1, где Х=СН, также получают путем катализируемой переходным металлом реакции перекрестного связывания, известной специалистам в данной области техники как, например, реакции Suzuki и Stille. Реакции могут проводиться между соединениями формулы 8:
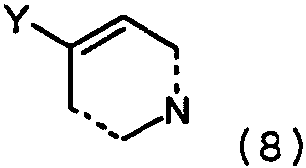
где Y представляет собой, например, диалкилборан, диалкенилборан или бороновую кислоту (например, BЕt2, B(OH)2 (пунктирные линии могут представлять собой двойные связи) или триалкиламин (например, SnMe3, SnBu3), и арил, замещенный уходящей группой формулы 7:
(определение Z смотри выше) в присутствии основания и катализатора в виде переходного металла с нулевой валентностью, такого как Pd или Ni, в соответствии с известными способами (Chem. Pharm. Bull., vol.33, 1985, 4755-4763, J. Am. Chem. Soc., vol.109, 1987, 5478-5486, Tetrahedron Lett., vol.33, 1992, 2199-2202). Кроме того, Y может представлять собой цинк- или магнийгалогенидную группу (например, ZnCl2, ZnBr2, ZnI2, MgBr2, MgI2) в соответствии с известными способами (Tetrаhedron Lett., vol.33, 1992, 5373-5374, Tetrahedron Lett., vol.37, 1996, 5491-5494). Катализатор, предпочтительно Pd, обладает способностью образовывать лигандный комплекс и подвергаться окислительному присоединению. Определение лигандов, оснований и растворителей представлено выше. Альтернативно, катализируемые переходным металлом реакции перекрестного связывания проводятся с противоположной картиной замещения:
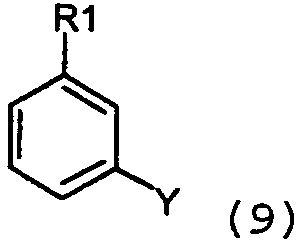
гетероарилом/алкенилом, замещенными уходящей группой формулы 10:
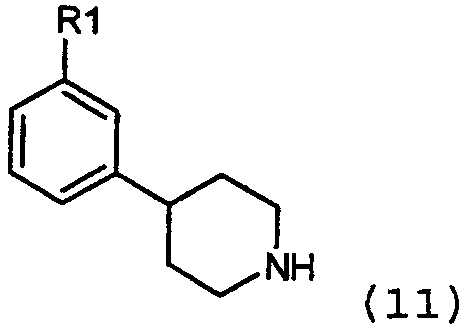
в присутствии основания и катализатора в виде переходного металла с нулевой валентностью, такого как Pd и Ni, в соответствии с известными способами, обсужденными в предыдущем разделе. Соединения формулы 11:
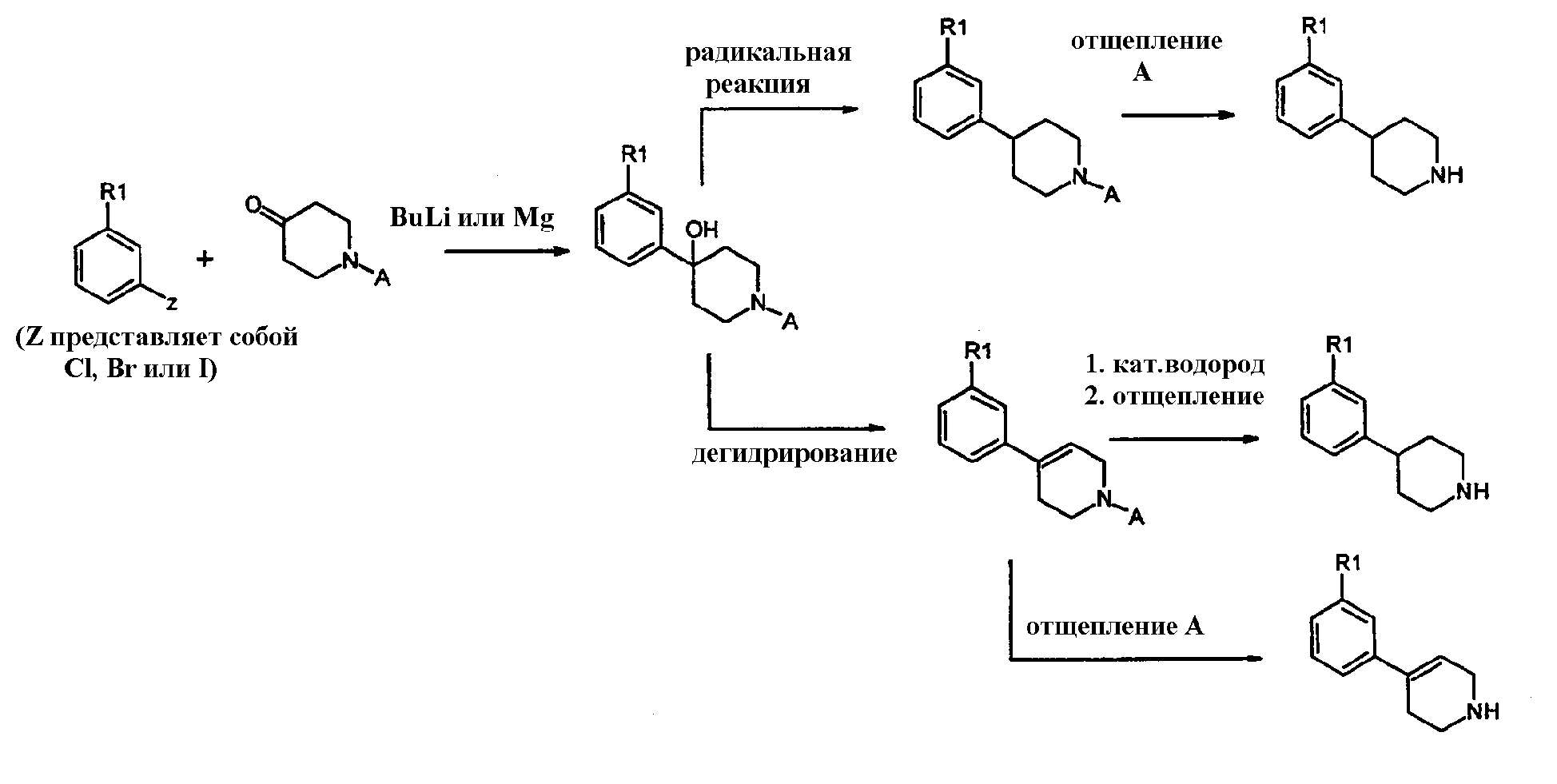
могут быть получены каталитическим гидрированием тетрагидропиридина или пиридина из предыдущего раздела, используя стандартный способ, известный в уровне техники, обычно с палладием на углероде, PtO2 или никелем Ренея в качестве катализатора. Реакцию проводят в инертном растворителе, таком как этанол или этилацетат, в присутствии или без протоновой кислоты, такой как уксусная кислота или НСl. Когда пиридиновое кольцо является кватернизованным алкильной группой, кольцо может быть частично восстановлено NaBH4 или NaCNBH4, давая тетрагидропиридиновый аналог, который может далее восстанавливаться каталитическим гидрированием. Другой удобный способ синтеза соединений формулы 1, где Х=СН, также осуществляется обработкой арилгалогенидов формулы 7:
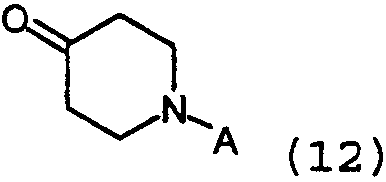
где Z представляет собой Cl, Br или I, с алкиллитиевыми реагентами, например, бутиллитием, втор-бутиллитием или трет-бутиллитием, предпочтительно бутиллитием, или Mg (реакция Гриньяра) в инертном растворителе. Подходящие растворители включают, например, эфир или тетрагидрофуран, предпочтительно тетрагидрофуран. Температура реакции находится в интервале от около -110°С до около 60°С. Образованные подобным образом промежуточные анионы лития или анионы магния могут затем далее взаимодействовать с подходящим электрофилом формулы 12:
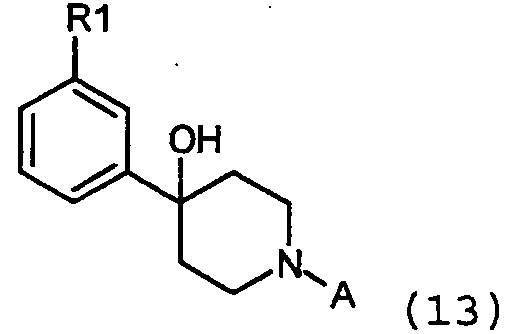
где А определяется, как защитная группа, подобная t-Boc (трет-бутоксикарбонил), Fmoc (флуоренилметоксикарбонил), Cbz (бензилоксикарбонил) или алкильная группа, подобная бензилу. Необходимо, чтобы гидроксигруппа образованных промежуточных веществ формулы 13:
была удалена таким образом, чтобы в результате получилось соединение формулы 1 (Х=СН). Данная стадия может проводиться одним из нескольких стандартных способов, известных в уровне техники. Например, тиокарбонильное производное (например, ксантат) может быть получено и удалено свободнорадикальным способом, известным в области техники. Альтернативно, гидрокси группа может быть удалена восстановлением источником гидрида, таким как триэтилсилан, в кислотных условиях, используя, например, трифторуксусную кислоту или бортрифторид. Реакция восстановления может проводиться без разведения или в растворителе, таком как метиленхлорид. Еще одним альтернативным способом является сначала превращение гидроксильной группы в подходящую уходящую группу, такую как тозилат или хлорид, используя стандартные способы. Уходящая группа затем удаляется с помощью нуклеофильного гидрида, такого как, например, литий алюминий гидрид. Эта последняя реакция проводится обычно в инертном растворителе, таком как эфир или тетрагидрофуран. Другой альтернативный способ удаления гидроксильной группы состоит сначала в дегидрировании спирта до олефина такими реагентами, как соль Бургеса (J. Org. Chem., vol.38, 1973, 26) с последующим каталитическим гидрированием двойной связи при стандартных условиях с таким катализатором, как палладий на углероде. Спирт также может быть дегидрирован до олефина обработкой кислотой, такой как пара-толуолсульфокислота или трифторуксусная кислота. Защитная группа А удаляется при стандартных условиях, известных специалистам в данной области. Например, t-Boc расщепление обычно проводится с трифторуксусной кислотой или чистой, или в сочетании с метиленхлоридом. F-moc обычно отщепляется простым основанием, таким как аммиак, пиперидин или морфолин, обычно в полярном растворителе, таком как ДМФ и ацетонитрил. Когда А представляет собой Cbz или бензил, они обычно отщепляются в условиях каталитического гидрирования. Бензильная группа также может отщепляться в условиях N-деалкилирования, такого как обработка Кроме того, возможно превратить радикал R1 в соединении формулы 1 в другой радикал R1, например, окислением метилсульфида в метилсульфон (например, m-хлорпероксибензойной кислотой), замещением трифлатной или галогенидной группы цианогруппой (например, катализируемым палладием цианированием), замещением трифлатной или галогенидной группы кетоном (например, катализируемой палладием Heck реакцией бутилвиниловым эфиром), замещением трифлатной или галогенидной группы карбоксамидом (например, катализируемым палладием карбонилированием) или расщеплением или, например, превращением метокси группы в соответствующее гидроксильное производное, которое может далее превращаться в соответствующий мезилат или трифлат. Термины мезилат и трифлат относятся к OSO2CH3, CH3SO3 или OSO2CF3, CF3SO3, соответственно. В целом можно сказать, что общий процесс получения настоящих соединений имеет шесть основных вариантов, которые могут быть вкратце описаны следующим образом: в соответствии со схемой 1:
или в соответствии со схемой 2:
или в соответствии со схемой 3:
или в соответствии со схемой 4:
или в соответствии со схемой 5:
или в соответствии со схемой 6:
Как используется здесь, термин С1-С4 алкил относится к алкилу, содержащему 1-4 атома углерода в любой изомерной форме. Различные углеродные фрагменты определяются следующим образом: алкил относится к алифатическому углеводородному радикалу и включает формы с разветвленной и неразветвленной цепью, такие как метил, этил, н-пропил, изо-пропил, н-бутил, изо-бутил, втор-бутил, трет-бутил. Термин циклоалкил относится к радикалу насыщенного циклического углеводорода, такого как циклопропил, циклобутил, циклопентил, циклогексил. Термин «больной», используемый здесь, относится к индивидууму, нуждающемуся в лечении в соответствии с изобретением. Термин «лечение», используемый здесь, относится как к лечению с целью излечивания или облегчения заболевания или состояния, так и к лечению с целью предупреждения развития заболевания или состояния. Лечение может проводиться как в острой, так и хронической форме заболевания. Как органические, так и неорганические кислоты могут использоваться для образования нетоксических фармацевтически приемлемых аддитивных солей кислот соединений по изобретению. Примерами кислот являются серная, азотная, фосфорная, хлористоводородная, лимонная, уксусная, молочная, винная, пальмовая, этандисульфоновая, сульфамовая, сукциновая, циклогексилсульфамовая, фумаровая, малеиновая и бензойная кислота. Эти соли легко получить способами, известными в области техники. Фармацевтические композиции, содержащие соединение по изобретению также могут содержать вещества, использующиеся для облегчения производства фармацевтического препарата или введения препаратов. Такие вещества хорошо известны специалистам в этой области и могут, например, быть фармацевтически приемлемыми адъювантами, носителями и консервантами. В клинической практике соединения, используемые в соответствии с настоящим изобретением, обычно вводятся орально, ректально или путем инъекций, или в форме фармацевтических препаратов, включающих активный ингредиент, или в форме свободного основания, или в виде фармацевтически приемлемой нетоксичной аддитивной соли кислоты, такой как хлористоводородная, молочная, уксусная, сульфаматная соль, в соединении с фармацевтически приемлемым носителем. Носитель может быть твердым, полутвердым или жидким. Обычно активное вещество составляет от 0,1 до 99% по весу от веса препарата, более характерно от 0,5 до 20% от веса препарата, предназначенного для инъекций, и от 0,2 до 50% от веса препарата, пригодного для орального введения. Для получения фармацевтических препаратов, содержащих соединение по изобретению, в виде единичной дозированной формы для орального введения выбранное соединение может быть смешано с твердым эксципиентом, например, лактозой, сахарозой, сорбитом, маннитом, крахмалом, таким как картофельный крахмал, кукурузный крахмал или амилопектин, производными целлюлозы, связывающим веществом, таким как желатин или поливинилпирролидин, и смазывающим агентом, таким как стеарат магния, стеарат кальция, полиэтиленгликоль, воски, парафин и им подобные, и затем спрессовано в таблетки. Если требуются таблетки с покрытием, содержимое, полученное как описано выше, может быть покрыто концентрированным раствором сахара, который может содержать, например, гуммиарабик, желатин, тальк, диоксид титана и им подобные. Альтернативно, таблетки могут быть покрыты полимером, известным специалистам в этой области, растворенным в легко летучем органическом растворителе или смешанным с органическими растворителями. Красители могут добавляться к этим покрывающим веществам для облегчения различия между таблетками, содержащими различные активные вещества или различные количества активного соединения. Для получения мягких желатиновых капсул активное вещество может быть смешано с, например, растительным маслом или полиэтиленгликолем. Твердые желатиновые капсулы могут содержать гранулы активного вещества, используя или упомянутые эксципиенты для таблеток, например, лактозу, сахарин, сорбит, маннит, крахмалы (например, картофельный крахмал, кукурузный крахмал или амилопектин), производные целлюлозы или желатин. Также жидкие или полужидкие лекарственные формы могут наполнять твердые желатиновые капсулы. Дозированные единицы для ректального введения могут быть растворами или суспензиями, или могут быть получены в форме суппозиториев, содержащих активное вещество в смеси с нейтральным жирным основанием, или желатиновых ректальных капсул, содержащих активное вещество в смеси с растительным маслом или парафиновым маслом. Жидкие препараты для орального введения могут быть в форме сиропов или суспензий, например, растворы, содержащие от 0,2% до около 20% по весу активного описанного здесь вещества, остальное составляет сахар и смесь этанола, воды, глицерина и пропиленгликоля. Необязательно такие жидкие препараты могут содержать красящие агенты, ароматизаторы, сахарин и карбоксиметилцеллюлозу в качестве загустителя или другие эксципиенты, известные специалистам в данной области. Растворы для парентерального введения путем инъекций могут быть получены в водном растворе водорастворимых фармацевтически приемлемых солей активного вещества, предпочтительно в концентрации от 0,5% до около 10% по весу. Эти растворы могут также содержать стабилизирующие агенты и/или буферные агенты и могут быть легко обеспечены в ампулах в виде различных дозированных единиц. Использование и введение больному, нуждающемуся в лечении в клинике очевидно для специалиста в данной области. При терапевтическом лечении эффективное количество или терапевтическое количество соединений по изобретению составляет от около 0,01 до около 500 мг/кг веса тела в день, предпочтительно 0,1-10 мг/кг веса тела в день. Соединения могут вводиться любым подходящим путем, таким как пероральный или парентеральный. Дневная доза предпочтительно вводится в индивидуальной дозировке 1-4 раза в день. Специалистам в данной области известно, что замещение водорода в незамещенном положении в ароматическом кольце атомом фтора может блокировать возможность ферментативного гидроксилирования, которое дает соединение с низкой оральной биодоступностью. Подобный вид обмена (H на F) редко изменяет фармакологический профиль. Так, в некоторых случаях для улучшения оральной биодоступности важно ввести атом фтора в незамещенное положение в ароматическом кольце соединения формулы 1. Далее изобретение иллюстрируется приведенными ниже примерами, которые никоим образом не предназначены для ограничения объема изобретения. Пример 1 1-(3-метансульфонилфенил)-4-пропилпиперазин Суспензию 1-(3-метансульфонилфенил)пиперазина (350 мг) и размолотого К3СО3 (403 мг) перемешивают в CH3CN (25 мл) при комнатной температуре. Добавляют 1-йодпропан (712 мкл). Смесь кипятят с обратным холодильником в течение ночи. Реакционную смесь фильтруют, и летучие вещества выпаривают в вакууме. Маслянистый остаток хроматографируют на колонке окиси кремния с MeOH:CH2Cl2 (1:30 (об/об)) в качестве элюента. Сбор фракций, содержащих чистый продукт, и выпаривание растворителя дает чистый 1-(3-метансульфонилфенил)-4-пропилпиперазин (220 мг). Амин превращают в НСl соль и перекристаллизовывают из этанола/диэтилового эфира: т.пл. 233°С. МС m/z (относительная интенсивность, 70 эВ) 282 (M+, 30), 254 (15), 253 (bp), 210 (17), 70 (21). Следующие соединения в соответствии с примерами 2-11 получают таким же способом, который описан в примере 1. Пример 2 1-пропил-4-(3-трифторметансульфонилфенил)пиперазин МС m/z (относительная интенсивность, 70 эВ) 336 (М+, 16), 307 (bp), 77 (18), 70 (38), 56 (23). Пример 3 1-[3-(4-пропилпиперазин-1-ил)фенил]этанон Исходя из 1-(3-пиперазин-1-ил-фенил)этанона и n-Pr-I: т.пл. 119°С (оксалат), МС m/z (относительная интенсивность, 70 эВ) 246 (M+, 10), 217 (33), 132 (18), 70 (bp), 56 (41); Rf 0,23 (EtOAc). Пример 4 1-пропил-4-(3-трифторметилфенил)пиперидин Исходя из 4-(3-трифторметилфенил)пиперидина и n-Pr-I: т.пл. 195°С (НСl), МС m/z (относительная интенсивность, 70 эВ) 271 (M+, 4), 243 (16), 242 (bp), 159 (13), 70 (49). Пример 5 1-бутил-4-(3-трифторметилфенил)пиперидин Исходя из 4-(3-трифторметилфенил)пиперидина и n-Bu-Br: т.пл. 222°С (НСl), МС m/z (относительная интенсивность, 70 эВ) 285 (M+, 3), 243 (12), 242 (bp), 70 (51), 56(17). Пример 6 4-(3-метансульфонилфенил)-1-пропилпиперидин Т.пл. 200°С (НСl), МС m/z (относительная интенсивность, 70 эВ) 281 (M+, 5), 252 (bp), 129 (20), 115 (20), 70 (25). Пример 7 4-(3-метансульфонилфенил)-1-пропил-1,2,3,6-тетрагидропиридин Исходя из 4-(3-метансульфонилфенил)-1,2,3,6-тетрагидропиридина и йодпропана: МС m/z (относительная интенсивность, 70 эВ) 279 (M+, 26), 250 (bp), 171 (6), 128 (12), 115 (8). Пример 8 4-(3-метансульфонилфенил)-1-этилпиперидин Исходя из 4-(3-метансульфонилфенил)пиперидина и йодэтана: т.пл.158°С (НСl). МС m/z (относительная интенсивность, 70 эВ) 267 (M+, 20), 252 (bp), 130 (10), 115 (12), 84 (20). Пример 9 1-изопропил-4-(3-метансульфонилфенил)пиперидин Исходя из 4-(3-метансульфонилфенил)пиперидина и изопропилбромида: т.пл. 220°С (НСl). МС m/z (относительная интенсивность, 70 эВ) 281 (M+, 4), 266 (bp), 187 (5), 129 (5), 115 (5). Пример 10 4-(3-метансульфонилфенил)-1-бутилпиперидин Исходя из 4-(3-метансульфонилфенил)пиперидина и n-BuCl. МС m/z (относительная интенсивность, 70 эВ) 295 (M+, 3), 252 (bp), 130 (5), 115 (3), 70 (8). Пример 11 1-изобутил-4-(3-метансульфонилфенил)пиперидин Исходя из 4-(3-метансульфонилфенил)пиперидина и изобутилбромида: т.пл. 212°С (НСl). МС m/z (относительная интенсивность, 70 эВ) 295 (M+, 1), 252 (80), 129 (40), 115 (50), 70 (bp). Пример 12 3-(1-пропилпиперидин-4-ил)бензонитрил Раствор 3-(1-пропилпиперидин-4-ил)бензамида (350 мг) и POCl3 (326 мкл) в сухом ДМФ (6 мл) нагревают при 80oС в течение 3 часов в атмосфере аргона. Выпаривание растворителя дает темный маслянистый остаток, который растворяют в воде. Раствор подщелачивают и экстрагируют СН2Сl2. Объединенные органические фазы сушат (MgSO4), фильтруют и выпаривают. Маслянистый остаток хроматографируют на колонке окиси кремния с МеОН:СН2Сl2 (1:19 (об/об)) в качестве элюента. Сбор фракций, содержащих чистый продукт, и выпаривание растворителя дает чистый 3-(1-пропилпиперидин-4-ил)бензонитрил (127 мг). Амин превращается в фумаратную соль и перекристаллизуется из этанола/диэтилэфира: т.пл. 122oС; МС m/z (относительная интенсивность, 70 эВ) 228 (M+, 2), 199 (42), 129 (26), 70 (bp), 56 (53). Пример 13 1-втор-бутил-4-(3-метансульфонилфенил)пиперидин 4-(3-метансульфонилфенил)пиперидина гидрохлорид (20 мг), ледяную уксусную кислоту (4,4 мг) и 2-бутанон (5,1 мг) смешивают в 1,2-дихлорэтане (5 мл). К раствору добавляют триацетоксиборгидрид (23,5 мг), и реакционную смесь перемешивают при комнатной температуре в атмосфере азота в течение 5 часов (G.L.C. анализ показывает завершение реакции). Реакцию гасят насыщенным водным NaHCO3, и продукт экстрагируют СН2Сl2. Объединенные органические фазы сушат (MgSO4), фильтруют, и растворитель выпаривают, получая 1-втор-бутил-(3-метансульфонилфенил)пиперидин в виде маслянистого остатка. Продукт хроматографируют на колонке окиси кремния с СН2Сl2:МеОН (9:1 (об/об)) в качестве элюента. Сбор фракций, содержащих чистый продукт, и выпаривание растворителя дают чистый амин (15 мг, 71%); МС m/z (относительная интенсивность, 70 эВ) 295 (M+, 1), 280 (7), 266 (bp), 187 (4), 129 (4). Пример 14 3-(1-пропилпиперидин-4-ил)фениловый сложный эфир метансульфоновой кислоты Раствор 3-(1-пропилпиперидин-4-ил)фенола (340 мг) и триэтиламина (187 мг) в 20 мл СН2Сl2 охлаждают до 0oС. Затем по каплям добавляют метансульфонилхлорид (194 мг), растворенный в 10 мл СН2Сl2. Реакционной смеси позволяют нагреться до комнатной температуры и затем перемешивают в течение 2,5 часов при 25oС. Наконец, реакцию гасят водой. Органические слои разделяют и промывают 10% НСl и затем 10% Na2CO3. После сушки (MgSO4) растворитель удаляют при пониженном давлении. Остаток хроматографируют на колонке окиси кремния, используя МеОН:СН2Сl2 (1:9 (об/об)) в качестве элюента. Фракции, содержащие чистый 3-(1-пропилпиперидин-4-ил)фениловый эфир метансульфоновой кислоты собирают, и растворитель удаляют в вакууме, получая 206 мг названного в заголовке соединения. (МС m/z (относительная интенсивность, 70 эВ) 297 (M+, 3), 268 (bp), 189 (24), 131 (13), 79 (16). Следующие соединения в примерах 15-19 получают таким же образом, как описано в примере 14. Пример 15 3-(1-этилпиперидин-4-ил)фениловый сложный эфир метансульфоновой кислоты Исходя из 3-(1-этилпиперидин-4-ил)фенола и метансульфонилхлорида: МС m/z (относительная интенсивность, 70 эВ) 283 (M+, 6), 268 (bp), 189 (54), 131 (20), 79 (70). Пример 16 3-(1-бутилпиперидин-4-ил)фениловый сложный эфир метансульфоновой кислоты Исходя из 3-(1-бутилпиперидин-4-ил)фенола и метансульфонилхлорида: МС m/z (относительная интенсивность, 70 эВ) 311 (M+, 3), 268 (bp), 189 (20), 131 (18), 79 (12). Пример 17 3-(4-пропилпиперазин-1-ил)фениловый сложный эфир метансульфоновой кислоты Исходя из 3-(4-пропилпиперазин-1-ил)фенола и метансульфонилхлорида: т.пл. 143-144°С (фумарат); МС m/z (относительная интенсивность, 70 эВ) 298 (M+, 35), 269 (95), 121 (25), 84 (30), 70 (bp); Пример 18 3-(1-пропилпиперидин-4-ил)фениловый сложный эфир трифторметансульфоновой кислоты Исходя из 3-(1-пропилпиперидин-4-ил)фенола и трифлата ангидрида: МС m/z (относительная интенсивность, 70 эВ) 351 (M+, 4), 322 (65), 189 (30), 131 (20), 69 (bp). Пример 19 3-(1-этилпиперидин-4-ил)фениловый сложный эфир трифторметансульфоновой кислоты Исходя из 3-(1-этилпиперидин-4-ил)фенола и ангидрида трифлата: МС m/z (относительная интенсивность, 70 эВ) 337 (M+, 4), 322 (65), 189 (30), 131 (20), 69 (bp). Пример 20 1-[3-(1-пропилпиперидин-4-ил)фенил]этанон К перемешиваемому раствору 3-(1-пропилпиперидин-4-ил)фенилового сложного эфира трифторметансульфоновой кислоты (300 мг) в ДМФ (4 мл) в атмосфере аргона при комнатной температуре последовательно добавляют NЕt3 (356 мкл), бутилвиниловый эфир (823 мкл) 1,3-бис(дифенилфосфино)пропан (50 мг) и Pd(OАc)2 (19 мг). Полученную в результате смесь затем нагревают до 80°С, и через 2 часа реакцию останавливают. Добавляют 5% раствор хлористоводородной кислоты (6 мл), и объединенную смесь перемешивают в течение 45 минут. Затем добавляют СН2Сl2, и фазы разделяют. Водный слой затем экстрагируют СН2Сl2. Объединенные органические фазы сушат (MgSO4), фильтруют и выпаривают досуха. Сырой продукт очищают флэш-хроматографией (МеОН:СН2Сl2 (1:9 (об/об)). Сбор фракций, содержащих чистый продукт, и выпаривание растворителя дает чистый 1-[3-(1-пропилпиперидин-4-ил)фенил]этанон (35 мг). МС m/z (относительная интенсивность, 70 эВ) 245 (M+, 4), 216 (bp), 100 (19), 70 (36), 57 (13). Пример 21 1-пропил-4-(3-трифторметилсульфонилфенил)-1,2,3,6-тетрагидропиридин 4-(3-трифторметилсульфонилфенил)пиридин (0,3 г) растворяют в 1-йодпропане (2 мл) и нагревают до 100°С в течение 2 часов. Затем летучие вещества выпаривают, и остаток растворяют в абс. EtOH (20 мл), и NaBH4 (340 мг) добавляют по порциям при -20°С. Смеси затем дают нагреться до комнатной температуры и перемешивают в течение ночи. К смеси добавляют 10% раствор Na2СO3 (20 мл). Водный слой экстрагируют СН2Сl2, и объединенные органические фазы сушат (MgSO4), фильтруют и выпаривают досуха. Сырой продукт очищают флэш-хроматографией (МеОН:СН2Сl2 (1:9 (об/об)). Сбор фракций, содержащих чистый продукт, и выпаривание растворителя дает чистый 1-пропил-4-(3-трифторметилсульфонилфенил)-1,2,3,6-тетрагидропиридин (150 мг). МС m/z (относительная интенсивность, 70 эВ) 333 (M+, 21), 305 (16), 304 (bp), 171 (14), 128 (14). Rf 0,55 (МеОН). Пример 22 1-пропил-4-(3-трифторметилсульфонилфенил)пиперидин Исходя из 1-пропил-4-(3-трифторметилсульфонилфенил)-1,2,3,6-тетрагидропиридина, 1-пропил-4-(3-трифторметилсульфонилфенил)пиперидин выделяют способом, описанным в препаративном примере 9. МС m/z (относительная интенсивность, 70 эВ) 335 (M+, 3), 307 (17), 306 (bp), 173 (26), 70 (10). Пример 23 1-аллил-4-(3-метансульфонилфенил)пиперидин Исходя из 4-(3-метансульфонилфенил)пиперидина и аллилбромида названное в заголовке соединение выделяют способом, описанным в примере 1. МС m/z (относительная интенсивность, 70 эВ) 279 (M+, 74), 96 (bp), 82 (98), 68 (74), 55 (93). Rf=0,42 (МеОН, 0,08 EtOAc). Пример 24 4-(3-метансульфонилфенил)-1-тетрагидрофуран-2-илметил)пиперидин Исходя из 4-(3-метансульфонилфенил)пиперидина и тетрагидрофурфурилхлорида названное в заголовке соединение выделяют способом, описанным в примере 1. МС m/z (относительная интенсивность, 70 эВ) 323 (M+, 1), 252 (bp), 129 (9), 115 (6), 70 (17). Rf=0,3 (МеОН, 0,03 EtOAc). Синтез промежуточных продуктов, используемых в вышеприведенных примерах, описан ниже в препаративных примерах. Препаративный пример 1 Трет-бутиловый сложный эфир 4-гидрокси-4-(3-метилсульфанилфенил)пиперидин-1-карбоновой кислоты 1-бром-3-метилсульфанилбензол (5,0 г, 24,6 ммоль) растворяют в сухом ТГФ (40 мл) и охлаждают до -78°С в потоке аргона (г). n-BuLi (12,8 мл, 2,5 М в гексане, 31,9 моль) добавляют по каплям через шприц, и реакционную смесь перемешивают еще 30 минут при -78°С, затем температуру повышают до 0°С в течение 5 минут и затем понижают до -78°С. Через шприц добавляют 1-трет-бутоксикарбонил-4-пиперидон (5,4 г, 27,06 ммоль), растворенный в сухом ТГФ (30 мл). Реакционную смесь доводят до комнатной температуры и затем перемешивают в течение 1 часа, и наконец, гасят насыщенным раствором хлорида аммония (30 мл). Смесь экстрагируют несколько раз EtOAc, и объединенные органические фазы сушат (MgSO4), фильтруют и выпаривают досуха. Маслянистый остаток хроматографируют на колонке окиси кремния, используя СН2Сl2:МеОН (19:1 (об/об)) в качестве элюента, получая трет-бутиловый сложный эфир 4-гидрокси-4-(3-метилсульфанилфенил)пиперидин-1-карбоновой кислоты (6 г, 76%). МС m/z (относительная интенсивность, 70 эВ) 323,1 (M+, 6), 223,0 (11), 178,0 (7), 152 (3), 57,0 (bp), 56 (30). Препаративный пример 2 1-бензил-4-(3-метоксифенил)пиперидин-4-ол Исходя из 3-броманизола (5 г) и 1-бензил-4-пиперидона (5,5 г), выделяют 4,58 г 1-бензил-4-(3-метоксифенил)пиперидин-4-ола способом, описанным в примере 1. МС m/z (относительная интенсивность, 70 эВ) 297 (M+, 8), 279 (13), 206 (28), 146 (17), 91 (bp). Препаративный пример 3 1-бензил-4-(3-трифторметилфенил)пиперидин-4-ол Исходя из 3-трифторметилйодбензола (3 г) и 1-бензил-4-пиперидона (2,1 г) выделяют 1,75 г названного в заголовке соединения способом, описанным в примере 1. МС m/z (относительная интенсивность, 70 эВ) 335 (M+, 29), 244 (22), 146 (19), 91 (bp), 56 (19). Препаративный пример 4 4-(3-метилсульфанилфенил)-1,2,3,6-тетрагидропиридин Трет-бутиловый сложный эфир 4-гидрокси-4-(3-метилсульфанилфенил)пиперидин-1-карбоновой кислоты (3,97 г) растворяют в СН2Сl2 (500 мл), и одной порцией добавляют трифторуксусную кислоту (80 мл). Смесь кипятят с обратным холодильником в течение 1 часа и затем промывают двумя порциями 10% Na2CO3, сушат (MgSO4), фильтруют и выпаривают досуха. Выход 2,07 г. МС m/z (относительная интенсивность, 70 эВ) 205 (M+, 73), 158 (44), 129 (95), 128 (80), 82 (bp). Препаративный пример 5 1-бензил-4-(3-метоксифенил)-1,2,3,6-тетрагидропиридин Исходя из 1-бензил-4-(3-метоксифенил)пиперидин-4-ола (4,5 г) и трифторуксусной кислоты (80 мл), 3,5 г 1-бензил-4-(3-метоксифенил)-1,2,3,6-тетрагидропиридина выделяют способом, описанным в препаративном примере 4. МС m/z (относительная интенсивность, 70 эВ) 279 (M+, 35), 145 (13), 115 (15), 91 (bp), 65 (22). Препаративный пример 6 1-бензил-4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин Исходя из 1-бензил-4-(3-трифторметилфенил)пиперидин-4-ола (1,74 г) выделяют 1,44 г названного в заголовке соединения способом, описанным в препаративном примере 4 (чистый СF3COOH). МС m/z (относительная интенсивность, 70 эВ) 317 (M+, 71), 226 (13), 172 (15), 91 (bp), 65 (17). Препаративный пример 7 Метиловый сложный эфир 4-(3-метилсульфанилфенил)-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты 4-(3-метилсульфанилфенил)-1,2,3,6-тетрагидропиридин (2 г) и NEt3 (1 г) растворяют в СН2Сl2 (75 мл) и охлаждают до 0oС. По каплям добавляют метилхлорформиат (0,96 г), растворенный в СН2Сl2 (20 мл), и реакционной смеси дают нагреться до комнатной температуры. Еще через два часа при комнатной температуре реакционную смесь промывают 10% раствором Na2CO3, сушат (MgSO4), фильтруют и концентрируют выпариванием. Маслянистый остаток хроматографируют на колонке окиси кремния, используя СН2Сl2:МеОН (19:1 (об/об)) в качестве элюента, получая метиловый сложный эфир 4-(3-метилсульфанилфенил)-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты (1,4 г). МС m/z (относительная интенсивность, 70 эВ) 263 (M+, 45), 248 (89), 129 (83), 128 (bp), 59 (96). Препаративный пример 8 Метиловый сложный эфир 4-(3-метансульфонилфенил)-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты Метиловый сложный эфир 4-(3-метилсульфанилфенил)-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты (1,4 г) растворяют в СН2Сl2 (150 мл) и охлаждают до 0oС. По порциям добавляют м-хлорпероксибензойную кислоту (2,48 г), и смесь перемешивают при комнатной температуре в течение трех часов. Полученный в результате прозрачный раствор промывают 10% раствором Na2CO3, сушат (MgSO4), фильтруют и концентрируют выпариванием, и получают маслянистый остаток (1,3 г). МС m/z (относительная интенсивность, 70 эВ) 295 (M+, 19), 280 (56), 129 (70), 128 (89), 59 (bp). Препаративный пример 9 Метиловый сложный эфир 4-(3-метансульфонилфенил)пиперидин-1-карбоновой кислоты Метиловый сложный эфир 4-(3-метансульфонилфенил)-3,6-дигидро-3Н-пиридин-1-карбоновой кислоты (2,0 г) растворяют в метаноле (40 мл). Добавляют концентрированную хлористоводородную кислоту (2 мл) и Pd/C (500 мг). Полученную в результате смесь гидрируют, под давлением, созданным газообразным водородом (50 фунт/кв. дюйм) в течение 8 часов и затем фильтруют через слой целита. Растворитель выпаривают в вакууме, и остаток очищают флэш-хроматографией (СН2Сl2:МеОН, 3:1 (об/об)). Выход 0,92 г. МС m/z (относительная интенсивность, 70 эВ) 297 (M+, 54), 282 (62), 238 (bp), 115 (92), 56 (93). Препаративный пример 10 4-(3-метоксифенил)пиперидин Исходя из 1-бензил-4-(3-метоксифенил)-1,2,3,6-тетрагидропиридина (5,1 г) и 900 мг Pd/C, 1,7 г 4-(3-метоксифенил)пиперидина выделяют способом, описанным в препаративном примере 9. Маслянистый остаток очищают флэш-хроматографией (SiO2, CH2Cl2:MeOH, 3:1 (об/об) с 1% NЕt3), получая чистое соединение, названное в заголовке. МС m/z (относительная интенсивность, 70 эВ) 191 (M+, 75), 160 (60), 83 (55), 57 (80), 56 (bp). Препаративный пример 11 4-(3-трифторметилфенил)пиперидин Исходя из 1-бензил-4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридина (1,44 г), 1 г названного в заголовке соединения в виде соли НС1 получают способом, описанным в препаративном примере 9. Т.пл. 202°С (НСl), МС m/z (относительная интенсивность, 70 эВ) 229 (M+, 44), 228 (33), 83 (12), 57 (54), 56 (bp). Препаративный пример 12 Метиловый сложный эфир 4-(3-метансульфонилфенил)пиперидин-1-карбоновой кислоты (0,92 г) растворяют в этаноле (15 мл) и 8 М НСl (40 мл) и кипятят с обратным холодильником в течение 12 часов. Смесь затем выпаривают в вакууме досуха. Выход составляет 0,85 г. МС m/z (относительная интенсивность, 70 эВ) 239 (M+, 59), 238 (50), 69 (20), 57 (79), 56 (bp). Препаративный пример 13 3-пиперидин-4-ил-фенол 4-(3-метоксифенил)пиперидин (1,7 г) растворяют в 48% HBr (60 мл) и перемешивают при 120oС в атмосфере аргона в течение 3 часов. Избыток HBr затем выпаривают, добавляют абсолютный этановый спирт и выпаривают. Эту процедуру повторяют несколько раз, получая сухие кристаллы 3-пиперидин-4-ил-фенола × HBr (2,3 г). МС m/z (относительная интенсивность, 70 эВ) 177 (M+, bp), 176 (23), 91 (14), 57 (44), 56 (60). Препаративный пример 14 3-(1-пропилпиперидин-4-ил)фенол × HBr Исходя из 3-пиперидин-4-ил-фенола × HBr (300 мг) и n-пропилйодида (200 мг), выделяют 340 мг 3-(1-пропилпиперидин-4-ил)фенола способом, описанным в примере 1. Получают соль HBr, чтобы обеспечить названное в заголовке соединение. МС m/z (относительная интенсивность, 70 эВ) 219 (M+, 21), 190 (bp), 119 (22), 91 (30), 70 (63); т.пл. 181-184°С (HBr). Препаративный пример 15 3-(1-этилпиперидин-4-ил)фенол Исходя из 3-пиперидин-4-ил-фенола × HBr (200 мг) и этилйодида (121 мг), выделяют 120 мг 3-(1-этилпиперидин-4-ил)фенола способом, описанным в примере 1. МС m/z (относительная интенсивность, 70 эВ) 205 (M+, 12), 190 (bp), 119 (36), 91 (22), 70 (87). Препаративный пример 16 3-(1-бутилпиперидин-4-ил)фенол Исходя из 3-пиперидин-4-ил-фенола × HBr (200 мг) и n-бутилхлорида (73 мг), выделяют 118 мг 3-(1-бутилпиперидин-4-ил)фенола способом, описанным в примере 1. МС m/z (относительная интенсивность, 70 эВ) 233 (M+, 6), 190 (bp), 119 (42), 91 (26), 70 (45). Препаративный пример 17 1-(3-метансульфонилфенил)пиперазин Смесь 1-бром-3-метансульфонилбензола (0,8 г), пиперазина (1 г), трет-бутоксида натрия (0,5 г), BINAP (42 мг) и [Pd2(dba)]3 (38 мг) в толуоле (7 мл) нагревают в атмосфере аргона при 80°С в течение 24 часов. После охлаждения до комнатной температуры растворитель выпаривают до сухого состояния. Сырой материал очищают флэш-хроматографией на силикагеле, используя EtOAc. Выход составляет 0,48 г: МС m/z (относительная интенсивность, 70 эВ) 240 (M+, 17), 199 (12), 198 (bp), 119 (9), 56 (7). Препаративный пример 18 1-(3-трифторметансульфонилфенил)пиперазин Исходя из 3-бромтрифторметансульфонилбензола и пиперазина названное в заголовке соединение получают способом, описанным в препаративном примере 17. МС m/z (относительная интенсивность, 70 эВ) 294 (M+, 22), 252 (bp), 119 (32), 104 (10), 56 (15). (45). Препаративный пример 19 1-(3-пиперазин-1-илфенил)этанон Исходя из 3-бромацетофенона и пиперазина названное в заголовке соединение получают способом, описанным в препаративном примере 17. МС m/z (относительная интенсивность, 70 эВ) 204 (M+, 5), 162 (35), 77 (30), 57 (35), 56 (bp). Препаративный пример 20 Метиловый сложный эфир 3-(1-пропилпиперидин-4-ил)бензойной кислоты Смесь трифторметансульфоновой кислоты 3-(1-пропилпиперидин-4-ил)фенилового сложного эфира (1,2 г), триэтиламина (0,9 г), МеОН (5,4 мл), Pd(OAc)2 (25 мг) и 1,3-бис(дифенилфосфино)пропана (45 мг) в 15 мл ДМСО перемешивают при комнатной температуре в течение 15 минут. Поток СО (г) пропускают через раствор в течение 4-5 минут, и затем реакционный сосуд помещают под слегка положительное давление СО (г). Температуру повышают до 70°С. Через 6 часов реакционной смеси дают охладиться до комнатной температуры. Затем добавляют воду, и водный раствор экстрагируют пятью порциями этилацетата, и объединенные органические фазы сушат (MgSO4) и выпаривают. Остаток хроматографируют на колонке окиси кремния, используя МеОН:СН2Сl2 (1:9 (об./об.)) в качестве элюента. Фракции, содержащие чистое названное в заголовке соединение собирают, и растворитель удаляют в вакууме, получая 650 мг названного в заголовке соединения. МС m/z (относительная интенсивность, 70 эВ) 261 (M+, 5), 233 (16), 232 (bp), 161 (5), 70 (20). Препаративный пример 21 3-(1-пропилпиперидин-4-ил)бензамид Раствор метилового сложного эфира 3-(пропилпиперидин-4-ил)бензойной кислоты (0,6 г) и формамида (320 мл) в ДМФ (9 мл) нагревают до 100oС под аргоном. По каплям добавляют метоксид натрия в метаноле (30%, 770 мкл), и через 1 час ГХ анализ выявляет полное отсутствие исходного вещества и показывает названное в заголовке соединение в виде единственного продукта. После охлаждения добавляют СН2Сl2, и полученный в результате раствор фильтруют через слой целита и выпаривают до сухого состояния. Остаток хроматографируют на колонке окиси кремния, используя МеОН:СН2Сl2 (1:3 (об./об.)) в качестве элюента. Фракции, содержащие чистое названное в заголовке соединение собирают, и растворитель удаляют в вакууме, получая 400 мг названного в заголовке соединения. Т.п. 182oС (оксалат). МС m/z (относительная интенсивность, 70 эВ) 246 (M+, 4), 217 (bp), 131 (19), 100 (22), 70 (63). Препаративный пример 22 4-(3-трифторметилсульфонилфенил)пиридин 1-Бром-3-трифторметилсульфонилбензол (580 мг) и 4-пиридинбороновую кислоту (275 мг) растворяют в толуоле (5 мл) и абс. EtOH (5 мл). К смеси затем добавляют Na2CO3 (424 мг) и Pd(PPh3)4 (119 мг) в атмосфере аргона. Полученную в результате смесь нагревают до 90°С в течение 18 часов. Затем добавляют СН2Сl2, и органическую фазу промывают водой и сушат (MgSO4), фильтруют и выпаривают досуха. Остаток затем используют без дальнейшей очистки. (МС m/z (относительная интенсивность, 70 эВ) 287 (M+,33), 218 (22), 154 (bp), 127 (56), 69 (27). Следующие тесты использовались для оценки соединений по изобретению. In vivo тест: Поведение Для тестирования поведенческой активности животных их помещали в отдельные коробки измерения подвижности 50×50×50 см, оборудованной множеством фотоячеек 16×16 (Digiscan монитор активности, RXYZM (16) TAO, Omnitech Electronics, США), соединенных с анализатором Omnitech Digiscan и компьютером Apple Macintosh, снабженным цифровой панелью управления (NB DIO-24, National Instruments, США). Данные поведенческой активности из каждой коробки измерения подвижности, представляющие положение (центр силы тяжести) животного в каждый момент времени, регистрировались с выбранной частотой 25 Гц и собирались, используя специально изготовленный письменный LABView In vivo тест: Нейрохимия После циклов определения поведенческой активности крыс, их подвергали декапитации, и их мозг быстро выделяли и помещали на охлажденные льдом чашки Петри. Лимбический передний мозг, полосатое тело, кору лобной доли и оставшиеся части полушарий каждой крысы иссекали и замораживали. Каждую часть мозга последовательно анализировали с точки зрения содержания в нем моноаминов и их метаболитов. Определяемые моноаминэргические стимуляторы представляли собой дофамин (DA), 3,4-дигидроксифенилуксусную кислоту (DOPAC), гомованилиновую кислоту (HVA), 3-метокситирамин (3-МТ), серотонин (5-НТ), 5-гидроксииндолуксусную кислоту (5-HIAA) и норадреналин (NA). Все моноаминэргические стимуляторы в иссеченных тканях анализировали с помощью ВЭЖХ с электрохимическим определением, как описано Svensson K, et al., 1986, Naunyn-Schmiedeberg’s Arch Pharmacol 334: 234-245 и в приведенных здесь ссылках. In vivo тест: Фаркмакокинетика у крыс Для определения оральной биодоступности (F) и плазменного времени полужизни (t1/2) проводились эксперименты на крысах с тестируемыми соединениями по изобретению. В первый день крысам имплантировали один катетер в яремную вену, и второй катетер в каротидную артерию под кетаминовой анестезией. На третий день вводили тестируемое соединение или орально, или через катетер в яремную вену. Образцы крови собирали в течение 8 часов из артериального катетера. Образцы крови гепаринизировали и центрифугировали. После центрифугирования образцов плазму собирали и замораживали. Уровни тестируемого соединения последовательно определяли в каждом образце посредством газовой хроматографии – масс-спектроскопии (Hewlett-Packard 5972MSD). Образцы плазмы, взятые от крыс линии Sprague-Dawley (0,5 мл), разводили водой (0,5 мл), и добавляли 25 мкл 30 пмоль (50 мкл) ((-)-S-3-(3-этилсульфонилфенил)-N-n-пропилпиперидином, как внутренний стандарт. рН доводили до 11,0 добавлением насыщенного Na2CO3. После смешивания образцы экстрагировали 4 мл дихлорметана встряхиванием в течение 20 минут. Органический слой после центрифугирования переносили в маленькие пробирки и выпаривали до сухого состояния в потоке азота и последовательно растворяли в 40 мкл толуола для анализа ГХ-МС. Получали стандартную кривую в диапазоне 1-500 пмоль добавлением соответствующего количества тестируемого соединения к свободным образцам плазмы. ГХ проводили на капиллярных колонках HP-Ultra 2 (12 м × 0,2 мм ID), и 2 мкл вводили непрерывным способом. Температуру ГХ поддерживали на 90oС в течение 1 минуты после введения и затем повышали на 30oС/мин до конечной температуры 290oС. Каждый образец дублировали. В общем, обнаружено, что самая низкая определяемая концентрация тестируемого соединения составляла 1 пмоль/мл.
Формула изобретения
1. Соединение 3-замещенного 4-(фенил-N-алкил)пиперазин формулы 1: 2. Соединение по п.1, где R1 выбран из группы, состоящей из OSO2CF3, OSO2CH3, SO2CH3, SO2CF3. 3. Соединение по п.1 или 2, где R2 выбран из н-пропила и этила. 4. Фармацевтическая композиция, обладающая допаминмодулирующим свойством, содержащая соединение по любому из пп.1-3 и один или более фармацевтически приемлемых носителей или разбавителей.
|
||||||||||||||||||||||||||
 ,
,
 -хлорэтилхлорформиатом (J. Org. Chem., vol.49, 1984, 2081-2082).
-хлорэтилхлорформиатом (J. Org. Chem., vol.49, 1984, 2081-2082).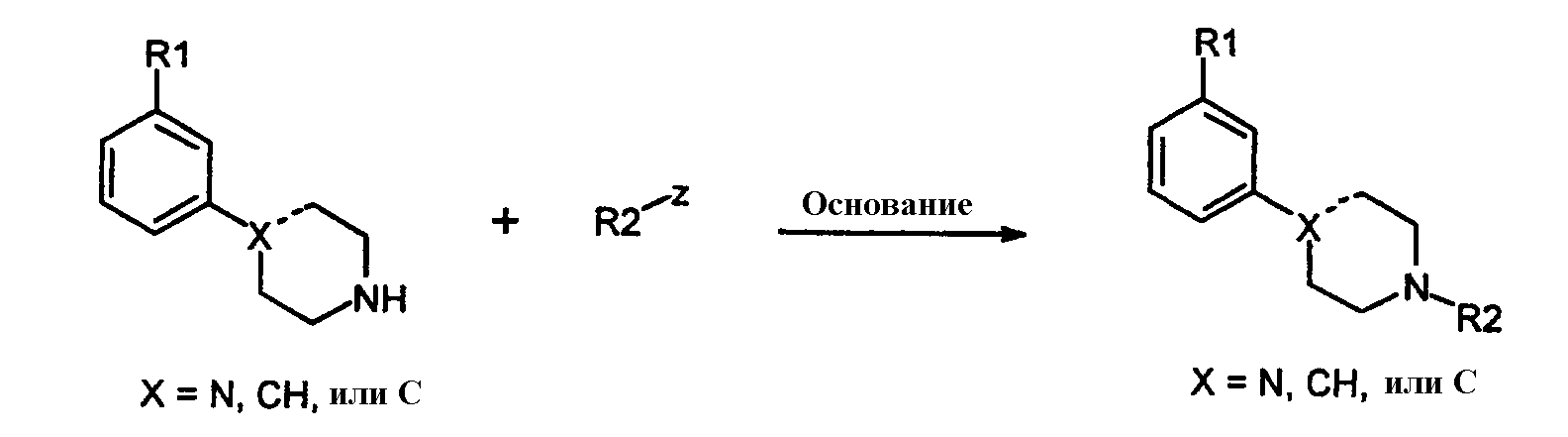
 аппликатор. Данные каждого цикла регистрации анализировали с точки зрения пройденного расстояния и мелкомасштабных движений, например, остановка в центре области регистрации поведения в течение цикла регистрации. Для определения остановки в центре, скорость в каждый момент времени рассчитывалась, как пройденное расстояние с предыдущего момента измерения, деленного на время, прошедшее с момента предыдущего измерения. Затем подсчитывали количество остановок, как число случаев изменения скорости с нулевого до нулевого значения. Количество остановок в центре области регистрации поведения рассчитывалось, как число остановок, происшедших по крайней мере на расстоянии десяти сантиметров от краев области регистрации. Для тестирования поведения приученных крыс, животных помещали в коробки измерения подвижности за 30 минут до введения тестируемого соединения. Каждый цикл регистрации поведения продолжался 60 или 30 минут, начиная сразу после введения тестируемого соединения. Подобную процедуру регистрации поведения проводили с неприрученными крысами, прирученными крысами и с крысами, предварительно обработанными лекарственным средством. Предварительно обработанные д-амфетамином крысы получали дозу 1,5 мг/кг подкожно за 5 минут до цикла регистрации измерения подвижности. Крысы, предварительно обработанные дизолципином (Мk-801), получали дозу 0,7 мг/кг интраперитонеально за 90 минут до цикла измерения подвижности.
аппликатор. Данные каждого цикла регистрации анализировали с точки зрения пройденного расстояния и мелкомасштабных движений, например, остановка в центре области регистрации поведения в течение цикла регистрации. Для определения остановки в центре, скорость в каждый момент времени рассчитывалась, как пройденное расстояние с предыдущего момента измерения, деленного на время, прошедшее с момента предыдущего измерения. Затем подсчитывали количество остановок, как число случаев изменения скорости с нулевого до нулевого значения. Количество остановок в центре области регистрации поведения рассчитывалось, как число остановок, происшедших по крайней мере на расстоянии десяти сантиметров от краев области регистрации. Для тестирования поведения приученных крыс, животных помещали в коробки измерения подвижности за 30 минут до введения тестируемого соединения. Каждый цикл регистрации поведения продолжался 60 или 30 минут, начиная сразу после введения тестируемого соединения. Подобную процедуру регистрации поведения проводили с неприрученными крысами, прирученными крысами и с крысами, предварительно обработанными лекарственным средством. Предварительно обработанные д-амфетамином крысы получали дозу 1,5 мг/кг подкожно за 5 минут до цикла регистрации измерения подвижности. Крысы, предварительно обработанные дизолципином (Мk-801), получали дозу 0,7 мг/кг интраперитонеально за 90 минут до цикла измерения подвижности.