Патент на изобретение №2384059
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) ТРАНСГЕННЫЕ КОПЫТНЫЕ ЖИВОТНЫЕ, ИМЕЮЩИЕ ПОНИЖЕННУЮ АКТИВНОСТЬ ПРИОННОГО БЕЛКА, И ИХ ПРИМЕНЕНИЯ
(57) Реферат:
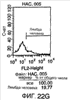
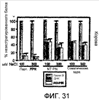
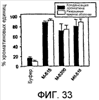
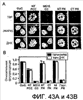
Изобретение относится к области генной инженерии и клонирования. Раскрыта трансгенная корова, у которой активность прионного белка снижена посредством одной или нескольких генетически сконструированных мутаций. Указанные трансгенные коровы также генетически модифицированы для экспрессии ксеногенных антител. Вследствие своей устойчивости к прионным заболеваниям, таким как губчатая энцефалопатия крупного рогатого скота, такие коровы являются безопасным источником человеческих антител для использования в фармацевтике и безопасным источником сельскохозяйственных продуктов. 5 н. и 7 з.п. ф-лы, 112 ил., 13 табл.
УРОВЕНЬ ТЕХНИКИ В общем случае, отличительным признаком изобретения являются клонированные трансгенные копытные животные (например, коровы), у которых активность прионного белка (PrP) снижена в результате одной или нескольких генетически сконструированных мутаций. Так как такие трансгенные коровы с пониженной активностью прионного белка должны быть резистентными к прионным заболеваниям, таким как губчатая энцефалопатия крупного рогатого скота (BSE, также известная как коровье бешенство), то они являются более безопасным и предпочтительным источником сельскохозяйственных и фармацевтических продуктов, таких как терапевтические антитела человека. С момента обнаружения первого случая BSE в Великобритании в 1986 г. это инфекционное заболевание распространилось в другие части мира, например в Японию. Опасность этого заболевания сильно повлияла на сельскохозяйственную и фармацевтическую области и ограничила применение крупного рогатого скота в этих отраслях. На основании исследований, проведенных в последние десять лет, в качестве основной причины этого инфекционного заболевания был идентифицирован прионный белок. Желательны животные с пониженной активностью прионного белка, так как они, как полагают, будут обладать резистентностью к прионным заболеваниям. До настоящего времени общеизвестными методами направленного воздействия на геном в эмбриональных стволовых (ЭС) клетках мышей, в которых возможна гомологичная рекомбинация, были созданы трансгенные мыши с пониженной активностью прионного белка. Однако сложно было получить, культивировать и генетически модифицировать ЭС клетки копытных животных, таких как крупный рогатый скот. При создании нокаутированных по приону овец эмбриональные фибробласты трансфицировали нокаутирующим вектором, не содержащим промотора (KO), так как ген прионного белка очень активно экспрессируется в эмбриональных фибробластах (Denning et al., Nature Biotech., 19:559-562, 2001). Используя такой тип нокаутирующего вектора, можно отобрать гомологичные целенаправленно измененные клоны, используя соответствующее лекарственное средство, такое как G418 или пуромицин, так как не содержащий промотора ген лекарственной резистентности (например, ген резистентности к неомицину или пуромицину) в нокаутированном векторе может экспрессироваться только тогда, когда он интегрирован в активно экспрессируемые локусы генов. Однако переносом ядра из этих гемизиготных целенаправленно измененных фибробластов овцы был получен только один живой ягненок, и ягненок умер примерно через 12 дней после рождения. В отличие от мышиных ЭС клеток с неограниченной продолжительностью жизни соматические фибробласты имеют ограниченную продолжительность жизни, что делает трудным направленное воздействие на соматические гены из-за продолжительности времени, необходимого для культивирования клеток в условиях жесткой селекции лекарственным средством, чтобы отобрать желаемые нокаутированные клетки. Кроме того, в общем случае, частота гомологичной рекомбинации в соматических фибробластах примерно в 10-100 раз меньше, чем в мышиных ЭС клетках. Эти ограничения в отношении соматических фибробластов, кроме низкой доли успешных попыток при обычных способах ядерного переноса, затрудняли получение жизнеспособного крупного рогатого скота с желаемой сайт-специфической мутацией. Предпринимались попытки уменьшить активность прионного белка у крупного рогатого скота путем направленного воздействия на геном. Насколько известно авторам, не сообщалось об успешном создании трансгенных коров с мутацией в прионном локусе, возможно, вследствие отсутствия соответствующих нокаутирующих векторов и/или способов ядерного переноса. Таким образом, необходимы улучшенные нокаутирующие векторы для получения мутаций в прионном локусе в клетках копытных животных (например, клетках коровы) с высокой эффективностью. Кроме того, требуются усовершенствованные способы создания трансгенных копытных животных (например, коров) из указанных генетически модифицированных донорных клеток. СУЩНОСТЬ ИЗОБРЕТЕНИЯ Признаком данного изобретения является конструирование нокаутирующих векторов, с помощью которых можно с высокой частотой осуществить гемизиготную или гомозиготную направленную интеграцию (так называемую гомологичную рекомбинацию) в прионный локус копытных животных (например, коров) в клетки доноров, такие как соматические фибробласты плода. Отличительным признаком изобретения также являются способы получения живых телят, имеющих гемизиготную или гомозиготную мутацию в прионном локусе, с использованием генетически модифицированных клеток донора в любом из способов переноса ядер, описанных здесь. Такой крупный рогатый скот может использоваться для получения фармацевтических и сельскохозяйственных продуктов, таких как терапевтические антитела человека для использования человеком. Способ по настоящему изобретению осуществляют с использованием нескольких методик, таких как (i) методика получения клеток, нокаутированных по гену, кодирующему прион, описанная здесь, (ii) способы клонирования млекопитающих, такие как перенос ядра или перенос хроматина, описанные в публикации PCT No. WO02/051997, и (iii) введение искусственной хромосомы человека (HAC), такой как дельта-HAC, копытным животным (публикация PCT No. WO02/70648; Kuroiwa et al., Nature Biotechnol. 20: 889-894, 2002). Нокаутированную по приону клетку копытного животного используют в качестве источника донорного генетического материала в способе клонирования млекопитающих, получая нокаутированное по приону (геми- или гомо-) потомство. Нокаутированные по приону копытные животные также могут обладать другим полезным свойством, таким как продукция антитела человека. Такие копытные животные могут быть созданы, используя сочетание указанных выше методик. Например, нокаутированное по приону и продуцирующее антитело человека животное может быть создано путем кроссбридинга нокаутированного по приону копытного животного и продуцирующего антитела человека копытного животного, как описано в публикации PCT No. WO02/70648. Последующую обработку эмбриональных фибробластов также можно использовать для создания такого копытного животного с или без скрещивания копытных животных. Последующая обработка эмбриональных фибробластов теленка включает повторение следующих стадий: (i) генетическая обработка фибробластов копытного животного (например, коров), (ii) клонирование млекопитающего, используя эти клетки, (iii) образование плода и (iv) выделение генетически модифицированного эмбрионального фибробласта. Например, нокаутированный по приону фибробласт может быть затем обработан для сохранения HAC и для инактивирования эндогенных генов Ig. Полученные моноклональные или поликлональные ксеногенные антитела могут иметь различное применение; например, их можно использовать в качестве ингредиентов в профилактических или терапевтических композициях против инфекции патогенными микроорганизмами, такими как бактерии или вирусы. Трансгенные копытные животные и клетки копытных В одном из аспектов изобретение относится к копытному животному (например, корове) или к клетке копытного животного (например, клетке коровы), имеющей неприродную мутацию (например, мутацию после кодона инициации ATC, такую как мутация в 10, 20, 50 или 100 нуклеотидах этого кодона) в одном или обоих аллелях эндогенной нуклеиновой кислоты, которая кодирует прионный белок. Предпочтительно, мутация снижает или по существу исключает экспрессию функционального прионного белка. В предпочтительных вариантах осуществления экспрессия функциональной части или всего прионного белка снижается по меньшей мере на 10, 20, 40, 60, 80, 90, 95 или 100%. Мутация может быть гемизиготной или гомозиготной. В некоторых вариантах осуществления мутация включает инсерцию маркера для позитивной селекции (например, ген устойчивости к антибиотику) в нуклеиновую кислоту приона. Предпочтительно, маркер для позитивной селекции оперативно связан с ксеногенным промотором. В случае копытных животных или клеток копытного животного с геном устойчивости к антибиотику, встроенным в оба аллеля нуклеиновой кислоты, которая кодирует прионный белок, каждая аллель может содержать один и тот же или разные гены устойчивости к антибиотику. В предпочтительном варианте осуществления маркер негативной селекции (например, DT-A или Tk) оперативно связан с ксеногенным промотором и присутствует в векторе, используемом для мутирования эндогенного аллеля приона. Мутация может содержать или не содержать делецию одного или нескольких нуклеотидов (например, следующих друг за другом нуклеотидов) в нуклеиновой кислоте приона. В предпочтительных вариантах осуществления указанного выше аспекта копытное животное (например, корова) или клетка копытного животного (например, клетка коровы) имеет один или несколько трансгенов и экспрессирует мРНК или белок (например, антитело), кодируемые трансгеном(ами). Предпочтительные копытные животные содержат природные участки хромосом человека (например, фрагменты хромосом человека) или искусственные хромосомы, которые содержат искусственно сконструированные фрагменты хромосом человека (т.е. фрагменты могут быть перегруппированы относительно генома человека). В некоторых вариантах осуществления в фрагменте хромосомы находится ксеногенная нуклеиновая кислота. Нуклеиновая кислота может быть интегрирована в хромосому копытного животного или может сохраняется в клетке копытного животного независимо от хромосомы хозяина. В различных вариантах осуществления нуклеиновая кислота находится в фрагменте хромосомы, таком как Предпочтительные копытные животные и клетки копытных животных имеют одну или несколько нуклеиновых кислот, содержащих локус гена ксеногенного антитела (например, нуклеиновую кислоту, кодирующую весь или часть ксеногенного гена иммуноглобулина (Ig), который подвергнут перестройке и экспрессирует по меньшей мере одну молекулу ксеногенного Ig) в одной или нескольких B-клетках. Предпочтительно, нуклеиновая кислота содержит не подвергнутые перестройке участки нуклеиновой кислоты легкой цепи антитела, в которой все участки нуклеиновой кислоты, кодирующие V-сегмент гена, отделены от всех участков нуклеиновой кислоты, кодирующих J-сегмент гена, одним или несколькими нуклеотидами. Другие предпочтительные нуклеиновые кислоты содержат не подвергнутые перестройке участки нуклеиновой кислоты тяжелой цепи антитела, в которых либо (i) все участки нуклеиновой кислоты, кодирующие V-сегмент гена, отделены от всех участков нуклеиновой кислоты, кодирующих D-сегмент гена, одним или несколькими нуклеотидами и/или (ii) все участки нуклеиновой кислоты, кодирующие D-сегмент гена, отделены от всех участков нуклеиновой кислоты, кодирующих J-сегмент гена, одним или несколькими нуклеотидами. Другие предпочтительные копытные животные имеют одну или несколько нуклеиновых кислот, кодирующих весь или часть подвергнутого перестройке ксеногенного гена иммуноглобулина (Ig), который экспрессирует по меньшей мере одну ксеногенную молекулу Ig. В других предпочтительных вариантах осуществления легкая цепь и/или тяжелая цепь ксеногенных антител кодируется нуклеиновой кислотой человека. В предпочтительных вариантах осуществления тяжелая цепь относится к любому классу тяжелой цепи, такому как В различных вариантах осуществления указанного выше аспекта копытное животное (например, корова) или клетка копытного животного (например, клетка коровы) имеет мутацию, которая снижает экспрессию эндогенного антитела. Предпочтительно, мутация снижает экспрессию функциональной части тяжелой цепи IgM или по существу исключает экспрессию функциональной части тяжелой цепи IgM. В других предпочтительных вариантах осуществления мутация снижает экспрессию функциональной части легкой цепи Ig или по существу исключает экспрессию функциональной части легкой цепи Ig. В еще других предпочтительных вариантах осуществления мутация снижает экспрессию функциональной части тяжелой цепи IgM и функциональной части легкой цепи Ig или мутация по существу исключает экспрессию функциональной части тяжелой цепи IgM и функциональной части легкой цепи Ig. Предпочтительно, копытное животное также имеет мутацию в одном или обоих аллелях эндогенной нуклеиновой кислоты, кодирующей альфа-(1,3)-галактозилтрансферазу и/или J-цепь. В других предпочтительных вариантах осуществления копытное животное имеет нуклеиновую кислоту, кодирующую экзогенную J-цепь, такую как J-цепь человека. Предпочтительно, мутация снижает или исключает экспрессию эндогенного фермента альфа-(1,3)-галактозилтрансферазы, эпитопа галактозил( Изобретение также относится к гибридомам, которые продуцируют ксеногенные (например, человеческие) антитела. В одном из таких аспектов изобретение относится к гибридоме, полученной слиянием B-клетки по изобретению с миеломной клеткой. Предпочтительно, антитело взаимодействует с интересующим антигеном. Способы получения трансгенных клеток копытных животных Признаком изобретения также являются способы создания клеток копытных животных (например, клеток коровы) с мутацией в одном или обоих аллелях нуклеиновой кислоты, которая кодирует прионный белок. Эти клетки можно использовать в качестве донорных клеток для создания трансгенных копытных животных (например, нокаутированных по приону копытных животных, относящихся к крупному рогатому скоту). Предпочтительно, мутация приводит к снижению количества функциональной части прионного белка и/или к снижению частоты возникновения прионных заболеваний или заболеваний, таких как BSE. Таким образом, в одном из таких аспектов признаком изобретения является способ получения трансгенного копытного животного (например, коровы). Этот способ заключается во введении первого вектора, который направленно воздействует на кодирующий прионный белок ген, в клетку копытного животного в условиях, при которых возможна гомологичная рекомбинация между первым вектором и первым аллелем эндогенной нуклеиновой кислоты, которая кодирует прионный белок в клетке, с введением, таким образом, гемизиготной мутации в клетку. Предпочтительно, первый вектор содержит первую гомологичную область с последовательностью, по существу идентичной первой области эндогенной нуклеиновой кислоты, которая кодирует прионный белок клетки, маркер позитивной селекции и вторую гомологичную область, имеющую последовательность по существу идентичную второй области нуклеиновой кислоты, которая кодирует прионный белок. В предпочтительном варианте осуществления одна гомологичная область по меньшей мере на 1, 2, 3, 4, 5, 6 или 8 тысяч пар оснований длиннее, чем другая гомологичная область. Предпочтительно, способ также включает в себя повторное введение первого вектора в клетку в условиях, при которых может осуществляться гомологичная рекомбинация между первым вектором и вторым аллелем эндогенной нуклеиновой кислоты, которая кодирует прионный белок в клетке, с введением, таким образом, гомозиготной мутации в клетку. В других вариантах осуществления способ также включает в себя введение в клетку второго вектора, мишенью которого является кодирующий прионный белок ген, который имеет другой ген устойчивости к антибиотику, отличный от гена первого вектора, в условиях, при которых возможна гомологичная рекомбинация между вторым вектором и вторым аллелем эндогенной нуклеиновой кислоты, которая кодирует прионный белок в клетке, с введением, таким образом, гомозиготной мутации в клетку. Предпочтительно, первый и/или второй вектор вводят в клетку в присутствии 1 мМ спермидина. Предпочтительные клетки включают эмбриональные фибробласты коров. В различных вариантах осуществления изобретения нуклеиновая кислота, используемая для мутирования эндогенной нуклеиновой кислоты, которая кодирует прионный белок (например, нокаутирующая кассета, которая содержит промотор, оперативно связанный с нуклеиновой кислотой, кодирующей селектируемый маркер, и оперативно связанный с нуклеиновой кислотой по существу идентичной последовательности нуклеиновой кислоты, кодирующей прионный белок), содержится не в вирусном векторе, таком как аденовирусный вектор или вектор на основе аденоассоциированного вируса. Например, нуклеиновая кислота может содержаться в плазмиде или искусственной хромосоме, которую встраивают в клетку копытного животного, используя стандартный способ, такой как трансфекция или липофекция, в котором не используется вирусное инфицирование клетки. В еще одном варианте осуществления нуклеиновая кислота, используемая для мутирования эндогенной нуклеиновой кислоты, которая кодирует прионный белок (например, нокаутирующая кассета, которая содержит промотор, оперативно связанный с нуклеиновой кислотой, кодирующей селектируемый маркер, и оперативно связанный с нуклеиновой кислотой, по существу идентичной последовательности с кодирующей прионный белок нуклеиновой кислотой) находится в вирусном векторе, таком как аденовирусный вектор или вектор на основе аденоассоциированного вируса. Согласно такому варианту осуществления вирус, содержащий вирусный вектор, используют для инфицирования клетки копытного животного, что приводит к инсерции части или всего вирусного вектора в клетку копытного животного. Способы получения трансгенных копытных животных Изобретение также относится к способам получения трансгенных копытных животных с одной или несколькими мутациями в эндогенных кодирующих прионный белок нуклеиновых кислотах. Один из таких способов заключается во встраивании клетки согласно любому указанному выше аспекту изобретения, хроматиновой массы клетки или ядра клетки в ооцит. Клетка имеет первую мутацию в эндогенной нуклеиновой кислоте, которая кодирует прионный белок. Ооцит или эмбрион, образованный из ооцита, переносят в матку копытного животного-хозяина в условиях, при которых возможно развитие ооцита или эмбриона в плод. Предпочтительно, плод развивается в жизнеспособного потомка. В предпочтительных вариантах осуществления первую мутацию вводят в клетку путем встраивания нуклеиновой кислоты, содержащей кассету, которая включает в себя промотор, оперативно связанный с нуклеиновой кислотой, кодирующей селектируемый маркер, и оперативно связанный с одной или несколькими нуклеиновыми кислотами по существу идентичной последовательности эндогенной нуклеиновой кислоты, кодирующей прионный белок, при этом кассета интегрируется в один эндогенный аллель нуклеиновой кислоты, которая кодирует прионный белок. В других предпочтительных вариантах осуществления мутацию вводят в клетку путем встраивания в клетку нуклеиновой кислоты, содержащей первую кассету, которая включает в себя первый промотор, оперативно связанный с нуклеиновой кислотой, кодирующей первый селектируемый маркер, и оперативно связанный с первой нуклеиновой кислотой, по существу идентичной последовательности эндогенной нуклеиновой кислоты, кодирующей прионный белок, при этом первая кассета интегрируется в первый эндогенный аллель нуклеиновой кислоты, которая кодирует прионный белок, с образованием первой трансгенной клетки. В первую трансгенную клетку встраивают нуклеиновую кислоту, которая содержит вторую кассету, которая включает в себя второй промотор, оперативно связанный с нуклеиновой кислотой, кодирующей второй селектируемый маркер, и оперативно связанный со второй нуклеиновой кислотой, по существу идентичной последовательности нуклеиновой кислоты, кодирующей прионный белок. Второй селектируемый маркер отличается от первого селектируемого маркера, и вторая кассета интегрируется во второй эндогенный аллель нуклеиновой кислоты, которая кодирует прионный белок, с образованием второй трансгенной клетки. В других предпочтительных вариантах осуществления клетку выделяют из эмбриона, плода или потомка, развившегося из плода, и вводят другую мутацию в нуклеиновую кислоту, которая кодирует прионный белок, или другую нуклеиновую кислоту (например, нуклеиновую кислоту тяжелой или легкой цепи антитела) клетки. Затем осуществляют второй раунд переноса ядер, используя полученную клетку, хроматиновую массу клетки или ядро клетки, с получением трансгенного копытного животного с двумя или более мутациями. Мутации находятся в одном и том же или разных аллелях гена или в разных генах. Клетка, используемая в первом или, необязательно, втором раунде переноса ядер, кодирует ксеногенное антитело. В конкретных вариантах осуществления клетка содержит одну или несколько нуклеиновых кислот, кодирующих весь или часть ксеногенного гена Ig, которая способна перестраиваться и экспрессировать одну или несколько ксеногенных молекул Ig в B-клетках. В предпочтительных вариантах осуществления клетка, которую подвергают мутациям, является фибробластом (например, эмбриональным фибробластом). Предпочтительно, мутируемый эндогенный ген оперативно связан с эндогенным промотором, который не активен в фибробластах. В других предпочтительных вариантах осуществления эндогенный промотор, оперативно связанный с мутируемым эндогенным геном, активен менее чем на 80, 70, 60, 50, 40, 30, 20, 10% по сравнению с эндогенным промотором, оперативно связанным с эндогенным геном домашнего хозяйства, таким как GAPDH. Активность промотора можно измерить, используя любой стандартный анализ, такой как анализ, в котором измеряют уровень мРНК или белка, кодируемого геном (смотри, например, Ausubel et al. Current Protocols in Molecular Biology, volume 2, p. 11.13.1-11.13.3, John Wiley and Sons, 1995). Преимуществом этого способа создания трансгенного копытного животного является возможность мутации гена, который не экспрессируется в донорной клетке (т.е. клетке, которая является источником генетического материала, используемого для переноса ядра). Предпочтительно, клетка, используемая для получения трансгенного копытного животного, имеет мутацию в одном или обоих аллелях эндогенной нуклеиновой кислоты, кодирующей альфа-(1,3)-галактозилтрансферазу и/или J-цепь. В других предпочтительных вариантах осуществления клетка содержит нуклеиновую кислоту, кодирующую экзогенную J-цепь, такую как J-цепь человека, или нуклеиновую кислоту, кодирующую ксеногенное антитело. Предпочтительно, ксеногенная нуклеиновая кислота кодирует весь или часть ксеногенного гена Ig, и ген способен перестраиваться и экспрессировать более одной ксеногенной молекулы Ig в B-клетках. В других предпочтительных вариантах осуществления антитело является поликлональным антителом. В следующих предпочтительных вариантах осуществления цепь иммуноглобулина или антитело экспрессируются в сыворотке и/или молоке. Авторами ранее были описаны некоторые улучшенные способы клонирования млекопитающих (например, копытных животных, таких как коровы), которые можно использовать для клонирования млекопитающих с одной или несколькими мутациями в нуклеиновой кислоте, которая кодирует прионный белок (смотри, например, публикацию патента США No. 2002-0046722 A1 и публикацию PCT No. WO02/051997). В некоторых из этих способов пермеабилизованную клетку инкубируют в средах для перепрограммирования (например, в клеточном экстракте) для добавления или удаления факторов из клетки и затем плазматическую мембрану пермеабилизованной клетки снова делают непроницаемой, чтобы сохранить внутри желаемые факторы и восстановить целостность мембраны клетки. Некоторые из этих способов также включают в себя конденсацию донорного ядра (например, изолированного ядра или ядра в пределах донорной клетки) в хроматиновую массу для обеспечения высвобождения ядерных компонентов, таких как факторы транскрипции, которые могут обеспечить транскрипцию генов, которые нежелательны для развития эмбриона с ядерным трансплантатом в жизнеспособное потомство. При желании стадии любого из этих способов можно повторять один или несколько раз или можно последовательно осуществлять различные способы перепрограммирования, чтобы повысить степень перепрограммирования, что приводит к большей выживаемости клонированных плодов. Один из способов создания трансгенного копытного животного (например, коровы) заключается в инкубировании пермеабилизованной клетки согласно любому из указанных выше аспектов изобретения (например, клетки, которая имеет одну или несколько мутаций в эндогенной нуклеиновой кислоте, которая кодирует прионный белок) в среде для перепрограммирования (например, в клеточном экстракте) в условиях, при которых происходит удаление фактора (например, ядерного или цитоплазматического компонента, такого как фактор транскрипции) из ядра, хроматиновой массы или хромосомы пермеабилизованной клетки или добавление фактора в ядро, хроматиновую массу или хромосому с образованием, таким образом, перепрограммированной клетки. Перепрограммированную клетку вводят в ооцит с удаленным ядром и полученный ооцит или эмбрион, образованный из ооцита, переносят в матку копытного животного-хозяина в условиях, при которых возможно развитие ооцита или эмбриона в плод. В предпочтительных вариантах осуществления пермеабилизованная клетка взаимодействует с одним или несколькими из следующих факторов в условиях, при которых возможно образование хроматиновой массы: экстрактом митотических клеток в присутствии или отсутствии анти-NuMA-антитела, раствором детергента и/или соли или раствором протеинкиназы. В еще одном предпочтительном варианте осуществления пермеабилизованную клетку инкубируют в интерфазных средах для перепрограммирования (например, в экстракте интерфазных клеток). В еще одном предпочтительном варианте осуществления ядро в пермеабилизованной клетке остается связанным с мембраной, и хромосомы в ядре не подвергаются конденсации во время инкубирования с интерфазными средами для перепрограммирования. В некоторых вариантах осуществления инкубация пермеабилизованной клетки в средах для перепрограммирования не вызывает репликации ДНК или вызывает репликацию ДНК только в менее чем 50, 40, 30, 20, 10 или 5% клеток. В других вариантах осуществления инкубация пермеабилизованной клетки в средах для перепрограммирования вызывает репликацию ДНК по меньшей мере в 60, 70, 80, 90, 95 или 100% клеток. В различных вариантах осуществления клетку пермеабилизуют путем инкубирования интактной клетки с протеазой, такой как трипсин, детергентом, таким как дигитонин, или бактериальным токсином, таким как стрептолизин O. В предпочтительном варианте осуществления перепрограммированную клетку не инкубируют в условиях, при которых мембрана перепрограммированной клетки может снова стать непроницаемой перед введением в ооцит. В еще одном варианте осуществления перепрограммированную клетку инкубируют в условиях, при которых мембрана перепрограммированной клетки может снова стать непроницаемой перед введением в ооцит. В других предпочтительных вариантах осуществления реконструированный ооцит или полученный эмбрион экспрессирует ламин A, ламин C или белок NuMA на уровне, который менее чем в 5 раз превосходит соответствующий уровень, экспрессируемый контрольным ооцитом или контрольным эмбрионом с таким же количеством клеток и из того же самого вида. В другом аспекте изобретение относится к другому способу создания трансгенного копытного животного (например, коровы). Этот способ включает в себя (a) инкубирование донорного ядра из клетки согласно изобретению (например, ядра, которое имеет одну или несколько мутаций в эндогенной нуклеиновой кислоте, которая кодирует прионный белок) в условиях, при которых возможно образование хроматиновой массы, не вызывая репликацию ДНК, (b) введение хроматиновой массы в лишенный ядра ооцит с образованием, таким образом, ооцита с перенесенным ядром и (c) перенос ооцита с перенесенным ядром или эмбриона, образованного из ооцита с перенесенным ядром, в матку копытного животного-хозяина в условиях, при которых возможно развитие ооцита с перенесенным ядром или эмбриона в плод. В предпочтительном варианте осуществления донорное ядро инкубируют в средах для перепрограммирования (например, в клеточном экстракте) в условиях, при которых возможно добавление или удаление из ядра или полученной хроматиновой массы ядерных или цитоплазматических компонентов, таких как факторы транскрипции, белки-репрессоры или ремоделирующие белки хроматина. Предпочтительно, донорное ядро контактирует с одним или несколькими из следующих факторов в условиях, при которых возможно образование хроматиновой массы: экстракт митотической клетки в присутствии или отсутствии анти-NuMA-антитела, раствор детергента и/или соли или раствор протеинкиназы. В других предпочтительных вариантах осуществления реконструированный ооцит или полученный эмбрион экспрессирует ламин A, ламин C или белок NuMA на уровне, который менее чем в 5 раз превосходит уровень, экспрессируемый контрольным ооцитом или контрольным эмбрионом с таким же количеством клеток и из того же самого вида. Предпочтительно, ядро имеет менее четырех наборов гомологичных хромосом (т.е. имеет менее двух пар полных хроматид). Предпочтительные способы создания химерных копытных животных Другие предпочтительные копытные животные являются химерными копытными животными, полученными с использованием клеток от двух или более эмбрионов. Например, клетки из эмбриона с перенесенным ядром (например, эмбриона, образованного введением клетки, ядра или хроматиновой массы в лишенный ядра ооцит) могут быть объединены с клетками из эмбриона, оплодотворенного in vitro, природного эмбриона или партеногенетически активированного эмбриона. Предпочтительно, большинство клеток и их потомство из эмбриона с перенесенным ядром введены в ткань плода полученного химерного эмбриона. По меньшей мере некоторые клетки и их потомство из второго эмбриона, предпочтительно, введены в плацентарную ткань и способствуют выживаемости полученного химерного эмбриона. В предпочтительных вариантах осуществления эмбрион с перенесенным ядром имеет мутацию в эндогенной нуклеиновой кислоте, которая кодирует прионный белок, и имеет нуклеиновую кислоту, кодирующую ксеногенное антитело. Таким образом, в одном аспекте изобретение относится к способу получения трансгенного копытного животного путем введения клетки, ядра или хроматиновой массы по изобретению (например, клетки, ядра или хроматиновой массы, имеющей мутацию в эндогенной нуклеиновой кислоте, которая кодирует прионный белок и, необязательно, содержащей одну или несколько нуклеиновых кислот, кодирующих ксеногенное антитело) в ооцит с образованием, таким образом, первого эмбриона. Одну или несколько клеток первого эмбриона подвергают взаимодействию с одной или несколькими клетками из второго эмбриона с образованием, таким образом, третьего эмбриона. Второй эмбрион является оплодотворенным in vitro эмбрионом, природным эмбрионом или партеногенетически активированным эмбрионом. Третий эмбрион переносят в матку копытного животного-хозяина в условиях, при которых возможно развитие третьего эмбриона в плод. В другом родственном аспекте изобретение относится к другому способу создания трансгенного копытного животного (например, коровы). Этот способ заключается в инкубировании пермеабилизованной клетки согласно изобретению (например, клетки, имеющей мутацию в эндогенной кодирующей прионный белок нуклеиновой кислоте и, необязательно, содержащей одну или несколько нуклеиновых кислот, кодирующих ксеногенное антитело) в средах для перепрограммирования (например, клеточном экстракте) в условиях, при которых возможно удаление фактора ядра, хроматиновой массы или хромосомы пермеабилизованной клетки или добавление фактора из сред для перепрограммирования в ядро, хроматиновую массу или хромосому с образованием, таким образом, перепрограммированной клетки. Перепрограммированную клетку вводят в лишенный ядра ооцит с образованием, таким образом, первого эмбриона. Одну или несколько клеток из первого эмбриона подвергают взаимодействию с одной или несколькими клетками из оплодотворенного in vitro эмбриона, природного эмбриона или партеногенетически активированного второго эмбриона, образуя третий эмбрион. Третий эмбрион переносят в матку копытного животного-хозяина в условиях, при которых возможно развитие третьего эмбриона в плод. В предпочтительном варианте осуществления пермеабилизованную клетку инкубируют в среде для перепрограммирования (например, в клеточном экстракте) в условиях, при которых возможно добавление или удаление из ядра или полученной хроматиновой массы ядерных или цитоплазматических компонентов, таких как факторы транскрипции. В других предпочтительных вариантах осуществления проводят взаимодействие пермеабилизованной клетки с одним или несколькими из следующих факторов в условиях, при которых возможно образование хроматиновой массы: экстрактом митотических клеток в присутствии или отсутствии анти-NuMA-антитела, раствором детергента и/или соли или раствором протеинкиназы. В еще одном предпочтительном варианте осуществления пермеабилизованную клетку инкубируют в интерфазных средах для перепрограммирования (например, в экстракте интерфазных клеток). В еще одном предпочтительном варианте осуществления ядро в пермеабилизованной клетке остается связанным с мембраной, и хромосомы в ядре не подвергаются конденсации во время инкубирования в интерфазных средах для перепрограммирования. В некоторых вариантах осуществления инкубирование пермеабилизованной клетки в средах для перепрограммирования не вызывает репликации ДНК или вызывает репликацию ДНК только в менее чем 50, 40, 30, 20, 10 или 5% клеток. В других вариантах осуществления инкубирование пермеабилизованной клетки в средах для перепрограммирования вызывает репликацию ДНК по меньшей мере в 60, 70, 80, 90, 95 или 100% клеток. В различных вариантах осуществления клетку пермеабилизуют при инкубировании интактной клетки с протеазой, такой как трипсин, детергентом, таким как дигитонин, или бактериальным токсином, таким как стрептолизин O. В предпочтительном варианте осуществления перепрограммируемую клетку не инкубируют в условиях, при которых мембрана перепрограммированной клетки снова может стать непроницаемой перед введением в ооцит. В еще одном варианте осуществления перепрограммированную клетку инкубируют в условиях, при которых мембрана перепрограммированной клетки снова может стать непроницаемой перед введением в ооцит. В следующих вариантах осуществления копытное животное создают в результате контактирования донорного ядра из клетки согласно изобретению (например, ядра, имеющего мутацию в эндогенной нуклеиновой кислоте, которая кодирует прионный белок, и, необязательно, содержащего одну или несколько нуклеиновых кислот, кодирующих ксеногенное антитело) в среде для перепрограммирования (например, в клеточном экстракте) в условиях, при которых возможно образование хроматиновой массы, и введения хроматиновой массы в лишенный ядра ооцит с образованием, таким образом, первого эмбриона. Одну или несколько клеток из первого эмбриона подвергают взаимодействию с одной или несколькими клетками из оплодотворенного in vitro, природного или партеногенетически активированного второго эмбриона с образованием, таким образом, третьего эмбриона. Третий эмбрион переносят в матку копытного животного-хозяина в условиях, при которых возможно развитие третьего эмбриона в плод. В предпочтительном варианте осуществления хроматиновая масса образуется в результате взаимодействия донорного ядра, которое имеет менее четырех наборов гомологичных хромосом, в среде для перепрограммирования в условиях, при которых возможно образование хроматиновой массы, не вызывая репликацию ДНК. Предпочтительно, донорное ядро подвергают взаимодействию с одним или несколькими факторами в условиях, при которых возможно образование хроматиновой массы: экстрактом митотических клеток в присутствии или отсутствии анти-NuMA-антитела, раствором детергента и/или соли или раствором протеинкиназы. В предпочтительных вариантах осуществления любого из указанных выше аспектов, относящихся к копытным животным, полученным с использованием клеток из двух эмбрионов, по меньшей мере один из первого эмбриона и второго эмбриона является компактным эмбрионом. В другом варианте осуществления первый эмбрион и второй эмбрион находятся на разных клеточных стадиях. Первый эмбрион и донорная клетка, используемая для получения второго эмбриона, могут быть из одного и того же вида или из разных родов или видов. Предпочтительно, по меньшей мере 10, 20, 30, 40, 50, 60, 70, 80, 90, 95 или 100% клеток в трофобласте или плацентарной ткани плода получены из второго эмбриона или по меньшей мере 30, 40, 50, 60, 70, 80, 90, 95 или 100% клеток во внутренней клеточной массе или эмбриональной ткани плода получены из первого эмбриона. В других предпочтительных вариантах осуществления первый эмбрион или третий эмбрион экспрессируют ламин A, ламин C или белок NuMA на уровне, который менее чем в 5 раз превышает соответствующий уровень экспрессии контрольного эмбриона с таким же количеством клеток и из того же самого вида. В других предпочтительных вариантах осуществления любого из указанных выше аспектов, касающихся копытных животных, полученных с использованием клеток из двух эмбрионов, часть или всю zona pellucida первого эмбриона или второго эмбриона удаляют перед взаимодействием клеток из каждого эмбриона. В одном варианте осуществления клетки из первого и второго эмбрионов подвергают взаимодействию, помещая их рядом друг с другом в растворе или на твердой подложке. В другом варианте осуществления используют стандартный способ для того, чтобы инъецировать клетки из первого эмбриона во второй эмбрион. Клетки можно инъецировать в любую область второго эмбриона, такую как область периферии эмбриона между zona pellucida и самим эмбрионом. Природные эмбрионы включают эмбрионы, которые хирургически или нехирургически извлечены из беременного копытного животного (например, коровы) с использованием стандартных способов. Оплодотворенные in vitro эмбрионы включают эмбрионы, созданные путем инъекции сперматозоида внутрь цитоплазмы с использованием стандартных способов. Также предполагается, что клетки из более чем двух эмбрионов (например, клетки из трех, четырех, пяти, шести или более эмбрионов) можно объединять с образованием химерного эмбриона для создания клонированного копытного животного. Предпочтительные варианты осуществления создания копытных животных В предпочтительных вариантах осуществления любого из указанных выше аспектов среды для перепрограммирования (например, клеточный экстракт) модифицируют путем обогащения или сокращения фактора, такого как ДНК-метилтрансфераза, деацетилаза гистонов, гистон, протамин, ядерный ламин, фактор транскрипции, активатор или репрессор. В других предпочтительных вариантах осуществления уровень экспрессии белка NuMA или AKAP95 в ооците или химерном эмбрионе по меньшей мере в 2-, 5-, 10- или 20 раз выше в ядре, чем в цитоплазме. В других вариантах осуществления по меньшей мере 30, 40, 50, 60, 70, 80, 90 или 100% белка AKAP95 в ооците или химерном эмбрионе экстрагируют с использованием раствора 0,1% тритона X-100, 1 мг/мл ДНКазы I и либо 100 мМ, либо 300 мМ NaCl. Предпочтительно, перед введением в лишенный ядра ооцит хроматиновую массу очищают от среды для перепрограммирования (например, экстракта). В другом предпочтительном варианте осуществления встраивание хроматиновой массы в лишенный ядра ооцит заключается в осуществлении контакта хроматиновой массы и ооцита с вызывающим слияние соединением в условиях, при которых возможно проникновение хроматиновой массы в ооцит. В еще одном предпочтительном варианте осуществления плод развивается в жизнеспособного потомка. Предпочтительно, по меньшей мере 1, 3, 5, 10, 20, 30, 40, 50, 60, 70, 80 или 90% ооцитов или эмбрионов с перенесенными ядрами развиваются в жизнеспособное потомство. В этом способе ооцит, содержащий хроматиновую массу, или перепрограммированную клетку можно культивировать в условиях, при которых происходит клеточное деление и одна из полученных клеток может быть повторно клонирована один или несколько раз. Донорное ядро, донорная хроматиновая масса или донорная клетка и ооцит, используемые в способе, могут быть из одного и того же вида или они могут быть из разных видов или родов. Желательно донорная клетка или полученное трансгенное копытное животное экспрессирует по меньшей мере на 10, 20, 40, 60, 80, 90, 95 или 100% меньше функционального или общего белка, чем соответствующая клетка или копытное животное естественного происхождения. Ооцит может быть оплодотворенным или неоплодотворенным. Предпочтительно, донорное ядро, хроматиновая масса или пермеабилизованная клетка взяты в клеточной фазе G1 или G0. Кроме того, геномная ДНК клонированного эмбриона, плода или копытного животного, предпочтительно, по существу идентична геномной ДНК донорной клетки. Также предполагается, что хроматиновая масса или перепрограммированная клетка могут быть введены в эмбрион для получения химерного эмбриона, плода или копытного животного, содержащего смесь клеток с ДНК, по существу идентичной ДНК хроматиновой массы или перепрограммированной клетки, и клеток с ДНК по существу идентичной ДНК клеток в природном эмбрионе. Также предполагается, что лишенный ядра ооцит можно использовать в способах по изобретению. Среды для перепрограммирования, используемые в любом из аспектов изобретения, могут содержать или не содержать экзогенные нуклеотиды. В других предпочтительных вариантах осуществления хроматиновая масса в средах для перепрограммирования или образованная в пермеабилизованной клетке, контактирует с вектором, содержащим нуклеиновую кислоту, кодирующую интересующий ген, в условиях, при которых возможно осуществление случайной интеграции или гомологичной рекомбинации между нуклеиновой кислотой в векторе и соответствующей нуклеиновой кислотой в геноме хроматиновой массы, приводящей к изменению генома хроматиновой массы. Вследствие отсутствия интактной плазматической мембраны и отсутствия ядерной мембраны хроматиновую массу в пермеабилизованной клетке или в растворе легче генетически модифицировать, чем в природной клетке. Примеры клеток, которые можно использовать для создания экстрактов для перепрограммирования, включают эмбриональные стволовые клетки и стволовые клетки взрослого организма из головного мозга, крови, костного мозга, поджелудочной железы, печени, кожи или любого другого органа или ткани. Другие иллюстративные клеточные экстракты для перепрограммирования включают экстракты ооцитов (например, экстракты ооцитов коровы или морского ежа) и экстракты мужских половых клеток (например, экстракты сперматогониев, сперматоцитов, сперматид или сперматозоидов позвоночных, беспозвоночных или млекопитающих, таких как бык). Донорная или пермеабилизованная клетки могут быть неиммортализованными или естественным образом, спонтанно или генетически иммортализованными. Донорная клетка, пермеабилизованная клетка, реципиентная клетка или цитопласт могут быть из источника любого возраста, такого как эмбрион, плод, молодое и взрослое млекопитающее (например, копытное животное). Клетки из более молодых источников могут иметь меньше приобретенных спонтанных мутаций и могут иметь более длительную продолжительность жизни после введения в ооцит. Способы размножения копытных животных В предпочтительных вариантах осуществления любого из описанных выше способов создания копытных животных или клеток копытных животных проводят спаривание копытного животного по изобретению с другим копытным животным (например, копытным животным с мутацией в нуклеиновой кислоте тяжелой или легкой цепи антитела или копытным животным с нуклеиновой кислотой, кодирующей ксеногенное антитело), чтобы получить плод или живое потомство с двумя или более генетическими модификациями. Предпочтительно, одну или несколько клеток выделяют из плода или потомства и вводят одну или несколько дополнительных генетических модификаций в изолированную клетку(ки). Способы получения антител Изобретение также относится к способу получения антител с использованием копытного животного по изобретению, которое экспрессирует ксеногенные антитела (например, антитела человека). Один такой способ заключается во введении одного или нескольких интересующих антигенов копытному животному по изобретению, имеющему нуклеиновую кислоту, кодирующую генный локус ксеногенного антитела. Сегменты нуклеиновой кислоты в генном локусе подвергаются перестройке, что приводит к синтезу антител, специфичных по отношению к антигену. Антитела выделяют из организма копытного животного. Антитела могут быть моноклональными или поликлональными и, предпочтительно, взаимодействующими с интересующим антигеном. Предпочтительно, антитела выделяют из сыворотки или молока копытного животного. В связанном аспекте изобретение относится к другому способу получения антител, который включает в себя выделение ксеногенных антител из организма копытного животного по изобретению, имеющего нуклеиновую кислоту, кодирующую генный локус ксеногенного антитела. Сегменты нуклеиновой кислоты в генном локусе подвергаются перестройке, что приводит к синтезу ксеногенных антител. Антитела могут быть моноклональными или поликлональными и, предпочтительно, реагируют с интересующим антигеном. Предпочтительно, антитела выделяют из сыворотки или молока копытного животного. Нуклеиновые кислоты Изобретение также относится к нуклеиновым кислотам (например, векторам), которые могут использоваться для мутирования гена, кодирующего прион, у копытных животных (например, коров). Одна такая нуклеиновая кислота имеет кассету, которая содержит по порядку от 5′- к 3′-концу первую гомологичную область по существу идентичной последовательности первой области эндогенной нуклеиновой кислоты, которая кодирует прионный белок клетки копытного животного, маркер позитивной селекции и вторую гомологичную область, по существу идентичную последовательности второй области нуклеиновой кислоты, которая кодирует прионный белок. В одном варианте осуществления первая гомологичная область (например, область длиной менее 3; 2; 1,5 или 1 тысяч пар оснований) короче второй гомологичной области (например, области длиной по меньшей мере 7, 8, 9 или 10 тысяч пар оснований), и кассета способна интегрироваться в эндогенную нуклеиновую кислоту, которая кодирует прионный белок клетки. В другом варианте осуществления первая гомологичная область длиннее, чем вторая гомологичная область. Предпочтительно, первая и/или вторая гомологичные области являются изогенными соответствующей области эндогенной нуклеиновой кислоты, которая кодирует прионный белок в мутируемой клетке. В связанном аспекте изобретение относится к нуклеиновой кислоте, которая имеет кассету, которая содержит первую гомологичную область, по существу идентичную последовательности первой области эндогенной нуклеиновой кислоты, которая кодирует прионный белок клетки копытного животного, маркер позитивной селекции, вторую гомологичную область, по существу идентичную последовательности второй области нуклеиновой кислоты, которая кодирует прионный белок, и маркер негативной селекции (например, DT-A или Tk) в ориентации, противоположной ориентации маркера позитивной селекции (например, маркер негативной селекции, промотор для которого связан с концом более короткой гомологичной области). Кассета способна интегрироваться в эндогенную кодирующую прионный белок нуклеиновую кислоту клетки. Желательно, чтобы одна гомологичная область имела длину по меньшей мере 7, 8, 9 или 10 тысяч пар оснований, а другая гомологичная область имела длину менее 3; 2; 1,5 или 1 тысячи пар оснований. В некоторых вариантах осуществления часть или весь маркер негативной селекции находится в более короткой гомологичной области. Предпочтительно, первая и/или вторая гомологичные области являются изогенными соответствующей области эндогенной нуклеиновой кислоты, которая кодирует прионный белок в мутируемой клетке. В другом аспекте изобретение относится к композиции, которая содержит нуклеиновую кислоту по данному изобретению и спермидин (например, от 0,1 до 10 мМ спермидина). Предпочтительные варианты осуществления указанных выше аспектов Предпочтительно, антисыворотка или молоко копытного животного содержит поликлональные иммуноглобулины человека. Предпочтительно, антисыворотка или молоко получены от коровы, овцы, свиньи или козы. В другом предпочтительном варианте осуществления Ig направлены против желаемого антигена. В предпочтительных вариантах осуществления антисыворотку используют в качестве внутривенно вводимого иммуноглобулина (IVIG) для лечения или профилактики заболевания у людей. В другом предпочтительном варианте осуществления интересующий антиген вводят копытному животному и копытное животное продуцирует Ig, направленные против антигена. Предпочтительно, сегменты нуклеиновой кислоты в генном локусе ксеногенного иммуноглобулина подвергаются перестройке и продуцируются ксеногенные антитела, реагирующие с интересующим антигеном. Предпочтительно, антисыворотка и/или молоко содержат по меньшей мере в 2, 5, 10, 20 или 50 раз больше ксеногенного антитела, чем эндогенного антитела, или не содержат эндогенного антитела. При желании можно получить гибридомы и моноклональные антитела, используя ксеногенные B-клетки, полученные из описанных выше трансгенных копытных животных (например, трансгенных коров). Также предполагается, что ксеногенные антитела (например, антитела человека), выделенные из копытных животных, могут быть затем химически модифицированы так, чтобы они были ковалентно связаны с токсином, терапевтически активным соединением, ферментом, цитокином, радиоактивной меткой, флуоресцирующей меткой или аффинной меткой. При желании флуоресцирующая или радиоактивная метка могут быть использованы для визуализации антитела in vitro или in vivo. Предпочтительно, мутация в нуклеиновой кислоте, которая кодирует прионный белок, уменьшает образование инфекционной формы прионного белка. В предпочтительных вариантах осуществления количество инфекционной формы прионного белка, продуцируемого копытным животным, составляет менее 80, 70, 60, 50, 40, 30, 20, 10 или 5% от количества, продуцируемого контрольным копытным животным без мутации. Предпочтительно, продукция прионного инфекционного белка отсутствует. В предпочтительных вариантах осуществления мутация является инсерцией части или всей кодирущей прионный белок нуклеиновой кислоты (например, кодирующей области) из другого животного (например, копытного животного другого вида или породы). Предпочтительно, этой ксеногенной кодирующей прионный белок нуклеиновой кислотой заменяют часть или всю эндогенную нуклеиновую кислоту, которая кодирует прионный белок. В предпочтительных вариантах осуществления ксеногенная нуклеиновая кислота, которая кодирует прионный белок, получена из вида или породы с повышенной устойчивостью к прионной инфекции, такого как овцы, которые резистентны к скрепи. Предпочтительно, процент копытных животных с мутацией, которые имеют прионную инфекцию, составляет менее 80, 70, 60, 50, 40, 30, 20, 10 или 5% от процента контрольных копытных животных без мутации, которые имеют прионную инфекцию. Предпочтительные копытные животные и донорные клетки Копытные животные включают представителей отряда Perissodactyla и Artiodactyla, таких как представитель рода Bos. Другие предпочтительные копытные животные включают овец, снежных баранов, коз, буйволов, антилоп, рогатый скот, лошадей, ослов, мулов, оленей, лосей, карибу, азиатских буйволов, верблюдов, лам, альпака, свиней и слонов. Предпочтительные донорные клетки включают дифференцированные клетки, такие как эпителиальные клетки, нервные клетки, эпидермальные клетки, кератиноциты, гематопоэтические клетки, меланоциты, хондроциты, B-лимфоциты, T-лимфоциты, эритроциты, макрофаги, моноциты, фибробласты и мышечные клетки; и недифференцированные клетки, такие как эмбриональные клетки (например, стволовые клетки и эмбриональные стволовые клетки). В другом предпочтительном варианте осуществления клетка получена из женской репродуктивной системы, такой как клетка молочной железы, кумулюса яичника, гранулезы или яйцевода. Другие предпочтительные клетки включают эмбриональные клетки и плацентарные клетки. Предпочтительные клетки также включают клетки из любого органа, такого как мочевой пузырь, головной мозг, пищевод, фаллопиева труба, сердце, кишечник, желчный пузырь, почка, печень, легкое, яичники, поджелудочная железа, предстательная железа, спинной мозг, селезенка, желудок, семенники, тимус, щитовидная железа, трахея, мочеточник, уретра и матка. Предпочтительно, донорная клетка, донорное ядро, донорная хроматиновая масса или реконструированный ооцит не являются тетраплоидными. В еще одном предпочтительном варианте осуществления ядро, пермеабилизованная клетка или хромосомы получены из трансгенной клетки или копытного животного или содержат мутацию, не обнаруженную в донорной клетке или в природной клетке. Предпочтительные трансгенные донорные ядра и донорные клетки кодируют белки, которые придают клонированному копытному животному повышенную устойчивость к заболеванию или паразитам. Альтернативно донорные ядра или донорные клетки можно конструировать так, чтобы клонированное копытное животное продуцировало рекомбинантный продукт, а именно чтобы происходила продукция белка человека в моче, крови или молоке коровы. Например, белки могут быть экспрессированы в моче крупного рогатого скота путем встраивания полинуклеотидной последовательности, кодирующей белок человека, под контролем промотора уроплакина. Терапевтические белки, которые могут быть продуцированы в молоке клонированных коров, включают факторы свертывания крови человека, такие как любой из факторов I-XIII (Voet and Voet, Biochemistry, John Wiley and Sons, New York, 1990). Указанные гетерологичные белки могут быть экспрессированы под контролем промотора пролактина или любого другого промотора, подходящего для экспрессии в молоке коров. Рекомбинантные белки из этих и других тканей или жидкостей можно очистить, используя стандартные способы очистки (смотри, например, Ausubel et al., выше). ОПРЕДЕЛЕНИЯ В этом описании под используемым термином «искусственная хромосома» понимают хромосому млекопитающего или ее фрагмент, который имеет искусственную модификацию, такую как добавление селектируемого маркера, добавление сайта клонирования, делеция одного или нескольких нуклеотидов, замена одного или нескольких нуклеотидов и тому подобное. Под «искусственной хромосомой человека» или «HAC» подразумевают искусственную хромосому, созданную из одной или нескольких хромосом человека. Искусственная хромосома может сохраняться в клетке-хозяине независимо от эндогенных хромосом клетки-хозяина. В этом случае HAC может стабильно реплицироваться и сегрегирует в направлении сегрегации эндогенных хромосом. Альтернативно она может быть подвергнута транслокации или инсерции в эндогенную хромосому клетки-хозяина. Две или более искусственные хромосомы можно ввести в клетку-хозяина одновременно или последовательно. Например, могут быть введены искусственные хромосомы, полученные из хромосомы человека Под «нуклеиновой кислотой в форме перед перестройкой или неперестроенной форме» подразумевается нуклеиновая кислота, которая не была подвергнута V(D)J-рекомбинации. В предпочтительных вариантах осуществления все сегменты нуклеиновой кислоты, кодирующие V-сегмент гена легкой цепи антитела, отделены от всех сегментов нуклеиновой кислоты, кодирующих J-сегмент гена, одним или несколькими нуклеотидами. Предпочтительно, все сегменты нуклеиновой кислоты, кодирующие V-сегмент гена тяжелой цепи антитела, отделены от всех сегментов нуклеиновой кислоты, кодирующих D-сегмент гена, одним или несколькими нуклеотидами, и/или все сегменты нуклеиновой кислоты, кодирующие D-сегмент гена тяжелой цепи антитела, отделены от всех сегментов нуклеиновой кислоты, кодирующих J-сегмент гена, одним или несколькими нуклеотидами. Предпочтительно, нуклеиновая кислота в своей неперестроенной форме в основном является человеческой. В других предпочтительных вариантах осуществления нуклеиновая кислота по меньшей мере на 70, 80, 90, 95 или 99% идентична соответствующей области природной нуклеиновой кислоты человека. «Уменьшение количества и/или активности эндогенного антитела» означает уменьшение количества эндогенных функциональных антител, продуцируемых B-клеткой или популяцией B-клеток, как описано, например, в U.S.S.N. 10/441503, поданной 19 мая 2003 г. Указанное снижение количества эндогенных антител может быть обусловлено уменьшением количества эндогенных антител, продуцируемых на B-клетку, уменьшением количества функциональных эндогенных B-клеток или сочетанием указанного. Предпочтительно, количество эндогенного антитела, секретируемого B-клеткой или экспрессируемого на поверхности B-клетки, экспрессирующей или секретирующей эндогенное антитело, уменьшается по меньшей мере на 25, 50, 75, 90 или 95%. В другом предпочтительном варианте осуществления количество эндогенных B-клеток в образце из млекопитающего-реципиента, таком как образец крови, снижено по меньшей мере на 25, 50, 75, 90 или 95%. «Бифункциональное антитело» означает антитело, которое включает антитело или фрагмент антитела, ковалентно связанный с другим антителом или другим фрагментом антитела. В одном предпочтительном варианте осуществления оба антитела или фрагмента связываются с разными эпитопами, экспрессированными на одном и том же антигене. Другие предпочтительные бифункцональные антитела связываются с двумя разными антигенами, например как легкая цепь антитела, так и тяжелая цепь антитела. Можно использовать стандартные способы молекулярной биологии, такие как способы, описанные в этой заявке, чтобы оперативно связать две нуклеиновые кислоты, так чтобы слитая нуклеиновая кислота кодировала бифункциональное антитело. «Фрагмент» означает полипептид, имеющий область следующих друг за другом аминокислот, которая идентична соответствующей области антитела по изобретению, но имеет длину, меньшую чем длина полной последовательности. Фрагмент обладает способностью связывать тот же самый антиген, который связывает соответствующее антитело, что определяется стандартными методами, такими как приведенные в этом описании. Предпочтительно, связывание фрагмента с антигеном составляет по меньшей мере 20, 40, 60, 80 или 90% от связывания соответствующего антитела. «Очищенный» означает отделенный от других компонентов, которые сопровождают его в природных условиях. Обычно фактор является в основном чистым, когда он по меньшей мере на 50% по массе не содержит белков, антител и природных органических молекул, с которым он ассоциирован в природе. Предпочтительно, фактор является чистым по меньшей мере на 75%, более предпочтительно по меньшей мере на 90% и наиболее предпочтительно по меньшей мере на 99% по массе. По существу чистый фактор может быть получен химическим синтезом, выделением фактора из природных источников или продуцированием фактора в рекомбинантной клетке-хозяине, которая не продуцирует фактор в природе. Белки, везикулы и органеллы могут быть очищены специалистом в данной области с использованием стандартных способов, таких как способы, описанные Ausubel et al. (см. выше). Фактор, предпочтительно, по меньшей мере в 2, 5 или 10 раз чище исходного вещества, что определяется с помощью электрофореза в полиакриламидном геле, хроматографии на колонке, измерений оптической плотности, анализа ВЭЖХ или вестерн-блот-анализа (Ausubel et al., см. выше). Предпочтительные способы очистки включают иммунопреципитацию, колоночную хроматографию, такую как иммуноаффинная хроматография, иммуноаффинная очистка на магнитных шариках и пэннинг с использованием связанного с планшетом антитела. Под «хроматиновой массой» подразумевается более чем одна хромосома, не окруженная мембраной. Предпочтительно, хроматиновая масса содержит все хромосомы клетки. Искусственно индуцированная хроматиновая масса, содержащая конденсированные хромосомы, может быть образована при экспозиции ядра в митотических перепрограммирующих средах (например, экстракте митотических клеток), как указано в этом описании. Альтернативно, искусственно индуцированная хроматиновая масса, содержащая деконденсированные или частично конденсированные хромосомы, может быть создана при воздействии на ядро одного из следующих условий, которые указаны в этом описании: экстракт митотических клеток, содержащий анти-NuMA-антитело, раствор детергента и/или соли или раствор протеинкиназы. Хроматиновая масса может содержать дискретные хромосомы, которые физически не соприкасаются друг с другом, или может содержать две или более хромосом, которые физически контактируют. При желании уровень конденсации хромосом можно определить, используя стандартные способы, путем измерения интенсивности окрашивания красителем ДНК DAPI. По мере того как хромосомы конденсируются, интенсивность такого окрашивания увеличивается. Таким образом, интенсивность окрашивания хромосом можно сравнить с интенсивностью окрашивания деконденсированных хромосом в интерфазе (определенных как 0%-конденсированные) и максимально конденсированных хромосом в митозе (определенных как 100%-конденсированные). На основании данного сравнения можно определить процент максимальной конденсации. Предпочтительные конденсированные хроматиновые массы конденсированы по меньшей мере на 50, 60, 70, 80, 90 или 100%. Предпочтительные деконденсированные или частично конденсированные хроматиновые массы конденсированы менее чем на 50, 40, 30, 20 или 10%. «Ядро» означает связанную с мембраной органеллу, содержащую большую часть или всю ДНК клетки. ДНК уложена в хромосомы в деконденсированной форме. Предпочтительно, мембрана, инкапсулирующая ДНК, содержит один или два липидных бислоя или имеет нуклеопорины. «Ядро, которое имеет менее четырех наборов гомологичных хромосом» означает ядро, которое имеет содержание ДНК менее чем 4n, где «n» означает количество хромосом, встречающееся в нормальном гаплоидном наборе хромосом млекопитающего конкретного рода или вида. Такое ядро не имеет четырех копий каждого гена или генетического локуса. Предпочтительно, ядро является диплоидным и, следовательно, имеет два набора гомологичных хромосом, но имеет менее двух полных пар хроматид. «Пронуклеус» означает гаплоидное ядро, полученное в результате мейоза или пронуклеус, полученный в результате переноса ядра. Женским пронуклеусом является ядро ооцита или яйцеклетки перед слиянием с мужским пронуклеусом. Мужским пронуклеусом является ядро сперматозоида после его вхождения в ооцит или яйцеклетку при оплодотворении, но до слияния с женским пронуклеусом. Пронуклеусом в результате переноса ядра является пронуклеус (например, диплоидный пронуклеус), который образуется после введения донорной клетки, ядра или хроматиновой массы в ооцит. Пронуклеус в результате переноса ядра имеет менее четырех наборов гомологичных хромосом. Под «донорной клеткой» подразумевается клетка, из которой получено ядро или хроматиновая масса, или пермеабилизованная клетка. Под «пермеабилизацией» подразумевается образование пор в плазматической мембране или частичное или полное удаление плазматической мембраны. Под «средами для перепрограммирования» подразумевается раствор, который делает возможным удаление фактора из клетки, ядра, хроматиновой массы или хромосомы или добавление фактора из раствора в клетку, ядро, хроматиновую массу или хромосому. Предпочтительно, добавление или удаление фактора увеличивает или уменьшает уровень экспрессии мРНК или белка в донорной клетке, хроматиновой массе или ядре или в клетке, содержащей перепрограммируемую хроматиновую массу или ядро. В другом варианте осуществления инкубация пермеабилизованной клетки, хроматиновой массы или ядра в средах для перепрограммирования изменяет фенотип пермеабилизованной клетки или клетки, содержащей перепрограммируемую хроматиновую массу или ядро, по сравнению с фенотипом донорной клетки. В еще одном варианте осуществления инкубация пермеабилизованной клетки, хроматиновой массы или ядра в средах для перепрограммирования приводит к тому, что пермеабилизованная клетка или клетка, содержащая перепрограммируемую хроматиновую массу или ядро, приобретает или теряет активность по сравнению с донорной клеткой. Примеры сред для перепрограммирования включают такие растворы, как буферы, которые не содержат таких биологических молекул, как белки или нуклеиновые кислоты. Такие растворы могут использоваться для удаления одного или нескольких факторов из ядра, хроматиновой массы или хромосомы. Другими предпочтительными средами для перепрограммирования являются экстракты, такие как клеточные экстракты из клеточных ядер, цитоплазмы клеток или их комбинация. Примеры клеточных экстрактов включают экстракты из ооцитов (например, ооцитов млекопитающих, позвоночных или беспозвоночных), мужских половых клеток (половых клеток млекопитающих, позвоночных или беспозвоночных, таких как сперматогоний, сперматоцит, сперматида или сперматозоид) и стволовых клеток (например, стволовых клеток взрослого организма или эмбриональных стволовых клеток). Другими средами для перепрограммирования являются растворы или экстракты, к которым был добавлен один или несколько природных или рекомбинантных факторов (например, нуклеиновые кислоты или белки, такие как ДНК-метилтрансферазы, деацетилазы гистонов, гистоны, протамины, ядерные ламины, факторы транскрипции, активаторы, репрессоры, белки, ремоделирующие хроматин, факторы роста, интерлейкины, цитокины или другие гормоны), или экстракты, из которых был удален один или несколько факторов. Следующие среды для перепрограммирования включают растворы детергента (например, 0,01%-0,1%, 0,1%-0,5% или 0,5%-2% ионогенный или неионогенный детергент, такой как один или несколько из следующих детергентов: SDS, тритон X-100, тритон X-114, CHAPS, Na-дезоксихолат, н-октилглюкозид, Nonidet P40, IGEPAL, твин 20, твин 40 или твин 80), соли (например, ~0,1; 0,15; 0,25; 0,5; 0,75; 1; 1,5 или 2 М NaCl или KCl), полиамин (например, ~1 мкМ, 10 мкМ, 100 мкМ, 1 мМ или 10 мМ спермин, спермидин, протамин или поли-L-лизин), протеинкиназу (например, циклин-зависимую киназу 1, протеинкиназу C, протеинкиназу A, MAP-киназу, кальций/кальмодулин-зависимую киназу, казеинкиназу CK1 или казеинкиназу CK2) и/или ингибитор фосфатаз (например, ~10 мкМ, 100 мкМ, 1 мМ, 10 мМ, 50 мМ, 100 мМ одного или нескольких из следующих ингибиторов: Na-ортованадат, Na-пирофосфат, Na-фторид, NIPP1, ингибитор 2, PNUTS, SDS22, AKAP149 или окадаевая кислота). В некоторых вариантах осуществления среда для перепрограммирования содержит анти-NuMA-антитело. При желании, множество сред для перепрограммирования можно использовать одновременно или последовательно, чтобы перепрограммировать донорную клетку, ядро или хроматиновую массу. Под «интерфазными перепрограммирующими средами» подразумеваются среды (например, экстракт интерфазной клетки), которые индуцируют деконденсацию хроматина и образование ядерной оболочки. Под «митотическими перепрограммирующими средами» подразумеваются среды (например, экстракт митотической клетки), которые индуцируют конденсацию хроматина и разрушение ядерной оболочки. «Перепрограммированная клетка» означает клетку, на которую воздействовали средами для перепрограммирования. Предпочтительно, в перепрограммированной клетке экспрессируются по меньшей мере 1, 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 300 или более молекул мРНК или белка, которые не экспрессируются в донорной или пермеабилизованной клетке. В другом предпочтительном варианте осуществления количество молекул мРНК или белка, которые экспрессируются в перепрограммированной клетке, но не экспрессируются в донорной или пермеабилизованной клетке, составляет от 1 до 5, от 5 до 10, от 10 до 25, от 25 до 50, от 50 до 75, от 75 до 100, от 100 до 150, от 150 до 200 или от 200 до 300 включительно. Предпочтительно, в донорной или пермеабилизованной клетке экспрессируются по меньшей мере 1, 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 300 или более молекул мРНК или белка, которые не экспрессируются в перепрограммированной клетке. В другом предпочтительном варианте осуществления количество молекул мРНК или белка, которые экспрессируются в донорной или пермеабилизованной клетке, но не экспрессируются в перепрограммированной клетке, составляет от 1 до 5, от 5 до 10, от 10 до 25, от 25 до 50, от 50 до 75, от 75 до 100, от 100 до 150, от 150 до 200 или от 200 до 300 включительно. В еще одном предпочтительном варианте осуществления указанные молекулы мРНК или белка экспрессируются как в донорной клетке (т.е. донорной или пермеабилизованной исходной клетке), так и в перепрограммированной клетке, но уровни экспрессии в этих клетках отличаются по меньшей мере в 2, 5, 10 или 20 раз, что измеряется с использованием стандартных анализов (смотри, например, Ausubel et al., выше). «Добавление фактора» означает связывание фактора с хроматином, хромосомой или компонентом ядерной оболочки, таким как ядерная мембрана или ядерный матрикс. Альтернативно фактор импортируют в ядро так, чтобы он был связан или инкапсулирован ядерной оболочкой. Предпочтительно, количество фактора, который связан с хромосомой или расположен в ядре, увеличивается по меньшей мере на 25, 50, 75, 100, 200 или 500%. «Удаление фактора» означает диссоциацию фактора из хроматина, хромосомы или компонента ядерной оболочки, такого как ядерная мембрана или ядерный матрикс. Альтернативно фактор экспортируют из ядра, с тем чтобы он не был больше связан или инкапсулирован ядерной оболочкой. Предпочтительно, количество фактора, который связан с хромосомой или расположен в ядре, снижается по меньшей мере на 25, 50, 75, 100, 200 или 500%. «Обогащение или сокращение фактора» означает добавление или удаление природного или рекомбинантного фактора по меньшей мере на 20, 40, 60, 80 или 100% от количества фактора, исходно присутствующего в средах для перепрограммирования (например, клеточном экстракте). Альтернативно может быть добавлен природный или рекомбинантный фактор, который не присутствует в природных условиях в средах для перепрограммирования. Предпочтительные факторы включают белки, такие как ДНК-метилтрансферазы, деацетилазы гистонов, гистоны, протамины, ядерные ламины, факторы транскрипции, активаторы и репрессоры; мембранные везикулы и органеллы. В одном предпочтительном варианте осуществления фактор очищают перед добавлением к перепрограммирующим средам, как описано ниже. Альтернативно, можно использовать один из способов очистки, описанных ниже, чтобы удалить нежелательный фактор из сред для перепрограммирования. «Повторно клонированный» означает использованный во втором раунде клонирования. В частности, клетку из эмбриона, плода или взрослого организма, созданного на основе способов по изобретению, можно инкубировать в митотических перепрограммирующих средах (например, экстракте митотических клеток), чтобы образовать хроматиновую массу для введения в лишенный ядра ооцит, как описано выше. Альтернативно клетку можно пермеабилизовать, инкубировать в средах для перепрограммирования и ввести в лишенный ядра ооцит, как описано выше. Осуществление двух или более раундов клонирования может приводить к дополнительному перепрограммированию донорной хроматиновой массы или донорной клетки, повышая при этом возможность создания жизнеспособного потомка после последнего раунда клонирования. Под «жизнеспособным потомком» подразумевается млекопитающее, которое выживает ex utero. Предпочтительно, млекопитающее является живым в течение по меньшей мере одной секунды, одной минуты, одного часа, одного дня, одной недели, одного месяца, шести месяцев или одного года, начиная с момента его выхода из материнского организма-хозяина. Млекопитающее не требует для выживания системы кровообращения окружения in utero. «Ооцит с перенесенным ядром» или «ооцит с ядерным трансплантатом» означает ооцит, в который введена или слита донорная клетка, ядро или хроматиновая масса. Эмбрион, образованный из ооцита, называют эмбрионом «с перенесенным ядром» или «с ядерным трансплантатом». «Эмбрион» или «эмбриональный» означает развивающуюся клеточную массу, которая не была имплантирована в мембрану матки материнского организма-хозяина. Следовательно, термин «эмбрион» может относиться к оплодотворенному ооциту; ооциту, содержащему донорную хроматиновую массу, ядро или перепрограммированную клетку; развивающейся клеточной массе на стадии перед бластоцистой; или любой другой развивающейся клеточной массе, которая находится на стадии развития перед имплантацией в мембрану матки материнского организма-хозяина и перед образованием генитального гребня. Эмбрион может представлять собой множественные стадии развития клеток. Например, одноклеточный эмбрион может называться зиготой; сплошная сферическая масса клеток, полученная из подвергнутого дроблению эмбриона, может быть названа морулой, и эмбрион, имеющий бластоцель, может быть назван бластоцистой. «Эмбриональная клетка» является клеткой, выделенной из эмбриона или находящейся в эмбрионе. «Клетки, полученные из эмбриона» означают клетки, которые получены в результате клеточного деления клеток в эмбрионе. «Химерный эмбрион» означает эмбрион, образованный из клеток от двух или более эмбрионов. Полученный плод или потомок может иметь клетки, которые получены только из одного из исходных эмбрионов, или клетки, полученные из более чем одного из исходных эмбрионов. При желании процент клеток из каждого эмбриона, которые включены в плацентарную ткань и в ткань плода, можно определить, используя стандартный FISH-анализ или анализ мембранного красителя, добавленного к одному эмбриону. «Химерное копытное животное» означает копытное животное, образованное из клеток из двух или более эмбрионов. Копытное животное может иметь клетки, которые получены только из одного из исходных эмбрионов, или клетки, полученные более чем из одного из исходных эмбрионов. При желании процент клеток из каждого эмбриона, которые включены в плацентарную ткань и в ткань плода, можно определить, используя стандартный FISH-анализ или анализ мембранного красителя, добавленного к одному эмбриону. Под «предкомпактным эмбрионом» подразумевается эмбрион перед компактизацией. Предкомпактный эмбрион по существу не экспрессирует E-кадхерина на поверхности его бластомеров. Предпочтительные предкомпактные эмбрионы экспрессируют по меньшей мере в 3, 5, 10, 20, 30 или 40 раз меньше E-кадхерина, чем полностью компактный эмбрион того же самого вида, или не экспрессируют E-кадхерин. Под «компактным эмбрионом» подразумевается эмбрион, подвергающийся компактизации, или эмбрион после компактизации. Бластомеры компактного эмбриона экспрессируют E-кадхерин на своей поверхности. Такую экспрессию E-кадхерина можно измерить, используя стандартные способы с антителом против E-кадхерина. E-кадхерин увеличивает сцепление между бластомерами. Предпочтительные компактные эмбрионы включают эмбрионы, в которых процесс компактизации завершен. Другие предпочтительные компактные эмбрионы экспрессируют по меньшей мере в 3, 5, 10, 20, 30 или 40 раз больше E-кадхерина, чем предкомпактный эмбрион того же самого вида. «Плод» означает развивающуюся клеточную массу, которая была имплантирована в мембрану матки материнского организма-хозяина. Плод может иметь определенные характерные признаки, такие как генитальный гребень, который легко идентифицируется специалистом в данной области. «Эмбриональной клеткой» является любая клетка, выделенная из плода или находящаяся в плоде. «Партеногенез» или «партеногенетическая активация» означает развитие ооцита или яйцеклетки без слияния ее ядра с мужским пронуклеусом с образованием зиготы. Например, ооцит может быть индуцирован к делению без оплодотворения. «Zona pellucida» означает полупрозрачный, эластичный неклеточный слой, окружающий ооцит или яйцеклетку многих млекопитающих. «Трофэктодерма» означает самый крайний слой клеток, окружающих бластоцель во время стадии бластоцисты в эмбриональном развитии млекопитающего. Трофэктодерма дает начало большей части или всей плацентарной ткани при дальнейшем развитии. «Внутренняя клеточная масса» означает клетки, окруженные трофэктодермой. Клетки внутренней клеточной массы дают начало большей части тканей плода при дальнейшем развитии. «мРНК или белок, специфичный по отношению к одному типу клеток» означает мРНК или белок, который экспрессируется в одном типе клеток на уровне, который по меньшей мере в 10, 20, 50, 75 или 100 раз выше, чем уровень экспрессии во всех других типах клеток. Предпочтительно, мРНК или белок экспрессируется только в одном типе клеток. «Мутация» означает изменение природной или эталонной последовательности нуклеиновой кислоты, такое как инсерция, делеция, мутация сдвига рамки, молчащая мутация, нонсенс-мутация или миссенс-мутация. Предпочтительно, аминокислотная последовательность, кодируемая последовательностью нуклеиновой кислоты, имеет по меньшей мере одно аминокислотное изменение по сравнению с природной последовательностью. Примеры способов рекомбинантной ДНК для изменения геномной последовательности клетки, эмбриона, плода или млекопитающего включают встраивание последовательности ДНК из другого организма (например, человека) в геном, делетирование одной или нескольких последовательностей ДНК и введение мутаций одного или нескольких оснований (например, сайт-направленные или случайные мутации) в последовательность ДНК мишени. Примеры способов получения указанных модификаций включают ретровирусную инсерцию, способы на основе искусственной хромосомы, генную инсерцию, случайную инсерцию с тканеспецифичными промоторами, гомологичную рекомбинацию, направленное изменение генов, транспозируемые элементы и любой другой способ введения чужеродной ДНК. Все указанные способы хорошо известны специалистам в области молекулярной биологии (смотри, например, Ausubel et al., выше). В способах по изобретению можно использовать хроматиновые массы, хромосомы и ядра из трансгенных клеток, содержащих модифицированную ДНК, или донорных трансгенных клеток. «Иммортализованная» означает способность подвергаться по меньшей мере на 25, 50, 75, 90 или 95% большему количеству клеточных делений, чем природная контрольная клетка того же самого клеточного типа, рода и вида, что иммортализованная клетка, или чем донорная клетка, из которой получена иммортализованная клетка. Предпочтительно, иммортализованная клетка способна подвергаться по меньшей мере в 2, 5, 10 или 20 раз большему количеству клеточных делений, чем контрольная клетка. Более предпочтительно, иммортализованная клетка способна подвергаться неограниченному количеству клеточных делений. Иммортализованные клетки включают клетки, которые естественным образом приобретают мутацию in vivo или in vitro, что изменяет нормальный процесс регуляции их роста. Еще одни предпочтительные иммортализованные клетки включают клетки, которые были генетически модифицированы так, чтобы экспрессировать онкоген, такой как ras, myc, abl, bcl2 или neu, или которые были инфицированы трансформирующим ДНК- или РНК-вирусом, таким как вирус Эпштейн-Барра или вирус SV40 (Kumar et al., Immunol. Lett. 65: 153-159, 1999; Knight et al., Proc. Nat. Acad. Sci. USA 85: 3130-3134, 1988; Shammah et al., J. Immunol. Methods 160-19-25, 1993; Gustafsson and Hinkula, Hum. Antibody Hybridomas 5: 98-104, 1994; Kataoka et al., Differentiation 62: 201-211, 1997; Chatelut et al., Scand. J. Immunol. 48: 659-666, 1998). Клетки также могут быть модифицированы так, чтобы они экспрессировали ген теломеразы (Roques et al., Cancer Res. 61: 8405-8507, 2001). «Неиммортализованная» означает не подвергнутая иммортализации, как описано выше. «Вызывающее слияние соединение» означает соединение, которое увеличивает вероятность того, что хроматиновая масса или ядро встраиваются в реципиентную клетку в том случае, если они расположены вблизи клетки. Например, вызывающее слияние соединение может увеличивать аффинность хроматиновой массы или ядра по отношению к плазматической мембране клетки. Вызывающее слияние соединение также может стимулировать объединение ядерной мембраны ядра с плазматической мембраной клетки. «По существу идентичная» означает имеющая последовательность, которая по меньшей мере на 60, 70, 80, 90 или 100% идентична другой последовательности. Идентичность последовательностей обычно измеряют, используя компьютерную программу для анализа последовательностей с установленными в ней параметрами по умолчанию (например, пакет компьютерных программ для анализа последовательностей Genetics Computer Group, University of Wisconsin Biotechnology Center, 1710 University Avenue, Madison, WI 53705). Указанная компьютерная программа сравнивает сходные последовательности, оценивая частоту различных замен, делеций и других модификаций. ПРЕИМУЩЕСТВА Данное изобретение предоставляет ряд преимуществ, связанных с получением трансгенных копытных животных (например, коров) с мутацией в одном или обоих аллелях гена, кодирующего прион. Например, нокаутирующий по приону вектор, способ культивирования клеток и способ трансфекции, приведенные в этом описании, инактивировали кодирующий прионный белок ген в клетках коровы с неожиданно высокой частотой. Большое количество целенаправленно измененных донорных клеток в комбинации с усовершенствованными способами переноса ядер по изобретению (например, перенос хроматина и SLOT) значительно облегчают создание клонированных трансгенных копытных животных (например, коров) с пониженной экспрессией или не экспрессирующие прионный белок. Такие полученные трансгенные копытные животные являются желательными источниками сельскохозяйственных продуктов (например, мяса) и фармацевтических продуктов (например, терапевтических антител человека, кодируемых одной или несколькими ксеногенными нуклеиновыми кислотами у трансгенных копытных животных). Другие отличительные признаки и преимущества изобретения будут очевидны из следующего подробного описания и формулы изобретения. КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ Заявка содержит чертежи, выполненные в цвете (фиг. 31, 34A-34C, 36A, 36B, 42A-42D, 45A, 46D, 47A, 47C, 48, 49B, 50A-50D и 51C-51E). Копии данного патента или заявки на выдачу патента с цветными чертежами будут представлены Ведомством при запросе и оплате необходимой пошлины. На фиг.1A представлен обзор способов, используемых для получения коровы, которая содержит Ig-нокаут и искусственную хромосому человека. Линия времени на фиг.1A основана на предполагаемых 18 месяцах для получения нокаутирующего по Ig вектора и создания нокаутированных клеток, 2 месяцах для создания плодов из нокаутированных клеток, 9 месяцах для осуществления последующих нокаутов, 9 месяцах периода беременности для того, чтобы родились телята, 12 месяцах до того, как можно будет получить эмбрионы от телочек, и 6 месяцах для осуществления переносов HAC. На фиг.1B представлен обзор способов, используемых для получения коровы, которая содержит мутацию в эндогенном гене Ig и содержит На фиг. 2A представлена нокаутирующая по мю (тяжелая цепь IgM) конструкция по изобретению. Фигура 2B является рестрикционной картой локусов иммуноглобулина крупного рогатого скота голштинской породы. На фиг. 3A-3B представлены схематичное изображение конструкций pSTneoB и pLoxP-StneoB, которые соответственно были использованы для получения мю-нокаутирующей конструкции ДНК. На фиг.3C представлена мю-нокаутирующая конструкция ДНК. На фиг.3D представлена полинуклеотидная последовательность области геномного локуса тяжелой цепи мю коровы длиной 1,5 т.п.о., которую использовали в качестве первой гомологичной области в мю-нокаутирующей конструкции (SEQ ID NO: 47). На фиг.3E представлена полинуклеотидная последовательность области геномного локуса тяжелой цепи мю коровы длиной 3,1 т.п.о., которую использовали в качестве второй гомологичной области в мю-нокаутирующей конструкции (SEQ ID NO: 48). В этой последовательности каждый «n» означает любой нуклеотид или отсутствие нуклеотида. Область следующих друг за другом нуклеотидов «n» представляет собой область длиной примерно от 0,9 до 1,0 т.п.о., полинуклеотидная последовательность которой не была определена. На фиг.3F представлена резистентная к пуромицину конструкция, нокаутирующая тяжелую цепь мю коровы. На фиг.3G представлена полинуклеотидная последовательность кДНК легкой цепи каппа коровы (SEQ ID NO: 60). Всю или часть этой последовательности можно использовать в конструкции, нокаутирующей по легкой цепи каппа. Кроме того, указанную легкую цепь каппа можно использовать для выделения геномной последовательности легкой цепи каппа для применения в конструкции, нокаутирующей по легкой цепи каппа. На фиг. 4 представлена схематично конструкция Фиг. 5 является фотографией агарозного геля, показывающей присутствие геномной ДНК, кодирующей тяжелую и легкую цепи человека, в плодах Фиг. 6 является фотографией агарозного геля, показывающей экспрессию Cмю-экзонов 3 и 4 человека в плоде Фиг. 7 является фотографией агарозного геля, показывающей перестройку эндогенной тяжелой цепи коровы в плоде Фиг. 8 является фотографией агарозного геля, показывающей экспрессию перестроенной тяжелой цепи человека в плоде Фиг. 9 является фотографией агарозного геля, показывающей экспрессию сплайсированной константной области из локуса тяжелой цепи человека в плоде Фиг. 10 является фотографией агарозного геля, показывающей экспрессию перестроенной тяжелой цепи человека в плоде На фиг. 11A представлена полинуклеотидная последовательность подвергнутого перестройке транскрипта тяжелой цепи человека из плода На фиг. 11B представлено выравнивание последовательностей области этой последовательности («запрос») с анти-пневмококковым антителом человека («объект») (SEQ ID NO: 50 и 51 соответственно). В случае запрашиваемой последовательности из плода На фиг.12A-12B изображены две дополнительные полинуклеотидные последовательности (SEQ ID NO: 52 и 54) и рассчитанные на их основе аминокислотные последовательности (SEQ ID NO: 53 и 55 соответственно) подвергнутых перестройке транскриптов тяжелой цепи человека из плода Фиг. 13 является фотографией агарозного геля, демонстрирующей, что плод Фиг. 14 является фотографией агарозного геля, демонстрирующей, что плоды Фиг. 15 является фотографией агарозного геля, показывающей экспрессию сплайсированной константной области мю из локуса тяжелой цепи человека в плоде Фиг. 16 является фотографией агарозного геля, показывающей перестройку и экспрессию локуса тяжелой цепи человека в плоде Фиг. 17 является фотографией агарозного геля, показывающей перестройку и экспрессию локуса лямбда Ig человека в плодах Фиг. 18 является фотографией агарозного геля, показывающей перестройку и экспрессию локуса лямбда Ig человека в плоде Фиг. 19 является фотографией агарозного геля, показывающей перестройку и экспрессию локуса лямбда Ig человека в плоде На фиг.20 представлена полинуклеотидная последовательность и соответствующая рассчитанная аминокислотная последовательность подвергнутого перестройке транскрипта легкой цепи человека из плода На фиг.21 представлена другая полинуклеотидная последовательность и соответствующая рассчитанная аминокислотная последовательность подвергнутого перестройке транскрипта легкой цепи человека из плода На фиг. 22A-22H представлены графики, полученные с помощью FACS-анализа экспрессии белков легкой цепи лямбда человека и тяжелой цепи коровы в плодах На фиг. 23 представлен схематично вектор, нокаутирующий по На фиг. 24 представлена схематичная иллюстрация BamHI-XhoI-фрагмента, содержащего экзоны 2, 3 и 4, который использовали в качестве остова AAV-вектора направленного воздействия. Маркер устойчивости к неомицину использовали для инсерционного мутагенеза локуса путем инсерции в экзон 4. Показано положение сайтов отжига для ПЦР-праймеров, которые использовали для последующего подтверждения соответствующего направленного воздействия. На фиг. 25 схематично представлено конструирование основанной на аденоассоциированном вирусе конструкции, предназначенной для удаления эндогенной последовательности IgH коровы. Фиг. 26 является фотографией агарозного геля, показывающей ПЦР-анализ отдельно трансдуцированных клонов в отношении соответствующих направленных событий. Вектор, используемый в этом эксперименте, показан на фиг.25. ПЦР-продукты, свидетельствующие о соответствующем попадании в мишень, отмечены звездочками. На фиг. 27 представлена таблица, в которой указаны частоты наступления беременности в случае эмбрионов, несущих HAC. Фиг. 28A-28J являются фотографиями, полученными в процессе FACS-анализа лимфоцитов периферической крови либо из экспериментальных плодов, инъецированных антителом против IgM коровы (das6), чтобы ингибировать развитие B-клеток, либо из контрольных плодов. Фигуры 28A-28E являются фотографиями FACS-анализа, осуществленного с использованием антитела против IgM коровы, чтобы выявить молекулы IgM, экспрессируемые на поверхности B-клеток. Как показано на фиг. 28A и фиг. 28B, примерно от 19,82 до 26,61% лимфоцитов периферической крови из контрольных плодов экспрессировали IgM. В отличие от этого 7,78, 11,80 или 3,95% лимфоцитов периферической крови из трех плодов, инъецированных антителом против IgM коровы, экспрессировали IgM (фиг. 28C-28E соответственно). Фигуры 28F-28J являются фотографиями FACS-анализа, осуществленного с использованием антитела против легкой цепи коровы (das10), чтобы выявить молекулы легкой цепи антитела, экспрессируемые на поверхности B-клеток. Как показано на фиг. 28F и фиг.28G, примерно от 12,43 до 29,47% лимфоцитов периферической крови из контрольных плодов экспрессировали молекулы легкой цепи антитела. В отличие от этого 2,54, 13,77 или 3,99% лимфоцитов периферической крови из трех плодов, инъецированных антителом против IgM коровы, экспрессировали молекулы легкой цепи антитела (фиг. 28H-28J соответственно). Фиг. 29A-29B иллюстрируют иммунодетекцию белков ядерной оболочки и ядерного матрикса в предимплантационных эмбрионах коровы. Фиг. 29A представляет собой изображение оплодотворенных in vitro эмбрионов коровы в стадии пронуклеусов и 8-клеточной стадии, исследованных с использованием тех же самых антител. Стрелками на фиг. 29A указано мечение анти-NuMA и анти-AKAP95 в женском пронуклеусе эмбрионов на стадии пронуклеусов. Вставки на фиг. 29A являются изображениями ДНК, меченой 0,1 мкг/мл Hoechst 33342 (масштаб столбиков 20 мкм). На фиг. 29B представлены результаты анализа с помощью иммуноблотинга фибробластов коровы (верхний ряд) и эмбрионов на стадии пронуклеусов (нижний ряд). Маркеры молекулярной массы показаны на фиг. 29B в кДа справа. Фиг. 30 является фотографией донорных фибробластов коровы (донорная клетка), эмбрионов с ядерными трансплантатами на стадии преждевременной конденсации хроматина (три часа после слияния), эмбрионов с ядерными трансплантатами на стадии пронуклеусов (19 часов после слияния) и партеногенетических эмбрионов на стадии пронуклеусов, активированных, как указано в этом описании. Разрушение донорного ядра и сборку нового пронуклеуса контролировали на стадии преждевременной конденсации хроматина через три часа после инъекции «hpi» («PCC») и через семь часов после инъекции («NT PN»), используя антитела против ламина B, ламинов A/C, NuMA и AKAP95. Женские пронуклеусы, образованные после активации партеногенеза ооцитов MII с использованием 10 мМ SrCl2, анализировали через пять часов после начала активирующей обработки («парт. PN»). Ламины A/C собирались в пронуклеусах эмбрионов коров с ядерными трансплантатами на стадии пронуклеусов. ДНК контрастно красили, используя 0,1 мкг/мл Hoechst 33342. ТРИТЦ относится к мечению вторыми антителами, конъюгированными с ТРИТЦ (масштаб столбиков 20 мкм). На фиг. 31 представлен график, демонстрирующий, что AKAP95 сильнее заякорен в пронуклеусах эмбрионов с ядерными трансплантатами по сравнению с партеногенетическими эмбрионами. Этот график показывает относительный процент неэкстрагированного ламина B, AKAP95 и мечения ДНК в пронуклеусе партенотов, эмбрионов с ядерными трансплантатами и соматических донорных ядрах после экстракции in situ с помощью 0,1% тритона X-100 и 1 мг/мл ДНКазы I вместе с 100 или 300 мМ NaCl в течение 30 минут при комнатной температуре перед фиксацией 3% параформальдегидом. Локализацию ламинов B-типа (красный) и AKAP95 (зеленый) исследовали методом двойной иммунофлуоресценции. Количественно оценивали интенсивность флуоресцентного мечения в каждом случае – красном (ламин B), голубом (ДНК) и зеленом (AKAP95). Эталонное значение (100% не экстрагированных) представляет собой относительные количества окрашивания ламинов B-типа, ДНК и AKAP95 в эмбрионах или клетках, пермеабилизованных с использованием 0,1% тритона X-100 только перед фиксацией. Исследовали примерно 30 эмбрионов в каждой группе. Фиг. 32 является фотографией эмбрионов коров с ядерными трансплантатами на стадии пронуклеусов, полученных слиянием фибробластов и активацией ооцитов либо 5 мкМ иономицином в течение четырех минут, затем 10 мкг/мл циклогексимидом/2,5 мкг/мл цитохалазином D в течение четырех часов (b’,-), иономицином/циклогексимидом/цитохалазином D как в (b’), затем дополнительным культивированием в течение девяти часов с 10 мкг/мл циклогексимида (b”, CHX) либо как в (b’) вместе с 1 мкг/мл актиномицина D в ходе полной активационной обработки (b”’). На тех же самых препаратах использовали антитела против ламина B (кроличьи поликлональные) и против ламинов A/C (мАт). Вставки являются изображениями мечения ДНК 0,1 мкг/мл Hoechst 33342 (масштаб столбиков 20 мкм). На фиг. 33 представлен график конденсации хромосом и разрушения ядерной оболочки в цитоплазматическом экстракте митотических клеток (M-S15), цитозольном экстракте митотических клеток (M-S200) и экстракте ооцитов (MII-S15) (n=300-400 ядер, исследованных в 3-5 повторах). На фиг. 34A-34C представлена серия снимков, полученных в ходе анализа иммунофлуоресценции очищенных исходных ядер фибробластов коровы (фиг. 34A) и конденсированного хроматина, полученного в цитозольном экстракте митотических клеток (фиг. 34B) и экстракте ооцитов (фиг. 34C). Исследовали указанные маркеры ядер. ДНК контрастно красили йодидом пропидия (красный) (масштаб столбиков 10 мкм). На фиг. 35 представлена серия фотографий, полученных в ходе иммунофлуоресцентного анализа конденсированного хроматина, получаемого в ооцитах в случае следования традиционным способам ядерных трансплантатов (NT) или инъекции ядер (NI) и после инъекции хроматиновых масс в ооциты (CT) с использованием способов по данному изобретению. По-видимому оба регистрируемых ламина B и A/C являются растворимыми (масштаб столбиков 10 мкм). На фиг. 36A-36B представлена серия фотографий иммунофлуоресцентного анализа пронуклеусов, полученных в результате переноса хроматина, ядерной трансплантации или инъекции ядер. Эмбрионы фиксировали через 19 часов после ядерной трансплантации, инъекции ядер или переноса хроматина и метили. Также исследовали контрольные партеногенетические пронуклеусы (парт.). На фиг. 36A показан анализ ламинов A/C и B. На фиг. 36B показан анализ AKAP95 и NuMA. Ламины A/C (зеленая метка) видны только в пронуклеусах после трансплантации ядер и инъекции ядер (масштаб столбиков 30 мкм). Фиг. 37 является схематичной иллюстрацией способа получения клонированных Tc-телят. Показано конструирование HAC с областями hChr22 и hChr14, содержащими гены Ig Фиг. 38A-38D иллюстрируют анализ Tc-плодов. G418-селекция регенерированной линии Tc-фибробластов и контрольных нетрансгенных фибробластов (фиг. 38A). Геномная ПЦР локусов IgH и Ig На фиг. 39A-39C представлены фотографии, иллюстрирующие анализ клонированных Tc-телят. Четыре клонированных Tc-теленка: теленок мужского пола ( Фиг. 40A-40B иллюстрируют распределение иммунофлуоресценции ламина B, ламина A/C, NuMA и AKAP95 в эмбриональных фибробластах коровы (фиг. 40A) и эмбрионах коровы на стадии пронуклеусов и стадии 8-16 клеток, культивированных in vitro (фиг. 40B). Стрелки указывают мечение NuMA и AKAP95 в женском пронуклеусе (наименьшее из двух пронуклеусов). На вставках – ДНК, меченая 0,1 мкг/мл Hoechst 33342. Фиг. 40C иллюстрирует анализ методом иммуноблоттинга ламина B, ламина A/C, NuMA и AKAP95 в коровьих фибробластах (фиб.) и эмбрионах на стадии пронуклеусов (PN). Масштаб столбиков 10 мкм (фиг. 40A), 50 мкм (фиг. 40B). Фиг. 41A-41D иллюстрируют динамику ядерной оболочки, NuMA и AKAP95 в коровьих NT-эмбрионах. Распад ядра донорного фибробласта и сборку нового пронуклеуса контролировали с помощью иммунофлуоресценции на стадии PCC и PN, используя индикаторные антитела (ТРИТЦ) (фиг. 41A). Также исследовали партеногенетические пронуклеусы после сходной процедуры активации (парт. PN). Показан процент (±с.о.) эмбрионов, меченых каждым антителом (n~20 эмбрионов/группу в трех повторах). Реципиентные ооциты активировали либо 5 мкМ иономицином в течение четырех минут, затем 10 мкг/мл CHX/2,5 мкг/мл цитохалазина D в течение четырех часов (b), иономицином/CHX/цитохалазином D, как в (b), затем 10 мкг/мл CHX в течение девяти часов (b’) или как в (b) вместе с 5 мкг/мл ActD в ходе активирующей обработки (b”) (фиг. 41B). ДНК (вставки в B) метили Hoechst 33342. Масштаб столбиков 10 мкм. На фиг. 41C представлена столбчатая диаграмма средней ± с.о. интенсивности иммунофлуоресценции ламинов A/C и B и NuMA в CHX- и ActD-обработанных NT-эмбрионах по сравнению с необработанными (-)NT-эмбрионами (100% флуоресценция; n=30 эмбрионов/группу в трех повторах). На фиг. 41D представлена столбчатая диаграмма внутриядерной ассоциации AKAP95 в пронуклеусах NT-эмбрионов. Партеноты (парт.) и NT-эмбрионы (NT) на стадии PN и донорные фибробласты (фиб.) экстрагировали 0,1% тритоном X-100/1 мг/мл ДНКазы I/300 мМ NaCl перед фиксацией. Ламин B и AKAP95 исследовали с помощью иммунофлуоресценции и ДНК красили Hoechst 33342. Данные показаны в виде средней ± с.о. интенсивности флуоресцентного мечения ламина B, ДНК и AKAP95 по сравнению с флуоресценцией в неэстрагированных партеногенетических или NT-эмбрионах или в соматических клетках (100% флуоресценция; n~30 эмбрионов/группу в трех повторах). Масштаб столбиков 10 мкм. Фиг. 42A-42D демонстрируют, что CT дает эмбрионы, лишенные ламина A/C и NuMA. Перераспределение ламинов A/C и B, NuMA и AKAP95 контролировали с помощью иммунофлуоресценции в ядрах фибробластов коров, инкубированных в экстракте из митотических фибробластов коровы (Chaudhary et al., J. Cell Biol. 122: 295- 306, 1993) (фиг. 42A). ДНК метили 0,1 мкг/мл йодида пропидия. Показано процентное содержание ядер, в которых наблюдается распад (исчезновение мечения) индикаторных маркеров (n > 200 ядер/маркер в трех повторах). PN-эмбрионы получали посредством CT, NT, NI или партеногенезом и исследовали распределение ламинов A/C и B, NuMA и AKAP95 с помощью иммунофлуоресценции через 14 часов после начала активации (Poccia et al., Trends Biochem. Sci. 17: 223-227, 1992 и Chaudhary et al., J. Cell Biol. 122: 295-306, 1993) (фиг. 42B). ДНК метили Hoechst 33342. Средняя ± с.о. интенсивность иммунофлуоресценции этих маркеров в CT-, NI- или партеногенетических эмбрионах по сравнению с интенсивностью флуоресценции в NT-эмбрионах на стадии PN (NT, 100% флуоресценция; n=15-20 эмбрионов/маркер в трех повторах) (фиг. 42C). Интенсивность иммунологического мечения ламина B, ДНК и AKAP95 в NT- и CT-эмбрионах на стадии PN после экстракции с использованием тритона X-100/ДНКазы I/NaCl (фиг. 42D). Значения выражены в виде средней ± с.о. интенсивности флуоресценции по отношению к неэкстрагированным NT- и CT-эмбрионам на стадии пронуклеусов, соответственно (n~15 эмбрионов/маркер/обработку в трех повторах). Масштаб столбиков 10 мкм (фиг. 42A), 20 мкм (фиг. 42B). Фиг. 43A-43B иллюстрирует распад донорного ядра in vitro и изменение локализации TBP в реконструированных пронуклеусах в случае CT. На фиг. 43B показано сравнение динамики TBP в ходе NT и CT, которую отслеживали методом иммунофлуоресцентного анализа донорных фибробластов (фиб.), конденсированного хроматина на стадии PCC (NT PCC), донорных хромосом, конденсированных в митотическом экстракте (MS15 CC), NT-пронуклеусов (NT PN) и CT-пронуклеусов (CT PN). ДНК метили Hoechst 33342. Масштаб столбиков 20 мкм. Отношения интенсивности флуоресценции TBP/AKAP95 и TBP/DNA на стадиях, показанных на фиг. 43A, и в партеногенетических пронуклеусах (парт. PN) (фиг. 43B). Показана средняя ± с.о. интенсивность флуоресценции в 10-13 эмбрионах на группу. Фиг. 44A является схематичной иллюстрацией нокаутирующего по прионному белку вектора по изобретению, содержащего ген резистентности к неомицину (вектор pBPrP(H)KOneo). Данная схема иллюстрирует, как идентифицировать целенаправленно измененные клоны. Фиг. 44B является схематичной иллюстрацией нокаутирующего по прионному белку вектора по изобретению, содержащего ген устойчивости к пуромицину (вектор pBPrP(H)KOpuro), и способа идентификации целенаправленно измененных клонов. На фиг. 44C представлена таблица, в которой указана частота гомологичной рекомбинации в локусе прионного белка при использовании нокаутирующего по прионному белку вектора по данному изобретению в разных клеточных линиях фибробластов коровы. На фиг. 45A представлена фотография, иллюстрирующая уровень ламина B, ламина A/C, NuMA, AKAP95 и ДНК в пронуклеусах из NT- и SLOT-эмбрионов. Фиг. 45B является графиком интенсивности флуоресценции в соответствии с фиг. 45A. На фиг. 46A-46D представлена динамика ядер фибробластов после NT. На фиг. 46A показано распределение иммунофлуоресценции ламина B, ламина A/C и TBP в фибробластах коровы и IVP-эмбрионах коровы на стадии пронуклеусов (PN) и стадии 8-16 клеток. Вставки, ДНК, меченая Hoechst 33342. Столбики, (слева) 10 мкм, (справа) 50 мкм. Фиг. 46B является фотографией иммуноблота ламина B, ламина A/C и TBP в эмбриональных фибробластах и эмбрионах на стадии пронуклеусов. Фиг. 46C является фотографией, полученной в ходе иммунофлуоресцентного анализа ламина B, ламина A/C и TBP в NT-эмбрионах на стадии PCC и стадии пронуклеусов (NT-PN). Столбики 10 мкм. Фиг. 46D является столбчатой диаграммой интенсивности иммунологического мечения указанных белков в мужских (MPN) и женских (FPN) пронуклеусах IVP-эмбрионов и в пронуклеусах NT-эмбрионов (NT-PN). Данные выражены в виде отношения (среднее ± с.о.) флуоресценции антител к флуоресценции Hoechst 33342 (ДНК). Анализировали более 20 эмбрионов на маркер в 3-5 повторах в (A, C, D). Фиг. 47A-47C иллюстрируют характеристику пронуклеусов в NT-эмбрионах. На фиг. 47A представлена столбчатая диаграмма интенсивности иммунного мечения (средняя ± с.о.) TBP и ДНК (Hoechst 33342) в IVP0- и NT-эмбрионах на стадии пронуклеусов и в фибробластах после экстракции in situ с использованием 1% тритона X-100/1 мг/мл ДНКазы I/300 мМ NaCl. Интенсивность мечения выражена относительно интенсивности мечения неэкстрагированных эмбрионов (n=30 эмбрионов или клеток на группу). Фиг. 47B является фотографией, иллюстрирующей экспрессию маркеров в NT-эмбрионах, активированных, как указано в этом описании в (b) или (b’) в присутствии 10 мкг/мл CHX или (b”) 5 мкг/мл ActD (n=30 эмбрионов/группу в 2-3 повторах). Вставки ДНК. Столбик 10 мкм. На фиг. 47C представлена столбчатая диаграмма отношения (среднего ± с.о.) интенсивности иммунного мечения указанных белков к флуоресценции Hoechst 33342 (ДНК) в эмбрионах, активированных, как показано на фиг. 47B (n=15-20 эмбрионов/обработку/маркер). Фиг. 48 иллюстрирует способ SLOT. На первой стадии донорные фибробласты делают обратимо пермеабилизованными в течение 30 минут с использованием 500 нг/мл SLO. На второй стадии пермеабилизованные клетки промывают и инкубируют в митотическом экстракте, содержащем АТФ-регенерирующую систему, чтобы вызвать конденсацию хромосом и стимулировать удаление ядерных компонентов (стрелки). На третьей стадии экстракт удаляют и клетки, необязательно, снова делают непроницаемыми в культуре с 2 мМ CaCl2 в течение двух часов. На четвертой стадии клетки сливают с лишенными ядер реципиентными ооцитами, и на пятой стадии ооциты активируют, как в случае NT, чтобы вызвать образование пронуклеусов и развитие. На фиг. 49A-49C показана индукция разрушения соматических ядер в митотических экстрактах. Фиг. 49A демонстрирует, что митотический экстракт не вызывает апоптоза. Интактные фибробласты коровы (дорожка 1), фибробласты, индуцированные к апоптозу с использованием 5 мкг/мл нокодазола в течение 20 часов (дорожка 2), или SLO-пермеабилизованные фибробласты, экспонированные с митотическим (дорожка 3) или интерфазным (дорожка 4) экстрактом в течение одного часа, подвергали иммуноблоттингу, используя анти-PARP-антитела (верхняя панель). Деградацию ДНК оценивали гель-электрофорезом в 0,8% агарозе и окрашиванием бромидом этидия (нижняя панель). Фиг. 49B является фотографией, полученной в ходе иммунофлуоресцентного анализа ламина B, ламинов A/C, TBP и AKAP95 после 45-минутного инкубирования пермеабилизованных фибробластов в митотическом экстракте с (+АТФ) или без (-АТФ) АТФ-регенерирующей системы. Столбики 10 мкм. Показано, что более 1000 клеток, анализированных в 4 повторах, демонстрировали такое же мечение. Фиг. 49C является изображением, полученным в ходе анализа методом иммуноблоттинга интерфазных фибробластов (дорожка 1), фибробластов, синхронизированных в митозе с использованием 1 мкг/мл нокодазола (дорожка 2), хроматина, выделенного из пермеабилизованных фибробластов, на которые воздействовали митотическим экстрактом в присутствии или в отсутствие АТФ-регенерирующей системы (дорожки 3, 4) или интерфазным экстрактом (дорожка 5), и целых фибробластов, на которые воздействовали митотическим экстрактом (дорожка 6). Блоты анализировали с использованием антител к указанным белкам. Гистон H4 использовали в качестве контроля загрузки белка на гель. Фиг. 50A-50D демонстрируют характеристику ядер SLOT-эмбрионов. Фиг. 50A иллюстрирует морфологию хроматина (стрелка) в пределах 30 минут введения в ооцит с помощью SLOT или NT. Пунктирной линией очерчена цитоплазма ооцита. На панелях справа показаны увеличения донорного хроматина в SLOT- и NT-эмбрионах. Столбик 20 мкм. Фиг. 50B иллюстрирует распределение ламина B, ламина A/C и TBP в эмбрионах на стадии пронуклеусов (две верхние панели) и стадии 8-16 клеток (две нижние панели), полученных посредством NT или SLOT. Столбики 20 мкм (n=20-30 эмбрионов/маркер в 3 повторах). На фиг. 50C представлена столбчатая диаграмма отношения (среднее ± с.о.) интенсивности иммунного мечения к интенсивности флуоресценции Hoechst 33342 (ДНК) для указанных белков в пронуклеусах NT- или SLOT-эмбрионов (n~20 эмбрионов/маркер в 3 повторах). На фиг. 50D представлена столбчатая диаграмма интенсивности иммунного мечения (среднее ± с.о.) TBP и ДНК (Hoechst 33342) в NT- и SLOT-эмбрионах на стадии пронуклеусов после экстракции с использованием 0,1% тритона X-100/1 мг/мл ДНКазы I/300 мМ NaCl относительно интенсивности мечения неэкстрагированных эмбрионов (n=15 эмбрионов/группу в 2 повторах). Фиг. 51A-51E иллюстрируют оценку развития и морфологическую оценку SLOT- и NT-клонов. На фиг.51A представлена столбчатая диаграмма степени развития in vivo NT- и SLOT-клонов. Частоту беременности оценивали ультразвуковым исследованием в указанные дни беременности. Также показан процент рожденных телят и живых телят через 24 часа после родов. Двенадцать SLOT-телят и 23 NT-теленка родились в результате 59 и 211 переносов эмбрионов соответственно, в течение восьми недель. Значения P получали с помощью точных критериев Фишера. На фиг.51B представлено изображение SLOT-клонов (идентичные самки от скрещивания симментальской x ангусской пород) в 5-6-недельном возрасте. На фиг. 51C представлен график оценки в баллах отдельных SLOT- и NT-телят и плацент в пределах 24 часов после рождения. Фиг.51D является графиком массы при рождении отдельных SLOT- и NT-клонов. На фиг.51C и 51D в SLOT- (кружки) или NT- (квадраты) клонах одним цветом указаны оценка в баллах теленка, оценка в баллах плаценты и масса при рождении для одного и того же индивидуума. На фиг. 51E представлены блочные диаграммы анализа оценки в баллах теленка, оценки в баллах плаценты и масс при рождении SLOT- и NT-клонов. ПОДРОБНОЕ ОПИСАНИЕ Данное изобретение частично основано на неожиданно высокой частоте инактивации прионного локуса коров с использованием способов, приведенных в этом описании. Большое количество целенаправленно измененных нокаутированных по приону клеток значительно облегчает создание нокаутированных по приону копытных животных (например, коров). Кроме того, изобретение относится к усовершенствованным способам повышения эффективности ядерного переноса, чтобы дополнительно упростить получение нокаутированных по приону копытных животных. Указанные полученные в результате трансгенные копытные животные являются желательным источником сельскохозяйственных продуктов (например, мяса) и фармацевтических продуктов (например, терапевтических человеческих антител, кодируемых одной или несколькими ксеногенными нуклеиновыми кислотами у трансгенных копытных животных). Нокаутирующий вектор, используемый для того, чтобы инактивировать прионный локус в донорных клетках коров, частично основан на последовательности локуса гена, кодирующего прионный белок коров, депонированного в Genbank (AJ298878). Источник геномной последовательности кодирующего прионный белок гена, используемой для конструирования нокаутирующего вектора, соответствует последовательности линии фибробластов, которые подвергали генетическому модифицированию. В частности геномную последовательность гена, кодирующего прионный белок, голштинской породы использовали для конструирования нокаутирующего вектора для эффективного направленного изменения фибробластов, полученных от животных голштинской породы. Нокаутирующий вектор содержал две области, гомологичные прионному локусу, которые являются смежными в прионном локусе, и вектор был сконструирован для встраивания последовательности терминации транскрипции (STOP) вместе с геном лекарственной резистентности, фланкированным двумя сайтами loxP, в прионный локус. Альтернативная методика заключается в использовании двух несмежных гомологичных областей в нокаутирующем векторе для того, чтобы делетировать область эндогенного гена, кодирующего прионный белок, между двумя гомологичными областями. Чтобы уменьшить активность прионного белка, STOP-последовательность встраивали сразу за инициирующим ATG-кодоном в экзоне 3. Для эффективной гомологичной рекомбинации в локусе одна гомологичная область была длиннее, чем другая гомологичная область. В частности, длина 3′-области составляла 8,3 т.п.о., что намного длиннее, чем гомологичная 5′-область длиной 1,1 т.п.о. Чтобы увеличить концентрацию гомологичных рекомбинантов с короткой гомологичной 5′-областью в противоположном направлении относительно гена, кодирующего прионный белок, связывали маркер негативной селекции, такой как ген DT-A. Нокаутирующий вектор линеаризовали перед трансфекцией соответствующим ферментом рестрикции так, чтобы основной плазмидный вектор, такой как pUC pBluescript, мог быть связан с концом маркера негативной селекции. Кроме того, чтобы усилить гомологичную рекомбинацию линеаризованный нокаутирующий вектор перед добавлением к фибробластам обрабатывали HBS (забуференный раствор соли Hepes), содержащим 1 мМ спермидин. К вектору направленного воздействия может быть добавлен полиамин и любое другое соединение, способствующее эффективной гомологичной рекомбинации. Предпочтительная концентрация полиамина составляет от 0,1 до 10 мМ; наиболее предпочтительно концентрация равна 1 мМ. Полиамин может быть выбран из спермидина, спермина, кадаверина и путресцина. Предпочтительным полиамином является спермидин. Другое соединение может быть выбрано из соединений, которые могут и образуют ионную связь с фосфатным остатком ДНК, такие как поли-L-лизин или поли-L-аргинин. Кроме того, эффективность гомологичной рекомбинации можно повысить, помещая маркер негативной селекции так, чтобы он не был экспонирован на конце линеаризованного вектора направленного воздействия. Предпочтительно, 5′- и 3′-концы функциональной единицы маркера негативной селекции расположены по меньшей мере на расстоянии 1 т.п.о, предпочтительно по меньшей мере 2 т.п.о. от 5′- и 3′-концов линеаризованного вектора направленного воздействия соответственно. Как правило, гомологичная геномная последовательность расположена с 5′- и 3′-стороны от маркера негативной селекции, обеспечивая достаточное расстояние маркера негативной селекции от концов линеаризованного вектора направленного воздействия. В линеаризованном векторе направленного воздействия другая сторона маркеров негативной селекции может быть сконструирована так, чтобы она вмещала по меньшей мере 1 т.п.о., предпочтительно по меньшей мере 2 т.п.о. дополнительной последовательности. Например, в качестве такой дополнительной последовательности в векторе направленного воздействия может быть использована последовательность вектора pUC. В одном конкретном способе полученный на основе голштинской породы нокаутирующий вектор путем электропорации вводили в эмбриональные фибробласты голштинских коров и получали гемизиготно направленно модифицированные клоны с неожиданно высокой частотой, составляющей более 50%. Большое количество целенаправленно модифицированных клонов должно быть достаточно для эффективного получения здоровых плодов и телят. Чтобы получить гомозиготно направленно модифицированные плоды и телят, гемизиготные нокаутированные фибробласты, выделенные от гемизиготных направленно модифицированных плодов, можно электропорировать таким же нокаутирующим вектором или нокаутирующим вектором с геном устойчивости к другому антибиотику, чтобы мутировать оставшийся аллель прионного локуса. Альтернативно гомозиготных нокаутированных телят можно получить спариванием гемизиготно нокаутированных представителей крупного рогатого скота женского и мужского пола. Способы получения копытных животных, которые экспрессируют ксеногенные антитела Гомозиготных нокаутированных по прионному белку коров, продуцирующих человеческие антитела (коровий прионный белок KO/hAb) можно создать путем введения локусов иммуноглобулина человека в фибробласты коров с гомозиготным нокаутированным по прионному белку генотипом, например, как описано в WO02/70648 и Kuroiwa et al. Nature Biotechnol. 20; 889-894, 2002. Подходящие способы включают, например, введение человеческой искусственной хромосомы (HAC), содержащей гены как тяжелой, так и легкой цепи иммуноглобулина копытному животному или введением B-клеток или предшественников B-клеток человека копытному животному во время его эмбриональной стадии или после его рождения (например, копытному животному с иммунодефицитом или с подавленным иммунитетом) (смотри, например, публикации PCT No. WO01/35735 и WO02/074938). Можно использовать стандартные способы для введения желаемой нуклеиновой кислоты, которая содержит гены (предпочтительно полные генные локусы) для продуцирования антител конкретного вида (например, человека), в донорную клетку для применения в создании трансгенного копытного животного, которое экспрессирует ксеногенные антитела. Предпочтительно используют искусственные хромосомы человека, такие как хромосомы, описанные в публикациях PCT No. WO97/07671 и WO00/10383. Указанные человеческие искусственные хромосомы также описаны в выданном патенте Японии JP 30300092. Каждая из указанных публикаций приведена здесь в качестве ссылки в полном объеме. Также конструирование искусственных человеческих хромосом или трансгенов, которые содержат и экспрессируют гены иммуноглобулинов человека, описано в Shen et al., Hum. Mol. Genet. 6: 1375-1382 (1997); Kuroiwa et al., Nature Biotechnol. 18: 1086-1090 (2000); Loupert et al., Chromosome 107: 255-259 (1998); публикациях PCT No. WO00/10383, W098/24893, WO96/33735, WO97/13852, WO98/24884 и WO97/07671 и патентах США No. 5877397, 5874299, 5814318, 5789650, 5770429, 5661016, 5633425, 5625126, 5569825 и 5545806, каждая из публикаций приведена здесь в качестве ссылки в полном объеме. Человеческие искусственные хромосомы также можно использовать для введения генов ксеногенных антител в животные клетки дикого типа; это осуществляют с использованием способов, описанных выше. Введение искусственной хромосомы в животные клетки, особенно эмбриональные фибробласты, можно осуществить слиянием микроклеток, как описано в этой заявке. В качестве альтернативы использования человеческой искусственной хромосомы можно интегрировать в хромосому нуклеиновые кислоты, кодирующие гены иммуноглобулинов, используя вектор YAC, вектор BAC или космидный вектор. Векторы, содержащие ксеногенные гены Ig (описанные, например, в публикациях PCT No. WO98/24893, WO96/33735, WO97/13852 и WO98/24884 и патентах США No. 5849992 и 5827690), можно ввести в эмбриональные фибробласты, используя известные способы, такие как электропорация, липофекция, слияние с дрожжевым сферопластом, содержащим вектор YAC, и тому подобные. Кроме того, векторы, содержащие ксеногенные гены Ig, могут быть нацелены на эндогенные локусы генов Ig эмбриональных фибробластов, в результате приводя к одновременному введению ксеногенного гена Ig и разрушению эндогенного гена Ig. Интеграцию нуклеиновой кислоты, кодирующей ксеногенный ген иммуноглобулина, также можно осуществить, как описано в патентах Lonberg et al. (см. выше). В конструкции «knockin», используемой для инсерции ксеногенных генов иммуноглобулинов в хромосому копытного животного-хозяина, один или несколько иммуноглобулиновых генов и ген устойчивости к антибиотику могут быть оперативно связаны с промотором, который является активным в типе клеток, трансфицированных конструкцией. Например, можно использовать конститутивно активный, индуцируемый или тканеспецифичный промотор, чтобы активировать транскрипцию интегрированного гена устойчивости к антибиотику, обеспечивая селекцию трансфицированных клеток на основе получаемой в результате резистентности данных клеток к антибиотику. Альтернативно может быть использована конструкция knockin, в которой кассета knockin, содержащая ген(гены) Ig и ген устойчивости к антибиотику, оперативно не связана с промотором. В этом случае клетки, в которых кассета knockin интегрируется ниже эндогенного промотора, могут быть отобраны на основе получаемой экспрессии маркера резистентности к антибиотику под контролем эндогенного промотора. Такие отобранные клетки можно использовать в способах переноса ядер, приведенных в этом описании, для создания трансгенного копытного животного, содержащего ксеногенный ген иммуноглобулина, интегрированный в хромосому хозяина. Используя подобную методологию можно получить и встроить искусственные хромосомы, содержащие гены для экспрессии Ig другого вида, такого как Ig собаки, кошки, других копытных животных, приматов, отличных от человека, наряду с другими видами. Как обсуждалось выше и хорошо известно в данной области, гены иммуноглобулинов разных видов имеют по существу гомологичные последовательности. После того как определено, что введенная искусственная хромосома, например искусственная хромосома человека, стабильно введена в клеточную линию, например эмбриональный фибробласт коровы, ее используют в качестве донора для переноса ядра. Это можно определить методами ПЦР. Полученные трансгенные телята содержат стабильно включенные нуклеиновые кислоты, такие как человеческая искусственная хромосома. После получения телят, которые содержат стабильно включенные нуклеиновые кислоты (например, искусственную хромосому человека), животных тестируют, чтобы определить, экспрессируют ли они гены Ig человека в ответ на иммунизацию и происходит ли созревание аффинности. Способы снижения продукции эндогенных антител у копытных животных, которые также экспрессируют ксеногенные антитела Также могут быть осуществлены модификации общего способа, описанного выше. Например, чтобы уменьшить количество эндогенного антитела, экспрессируемого полученным трансгенным копытным животным, в донорной клетке можно инактивировать один или несколько эндогенных генов Ig перед или после введения ксеногенной нуклеиновой кислоты. Кроме того, животное, несущее ксеногенные гены Ig, может быть скрещено с животным, у которого эндогенный ген Ig инактивирован. Примерная схема последовательных манипуляций с донорными клетками (например, фибробластами коровы) описана ниже. Во-первых, создают гомозиготные нокаутированные по прионному белку эмбриональные фибробласты. Затем создают гемизиготные нокаутированные по тяжелой цепи иммуноглобулина коровы (BIgH) фибробласты путем направленного изменения генов и получают плоды, используя перенос ядер (например, SLOT или перенос хроматина). Гомозиготные нокаутированные по BIgH фибробласты получают вторым направленным изменением генов и затем получают плоды, используя ядерный перенос. Гемизиготные нокаутированные по легкой цепи иммуноглобулина коровы (BIgL) фибробласты получают направленным изменением генов с последующим получение плодов с помощью ядерного переноса. Гомозиготные нокаутированные по BIgL фибробласты создают посредством второго направленного изменения генов и получают плоды с помощью ядерного переноса. Затем локусы генов иммуноглобулинов человека вводят в описанные выше последовательно нокаутированные фибробласты, имеющие гомозиготный генотип, нокаутированы по прионному белку/BIgH/BIgL и получают плоды и телят с помощью ядерного переноса. Примерные способы создания копытных животных по изобретению Предпочтительные способы создания копытного животного, имеющего мутацию в эндогенной нуклеиновой кислоте, которая кодирует прионный белок, и, необязательно, имеющего одну или несколько нуклеиновых кислот, кодирующих ксеногенное антитело, заключаются в ремоделировании донорного генетического материала перед его введением в реципиентный ооцит для образования трансгенного копытного животного. Ремоделирование относится к любому морфологическому изменению, которое улучшает развитие полученного ооцита с ядерным трансплантатом по сравнению с ооцитом, полученным либо на основе переноса целых клеток, либо интактных ядер в реципиентный ооцит. Перепрограммирование достигается инкубацией донорного ядра, которое имеет мутацию в эндогенной нуклеиновой кислоте, которая кодирует прионный белок, и, необязательно, имеет одну или несколько нуклеиновых кислот, кодирующих ксеногенное антитело, в перепрограммирующих средах (например, митотическом экстракте, растворе детергента и/или соли или растворе протеинкиназы), приводящей к распаду ядерной оболочки и, возможно, конденсации хроматина. Это разрушение ядерной оболочки и конденсация хроматина приводит к высвобождению белков, регулирующих транскрипцию, которые связаны с хромосомами и которые в противном случае стимулировали бы транскрипцию генов, нежелательных для развития ооцита, эмбриона или плода. Дополнительные регуляторные белки могут быть удалены при очистке хроматиновой массы перед переносом ее в реципиентный ооцит. Альтернативно специфичные регуляторные белки, которые высвобождаются из хромосом, могут быть иммунологически элиминированы или иным образом удалены из перепрограммирующих сред (например, клеточного экстракта), чтобы предотвратить их повторное связывание с хромосомами. После ядерного переноса новые белки из цитоплазмы ооцита могут быть связаны с хромосомами во время деконденсации хроматина и образования ядерной оболочки в ооците. Указанные белки стимулируют транскрипцию генов, которые позволяют ооциту развиваться в жизнеспособного потомка. Другой способ клонирования, который можно использовать для получения трансгенного копытного животного, заключается в перепрограммировании пермеабилизованной клетки (т.е. клетки, имеющей мутацию в эндогенной нуклеиновой кислоте, которая кодирует прионный белок, и, необязательно, имеющей одну или несколько нуклеиновых кислот, кодирующих ксеногенное антитело) посредством ее инкубирования в перепрограммирующих средах (например, клеточном экстракте), чтобы произошло добавление или удаление факторов из клетки. Плазматическую мембрану пермеабилизованной клетки, предпочтительно, вновь делают непроницаемой, чтобы заключить в клетке требуемые факторы и восстановить целостность мембраны клетки. Затем перепрограммированную клетку переносят в реципиентный ооцит для получения клонированного млекопитающего. Указанный способ клонирования использовали для получения плодов без ксеногенной нуклеиновой кислоты иммуноглобулина, которые остаются в живых после 60 дней. Предварительные результаты показывают, что выживаемость плодов от 40 дня до 60 дня выше в случае плодов, образованных с использованием данного способа (7/10; 70%), чем в случае плодов с традиционным ядерным переносом (8/16; 50%). Подобные результаты ожидаются для плодов, имеющих мутацию в эндогенной нуклеиновой кислоте, которая кодирует прионный белок, и/или имеющих одну или несколько нуклеиновых кислот, кодирующих ксеногенное антитело. В альтернативных способах создают химерные эмбрионы, в которых основная часть плацентарной ткани происходит из одного генетического источника, а основная часть эмбриональной ткани происходит из другого генетического источника. Такие химерные эмбрионы могут иметь меньше аномалий плаценты и, таким образом, могут иметь повышенную частоту выживания. В одном таком способе клетки из оплодотворенного in vitro эмбриона или природного эмбриона подвергаются взаимодействию с клетками из эмбриона, который имеет мутацию в эндогенной нуклеиновой кислоте, которая кодирует прионный белок, и, необязательно, имеет одну или несколько нуклеиновых кислот, кодирующих ксеногенное антитело, и который получен с использованием традиционных способов ядерного переноса или любых других способов клонирования, приведенных здесь. Например, клетки из оплодотворенного in vitro эмбриона могут быть инъецированы в периферическую область эмбриона с перенесенным ядром (например, между zona pellucida и самим эмбрионом). Этот способ использовали для получения химерных эмбрионов без генов ксеногенных антител, которые имели 67% частоту выживаемости на 40 день по сравнению с 25% частотой выживаемости в случае контрольных эмбрионов с перенесенным ядром. Сходные результаты ожидаются в случае использования клеток из эмбрионов с перенесенным ядром, имеющих мутацию в эндогенной нуклеиновой кислоте, которая кодирует прионный белок, и/или имеющих одну или несколько нуклеиновых кислот, кодирующих ксеногенное антитело. В альтернативном способе клетки из предкомпактного оплодотворенного in vitro эмбриона или природного эмбриона инкубируют с клетками из предкомпактного эмбриона с перенесенным ядром в условиях, при которых возможна реорганизация клеток из каждого эмбриона с получением одного химерного эмбриона (Wells and Powell, Cloning 2: 9-22, 2000). В обоих способах клетки из оплодотворенного in vitro эмбриона или природного эмбриона, предпочтительно, включаются в плаценту, а клетки, полученные путем ядерного переноса, предпочтительно, включаются в ткань плода. УРОВЕНЬ ТЕХНИКИ ДАННОГО ИЗОБРЕТЕНИЯ Хотя Ig человека был экспрессирован у мышей, нельзя было предсказать, будет ли Ig человека подвергаться частичной перестройке и экспрессироваться у коров или других копытных животных вследствие различий в структуре генов антител, механизма продуцирования антител и функции B-клеток. В частности, в отличие от мышей крупный рогатый скот и овцы отличаются от людей по своей иммунофизиологии (Lucier et al., J. Immunol. 161: 5438, 1998; Parng et al., J. Immunol. 157: 5478, 1996; и Butler, Rev. Sci. Tech. 17: 43, 2000). Например, разнообразие генов антител у коров и овец гораздо больше основано на конверсии генов, чем на перестройке генов, как у человека и мышей. Также первичным местом локализации B-клеток у людей и мышей является костный мозг, тогда как у коров и овец B-клетки расположены в Пейеровых бляшках подвздошной кишки. Следовательно, до настоящего изобретения было бы трудно или невозможно предсказать, будет ли иметь место перестройка иммуноглобулинов и разнообразие локусов иммуноглобулинов человека в B-клеточной линии коровы (или другого копытного животного). Кроме того, также было бы невозможно предсказать, будет ли корова способна выживать, т.е. будут ли у нее вызваны нормальные иммунные функции в отсутствие ее эндогенного Ig или в случае помех со стороны человеческих антител. Например, не было определено, будут ли коровьи B-клетки, экспрессирующие Ig человека, правильно мигрировать к Пейровой бляшке подвздошной кишки у коров, так как это не происходит у человека. Также было неясно, будет ли нормальной в коровьей системе функция Fc-рецептора человека, которая опосредует активацию комплемента, индукцию высвобождения цитокинов и удаление антигена. Нельзя было предсказать, могли ли такие копытные животные выживать, так как было неясно, будут ли Ig человека функционально экспрессироваться или экспрессироваться в достаточных количествах, чтобы обеспечить адекватные иммунные ответы. Также было неясно, могли ли хромосомы человека стабильно сохраняться у трансгенных копытных животных. Хотя подходы, используемые в изобретении, описаны выше, способы, которые используются, описаны более подробно ниже. Данные примеры представлены для иллюстрации изобретения и их не следует рассматривать как ограничивающие. В частности, хотя указанные примеры сфокусированы на трансгенных коровах, описанные способы можно использовать для получения и тестирования любого трансгенного копытного животного. ПРИМЕР 1: Трансгенные копытные животные, обладающие пониженной активностью прионного белка Клеточные клоны фибробластов коровы, в которых мутированы оба аллеля локуса PrP, создавали последовательной гомологичной рекомбинацией. Конструкцию ДНК для создания гомозиготных нокаутированных по PrP клеток использовали для предотвращения транскрипции функциональной полноразмерной мРНК PrP посредством встраивания как гена устойчивости к неомицину, так и гена устойчивости к пуромицину (neo или puro, описанные в примере 3) и кассеты терминации транскрипции (STOP) в экзон 3 сразу после начального кодона ATG. Таким образом, в полученных незрелых транскриптах PrP отсутствовал функциональный кодирующий домен. Конструкцию ДНК, содержащую ген neo (т.е. вектор PrPKOneo), сначала путем электропорации вводили в клеточные линии фибробластов коровы и затем выделяли резистентные к неомицину колонии. На основании анализа ПЦР – в некоторых колониях происходила гомологичная рекомбинация в экзоне 3. Таким образом создавали клеточные линии фибробластов коровы, в которых один аллель локуса PrP мутирован. Из гемизиготных мутантных (геми-PrPKO) фибробластов создавали три плода, в которых мутирован один аллель локуса PrP, и повторно получали клеточную линию фибробластов геми-PrPKO. Затем другую конструкцию ДНК, содержащую ген puro (т.е. вектор PrPKOpuro) путем второй электропорации вводили в фибробласты геми-PrPKO и затем выделяли резистентные к пуромицину колонии. На основании анализа ПЦР в некоторых клонах происходила гомологичная рекомбинация в экзоне 3 оставшегося аллеля с частотой ~5%. Таким образом были получены клоны фибробластов коровы, в которых оба аллеля локуса PrP мутированы (гомо-PrPKO). Из гомозиготных мутантных (гомо-PrPKO) фибробластов повторно получали десять плодов, в которых оба аллеля локуса PrP мутированы, и повторно создавали клеточные линии фибробластов гомо-PrPKO. В указанных фибробластах гомо-PrPKO полное отсутствие экспрессии PrP подтверждали анализом ОТ-ПЦР. Фибробласты гомо-PrPKO также могут быть созданы путем сочетания такого же нокаутирующего вектора и более высокой концентрации антибиотика, чтобы отобрать гомозиготные нокаутированные клетки. При использовании переноса ядер будут созданы телята гомо-PrPKO из фибробластов гомо-PrPKO. Указанные способы описаны далее. Конструирование нокаутирующего по прионному белку вектора Нокаутирующий по прионному белку вектор создавали следующим образом (фиг. 44A). Чтобы выделить геномную ДНК вокруг экзона 3 гена, кодирующего прионный белок, ДНК-зонд амплифицировали в ПЦР, используя следующую пару праймеров 5′-CCA CAT AGG CAG TTG GAT CC-3′ (SEQ ID NO: 69) и 5′-ATA AGA GGC CTG CTC ATG GC-3′ (SEQ ID NO: 70). Используя этот зонд, проводили скрининг геномной библиотеки коровы (голштинской породы) на основе фага Способы трансфекции/нокаута Трансфекцию клеточных линий эмбриональных фибробластов быков голштинской породы осуществляли, используя стандартный протокол электропорации, следующим образом. Используемая для культивирования бычьих эмбриональных фибробластов среда содержала 500 мл Alpha MEM (Gibco, 12561-049), 50 мл фетальной телячьей сыворотки (Hy-Clone Скрининг направленных интеграций Как описано выше, геномную ДНК экстрагировали из каждой из 24 лунок независимо, используя набор для выделения ДНК PUREGENE (Gentra Systems), согласно протоколу производителя. Каждый образец геномной ДНК ресуспендировали в 20 мкл 10 мМ трис-Cl (pH 8,0) и 1 мМ EDTA (EDTA). Скрининг с помощью ПЦР осуществляли, используя следующую пару праймеров F7 (5′-TGT CAA AGA GAC ACT CCT CAT TTG TCT TCC-3′, SEQ ID NO: 71) и R7 (5′-TCA TAG CCG AAT AGC CTC TCC ACC C-3′, SEQ ID NO: 72). Последовательность одного праймера расположена в нокаутирующем векторе, а последовательность другого праймера расположена непосредственно с внешней стороны интегрированного вектора в направленно измененном эндогенном локусе (фиг. 44A). Следовательно, ожидаемый ПЦР-продукт должен регистрироваться только в том случае, когда нокаутирующий вектор интегрируется в являющийся мишенью локус посредством гомологичной рекомбинации. Смеси для реакции ПЦР содержали 17,9 мкл воды, 3 мкл 10Х буфера для ПЦР LA II (плюс Mg2+), 4,8 мкл смеси dNTP, 10 пмоль прямого праймера, 10 пмоль обратного праймера, 2 мкл геномной ДНК и 0,3 мкл Taq LA. Тридцать циклов ПЦР осуществляли путем инкубирования реакционных смесей в следующих условиях: 85°C в течение трех минут, 94°C в течение одной минуты, 98°C в течение 10 секунд и 68°C в течение 5 минут. После ПЦР реакционные смеси анализировали электрофорезом. ПЦР-продукты ожидаемого размера получали примерно в 50% из 94 подвергнутых скринингу резистентных к неомицину клонов (фиг. 44B). Эти позитивные клоны также подтверждали посредством использования медода ПЦР-амплификации с другой парой праймеров F10 (5′-TGA AGA TGT CCC AGT GCT GGG GAT-3′, SEQ ID NO: 73) и R10 (5′-GAG TTA GGG GCG GGA CTA TGG TTG C-3′, SEQ ID NO: 74), получая ПЦР-продукты ожидаемого размера 2,7 т.п.н. Высокая частота гомологичной рекомбинации, составляющая более 50%, была воспроизведена в другой клеточной линии фибробластов, клеточной линии особей женского пола F1 (Голштин x Джерси) (фиг. 44C). Таким образом были успешно созданы клеточные линии фибробластов коров, в которых один аллель прионного локуса мутирован нокаутирующим вектором, независимо от исходной используемой клеточной линии. Перенос хроматина Гемизиготные нокаутированные по приону клетки или ядра из этих клеток можно использовать в любом из способов переноса ядер, приведенных в этом описании, чтобы создать гемизиготное нокаутированное по приону копытное животное (например, нокаутированного теленка). При желании клетки из нокаутированного плода или живого копытного животного можно использовать, как описано ниже, для создания гомозиготных нокаутированных по приону клеток. В одном конкретном способе фибробласты (например, первичные эмбриональные фибробласты коровы) синхронизировали в митозе с использованием 1 мкг/мл нокодазола в течение 18 часов, собирали стряхиванием митотических клеток и два раза промывали в фосфатно-солевым буфере и один раз в буфере для лизирования клеток (20 мМ Hepes, pH 8,2, 5 мМ MgCl2, 10 мМ EDTA, 1 мМ DTT и ингибиторы протеаз). Осажденные клетки ресуспендировали в одном объеме ледяного буфера для лизирования клеток, давали возможность набухнуть на льду в течение одного часа и гомогенизировали в гомогенизаторе Даунса, используя плотно притертый стеклянный пестик. Лизат центрифугировали при 15000 × g в течение 15 минут при 4 Из созревших in vitro ооцитов удаляли ядра через 20 час после созревания (hpm). Трансфицированные эмбриональные фибробласты коровы из отобранных колоний промывали в не содержащем Ca+2/Mg2+ сбалансированном растворе солей Хенкса (HBSS) и пермеабилизовали посредством инкубирования клеток в суспензии с 31,2 ед. стрептолизина O (SLO; Sigma) в 100 мкл HBSS в течение 30 минут на водяной бане при температуре примерно 37°C. Пермеабилизацию оценивали по поглощению непроходящей через мембрану ДНК, окрашенной йодидом пропидия (0,1 мкг/мл). Пермеабилизованные фибробласты осаждали, промывали и инкубировали в 40 мкл митотического экстракта, содержащего АТФ-генерирующую систему (1 мМ АТФ, 10 мМ креатинфосфат и 25 мкг/мл креатинкиназы), в течение 30-45 минут примерно при 37°C. Аликвоты метили 0,1 мкг/мл Hoechst 33342, чтобы наблюдать за конденсацией хроматина. В конце инкубирования реакционную смесь разбавляли 500 мкл Alpha MEM/10% фетальной телячьей сывороткой (Hyclone). Клетки сливали с лишенными ядер ооцитами; ооциты активировали через 28 hpm и эмбрионы культивировали до стадии бластоцисты in vitro. Переносили по два эмбриона на реципиентную самку. Беременность контролировали ультразвуковым исследованием и делали C-сечения у реципиентов, чтобы извлечь плоды для получения клеточной линии. Способы второй трансфекции/нокаута Трансфекцию клеточных линий эмбриональных фибробластов геми-PrPKO, в которых первый вектор KO уже интегрирован в «аллель A», осуществляли следующим образом. Среда, используемая для культивирования эмбриональных фибробластов коровы, содержала 500 мл Alpha MEM (Gibco, 12561-049), 50 мл фетальной телячьей сыворотки (Hy-Clone Скрининг гомозиготных целенаправленных интеграций Как описано выше, геномную ДНК экстрагировали из каждой из 24 лунок независимо, используя набор для выделения ДНК PUREGENE (Gentra Systems), согласно протоколу производителя. Каждый образец геномной ДНК ресуспендировали в 20 мкл 10 мМ трис-Cl (pH 8,0) и 1 мМ EDTA (EDTA). Скрининг с помощью ПЦР осуществляли, используя следующую пару праймеров F14; 5′- TTG CTC CAC CTT CCG TCT TCT GTC-3′ (SEQ ID NO: 3) и R14; 5′- GTT GGC GCC TAC CGG TGG ATG TG-3′ (SEQ ID NO: 4). Последовательность одного праймера расположена в KO-векторе, а последовательность другого праймера расположена непосредственно с внешней стороны интегрированного вектора в направленно измененном эндогенном локусе (фиг. 1). Следовательно, ожидаемый ПЦР-продукт должен регистрироваться только в том случае, когда KO-вектор интегрируется в являющийся мишенью локус посредством гомологичной рекомбинации. Смеси для ПЦР содержали 17,9 мкл воды, 3 мкл 10Х буфера для ПЦР LA II (плюс Mg2+), 4,8 мкл смеси dNTP, 10 пмоль прямого праймера, 10 пмоль обратного праймера, 2 мкл геномной ДНК и 0,3 мкл Taq LA. Тридцать циклов ПЦР осуществляли путем инкубирования реакционных смесей в следующих условиях: 85°C в течение трех минут, 94°C в течение одной минуты, 98°C в течение 10 секунд и 68°C в течение 5 минут. После ПЦР реакционные смеси анализировали электрофорезом. В результате скрининга 332 клонов 15 клонов ( В таблице 1 суммированы данные, относящиеся к беременности после переноса хроматина с использованием гомозиготных PrP KO-клонов, полученных из гемизиготных клеточных линий PrP KO.
Подтверждение отсутствия экспрессии PrP в плодах гомо-PrP Из некоторых плодов были получены фибробласты и выделена суммарная РНК с использованием набора RNeasy Mini (QIAGEN). С помощью одного микролитра суммарной РНК осуществляли синтез первой нити кДНК (система синтеза первой нити SuperScript для ОТ-ПЦР; Invitrogen) с последующей ОТ-ПЦР. ОТ-ПЦР осуществляли, как описано ниже. Используемые праймеры представляли собой 5′-AAG AAG CGA CCA AAA CCT GG-3′ и 5′-GTA ACG GTG CAT GTT TTC ACG-3′. Смеси для реакции ПЦР содержали 32,5 мкл воды, 5 мкл 10X буфера Ex Taq (TAKARA), 8 мкл смеси dNTP, 10 пмоль прямого праймера, 10 пмоль обратного праймера, 2 мкл кДНК первой нити и 0,5 мкл Ex Taq (TAKARA). Тридцать пять циклов ПЦР осуществляли посредством инкубирования реакционных смесей в следующих условиях: 85°C в течение трех минут, 94°C в течение одной минуты, 98°C в течение 10 секунд, 62°C в течение 30 секунд и 72°C в течение 1 минуты. После ПЦР реакционные смеси анализировали электрофорезом. Несмотря на то, что отчетливо видимые ПЦР-продукты регистрировали как в клетках дикого типа, так и в клетках геми-PrP KO, не было позитивных ПЦР-продуктов ни в одной из десяти клеточных линий гомо-PrP KO. Этот результат подтверждает отсутствии экспрессии PrP в клеточных линиях гомо-PrP KO. ПРИМЕР 2: Введение и перестройка HAC Сущность способов инсерции HAC Для создания копытных животных (например, коров), которые имеют мутацию в одном или обоих аллелях гена, кодирующего прион и которые экспрессируют ксеногенное антитело, можно использовать стандартные способы встраивания нуклеиновой кислоты, кодирующей ксеногенное антитело (например, HAC), в клетку. Например, HAC может быть введена до или после того, как инактивируют один или оба аллеля гена, кодирующего прион. При желании один или несколько эндогенных генов антител в клетке также могут быть мутированы. Затем клетку используют в стандартных способах переноса ядер, чтобы получить желаемое трансгенное копытное животное, как подробно описано ниже. По существу, клеточные линии эмбриональных фибробластов быков и коров, содержащие последовательности искусственной хромосомы человека (например, Например, HAC, полученные из хромосомы человека Перенос этих хромосомных фрагментов тестировали путем скрещивания животного Также хромосомные фрагменты Обоснование Для введения HAC животным (например, животным, нокаутированным по Ig) и размножения животных в продуктивные стада можно использовать перенос HAC в зародышевую линию. Проблемой в передачи HAC в зародышевую линию является неполное образование пар хромосомного материала при мейозе. Однако передача в зародышевую линию была успешной у мышей, как показано Tomizuka et al. (Proc. Natl. Acad. Sci. USA, 97: 722, 2000). Методика, изображенная на фиг. 1A, заключается во встраивании Дизайн эксперимента Клетки получают на основании исходного скрининга клеточных линий. Это могут быть линии голштинской породы или другие линии, отличные от линий, используемых выше. Это дает возможность скрещивания при сохранении генетической изменчивости в стаде насколько это возможно. Затем осуществляют введение HAC в клеточные линии и селекцию позитивных клеточных линий. Отобранные линии клеток используют для переноса ядер и получают телят. Начиная с 12-месячного возраста, собирают сперму и яйцеклетки, проводят оплодотворение и переносят реципиентным животным. Берут образцы клеток для анализа ДНК-маркера и кариотипирования. Начиная с рождения, берут образцы крови и анализируют на присутствие белков Ig человека. Как указано выше, HAC также, необязательно, переносят в линии клеток Homo H/L, используя способы, разработанные в описанных выше экспериментах. Дополнительно представлены следующие примеры для дальнейшей иллюстрации изобретения. Примерные способы встраивания HAC Осуществляли дополнительные эксперименты для того, чтобы показать, что ксеногенные (например, человеческие) тяжелая цепь иммуноглобулина (мю) и легкая цепь лямбда могут быть продуцированы хозяином-коровой либо отдельно, либо в комбинации. Кроме того, данные эксперименты показали, что цепи иммуноглобулина человека подвергались перестройке и что получалась поликлональная сыворотка. В данных способах гены, экспрессирующие иммуноглобулин, вводили в фибробласты коровы, используя искусственные хромосомы человека. Затем фибробласты использовали для переноса ядер, получали плоды и анализировали в отношении продукции антител. Указанные способы и результаты более подробно описаны ниже. Сходные результаты ожидаются в случае фибробластов, которые выделяют из плода с мутацией в одном или обоих аллелях гена, кодирующего прион, и которые используют в способах, описанных ниже, для введения HAC. Альтернативно можно ввести HAC в клетку, а затем можно мутировать (i) кодирующий прионный белок ген клетки с HAC или (ii) кодирующий прионный белок ген клетки из клонированного плода, созданного с использованием клетки с HAC. Конструкции HAC Искусственные хромосомы человека (HAC) конструировали с использованием ранее описанной системы клонирования хромосом (Kuroiwa et al., Nature Biotech. 18: 1086-1090, 2000). Коротко, конструкцию Подобным образом также конструировали Функциональность Клетки DT40 цыпленка, несущие указанные HAC, депонированы по условиям Будапештского договора 9 мая 2001 г. в International Patent Organism Depository, National Institute of Advanced Industrial Science and Technology, Tsukuba Central 6, 1-1, Higashi 1-Chome Tsukuba-shi, Ibaraki-ken, 305-8566 Japan. Присвоены следующие депозитарные номера: Вставка hChr22 длиной 2,5 миллионов оснований (м.о.) в Создание эмбриональных фибробластов коровы Чтобы создать эмбриональные фибробласты коровы, собирали плоды с 45 по 60 день от тестированных на заболевание коров голштинской породы и породы Джерси, содержащихся в Trans Ova (Iowa), у которых родословная родителей по мужской и женской линии подтверждена документами в трех последовательных поколениях. Собранные плоды перевозили на льду в Hematech’s Worcester Molecular Biology Division для создания первичных эмбриональных фибробластов. После доставки плоды переносили в 100-мм пластиковые чашки Петри, качества, не предназначенного для культуры ткани, в бокс для культуры ткани. Используя стерильные пинцет и ножницы, с плода удаляли внезародышевую оболочку и пуповину. После переноса плода в новую пластиковую чашку Петри удаляли голову, конечности и внутренние органы. Плод с извлеченными внутренностями переносили в третью чашку Петри, содержащую примерно 10 мл раствора для промывки плода, состоящего из: 125 мл 1X Дульбекко-PBS (D-PBS) с Ca2+ и Mg2+ (Gibco-BRL, Каждый плод дополнительно промывали три раза раствором для промывки плода, чтобы удалить следы крови, переносили в коническую пробирку для культуры ткани объемом 50 мл и наконец измельчали на небольшие кусочки стерильным скальпелем. Кусочки ткани один раз промывали 1X D-PBS с Ca2+ и Mg2+ (Gibco-BRL, Первичные фибробласты размножали при 38,5°C/5% CO2 в полной среде для фибробластов при плотности клеток 1 x 106 живых клеток на флакон для культуры ткани T75 см2. Через 3 дня культивирования или перед достижением слияния фибробласты собирали, промывая флакон один раз 1X D-PBS (без Ca2+ и Mg2+) и инкубируя с 10 мл буфера для диссоциации клеток в течение от 5 до 10 минут при комнатной температуре. Открепление клеток контролировали визуально, используя инвертированный микроскоп. На данной стадии важно было, чтобы скопления клеток были дезагрегированы трипсинизацией. После промывки и подсчета диссоциированные фибробласты были готовы для применения в экспериментах по направленному воздействию на геном. Указанные клетки также могут быть помещены при низких температурах для долговременного хранения. Введение HAC в эмбриональные фибробласты коровы
Эмбриональные фибробласты коровы культивировали в среде Подобным образом Перенос ядер, активация и культура эмбрионов Процедуру переноса ядер осуществляли по существу, как описано ранее (Cibelli et al., Science 1998: 280: 1256-1258). Из созревших in vitro ооцитов удаляли ядра примерно через 18-20 часов после созревания (hpm) и удаление хромосом подтверждали мечением бис-бензимидом (Hoechst 33342, Sigma) в УФ-свете. Компоненты пары цитопласт-донорная клетка сливали, используя одиночный электрический импульс 2,4 кВ/см в течение 20 мксек (манипулятор Electrocell 200, Genetronics, San Diego, CA). Через 3-4 час случайную подгруппу, составляющую 25% от общего количества пар с переносом, выделяли и слияние подтверждали на основе мечения бис-бензимидом перенесенного ядра. Через 30 hpm реконструированные ооциты и контроли активировали кальциевым ионофором (5 мкМ) в течение 4 минут (Cal Biochem, San Diego, CA) и с добавлением 10 мкг циклогексимида и 2,5 мкг цитохалазина D (Sigma) в культуральной среде ACM в течение 6 часов, как описано ранее (Lin et al., Mol. Reprod. Dev. 1998: 49: 298-307; Presicce et al., Mol. Reprod. Dev. 1994: 38: 380-385). После активации яйцеклетки пять раз промывали в забуференной HEPES среде для культивирования эмбрионов хомячка (HECM-Hepes) и помещали в культуру в 4-луночные чашки для культуры ткани, содержащие облученные эмбриональные фибробласты мыши и 0,5 мл среды для культивирования эмбрионов, покрытые 0,2 мл минерального масла, проверенного на эмбрионах (Sigma). От двадцати до 50 эмбрионов помещали в каждую лунку и инкубировали при 38,5°C в воздушной атмосфере с 5% CO2. На четвертый день к среде для культивирования добавляли 10% FCS. Перенос эмбрионов На 7 и 8 день бластоцисты с перенесенными ядрами переносили синхронизированным на 6 и 7 день реципиентным телочкам соответственно. Реципиентных животных синхронизировали, используя однократную инъекцию лутализа (Pharmacia and Upjohn, Kalamazoo, MI) с последующей регистрацией течки. Реципиентов обследовали на 30 день и 60 день после переноса эмбрионов ультрасонографией в отношении присутствия оплодотворенного яйца, а после этого каждые 30 дней ректальной пальпацией вплоть до 270 дней. Сохранение HAC у данных плодов коров суммировано в таблице 2 и более подробно описано в параграфах далее.
Введение HAC, содержащей фрагмент хромосомы Фрагмент SC20, фрагмент хромосомы человека Состояние беременности 28 реципиентов, которым переносили клонированные эмбрионы из клеток, содержащих hchr.14fg, проверяли ультрасонографией. Результаты суммированы в таблице 3. При желании клетку из полученного HAC-плода или HAC-потомка можно использовать во втором раунде переноса ядер, чтобы создать дополнительное клонированное потомство. Клетки из первичного HAC-плода или HAC-потомка также можно заморозить для получения клеточной линии, используемой в качестве источника донорных клеток для создания дополнительных копытных животных с HAC.
Частота беременности была ниже, чем ожидалось. Предполагается, что это можно объяснить чрезвычайно аномально жаркой погодой во время переноса эмбрионов. Как показано на фиг. 27, частота беременности в случае несущих HAC эмбрионов, по-видимому, эквивалентна частоте беременности в случае нетрансгенных клонированных эмбрионов. Одна реципиентная корова, несущая Демонстрация перестройки и экспрессии локуса тяжелой цепи человека у плода коровы Клонированные Наличие тяжелой и/или легкой цепи человека у Чтобы определить, сохраняются ли тяжелая и легкие цепи человека у Для выявления геномной ДНК тяжелой цепи использовали следующие праймеры: VH3-F 5′-AGT GAG ATA AGC AGT GGA TG-3′ (SEQ ID NO: 1) и VH3-R 5′-CTT GTG CTA CTC CCA TCA CT-3′ (SEQ ID NO: 2). Праймеры, используемые для выявления ДНК легкой цепи лямбда, представляли собой IgL-F 5′-GGA GAC CAC CAA ACC CTC CAA A-3′ (SEQ ID NO: 3) и IgL-R 5′-GAG AGT TGC AGA AGG GGT YGA CT-3′ (SEQ ID NO: 4). Смеси для реакции ПЦР содержали 18,9 мкл воды, 3 мкл 10X буфера Ex Taq, 4,8 мкл смеси dNTP, 10 пмоль прямого праймера и 10 пмоль обратного праймера, 1 мкл геномной ДНК и 0,3 мкл Ex Taq. Тридцать восемь циклов ПЦР осуществляли путем инкубирования реакционных смесей в следующих условиях: 85°C в течение трех минут, 94°C в течение одной минуты, 98°C в течение 10 секунд, 56°C в течение 30 секунд и 72°C в течение 30 секунд. Как показано на фиг. 5, каждый из плодов Наличие экзонов Cмю человека в Праймеры, специфичные по отношению к транскрипту мРНК, включающему в себя части Cмю 3 и Cмю 4, использовали для определения присутствия Для этого ОТ-ПЦР-анализа геномной константной области тяжелой цепи мю человека использовали праймеры «CH3-F1» (5′-acc acc tat gac agc gtg ac-3′, SEQ ID NO: 5) и «CH4-R2» (5′-gtg gca gca agt aga cat cg-3′, SEQ ID NO: 6), чтобы создать ОТ-ПЦР-продукт длиной 350 пар оснований. Данную ПЦР-амплификацию осуществляли посредством начального денатурирующего инкубирования при 95°C в течение пяти минут. Затем 35 циклов денатурации, отжига и амплификации осуществляли путем инкубирования при 95°C в течение одной минуты, 59°C в течение одной минуты и 72°C в течение двух минут. Затем реакционные смеси инкубировали при 72°C в течение 10 минут. Подвергнутую перестройке тяжелую цепь коровы выявляли с использованием праймеров I7L и P9, как описано ниже (фиг. 7). В качестве внутреннего контроля регистрировали РНК GAPDH, используя праймеры «GAPDH прямой» (5′-gtc atc atc tct gcc cct tct g-3′, SEQ ID NO: 7) и «GAPDH обратный» (5′-aac aac ttc ttg atg tca tca t-3′, SEQ ID NO:8). Для указанной амплификации РНК GAPDH образцы инкубировали при 95°C в течение пяти минут с последующими 35 циклами инкубирования при 95°C в течение одной минуты, 55°C в течение одной минуты и 72°C в течение двух минут. Затем смеси инкубировали при 72°C в течение семи минут. Этот исследование показало, что ОТ-ПЦР-анализ селезенки плода Перестройка локуса тяжелой цепи коровы к 77 дню беременности
Дорожка 5 на фиг. 7 показывает, что был получен продукт ожидаемого размера для амплификации подвергнутой перестройке тяжелой цепи коровы (450 пар оснований). Этот продукт мигрировал до положения, эквивалентного положению контрольной кДНК тяжелой цепи Cмю коровы, которая, как известно, содержит последовательности, соответствующие подвергнутым перестройке транскриптам тяжелой цепи коровы (дорожка 7). Как предполагалось, перестроенная тяжелая цепь экспрессировалась в селезенке (дорожка 5), но отсутствовала в головном мозге (дорожка 2) и печени (дорожка 3) в данный момент развития. Перестройка и экспрессия локуса тяжелой цепи человека в Перестройку и экспрессию локуса тяжелой цепи человека показали посредством амплификации участка ДНК, содержащего части областей Cмю и VH. Использовали праймеры, специфичные по отношению к транскриптам РНК, содержащим части Cмю (Cмю1) и VH (VH3-30), чтобы определить, присутствуют ли транскрипты РНК, содержащие перестроенные последовательности VDJ Cмю человека (фиг. 8). Для данного анализа ОТ-ПЦР использовали праймеры «Cмю1» (5′-cag gtg cag ctg gtg gag tct gg-3′, SEQ ID NO: 11) и «VH3-30» (5′-cag gag aaa gtg atg gag tc-3′, SEQ ID NO: 12), чтобы получить продукт ОТ-ПЦР длиной 450 пар оснований. Данную ОТ-ПЦР осуществляли посредством инкубирования реакционных смесей при 95° в течение 3 минут с последующими 40 циклами инкубирования при 95° в течение 30 минут, 69° в течение 30 минут и 72° в течение 45 минут и одним циклом инкубирования при 72° в течение 10 минут. Затем полученный продукт ОТ-ПЦР повторно амплифицировали с такими же праймерами посредством одного цикла инкубирования при 95°C в течение трех минут, 40 циклов инкубирования при 95°C в течение одной минуты, 59°C в течение одной минуты, 72°C в течение одной минуты и одного цикла инкубирования при 72°C в течение 10 минут. В качестве внутреннего контроля осуществляли ОТ-ПЦР-амплификацию GAPDH, как описано выше. Фотография геля на фиг. 8 показывает, что анализ ОТ-ПЦР селезенки из плода Перестройка и экспрессия области тяжелой цепи человека в плоде Как показано на дорожках 6 и 7 на фиг. 9, амплифицированная последовательность из селезенки плода VDJ-перестройка локуса тяжелой цепи человека у Анализ ОТ-ПЦР также осуществляли для того, чтобы дополнительно показать VDJ-перестройку в локусе тяжелой цепи у Как показано на дорожках 6 и 7 на фиг. 10, анализ ОТ-ПЦР селезенки плода Подтверждение перестройки кДНК, полученную обратной транскрипцией РНК из селезенки Последовательность из данного Также получали дополнительные последовательности из перестроенных тяжелых цепей человека посредством ОТ-ПЦР-анализа селезенки плода Идентифицировали по меньшей мере два перестроенных транскрипта тяжелой цепи человека, которые представляли собой VH3-11/D7-27/JH3/C Перестройка и экспрессия локусов тяжелой и легкой цепей человека в Клонированные плоды, полученные из эмбриональных фибробластов коровы, в которые перенесены хромосомы Присутствие локусов тяжелой и легкой цепи человека в Чтобы определить сохраняют ли Как показано на фиг. 13 и 14, позитивный контроль 58-дневный плод Перестройка и экспрессия локуса тяжелой цепи человека у ОТ-ПЦР использовали для выявления экспрессии перестроенных транскриптов РНК тяжелой цепи человека у Дорожки 4 и 5 на фиг. 15 содержали амплифицированные сплайсированные последовательности константной области тяжелой цепи мю из селезенки плода Перестройка и экспрессия локуса тяжелой цепи человека в ОТ-ПЦР использовали для выявления экспрессии перестроенных РНК-транскриптов тяжелой цепи человека в селезенке Дорожка 3 на фиг. 16 содержит ОТ-ПЦР-продукт указанного анализа Перестройка и экспрессия локуса лямбда человека в Праймеры, специфичные для амплификации транскрипта, содержащего части цепи лямбда человека, использовали для выявления транскриптов РНК из перестроенного локуса легкой цепи лямбда человека. Для анализа ОТ-ПЦР, показанного на фиг. 17, использовали эквимолярную смесь праймеров C Как показано на фиг. 18, данный анализ ОТ-ПЦР также осуществляли с использованием эквимолярной смеси праймеров V Условия реакции ОТ-ПЦР были такими же, как условия, описанные выше для фиг. 7. Дорожки 6 и 7 на фиг. 17 и дорожки 4 и 5 на фиг. 18 содержали продукты ОТ-ПЦР из селезенки плода Перестройка и экспрессия локуса лямбда человека у Транскрипты РНК из локуса легкой цепи лямбда человека также выявляли у Данный анализ показал, что кДНК селезенки из Подтверждение перестройки Анализ ОТ-ПЦР осуществляли на образце селезенки из плода Идентифицировали по меньшей мере два перестроенных транскрипта легкой цепи лямбда человека (V1-17/JL3/C FACS-анализ экспрессии легкой цепи лямбда человека и тяжелой цепи коровы в Лимфоциты селезенки из Экспрессия белка антитела человека у HAC-телят Как описано выше, разработан новый способ получения телят с перенесенными хромосомами (Tc) (фиг. 37). Этот способ преодолел ограничения вследствие ограниченной продолжительности жизни, составляющей только примерно 35 удвоений популяции первичных фибробластов коров, и необходимости введения и сохранения вставки крупной ДНК в донорных клетках. В частности, использовали вектор на основе искусственной хромосомы человека (HAC), чтобы ввести полные не подвергнутые перестройке локусы как тяжелой цепи Ig (IgH), так и легкой цепи лямбда (Ig HAC вводили в эмбриональные фибробласты коров из CHO-клонов, используя методику MMCT, описанную в данной заявке. Фибробласты высевали в условиях селекции в отношении маркерного гена neo в векторе HAC с использованием G418 (700 мкг/мл) вплоть до того, как начинали появляться колонии. Полную селекцию по отношению к антибиотику и основанный на ДНК скрининг на данной стадии избегали, чтобы минимизировать деления клеток перед переносом ядер. Колонии отбирали на основе роста и морфологии, и перенос ядер осуществляли, как описано ранее (Lucier et al., J. Immunol. 161: 5438-5444, 1998). Так как клетки могли быть использованы для переноса ядер только в течение нескольких дней, окончательную селекцию осуществляли после омоложения и размножения клеток при получении клонированных плодов. Развитие до стадии бластоцисты было в пределах от 17 до 21%, а беременность на 40 день в пределах от 22 до 50%, при этом не было различий между линиями клеток. На 56-58 дни извлекали четыре Из повторно клонированных линий Результаты данного исследования показывают, что сочетание способов хромосомного клонирования, переноса хромосом и повторного клонирования соматических клеток можно использовать для получения здоровых телят, у которых сохраняется HAC-вектор, несущий геномные трансгены размером в миллионы оснований. Кроме того, данные методики используют для того, чтобы продемонстрировать перенос и сохранение полных локусов как генов IgH, так и генов Ig
ПРИМЕР 3: Трансгенные копытные животные, продуцирующие ксеногенные антитела, которые имеют мутацию в одном или нескольких эндогенных антителах У копытного животного, которое экспрессирует ксеногенные антитела и которое имеет мутацию в гене приона, как описано в примере 2, экспрессия эндогенных антител, необязательно, может быть снижена введением мутации в один или несколько эндогенных генов антител. Посредством увеличения количества функциональных ксеногенных генов тяжелой или легкой цепи иммуноглобулина по сравнению с количеством функциональных эндогенных генов тяжелой или легкой цепи процент B-клеток, экспрессирующих желаемые ксеногенные антитела (например, человеческие терапевтические антитела), может быть увеличен. Для создания указанных трансгенных копытных животных Конструкции ДНК Могут быть использованы описанные выше конструкции, нокаутирующие тяжелую цепь мю (фиг. 2A), легкую цепь лямбда, легкую цепь каппа, альфа-(1,3)-галактозилтрансферазу, прион и/или цепь J. Альтернативно можно использовать конструкцию тяжелой цепи мю с устойчивостью к пуромицину, описанную ниже (фиг. 3F). Эту нокаутирующую конструкцию конструировали так, чтобы удалить 4 основных кодирующих экзона локуса тяжелой цепи мю коров, но оставить интактным трансмембранный домен, что приводит к инактивации локуса тяжелой цепи мю. Резистентную к пуромицину конструкцию собирали следующим образом. XhoI-фрагмент длиной 4,4 тысяч пар оснований, содержащий область, расположенную непосредственно проксимально по отношению к кодирующему экзону 1, встраивали в XhoI-сайт pBluescript II SK+. Плазмиду pPGKPuro, которая содержит ген устойчивости к пуромицину, получали от д-ра Peter W. Laird, Whitehead Institute, USA. XhoI-фрагмент длиной 1,7 т.п.о., содержащий ген устойчивости к пуромицину, субклонировали вблизи или ниже фрагмента длиной 4,4 т.п.о в SalI-сайте, присутствующем в области полилинкера. Этот маркер устойчивости к пуромицину длиной 1,7 т.п.о. заменял кодирующие экзоны CH1, CH2, CH3 и CH4 локуса тяжелой цепи иммуноглобулина коровы. XbaI-фрагмент, содержащий область длиной 4,6 т.п.о. локуса мю, который в геномной последовательности дикого типа расположен ниже этих четырех экзонов, добавляли к этой конструкции для использования в качестве второй гомологичной области. Чтобы получить конечную конструкцию направленного воздействия, создавали субклон этой конструкции разрезанием трех собранных фрагментов с помощью NotI и MluI. Рестрикционное расщепление MluI укорачивает фрагмент длиной 4,6 т.п.о. до 1,4 т.п.о. Сайт NotI лежит в полилинкере и не делает разреза в самой субклонированной ДНК. Сайт MluI заполняли фрагментом Кленова, чтобы создать тупой конец, и NotI/заполненный MluI-фрагмент субклонировали в новый вектор pBluescript II SK+, используя сайты NotI и SmaI, присутствующие в векторе pBluescript. Для получения направленного воздействия на ген конечный вектор линеаризовали NotI. Воздействие на гены путем электропорации и селекция лекарственными средствами трансфицированных фибробластов Для электропорации клеточную суспензию 1 × 107 эмбриональных фибробластов коров (например, фибробластов, полученных, как описано в примере 2, из трансгенного Сбор резистентных к лекарственным средствам колоний фибробластов и размножение клеток После завершения стадии селекции лекарственными средствами (обычно от 7 до 14 дней) резистентные к лекарственным средствами колонии видны под микроскопом и готовы для переноса на 48-луночные планшеты для культуры ткани для размножения. Чтобы облегчить процесс переноса, отдельные колонии очерчивают на дне чашки для культивирования клеток, используя красящий маркер (Sharpie). Чашки для культуры ткани, содержащие колонии, промывают 2 раза 1X D-PBS (без Ca2+ и Mg2+) и затем в чашки добавляют по 5 мл буфера для диссоциации клеток в разведении 1:5. После стадии инкубирования при комнатной температуре в течение от трех до пяти минут отдельные колонии начинают открепляться от дна чашки для культуры ткани. До того как колонии открепятся, их по отдельности переносят в отдельную лунку 48-луночного планшета для культуры ткани, используя пипетку P200 и наконечник для пипетки с фильтрами против аэрозоля (200 или 250 мкл). После переноса колонию подвергают полной диссоциации пипетированием вверх-вниз и добавляют 1 мл полной среды для фибробластов. Чтобы удостовериться, что клетки резистентны к лекарственному средству, лекарственную селекцию продолжают на стадии 48-луночного культивирования. Перенесенные колонии культивируют при 38,5°C/5% CO2 и контролируют визуально в инвертированном микроскопе. Спустя от двух до семи дней лунки, в которых клетки слились, два раза промывают 1X D-PBS (без Ca2+ и Mg2+) и клетки открепляют от дна лунки добавлением 0,2 мл буфера для диссоциации клеток с последующей стадией инкубирования при комнатной температуре в течение пяти минут. После открепления клетки дополнительно подвергают диссоциации посредством пипетирования вверх-вниз, используя пипетку P1000 и защищенный от аэрозоля наконечник пипетки (1000 мкл). Примерно 75% диссоциированных фибробластов переносят в отдельную лунку 24-луночного планшета для культуры ткани, чтобы далее размножить для последующего ПЦР-анализа, а остальные 25% переносят в отдельную лунку второго 24-луночного планшета для размножения и в конечном итоге используют для экспериментов по переносу ядер соматических клеток. Когда клетки в планшете, содержащем 75% исходных клеток, размножаются почти до слияния в слой, ДНК из данного клона выделяют для генетического анализа. Приготовление ДНК Способ, используемый для выделения ДНК для генетических анализов, является адаптированным способом Laird et al, Nucleic Acids Research, 1991, Volume 19, No. 15. В частности, после того как клон почти достиг слияния в слой в одной лунке 24-луночного планшета, культуральную среду отсасывают из данной лунки и прикрепленные клетки дважды промывают PBS. PBS отсасывают и заменяют 0,2 мл буфера для лизиса клеток и расщепления избытка белка в выделяемой ДНК. Этот буфер состоит из 100 мМ трис-HCl (pH 8,5), 5 мМ EDTA, 0,2% SDS, 200 мМ NaCl и 100 мкг/мл протеиназы K. 24-Луночный планшет возвращают в инкубатор для культуры ткани минимум на три часа, чтобы обеспечить возможность высвобождения ДНК и расщепления белка. Вязкий продукт, полученный в указанной процедуре, переносят в микроцентрифужную пробирку объемом 1,5 мл и добавляют 0,2 мл изопропанола, чтобы осадить ДНК. Осадок выделяют центрифугированием, осадок ДНК промывают 70% этанолом и после сушки на воздухе осадок ресуспендируют в 25-50 мкл буфера, содержащего 10 мМ трис, pH 8 и 1 мМ EDTA. Полученную ДНК используют для ПЦР-анализов клонов. Скрининг клонов Для скрининга клонов используют два разных способа, оба с применением полимеразной цепной реакции (ПЦР). Все способы, описанные в этом разделе, можно адаптировать для направленного воздействия на любой другой ген, при этом различие будет только в последовательностях праймеров, используемых для генетического анализа. Согласно первому способу две отдельных пары праймеров используют для того, чтобы независимо амплифицировать продукты стабильной трансфекции. Одну пару праймеров используют для выявления наличия вектора направленного воздействия в геноме клона независимо от сайта интеграции. Конструируют праймеры для отжига с последовательностями ДНК, которые обе присутствуют в векторе направленного воздействия. Интенсивность ПЦР-продукта данной ПЦР-реакции может коррелировать с количеством копий вектора направленного воздействия, который был интегрирован в геном. Таким образом, клетки, содержащие только одну копию вектора направленного воздействия, имеют тенденцию давать менее интенсивные полосы в результате ПЦР. Другую пару праймеров конструируют, чтобы выявить только те копии вектора, которые интегрированы в желаемый локус. В этом случае один праймер конструируют для отжига с вектором направленного воздействия, а другой конструируют для отжига с последовательностями, специфичными для локуса, на который направленно воздействуют, которые не присутствуют в векторе направленного воздействия. В этом случае ПЦР-продукт выявляют только, если вектор направленного воздействия интегрирован непосредственно рядом с сайтом, не присутствующим в векторе направленного воздействия, что свидетельствует о желаемом событии направленного воздействия. Если продукт выявляют, то клон используют для переноса ядер. В случае резистентной к неомицину конструкции, нокаутирующей тяжелую цепь, используют праймеры Neo1 (5′-CTT GAA GAC GAA AGG GCC TCG TGA TAC GCC-3′; SEQ ID NO: 42) и IN2521 (5′-CTG AGA CTT CCT TTC ACC CTC CAG GCA CCG-3′; SEQ ID NO: 43), чтобы выявить присутствие в клетках вектора направленного воздействия, независимо от положения интеграции. Праймеры Neo1 и OUT3570 (5′-CGA TGA ATG CCC CAT TTC ACC CAA GTC TGT C-3′; SEQ ID NO: 44) используют для того, чтобы специфично амплифицировать только те копии конструкции направленного воздействия, которые интегрированы в локус тяжелой цепи мю. В случае этих ПЦР для анализа интеграции резистентной к неомицину конструкции, нокаутирующей тяжелую цепь, используют набор для ПЦР Qiagen. Смесь для реакции ПЦР содержит 1 пмоль каждого праймера, 5 мкл 10X-буфера реакции, 10 мкл раствора Q, 5 мкл ДНК и 1 мкл раствора dNTP. Реакционную смесь доводят до общего объема 50 мкл, используя H2O. Такую ПЦР-амплификацию осуществляют с использованием исходного денатурирующего инкубирования при 94°C в течение двух минут. Затем осуществляют 30 циклов денатурации, отжига и амплификации посредством инкубирования при 94°C в течение 45 секунд, 60°C в течение 45 секунд и 72°C в течение двух минут. Затем реакционную смесь инкубируют при 72°C в течение пяти минут и при 4°C вплоть до извлечения смеси из устройства для ПЦР. В альтернативном способе используют один набор праймеров, чтобы амплифицировать направленно измененный локус, и размер ПЦР-продуктов является диагностическим признаком в отношении точного попадания в мишень. Один праймер конструируют для отжига с областью локуса, не присутствующей в векторе направленного воздействия, а другой праймер конструируют для отжига с сайтом, присутствующим в векторе направленного воздействия, но также присутствующем в локусе дикого типа. В этом случае не выявляется вектор направленного воздействия, который был интегрирован в нежелательные сайты генома. Так как область, делетированная вектором направленного воздействия, отличается по размеру от маркера для селекции по отношению к лекарственному средству, встроенного на ее место, то размер продукта зависит от того, имеет ли амплифицированный локус генотип дикого типа или направленно измененный генотип. Амплификация ДНК из клонов, содержащих неправильные инсерции или совсем не содержащих инсерций вектора направленного воздействия, приводит к единственному ПЦР-продукту с размером, ожидаемым для локуса дикого типа. Амплификация ДНК из клонов, содержащих направленно измененный («нокаутированный») аллель, приводит к образованию двух ПЦР-продуктов, одного, представляющего собой амплификацию аллеля дикого типа, и одного, измененного, имеющего предсказуемый размер вследствие замены какой-либо последовательности в аллеле дикого типа маркером устойчивости к лекарственному средству, который отличается по длине от замененной им последовательности. В случае резистентной к пуромицину конструкции, нокаутирующей тяжелую цепь, используют праймеры Shortend (5′-CTG AGC CAA GCA GTG GCC CCG AG-3′; SEQ ID NO: 45) и Longend (5′-GGG CTG AGA CTG GGT GAA CAG AAG GG-3′; SEQ ID NO: 46). Данная пара праймеров амплифицирует как локус тяжелой цепи дикого типа, так и локусы, которые были соответствующим образом направленно изменены пуромициновой конструкцией. Различие по размеру между двумя полосами составляет примерно 0,7 т.п.о. Присутствие более короткой полосы является показателем соответствующего попадания в мишень. В случае данной ПЦР для анализа интеграции резистентной к пуромицину конструкции, нокаутирующей тяжелую цепь, используют набор Promega Master Mix. Смесь для реакции ПЦР содержит 1 пмоль каждого праймера, 2,5 мкл ДНК и 25 мкл 2X Promega Master Mix. Реакционную смесь доводят до общего объема 50 мкл с использованием H2O. Данную ПЦР-амплификацию осуществляют, используя начальную денатурирующую инкубацию при 94°C в течение двух минут. Затем 30 циклов денатурации, отжига и амплификации осуществляют посредством инкубирования при 94°C в течение 45 секунд, 60°C в течение 45 секунд и 72°C в течение двух минут. Затем реакционную смесь инкубируют при 72°C в течение пяти минут и при 4°C вплоть до извлечения смеси из устройства для ПЦР. Первый раунд переноса ядер Отобранные фибробласты, в которых был инактивирован ген иммуноглобулина, могут быть использованы для переноса ядер, как описано в примере 2, чтобы создать трансгенное копытное животное, содержащее мутацию в эндогенном гене иммуноглобулина и содержащее HAC, кодирующую ксеногенный ген иммуноглобулина. Альтернативно перенос ядер можно осуществить, используя стандартные способы для того, чтобы встроить ядро или хроматиновую массу (т.е. одну или несколько хромосом, не заключенных в мембрану) из отобранного трансгенного фибробласта в лишенный ядра ооцит (описано, например, в U.S.S.N. 60/258151; поданном 22 декабря 2000 г.). Такие способы также можно использовать для клеток, в которых была мутирована эндогенная нуклеиновая кислота, кодирующая альфа-(1,3)-галактозилтрансферазу, прион и/или цепь J. Второй раунд мутагенеза и переноса ядер При желании клетку (например, эмбриональный фибробласт) можно получить из трансгенного копытного животного, созданного в первом раунде переноса ядер. Можно осуществить другой раунд направленного изменения гена, как описано выше, чтобы инактивировать второй аллель гена, инактивируемого в первом раунде направленного изменения. Альтернативно в этом раунде целенаправленного изменения можно инактивировать другой ген иммуноглобулина (например, тяжелой цепи мю, легкой цепи лямбда, легкой цепи каппа или цепи J), альфа-(1,3)-галактозилтрансферазы или приона. Для данного второго раунда направленного изменения можно использовать либо более высокую концентрацию антибиотика, либо нокаутирующую конструкцию с другим маркером устойчивости к антибиотику. Резистентные к антибиотику клетки можно отобрать, как описано выше. Отобранные клетки можно использовать во втором раунде переноса ядер, как описано выше, чтобы создать, например, трансгенное копытное животное, содержащее две мутации в эндогенных генах иммуноглобулина и содержащее HAC, кодирующую ксеногенный ген иммуноглобулина. Альтернативно отобранные резистентные к антибиотику клетки сначала могут быть обработаны, чтобы выделить клетки в G1-фазе, как описано ниже, которые используют для второго раунда переноса ядер. Чтобы выделить клетки G1 для переноса ядер, 5,0 x 105 клеток высевают в чашки для культуры ткани диаметром 100 мм, содержащие 10 мл ПРИМЕР 4: Дополнительные способы введения мутаций в эндогенные гены иммуноглобулина В некоторых вариантах осуществления данного способа продукция ксеногенного иммуноглобулина у копытного животного с одной или несколькими мутациями в гене, кодирующем прион, осуществляется, по существу, в результате применения сочетания способов гомологичной рекомбинации, введения искусственных хромосом, несущих полные ксеногенные локусы Ig, переноса ядер и введения антитела, чтобы элиминировать эндогенное антитело. Более конкретно, способ, предпочтительно, включает в себя направленное разрушение одного или обоих аллелей гена тяжелой цепи IgM и, необязательно, одного или обоих аллелей гена легкой цепи Ig, хотя продуцирование ксеногенного антитела также может быть осуществлено у животных без мутаций в нуклеиновых кислотах Ig. Нокауты генов могут быть выполнены последовательной гомологичной рекомбинацией, затем еще одной процедурой скрещивания. В предпочтительном варианте осуществления сначала выполняют направленное разрушение одного аллеля гена тяжелой цепи IgM эмбрионального фибробласта копытного животного мужского или женского пола (например, крупного рогатого скота) в культуре ткани с использованием подходящего вектора для гомологичной рекомбинации. Применение эмбриональных фибробластов предпочтительно по сравнению с некоторыми другими соматическими клетками, так как указанные клетки легко размножать и проводить генетические манипуляции в культуре ткани. Однако применение эмбриональных фибробластов не является существенно важным для изобретения и в действительности они могут быть заменены другими клеточными линиями с эквивалентными результатами. Конечно данный способ включает в себя создание ДНК-конструкции, имеющей гомологичные области с являющимся мишенью аллелем тяжелой цепи IgM, так чтобы конструкция при интеграции в аллель тяжелой цепи IgM в геноме копытного животного нарушала его экспрессию. Типичный вектор для осуществления такого целенаправленного разрушения аллеля IgM описан в примере, который следует далее. Способы конструирования векторов, которые обеспечивают гомологичную рекомбинацию в сайте-мишени, хорошо известны специалистам в данной области. Кроме того, в этом случае конструирование подходящего вектора возможно на уровне развития данной области, особенно учитывая, что последовательности генов тяжелой цепи IgM и легкой цепи лямбда Ig коровы известны, как и последовательности генов иммуноглобулинов из других копытных животных (смотри ниже). Для того чтобы облегчить гомологичную рекомбинацию, векторы, используемые для осуществления гомологичной рекомбинации и инактивации гена IgM, соответственно содержат части ДНК, по существу идентичные последовательностям генов тяжелой цепи IgM и легкой цепи Ig копытного животного. Предпочтительно, указанные последовательности обладают по меньшей мере 98% идентичностью последовательностей, более предпочтительно по меньшей мере 99% идентичностью последовательностей и еще более предпочтительно будут изогенными локусам генов-мишеней, чтобы облегчить гомологичную рекомбинацию и направленное делетирование или инактивацию. Обычно и предпочтительно, конструкция будет содержать маркерный ген, который обеспечивает селекцию желаемых гомологичных рекомбинантов, например клеток фибробластов, в которых ген тяжелой цепи IgM и/или ген легкой цепи Ig был эффективно поврежден. Примеры маркерных генов наряду с прочим включают маркеры устойчивости к антибиотикам, маркеры лекарственной устойчивости и зеленый флуоресцирующий белок. Предпочтительная конструкция показана на фиг. 2A, а исходные материалы, используемые для получения такой конструкции, показаны на фиг. 3A и 3B. Другие конструкции, содержащие две гомологичные области с эндогенным геном иммуноглобулина, которые фланкируют маркер для позитивной селекции (например, ген устойчивости к антибиотику), который оперативно связан с промотором, могут быть созданы с использованием стандартных способов молекулярной биологии и использованы в способах по данному изобретению. Конструкцию, нокаутирующую по мю, показанную на фиг. 2A и 3C, конструировали так, чтобы удалить экзоны, кодирующие константную область тяжелой цепи иммуноглобулина коровы, называемые «экзонами C-мю 1-4», и два экзона, кодирующих трансмембранный домен, называемые «TM-экзонами». Чтобы сконструировать данный вектор, область, называемую «1», XbaI-XhoI-фрагмент из геномной последовательности тяжелой цепи мю коровы, субклонировали в коммерческий ДНК-вектор pBluescript (Stratagene, LaJolla, California), предварительно разрезанный ферментами XbaI и XhoI. После клонирования данного фрагмента имелась последовательность узнавания ферментом рестрикции NotI вблизи сайта XbaI, используемая для инсерции NotI-фрагмента длиной примерно 3,5 т.п.о. Данный фрагмент содержит маркер устойчивости к неомицину, описанный далее. При желании можно сконструировать другие конструкции, нокаутирующие мю, с использованием геномной последовательности тяжелой цепи мю из другой породы, вида или рода копытного животного (например, последовательности тяжелой цепи мю, депонированной в Genbank с номером доступа U63637, из гибрида швейцарской/голштинской пород). После того как фрагмент «1» и маркер устойчивости к неомицину соединяли вместе в pBluescript, оставался сайт SacI вблизи маркера устойчивости к неомицину. Новую конструкцию линеаризовали SacI и концы делали тупыми, заполняя липкие концы, оставшиеся от расщепления SacI, используя ДНК-полимеразу. Фрагмент, обозначенный «2», выделяли в виде XhoI-BstI107l-фрагмента и превращали во фрагмент с тупыми концами, заполняя липкие концы, оставшиеся от действия ферментов Xho0I и BstI107l, используя ДНК-полимеразу. После завершения конечная конструкция содержала область 2, маркер устойчивости к неомицину и область 1 соответственно. Для трансфекции фибробластов коровы конструкцию расщепляли ферментом рестрикции KpnI (два сайта KpnI показаны на диаграмме) и ДНК-фрагмент использовали для гомологичной рекомбинации. Конструкцию устойчивости к неомицину собирали следующим образом. Конструкцию, названную «pSTneoB» (Katoh et al., Cell Struct. Funct. 12: 575, 1987; Japanese Collection of Research Biologicals (JCRB), номер депозита VE039), конструировали так, чтобы она содержала ген устойчивости под контролем промотора SV40 и энхансер TK выше кодирующей области. Ниже кодирующей области находится последовательность терминатора SV40. Кассету neo вырезали из «pSTneoB» в виде XhoI-фрагмента. После превращения концов фрагмента в тупые концы с использованием стандартных способов молекулярной биологии фрагмент с тупыми концами клонировали в сайт EcoRV в векторе pBS246 (Gibco/Life Technologies). Указанный сайт фланкирован сайтами loxP. Новую конструкцию, названную «pLoxP-STNeoR», использовали для создания ДНК-конструкции, нокаутирующей мю. Желаемый фрагмент этой конструкции фланкирован сайтами loxP и сайтами NotI, которые исходно присутствуют в клонирующем векторе pBS246. Желаемый NotI-фрагмент, который содержит loxP-neo-loxP, использовали для замены экзонов константной области мю иммуноглобулина. Промотор SV40, оперативно связанный с геном устойчивости к неомицину, активирует транскрипцию гена устойчивости к неомицину, позволяя отбирать клетки, в которых желаемый NotI-фрагмент заменил экзоны константной области мю, на основе возникающей у них устойчивости к антибиотику. После получения клеточной линии, в которой аллель тяжелой цепи IgM эффективно поврежден, ее используют в качестве донора для переноса ядер, чтобы получить клонированный плод копытного животного (например, клонированный плод коровы) и в конечном итоге плод или животное, у которого один из аллелей тяжелой цепи IgM поврежден. После этого может быть осуществлен второй раунд направленного повреждения гена с использованием полученных от них соматических клеток, например фибробластов, чтобы получить клетки, в которых инактивирован второй аллель тяжелой цепи IgM, используя сходный вектор, но содержащий другой селектируемый маркер. Предпочтительно, одновременно с первым направленным повреждением гена также генетически модифицируют вторую линию соматических клеток копытного животного (например, коровы), которая в равной мере может происходить от животного мужского или женского пола. Если первая подвергаемая обработке клеточная линия относится к мужскому полу, то предпочтительно модифицировать линию женских клеток; и наоборот, если первая подвергаемая обработке клеточная линия относится к женскому полу, то предпочтительно следует выбрать линию мужских клеток. Опять-таки, предпочтительно, подвергаемые обработке клетки являются эмбриональными фибробластами копытного животного (например, коровы). В предпочтительном варианте осуществления эмбриональный фибробласт женского организма генетически модифицируют так, чтобы ввести направленное повреждение одного аллеля гена легкой цепи лямбда Ig. Этот способ осуществляют сходным образом, используя вектор, имеющий гомологичные области с легкой цепью лямбда Ig копытного животного (например, коровы) и селектируемый маркер, и такую конструкцию ДНК создают так, чтобы при интеграции и гомологичной рекомбинации с эндогенной легкой цепью Ig она приводила к повреждению (инактивации) гена легкой цепи лямбда Ig, являющегося мишенью. После того как отбирают клетку женского фибробласта, имеющую желаемое направленное повреждение, ее подобным образом используют в качестве донорной клетки для переноса ядра или ДНК из такой клеточной линии, используя в качестве донора для переноса ядер. Альтернативно, такая клетка может быть подвергнута второму раунду гомологичной рекомбинации, чтобы инактивировать вторую легкую цепь лямбда Ig, используя конструкцию ДНК, сходную с конструкцией, используемой для повреждения первого аллеля, но содержащую другой селектируемый маркер. Способы осуществления переноса ядер и, в частности, для получения клонированных коров и клонированных трансгенных коров сообщались и описаны в патенте США No. 5945577. Альтернативно можно использовать способы переноса ядер, раскрытые в любой из публикаций PCT No. WO95/16670, WO96/07732, WO97/07669 или WO97/07668 (вместе способы Roslin). Способы Roslin отличаются от способов, разработанных в университете Массачусетса тем, что в них используют покоящиеся, а не пролиферирующие донорные клетки. Все указанные патенты приведены здесь в качестве ссылки в полном объеме. Такие способы переноса ядер будут давать трансгенный клонированный плод, который можно использовать для получения клонированного трансгенного потомства коров, например потомства, которое содержит направленное повреждение по меньшей мере одного аллеля гена легкой цепи Ig и/или гена IgM. После создания таких клеточных линий они могут быть использованы для получения гемизиготно нокаутированного по тяжелой и легкой цепи плода и потомства мужского и женского пола (M и F геми-H/L). Кроме того, указанные способы не ограничены применением для получения трансгенных коров; указанные выше способы также могут быть использованы для переноса ядер других копытных животных. После переноса ядер может быть осуществлено получение желаемых животных либо скрещиванием копытных животных, либо вторым направленным изменением гена с использованием гомологичного вектора направленного воздействия, описанного выше. Как указано ранее, следующей целью изобретения является создание гемизиготных нокаутированных по тяжелой и легкой цепи организмов мужского и женского пола, при этом такие гемизиготные нокаутированные организмы получают с использованием клеточных линий, которые уже описаны. Это можно осуществить либо скрещиванием потомства, полученного согласно описанным выше способам, при котором потомка, который содержит поврежденный аллель гена тяжелой цепи IgM, скрещивают с другим потомком, который содержит поврежденный аллель легкой цепи Ig. Альтернативно это можно осуществить посредством второго направленного изменения гена при обработке клетки, которую получают от потомства, полученного согласно описанным выше способам. Это включает осуществление посредством гомологичной рекомбинации направленного повреждения аллеля гена тяжелой цепи IgM или аллеля легкой цепи Ig. После получения клеточной линии, которая содержит гемизиготные нокаутированные по тяжелой и легкой цепи мужские и женские клетки (M и F геми-H/L), клеточную линию используют для получения плода или теленка, который содержит такой нокаут. Как указано, это осуществляют либо скрещиванием, либо вторым направленным воздействием на ген. После получения гемизиготных нокаутированных по тяжелой и легкой цепи организмов мужского и женского пола клетки от таких животных можно использовать для создания гомозиготных нокаутированных (гомо-H/L) плодов. Это осуществляют также либо последовательным направленным изменением гена, либо скрещиванием. По существу, в случае осуществления скрещиванием это будет заключаться в скрещивании гемизиготного нокаутированного по тяжелой и легкой цепи организма мужского пола с гемизиготным нокаутированным по тяжелой и легкой цепи организмом женского пола и отбором потомства, которое содержит гомозиготный нокаут. Альтернативно клетки от гемизиготных нокаутированных животных, описанных выше, могут быть подвергнуты манипуляциям в культуре ткани, так чтобы нокаутировать другой аллель гена IgM или гена легкой цепи (лямбда) Ig. Второе направленное изменение гена может быть предпочтительным по сравнению со скрещиванием, так как может давать более быстрые результаты, особенно принимая во внимание, что период беременности копытных животных, таких как коровы, является относительно длинным. В ходе данного последовательного нокаута и/или методики скрещивания один или оба аллеля гена, кодирующего прион, могут быть мутированы в любой донорной клетке, полученной в ходе этого процесса (например, донорной клетке с или без мутации в эндогенной нуклеиновой кислоте Ig и с или без ксеногенной нуклеиновой кислоты Ig). Таким образом, копытные животные, созданные с использованием способов, приведенных в этом описании, могут иметь некоторое количество желаемых мутаций в гене приона, гене тяжелой цепи Ig, гене легкой цепи Ig, гене альфа-(1,3)-галактозилтрансферазы и/или гене цепи J и могут иметь один или несколько ксеногенных генов Ig и/или цепи J. Способы нокаута для получения трансгенных копытных животных, которые экспрессируют Ig человека Способы получения плодов или телят гомо-H/L суммированы на фиг. 1. На фигуре изображены три схемы. Первая основана на успешных нокаутах в регенерированных эмбриональных клеточных линиях. Этот способ технически является наиболее трудным и имеет самый высокий уровень риска, но, как указано выше, потенциально дает более быстрые результаты, чем способы на основе скрещивания. Две другие схемы основаны на скрещивании животных. В случае второй схемы требуются только однократные нокауты генов тяжелой и легкой цепи в линиях мужских и женских клеток соответственно. Данная схема не основана на регенерации клеточных линий и технически является самым простым способом, но требует самого длительного периода времени для полного завершения. Схема 3 является промежуточной между схемами 1 и 2. Во всех схемах создают только гомо-H/L-плоды из-за потенциальных трудностей, связанных с жизнеспособностью и поддержанием нокаутированных телят гомо-H/L. При необходимости можно использовать пассивную иммунотерапию, чтобы повысить жизнеспособность нокаутированных телят гомо-H/L. Дизайн эксперимента Данное изобретение, предпочтительно, включает в себя получение мужских гемизиготных нокаутов по тяжелой цепи (M геми-H) и женских гемизиготных нокаутов по легкой цепи (F геми-L) и получение 40-дневных плодов на основе таких направленных делеций. Клетки из эмбрионов собирают и один аллель локуса легкой цепи подвергают направленному воздействию в клетках M геми-H, а один аллель локуса тяжелой цепи подвергают направленному воздействию в клетках F геми-L, получая клетки с гемизиготными делециями в обоих локусах H и L (геми-H/L). Эти клетки используют для получения 40-дневных плодов, из которых выделяют фибробласты. Фибробласты M геми-H/L подвергают направленному изменению в отношении другого аллеля цепи H, чтобы создать M гомо-H/геми-L, и F геми-H/L подвергают целенаправленному изменению в отношении другого аллеля цепи L, чтобы создать F гомо-L/геми-H. Чтобы создать гомозиготные делеции, используют более высокие концентрации лекарственного средства для стимулирования гомозиготного целенаправленного изменения. Однако возможно, что данный способ может оказаться успешным и скрещивание может не потребоваться. Пример способа, который основан на опосредованном cre/lox направленном воздействии кассеты, селекции, позволяет использовать одну и ту же систему селекции для более чем одной направленной делеции. Указанные фибробласты клонируют, собирают 40-дневные плоды и выделяют фибробласты. Эмбриональные клетки, полученные в результате указанного клонирования, подвергают направленному изменению, чтобы получить гомозиготные делеции либо локуса H, либо локуса L, результатом которых являются эмбриональные фибробласты M гомо-H/L и F гомо-H/L. Указанные фибробласты клонируют и получают 40-дневные плоды, и выделяют фибробласты. Эмбриональные фибробласты гомо-H/L затем используют для введения HAC, необязательно, с применением способов скрещивания. Конструирование библиотеки Эмбриональные фибробласты используют для конструирования геномной библиотеки. Хотя сообщается о важности того, чтобы конструкция направленного воздействия была изогенной с клетками, используемыми для клонирования, это не существенно для изобретения. Например, можно использовать изогенную, по существу изогенную или неизогенную конструкции, чтобы получить мутацию в эндогенном гене иммуноглобулина. В одном возможном способе используют крупный рогатый скот голштинской породы, который генетически содержит высокий уровень инбридинга по сравнению с другими породами. Авторы не обнаружили какого-либо полиморфизма в генах иммуноглобулинов среди других животных. Это свидетельствует о том, что гомология последовательностей должна быть высокой и что направленное изменение с помощью неизогенных конструкций может быть успешным. Библиотеку конструируют на основе одной мужской клеточной линии и одной женской клеточной линии при одновременном проведении тестирования «способности к клонированию». Предполагается, что к концу процесса будет получена библиотека и будет проведено тестирование ряда различных линий эмбриональных клеток и выбрана одна клеточная линия, как наилучшая линия в целях клонирования. Геномные библиотеки конструируют с использованием ДНК с высокой молекулярной массой, выделенной из эмбриональных фибробластов. ДНК фракционируют по размеру и высокомолекулярную ДНК длиной 20-23 т.п.о. встраивают в вектор на основе фага лямбда LambdaZap или LambdaFix. Авторы изобретения получили хорошие результаты в случае библиотек, полученных в Stratagene. Поэтому ДНК выделяют и отобранную по размеру ДНК посылают в Stratagene для получения библиотеки. Чтобы выделить клоны, содержащие тяжелую и легкую цепи коровы, используют радиоактивно меченую кДНК IgM и радиоактивно меченую кДНК легкой цепи. Кроме того, геномные клоны легкой цепи выделяют в случае, если необходимо делетировать локус. Каждую библиотеку эмбриональных клеток подвергают скринингу в отношении клонов, содержащих тяжелую и легкую цепь коровы. Ожидается, что скрининг примерно 105 – 106 бляшек должен приводить к выделению клонов, содержащих локус либо тяжелой цепи, либо легкой цепи. После выделения оба локуса субклонируют в pBluescript и подвергают рестрикционному картированию. Рестрикционная карта этих локусов у животных голштинской породы представлена на фиг. 2B (Knight et al. J. Immunol. 140: 3654-3659, 1988). Кроме того, создают карту полученных клонов и используют для сборки конструкции направленного воздействия. Получение конструкций направленного воздействия После выделения генов тяжелой и легкой цепи создают конструкции. Конструкцию IgM создают делетированием мембранного домена константной области IgM. Как показано Rajewsky с соавторами, у мышей делеция мембранного домена IgM приводит к блокированию развития B-клеток, так как поверхностный IgM является необходимым сигналом для непрерывного развития B-клеток (Kitamura et al., Nature 350: 423-426). Таким образом, у гомозиготного по IgM крупного рогатого скота отсутствуют B-клетки. Это не должно создавать проблему, так как в этом способе не нужны живорожденные животные с отсутствующим функциональным Ig. Однако при необходимости можно использовать пассивную иммунотерапию, чтобы повысить жизнеспособность животных вплоть до последней стадии, когда вводятся локусы Ig человека. Пример конструкции направленного воздействия, используемой для осуществления нокаута аллеля тяжелой цепи IgM, показан на фиг. 2A. В случае тяжелой цепи мембранный домен IgM заменяют неомициновой кассетой, фланкированной сайтами lox P. Связанный мембранный домен сплайсируется вместе с neo-кассетой, так что мембранный домен имеет стоп-кодон TAG, встроенный непосредственно с 5′-стороны от сайта lox P, обеспечивающий инактивацию мембранного домена. Его помещают на 5′-конце конструкции направленного воздействия примерно с 5-6 тысячами пар оснований 3′-хромосомной ДНК. Если возрастающие концентрации лекарственного средства не позволяют делетировать второй аллель либо тяжелой цепи IgM, либо легкой цепи, используют систему cre/lox (описанную в обзоре Sauer, 1998, Methods 14: 381-392), чтобы делетировать селектируемый маркер. Как описано ниже, система cre/lox создает возможность для направленной делеции селектируемого маркера. Все селектируемые маркеры фланкируют последовательностями loxP, чтобы облегчить делецию данных маркеров, если это будет необходимо. Конструкция легкой цепи содержит константную область цепи лямбда коровы (например, константную область легкой цепи лямбда, имеющуюся в GenBank с номером доступа AF396698, или любую другую константную область легкой цепи лямбда копытного животного) и кассету гена устойчивости к пуромицину, фланкированную сайтами lox P, и будет заменять ген коровы пуромициновой кассетой, фланкированной сайтами lox P. Примерно 5-6 тысяч пар оснований ДНК, расположенных с 3′-стороны от гена константной области лямбда, будет заменено с 3′-стороны от гена устойчивости к пуромицину. Ген устойчивости к пуромицину будет нести сайты lox P на обоих 5′- и 3′- концах, чтобы при необходимости обеспечить делецию. Вследствие высокой степени гомологии между генами антител копытных животных предполагается, что последовательность легкой цепи лямбда коровы в GenBank с номером доступа AF396698 гибридизуется с геномной последовательностью легкой цепи лямбда множества копытных животных и таким образом может быть использована в стандартных способах, чтобы выделять различные геномные последовательности легкой цепи лямбда копытных животных. Такие геномные последовательности можно использовать в стандартных способах, таких как способы, приведенные в этом описании, чтобы создать нокаутирующие конструкции для инактивации эндогенных легких цепей лямбда в любом копытном животном. Конструкция, нокаутирующая легкую цепь каппа, может быть сконструирована сходным образом с использованием последовательности легкой цепи каппа коровы, показанной на фиг. 3G, или любой другой последовательности легкой цепи каппа копытного животного. Данную легкую цепь каппа коровы можно использовать в качестве зонда для гибридизации, чтобы выделить геномные последовательности легкой цепи каппа из множества копытных животных. Такие геномные последовательности можно использовать в стандартных способах, таких как способы, приведенные в этом описании, чтобы создать нокаутирующие конструкции для инактивации эндогенных легких цепей каппа у любого копытного животного. Необязательно, могут быть мутированы или инактивированы дополнительные гены копытного животного. Например, может быть нокаутирован эндогенный ген J-цепи Ig копытного животного, чтобы предотвратить потенциальную антигенность J-цепи Ig копытного животного в антителах по изобретению, которые вводят человеку. Для конструирования вектора направленного воздействия можно использовать последовательность кДНК области J-цепи Ig коровы, имеющуюся в Genbank под номером доступа U02301. Такую последовательность кДНК можно использовать в качестве зонда для выделения геномной последовательности J-цепи Ig коровы из BAC-библиотеки, такой как RPC1-42 (BACPAC in Oakland, CA), или для выделения геномной последовательности J-цепи из любого другого копытного животного. Кроме того, кодирующую последовательность J-цепи человека можно ввести копытному животному по данному изобретению для функциональной экспрессии молекул IgA и IgM человека. Последовательность кДНК J-цепи человека доступна из GenBank с номерами доступа AH002836, M12759 и M12378. Данная последовательность может быть встроена в эмбриональный фибробласт копытного животного с использованием стандартных способов, таких как способы, приведенные в этом описании. Например, нуклеиновая кислота J-цепи человека в HAC, YAC-векторе, BAC-векторе, космидном векторе или нокаутирующей конструкции может быть интегрирована в эндогенную хромосому копытного животного или сохраняться независимо от эндогенных хромосом копытного животного. Полученные трансгенные клетки копытного животного можно использовать в способах переноса ядер, приведенных в этом описании, чтобы создать желаемых копытных животных, которые имеют мутацию, которая снижает или исключает экспрессию функциональной J-цепи копытного животного, и которые содержат ксеногенную нуклеиновую кислоту, которая экспрессирует J-цепь человека. Кроме того, может быть мутирован ген При желании кодирующий прионный белок ген копытного животного может быть мутирован или инактивирован, чтобы уменьшить возможный риск инфекции, такой как губчатая энцефалопатия крупного рогатого скота (BSE). Для конструирования вектора направленного воздействия можно использовать геномную последовательность ДНК кодирующего прион гена коровы (номер доступа в GenBank AJ298878), как описано в примере 1. Альтернативно эту геномную последовательность приона можно использовать для выделения геномной последовательности приона из других копытных животных. Ген приона может быть инактивирован с использованием способов, приведенных в этом описании. Альтернативно ранее описанные способы мутирования гена альфа-(1,3)-галактозилтрансферазы или кодирующего прион гена у овец (Denning et al., Nature Biotech., 19: 559-562, 2001) можно адаптировать на основе приведенных в этом описании инструкций. Например, можно использовать усовершенствованные способы, которые приведены в этом описании для культивирования донорных клеток (например, эмбриональных фибробластов), создания нокаутирующих векторов, трансфекции донорных клеток нокаутирующими векторами (например, трансфекции в присутствии спермидина) и/или переноса генетического материала из нокаутированных клеток в ооцит для создания трансгенного копытного животного. Для направленного изменения второго аллеля каждого локуса может быть необходима сборка новой конструкции направленного воздействия, содержащей другой селектируемый маркер, если первый селектируемый маркер сохраняется в клетке. Как описано в таблице 5, имеется множество способов селекции и их можно сравнить и выбрать подходящую систему селекции. Сначала второй аллель направленно изменяют повышением концентрации лекарственного средства (например, удвоением концентрации лекарственного средства). Если это не дает успеха, можно использовать новую конструкцию направленного воздействия.
Дополнительные мутации или инактивация генов, указанные выше, могут быть введены копытным животным по данному изобретению с использованием различных методик. После создания трансгенной линии клеток копытного животного для каждой желаемой мутации можно использовать кроссбридинг для введения дополнительных мутаций копытным животным по данному изобретению. Альтернативно эмбриональные фибробласты, которые имеют дополнительные мутации, можно использовать в качестве исходного материала для нокаута эндогенных генов Ig и/или введения ксеногенных генов Ig. Также эмбриональные фибробласты, имеющие нокаутирующую мутацию в эндогенных генах Ig и/или содержащие ксеногенные гены Ig, можно использовать в качестве исходного материала для таких дополнительных мутаций или инактиваций. Целенаправленная делеция локусов Ig Конструкции направленного воздействия вводят в эмбриональные фибробласты, например, электропорацией. Клетки, которые включают вектор направленного воздействия, отбирают, используя соответствующий антибиотик. Клоны, которые являются резистентными к выбранному лекарственному средству, будут подвергнуты селекции в отношении роста. Затем указанные клоны подвергают негативной селекции ганцикловиром, который будет отбирать те клоны, которые имеют соответствующие интеграции. Альтернативно клоны, которые выживают при лекарственной селекции, подвергают отбору с помощью ПЦР. Рассчитано, что будет необходимо провести скрининг по меньшей мере 500-1000 клонов, чтобы найти соответствующим образом направленно измененный клон. Оценка авторов изобретения основана на данных Kitamura (Kitamura et al., Nature 350: 423-426, 1991), который обнаружил, что при направленном воздействии на мембранный домен константной области тяжелой цепи IgM примерно 1 из 300 neo-резистентных клонов были правильным образом направленно изменены. Таким образом предлагается объединить клоны в группы из 10 клонов в 96-луночном планшете и провести скрининг пулов из 10 клонов в отношении целенаправленно измененных предпочтительных клонов. После идентификации позитивного сигнала будет проведен скрининг отдельных клонов, выделенных из объединенного клона. Указанная методика может обеспечить возможность идентификации направленно измененного клона. Поскольку фибробласты передвигаются в культуре, то трудно отличить отдельные клоны, когда на чашке образуется примерно более десяти клонов. Кроме того, может быть разработана методика для клонального размножения с высокоэффективной трансфекцией. Может быть использовано несколько подходящих методик, таких как клонирование с разбавлением. Cre/Lox-эксцизия маркера устойчивости к лекарственному средству Как показано выше, иллюстративные конструкции направленного воздействия содержат селектируемые маркеры, фланкированные сайтами loxP, чтобы обеспечить эффективную делецию маркера с использованием системы cre/lox. Эмбриональные фибробласты, несущие вектор направленного воздействия, трансфицируют посредством электропорации Cre-содержащей плазмидой. Можно использовать недавно описанную Cre-плазмиду, которая содержит слитый ген GFPcre (Gagneten et al., Nucleic Acids Res. 25: 3326-31, 1997). Это создает возможность для быстрой селекции всех клонов, которые содержат белок Cre. Эти клетки отбирают либо FACS-сортировкой, либо ручным сбором зеленых флуоресцирующих клеток с помощью микроманипулирования. Предполагается, что клетки, которые являются зелеными, несут активно транскрибируемую Cre-рекомбиназу и, следовательно, делетируют маркер устойчивости к лекарственному средству. Клетки, отобранные в отношении экспрессии Cre, клонируют и клоны анализируют в отношении делеции маркера устойчивости к лекарственному средству с помощью ПЦР-анализа. Те клетки, которые на основании определения подверглись эксцизии, выращивают до небольших клонов, делят и одну аликвоту тестируют в селективной среде, чтобы с уверенностью убедиться в том, что ген устойчивости к лекарственному средству был делетирован. Другую аликвоту используют для следующего раунда целенаправленной делеции. Применение методики направленного воздействия для изменения генов иммуноглобулинов других копытных животных Чтобы изменить гены иммуноглобулинов других копытных животных конструируют векторы направленного воздействия так, чтобы они содержали три основных области. Первая область гомологична локусу, подвергаемому направленному изменению. Вторая область является маркером для лекарственной селекции, который специфично заменяет часть направленно изменяемого локуса. Третья область подобно первой области гомологична направленно изменяемому локусу, но не является смежной с первой областью в геноме дикого типа. Гомологичная рекомбинация между вектором направленного воздействия и желаемым локусом дикого типа приводит к делеции последовательностей локуса между двумя гомологичными областями, представленными в векторе направленного воздействия, и замене этой последовательности маркером устойчивости к лекарственному средству. В предпочтительных вариантах осуществления общий размер двух гомологичных областей составляет примерно 6 тысяч пар оснований, а размер второй области, которая заменяет часть целенаправленно изменяемого локуса, составляет примерно 2 тысячи пар оснований. Такая методика направленного изменения применима для широкого круга видов – от прокариотической клетки до клетки человека. Уникальность каждого используемого вектора заключается в локусе, выбранном для способов направленного изменения генов, и используемых в данной методике последовательностях. Этот способ можно использовать для всех копытных животных, включая без ограничения коз (Capra hircus), овец (Ovis aries) и свиней (Sus scrufa), также крупный рогатый скот (Bos taurus). Применение электропорации для направленного изменения специфичных генов в клетках копытных животных также может быть широко использовано у копытных животных. Общий способ, приведенный в этом описании, может быть адаптирован по отношению к введению направленных мутаций в геномы других копытных животных. Модификацию условий электропорации (напряжения и емкости) можно использовать для оптимизации количества трансфектантов, получаемых в случае других копытных животных. Кроме того, способ, используемый в данном изобретении для направленного изменения локуса тяжелой цепи у крупного рогатого скота (например, удаление всех кодирующих экзонов и вставочных последовательностей с использованием вектора, содержащего области, гомологичные областям, непосредственно фланкирующим удаляемые экзоны), также можно в равной мере успешно использовать у других копытных животных. Например, был проведен всесторонний анализ последовательностей для локуса тяжелой цепи иммуноглобулина овец (Ovis aries), и локус овец в высокой степени подобен локусу коров как по структуре, так и по последовательности (номера доступа в GenBank Z71572, Z49180-Z49188, M60441, M60440, AF172659-AF172703). Кроме большого количества последовательностей кДНК, описанных для перестроенных цепей иммуноглобулина Ovis aries, представлена информация о геномных последовательностях локуса тяжелой цепи, включая 5′-энхансер тяжелой цепи (номер доступа в GenBank Z98207), 3′-область переключения мю (Z98680) и 5′-область переключения мю (Z98681). Полная последовательность мРНК для секретируемой формы тяжелой цепи овец депонирована с номером доступа X59994. Указанный депозит содержит полную последовательность четырех кодирующих экзонов, которая в высокой степени гомологична соответствующей последовательности коровы. Информацию о локусе овец получали из GenBank и использовали для определения гомологичных областей с последовательностью коровы для конструирования праймеров, используемых для ПЦР-анализа. Так как для целенаправленного изменения клеток коров использовали неизогенную ДНК, нахождение областей высокой гомологии с последовательностью овец использовали в качестве показателя того, что возможен сходный консерватизм последовательностей между породами коров. Принимая во внимание сходство между последовательностями и структурами локусов иммуноглобулина коров и овец, можно ожидать, что способы направленного изменения, используемые для удаления локусов иммуноглобулина коров, могут быть успешно применены к овечьей системе. Кроме того, существующая информация в отношении свиньи (Sus scrofa, номер доступа S42881) и козы (Capra hircus, номер доступа AF140603) показывает, что локусы иммуноглобулина обоих указанных видов также достаточно сходны с локусами коров для того, чтобы использовать данную методику направленного изменения. ПРИМЕР 5: Нокаут IgM коров Следующие способы использовали для создания клеточных линий фибробластов коровы, в которых один аллель локуса тяжелой цепи иммуноглобулина (мю) поврежден в результате гомологичной рекомбинации. Конструкцию ДНК для осуществления нокаута IgM создавали удалением экзонов 1-4 локуса мю (соответствует гену тяжелой цепи IgM), которые заменяли копией гена устойчивости к неомицину. Используя данную конструкцию, получили резистентные к неомицину линии клеток, которые успешно использовали в способах переноса ядер, и бластоцисты из этих линий клеток имплантировали коровам-реципиентам. Кроме того, некоторые из этих бластоцист тестировали с использованием способов ПЦР, чтобы подтвердить, что направленная инсерция происходила соответствующим образом в локусе мю. Бластоцисты, полученные в результате процедур переноса ядер из нескольких полученных клеточных линий, показали, что при беременности имелись гетерозиготные нокаутированные по IgM плоды. Кроме того, были получены как мужские, так и женские линии клеток, которые содержат один нокаут тяжелой цепи IgM (мю). Предполагается, что скрещивание животных, клонированных из этих линий клеток, будет давать потомство, у которого инактивированы обе копии мю. Как указано в этом описании, потомство может быть скрещено с нокаутированными по приону копытными животными, чтобы получить потомство с мутациями в генах IgM и приона. Альтернативно клетку из потомства можно генетически модифицировать, чтобы инактивировать один или оба аллеля кодирующего прион гена, как описано в примере 1. Указанные способы более подробно обсуждаются ниже. Конструкция ДНК ДНК, используемую во всех трансфекциях, описанных в этом документе, создавали следующим образом. Четыре основных экзона (включая экзоны трансмембранного домена), CH1-4, фланкировали сайтом рестрикции XhoI на нижнем (CH4) конце и сайтом XbaI на верхнем (CH1) конце. Конструкция, используемая для процедуры трансфекции, состояла из 1,5 т.п.о. геномной последовательности ниже сайта XhoI и 3,1 т.п.о. геномной последовательности выше сайта XbaI (фиг. 3D и 3E). Эти последовательности выделяли, как указано в этом описании, из коровы голштинской породы из молочного стада в Массачусетсе. Маркер устойчивости к неомицину встраивали между указанными двумя фрагментами во фрагменте длиной 3,5 т.п.о., заменяющем 2,4 т.п.о. ДНК, исходно содержащей CH1-4, из исходной геномной последовательности. Остов вектора представлял собой pBluescriptII SK+ (Stratagene), и вставку длиной 8,1 т.п.о. очищали и использовали для трансфекции эмбриональных фибробластов коровы. Данная конструкция показана на фиг. 3A-3C. Другие нокаутирующие мю конструкции, содержащие другие гомологичные области и/или содержащие другой ген устойчивости к антибиотику, также можно сконструировать, используя стандартные способы, и использовать для введения мутации в эндогенный ген тяжелой цепи мю. Способы трансфекции/нокаута Трансфекцию эмбриональных клеток коровы осуществляли с использованием коммерческого реагента для трансфекции Superfect (Qiagen, Valencia, CA, USA). Фибробласты коровы от тестированного в отношении заболеваний крупного рогатого скота Charolais мужского пола получали на предприятии Hematech Kansas и посылали в лабораторию молекулярной биологии Hematech Worcester для использования во всех описанных экспериментах. В качестве источника донорных клеток можно использовать любую другую породу, род или вид копытного животного (например, соматических клеток, таких как эмбриональные фибробласты). Донорные клетки генетически модифицируют так, чтобы они содержали мутацию, которая снижает или исключает экспрессию функционального эндогенного Ig. Среда, используемая для культивирования эмбриональных фибробластов коровы, состояла из следующих компонентов: 500 мл альфа-MEM (Bio-Whittaker За день до процедуры трансфекции клетки высевали в чашках для культуры ткани диаметром 60 мм с целью достичь смыкания монослоя на 40-80%, которое определяли микроскопическим исследованием. В день трансфекции 5 мкг ДНК, доведенной до общего объема 150 мкл в бессывороточной не содержащей антибиотика среде, смешивали с 20 мкл реагента для трансфекции Superfect и давали возможность отстояться при комнатной температуре в течение 5-10 минут для образования комплекса ДНК-Superfect. Пока происходило образование комплекса, среду удаляли из чашек для культуры ткани диаметром 60 мм, содержащих фибробласты коровы, подлежащие трансфекции, и клетки один раз промывали 4 мл фосфатно-солевого буфера. Один миллилитр ростовой среды добавляли к 170 мкл смеси ДНК/Superfect и сразу же переносили к клеткам в чашки диаметром 60 мм. Клетки инкубировали при 38,5°C, 50% углекислого газа в течение 2,5 часов. После инкубирования клеток с комплексами ДНК/Superfect среду отсасывали и клетки четыре раза промывали 4 мл PBS. Добавляли пять мл полной среды и культуры инкубировали в течение ночи при 38,5°C, 5% CO2. Затем клетки один раз промывали PBS и инкубировали в одном мл 0,3% трипсина в PBS при 37°C вплоть до того, как клетки откреплялись от чашки, что определяли наблюдением под микроскопом. Клетки из каждой 60 мм-чашки делили на 24 лунки 24-луночного планшета для культуры ткани (41,7 мкл/лунку). В каждую лунку добавляли один миллилитр среды для культуры ткани и планшеты инкубировали в течение 24 часов при 38,5°C и 5% CO2 24 часа. В ходе всех процедур трансфекции проводили ложные трансфекции, используя смесь Superfect/PBS, не содержащую ДНК, так что можно было ожидать, что ни одна из таких клеток не будет содержать гена устойчивости к неомицину, и можно было ожидать, что все клетки погибнут после добавления G418 к среде для культуры ткани. Это служило негативным контролем для позитивной селекции клеток, которые получили ДНК. После 24-часового инкубирования в каждую лунку добавляли еще один миллилитр среды для культуры ткани, содержащей 400 мкг G418, доводя конечную концентрацию G418 до 200 мкг/мл. Клетки возвращали в инкубатор для 7 дней селекции на G418. В течение указанного периода за трансфицированными и ложно трансфицированными планшетами вели наблюдение в отношении гибели клеток, и в течение 7 дней подавляющее большинство лунок с ложными трансфекциями содержали немного или не содержали живых клеток, тогда как в планшетах, содержащих клетки, которые получили ДНК, наблюдали прекрасный рост клеток. После 7-дневного периода селекции клетки из лунок при 90-100% смыкании в монослой открепляли, используя 0,2 мл 0,3% трипсина в PBS, и переносили в чашки для культуры ткани диаметром 35 мм для размножения и инкубировали вплоть до того, как они смыкались по меньшей мере на 50%, и в этот момент клетки трипсинизировали 0,6 мл 0,3% трипсина в PBS. Из каждой 35-мм чашки для культуры ткани 0,3 мл из 0,6 мл суспензии клеток переносили во флаконы для культуры ткани 12,5 см2 для дальнейшего размножения. Остальные 0,3 мл повторно высевали на 35-мм чашки и инкубировали вплоть до того, как они достигали минимального смыкания в монослой примерно на 50%, в этот момент клетки из этих чашек обрабатывали для экстракции ДНК для ПЦР-анализа. Флаконы из каждой линии оставляли к инкубаторе до тех пор, пока они не были подвергнуты этому анализу и либо прекращали их культивировать, если они не содержали желаемой интеграции ДНК, либо оставляли для будущего переноса ядер и криоконсервации. Скрининг в отношении направленных интеграций Как описано выше источником ДНК для скрининга трансфектантов, содержащих конструкцию ДНК, была чашка для культуры ткани диаметром 35 мм, содержащая пассаж анализируемых клеток. ДНК получали следующим образом и адаптировали для способа, опубликованного Laird et al., Nucleic Acids Res., 19: 4293). Коротко, ДНК получали следующим образом. Готовили буфер для лизиса клеток со следующими компонентами: 100 мМ трис-HCl-буфер, pH 8,5; 5 мМ EDTA, pH 8,0; 0,2% додецилсульфат натрия; 200 мМ NaCl и 100 мкг/мл протеиназы K. Среду отсасывали из каждой 35-мм чашки для культуры ткани и заменяли 0,6 мл описанного выше буфера. Чашки вновь помещали в инкубатор на три часа и в течение указанного периода времени давали возможность происходить лизису клеток и расщеплению белков. После данного инкубирования лизат переносили в микроцентрифужную пробирку объемом 1,5 мл и добавляли 0,6 мл изопропанола для преципитации ДНК. Пробирки осторожно встряхивали переворачиванием и давали возможность отстояться при комнатной температуре в течение 3 часов, после чего преципитаты ДНК осаждали центрифугированием в микроцентрифуге при 13000 об/мин в течение десяти минут. Супернатант из каждой пробирки отбрасывали, а осадки промывали один раз 70% этанолом. 70%-Этанол отсасывали и осадкам ДНК давали возможность сушиться на воздухе. После подсушивания каждый осадок ресуспендировали в 30-50 мкл буфера трис (10 мМ)-EDTA (1 мМ), pH 7,4, и позволяли гидратироваться и растворяться в течение ночи. Для каждой процедуры полимеразной цепной реакции (ПЦР) использовали 5-7 микролитров каждого раствора ДНК. Для анализа трансфектантов использовали два отдельных способа ПЦР. В первом способе использовали два праймера, которые предположительно отжигаются с сайтами, которые оба расположены в ДНК, используемой для трансфекции. Последовательность первого праймера гомологична кассете устойчивости к неомицину конструкции ДНК, а вторая располагается примерно на расстоянии 0,5 т.п.о., давая короткий ПЦР-продукт 0,5 т.п.о. В частности, использовали праймеры Neo1 (5′-CTT GAA GAC GAA AGG GCC TCG TGA TAC GCC-3′; SEQ ID NO: 42) и IN2521 (5′-CTG AGA CTT CCT TTC ACC CTC CAG GCA CCG-3′; SEQ ID NO: 43). Для данной реакции ПЦР использовали набор для ПЦР Qiagen. Смесь для реакции ПЦР содержала 1 пмоль каждого праймера, 5 мкл 10X буфера для реакции, 10 мкл раствора Q, 5 мкл ДНК и 1 мкл раствора dNTP. Реакционную смесь доводили до общего объема 50 мкл, используя H2O. Такую ПЦР-амплификацию осуществляли, используя начальную денатурирующую инкубацию при 94°C в течение двух минут. Затем 30 циклов денатурации, отжига и амплификации осуществляли посредством инкубирования при 94°C в течение 45 секунд, 60°C в течение 45 секунд и 72°C в течение двух минут. Затем реакционную смесь инкубировали при 72°C в течение пяти минут и при 4°C вплоть до извлечения смеси из устройства для ПЦР. Альтернативно в стандартной ПЦР-реакции при соответствующих условиях реакции можно использовать любые другие праймеры, которые гомологичны области нокаутирующей конструкции, которая интегрирует в геном клетки, чтобы подтвердить, что клетки, выживающие при селекции на G418, были резистентными в результате интеграции конструкции ДНК. Поскольку можно ожидать, что только небольшой процент трансфектантов содержит интеграцию ДНК в желаемом положении (локус Mu), использовали другую пару праймеров, чтобы определить не только то, что введенная ДНК присутствует в геноме трансфектантов, но также что она интегрирована в желаемом положении. Для выявления соответствующей интеграции осуществляли ПЦР с использованием одного праймера, локализованного в кассете устойчивости к неомицину конструкции ДНК, и одного праймера, который, предположительно, может отжигаться на расстоянии 1,8 т.п.о, но только если ДНК интегрирована в соответствующий сайт локуса IgM (так как гомологичная область была вне области, включенной в конструкцию ДНК, используемую для трансфекции). Праймер конструировали для отжига с последовательностью ДНК непосредственно вблизи последовательностей, представленных в конструкции ДНК, в том случае, если бы она интегрировала в желаемое положение (последовательность ДНК локуса как в области, присутствующей в конструкции ДНК, так и в смежной с ней в геноме определяли предварительно). В частности, для данного анализа использовали праймеры Neo1 и OUT3570 (5′-CGA TGA ATG CCC CAT TTC ACC CAA GTC TGT C-3′; SEQ ID NO: 44). Данную реакцию ПЦР осуществляли с использованием набора для ПЦР Qiagen, как описано выше для первой реакции ПЦР, чтобы подтвердить интеграцию конструкции направленного воздействия в клетки. Альтернативно, такой ПЦР-анализ можно осуществить с использованием любых подходящих условий реакции с любым другим праймером, который гомологичен области нокаутирующей конструкции, которая интегрирует в геном клеток, и любым другим праймером, который гомологичен области генома клеток, которая находится выше или ниже сайта интеграции. Используя указанные способы, проводили скрининг 135 независимых чашек диаметром 35 мм в отношении направленной интеграции конструкции ДНК в соответствующий локус. Было определено, что ДНК из восьми чашек содержит соответствующим образом направленную в мишень конструкцию ДНК, три из них отбирали для применения в способах переноса ядер. Указанные клеточные линии обозначены «8-1C», «5-3C» и «10-1C». Оставшиеся бластоцисты, не используемые для переноса реципиентным коровам, использовали для экстракции ДНК, которую подвергали дополнительному ПЦР-анализу. Этот анализ осуществляли с использованием способа гнездовой ПЦР, используя праймеры, которые также использовали для начального скрининга трансфицированных линий. Как указано выше, создавали три линии клеток, используя конструкцию для направленного изменения гена, сконструированную так, чтобы удалять экзоны 1-4 локуса мю. Все указанные линии при тестировании в отношении направленных инсерций с использованием теста на основе ПЦР были позитивными и их использовали для переноса ядер. Оставшиеся бластоцисты, полученные в результате этих переносов ядер, подвергали скринингу посредством ПЦР-тестирования соответствующим образом направленно введенной конструкции. Получили следующие частоты позитивных бластоцист:
Хотя на сороковой день беременности посредством ультразвука регистрировали 11 общих случаев беременности, на 60 день семь плодов погибли. Оставшиеся четыре плода обрабатывали для регенерации новых эмбриональных фибробластов, а остальные органы использовали для получения образцов тканей для ПЦР-анализа. Результаты анализов приведены ниже: Линия 8-1C: два плода, один плод позитивный в отношении направленной инсерции при ПЦР; Линия 10-1C: один плод, позитивный в отношении направленной инсерции при ПЦР; Линия 5-3C: один плод, негативный в отношении направленной инсерции при ПЦР. Неожиданно, хотя частота бластоцист 10-1C, тестируемых позитивно в отношении направленной инсерции, составляла только 2/16, один жизнеспособный 60-дневный плод, полученный из данной линии клеток, был позитивным на основании определения в ПЦР. Также получали позитивный плод из 8-1C. Проводится Саузерн-блот-анализ ДНК всех образцов тканей, чтобы подтвердить, что конструкция целенаправленно встроена не только на одном конце (которые определяют с помощью ПЦР более короткой гомологичной области, присутствующей в исходной конструкции), но также и на другом конце. На основании полученных к настоящему времени результатов, предполагается, что получены два нокаутированных по тяжелой цепи плода в результате двух независимых событий интеграции. Также, поскольку указанные плоды были получены из двух разных линий, по меньшей мере, один вероятно имеет конструкцию, корректно интегрированную на обоих концах. После подтверждения Саузерн-блот-анализами соответствующего направленного встраивания на обоих концах конструкции направленного воздействия осуществляют следующие переносы ядер, чтобы создать дополнительные плоды, которые вынашиваются до рождения. Перенос ядер и перенос эмбрионов Переносы ядер осуществляли с использованием линии клеток K/O (8-1-C (18)) и получили восемь эмбрионов. Всего шесть эмбрионов из данной партии переносили трем не имеющим заболеваний реципиентам в Trans Ova Genetics («TOG»; Iowa). Замороженные эмбрионы переносили десяти не имеющим заболеваний реципиентам, чтобы получить не пораженные заболеванием клеточные линии женских фибробластов. Извлечения плодов осуществляли по графику после подтверждения беременностей на 35-40 день. Диагностика беременности и извлечение плодов Статус беременности восемнадцати реципиентов, которым переносили эмбрионы, клонированные из нокаутированных эмбриональных клеток, проверяли посредством ультразвукового исследования. Результаты суммированы в таблице 6.
Диагностика беременности Исследовали статус беременности трех реципиентов, которым переносили клонированные эмбрионы из нокаутированных клеток (8-1C); один был обнаружен, а два других требовали подтверждения через один месяц. Извлечение плодов и закладка клеточных линий Получили одиннадцать беременностей с K/O-эмбрионами на 40 день. Из них четыре живых плода извлекали на 60 день. Клеточные линии закладывали из всех четырех и криоконсервировали для будущего применения. Авторы также собирали и мгновенно замораживали образцы тканей из плодов и посылали их в лабораторию молекулярной биологии Hematech для ПЦР/Саузерн-блот-анализа. Все четыре клеточных линии были мужскими. Для того чтобы обеспечить женскую линию клеток, закладывали и криоконсервировали для будущей закладки клеточные линии K/O-клеток из плодов (шести), собранных на 55 день беременности от беременных животных, полученных в Trans Ova Genetics с использованием не имеющих заболеваний реципиентов. Недавно подтвердили существование женской линии клеток, содержащих нокаут мю. Эта женская линия клеток может быть использована для получения клонированных животных, которые могут быть спарены с животными, созданными на основе мужских линий клеток, и проведен скрининг потомства в отношении животных, которые содержат двойной нокаут мю. При желании клетку из полученного нокаутированного плода или нокаутированного потомка можно использовать во втором раунде переноса ядер, чтобы создать дополнительное клонированное потомство. Клетки из исходного нокаутированного плода или нокаутированного потомка также могут быть заморожены для создания клеточной линии, чтобы использовать ее в качестве источника донорных клеток для создания дополнительных нокаутированных копытных животных. ПРИМЕР 6: Трансгенные копытные животные, имеющие пониженную активность При необходимости могут быть созданы трансгенные копытные животные, у которых Данные способы дополнительно описаны ниже. Конструирование нокаутирующего по Нокаутирующий по Способы трансфекции/нокаута Трансфекцию трех клеточных линий эмбриональных фибробластов (две от крупного рогатого скота Джерси мужского пола и одна от крупного рогатого скота Джерси женского пола) осуществляли, используя стандартный протокол электропорации, следующим образом. Среда, используемая для культивирования эмбриональных фибробластов коров, содержала 500 мл альфа-MEM (Gibco, 12561-049), 50 мл фетальной телячьей сыворотки (Hy-Clone Скрининг направленных интеграций Как описано выше, геномную ДНК экстрагировали из каждой из 24 лунок независимо, используя набор для выделения ДНК PUREGENE (Gentra Systems), согласно протоколу производителя. Каждый образец геномной ДНК ресуспендировали в 20 мкл 10 мМ трис-Cl (pH 8,0) и 1 мМ EDTA (EDTA). Скрининг с помощью ПЦР осуществляли, используя следующую пару праймеров 5′-aag aag aga aag gta gaa gac ccc aag gac-3′ (SEQ ID NO: 63) и 5′-cct ggg tat aga cag gtg ggt att gtg c-3′ (SEQ ID NO: 64). Последовательность одного праймера расположена в векторе, нокаутирующем по альфа-1,3-галактозилтрансферазе, а последовательность другого праймера расположена непосредственно с внешней стороны интегрированного вектора в направленно измененном эндогенном локусе (фиг. 23). Следовательно, ожидаемый ПЦР-продукт должен регистрироваться только в том случае, когда нокаутирующий вектор интегрируется в являющийся мишенью локус посредством гомологичной рекомбинации. Смеси для реакции ПЦР содержали 18,9 мкл воды, 3 мкл 10Х буфера для ПЦР LA II (плюс Mg2+), 4,8 мкл смеси dNTP, 10 пмоль прямого праймера, 10 пмоль обратного праймера, 1 мкл геномной ДНК и 0,3 мкл Taq LA. Сорок циклов ПЦР осуществляли путем инкубирования реакционных смесей в следующих условиях: 85°C в течение трех минут, 94°C в течение одной минуты, 98°C в течение 10 секунд и 68°C в течение 15 минут. После ПЦР реакционные смеси анализировали электрофорезом. Отбирали резистентные к пуромицину клоны, которые давали ПЦР-продукты ожидаемого размера (фиг. 23). Таким образом успешно создавали клеточные линии фибробластов коров, в которых один аллель локуса ПРИМЕР 7: Альтернативный способ получения трансгенных копытных животных с использованием аденоассоциированных вирусов для введения мутации в эндогенный ген Аденоассоциированный вирус (AAV) можно использовать для специфичной замены последовательностей-мишеней, присутствующих в геноме клеток (Inoue et al., Mol. Ther. 3:526-530, 2001; Hirata et al., J. Virol. 74: 16536-16542, 2000; Inoue et al., J. Virol. 73: 7376-7380, 1999 и Russell et al., Nat. Genet. 18: 325-330, 1998). Интенсивность направленного воздействия на гены является высокоэффективной по сравнению с более традиционными способами направленного изменения генов. AAV имеет широкий диапазон специфичности по отношению к хозяевам и тканям, включая специфичность по отношению к фибробластам кожи как коровы, так и человека. Таким образом, AAV может быть использован для получения трансгенных клеток копытного животного, содержащих одну или несколько мутаций в эндогенном гене иммуноглобулина (например, тяжелой цепи мю, легкой цепи лямбда, легкой цепи каппа или цепи J), альфа-(1,3)-галактозилтрансферазы или приона. Такие трансгенные клетки затем можно использовать в способах переноса ядер, приведенных в этом описании, для получения трансгенных копытных животных по данному изобретению. Использование AAV приводило к гомологичной рекомбинации локуса тяжелой цепи иммуноглобулина коровы с более высокой частотой, чем получали ранее, используя традиционные способы направленного изменения генов (т.е. способы электропорации и липофекции). В первом раунде экспериментов по целенаправленному изменению генов получили пять клонов соответствующим образом направленно измененных фибробластов из 73 стабильных трансдуктантов, содержащих ДНК, введенную посредством AAV-вектора. Указанные эксперименты осуществляли следующим образом. Нокаутирующие векторы на основе AAV Конструкции AAV могут нарушать ген либо простой инсерцией чужеродных последовательностей, либо заменой эндогенных последовательностей новой последовательностью, присутствующей в AAV-векторе. На фигуре 24 показана конструкция AAV, в которой все четыре кодирующих экзона константной области тяжелой цепи мю иммуноглобулина коровы присутствуют в BamHI-XhoI-фрагменте длиной 2822 пар оснований. Фрагмент размером 1,16 т.п.о., содержащий маркер устойчивости к неомицину, присутствующий в коммерчески доступном векторе, pMC1Neo, встраивали в SacII-сайт, присутствующий в экзоне 4 локуса тяжелой цепи мю из коров голштинской породы. Этот локус представляет собой локус, находящийся в фаговом клоне, выделенном для создания нокаутирующего вектора, описанного в примере 3. Чтобы создать AVV-вектор, сайт SacII в локусе тяжелой цепи мю заполняли, чтобы создать тупые концы, которые затем лигировали с тупыми SalI-линкерами (New England Biolabs). Затем XhoI-фрагмент pMC1Neo, который содержит ген устойчивости к неомицину, лигировали с сайтом SalI, добавленным к локусу через SalI-линкер. Такое лигирование может быть осуществлено, так как сайты рестрикции XhoI и SalI имеют совместимые концы. Этот нокаутирующий вектор вызывает разрывающую инсерцию гена устойчивости к неомицину в эндогенный ген тяжелой цепи мю, инактивируя при этом ген тяжелой цепи мю. Указанная инактивация гена происходит без делеции областей эндогенного локуса мю. Альтернативный вектор конструировали для удаления экзонов 3 и 4 из эндогенного локуса во время направленного воздействия, приводящего к замене этих двух экзонов функциональной копией гена устойчивости к неомицину (фиг. 25). Такую конструкцию создавали с использованием ПЦР-амплификации геномной ДНК из крупного рогатого скота Джерси женского пола. В частности гомологичную 3′-область амплифицировали с использованием следующих праймеров: 5′-GGG GTC TAG Agc aga cac tac act gat ggg ccc ttg gtc c-3′ (SEQ ID NO: 65), который добавляет сайт рестрикции XbaI, и 5′- GGG GAA GCT Tcg tgt ccc tgg tcc tgt ctg aca cag-3′ (SEQ ID NO: 66), который добавляет сайт рестрикции HindIII. Гомологичную 5′-область амплифицировали с праймерами 5′-GGG GCT CGA Ggt cgg cga agg atg ggg gga ggt g-3′ (SEQ ID NO: 67), который добавляет сайт рестрикции XhoI, и 5′-GGG GGG TAC Cgc tgg gct gag ctg ggc aga gtg gg-3′ (SEQ ID NO: 68), который добавляет сайт рестрикции KpnI. Напечатанные прописными буквами нуклеотиды в этих последовательностях праймеров являются нуклеотидами, которые не отжигаются с локусом тяжелой цепи мю, но включены в праймеры, чтобы добавить сайты рестрикции для облегчения более поздних стадий субклонирования. Первые четыре гуанина добавляют, чтобы отделить сайты рестрикции от самого конца праймеров, так как ферменты рестрикции не разрезают сайты, которые находятся на самом конце праймеров, также как внутренние сайты. Гомологичная 5′-область имеет длину 1,5 т.п.о. и содержит экзоны 1 и 2. Гомологичная 5′-область также содержит первые 25 нуклеотидов экзона 3, чтобы сохранить акцепторный сайт сплайсинга экзона 3. Акцепторный сайт сплайсинга позволяет использовать для сплайсинга экзон 3 и таким образом предотвращает возможный сплайсинг экзонов 1 и 2 до расположенного ниже трансмембранного домена с образованием аберрантного связанного с мембраной продукта. Гомологичная 3′-область имеет длину 1,24 т.п.о. и содержит область, расположенную непосредственно ниже экзона 4. Для конструкции, показанной на фиг. 24, кассету направленного воздействия встраивают в AAV-вектор, описанный Ryan et al. (J. Virol. 70: 1542-1553, 1996), который содержит последовательности длинных концевых повторов (LTR) вирусов, используя стандартные способы. AAV-вектор упаковывают в капсиды, используя линию пакующих клеток TtetA2, как описано ранее (Inoue and Russell, J. Virol. 72: 7024-7031, 1998), очищают, как описано ранее (Zolotukhin et al., Gene Therapy, 6: 973-985, 1999). Для конструкции, показанной на фиг. 25, можно использовать описанный выше способ или любой другой стандартный способ, чтобы встроить кассету направленного воздействия в AAV-вектор, описанный Ryan et al., или любой другой AAV-вектор (такой как коммерчески доступный вектор из Stratagene) и создать вирусы, содержащие вектор. Способы трансдукции Фибробласты крупного рогатого скота Джерси женского пола высевали в одну лунку 48-луночного планшета для культуры ткани в количестве 40000 клеток на лунку и культивировали в полной среде при 38,5°C и 5% CO2 вплоть до прикрепления клеток к поверхности дна лунки. После прилипания клеток среду удаляли и заменяли 0,2 мл свежей среды, содержащей частицы AAV с вектором, показанным на фиг. 24, при множественности инфекции (MOI) 500-20000 частиц/клетку. MOI выбирали на основе предварительных экспериментов, в которых определяли получаемое количество колоний и расстояния между колониями во время фазы лекарственной селекции. Планшеты инкубировали в течение ночи. После этого инкубирования лунки с трансдуцированными клетками промывали не содержащим кальция и магния PBS и открепляли от лунок, используя либо трипсин, либо буфер для диссоциации клеток, описанный выше. Однородную суспензию клеток получали осторожным пипетированием открепленных клеток, и клетки из лунки перераспределяли на десять чашек для культуры ткани диаметром 100 мм. Чашки инкубировали с полной средой в течение ночи. После указанного инкубирования 100 мм-чашек среду заменяли селективной средой, содержащей G418 в концентрации 350 микрограмм/мл. Селективную среду меняли каждые 2-3 дня до тех пор, пока колонии не становились видны на поверхности чашки. На этой стадии отдельные колонии собирали и переносили в собственные сосуды. Области, содержащие колонии, метили на наружной поверхности чашки для культуры ткани. После того как все колонии были обведены, среду отсасывали из чашек и чашки три раза промывали не содержащим кальция и магния PBS. После промывки чашки заливали разведением 1X трипсина 1:25 и давали возможность отстояться при комнатной температуре вплоть то того, как колонии видимо начинали открепляться от поверхности чашки. Чашки держали стационарно, чтобы предотвратить уплывание открепленных колоний в другое положение на чашке. Наконечник пипетки использовали для сбора комочков клеток в объем 50 микролитров, и содержимое наконечника пипетки переносили в одну лунку 24-луночного планшета для культуры ткани. После переноса всех колоний добавляли полную среду, содержащую G418, и изолированным клонам давали возможность пролиферировать почти до смыкания в монослой. Когда отдельная лунка была близка к смыканию клеток, ее два раза промывали не содержащим кальция и магния PBS. Клетки открепляли, используя 0,2 мл буфера для диссоциации клеток. Из этой клеточной суспензии 20 мкл переносили в новый 24-луночный планшет, а оставшимся клеткам давали возможность вновь прикрепиться к поверхности исходного 24-луночного планшета после добавления 2,0 мл полной среды. Исходный планшет инкубировали до 100% смыкания в монослой. Новый планшет служит в качестве источника соответствующим образом направленно измененных клеток для будущих процедур клонирования коров. Когда в лунке в исходном 24-луночном планшете достигалось 100% смыкание клеток среду удаляли и клетки один раз промывали PBS. PBS удаляли и заменяли буфером для лизиса клеток, заимствованным из Laird et al. (Nucleic Acids Res. 19: 4293, 1991). Коротко, в лунку добавляли 0,2 мл лизирующего буфера, содержащего 200 мМ NaCl, 100 мМ трис-HCl, pH 8,5, 5 мМ EDTA, 0,2% SDS и 100 мкг/мл протеиназы K. Планшет возвращали в инкубатор на период времени от трех часов до всей ночи. Затем вязкий клеточный лизат переносили в микроцентрифужную пробирку. Добавляли равный объем изопропанола, чтобы преципитировать ДНК. После 10 минут центрифугирования в микроцентрифуге супернатант отбрасывали, а осадок промывали один раз 0,5 мл 70% этанола. После удаления этанола осадок ДНК сушили на воздухе и ресуспендировали в 35 микролитрах буфера TE (10 мМ трис pH 8 и 1 мМ EDTA). Аликвоты объемом 3 мкл использовали для ПЦР-анализа. ПЦР-анализ Образцы ДНК из резистентных к лекарственному средству клонов, трансдуцированных частицами AAV, подвергали скринингу в отношении соответствующего направления воздействия вектора, используя ПЦР-анализ. В данной методике скрининга использовали один праймер, который отжигается с ДНК, кодирующей маркер лекарственной селекции, и другой праймер, который отжигается с локусом, являющимся мишенью, но с внешней стороны последовательности, присутствующей в частицах AAV направленного воздействия. ПЦР-продукты выявляются только в том случае, если ДНК AAV направленного воздействия интегрирована в желаемом положении эндогенного генома. Результаты одного эксперимента по направленному изменению с использованием частиц AAV показаны на фиг. 26. На основании данного анализа пять из 73 независимых клонов содержали соответствующим образом направленную в мишень ДНК вектора. Этот способ также можно использовать с вектором AAV, показанным на фиг. 25, или любым другим подходящим аденовирусным вектором, или вектором на основе аденоассоциированного вируса. При желании можно вести мутацию во второй аллель тяжелой цепи мю в изолированных колониях путем их трансдукции вектором AAV с геном устойчивости к другому антибиотику (т.е. другим геном, отличным от гена устойчивости к неомицину). Чтобы отобрать полученные гомозиготные нокаутированные клетки, инфицированные клетки культивируют в присутствии соответствующего антибиотика. Альтернативно изолированные колонии можно трансдуцировать вектором AAV, содержащим ген устойчивости к неомицину, и культивировать в присутствии высокой концентрации антибиотика (т.е. концентрации антибиотика, которая убивает гетерозиготные нокаутированные клетки, но не гомозиготные нокаутированные клетки). ПРИМЕР 8: Доказательство недостаточности перепрограммирования ядер в эмбрионах коров с традиционными ядерными трансплантатами Традиционные способы получения ядерных трансплантатов обычно дают низкий процент живых новорожденных. Как описано ниже, такая эффективность может быть результатом, по меньшей мере частично, неспособности реконструированного ооцита перепрограммировать донорную клетку или донорное ядро так, чтобы стимулировать транскрипцию генов, требуемых для развития ооцита, и ингибировать транскрипцию генов, нежелательных для развития. Примеры 11 и 12 описывают усовершенствованные способы клонирования, которые можно использовать для получения трансгенных копытных животных с мутацией в гене, кодирующем прион, и, необязательно, с геном ксеногенного Ig, в способах по данному изобретению. Распределение ядерной оболочки, ядерного матрикса и компонентов области контакта хроматин-матрикс в ходе предимплантационного развития коров Чтобы определить распределение ядерной оболочки (ламины B-типа и A/C-типа), ядерного матрикса (NuMA) и компонентов области контакта хроматин-матрикс (AKAP95) в предимплантационных эмбрионах получали эмбрионы коров посредством оплодотворения in vitro (IVF) и исследовали с помощью иммунофлуоресцентного анализа. Оплодотворение для коров in vitro осуществляли, как описано ранее (Collas et al., Mol. Reprod. Devel. 34: 212-223, 1993). Коротко, замороженную и оттаянную сперму крупного рогатого скота от одного быка наносили на 45-90% градиент перкола и центрифугировали в течение 30 минут при 700 × g. Определяли концентрацию сперматозоидов в осадке и сперму разбавляли так, чтобы конечная концентрация при оплодотворении составляла 106 сперматозоидов/мл. Через 22 часа после созревания ооциты три раза промывали в TL HEPES и помещали в 480 мкл среды для оплодотворения. К 50 ооцитам добавляли двадцать микролитров суспензии сперматозоидов в концентрации 106 сперматозоидов/мл. Эмбрионы помещали в культуру в четырех-луночные чашки для культуры ткани, содержащие монослой мышиных эмбриональных фибробластов в 0,5 мл среды для культивирования эмбрионов, покрытой 0,3 мл проверенного на эмбрионах минерального масла (Sigma). От 25 до 50 эмбрионов помещали в каждую лунку и инкубировали при 38,5°C в атмосфере воздуха с 5% CO2. Частота оплодотворения составляла более 90%, что определяли по развитию пронуклеуса. Для иммунофлуоресцентного анализа этих оплодотворенных in vitro эмбрионов коров антитела против ламина B человека получали от д-ра Jean-Claude Courvalin, CNRS, Paris, France. Моноклональные антитела против ламинов A/C приобретали у Santa-Cruz Biotechnology и анти-NuMA-антитела получали от Transduction Laboratories. Аффинно очищенные кроличьи поликлональные антитела против AKAP95 крыс получали от Upstate Biotechnologies. Оплодотворенные in vitro эмбрионы коров осаждали на покрытые поли-L-лизином стеклянные покровные стекла, фиксировали 3% параформальдегидом в течение 15 минут и пермеабилизовали 0,1% тритоном X-100 в течение 15 минут (Collas et al., J. Cell Biol. 135: 1715-1725, 1996). Белки блокировали 2% БСА в PBS/0,01% твин 20 (PBST) в течение 15 минут. Первые антитела (анти-AKAP95, анти-ламин B, анти-LBR, анти-NuMA и анти-ламины A/C) и вторые антитела инкубировали в течение 30 минут и использовали в разведении 1:100 в PBST-БСА. ДНК контрастно красили 0,1 мкг/мл Hoechst33342, включенным в среду для заливки против обесцвечивания. Образцы помещали на предметные стекла и покровные стекла приклеивали лаком для ногтей. Исследования иммунофлуоресценции проводили на эпифлуоресцентном микроскопе Olympus BX60, и фотографии делали, используя камеру JVC CCD и компьютерную программу AnalySIS. Изображения обрабатывали, используя компьютерную программу Aldus Photostyler. Относительную количественную оценку сигналов флуоресценции осуществляли, используя программу для количественной оценки AnalySIS. Данные выражали в виде средних интенсивностей относительной флуоресценции. Иммунофлуоресцентный анализ эмбрионов коров показал, что ламины типа B выявлялись на периферии ядра (фиг. 29A). Однако ламины A/C не выявлялись в пронуклеусах или на стадии 8 клеток. Такая неспособность выявить ламины A/C на таких ранних клеточных стадиях, вероятно, является маркером дифференцированных клеток (Guilli et al., EMBO J. 6: 3795-3799, 1987). Структурный белок ядерного матрикса, NuMA, выявляли на всех стадиях, которые исследовали (фиг. 29A). Однако в эмбрионах коров на стадии пронуклеусов мечение NuMA было ограничено женским пронуклеусом (FPN), наименьшим из двух пронуклеусов (фиг. 29A, стрелки). AKAP95, который недавно был охарактеризован в ранних эмбрионах мышей (Bomar et al., 2002 рукопись представлена на рассмотрение) и выявлен с использованием аффинно очищенных антител против AKAP95 крыс, также был ограничен женским пронуклеусом (Фиг. 29A). Тем не менее, внутриядерное распределение AKAP95 наблюдали в ядрах всех бластомеров на последующих стадиях развития (фиг. 29A). Специфичность иммунофлуоресцентного мечения подтверждали Вестерн-блот-анализом первичных эмбриональных фибробластов коровы и оплодотворенных in vitro эмбрионов на стадии пронуклеусов (фиг. 29B). Для этого анализа белки разделяли в 10% SDS-PAGE при 40 мА на гель. Белки электрофоретически переносили на нитроцеллюлозную мембрану в буфере для переноса (25 мМ трис-HCl, pH 8,3, 192 мМ глицин, 20% метанол и 0,1% SDS) при 100 В в течение одного часа. Мембраны промывали в течение 10 минут трис-забуференным раствором соли (TBS; т.е. 140 мМ NaCl, 2,7 мМ KCl и 25 мМ трис-HCl при pH 8,0), блокировали в течение одного часа TBST (TBS с 0,05% твина-20), содержащим 5% молока, и инкубировали в течение 1,5 часов со следующими первыми антителами: анти-AKAP95 (разведение 1:250), анти-ламин B (1:1000), анти-LBR (1:500), анти-NuMA (1:500) и анти-ламины A/C (1:500). Блоты промывали дважды по 10 минут в TBST и инкубировали в течение одного часа со вторыми антителами, конъюгированными с пероксидазой хрена. Блоты промывали дважды по 10 минут в TBS и проявляли, используя усиленную хемилюминесценцию (ECL, Amersham). Все белки выявляли при ожидаемых кажущихся Mr: 68 кДа (ламины B-типа), 70 и 60 кДа (ламины A и C соответственно), ~180 кДа (NuMA) и 95 кДа (AKAP95). В общем случае полученные результаты свидетельствуют, что предимплантационные эмбрионы коров экспрессируют ядерные структурные белки, которые можно выявить с помощью перекрестно реагирующих антител. Заслуживает внимания, что ламины A/C не выявляются иммунологически в предимплатационных эмбрионах коров. Так как ламины A/C экспрессируются в соматических клетках (фиг. 29B), то они потенциально представляют собой молекулярные маркеры перепрограммирования ядер в эмбрионах с ядерными трансплантатами. Динамика ядерной оболочки, NuMa и AKAP95 в эмбрионах коров с ядерными трансплантатами Динамику структур ядерной оболочки и ядерного матрикса исследовали в ходе осуществления традиционного способа трансплантации ядер у коров. Указанные структуры исследовали, используя антитела к ламинам A/C и B, NuMA и AKAP95 соответственно. Чтобы определить динамику этих маркеров в ходе ядерного ремоделирования, получали эмбрионы коров с ядерными трансплантатами, используя первичные эмбриональные фибробласты, которые выделяли, как описано ранее, в качестве донорных клеток (Kasinathan et al., Biol. Reprod. 64: 1487-1493, 2001). Коротко, клетки собирали из плодов коров трипсинизацией, используя 0,08% трипсин и 0,02% EDTA в PBS (трипсин-EDTA). Клетки высевали во флаконы для культивирования T75 (Corning) в Перенос ядер у коров осуществляли, как описано ранее (Kasinathan et al., Biol. Reprod. 64: 1487-1493, 2001). Созревшие in vitro ооциты лишали ядер через 18-20 часов после созревания. После переноса донорных клеток в фазе G1 в перивителлиновое пространство их сливали, используя единичный электрический импульс 2,4 кВ/см в течение 20 микросекунд (Electrocell Manipulator 200, Genetronics). Через 28-30 часов после созревания (т.е. через 28-30 часов после того, как ооциты помещали в среду для созревания после сбора из яичников и, по меньшей мере, через два часа после слияния с донорными клетками) реконструированные ооциты и партеногенетические контроли активировали кальциевым ионофором (5 мкМ) в течение четырех минут (Cal Biochem), затем 10 мкг циклогексимида и 2,5 мкг цитохалазина D (Sigma) в среде ACM (100 мМ NaCl, 3 мМ KCl, 0,27 мМ CaCl2, 25 мМ NaHCO3, 1 мМ лактат натрия, 0,4 мМ пируват, 1 мМ L-глутамин, 3 мг/мл БСА, 1% аминокислоты BME и 1% заменимых аминокислот MEM), в течение пяти часов (Liu et al., Mol. Reprod. Dev. 49: 298-307, 1998). После активации эмбрионы или ооциты с ядерными трансплантатами промывали пять раз и культивировали совместно с мышиными эмбриональными фибробластами при 38,5°C в атмосфере 5% CO2. Реконструированные эмбрионы активировали, используя стандартные способы, и через три часа после слияния эмбрионы в стадии преждевременной конденсации хроматина (PCC) фиксировали параформальдегидом и анализировали методом иммунофлуоресценции, используя антитела к ламинам A/C, ламину B, NuMA и AKAP95 (фиг. 30, PCC). Кроме того, подобным образом анализировали группы эмбрионов с ядерными трансплантатами, которым давали возможность прогрессировать до стадии пронуклеусов (PN) (т.е. эмбрионы коров через 15 часов после слияния) (фиг. 30, PN с ядерными трансплантатами). В качестве контролей партеногенетические ооциты, активированные, как указано здесь, также исследовали на стадии пронуклеусов (фиг. 30, парт. PN). Как предполагалось, соматические донорные клетки (эмбриональные фибробласты коров, фиг. 30) экспрессировали все маркеры с распределением, ожидаемым на основании литературных данных. На стадии преждевременной конденсации хроматина отчетливые конденсированные хроматиновые массы подтверждали с помощью окрашивания ДНК Hoechst 33342. Ламины A/C и B на конденсированных хромосомах или вблизи них не выявляли (фиг. 30, PCC) предположительно в результате из рассеивания в цитоплазме яйцеклетки. Выявляли некоторое количество меченого NuMA; указанный NuMA, предположительно, был связан с полюсами веретена, удерживающими конденсированные хромосомы. В отличие от этого AKAP95 был связан с конденсированными (PCC) хромосомами. Полученный результат напоминает мечение AKAP95 в митотических клетках человека (Collas et al., J. Cell Biol. 147: 1167-1180, 1999; Steen et al., J. Cell Biol. 150: 1251-1262, 2000). На стадии пронуклеусов выявляли все маркеры. Ламины A/C присутствовали в оболочке пронуклеуса (фиг. 2, PN с ядерными трансплантатами). Это расходилось с их отсутствием в оболочке пронуклеусов контрольных партенотов (фиг. 30) и оболочке оплодотворенных пронуклеусов (фиг. 29A). Ламин B выявляли в пронуклеусах с ядерными трансплантатами, так же как в контрольных пронуклеусах. Также NuMA и AKAP95 окрашивали внутреннюю часть ядра за исключением ядрышек. Мечение NuMA было соответственно ярче в пронуклеусах с ядерными трансплантатами, чем в контрольных партеногенетических пронуклеусах (сравните PN с ядерными трансплантатами и парт. PN, фиг. 30). Вместе указанные наблюдения свидетельствуют, что в пронуклеусах эмбрионов с ядерными трансплантатами вновь происходит сборка соматических ядерных маркеров ламинов A и C и в них наблюдается сильное окрашивание NuMA. Дифференциальное заякоривание AKAP95 в пронуклеусах партеногенетических эмбрионов и эмбрионов с ядерными трансплантатами A-киназный заякоривающий белок AKAP95 является ядерным белком, вовлеченным в конденсацию митотических хромосом. Для использования в качестве другого молекулярного маркера, влияющего на перепрограммирование соматических ядер после ядерной трансплантации, были охарактеризованы свойства заякоривания внутри ядра белка AKAP95 в эмбрионах коров с ядерными трансплантатами на стадии пронуклеусов, образованных из эмбриональных фибробластов. Также исследовали заякоривание AKAP95 в пронуклеусах из партеногенетических эмбрионов и ядрах соматических донорных клеток. Внутриядерное заякоривание AKAP95 в эмбрионах на стадии пронуклеусов исследовали in situ посредством экстракции эмбрионов 0,1% тритоном X-100, 1 мг/мл ДНКазы I и либо 100, либо 300 мМ NaCl в течение 30 минут при комнатной температуре. Как указано выше, мужские пронуклеусы совсем не содержат AKAP95. В отличие от этого имелось значительное количество AKAP95, и ДНК была резистентной к ДНКазе I и 300 мМ NaCl в пронуклеусах эмбрионов с ядерными трансплантатами и в донорных ядрах коров (фиг. 31). Ламины B-типа не экстрагировались ДНКазой I и 300 мМ NaCl в пронуклеусах партенотов или ядерных трансплантатов (фиг. 31), что свидетельствует об изменениях в распределении AKAP95 и ДНК не являются результатом крупных изменений в архитектуре ядер. Полученные данные свидетельствуют, что в соматических ядрах AKAP95 сильнее заякорен на внутриклеточных структурах в пронуклеусах ядерных трансплантатов, чем в партеногенетических пронуклеусах у коров. Остается определить, накладывает ли данная ассоциация ограничения на организацию ДНК или возникает в результате измененной организации генома в эмбрионах с ядерными трансплантатами. Так как резистентная к ДНКазе I ДНК является транскрипционно молчащей, неполное ремоделирование заякоривания AKAP95 после трансплантации ядер по-видимому нарушает экспрессию важных для развития генов. Неправильная регуляция транскрипции ламинов A/C в эмбрионах коров с ядерными трансплантатами Поразительным наблюдением явилось то, что ламины A/C снова подвергаются сборке на периферии пронуклеусов в эмбрионах коров с ядерными трансплантатами, в то время как данный специфичный для соматических клеток маркер исчезает в оплодотворенных in vitro и партеногенетических пронуклеусах. Таким образом авторы исследовали, является ли повторная сборка ламинов A/C результатом (i) повторного направленного воздействия соматических ламинов после их разборки на стадии преждевременной конденсации хроматина (фиг. 30), (ii) трансляции и сборки ламинов из пула материнской мРНК ламина A/C или (iii) транскрипции de novo соматического гена ламина A (LMNA) в пронуклеусах ядерных трансплантатов. Чтобы различить указанные возможности, эмбрионы коров с ядерными трансплантатами получали либо «традиционным» способом трансплантации ядер, который приведен в этом описании, трансплантацией ядер с последующей активацией реконструированных эмбрионов ингибитором синтеза белка циклогексимидом (CHX), либо трансплантацией ядер с последующей активацией в присутствии ингибитора РНК-полимеразы II (PolII) актиномицина D (ActD), чтобы ингибировать транскрипцию de novo. В случае культивирования эмбрионов коров с ядерными трансплантатами в циклогексимиде ооциты активировали после переноса ядер, как описано выше, за исключением того, что ооциты инкубировали в течение 14 часов в циклогексимиде (CHX). Через 14 часов после активации ооциты промывали пять раз и помещали в культуральную среду ACM, содержащую 15 мкг/мл Hoechst 33342 (Sigma), на один час. После инкубирования исследовали развитие пронуклеусов эпифлуоресцентной микроскопией. Эмбрионы на стадии пронуклеусов затем фиксировали в 3% параформальдегиде в PBS, промывали и помещали на предметные стекла. В случае культивирования ооцитов коров с ядерными трансплантатами в актиномицине D ооциты активировали после переноса ядер, как описано выше, за исключением того, что добавляли 5 мкг/мл актиномицина D (ActD) на стадии инкубирования с циклогексимидом. Через пять часов яйцеклетки промывали пять раз и помещали в культуральную среду ACM, содержащую 5 мкг/мл актиномицина D. Через 14 часов после активации яйцеклетки пять раз промывали и помещали в культуральную среду ACM, содержащую 15 мкг/мл Hoechst 33342 (Sigma), на один час. После инкубирования наблюдали развитие пронуклеусов эпифлуоресцентной микроскопией. Эмбрионы на стадии пронуклеусов фиксировали в 3% параформальдегиде в PBS, промывали и помещали на предметные стекла. На сборку ламина B вокруг пронуклеуса ядерного трансплантата не влияло ни ингибирование синтеза белка, ни ингибирование синтеза РНК. Полученный результат свидетельствует, что ламин B повторно собирался или из ранее подвергнутого разборке соматического пула и/или из большого пула ламина B в цитоплазме ооцита. Ламины A/C, которые выявлялись в пронуклеусах ядерных трансплантатов (фиг. 30), отсутствовали в ядрах, вновь образованных после активации циклогексимидом. Полученный результат свидетельствует, что сборка ламинов A/C требует синтеза белка de novo и что указанные ламины не подвергаются повторному направленному воздействию из соматического пула, подвергнутого разборке, внесенного в ооцит при инъекции донорного ядра или при слиянии клеток. Кроме того, ламины A/C не подвергаются повторной сборке, когда эмбрионы активируют в присутствии актиномицина D. Полученный результат свидетельствует о том, что повторная сборка ламинов A/C в пронуклеусах ядерных трансплантатов является результатом транскрипции de novo гена LMNA в реконструированном пронуклеусе. NuMA, который выявляли в пронуклеусах ядерных трансплантатов, не подвергается повторной сборке в пронуклеусах эмбрионов с ядерными трансплантатами, активированных циклогексимидом, но едва различимо выявляется в пронуклеусах обработанных актиномицином D эмбрионов с ядерными трансплантатами. Такие данные строго свидетельствуют о том, что повторная сборка NuMA в пронуклеусах ядерных трансплантатов требует трансляции de novo, которая происходит, по меньшей мере частично, из пула материнской мРНК NuMA. Согласующееся с этим наблюдение, что мечение анти-NuMA является более слабым в пронуклеусах обработанных актиномицином D эмбрионов с ядерными трансплантатами по сравнению с контрольными необработанными эмбрионами с ядерными трансплантатами (b’ и b Вместе эти результаты показывают, что ген LMNA не выключается при ядерном ремоделировании после трансплантации ядер. Подобным образом ген NuMA, по-видимому, остается активным в эмбрионах с ядерными трансплантатами на стадии пронуклеусов. Вероятно, временная инактивация этих генов имеет место во время преждевременной конденсации хроматина, что можно ожидать из-за высококонденсированной природы хроматина (фиг. 30). Полученные результаты ясно иллюстрируют неполное ядерное перепрограммирование в эмбрионах с ядерными трансплантатами, полученными в условиях, приведенных в этом описании. Как обсуждалось ранее в случае AKAP95, авторы предположили, что сохранение ламинов A/C в пронуклеусах ядерных трансплантатов влияет на экспрессию генов, такую как экспрессия важных для развития генов. Описанные ранее взаимодействия ламинов A и C с белками хроматина и ДНК и ассоциация этих ламинов с факторами транскрипции также подтверждают данную гипотезу. ПРИМЕР 9: Характерная недостаточность перепрограммирования ядер в эмбрионах коров с традиционными ядерными трансплантатами Ниже также описаны характерные различия между природными эмбрионами и эмбрионами с традиционными ядерными трансплантатами. Такие различия включают различия в сборке в пронуклеусах специфичных для дифференцированных клеток ядерных ламинов A-типа, повышенные концентрации в пронуклеусах NuMA и TATA-связывающего белка и повышенную чувствительность компонента области контакта ядерный матрикс-хроматин AKAP95 и ДНК к экстракции детергентом, ДНКазой и солью. Для данных исследований закладывали клеточные линии эмбриональных фибробластов коров, как описано ранее (Kasinathan et al., Nat. Biotechnol. 19: 1176-1178, 2001 и Kasinathan et al., Biol. Reprod. 64: 1487-1493, 2001). Дублеты фибробластов в G1-фазе выделяли из культур, используя ранее описанный способ стряхивания (Kasinathan et al., Nat. Biotechnol. 19: 1176-1178, 2001). Оплодотворение in vitro с использованием созревших in vitro ооцитов осуществляли, как описано ранее (Collas et al., Mol. Reprod. Dev. 34: 224-231, 1993). Для ядерной трансплантации (NT) и активации ооцитов осуществляли трансплантацию ядер, используя донорные клетки в G1-фазе через ~20 часов после созревания (hpm), как сообщалось ранее (Kasinathan et al., Nat. Biotechnol. 19: 1176-1178, 2001 и Kasinathan et al., Biol. Reprod. 64: 1487-1493, 2001). Реконструированные эмбрионы активировали на стадии 28-30 hpm (T=0), используя 5 мкМ кальциевого ионофора, в течение четырех минут, затем 10 мкг/мл CHX и 2,5 мкг/мл цитохалазина D в течение пяти часов. Эмбрионы промывали и культивировали совместно с эмбриональными фибробластами мышей (Kasinathan et al., Biol. Reprod. 64: 1487-1493, 2001). Когда реконструированные эмбрионы культивировали в CHX, ооциты активировали, как описано выше, и культивировали с 2,5 мкг/мл CHX еще дополнительно в течение девяти часов (всего 14 часов в CHX). Эмбрионы осторожно промывали и культивировали, как описано (Kasinathan et al., Biol. Reprod. 64: 1487-1493, 2001). Когда на реконструированные эмбрионы воздействовали ActD, ооциты активировали, как описано выше, за исключением того, что добавляли 5 мкг/мл ActD на стадии пятичасового инкубирования с CHX. Для иммунологического анализа клетки, ооциты, эмбрионы, ядра (пример 14) и хроматиновые массы (пример 14) помещали на покрытые поли-L-лизином покровные стекла, фиксировали 3% параформальдегидом в течение 15 минут и пермеабилизовали 0,1% тритоном X-100 в течение 15 минут. Белки блокировали, используя PBS/2% БСА/0,01% твин 20. Первые и вторые антитела (разведение 1:100) инкубировали по 30 минут каждое. В частности, использовали поликлональные кроличьи антитела против пептида ламина B человека (Chaudhary et al., J. Cell Biol. 122: 295-306, 1993). Также использовали козьи поликлональные антитела против ламина B, моноклональные антитела против ламина A/C и анти-TBP-антитела от Santa-Cruz Biotechnology. Моноклональные анти-NuMA антитела были от Transduction Laboratories и аффинно очищенные поликлональные антитела против AKAP95 крыс были от Upstate Biotechnologies. ДНК контрастно красили 0,1 мкг/мл Hoechst 33342. Фотографии делали с помощью камеры JVC CCD и количественную оценку интенсивности иммунофлуоресценции осуществляли, используя компьютерную программу AnalySIS. Данные выражали в виде интенсивности (средней ± с.о.) флуоресценции относительно контроля по меньшей мере в трех повторах. Для экстракций in situ эмбрионы и клетки, помещенные на покровные стекла, инкубировали в течение 15 минут с 0,1% тритоном X-100, 1 мг/мл ДНКазы I и 300 мМ NaCl в трис-HCl (pH 7,2) перед иммунофлуоресцентным анализом. Для иммуноблоттинга 100 эмбрионов растворяли в 20 мкл буфера для образцов с SDS, белки разделяли в 10% SDS-PAGE и анализировали со следующими антителами: анти-ламин B, 1:1000; анти-ламин A/C, 1:250; анти-NuMA, 1:500; анти-AKAP95, 1:250. На основании вышеописанного иммунологического анализа было охарактеризовано распределение ламинов A/C- и B-типа, NuMA и AKAP95 в эмбриональных фибробластах коров, обычно используемых для NT (фиг. 40). В полученных in vitro предимплантационных эмбрионах коров ламин B выявляли на периферии ядер в такой ранней стадии, как стадия пронуклеусов (PN) (фиг. 40). Как и предполагалось, ламин A/C, являясь маркером дифференцированных клеток, отсутствовал. NuMA и AKAP95 были ограничены женским пронуклеусом на стадии пронуклеусов, но присутствовали во всех ядрах на последующих стадиях. Специфичность данных иммунофлуоресцентного анализа подтверждал иммуноблоттинг (фиг. 40). Динамику ламинов A/C и B, NuMA и AKAP95 исследовали во время морфологического ядерного ремоделирования, связанного с трансплантацией фибробластов коровы в лишенные ядер ооциты посредством электрослияния (Kasinathan et al. Biol. Reprod. 64: 1487-1493, 2001). Донорные ядра подвергались преждевременной конденсации хроматина (PCC) в пределах трех часов слияния (фиг. 41A). Ядерные ламины и NuMA перераспределялись в цитоплазме ооцита и исчезали из PCC-хромосом (фиг. 41A). AKAP95 был ассоциирован с PCC-хромосомами, свойство, напоминающее митотические клетки. Через четырнадцать часов после начала активационной обработки реципиентных ооцитов NT-эмбрионы демонстрировали полностью развившиеся пронуклеусы (фиг. 41A, NT PN). Однако в отличие от пронуклеусов партеногенетических или оплодотворенных эмбрионов, по существу, все NT-пронуклеусы проявляли сильную иммунореактивность ламина A/C и NuMA, две характерные особенности соматических донорных клеток (фиг. 41A). Активация реципиентных ооцитов в присутствии 10 мкг/мл ингибитора синтеза белка циклогексимида (CHX) или 5 мкг/мл ингибитора РНК-полимеразы (Pol) II актиномицина D (ActD), и то, и другое совместимо с образованием пронуклеусов, ингибировала сборку ламина A/C пронуклеусов (фиг. 41B и 41C). Полученный результат свидетельствует о том, что сборка этих соматических ламинов является результатом транскрипции гена соматического ламина A (LMNA). CHX или ActD не нарушает сборку ламина B, что свидетельствует о том, что он был опосредован диссоциированным соматическим пулом и/или материнским пулом ламинов B-типа (фиг. 41B и 41C). По существу не выявляли NuMA после экспозиции с CHX; однако 40% иммунореактивности NuMA в NT-пронуклеусах выявляли после обработки ActD (фиг. 41B и 41C). Полученный результат свидетельствует о том, что NuMA собирается в результате трансляции из материнской мРНК и транскрипции de novo. Так как ламин A/C и NuMA аномально транскрибируются в NT-пронуклеусах, указанные белки можно использовать в качестве маркеров для определения способности способов переноса ядер перепрограммировать донорный генетический материал. Как обсуждалось в примере 8, свойства внутриядерного заякоривания AKAP95, структурного мультивалентного белка области контакта матрикс-хроматин (Collas et al., J. Cell Biol. 147: 1167-1179, 1999), которым обогащен гипоацетилированный хроматин, также можно использовать в качестве маркера перепрограммирования донорного генетического материала. AKAP95 был единственным исследованным маркером, который был выявлен в соматических донорных ядрах на PCC-хромосомах и в NT-пронуклеусах с интенсивностью мечения, сходной с интенсивностью мечения партенотов (фиг. 41A) и PN-эмбрионов (фиг. 40B). Ассоциация AKAP95 с NT-пронуклеусами сохранялась при ингибировании синтеза белка или РНК. Таким образом, основная фракция PN-AKAP95 в NT-эмбрионах имеет соматическое происхождение. Заякоривание AKAP95 исследовали экстракцией NT-эмбрионов, партенотов или донорных фибробластов 0,1% тритоном X-100, 1 мг/мл ДНКазы I и 300 мМ NaCl. У партенотов экстрагировали ~90% AKAP95 и ДНК; однако 35% AKAP95 в NT-пронуклеусах не поддавалось экстракции (фиг. 41D). Чувствительность AKAP95 и ДНК к ДНКазе I и NaCl было похоже на чувствительность ядер фибробластов (фиг. 41D). Ламин B не экстрагировался при данных условиях, что свидетельствует о том, что различия в способности к экстракции AKAP95 и ДНК не было результатом крупных изменений в архитектуре ядра. Такие результаты свидетельствуют о том, что NT-пронуклеусы характеризуются сильным заякориванием AKAP95 и ограниченной доступностью ДНК для ДНКазы I. Таким образом, пронуклеусы, полученные соматическим NT, по-видимому, проявляют структурные аномалии в результате неполного морфологического ремоделирования донорных ядер и/или неправильной регуляции транскрипции соматических генов. ПРИМЕР 10: Характерная недостаточность перепрограммирования ядер в эмбрионах коров с традиционными ядерными трансплантатами Как описано в примере 9, картины экспрессии в эмбрионах с ядерными трансплантатами (NT) сравнивали с картинами экспрессии в полученных in vitro (IVP) предимплантационных эмбрионах. Для этого сравнения осуществляли оплодотворение in vitro и эмбрионы культивировали, как описано ранее (Collas et al., Mol. Reprod. Dev. 34: 224-231, 1993 и Kasinathan et al., Biol. Reprod. 64: 1487-1493, 2001). NT осуществляли слиянием донорных эмбриональных фибробластов коров с лишенными ядер ооцитами (Kasinathan et al., Nat. Biotechnol. 19: 1176-1178, 2001 и Kasinathan et al., Biol. Reprod. 64: 1487-1493, 2001). Реципиентные ооциты активировали через 28-30 часов после созревания (hpm) 5 мкМ кальциевым ионофором в течение 4 минут, затем 10 мкг/мл CHX и 2,5 мкг/мл цитохалазином D в течение 5 часов и промывали. Эмбрионы культивировали совместно с эмбриональными фибробластами мыши (Kasinathan et al., Biol. Reprod. 64: 1487-1493, 2001). В случае обработки CHX ооциты активировали, как описано выше, и эмбрионы культивировали с 2,5 мкг/мл CHX еще дополнительно в течение девяти часов перед культивированием. В случае обработки ActD ооциты активировали, как описано выше, за исключением того, что добавляли 5 мкг/мл ActD на пятичасовой стадии инкубирования с CHX и эмбрионы поддерживали в 5 мкг/мл ActD еще дополнительно в течение девяти часов перед культивированием. NT-эмбрионы культивировали до стадии бластоцисты in vitro и переносили два эмбриона одной самке реципиенту. Беременности контролировали ультразвуковым исследованием и делали C-сечения. Телята оценивались ветеринарами в течение 24 часов после рождения. Для анализа уровней белков в NT- и IVP-эмбрионах использовали поликлональные антитела против ламина B, моноклональные антитела против ламина A/C и поликлональные или моноклональные антитела против TBP от Santa-Cruz Biotechnology. Анти-AKAP95 антитела были от Upstate Biotechnologies. Анализ иммунофлуоресценции осуществляли, как описано (Bomar et al., J. Cell Sci. 115: 2931-2940, 2002). Коротко, клетки и эмбрионы осаждали на покрытые поли-L-лизином покровные стекла, фиксировали 3% параформальдегидом в течение 15 минут, пермеабилизовали 0,1% тритоном X-100 в течение 15 минут и белки блокировали в PBS/2% БСА/0,01% твине 20. Образцы инкубировали с первыми и вторыми антителами (разведение 1:100) по 30 минут с каждым. ДНК контрастно красили 0,1 мкг/мл Hoechst 33342. Фотографии делали камерой JVC CCD и количественное определение интенсивности флуоресценции осуществляли, используя компьютерную программу AnalySIS. В указанных случаях образцы экстрагировали на покровных стеклах смесью 1% тритона X-100, 1 мг/мл ДНКазы I и 300 мМ NaCl в трис-HCl (pH 7,2) в течение 15 минут перед анализом иммунофлуоресценции. Для иммуноблоттинга образцы белков (30 мкг) разделяли в 10% SDS-PAGE, с помощью блоттинга переносили на нитроцеллюлозу и исследовали с использованием указанных антител. Динамика донорных ядер в эмбрионах с ядерными трансплантатами Чтобы исследовать динамику соматических ядер во время трансплантации ядер коров (NT), определяли распределение двух структурных компонентов ядерной оболочки, повсеместно экспрессируемых ламинов B-типа (называемых ламином B) и специфичных для дифференцированных клеток ламинов A-типа (ламин A/C) (Gruenbaum et al., J. Struct. Biol. 129: 313-323, 2000). Ядерные ламины служат якорем ядерным мембранам к хроматину, и высказано предположение, что они стимулируют размножение ядер после восстановления ядер (пронуклеусов) in vitro. Также показано, что ламины важны для жизнеспособности клетки, так как неспособность к сборке ламинов B-типа приводит к гибели клетки. В качестве маркера аппарата транскрипции анализировали динамику TATA-связывающего белка, TBP, фактора транскрипции практически для всех генов. Перинуклеарное распределение ламина B и совместная локализация TBP с ДНК в эмбриональных фибробластах коров и в полученных in vitro (IVP) предимплантационных эмбрионах согласуются с наблюдениями на других видах (Holy et al., Dev. Biol. 168: 464-478, 1995; Houliston et al., Development 102: 271-278, 1988; и Worrad et al., Development 120: 2347-2357, 1994) (фиг. 46A). Как и ожидалось, ламин A/C, являясь маркером дифференцированных клеток, не выявлялся во время предимплантационного развития (фиг. 46A). Специфичность иммунофлуоресцентного мечения подтверждали на иммуноблотах (фиг. 46B). После трансплантации ядер фибробластов в лишенные ядер ооциты как ламины A/C, так и ламин B подвергались диссоциации из преждевременно конденсированных хромосом (PCC; через три часа после слияния), в то время как TBP оставался связанным с хромосомами (фиг. 46C, PCC). Через четырнадцать часов после начала активации реципиентных ооцитов все NT-эмбрионы содержали полностью развившиеся пронуклеусы с перинуклеарным мечением ламина B и TBP, локализованным совместно с ДНК (фиг. 46C, NT-PN). Однако в отличие от IVP-эмбрионов в 95-99% NT-эмбрионов наблюдали экспрессию ламина A/C в такой ранней стадии, как стадия пронуклеусов (фиг. 46C), и экспрессия сохранялась во время раннего развития (смотри ниже). Относительные количества иммунологически меченого ламина B, ламина A/C и TBP в пронуклеусах NT- и IVP-эмбрионов оценивали путем измерения отношения интенсивности флуоресценции второго антитела к интенсивности флуоресценции ДНК (Hoechst 33342), чтобы рассчитать содержание ДНК (гаплоидное или диплоидное) в исследуемых ядрах. В то время как относительные количества ламина B были сходны в пронуклеусах NT- и IVP-эмбрионов, относительные количества ламина A/C и TBP были выше в NT-пронуклеусах, чем в мужских (MPN) или женских (FPN) пронуклеусах в IVP-эмбрионах (фиг. 46D). TBP, ДНК и ламины A-типа в пронуклеусах NT-эмбрионов Более высокие количества TBP в NT-пронуклеусах связаны с более высокой устойчивостью к экстракции in situ комбинацией детергента (1% тритон X-100), нуклеазы (1 мг/мл ДНКазы I) и соли (0,3 М NaCl). Количественное определение интенсивности иммунофлуоресцентного мечения в экстрагированных эмбрионах по сравнению с не подвергнутыми экстракции контролями показывает, что ~35% TBP оставалось неэкстрагрованным в мужских (MPN) или женских (FPN) пронуклеусах IVP-эмбрионов (фиг. 47A). Однако TBP NT-пронуклеусов проявлял чрезвычайную устойчивость к экстракции, как и в ядрах фибробластов (фиг. 47A). Также наблюдали 4,5-кратное увеличение устойчивости ДНК NT-пронуклеусов к экстракции в данных условиях по сравнению с пронуклеусами IVP-эмбрионов (фиг. 47A, ДНК), что свидетельствует о более компактной организации хроматина. Чтобы определить происхождение ламина B, ламина A/C и TBP в пронуклеусах NT-эмбрионов реципиентные ооциты активировали в присутствии ингибитора РНК-полимеразы (Pol) II актиномицина D (ActD; 5 мкг/мл) или ингибитора синтеза белка циклогексимида (CHX, 10 мкг/мл) (Knott et al., Biol. Reprod. 66: 1095-1103, 2002), как указано в этом описании. Сборку ламина A/C, ламина B и TBP оценивали денситометрическим анализом иммунофлуоресцентно меченых эмбрионов на стадии пронуклеусов. Оба ингибитора предотвращали сборку ламина A/C в пронуклеусах (фиг. 47B и 47C), свидетельствуя о том, что сборка соматических ламинов в NT-эмбрионах являлась следствием транскрипции гена ламина A на стадии пронуклеусов. Сборка ламина B не нарушалась обработкой CHX или ActD (фиг. 47B и 47C), свидетельствуя о том, что ламин B подвергался сборке из соматических ламинов, солюбилизированных в цитоплазме ооцита после NT, и/или из материнского пула ламинов. Сходные количества TBP выявляли в необработанных эмбрионах или после ингибирования синтеза РНК или белка (фиг. 47B и 47C). Так как TBP ассоциирован с конденсированными хромосомами во время PCC и цитоплазма ооцитов в метафазе II лишена регистрируемого TBP, вероятно TBP соматического происхождения остается связанным с донорным геномом во время NT. Таким образом, кроме экспрессии ламинов A-типа в пронуклеусах NT-эмбрионов, наблюдаются более высокие количества и усиленное внутриклеточное заякоривание TBP. ПРИМЕР 11: Применение перепрограммированных донорных хроматиновых масс для клонирования млекопитающих Чтобы преодолеть проблему неполного перепрограммирования в эмбрионах с традиционным ядерным переносом, которая была показана в примерах 8-10, разрабатывали новые способы для более эффективного перепрограммирования донорного хроматина перед переносом ядер (PCT/USO1/50406, подана 21 декабря 2001 г.). Эти способы заключаются в инкубировании ядра (например, ядра, которое кодирует ксеногенное антитело) из донорной клетки в средах для перепрограммирования (например, клеточном экстракте), что приводит к растворению ядерной оболочки и возможно конденсации хроматина. Указанное разрушение ядерной оболочки и конденсация хроматина позволяют высвобождать белки, регулирующие транскрипцию, которые были связаны с хромосомами и которые при других обстоятельствах стимулируют транскрипцию генов, не желательных для развития ооцита, эмбриона или плода. Кроме того, регуляторные белки из перепрограммирующих сред могут связываться с хроматиновой массой и стимулировать транскрипцию генов, желательных для развития. Чтобы создать копытное животное с мутацией в гене, кодирующем прион, и, необязательно, с ксеногенным геном Ig, донорное ядро или хроматиновую массу можно модифицировать до, во время или после перепрограммирования путем мутации одного или более аллелей гена, кодирующего прион и, необязательно, инсерцией одной или нескольких нуклеиновых кислот, кодирующих ксеногенное антитело. При желании клетку из клонированного плода или клонированного потомка можно использовать во втором раунде переноса ядер, чтобы создать дополнительное клонированное потомство. Клетки из исходного клонированного плода или клонированного потомка также можно заморозить, чтобы образовать линию клеток, используемую в качестве источника донорных клеток для создания дополнительных клонированных копытных животных. Массовое получение донорных ядер для применения в клонировании До нескольких миллионов ядер можно выделить из синхронизированных и несинхронизированных популяций клеток в культуре. Популяции клеток можно синхронизировать естественным образом или химически. Предпочтительно, по меньшей мере 40, 60, 80, 90 или 100% клеток в популяции задерживают в фазе G0 или G1. Чтобы это осуществить, клетки можно инкубировать, например, при низком содержании сыворотки, таком как 5%, 2% или 0% сыворотки в течение 1, 2, 3 или большего количества дней, чтобы увеличить процент клеток в фазе G0. Чтобы синхронизировать клетки в G1, клетки можно выращивать до смыкания в монослой в виде прикрепленных клеток и затем инкубировать в 0,5-1 мкг/мл нокодазола (Sigma Chemicals, St. Louis, MO) в течение 17-20 часов, как описано ранее (смотри, например, Collas et al., J. Cell Biol. 147: 1167-1180, 1999 и ссылки, представленные здесь). Флаконы, содержащие прикрепленные клетки, энергично встряхивают, многократно тряся флаконы одной рукой, таким образом приводя к откреплению митотических клеток и дублетов в фазе G1. Дублеты в фазе G1 представляют собой пары растягивающихся клеток в конце процесса деления, которые еще связаны тонким мостиком. Открепленные дублеты в фазе G1 можно выделить из среды, основываясь на их характерной дублетной структуре. Дублеты в фазе G1 могут оставаться прикрепленными и могут делиться на две отдельные клетки после выделения. Синхронизированные или несинхронизированные клетки собирают в фосфатно-солевом буфере (PBS), используя стандартные способы, и осуществляют несколько стадий промывки, чтобы перенести клетки из их исходных сред в гипотонический буфер (10 мМ HEPES, pH 7,5, 2 мМ MgCl2, 25 мМ KCl, 1 мМ DTT, 10 мкМ апротинин, 10 мкм лейпептин, 10 мкМ пепстатин A, 10 мкМ соевый ингибитор трипсина и 100 мкМ PMSF). Например, клетки можно промыть 50 мл PBS и осадить центрифугированием при 500 × g в течение 10 минут при 4°C. Супернатант PBS отбрасывают, а осажденные клетки ресуспендируют в 50 мл PBS и центрифугируют, как описано выше. После указанного центрифугирования осажденные клетки ресуспендируют в 20-50 объемах ледяного гипотонического буфера и центрифугируют при 500 × g в течение 10 мин при 4°C. Супернатант снова отбрасывают и к осадку клеток добавляют примерно 20 объемов гипотонического буфера. Клетки осторожно ресуспендируют в этом буфере и инкубируют на льду по меньшей мере в течение одного часа, получая постепенное набухание клеток. Чтобы обеспечить выделение ядер из клеток, клетки лизируют, используя стандартные способы. Например, 2-5 мл суспензии клеток можно перенести в стеклянный гомогенизатор и гомогенизировать в гомогенизаторе Даунса, используя первоначально 10-20 ударов плотно подогнанного пестика. Альтернативно суспензию клеток гомогенизируют, используя миксер с двигателем (например, Ultraturrax). При желании лизис клеток можно контролировать, используя метод фазово-контрастной микроскопии при 40-кратном увеличении. В ходе указанной гомогенизации ядра должны оставаться интактными и большая часть или, предпочтительно, все исходно прикрепленные цитоплазматические компоненты, такие как везикулы, органеллы и белки, должны высвобождаться из ядер. При необходимости можно добавить 1-20 мкг/мл ингибиторов цитоскелета, цитохалазина B или цитохалазина D к указанному выше гипотоническому буферу, чтобы облегчить данный процесс. Гомогенизацию продолжают сколько необходимо, чтобы лизировать клетки и высвободить цитоплазматические компоненты из ядер. Для некоторых типов клеток может потребоваться до 100, 150 или более ударов. Затем лизат переносят в коническую пробирку объемом 15 мл на лед и процедуру лизиса клеток повторяют с остальной частью суспензии набухших клеток. К лизату клеток добавляют сахарозу из 2 М маточного раствора в гипотоническом буфере (например, к лизату добавляют 1/8 объема 2 М маточного раствора), получая конечную концентрацию 250 мМ сахарозы. Этот раствор перемешивают переворачиванием и ядра осаждают центрифугированием при 400 × g в откидном роторе в течение 10-40 минут при 4°C. Затем супернатант отбрасывают, а осажденные ядра ресуспендируют в 10-20 объемах буфера для ядер (10 мМ HEPES, pH 7,5, 2 мМ MgCl2, 250 мМ сахароза, 25 мМ KCl, 1 мМ DTT, 10 мкм апротинин, 10 мкМ лейпептин, 10 мкМ пепстатин A, 10 мкМ соевый ингибитор трипсина и 100 мкМ PMSF). Ядра осаждают и ресуспендируют в 1-2 объемах буфера для ядер, как описано выше. Только что выделенные ядра могут быть либо сразу использованы для перепрограммирования in vitro и переноса ядер, как описано ниже, либо могут храниться для более позднего использования. Для хранения ядра разбавляют в буфере для ядер до концентрации примерно 106/мл. Добавляют глицерин (2,4 объема 100% глицерина) и хорошо перемешивают осторожным пипетированием. Суспензию делят на аликвоты объемами по 100-500 мкл в пробирки на 1,5 мл на льду, сразу замораживают в бане со смесью метанол-сухой лед и хранят при -80°C. Перед применением аликвоты ядер размораживают на льду или при комнатной температуре. Добавляют один объем ледяного буфера для ядер и раствор центрифугируют при 1000 × g в течение 15 минут в откидном роторе. Осажденные ядра ресуспендируют в 100-500 мкл буфера для ядер и центрифугируют, как описано выше. Затем осажденные ядра ресуспендируют в минимальном объеме буфера для ядер и хранят на льду до использования. Приготовление митотического экстракта или сред для применения в перепрограммировании донорного генетического материала Для приготовления митотического экстракта линию соматических клеток (например, фибробластов) синхронизируют в митозе инкубацией в 0,5-1 мкг/мл нокодазола в течение 17-20 часов (смотри, например, Collas et al., J. Cell Biol. 147: 1167-1180, 1999 и ссылки в этой работе) и митотические клетки открепляют энергичным встряхиванием, как описано выше. Открепленные дублеты в фазе G1 могут быть отброшены или они могут оставаться с митотическими клетками, которые составляют большинство открепленных клеток (обычно по меньшей мере 80%). Собранные открепленные клетки центрифугируют при 500 × g в течение 10 минут в конической пробирке объемом 10 мл при 4°C. Несколько осадков клеток объединяют, ресуспендируют в общем объеме 50 мл холодного PBS и центрифугируют при 500 × g в течение 10 минут при 4°C. Эту стадию промывки PBS повторяют. Осадок клеток ресуспендируют примерно в 20 объемах ледяного буфера для лизиса клеток (20 мМ HEPES, pH 8,2, 5 мМ MgCl2, 10 мМ EDTA, 1 мМ DTT, 10 мкМ апротинин, 10 мкМ лейпептин, 10 мкМ пепстатин A, 10 мкМ соевый ингибитор трипсина, 100 мкМ PMSF и, необязательно, 20 мкг/мл цитохалазин B), и клетки осаждают центрифугированием при 800 × g в течение 10 минут при 4°C. Супернатант отбрасывают и осадок клеток осторожно ресуспендируют не более чем в одном объеме буфера для лизиса клеток. Клетки инкубируют на льду в течение одного часа, позволяя клеткам набухнуть. Клетки лизируют либо озвучиванием с использованием ультразвукового аппарата с наконечником, либо гомогенизацией в гомогенизаторе Даунса с использованием стеклянной ступки и пестика. Лизис клеток осуществляют вплоть до лизиса по меньшей мере 90% клеток и ядер, что можно оценить с использованием фазово-контрастной микроскопии. Время озвучивания, необходимое для лизиса по меньшей мере 90% клеток и ядер, может варьировать в зависимости от типа клетки, используемой для приготовления экстракта. Лизат клеток помещают в центрифужную пробирку объемом 1,5 мл и центрифугируют при 10000-15000 × g в течение 15 минут при 4°C, используя настольную центрифугу. Пробирки извлекают из центрифуги и сразу помещают на лед. Супернатант осторожно собирают, используя наконечник пипетки на 200 мкл, и супернатант из нескольких пробирок объединяют и помещают на лед. Полученный супернатант представляет собой «митотический цитоплазматический» или «MS15» экстракт. Полученный клеточный экстракт можно разделить на аликвоты объемом 50 мкл или 10 мкл экстракта на пробирку на льду в зависимости от того, будет ли использован обычный способ или микроспособ для создания хроматиновых масс. Экстракты сразу же мгновенно замораживают в жидком азоте и хранят при -80°C вплоть до применения. Альтернативно клеточный экстракт помещают в ультрацентрифужную пробирку на льду (например, подобранную для ротора SW55 Ti; Beckman). При необходимости пробирку сверху покрывают минеральным маслом. Экстракт центрифугируют при 200000 × g в течение трех часов при 4°C, чтобы осадить мембранные пузырьки, находящиеся в экстракте MS15. В конце центрифугирования масло отбрасывают. Супернатант осторожно собирают, при необходимости объединяют и помещают в холодную пробирку объемом 1,5 мл на льду. Полученный супернатант называют «MS200» или «митотический цитоплазматический» экстракт. Экстракт делят на аликвоты и замораживают, как описано для экстракта MS15. При желании экстракт можно обогатить дополнительными ядерными факторами. Например, ядра можно очистить из клеток клеточного типа, из которого получают экстракт для перепрограммирования, или из клеток любого другого клеточного типа и лизировать озвучиванием, как описано выше. Ядерные факторы экстрагируют 10-60-минутной инкубацией в буфере для ядер, содержащем NaCl или KCl в концентрации 0,15-800 мМ, при встряхивании. Лизат центрифугируют, чтобы осадить неэкстрагируемые компоненты. Супернатант, содержащий интересующие экстрагированные факторы, диализуют, чтобы удалить NaCl или KCl. Диализованный ядерный экстракт делят на аликвоты и хранят в замороженном виде. Полученный ядерный экстракт добавляют в разных концентрациях к целому клеточному экстракту, описанному выше, перед добавлением ядер для перепрограммирования. Митотические экстракты также могут быть приготовлены из половых клеток, таких как ооциты или мужские половые клетки. Например, ооциты в метафазе II, которые естественным образом задержаны на данной стадии, можно собрать, промыть и лизировать, как описано выше, для создания экстракта ооцитов. Чтобы получить экстракт мужских половых клеток, половые клетки выделяют из семенников, полученных со скотобойни, измельчением органа и дифференциальным центрифугированием собранных клеток в градиенте сахароза или перкола. Половые клетки отделяют от соматических клеток (Лейдига и Сертоли), промывают суспендированием и осаждением в PBS. Затем клетки один раз промывают в ледяном буфере для лизиса клеток, как описано выше, и лизируют озвучиванием. Лизат осветляют центрифугированием при 15000 × g в течение 15 минут при 4°C и супернатант (т.е. экстракт половых клеток) делят на аликвоты и мгновенно замораживают в жидком азоте. В качестве альтернативы клеточному экстракту среды для перепрограммирования также могут быть образованы добавлением одного или нескольких природных или рекомбинантных факторов (например, нуклеиновых кислот или белков, таких как ДНК-метилтрансферазы, деацетилазы гистонов, гистоны, протамины, ядерные ламины, факторы транскрипции, активаторы, репрессоры, белки ремоделирования хроматина, ростовые факторы, интерлейкины, цитокины или другие гормоны) к раствору, такому как буфер. Предпочтительно, один или несколько факторов являются специфичными для ооцитов или стволовых клеток. Образование конденсированных хроматиновых масс Аликвоту экстракта MS15 или MS200 или митотических сред оттаивают на льду. Добавляют АТФ-генерирующую систему (0,6 мкл) к 20 мкл экстракта или среды и смешивают встряхиванием. Для приготовления АТФ-генерирующей системы равные части 100 мМ маточного раствора АТФ, 1 М креатинфосфата и 2,5 мг/мл креатинкиназы в виде маточных растворов (100x), приготовленных в H2O, смешивают и хранят на льду вплоть до использования. После добавления АТФ-генерирующей системы к экстракту конечные концентрации становятся 1 мМ ATP, 10 мМ креатинфосфата и 25 мкг/мл креатинкиназы. Суспензию ядер добавляют к экстракту или среде в концентрации 1 мкл ядер на 10 мкл экстракта или среды, хорошо перемешивают пипетированием и инкубируют на водяной бане с температурой 30, 33, 35, 37 или 39°C. По пробирке, содержащей смесь, осторожно постукивают с регулярными интервалами, чтобы предотвратить образование комочков хромосом на дне пробирки. Разрушение ядерной оболочки и конденсацию хромосом контролируют через равные примежутки времени, а именно каждые 15 минут, под микроскопом. Когда ядерная оболочка разрушается и хромосомы начинают конденсироваться, производят процедуру выделения хроматиновых масс из экстракта или среды. Альтернативно можно образовать хроматиновые массы, которые не конденсированы или только частично конденсированы, осуществляя описанный выше способ после предварительной нагрузки изолированных ядер антителом к белку ядерного матрикса NuMA (Steen et al., J. Cell Biol. 149, 531-536, 2000). Указанный способ позволяет удалять ядерные компоненты из хроматина разрушением ядерной мембраны, окружающей донорные ядра; однако стадию конденсации ингибируют добавлением анти-NuMA-антитела. Предотвращение конденсации хромосом может снижать риск разрывов или потерь хромосом, когда хромосомы инкубируют в митотическом экстракте. Для данного способа очищенные клеточные ядра (2000 ядер/мкл) пермеабилизуют в 500 мкл буфера для ядер, содержащего 0,75 мкг/мл лизолецитина, в течение 15 минут при комнатной температуре. Избыток лизолецитина гасят добавлением 1 мл 3% БСА, приготовленного в буфере для ядер, и инкубацией в течение 5 минут на льду. Затем ядра осаждают и промывают один раз в буфере для ядер. Ядра ресуспендируют в концентрации 2000 ядер/мкл в 100 мкл буфера для ядер, содержащего анти-NuMA-антитело (разведение 1:40; Transduction Laboratories). После одночасового инкубирования на льду при осторожном встряхивании ядра осаждают при 500 × g через 1 М сахарозу в течение 20 минут. Затем ядра ресуспендируют в буфере для ядер и добавляют к митотическому экстракту или среде, содержащей АТФ-регенерирующую систему, которая описана в предыдущем разделе. Необязательно, к экстракту или среде может быть добавлено анти-NuMA-антитело, чтобы дополнительно предотвратить конденсацию хромосом. Также можно образовать хроматиновые массы, которые не конденсированы или конденсированы только частично, воздействием детергентом или протеинкиназой. Детергент можно использовать для растворения ядерных компонентов, которые либо не связаны, либо слабо связаны с хромосомами в ядре, что приводит к удалению ядерной оболочки. В этом способе очищенные клеточные ядра (2000-10000 ядер/мкл) инкубируют в буфере для ядер с добавлением детергента, такого как 0,1%-0,5% тритон X-100 или NP-40. Чтобы облегчить удаление ядерной оболочки, к буферу можно дополнительно добавить соль, такую как NaCl, в концентрации примерно 0,1, 0,15, 0,25, 0,5, 0,75 или 1 М. После 30-60-минутного инкубирования на льду при осторожном встряхивании ядра осаждают центрифугированием при 1000 × g в откидном роторе в течение 10-30 минут в зависимости от общего объема. Осажденные ядра ресуспендируют в 0,5-1 мл буфера для ядер и осаждают, как описано выше. Такую процедуру промывки повторяют дважды, чтобы гарантировать полное удаление детергента и дополнительной соли. Альтернативно ядерную оболочку можно удалить с использованием рекомбинантных или природных протеинкиназ, отдельно или в сочетании. Предпочтительно, протеинкиназы очищают, используя стандартные способы, или получают в очищенной форме из коммерческих источников. Такие киназы могут фосфорилировать компоненты ядерной мембраны, ядерного матрикса или хроматина, что приводит к удалению ядерной оболочки (смотри, например, Collas and Courvalin, Trends Cell Biol. 10: 5-8, 2000). Предпочтительные киназы включают циклинзависимую киназу 1 (CDK1), протеинкиназу C (PKC), протеинкиназу A (PKA), MAP-киназу, кальций/кальмодулинзависимую киназу (CamKII) и казеинкиназу CK1 или казеинкиназу CK2. В этом способе примерно 20000 очищенных ядер инкубируют в 20 мкл буфера для фосфорилирования при комнатной температуре в центрифужной пробирке объемом 1,5 мл. Предпочтительный буфер для фосфорилирования в случае CDK1 (Upstate Biotechnology) содержит 200 мМ NaCl, 50 мМ трис-HCl (pH 7,2-7,6), 10 мМ MgSO4, 80 мМ Реакцию фосфорилирования инициируют добавлением протеинкиназы до конечного количества 25-100 нг. Реакционную смесь инкубируют при комнатной температуре до одного часа. Разрушение ядерной оболочки в ходе этого инкубирования можно контролировать микроскопией, а именно с 15-минутными интервалами. После разрушения ядерной оболочки ядра три раза промывают, как описано выше, для удаления раствора детергента. Выделение хроматиновых масс Экстракт или раствор, содержащий конденсированные, частично конденсированные или неконденсированные хроматиновые массы, помещают под равный объем 1 М раствора сахарозы, приготовленного в буфере для ядер. Хроматиновые массы осаждают центрифугированием при 1000 × g в течение 10-30 минут в зависимости от объема образца в откидном роторе при 4°C. Супернатант отбрасывают, а осажденные хроматиновые массы осторожно ресуспендируют пипетированием в 0,1-1,0 мл буфера для ядер или буфера для липослияния (150 мМ NaCl, 10 мкм апротинин, 10 мкМ лейпептин, 10 мкМ пепстатин A, 10 мкМ соевый ингибитор трипсина и 100 мкМ PMSF либо в 20 мМ HEPES около pH 7,0 или pH 7,5, либо 20 мМ MES около pH 6,2) и центрифугируют при 1000 × g в течение 10-30 минут. Супернатант отбрасывают и осажденные хроматиновые массы ресуспендируют в буфере для ядер или буфере для липослияния и хранят на льду вплоть до использования. Каждую хроматиновую массу переносят в каплю 20 мкл забуференной HEPES среды под масло в чашку для микроманипуляций. Одну хроматиновую массу водят в каждый лишенный ядра ооцит, как описано ниже. Микроспособ приготовления хроматиновых масс Каплю в количестве 10-20 мкл экстракта MS15 или MS200 или митотической среды, содержащей АТФ-генерирующую систему, раствора детергента и/или соли или раствора протеинкиназы, которые описаны выше, помещают на чашку Петри. Затем добавляют каплю в количестве 50 мкл выделенных дублетов клеток в фазе G1 или клеток в фазе G0 в культуральной среде, отдельную «лизирующую» каплю в количестве 50 мкл забуференной HEPES или бикарбонатом среды, содержащей 0,1% тритона X-100 или NP-40, применяемой для обеспечения лизиса клеток, и каплю в количестве 50 мкл среды для инъекции ооцитов. Каждую из этих капель покрывают уравновешенным CO2 минеральным маслом. На чашку Петри также добавляли «каплю для промывки» культуральной среды объемом 50 мкл, применяемую для промывки лизированных клеток или ядер. Клетки переносят в лизирующую каплю, используя микропипетку. Мембраны клеток лизируют в пипетке путем осторожных повторных аспираций. Когда клетки лизированы, лизат осторожно выводят в каплю для промывки и ядро сразу же повторно отсасывают, чтобы удалить детергент. Необязательно, ядра можно пермеабилизовать и инкубировать с анти-NuMA-антителами перед их добавлением к митотическому экстракту или среде. Затем ядро выталкивают в каплю MS15, MS200 или среды, раствора детергента и/или соли или раствора протеинкиназы. Разрешение ядер и конденсацию хромосом контролируют, как описано выше. После того как ядерная оболочка разрушена и, если использовали митотический экстракт без анти-NuMA-антител, хромосомы начали конденсироваться, отдельную интактную хроматиновую массу выделяют с помощью микропипетки и переносят в лишенный ядра реципиентный ооцит, как описано ниже. Удаление ядер из ооцитов Предпочтительно, реципиентный ооцит является ооцитом в стадии метафазы II. На данной стадии ооцит может быть активирован или уже является в достаточной степени активированным, чтобы воздействовать на введенную хроматиновую массу, так как он воздействует на оплодотворяющий сперматозоид. В случае удаления ядра из ооцита часть или, предпочтительно, всю ДНК в ооците удаляют или инактивируют. Такое разрушение или удаление ДНК в реципиентном ооците предотвращает внесение генетическим материалом ооцита вклада в рост и развитие клонированного млекопитающего. Одним из способов разрушения пронуклеуса ооцита является воздействие ультрафиолетовым светом (Gurdon, в Methods in Cell Biology, Xenopus Laevis: Practical Uses in cell and Molecular Biology, Kay and Peng, eds., Academic Press, California, volume 36: pages 299-309, 1991). Альтернативно пронуклеус ооцита можно удалить хирургически любым стандартным способом (смотри, например, McGrath and Solter, Science 220: 1300-1319, 1983). В одном возможном способе иглу вводят в ооцит и ядро отсасывают во внутреннее пространство иглы. Затем иглу удаляют из ооцита, не разрывая плазматической мембраны (патенты США номер 4994384 и 5057420). Липослияние для введения хроматиновых масс в ооциты Хроматин может быть введен в реципиентные ооциты посредством липослияния, как описано ниже, или стандартными способами микроинъекции или электрослияния (смотри, например, патенты США No. 4994384 и 5945577). Следующий способ липослияния также можно использовать в других случаях, чтобы встроить хромосомы в другие реципиентные клетки. Хроматиновые массы выделяют из митотического экстракта, раствора детергента и/или соли или раствора протеинкиназы центрифугированием и затем промывают буфером для липослияния, как описано выше. Хроматиновые массы можно хранить в ледяном буфере для липослияния вплоть до использования. Альтернативно хроматиновые массы делят на аликвоты, замораживают в жидком азоте или на бане со смесью метанол-сухой лед и хранят замороженными при -80°C. Раствор для липослияния готовят смешиванием одного или нескольких вызывающих слияние реагентов с буфером для липослияния в соответствующих пропорциях в диапазоне примерно от 5:1 до 1:10. Реагенты, вызывающие слияние, включают без ограничения полиэтиленгликоль (ПЭГ) и липофильные соединения, такие как Lipofectin®, Lipofectamin®, DOTAP® {метилсульфат N-[1-(2,3-диолеоилокси)пропил]-N,N,N-триметиламмония; C43H83NO8S}, DOSPA® {трифторацетат 2,3-диолеилокси-N-[2-(сперминкарбоксамидо)этил]-N,N-диметил-1-пропанаммония} и DOPED® (диолеоилфосфатидилэтаноламин). Другие предпочтительные липиды включают нейтральные и моновалентные или поливалентные катионные липиды, такие как липиды, содержащие группы четвертичного аммония. Дополнительные предпочтительные липиды имеют остаток холестерина, такой как остаток, образованный в результате реакции гидроксильной группы в холестерине с группой в липиде. Другие предпочтительные липиды имеют насыщенную или ненасыщенную жирную кислоту, которая, предпочтительно, содержит от 5 до 10, от 10 до 15, от 15 до 20 или от 20 до 30 атомов углерода включительно. Такие липиды могут быть синтезированы с использованием стандартных способов химического синтеза, получены из природных источников или могут быть коммерчески доступными (Summers et al., Biophys. J. 71: 3199-3206, 1996; Nabekura et al., Pharm. Res. 13: 1069-1072, 1996; Walter et al., Biophys. J. 66: 366-376, 1994; Yang et al., Biosci. Rep. 13: 143-157, 1993; Walter and Siegel, Biochemistry. 6:32: 3271-3281, 1993). Другие предпочтительные вызывающие слияние соединения являются фосфолипидами, такими как фракции мембранных пузырьков из яиц морского ежа или любого другого источника (Collas and Poccia, J. Cell Science 109: 1275-1283, 1996). Предпочтительно, контактирование хромосом с фракцией мембранных пузырьков не приводит к инкапсулированию хромосом интактной мембраной. Например, можно использовать катионный липид, такой как DOTAP®, в концентрации примерно от 0,1 до 30 мкг/мл в буфере для липослияния. Альтернативно можно использовать липосомный препарат, состоящий из смеси катионного липида и нейтрального липида, такого как DOPE®. Хроматиновые массы, либо свежеприготовленные, либо замороженные и оттаянные, смешивают с раствором для липослияния, так чтобы обеспечить покрывание хроматиновых масс соединением. Инкубирование происходит при температуре 20-30°C в течение примерно 10-30 минут. Микрокапли, содержащие хроматиновые массы в растворе для липослияния, помещают под уравновешенное CO2 минеральное масло. Также готовят каплю, содержащую лишенные ядра реципиентные ооциты. Хроматиновые массы, покрытые реагентом для липослияния, собирают в микропипетку и вводят в перивителлиновое пространство между цитоплазмой ооцита и zona pellucida. Хроматиновую массу помещают рядом с мембраной ооцита, чтобы обеспечить контакт с ооцитом. Комплексы хроматиновая масса-ооцит выдерживают при температуре 20-30°C и слияние контролируют под микроскопом. После того как происходит слияние, реконструированные ооциты активируют, как описано ниже. Активация, культивирование и трансплантация реконструированных ооцитов Чтобы предотвратить экструзию полярных телец и потерю хромосом, ооцит может быть активирован в присутствии цитохалазина B, или цитохалазин B может быть добавлен сразу после активации (Wakayama et al., PNAS 96: 14984-14989, 1999; Wakayama et al., Nature Genetics 24: 108-109, 2000). Для активации реконструированных ооцитов можно использовать либо электрические, либо неэлектрические способы. Электрические способы активации клеток хорошо известны в данной области (смотри, например, патенты США No. 4994384 и 5057420). Неэлектрические способы активации клеток могут включать любой способ, известный в данной области, который увеличивает вероятность клеточного деления. Примеры неэлектрических способов активации ооцита включают инкубацию ооцита в присутствии этанола; инозитолтрифосфата; Ca++-ионофора и ингибиторов протеинкиназ; ингибитора синтеза белка; форболовых эфиров; тапсигаргина или любого компонента спермы. Другие неэлектрические способы активации включают способы, при которых ооцит подвергают холодовому шоку или механическому стрессу. Альтернативно через один-три часа после переноса ядер ооциты можно инкубировать в течение примерно шести часов в среде, содержащей Sr2+, чтобы их активировать, и цитохалазин B, чтобы предотвратить цитокинез и экструзию полярных телец (Wakayama et al., PNAS 96: 14984-14989, 1999; Wakayama et al., Nature Genetics 24: 108-109, 2000). В зависимости от типа клонируемого млекопитающего предпочтительная продолжительность активации может варьировать. Например, у домашних животных, таких как крупный рогатый скот, период активации ооцита обычно находится в пределах примерно 16-52 часов или, предпочтительно, примерно 28-42 часов. После активации ооцит помещают в среду для культивирования на соответствующий период времени, чтобы обеспечить возможность развития полученного эмбриона. На стадии двух клеток или на более поздней стадии эмбрион переносят приемной реципиентной самке для развития до родов. В случае вида коров эмбрионы обычно культивируют до стадии бластоцисты (например, в течение примерно 6-8 дней) перед переносом в материнский организм-хозяин. В случае других клонированных животных подходящая продолжительность культивирования in vitro известна специалисту в данной области и может быть определена обычным экспериментированием. Способы имплантации эмбрионов в матку млекопитающего также известны в данной области. Предпочтительно, стадия развития эмбриона коррелирует с эстральным циклом млекопитающего-хозяина. После помещения эмбриона в матку млекопитающего эмбрион может развиваться до родов. Альтернативно эмбриону дают возможность развиваться в матке до выбранного времени и затем эмбрион (или плод) извлекают, используя стандартные хирургические способы, чтобы определить состояние его здоровья и жизнеспособность. Эмбрионы от одного вида можно поместить в условия матки животного другого вида. Например, эмбрионы коровы могут развиваться в яйцеводах овец (Stice and Keefer, Biology of Reproduction 48: 715-719, 1993). Любая межвидовая взаимосвязь между эмбрионом и маткой может быть использована в способах по изобретению. Липослияние ядер с ооцитами или другими реципиентными клетками Раствор для липослияния готовят смешиванием одного или нескольких вызывающих слияние реагентов с буфером для липослияния в соответствующих пропорциях в пределах примерно от 5:1 до 1:10, как описано выше. Ядра либо свежеприготовленные, либо замороженные и оттаянные, как описано выше, смешивают с раствором для липослияния, так чтобы обеспечить покрывание ядер соединением. Инкубация происходит при температуре 20-30°C в течение примерно 10-30 минут. Микрокапли, содержащие ядра в растворе для липослияния, помещают под уравновешенное CO2 минеральное масло. Также готовят каплю, содержащую реципиентную клетку, предпочтительно лишенную ядра клетку. Лишенные ядер реципиентные клетки готовят посредством физического удаления хромосом или ядер путем микроманипуляций или посредством повреждения генетического материала при воздействии УФ-света, как описано выше. Для введения в ооциты ядра, покрытые реагентом для липослияния, собирают в микропипетку и вводят в перивителлиновое пространство между цитоплазмой ооцита и zona pellucida. Для инсерции в другие реципиентные клетки покрытые ядра, предпочтительно, помещают рядом с клеточной мембраной, чтобы обеспечить контакт с клеткой. Комплексы ядро-клетка выдерживают при температуре 20-30°C и слияние контролируют, используя микроскоп. После того как происходит слияние, реконструированные ооциты активируют, как описано ниже. ПРИМЕР 12: Применение перепрограммированных пермеабилизованных клеток для клонирования млекопитающих Клетки также можно перепрограммировать без необходимости выделения ядер или хроматиновых масс из клеток. В этом способе клетки пермеабилизуют и затем инкубируют в интерфазных или митотических перепрограммирующих средах в условиях, при которых возможен обмен факторов между средами (например, в клеточном экстракте) и клетками. Если используют интерфазную среду, то ядра в клетках остаются связанными с мембранами; если используют митотическую среду, то может происходить разрушение ядерной оболочки и конденсация хроматина. После того как ядра перепрограммируют инкубацией в данной среде, плазматическую мембрану, предпочтительно, снова делают непроницаемой с образованием интактной перепрограммированной клетки, которая содержит желаемые факторы из среды. При желании среда может быть обогащена дополнительными ядерными факторами, как описано в примере 11. Затем перепрограммированные клетки сливают с реципиентными ооцитами и эмбрионы, образованные из воссозданных ооцитов, вводят в материнские организмы млекопитающих-реципиентов для создания клонированных млекопитающих. Для получения копытного животного с мутацией в гене, кодирующем прион, и, необязательно, с геном ксеногенного Ig донорные клетки модифицируют до, во время или после перепрограммирования посредством мутации в одном или более аллелях гена, кодирующего прион, и, необязательно, посредством инсерции одной или нескольких нуклеиновых кислот, кодирующих ксеногенное антитело. При желании клетку из клонированного плода или клонированного потомка можно использовать во втором раунде переноса ядер, чтобы создать дополнительное клонированное потомство. Клетки из исходного клонированного плода или клонированного потомка также можно заморозить с образованием клеточной линии, используемой в качестве источника донорных клеток для создания дополнительных клонированных копытных животных. Пермеабилизация клеток Клетки, которые можно перепрограммировать с использованием данного способа, включают несинхронизированные клетки и клетки, синхронизированные в фазе G0, G1, S, G2 или M или в сочетании этих фаз. Клетки пермеабилизуют, используя любой стандартный способ, такой как пермеабилизация с использованием дигитонина или стрептолизина O. Коротко, клетки собирают, используя стандартные способы, и промывают PBS. Для пермеабилизации дигитонином клетки ресуспендируют в культуральной среде, содержащей дигитонин в концентрации примерно 0,001-0,1%, и инкубируют на льду в течение 10 минут. Для пермеабилизации стрептолизином O клетки инкубируют в растворе стрептолизина O (смотри, например, Maghazachi et al., FASEB J. 11: 765-774, 1997 и ссылки здесь) в течение ~15, 30 или 60 минут при комнатной температуре. После любой из инкубаций клетки промывают центрифугированием при 400 × g в течение 10 минут. Такую стадию промывки повторяют дважды ресуспендированием и осаждением в PBS. Клетки хранят в PBS при комнатной температуре вплоть до использования. Предпочтительно, пермеабилизованные клетки сразу же добавляют к интерфазной или митотической среде для перепрограммирования, которая описана ниже. Приготовление перепрограммирующей среды Чтобы получить интерфазный экстракт для перепрограммирования, культивируемые клетки в интерфазе собирают, используя стандартные способы, и промывают центрифугированием при 500 × g в течение 10 минут в конической пробирке объемом 10 мл при 4°C. Супернатант отбрасывают, а осадок клеток ресуспендируют в общем объеме 50 мл холодного PBS. Клетки центрифугируют при 500 × g в течение 10 минут при 4°C. Эту стадию промывки повторяют и осадок клеток ресуспендируют примерно в 20 объемах ледяного буфера для лизиса интерфазных клеток (20 мМ HEPES, pH 8,2, 5 мМ MgCl2, 1 мМ DTT, 10 мкМ апротинин, 10 мкМ лейпептин, 10 мкМ пепстатин A, 10 мкМ соевый ингибитор трипсина, 100 мкМ PMSF и, необязательно, 20 мкг/мл цитохалазина B). Клетки осаждают центрифугированием при 800 × g в течение 10 минут при 4°C. Супернатант отбрасывают, а осадок клеток осторожно ресуспендируют не более чем в одном объеме буфера для лизиса интерфазных клеток. Клетки инкубируют на льду в течение одного часа, чтобы позволить клеткам набухнуть. Клетки лизируют либо обработкой ультразвуком с использованием ультразвукового аппарата с наконечником, либо гомогенизацией в гомогенизаторе Даунса с использованием стеклянной ступки и пестика. Лизис клеток осуществляют вплоть до лизиса по меньшей мере 90% клеток и ядер, что можно оценить с использованием фазово-контрастной микроскопии. Время озвучивания, необходимое для лизиса по меньшей мере 90% клеток и ядер, может варьировать в зависимости от типа клетки, используемой для приготовления экстракта. Лизат клеток помещают в центрифужную пробирку объемом 1,5 мл и центрифугируют при 10000-15000 × g в течение 15 минут при 4°C, используя настольную центрифугу. Пробирки извлекают из центрифуги и сразу помещают на лед. Супернатант осторожно собирают, используя наконечник пипетки на 200 мкл, и супернатант из нескольких пробирок объединяют и помещают на лед. Полученный супернатант представляет собой «интерфазный цитоплазматический» или «IS15» экстракт. Полученный клеточный экстракт может быть разделен на аликвоты объемом 20 мкл экстракта на пробирку на льду и сразу же мгновенно заморожен в жидком азоте и храниться при -80°C вплоть до применения. Альтернативно клеточный экстракт помещают в ультрацентрифужную пробирку на льду (например, подобранную для ротора SW55 Ti; Beckman). При необходимости пробирку сверху покрывают минеральным маслом. Экстракт центрифугируют при 200000 × g в течение трех часов при 4°C, чтобы осадить мембранные пузырьки, находящиеся в экстракте IS15. В конце центрифугирования масло отбрасывают. Супернатант осторожно собирают, при необходимости объединяют и помещают в холодную пробирку объемом 1,5 мл на лед. Полученный супернатант называют «IS200» или «интерфазный цитоплазматический» экстракт. Экстракт делят на аликвоты и замораживают, как описано для экстракта IS15. При желании экстракт можно обогатить дополнительными ядерными факторами. Например, ядра можно очистить из клеток клеточного типа, из которого получают экстракт для перепрограммирования, или из клеток любого другого клеточного типа, и лизировать озвучиванием, как описано выше. Ядерные факторы экстрагируют 10-60-минутной инкубацией в буфере для ядер, содержащем NaCl или KCl в концентрации 0,15-800 мМ, при встряхивании. Лизат центрифугируют, чтобы осадить неэкстрагируемые компоненты. Супернатант, содержащий интересующие экстрагированные факторы, диализуют, чтобы удалить NaCl или KCl. Диализованный ядерный экстракт делят на аликвоты и хранят в замороженном виде. Полученный ядерный экстракт добавляют в разных концентрациях к целому клеточному экстракту, описанному выше, перед добавлением клеток для перепрограммирования. Интерфазные экстракты также могут быть приготовлены из половых клеток, таких как ооциты или мужские половые клетки. Например, ооциты активируют, как описано выше, и культивируют в течение пяти часов, чтобы создать возможность для их вступления в интерфазу. Затем ооциты обрабатывают, как описано в примере 11 для экстрактов ооцитов в метафазе II за исключением того, что EDTA исключают из лизирующего буфера. Экстракты мужских половых клеток могут быть приготовлены, как описано в примере 11. В качестве альтернативы клеточному экстракту среды для перепрограммирования также могут быть образованы добавлением одного или нескольких природных или рекомбинантных факторов (например, нуклеиновых кислот или белков, таких как ДНК-метилтрансферазы, деацетилазы гистонов, гистоны, протамины, ядерные ламины, факторы транскрипции, активаторы, репрессоры, белки ремоделирования хроматина, ростовые факторы, интерлейкины, цитокины или другие гормоны) к раствору, такому как буфер. Предпочтительно, один или несколько факторов являются специфичными для ооцитов или стволовых клеток. Перепрограммирование клеток в среде Пермеабилизованные клетки суспендируют в интерфазной перепрограммирующей среде, описанной выше, или одной из митотических перепрограммирующих сред, описанных в примере 11, в концентрации примерно 100-1000 клеток/мкл. К экстракту добавляют АТФ-генерирующую систему и ГТФ, как описано выше, и реакционную смесь инкубируют при 30-37°C до двух часов, чтобы стимулировать перенос факторов из экстракта в клетку и активировать захват факторов ядрами или их связывание с хромосомами. Перепрограммированные клетки центрифугируют при 800 × g, промывают ресуспендированием и центрифугируют при 400 × g в PBS. При желании клетки ресуспендируют в культуральной среде, содержащей 20-30% фетальной телячьей сыворотки (FCS), RPMI1640, содержащей 2 мМ CaCl2 (добавляемый из 1 М маточного раствора в H2O), или в среде Альтернативный способ перепрограммирования пермеабилизованных клеток Альтернативно клетки можно пермеабилизовать при помещении их на покровные стекла, чтобы минимизировать манипуляции с клетками и исключить центрифугирование клеток, таким образом делая максимальной выживаемость клеток. Клетки (например, фибробласты) выращивают на 16-мм покрытых поли-L-лизином покровных стеклах в RPMI1640 до 50000-100000 клеток/покровное стекло в 12-луночных планшетах. Клетки пермеабилизуют в 200 нг/мл стрептолизина O в не содержащем Ca2+ сбалансированном растворе солей Хенкса (Gibco-BRL) в течение 50 минут при 37°C в обычной атмосфере. При необходимости можно измерить процент клеток, которые были пермеабилизованы в данных условиях, на основе поглощения йодида пропидия. Стрептолизин O отсасывают; на покровные стекла наслаивают 80-100 мкл среды для перепрограммирования; и клетки инкубируют в течение от тридцати минут до одного часа при 37°C в атмосфере CO2. Среда для перепрограммирования, предпочтительно, содержит АТФ-генерирующую систему и 1 мМ каждого из АТФ, ЦТФ, ГТФ и УТФ. К плазматическим мембранам, необязательно вновь сделанными непроницаемыми, в каждую лунку добавляют среду Влияние обработки стрептолизином O на процент пермеабилизованных и вновь возвращенных к непроницаемому состоянию клеток Чтобы оценить процент пермеабилизованных и вновь возвращенных к непроницаемому состоянию клеток, осуществляли дозовые и временные титрования инкубирования со стрептолизином O (таблица 7). Пермеабилизованные клетки оценивали по поглощению 0,1 мкг/мл ДНК, окрашенной йодидом пропидия, в конце обработки стрептолизином O. Возвращение к непроницаемому состоянию оценивали сходным образом в конце обработки по возвращению к непроницаемому состоянию в отдельной группе клеток.
Оценка жизнеспособности фибробластов коров, пермеабилизованных обработкой стрептолизином O и экспонированных с митотическим экстрактом Проводили TUNEL-анализ, чтобы оценить апоптоз в клетках, пермеабилизованных с помощью 0 или 500 нг/мл стрептолизина O и возвращенных к непроницаемому состоянию, или в клетках, пермеабилизованных стрептолизином O, экспонированных с экстрактом митотических клеток в течение 30 или 60 минут и возвращенных к непроницаемому состоянию. TUNEL-позитивные клетки представляют собой клетки, подвергающиеся апоптозу (т.е. гибели клеток). Данные показывают, что стрептолизин O сам по себе не индуцирует апоптоз (таблица 8). Экспонирование обработанных стрептолизином O клеток с митотическим экстрактом в течение 60 минут, но не 30 минут, индуцирует 10% увеличение частоты апоптоза на основании TUNEL-анализа (таблица 8). На основании полученных данных 30-минутная инкубация донорных клеток в экстракте более предпочтительная, чем 60-минутная инкубация. Иммунофлуоресцентным анализом клеток показано, что тридцатиминутные инкубирования индуцируют разрушение ядерной оболочки в большинстве исследованных ядер (~90%, n > 100). Кроме того, очищенные ядра, инкубированные в экстракте и промытые в буфере N или TL-HEPES и сахарозе, как описано в примере 11 для способа переноса хроматина, не подвергаются апоптозу (2/34 и 3/47 TUNEL-позитивных соответственно).
Способ пермеабилизации, выбранный для этих способов клонирования, представлял собой 500 нг/мл SLO в течение 30 минут при 38°C. Способ возвращения к непроницаемому состоянию, выбранный для формирования интактной мембраны вокруг перепрограммированной клетки, представлял собой двухчасовую инкубацию в среде Альтернативный способ пермеабилизации клеток с использованием протеазы В приведенном в качестве примера способе пермеабилизации клеток с использованием протеазы клетки (например, фибробласты) выращивают до смыкания в монослой на чашкам диаметром 35 мм в течение ночи. Фибробласты отделяют от чашки, используя обычный способ обработки на основе трипсина-EDTA (0,5%-5,3 мМ) в течение пяти минут. Фибробласты промывают в PBS и затем ресуспендируют в 1 мл TL Hepes. Добавляют один мл раствора 3 мг/мл протеазы в TL Hepes к 1 мл суспензии клеток (конечная концентрация протеазы составляет 1,5 мг/мл) и инкубируют при комнатной температуре в течение одной минуты. Клетки промывают в 10 мл HBSS и ресуспендируют в 1 мл HBSS и подсчитывают. Примерно 105 клеток в минимальном объеме используют для инкубирования с митотическим экстрактом. Сорок мкл митотического экстракта MS-15 помещают на клетки на 30 минут при 37°C. Затем клетки промывают в TL Hepes и используют в одном из способов переноса ядер, приведенных в этом описании. Такие клетки проявляют признаки пермеабилизации, так как после инкубирования в митотическом экстракте 70-80% клеток подвергаются разрушению ядерной оболочки и преждевременной конденсации хромосом, что свидетельствует о том, что MPF (фактор, стимулирующий созревание) из экстракта или какой-либо компонент MPF прошел через мембрану. Этот способ пермеабилизации клеток можно использовать с множеством протеаз, таких как трипсин, и с разными половыми, соматическими, эмбриональными клетками плода, клетками взрослого организма, дифференцированными и недифференцированными клетками. Образование, активация, культивирование и трансплантация реконструированных ооцитов Перепрограммированные клетки вводят в реципиентные ооциты или сливают с ними, используя стандартные способы микроинъекции или электрослияния (смотри, например, патенты США No. 4994384 и 5945577). Например, клетки могут быть помещены рядом с ооцитами в стандартной среде для клеток в присутствии или отсутствии сахарозы (например, 2,5% сахарозы) и клетки могут быть втянуты в пипетку для инъекции. Затем пипеткой несколько раз проводят прокачивание, чтобы лизировать клетки и удалить цитоплазматические компоненты из ядра, которое затем инъецируют в ооцит. Реконструированные ооциты затем активируют, культивируют и трансплантируют материнским млекопитающим-реципиентам, используя стандартные способы, такие как способы, описанные в примере 11, чтобы получить клонированных млекопитающих. ПРИМЕР 13: Доказательство более полного перепрограммирования ядер с использованием переноса хроматина и переноса с использованием стрептолизина O Как показано в примерах 8-10, происходит неполное ремоделирование и перепрограммирование ядер в эмбрионах на стадии пронуклеусов с традиционными ядерными трансплантатами. Это открытие продемонстрировано на основании сборки ламинов A/C в ядерной оболочке эмбрионов с ядерными трансплантатами на стадии пронуклеусов и избытка иммунофлуоресцентного мечения NuMA. Более полного перепрограммирования ядер достигали с использованием способа переноса хроматиновой массы, описанного в примере 11, и способа пермеабилизации и перепрограммирования клеток (также называемого SLOT), описанного в примере 12. В частности, способы клонирования по данному изобретению давали эмбрионы с картинами экспрессии белка, которые больше были похожи на оплодотворенные in vitro эмбрионы, чем на клонированные эмбрионы, полученные с использованием традиционных способов клонирования. Как показано в примерах 8-10, эмбрионы с перенесенным хроматином экспрессировали намного меньше белка ламина A/C, чем эмбрионы с традиционным переносом ядер. Ламины A/C являются специфичными для соматических клеток компонентами ядерной пластинки, которые в природе экспрессируются в дифференцированных клетках, но не экспрессируются в эмбрионах. Вследствие описанного взаимодействия ламинов с факторами транскрипции, белками хроматина и ДНК вероятно, что экспрессия ламинов A/C в эмбрионах с традиционным переносом ядер стимулирует экспрессию белков, специфичных для соматических клеток, которые нежелательны для развития эмбрионов. Таким образом, эмбрионы с перенесенным хроматином по данному изобретению могут экспрессировать меньше нежелательных специфичных для соматических клеток белков, чем эмбрионы с традиционным переносом ядер. Кроме того, эмбрионы с перенесенным хроматином имеют картины экспрессии для NuMA, основного компонента ядерного матрикса, который вовлечен в регуляцию транскрипции, которые больше напоминают картины для оплодотворенных in vitro эмбрионов, чем эмбрионов с традиционным переносом ядер. Полученный результат также свидетельствует, что эмбрионы с перенесенным хроматином более эффективно перепрограммируются, чем эмбрионы с традиционным переносом ядер. Оценка разрушения ядер in vitro в случае ядер фибробластов коров Экстракты, приготовленные из митотических фибробластов коров, постоянно поддерживали разрушение ~80% исходных очищенных ядер фибробластов (фиг. 33). Экстракт из ооцитов в метафазе II (т.е. экстракт из ооцитов, естественным образом задержанных в метафазе II перед оплодотворением) также успешно поддерживал разрушение ядер (75% ядер в течение 30 минут). Исходные интерфазные ядра (фиг. 34A), хроматиновые массы, полученные из ядер, инкубированных в митотическом экстракте MS15 (фиг. 34B), и хроматиновые массы, полученные из ядер, инкубированных в экстракте ооцитов (фиг. 34C), исследовали в отношении экспрессии следующих маркеров: рецептор ламина B (LBR), интегральный белок внутренней ядерной мембраны (мембранный маркер); ламин B, повсеместный компонент ядерной пластинки; ламины A/C, специфичный для соматических клеток компонент ядерной пластинки, присутствующий только в дифференцированных клетках и отсутствующий в эмбрионах; NuMA, основной компонент ядерного матрикса; AKAP95, PKA – заякоривающий белок ядра и ДНК. Оба экстракта, соматический цитозольный экстракт MS15 и экстракт ооцитов MS15, индуцировали солюбилизацию ламина B, ламинов A/C, LBR и NuMA в ~100% исследованных единиц хроматина (фиг. 34B и 34C). Как предполагалось, AKAP95 оставался связанным с хромосомами, так же как наблюдалось в митотических клетках человека (Collas et al., J. Cell Biol. 147: 1167-1180, 1999). Этот результат был также описан в примере 8 для эмбрионов коров с ядерными трансплантатами на стадии преждевременной конденсации хроматина. Как митотический экстракт, так и экстракт ооцитов, по-видимому, были также эффективны, как интактные ооциты в стимулировании солюбилизации ядерной оболочки, независимо от применяемого способа, т.е. традиционного ядерного трансплантата, ядерной инъекции (NI) или переноса хроматина (фиг. 35). Сравнение эмбрионов на стадии пронуклеусов, полученных переносом хроматина, и пронуклеусов из эмбрионов с ядерными трансплантатами и ядерной инъекцией Чтобы создать эмбрионы с перенесенным хроматином из созревших in vitro ооцитов, удаляли ядра примерно через 18-20 часов после созревания. Ядра из интерфазных эмбриональных фибробластов коров инкубировали в митотическом экстракте MS15, который готовили из эмбриональных клеток коров, как указано в этом описании. Хроматиновые массы выделяли из экстрактов после того, как произошло разрушение ядерной оболочки и до завершения конденсации хроматина. В частности, хроматиновые массы выделяли, когда хроматин был конденсирован примерно на 50-60% по сравнению с уровнем конденсации хромосом в интерфазе (обозначаемым 0% конденсации) и максимальным уровнем конденсации хромосом в митозе (обозначаемым 100% конденсации). На данной стадии отдельные хромосомы в хроматиновой массе нельзя было различить и края хроматиновой массы имели неправильную форму. Хроматиновые массы, которые были выделены из митотического экстракта, помещали в микрокаплю TL HEPES с 2,5% сахарозы вместе с лишенными ядра ооцитами. Сахарозу добавляли к буферу, чтобы минимизировать повреждение ооцитов в результате последующей процедуры инъекции. Хроматиновые массы инъецировали в ооциты, используя скошенную на конус пипетку для микроинъекций, с использованием пьезо-сверла Burleigh (Fishers, NY) (частота 2 Гц в течение 75 микросекунд при амплитуде 70 В). Обычно проводили множественные импульсы, а именно 2, 3, 4 или 5 импульсов, так чтобы игла достаточно проникла в ооцит для инъекции. После инъекции ооциты промывали в серийных разведениях TL HEPES в сахарозе, чтобы минимизировать осмотический шок. Через 28-30 часов после созревания (т.е. 28-30 часов после того, как ооциты помещали в среду для созревания после сбора из яичников, что составляет также по меньшей мере два часа после инъекции хроматиновых масс) реконструированные ооциты и контроли партеногенетического развития активировали кальциевым ионофором (5 мкМ) в течение четырех минут (Cal Biochem, San Diego, CA) и 10 мкг/мл циклогексимида и 2,5 мкг/мл цитохалазина D (Sigma) в среде для культивирования ACM (100 мМ NaCl, 3 мМ KCl, 0,27 мМ CaCl2, 25 мМ NaHCO3, 1 мМ лактат натрия, 0,4 мМ пируват, 1 мМ L-глутамат, 3 мг/мл БСА (не содержащий жирных кислот), 1% аминокислот BME и 1% заменимых аминокислот MEM (Sigma)) в течение пяти часов, как описано ранее (Liu et al., Mol. Reprod. Dev. 49: 298-307, 1998). После активации яйцеклетки промывали пять раз и помещали в культуру в четырехлуночные чашки для культуры ткани, содержащие эмбриональные фибробласты мышей и 0,5 мл среды для культивирования эмбрионов, покрытые 0,3 мл тестированного на эмбрионах минерального масла (Sigma). В каждую лунку помещали от 25 до 50 эмбрионов и инкубировали при 38,5°C в воздушной атмосфере с 5% CO2. При необходимости к среде для культивирования можно добавить кальций (например, ~ 0,5, 1,0, 1,5, 2,0, 2,5, 3, 3,5, 5 мМ или более CaCl2) на ~ 0,5, 1,0, 1,5, 2,0, 2,5, 3,0 или более часов, чтобы стимулировать возвращение ооцитов к непроницаемому состоянию после инъекции. Возвращенные к непроницаемому состоянию ооциты, по-видимому, обладают повышенной степенью жизнеспособности благодаря интактному слою, окружающему ооциты, когда их имплантируют реципиентному млекопитающему, используя стандартные способы, приведенные здесь. Получение эмбрионов с инъекцией ядер производили, как описано выше для эмбрионов с перенесенным хроматином, за исключением того, что вместо хроматиновых масс в ооциты инъецировали ядра интерфазных эмбриональных фибробластов коров, которые не инкубировали в экстракте. Эмбрионы с ядерными трансплантатами создавали с использованием обычных способов, описанных в примере 8. В пронуклеусах при ядерной трансплантации, инъекции ядер и переносе хроматина, как и предполагалось, повторно происходит сборка ламина B (фиг. 36A, красная метка) и AKAP95 (фиг. 36B, красная метка). В пронуклеусах при ядерной трансплантации и инъекции ядер также повторно происходит сборка ламинов A/C, компонента, специфичного для соматических клеток (фиг. 36A, зеленая метка), что согласуется с результатами, описанными выше для эмбрионов с ядерными трансплантатами. Однако в пронуклеусах в случае переноса хроматина и контрольных пронуклеусах партенотов не происходит повторная сборка ламинов A/C (фиг. 36A). Пронуклеусы в случае ядерных трансплантатов также содержат NuMA (зеленая метка) в отличие от большинства пронуклеусов в случае переноса хроматина и пронуклеусов партенотов (фиг. 36B, зеленая метка). В некоторой части ядер партенотов и ядер в случае переноса хроматина на низком уровне происходит сборка NuMA, как описано выше. Происходящая in vitro разборка ядер с последующим переносом хроматина приводит к образованию пронуклеусов, которые морфологически сходны с контрольными пронуклеусами партенотов. В отличие от этого пронуклеусы в случае ядерных трансплантатов и инъекции ядер несут специфичные для соматических клеток компоненты (ламины A/C и обширное мечение NuMA). Этот результат является показателем неполного ремоделирования ядер после традиционных способов ядерной трансплантации и инъекции ядер. Как описано выше, ламины A/C, выявляемые в пронуклеусах ядерных трансплантатов и пронуклеусах при инъекции ядер, происходят из ламинов, транскрибированных de novo на стадии пронуклеусов. Так как ядерные ламины и возможно NuMA вовлечены в регуляцию транскрипции и заболевание у людей, сохранение ламинов A/C в пронуклеусах обычных ядерных трансплантатов может быть показателем неправильного функционального перепрограммирования. Авторы делают вывод, что разборка ядер in vitro и перенос хроматина дает более нормальные пронуклеусы, чем традиционные способы ядерной трансплантации или инъекции ядер. Эффективность клонирования с использованием перепрограммированных хроматиновых масс или пермеабилизованных клеток в качестве донорного источника Как описано в примере 12, был разработан способ клонирования, названный «SLOT», который заключается в индуцированной стрептолизином O (SLO) пермеабилизации первичных эмбриональных фибробластов коров, экспозиции пермеабилизованных клеток со средой для перепрограммирования (например, митотическим экстрактом) в течение 30 минут, необязательно, восстановлении непроницаемого состояния фибробластов с использованием 2 мМ кальция в культуре и переносе хроматина в ооциты с использованием стандартных способов слияния клеток. Для данного способа клонирования флакон стрептолизина O (Sigma S-5265; 25000 единиц, который хранят в порошкообразной форме для хранения при 4°C) растворяли в 400 мкл H2O и хорошо перемешивали. Все содержимое переносили в коническую пробирку объемом 15 мл и затем добавляли 3,6 мл H2O и смешивали встряхиванием. Аликвоты по 10 мкл замораживали при -20°C в концентрации маточного раствора 0,062 ед./мкл. Клетки (~100000) суспендировали в 100 мкл HBSS (Gibco BRL, кат. No. 14170-120) при комнатной температуре. Такие клетки были в состоянии смыкания в монослой и соответственно ~80-85% клеток было в фазе G1, а основанная масса других клеток была в фазе S. Добавляли маточный раствор стрептолизина O (5 мкл) (т.е. конечная концентрация 500 нг/мл или 0,3 ед./мл) и смесь инкубировали при 38°C в течение 25 минут на водяной бане. Пробирку осторожно постукивали 2-3 раза во время инкубирования, чтобы обеспечить поддержание клеток в суспензии. Добавляли PBS комнатной температуры (200 мкл) и хорошо перемешивали осторожным пипетированием. Клетки центрифугировали при 5000 об/мин в течение пяти минут при комнатной температуре в настольной центрифуге. Весь супернатант отбрасывали. На данной стадии осадок маленький и может не быть четко видимым. Добавляли митотический экстракт, содержащий АТФ-генерирующую систему (40 мкл, «MS15»), и хорошо перемешивали. Экстракт готовили во время центрифугирования клеток посредством оттаивания одного флакона с 40 мкл экстракта и добавления 1,2 мкл АТФ-генерирующей системы с хорошим перемешиванием и инкубацией при комнатной температуре. Этот митотический экстракт был таким же экстрактом, который использовали для создания хроматиновых масс в приведенном выше разделе. Смесь инкубировали при 38°C на водяной бане в течение 30 минут и пробирку иногда осторожно постукивали. Добавляли среду для восстановления непроницаемого состояния комнатной температуры (RM, 500 мкл) (полная среда Развитие эмбрионов, образованных с использованием способа SLOT и способа переноса хроматина по данному изобретению, суммировано в таблице 7. Развитие до стадии бластоцисты было несколько меньше в случае SLOT-эмбрионов по сравнению с эмбрионами в случае традиционного переноса ядер. Различия между развитием при SLOT и ядерном переносе на стадии бластоцисты может быть следствием использования большего процента клеток в фазе G1 клеточного цикла в случае переноса ядер, чем при SLOT. Степень выживаемости была ниже в случае эмбрионов с перенесенным хроматином, как и ожидалось для инвазивного способа. Частоты беременности были сравнимы для эмбрионов в случае переноса ядер и SLOT на 40 день беременности (таблица 9). Наблюдали тенденцию, что выживаемость с 40 дня по 60 день беременности была более высокой для SLOT-эмбрионов, чем для эмбрионов в случае переноса ядер, полученных с использованием традиционных способов.
Как указано выше, степень выживаемости эмбрионов в случае переноса хроматина может быть увеличена инкубацией реконструированных ооцитов в кальции в течение нескольких часов, чтобы позволить ооцитам возвратиться к непроницаемому состоянию перед введением реципиентным млекопитающим. Степени выживаемости SLOT-эмбрионов также можно повысить уменьшением количества времени от момента, когда клетки берут из культуры, и до момента, когда их сливают с ооцитами. Например, продолжительность инкубирования в стрептолизине O, инкубирования в среде для перепрограммирования и/или инкубирования в среде для восстановления непроницаемого состояния может быть уменьшена. В частности, инкубирование в среде для восстановления непроницаемого состояния может быть уменьшена примерно до одного часа или меньше. Такую укороченную обработку для восстановления непроницаемого состояния можно осуществить в присутствии 2 мМ кальция, как описано выше, или в присутствии более высоких концентраций кальция (например, ~ 2,5, 3,0, 3,5, 4,0, 4,5, 5,0 или 6,0 мМ кальция), чтобы повысить степень возращения к непроницаемому состоянию. При уменьшении количества времени, в течение которого клетки обрабатывают перед слиянием с ооцитами, менее вероятно, что клетки вступят в S-фазу и начнется репликация ДНК, которая снижает степень выживаемости реконструированных ооцитов. ПРИМЕР 14: Доказательство более полного перепрограммирования ядер с использованием переноса хроматина Как описано в примере 11, разработан способ усиления ремоделирования донорных ядер, стимулирования репрессии соматических генов и получения эмбрионов с архитектурой пронуклеусов, сходной с архитектурой оплодотворенных зигот. В конкретном примере данного общего способа переноса хроматина выделенные интактные ядра фибробластов коров инкубировали в цитоплазматическом экстракте митотических фибробластов коров в присутствии АТФ-генерирующей системы, которая требовалась для запуска разборки ядра. Чтобы получить митотический перепрограммирующий экстракт для превращения донорных ядер в хроматиновые массы, фибробласты синхронизировали в митозе, используя 0,5 мкг/мл нокодазола, в течение ~18 часов, собирали стряхиванием митотических клеток и дважды промывали в ледяном PBS и один раз в ледяном буфере для лизиса клеток (20 мМ Hepes, pH 8,2, 5 мМ MgCl2, 10 мМ EDTA, 1 мМ DTT и ингибиторы протеаз) (Collas et al., J. Cell Biol. 147: 1167-1179, 1999). Уплотненные клетки ресуспендировали в одном объеме буфера для лизиса клеток, давали возможность набухать на льду в течение одного часа и гомогенизировали в гомогенизаторе Даунса на льду, используя плотно подогнанный пестик, до тех пор, пока не произойдет лизис всех клеток. Лизат центрифугировали при 15000 × g в течение 15 минут при 4°C и супернатант (митотический экстракт) собирали, делили на аликвоты и мгновенно замораживали в жидком азоте и хранили при -80°C. Для образования донорного хроматина несинхронизированные фибробласты в состоянии смыкания в монослой собирали, промывали и ресуспендировали в ~20 объемах ледяного гипотонического буфера для выделения ядер (10 мМ Hepes, pH 7,5, 2 мМ MgCl2, 25 мМ KCl, 1 мМ DTT и смесь ингибиторов протеаз) (Collas et al., J. Cell Biol. 147: 1167-1179, 1999). После одного часа на льду клетки гомогенизировали в гомогенизаторе Даунса с плотно притертым пестиком. Добавляли сахарозу из 2 М маточного раствора до концентрация 250 мМ и ядра осаждали при 400 × g в течение 10 минут при 4°C. Ядра промывали в буфере для выделения ядер (такой же буфер, как описано выше, но с 250 мМ сахарозой) и либо использовали свежими, либо замораживали в буфере для выделения ядер/70% глицерине (Collas et al., J. Cell Biol. 147: 1167-1179, 1999). Для перепрограммирования выделенные ядра фибробластов инкубировали в 40 мкл митотического экстракта, содержащего АТФ-генерирующую систему (1 мМ АТФ, 10 мМ креатинфосфат и 25 мкг/мл креатинкиназы), в концентрации 4000 ядер/мкл в течение 30 минут на H2O-бане при 38°C. Разрушение ядерной оболочки и конденсацию хроматина контролировали фазово-контрастной микроскопией. В конце инкубирования реакционную смесь разбавляли 500 мкл TL Hepes, содержащего 2,5% сахарозы, и хроматин выделяли осаждением при 2000 × g в течение пяти минут. Хроматиновые массы ресуспендировали в TL Hepes/сахарозе и переносили в TL Hepes/сахарозу под минеральным маслом вместе с лишенными ядер ооцитами. Отдельные хроматиновые массы инъецировали в созревшие in vitro ооциты, лишенные ядер, на стадии 20 hpm с помощью скошенной на конус пипетки для микроинъекции, используя пьезо-сверло Burleigh (Fishers, NY) (2 Гц, 2 мксек, 70 В). После инъекции ооциты промывали в серийных разведениях сахарозы в TL HEPES, чтобы минимизировать осмотический шок, и культивировали. На стадии 28 hpm реконструированные ооциты и партеногенетические контроли активировали, как описано для NT, и культивировали (Kasinathan et al., Nat. Biotechnol. 19: 1176-1178, 2001 и Kasinathan et al., Biol. Reprod. 64: 1487-1493, 2001). Для ядерных инъекций (NI) очищенные ядра фибробластов экспонировали с буфером для лизиса клеток вместо митотического экстракта и инъецировали в лишенные ядер ооциты, как в случае CT. В результате перепрограммирования ламин A/C, ламин B и NuMA быстро подвергались разборке, в то время как AKAP95 оставался связанным с конденсированными хромосомами (фиг. 42A). TUNEL-анализ показал, что в экстракте не происходит апоптоза. Сходные исследования разрушения ядер, проведенные с ядрами клеток HeLa и митотическими экстрактами, показали, что конденсированные хроматиновые массы способны поддерживать реконструирование ядер (Steen et al., J. Cell Biol. 150: 1251-1562, 2000) и транскрипцию в интерфазной цитоплазме. Таким образом, разборка ядер in vitro дает конденсированный хроматин, способный вновь образовывать функциональные ядра. Конденсированный хроматин фибробластов выделяли осаждением, и отдельные хроматиновые массы инъецировали в лишенные ядер реципиентные ооциты. После возвращения в культуру ооциты активировали, как описано в примере 9 для NT-ооцитов. Для контроля артефактов, возникающих при манипуляциях с ядрами, также инъецировали интактные ядра, экспонированные только с буфером для экстракта. Результаты CT и контрольных инъекций ядер (NI) показаны на фиг. 42A-42D. NI давали пронуклеусы, которые подобно пронуклеусам NT содержали ламины A/C и B, NuMA и AKAP95 (фиг. 42B и 42C). CT приводил к образованию PN в более чем 80% эмбрионов, которые переживали инъекцию и активацию (~50% инъецированных ооцитов, n > 2000). Однако в пронуклеусах при CT не наблюдалось регистрируемого ламина A/C и наблюдалось 3-кратное снижение иммунологического мечения анти-NuMA по сравнению с пронуклеусами NT и NI (фиг. 42B и 42C). Такая картина сходна с картиной в случае партеногенетических пронуклеусов. Способность AKAP95 и ДНК подвергаться экстракции с использованием тритона X-100/ДНКазы I/NaCl, как описано выше, усиливалась в 8 раз в пронуклеусах CT по сравнению с пронуклеусами NT, отражая полезное действие CT на заякоривание AKAP95 в пронуклеусах и доступность для ДНКазы I (фиг. 42D). Так как связанный с хроматином AKAP95 фракционируется совместно с первично транскрипционно репрессированным (гипоацетилированным) хроматином, то полученный результат свидетельствует о том, что CT усиливает образование эухроматина при сборке PN. Динамику TATA-связывающего белка, TBP, исследовали в ходе ремоделирования донорных ядер фибробластов способами NT и CT, осуществляемыми параллельно. TBP облегчает сборку общего аппарата транскрипции практически для всех генов (Sharp et al., Cell 68: 819-821, 1992). TBP локализуется совместно с AKAP95 и ДНК ядрах донорных фибробластов (фиг. 43A). PCC-хромосомы, полученные после NT, содержали в 5 раз больше TBP, чем хромосомы, конденсированные в митотическом экстракте, как показано на основании отношений интенсивности флуоресценции TBP/AKAP95 и TBP/ДНК (фиг. 43B). Интенсивность мечения TPB хромосом, конденсированных in vitro, была похожа на интенсивность мечения митотических фибробластов. На PN-стадии TBP едва выявлялся в CT-эмбрионах, но демонстрировал сильную иммунореактивность в NT- и NI-эмбрионах (фиг. 43A). TBP пронуклеусов в NT-эмбрионах имел соматическое происхождение, так как ингибирование транскрипции или трансляции сохраняло интенсивное мечение TBP. Было показано, что концентрация TBP в пронуклеусах у мыши увеличивается при прохождении через интерфазу (Worrad et al., Development 120: 2347-2357, 1994). Однако сходство кинетики образования PN из PCC- или in vitro-конденсированного хроматина между реконструированными NT- и CT-эмбрионами, показало, что повышенная концентрация TBP в NT-пронуклеусах не является следствием более продвинутой стадии клеточного цикла. Таким образом, разборка in vitro донорных ядер стимулирует диссоциацию TBP из хроматина, так что полученные CT-пронуклеусы содержат в ~10 раз меньше TBP, чем NT-пронуклеусы на той же стадии реконструирования. Таким образом, пять структурных и функциональных маркеров неполного перепрограммирования при NT включают сборку в пронуклеусах ламинов A/C, повышенные концентрации NuMA и TBP в пронуклеусах и повышенную чувствительность AKAP95 и ДНК к экстракции детергентом, ДНКазой и солью. Напротив, ламины B-типа, важные для правильного образования ядра заново и для жизнеспособности клеток (Steen et al., J. Cell Biol. 153: 621-626, 2001), по-видимому, собираются нормально в NT-пронуклеусах, хотя авторы не могли определить, был ли пул собранных ламинов B-типа соматическим (и, следовательно, повторно направленно воздействующим) или имел материнское происхождение. Ген LA остается активным в NT-пронуклеусах, что приводит к сборке специфичных для дифференцированных клеток ламинов типа A. Было сделано предположение, что благодаря взаимодействиям с хроматином и аппаратом транскрипции ядерные ламины принимают участие в регуляции транскрипции (Cohen et al., Trends Biochem. Sci. 26: 41-47, 2001); таким образом, сборка правильного набора (наборов) ядерных ламинов вероятно является наиболее важной для правильного функционирования пронуклеусов в NT-эмбрионах. В данных способах соматические ядерные компоненты диспергируются в экстракте и обычно не вступают в контакт в цитоплазмой ооцита. Это препятствует повторному направлению специфичных для соматических клеток молекул в развивающееся ядро, таких как ламины B-типы, состав которых может отличаться от материнских ламинов (Cohen et al., Trends Biochem. Sci. 26: 41-47, 2001). Конденсация хроматина in vitro также может стимулировать высвобождение ДНК-связанных факторов, таких как ферменты ремоделирования хроматина (Sif et al., Genes Devel. 12: 2842-2851, 1998) и факторы транскрипции, таким образом «очищая» донорный геном от потенциальных ингибирующих соматических компонентов. В частности, удаление TBP может приводить к инактивации специфичных для соматических клеток генов в реконструированных CT-пронуклеусах и удваивать низкую транскрипционную активность мужского пронуклеуса после оплодотворения (Poccia et al., Trends Biochem. Sci. 17: 223-227, 1992). Вовлечение удаления факторов из донорного ядра может облегчить введение материнских компонентов в хроматин и ремоделирование в физиологический пронуклеус. CT увеличивает чувствительность AKAP95 и ДНК к нуклеазам и соли. Резистентная к ДНКазе I ДНК является главным образом транскрипционно молчащей; таким образом, неполное ремоделирование заякоривания AKAP95 при NT может нарушать экспрессию важных для развития генов, таких как гены, вовлеченные в развитие плаценты, поддержание поздней беременности и постнатальную жизнеспособность клонированных животных. Следующие характеристики CT-ооцитов показывают, что ядерный перенос хроматиновой массы, инкубированной в митотическом экстракте, значительно улучшает функциональные характеристики полученного реконструированного ооцита. Белок ядерного матрикса, который экспрессируется на высоких уровнях в соматических донорных клетках, NuMA, экспрессировался в 3 раза меньше в CT-пронуклеусах (т.е. пронуклеусах, образованных после введения хроматиновой массы в ооцит), чем в NT-пронуклеусах. Этот результат показывает, что уровень NuMA в CT-ооцитах больше похож на уровень NuMA в природных ооцитах, чем в NT-ооцитах. CT-пронуклеусы также экспрессировали в 10 раз меньше общего фактора транскрипции TBP, чем NT-пронуклеусы. Такое удаление TBP из донорной хроматиновой массы при инкубировании в митотическом экстракте может приводить к инактивации нежелательных специфичных для соматических клеток генов в полученном CT-ооците. Ламин A/C, белок ядерной оболочки, который является специфичным для дифференцированных клеток и не выявляется в оплодотворенных in vitro или партеногенетически активированных ооцитах, также не выявлялся в CT-пронуклеусах, но выявлялся в NT пронуклеусах. Так как ядерные ламины могут регулировать транскрипцию, отсутствие регистрируемого ламина A/C в CT-пронуклеусах может приводить к более подходящей регуляции транскрипции в CT-ооцитах, чем в NT-ооцитах. Чувствительность AKAP95, структурного белка области контакта ядерный матрикс-хроматин, которым обогащен транскрипционно репрессированный (гипоацетилированный) хроматин, и ДНК к экстракции детергентом, ДНКазой и солью измеряли в пронуклеусах, чтобы охарактеризовать заякоривание AKAP95 в ДНК и чтобы охарактеризовать доступность ДНК в пронуклеусах. Способность подвергаться экстракции повышалась в 8 раз в CT-пронуклеусах по сравнению с NT-пронуклеусами, отражая полезное влияние инкубирования донорной хроматиновой массы в митотическом экстракте на заякоривание AKAP95 и морфологическое ремоделирование донорной ДНК. Так как AKAP95 и резистентная к ДНКазе I ДНК связаны с транскрипционно репрессированной или молчащей ДНК, то повышение чувствительности AKAP95 и ДНК к нуклеазам, соли и детергенту свидетельствует о том, что CT-ооциты могут иметь повышенную экспрессию важных для развития генов. Сходные результаты ожидаются в случае донорных ядер из клеток с одной или несколькими мутациями в гене, кодирующем прион. ПРИМЕР 15: Доказательство более полного перепрограммирования ядер с использованием стрептолизин O-переноса Перепрограммирование хроматиновой массы во время инкубирования пермеабилизованной клетки в митотическом экстракте с использованием SLOT также показано на фиг. 45A и 45B. В этом исследовании экспрессию белка ядерного матрикса, который экспрессируется на высоких уровнях соматическими донорными клетками, NuMA, и экспрессию белка ядерной оболочки, который специфичен для дифференцированных клеток, ламина A/C, сравнивали для ооцитов, полученных с использованием SLOT, и ооцитов, полученных с использованием традиционного ядерного переноса клетки, содержащей ядро, которую не инкубировали в экстракте («NT»). Через четырнадцать часов после активации SLOT-пронуклеусы (т.е. пронуклеусы, образованные после введения донорной клетки, содержащей хроматиновую массу, в ооцит) экспрессировали значительно меньше NuMA и ламина A/C, чем NT-пронуклеусы (n=15-20 эмбрионов/маркер в трех повторах). Полученные результаты показывают, что реконструированные SLOT-ооциты перепрограммированы более эффективно, чем NT-ооциты и больше похожи на природные оплодотворенные ооциты. Так как ядерные ламины могут регулировать транскрипцию, то значительно более низкий уровень ламина A/C в SLOT-пронуклеусах может приводить к более подходящей регуляции транскрипции в SLOT-ооцитах, чем в NT-ооцитах. Кроме снижения экспрессии нежелательных факторов, в полученном ооците SLOT увеличивает жизнеспособность полученных плодов по сравнению с традиционными способами ядерного переноса (таблица 10). Таким образом, многие структурные и функциональные отличия SLOT-донорных клеток по существу повышают способность реконструированных ооцитов образовывать эмбрионы организмов, отличных от человека, и млекопитающих, отличных от человека.
ПРИМЕР 16: Пример доказательства более полного перепрограммирования ядер с использованием стрептолизин O-переноса (SLOT) Как описано в примерах 12 и 15, разработана новая система ремоделирования ядер in vitro. Система заключается в пермеабилизации донорных клеток, индукции конденсации хроматина в экстракте митотических клеток и промывке пермеабилизованной клетки, чтобы удалить ядерные факторы, солюбилизированные во время конденсации хроматина. Пронуклеусы эмбрионов коров с трансплантатами хроматина проявляют характер экспрессии нескольких маркеров, который очень похож на характер экспрессии нормальных эмбрионов, в противоположность эмбрионам с ядерными трансплантатами, которые сходны с соматическими клетками. Получили восемь здоровых телят с использованием системы переноса хроматина. В случае переноса хроматина наблюдалась тенденция к повышенной выживаемости до рождения, более низкой частоте появления крупных телят и значительно повышенной жизнеспособности после рождения. Результаты демонстрируют успешную обработку соматических донорных ядер перед трансплантацией. Как описано ниже, разборка соматического ядра в митотическом экстракте с последующим переносом конденсированного хроматина в ооцит усиливает ремоделирование ядер и демонстрирует доказательство улучшенного развития и жизнеспособности клонов. Этот способ при необходимости можно использовать для дальнейшей характеристики механизма перепрограммирования ядер. Способ переноса хроматина Чтобы ослабить дефекты, идентифицированные в NT-эмбрионах на стадии пронуклеусов, ядра фибробластов обрабатывали in vitro перед переносом в реципиентные ооциты. Указанная SLOT-система представлена на фиг. 48. Для образования митотического экстракта фибробласты синхронизировали в митозе с использованием 1 мкг/мл нокодазола в течение 18 часов, собирали стряхиванием митотических клеток и два раза промывали в фосфатно-солевом буфере и один раз в буфере для лизирования клеток (20 мМ Hepes, pH 8,2, 5 мМ MgCl2, 10 мМ EDTA, 1 мМ DTT и ингибиторы протеаз). Осажденные клетки ресуспендировали в одном объеме ледяного буфера для лизирования клеток, давали возможность набухнуть на льду в течение одного часа и гомогенизировали в гомогенизаторе Даунса, используя плотно притертый стеклянный пестик. Лизат центрифугировали при 15000 × g в течение 15 минут при 4°C, и супернатант (митотический экстракт) делили на аликвоты, замораживали в жидком азоте и хранили при -80°C. Использовали свежие и замороженные экстракты без заметных различий в эффективности разрушения ядер. Эмбриональные фибробласты коровы из культур, достигших смыкания в монослой, промывали в не содержащем Ca+2/Mg2+ сбалансированном растворе солей Хенкса (HBSS) и пермеабилизовали посредством инкубирования 100000 клеток в суспензии с 31,2 ед. стрептолизина O (SLO; Sigma) в 100 мкл HBSS в течение 30 минут на водяной бане при температуре примерно 38,5°C. Пермеабилизацию оценивали по поглощению непроходящей через мембрану ДНК, окрашенной йодидом пропидия (0,1 мкг/мл). Пермеабилизованные фибробласты осаждали, промывали и инкубировали в 40 мкл митотического экстракта, содержащего АТФ-генерирующую систему (1 мМ АТФ, 10 мМ креатинфосфат и 25 мкг/мл креатинкиназы), в течение 30-45 минут примерно при 38,5°C, чтобы стимулировать разборку ядер и удаление ядерных компонентов. Аликвоты метили 0,1 мкг/мл Hoechst 33342, чтобы наблюдать за конденсацией хроматина. После инкубирования фибробласты выделяли из экстракта осаждением и промывали. Реакционную смесь разбавляли 500 мкл Alpha MEM/10% фетальной телячьей сывороткой (Gibco-BRL), содержащей 2 мМ CaCl2 для возвращения мембран к непроницаемому состоянию, и клетки культивировали в течение двух часов при 38,5°C (Hakelien et al., Nat. Biotechnol. 20: 460-466, 2002). Возвращение к непроницаемому состоянию контролировали по поглощению йодида пропидия. Возвращенные к непроницаемому состоянию клетки сливали с созревшими in vitro ооцитами, из которых были удалены ядра на стадии 20 hpm; ооциты активировали в момент времени 28 hpm и эмбрионы культивировали, как описано для NT в примере 10. SLOT-эмбрионы культивировали до стадии бластоцисты in vitro и переносили по два эмбриона на реципиентную самку. Беременность контролировали ультразвуковым исследованием и делали C-сечения. Телят оценивали ветеринары в пределах 24 часов после рождения. Разрушение ядер фибробластов в митотическом экстракте Митотический экстракт состоял из надосадка 15000 × g из лизата митотических фибробластов коров и сдержал АТФ-регенерирующую систему. Экстракт не индуцировал апоптоз, что наблюдали по отсутствию протеолиза поли(АДФ)рибозилполимеразы (PARP) и фрагментации ДНК, характерной для апоптозных фибробластов (фиг. 49A). Таким образом, экстракт был подходящим для стимулирования ремоделирования соматических ядер. Экстракт вызывал АТФ-зависимую конденсацию хромосом, разборку ламинов A/C- и B-типа из хроматина, о чем судили по цитоплазматическому мечению этих ламинов и удалению TBP из хроматина (фиг. 49B). Указанные события подтверждали анализом на основе иммуноблоттинга конденсированного хроматина, очищенного из фибробластов после выделения из митотического экстракта (фиг. 49C; сравни дорожки 1 и 3). В этом эксперименте A-киназный заякоривающий белок AKAP95 использовали в качестве маркера ядерного компонента, который остается связанным с конденсированными хромосомами, как это происходит в норме в митозе (Collas et al., J. Cell Biol. 147: 1167-1180, 1999). Гистон H4 использовали в качестве контроля нагрузки белка в геле (фиг. 49C). Разборка ядерных ламинов и TBP из хроматина в митотическом экстракте зависела от АТФ-регенерирующей системы (фиг. 49B и 49C, дорожки 3 и 4) и напоминала разборку, происходящую в митотических клетках (фиг. 49C, дорожка 2). Кроме того, анализ на основе иммуноблоттинга целых пермеабилизованных фибробластов (по сравнению с изолированным хроматином) после экспозиции с митотическим экстрактом показал, что часть солюбилизированного ламина A/C и весь регистрируемый TBP удалялись из клеток и/или подвергались протеолизу (фиг. 49C, дорожка 6). Наконец контрольный экстракт из интерфазных фибробластов (фиг. 49C, дорожка 5) или только буфер для лизиса клеток, оба содержащие АТФ-регенерирующую систему, не могли стимулировать разборку ядер, свидетельствуя о том, что разрушение было АТФ-зависимым и специфичным для митотического экстракта. Пермеабилизованные фибробласты, экспонированные с митотическим экстрактом и возвращенные к непроницаемому состоянию с использованием CaCl2, можно было культивировать в течение нескольких пассажей, что свидетельствует о том, что пермеабилизация мембран, инкубация пермеабилизованных клеток в экстракте и возвращение мембран к непроницаемому состоянию являются способами, сохраняющими жизнеспособность. Характеристика ядер в эмбрионах, полученных переносом хроматина Слияние возвращенных к непроницаемому состоянию фибробластов с реципиентными ооцитами происходило также эффективно (более 70%), как и с непермеабилизованными клетками. Донорный хроматин был в конденсированной форме во время введения в ооциты (фиг. 50A, SLOT). Напротив, хроматин фибробластов, используемых для NT, был еще деконденсированным в течение 30 минут слияния (фиг. 50A, NT). Таким образом, возвращение к непроницаемому состоянию обработанных митотическим экстрактом фибробластов с использованием CaCl2 перед переносом в ооциты не стимулировало повторного образования ядра в донорных клетках. Данное наблюдение подтверждается отсутствием ядерной оболочки вокруг конденсированного хроматина в SLOT-эмбрионах сразу после слияния, о чем можно судить по анализу иммунофлуоресценции нескольких белков ядерной пластинки и внутренних белков ядерной мембраны. Результаты исследования с помощью иммунологического мечения ядерных ламинов и TBP в ядрах SLOT- и NT-эмбрионов и интенсивность иммунологического мечения этих маркеров относительно интенсивности флуоресценции ДНК показаны на фиг. 50B и 50C. Интенсивность мечения перинуклеарного ламина B была сходной в SLOT- и NT-пронуклеусах. Однако удивительно, что в отличие от NT-пронуклеусов ламин A/C не выявлялся в пронуклеусах и по меньшей мере до стадии 8-16 клеток в SLOT-эмбрионах (фиг. 50B). В SLOT-пронуклеусах также наблюдали 4-кратное снижение мечения TBP по сравнению с NT-пронуклеусами (фиг. 50C). Было показано, что концентрация TBP в пронуклеусах у мыши увеличивается по мере прохождения через интерфазу (Worrad et al., Development 120: 2347-2357, 1994). Однако, так как кинетики образования пронуклеусов из PCC- или in vitro-конденсированного хроматина сходны в NT- и SLOT-эмбрионах, мало вероятно, хотя формально и не исключено, что повышенная концентрация TBP в NT-пронуклеусах была следствием более продвинутой стадии клеточного цикла. Устойчивость TBP к экстракции 1% тритоном X-100, 1 мг/мл ДНКазы I и 0,3 М NaCl уменьшалась более чем в 2 раза (фиг. 50D), что свидетельствует о более слабой связи TBP с внутриядерными лигандами. Подобным образом устойчивость ДНК к ДНКазе I уменьшалась почти в 4 раза в SLOT-пронуклеусах, свидетельствуя о том, что SLOT способствует установлению более свободной конфигурации хроматина в пронуклеусах (фиг. 50D). Таким образом, разборка ядер фибробластов в митотическом экстракте с последующим переносом конденсированного хроматина в ооциты усиливает морфологическое ремоделирование донорных ядер и ослабляет дефекты, выявляемые в пронуклеусах NT-эмбрионов. Перенос хроматина воспроизводит здоровые клоны SLOT приводил к развитию до рождения клонированных эмбрионов (фиг. 51A и 51B). Частоты беременности после переноса бластоцист реципиентным самкам были значительно выше на 40 день в случае SLOT-эмбрионов, чем в случае NT-эмбрионов (P = 0,02; критерий Фишера), и тенденция сохранялась при развитии вплоть до рождения (12/59 и 23/211 рожденных телят соответственно; P = 0,05) (фиг. 51A). Кроме того, SLOT повышал степень выживаемости клонов свыше 24 часов после родов (10/59 и 17/211 соответственно; P = 0,04) (фиг. 51A и 51B). Состояние здоровья рожденных NT-клонов (пример 10) и SLOT-клонов оценивали по баллам в отношении животных и плацент по шкале от 1 (нормальный) до 5 (сильно аномальный). Оценка плаценты включала такие параметры, как отек плаценты, количество котиледонов, размер и морфология, окраска амниотической жидкости, морфология матки и пупок. Оценка животных включала функциональную оценку и общий вид дыхательной, сердечно-сосудистой, пищеварительной, мочевой, мышечной, скелетной и нервной систем. Данные для отдельных телят показаны на фиг. 51C. Анализ графиков в виде прямоугольной диаграммы показывает, что оценки животных и плацент, полученных NT, были более разбросаны, чем оценки, полученные при SLOT (фиг. 51E). Средняя масса при рождении SLOT- и NT-клонов различалась не существенно; однако доля SLOT-телят свыше 45 кг при рождении была ниже (P = 0,02; Фишер), чем доля таких NT-животных (фиг. 51D и 51E). Вместе полученные результаты свидетельствуют о том, что перенос хроматина дает живое потомство, и демонстрируют доказательство улучшенного развития и жизнеспособности. В сравнении с NT (пример 10) и несмотря на ограниченное количество полученного SLOT-потомства (n=8), SLOT значительно повышает частоту беременности на 40 день (P = 0,02) и жизнеспособность рожденных телят свыше 24 часов (P = 0,04). Тенденция к улучшению состояния здоровья животных, полученных посредством SLOT, также отражается в анализе графиков в виде прямоугольных диаграмм. Кроме того, более низкая частота крупных (более 45 кг) телят, получаемых посредством SLOT, имеет практические и экономические последствия для содержания животных. В NT-эмбрионах идентифицировано несколько ядерных дефектов, включая сборку ламина A/C, повышенное содержание TBP в пронуклеусах и повышенную устойчивость ДНК к ДНКазе I. Эти дефекты могут быть результатом неполного ремоделирования ядер фибробластов и/или результатом неправильной регуляции экспрессии специфичных для дифференцированных клеток генов (например, ламина A). Ремоделирование ядер in vitro и трансплантация конденсированного хроматина в ооциты ослабляет указанные дефекты. Ремоделирование ядер посредством SLOT повышает чувствительность ДНК к ДНКазе I и может стимулировать образование транскрипционно активного (или потенциально активного) хроматина. Этот эффект может в свою очередь облегчать экспрессию важных для развития генов, таких как гены, вовлеченные в развитие плаценты, сохранение поздней беременности и постнатальную жизнеспособность. SLOT также индуцирует репрессию экспрессии гена ламина A в клонированных эмбрионах. Манипуляции in vitro и in vivo с составом ядерной пластинки показал, что неспособность к сборке правильного набора ламинов без исключения приводит к апоптозу (Steen et al., J. Cell Biol. 153: 621-626, 2001). Кроме того, так как ламины взаимодействуют с ДНК, хроматином и аппаратом транскрипции, то правильная реконструкция ядерной пластинки, вероятно, важна для нормального функционирования ядра (Gruenbaum et al., J. Struct. Biol. 129: 313-323, 2000 и Cohen et al., Trends Biochem. Sci. 26: 41-47, 2001) в клонированных эмбрионах. Конденсация хроматина в митозе или in vitro связана с высвобождением ДНК-связанных компонентов, таких как ферменты ремоделирования хроматина, факторы транскрипции (например, TBP) или другие потенциально ингибирующие соматические компоненты. Удаление TBP из донорного соматического хроматина может способствовать репрессии или понижающей регуляции специфичных для соматических клеток генов в SLOT-эмбрионах, которые могут нарушать развитие. Вовлечение удаления факторов из донорного ядра является условием, которое может облегчить введение материнских компонентов в хроматин и последующее ремоделирование в физиологический пронуклеус. В заключение, существует возможность прямо ремоделировать соматическое ядро в клеточном экстракте и получать живое потомство. Обработка ядер in vitro в целях клонирования или трансдифференцировки (Landsverk et al., EMBO Rep. 3: 384-389, 2002 и Hakelien et al., Nat. Biotechnol. 20: 460-466, 2002) является полезным средством для необязательного дальнейшего исследования механизмов перепрограммирования ядер. Перенос хроматина представляет доказательство улучшенного развития до родов и жизнеспособность клонов. Дополнительная обработка системы может привести к дальнейшему повышению эффективности клонирования млекопитающих. ПРИМЕР 17: Дополнительное доказательство более полного перепрограммирования ядер с использованием стрептолизин O-переноса (SLOT) Способ SLOT, описанный в примере 12, давал улучшенные результаты в том случае, когда донорные пермеабилизованные клетки не возвращали к непроницаемому состоянию перед генетическим переносом. Кроме того, применение донорных клеток в фазе G1 вместо клеток на стадии смыкания в монослой может приводить к повышенной жизнеспособности реконструированного ооцита и полученного эмбриона. Указанные эксперименты описаны ниже. Приготовление буферов 1-Молярный раствор DTT готовили, помещая 1 мл H2O в пробирку Эппендорф, добавляя 154 мг охлажденного DTT и хорошо перемешивая встряхиванием. Раствор делили на аликвоты объемом 10 мкл и замораживали при -20°C. Аликвоты можно оттаивать и повторно замораживать несколько раз. Объем 100 мл буфера для лизиса клеток в случае приготовления клеточных экстрактов для анализа сборки/разборки ядер содержал NaCl (50 мМ, 1 мл 5 М маточного раствора), MgCl2 (5 мМ, 0,5 мл 1 М маточного раствора), Hepes, pH 8,2 (20 мМ, 2 мл 1 М маточного раствора, pH 8,2) и H2O (96,5 мл). Буфер делили на аликвоты и хранили при 4°C. При приготовлении лизата имело место падение pH на ~1 единицу pH. Для приготовления митотических экстрактов добавляли 10 мМ EGTA (1 мл 1 М маточного раствора) и добавляли 95,5 мл H2O вместо 96,5 мл. Перед применением делали следующие добавки: DTT (1 мкл/мл раствора 1 М маточного раствора при -20°C, конечная концентрация 1 мМ), PMSF (10 мкл/мл раствора 100 мМ маточного раствора, конечная концентрация 1 мМ), смесь CAL (10 мкл смеси CAL при -20 Чтобы приготовить маточный раствор стрептолизина O (SLO), флакон SLO (Sigma S-5265; 25000 единиц, который хранят в виде порошка при 4°C) растворяли в 400 мкл H2O и хорошо перемешивали. Все содержимое переносили в коническую пробирку объемом 1,5 мл, делили на аликвоты по 10 мкл и замораживали при -20°C. Концентрация маточного раствора была «10X». Чтобы приготовить раствор протеазы, 3 мл TL Hepes и 9 мг протеазы (Sigma P-8811) добавляли в коническую пробирку объемом 15 мл и перемешивали встряхиванием. Раствор фильтровали через фильтр 0,22 мкм для шприца непосредственно в TL Hepes. Так как клетки удваивали объем, конечная концентрация составляла 1,5 мг/мл. Для HECM Hepes объединяли NaCl (114 мМ, 6,662 г), KCl (3,2 мМ, 0,239 г), CaCl2·2H2O (2,0 мМ, 0,294 г), MgCl2·6H2O (0,5 мМ, 0,102 г), Pen/Strep (10 мл, Sigma P3539 Pen/Strep, конечная концентрация 100 ед./мл и 100 мкг/мл), феноловый красный (5 мкг/мл, 1 мл) и H2O (в достаточном количестве, чтобы увеличить общий объем до 990 мл). Затем добавляли 100X A.A. (10 мл), лактат Na (10 мМ, 1,44 мл), пируват Na (0,1 мМ, 0,011 г), NaHCO3 (2 мМ, 0,168 г) и HEPES (10 мМ, 2,38 г). Конечный раствор имел осмомолярность 260-270 мосмоль. Затем добавляли 3 г бычьего сывороточного альбумина (фракция V) и pH доводили до 7,4. Раствор фильтровали через фильтр 0,22 мкм и хранили при 4°C. Для приготовления маточного раствора АТФ (Sigma A3377: 100 мМ маточный раствор, 100x) объединяли H2O (1 мл) и АТФ (0,055 г) и аликвоты по 10 мкл замораживали при -20°C. Для приготовления креатинфосфата (Sigma P7936: 1 М маточный раствор, 100x) объединяли H2O (1 мл) и креатинфосфат (0,255 г) и аликвоты по 10 мкл замораживали при -20°C. Чтобы приготовить креатинкиназу (Sigma C3755: маточный раствор 2,5 мг/мл, 100x) объединяли H2O (1 мл) и креатинкиназу (0,0025 г) и аликвоты по 10 мкл замораживали при -20°C. Для приготовления АТФ-генерирующей системы готовили равные части 100 мМ маточного раствора АТФ, маточных растворов 1 М креатинфосфата и 2,5 мг/мл креатинкиназы (100x), используя H2O, смешивали и хранили в виде замороженного раствора. АТФ-генерирующую систему хранили на льду вплоть до применения. АТФ-генерирующую систему (1,2 мкл) добавляли к экстракту (40 мкл) и раствор смешивали встряхиванием. Конечная концентрация АТФ-генерирующей системы в экстракте составляла 1 мМ АТФ, 10 мМ креатинфосфат и 25 мкг/мл креатинкиназы. Приготовление митотического экстракта Один флакон, содержащий от 1,5 до 2 миллионов клеток, размораживали. Клетки делили на два флакона T75 и выращивали в течение двух дней или вплоть до их смыкания в монослой. Клетки пересевали в 6-8 флаконов T75 и выращивали до смыкания. Клетки обрабатывали трипсином и подсчитывали, используя гемоцитометр, и 3 миллиона клеток добавляли в каждый из такого количества флаконов T150, которое можно было использовать с имеющимся количеством клеток. Клеточную линию (например, фибробласты, такие как первичные фибробласты, эпителиальные клетки или иммортализованные и не пораженные заболеванием клетки, такие как клетки MDBK) синхронизировали при смыкании на 70-80% в митозе с использованием 0,5-1 мкг/мл нокодазола в течение 17-20 часов, используя стандартные способы (например, Collas et al., J. Cell Biol. 147: 1167-1180, 1999 и представленные здесь ссылки). Синхронизированные клетки собирали стряхиванием митотических клеток. Каждый флакон, содержащий клетки, энергично встряхивали многократно, тряся одной рукой. Митотические клетки откреплялись и всплывали в культуральную среду. Собранные клетки центрифугировали при 500 × g в течение 10 минут в конической пробирке объемом 50 мл при 4°C. Супернатант отбрасывали и клеточные осадки ресуспендировали в общем объеме 50 мл холодного фосфатно-солевого буфера, не содержащего Ca/Mg (PBS). При необходимости несколько клеточных осадков можно объединить в одной 50-мл пробирке. Клетки центрифугировали при 500 × g в течение 10 минут при 4°C и повторяли описанную выше стадию промывки. Определяли объем клеточного осадка и клеточный осадок ресуспендировали примерно в 20 объемах ледяного буфера для лизиса клеток, содержащего ингибиторы протеаз (т.е. DTT и PMSF). Затем клетки осаждали при 500 × g в течение пяти минут при 4°C. Супернатант отбрасывали и определяли объем клеточного осадка. Клеточный осадок ресуспендировали не более чем в одном объеме буфера для лизиса клеток, содержащего все ингибиторы протеаз. Клетки инкубировали на льду в течение одного часа, чтобы позволить клеткам набухнуть. Используя ультразвуковой аппарат с наконечником, суспензию клеток озвучивали вплоть до того, как все клетки были вскрыты. Лизис клеток контролировали в фазово-контрастном микроскопе. Желательно, перед переходом к следующей стадии лизировали 90% клеток. Озвучивание можно продолжать сколько это необходимо; время озвучивания варьирует в зависимости от типа клеток, используемых для приготовления экстракта. Альтернативно клетки лизируют гомогенизацией по Даунсу, используя стеклянную ступку и пестик (гомогенизатор), желательно вплоть до лизиса по меньшей мере 90% клеток. Лизат клеток помещали в центрифужную пробирку объемом 1,5 мл и центрифугировали при ~15000 × g в течение 15 минут при 4°C, используя настольную центрифугу с охлаждением. Центрифугу помещали в холодной комнате или в холодильнике и давали возможность для уравновешивания температуры. Пробирки извлекали из центрифуги и сразу же помещали на лед. Супернатант осторожно собирали, используя наконечник пипетки на 200 мкл. Этот супернатант является митотическим цитоплазматическим экстрактом. Экстракт помещали в другую пробирку на лед. Экстракты, собранные из нескольких пробирок, объединяли. На данной стадии существует две альтернативы. Клеточный экстракт делили на аликвоты в пробирки на льду, 41 мкл экстракта на пробирку. Экстракт сразу же мгновенно замораживали в жидком азоте и хранили в холодильнике при -80°C вплоть до использования. Такие экстракты, приготовленные центрифугированием при 15000 × g, названы MS15 (или митотический цитоплазматический экстракт). Альтернативно экстракт MS15 помещают в ультрацентрифужную пробирку на льду (например, подходящую для ротора SW55 Ti; Beckman). Сверху пробирки наслаивают минеральное масло, если необходимо предотвратить сплющивание пробирки при ультрацентрифугировании. Экстракт центрифугировали при 200000 × g в течение трех часов при 4°C, чтобы осадить мембранные пузырьки, находящиеся в MS15. В конце центрифугирования масло отбрасывали. Супернатант осторожно собирали и помещали в холодную пробирку объемом 1,5 мл на льду. При необходимости использовали несколько супернатантов. Этот супернатант называют MS200 (или митотический цитозольный экстракт). Экстракт MS200 делили на аликвоты и замораживали, как описано для экстракта MS15. Перенос хроматина За день до процедуры клонирования готовили одну чашку Nunc диаметром 35 мм с 1 миллионом фибробластов или готовили шесть флаконов T75, содержащих 500000, для стряхивания. Утром в день клонирования полную среду альфа-MEM с 15% облученной FBS во всех флаконах заменяли, чтобы удалить обломки и погибшие клетки. Перед осуществлением пермеабилизации клеток и взаимодействием с митотическим экстрактом наименьшую концентрацию SLO, необходимую для того, чтобы пермеабилизовать 80-90% клеток, определяли посредством серийного разведения маточного раствора SLO (например, разведения 1X, 0,5X, 0,3X и 0,1X), инкубирования в течение 30 минут примерно при 38,5°C на водяной бане и затем окраски йодидом пропидия. Клетки подвергали диссоциации из культуры, слитой в монослой, или культуры вновь трансфицированных клеток, используя трипсин или буфер для диссоциации клеток, помещали в коническую пробирку объемом 15 мл и промывали один раз центрифугированием, используя сбалансированный солевой раствор Хенкса, не содержащий Ca/Mg (HBSS). Осадок клеток ресуспендировали в 1 мл HBSS и клетки подсчитывали, чтобы определить концентрацию. Примерно 50000-100000 клеток суспендировали в 100 мкл HBSS (Gibco BRL, кат. No. 14170-120) при комнатной температуре. Добавляли 5 мкл маточного раствора SLO с предварительно определенной концентрацией. Смесь инкубировали примерно при 38,5°C в течение 30 минут на водяной бане. Пробирку осторожно постукивали 2-3 раза во время инкубирования, чтобы гарантировать, что клетки оставались в суспензии. Добавляли 200 мкл объем PBS (не содержащего Ca/Mg) комнатной температуры и хорошо перемешивали осторожным пипетированием. Клетки центрифугировали при 500 × g в течение пяти минут при комнатной температуре в настольной центрифуге. Весь супернатант отбрасывали. Осадок был маленьким и мог быть неясно видимым. Добавляли 40 мкл объем митотического экстракта, содержащего АТФ-генерирующую систему, и раствор хорошо перемешивали. Экстракт готовили в ходе 30-минутного инкубирования, описанного выше. Один флакон с 40 мкл экстракта размораживали и добавляли к 1,2 мкл АТФ-генерирующей системы. Раствор хорошо перемешивали и хранили при комнатной температуре. Смесь инкубировали при 38,5°C на водяной бане в течение 30 минут и пробирку иногда осторожно постукивали. Добавляли 500 мкл объем полной среды комнатной температуры (альфа-MEM + 15% фетальной телячьей сыворотки) и хранили в инкубаторе примерно при 38,5°C с открытой крышкой вплоть до использования. Клетки центрифугировали при 500 × g в течение пяти минут при комнатной температуре в настольной центрифуге. Осадок клеток ресуспендировали в 1 мл TL Hepes, имеющего комнатную температуру, и переносили в коническую пробирку объемом 15 мл. Добавляли 1 мл объем раствора 3 мг/мл протеазы в TL Hepes (профильтрованного) до конечной концентрации протеазы в клетках 1,5 мг/мл, и смесь инкубировали в течение 30 секунд. Добавляли TL Hepes, чтобы наполнить коническую пробирку объемом 15 мл, и пробирку закрывали и центрифугировали при 2300 об/мин в течение пяти минут. TL Hepes удаляли из клеток и к клеткам добавляли 150 мкл TL Hepes. На пробирке помечали соответствующую линию клеток и помещали на подогревающую платформу. Во время подготовки клеток готовили рабочее место для переноса клеток с использованием пипетки для переноса клеток соответствующего размера. Примерно 50 лишенных ядер ооцитов помещали в каплю TL Hepes объемом 50 мкл под вязким минеральным маслом в чашке диаметром 100 мм. Промытые клетки помещали в каплю с лишенными ядер ооцитами. Использовали количество клеток, достаточное для генетического переноса во все ооциты. Важно было избежать такого большого количества клеток, когда они стерически блокируют средства для манипуляций. Каждый лишенный ядра ооцит помещали на держателе и одну клетку помещали в перивителлиновое пространство под zona, так чтобы клетка соприкасалась с плазматической мембраной ооцита. Чтобы облегчить слияние, клетку помещали вблизи или напротив полярного тельца. После того как во всех лишенных ядер ооцитах были перенесенные к ним клетки, ооциты извлекали из микрокапли и помещали в предварительно нагретую чашку диаметром 35 мм с HECM Hepes для слияния. Результаты Сравнение результатов в таблице 12 ниже для донорных клеток, в которых мембраны не были возвращены к непроницаемому состоянию перед генетическим переносом, с результатами в таблице 11 для донорных клеток, в которых мембрану возвращали к непроницаемому состоянию перед переносом генетического материала, свидетельствует о том, что можно повысить эффективность клонирования, не возвращая пермеабилизованные клеточные мембраны к непроницаемому состоянию. Кроме того, таблица 12 показывает, что применение донорных клеток в фазе G1 вместо клеток в состоянии смыкания в монослой может приводить к повышенной жизнеспособности реконструированного ооцита и полученного эмбриона. Данные в таблице 11 получали с использованием экстракта из первичных фибробластов коров и донорных эмбриональных фибробластов коров. Клетки MDBK также успешно использовали для образования экстрактов для перепрограммирования.
ПРИМЕР 18: Способы создания химерных млекопитающих Предполагается, что многие спонтанные выкидыши, которые происходят в случае использования традиционных способов клонирования млекопитающих, являются результатом аномалий плаценты, а не проблем, связанных с плодом. Соответственно разработаны способы получения химерных эмбрионов с плацентарной тканью главным образом из одного источника (например, из эмбриона, оплодотворенного in vitro, природного или партеногенетически активированного эмбриона) и тканью плода в основном из другого источника (например, эмбриона с перенесенным ядром, кодирующего ксеногенное антитело). Химерные эмбрионы с плацентарной тканью, главным образом полученной из клеток от оплодотворенных in vitro, природных или партеногенетически активированных эмбрионов, могут быть больше похожи на природную плацентарную ткань и обеспечивают повышенное продуцирование жизнеспособного потомства. Предпочтительно, большинство клеток потомства получены из клеток эмбриона с перенесенным ядром и, таким образом, имеют геном, который по существу идентичен геному донорной клетки, используемой для создания эмбриона с перенесенным ядром. В одном таком способе клетки из оплодотворенного in vitro эмбриона инъецируют в периферию компактного эмбриона, кодирующего ксеногенное антитело (например, между zona pellucida и самим эмбрионом), который получают с использованием традиционных способов переноса ядер или любым другим способом клонирования из приведенных в этом описании способов. В альтернативном способе клетки из предкомпактного оплодотворенного in vitro эмбриона инкубируют с клетками из предкомпактного эмбриона, кодирующего ксеногенное антитело, полученного с использованием одного из способов клонирования по данному изобретению (например, с использованием в качестве донорного источника перепрограммированной хроматиновой массы или пермеабилизованной клетки), в условиях, при которых возможна реорганизация клеток из каждого эмбриона с образованием одного химерного эмбриона (Wells and Powell, Cloning 2: 9-22, 2000). В обоих способах клетки из оплодотворенного in vitro эмбриона, предпочтительно, включаются в плаценту, а клетки, полученные способом переноса ядер, предпочтительно, включаются в ткань плода. Эти способы дополнительно описаны ниже. Указанные результаты получали с использованием эмбрионов с перенесенными ядрами, которые не содержат гена ксеногенного антитела; однако сходные результаты ожидаются для эмбрионов с перенесенными ядрами, содержащих ген ксеногенного антитела и/или содержащих мутацию в гене, кодирующем прионный белок. Выделение G1-фибробластов Для выделения G1-фибробластов в качестве донорных клеток для получения эмбрионов с перенесенными ядрами использовали описанный ранее способ «стряхивания» (Kasinathan et al., Nature Biotech. 19: 1176-1178, 2001). Коротко, за 24 часа до выделения 5,0 x 105 клеток высевали в чашки для культуры ткани диаметром 100 мм, содержащие 10 мл Трансплантация ядер, активация и культивирование эмбрионов Способ переноса ядер с использованием изолированных G1-фибробластов осуществляли по существу, как описано ранее (Cibelli et al., Nature Biotech. 16: 642-646, 1998; Kasinathan et al., Biol. Reprod. 64: 1487-1493, 2000). Из созревших in vitro ооцитов удаляли ядра примерно через 18-20 часов после созревания и удаление хромосом подтверждали мечением бис-бензимидом (Hoechst 33342, Sigma) в УФ-свете. Компоненты пары цитопласт-донорная клетка сливали, используя одиночный электрический импульс 2,4 кВ/см в течение 20 микросекунд (манипулятор Electrocell 200, Genetronics, San Diego, CA). Через 30 часов после созревания реконструированные ооциты и контроли активировали кальциевым ионофором (5 мкМ) в течение четырех минут (Cal Biochem, San Diego, CA) и 10 мкг циклогексимида и 2,5 мкг цитохалазина D (Sigma) в культуральной среде ACM (100 мМ NaCl, 3 мМ KCl, 0,27 мМ CaCl2, 25 мМ NaHCO3, 1 мМ лактат натрия, 0,4 мМ пируват, 1 мМ L-глутамин, 3 мг/мл БСА (не содержащий жирных кислот), 1% аминокислот BME и 1% заменимых аминокислот MEM; все реагенты из Sigma) в течение шести часов, как описано ранее (Liu et al., Mol. Reprod. Dev. 49: 298-307, 1998; Presicce et al., Mol. Reprod. Dev. 38: 380-385, 1994). После активации яйцеклетки пять раз промывали в забуференной HEPES среде для культивирования эмбрионов хомячка (HECM-HEPES, 114 мМ NaCl, 3,2 мМ KCl, 2 мМ CaCl2, 10 мМ лактат натрия, 0,1 мМ пируват натрия, 2 мМ NaHCO3, 10 мМ HEPES и 1% аминокислот BME; Sigma) и помещали в культуру в 4-луночные чашки для культуры ткани, содержащие эмбриональные фибробласты мыши и 0,5 мл среды для культивирования эмбрионов, покрытые 0,2 мл минерального масла, проверенного на эмбрионах (Sigma). От двадцати пяти до 50 эмбрионов помещали в каждую лунку и инкубировали при 38,5°C в атмосфере воздуха с 5% CO2. На четвертый день к среде для культивирования добавляли 10% FCS. На седьмой и восьмой день регистрировали развитие до стадии бластоцисты. Оплодотворение коров in vitro Оплодотворение коров in vitro осуществляли, как описано ранее, для получения оплодотворенных in vitro эмбрионов коров (Collas et al., Mol. Reprod. Devel. 34: 224-231, 1993). Готовили градиент 45% и 90% изотоничного перкола, используя маточный раствор TL для спермы (Parrish et al., Theriogenology 24: 537-549, 1985). Замороженную и оттаянную сперму крупного рогатого скота от одного быка наносили на градиент и центрифугировали в течение 30 минут при 700 × g (2000 об/мин с использованием радиуса у вершины 6,37 дюймов). Определяли концентрацию сперматозоидов в осадке и сперматозоиды разбавляли в TL для сперматозоидов (маточный раствор TL для сперматозоидов, 1 мМ пируват, 6 мг/мл БСА и 1% PS) так, чтобы конечная концентрация при оплодотворении составляла 106 сперматозоидов/мл. Через 22 часа после созревания ооциты три раза промывали в TL HEPES и помещали в 480 мкл среды для оплодотворения TL (Bavister et al., Biol. Reprod. 28: 235-247, 1983) в лунки Nunc, содержащие 6 мг/мл БСА, 0,2 мМ пируват, 20 мкМ пеницилламин, 10 мкМ гипотаурин, 1 мМ эпинефрин (Leibfried et al., J. Reprod. Fertil. 66: 87-93, 1982) и 0,004 мкг/мл гепарина. Добавляли двадцать микролитров сперматозоидов, чтобы создать конечную концентрацию 106 сперматозоидов/мл на 50 ооцитов. Условия культивирования были такими же, как условия, описанные выше для переноса ядер. Частоты оплодотворения составляли более 90% на основании развития пронуклеусов. Химерные эмбрионы с перенесенными ядрами Оплодотворенные in vitro эмбрионы на стадии 8 клеток (6-12 бластомеров) собирали примерно через 96 часов после оплодотворения, перед компактизацией. Zona pellucida удаляли протеазой (3 мг/мл в TL-HEPES). За распадом zona внимательно наблюдали, используя препаровальную лупу. Когда появлялись первые признаки того, что zona растворяется (~ две минуты), эмбрионы извлекали и промывали в TL-HEPES и переносили чашки в Петри диаметром 30 мм, содержащие раствор сбалансированных солей Хенкса, и инкубировали при 37,5°C в течение 30 минут. Бластомеры из этих предкомпактных эмбрионов переносили в микрокапли (50 мкл) TL-HEPES под минеральным маслом в чашку Петри диаметром 100 мм. На четвертый день отбирали эмбрионы с перенесенными ядрами в стадии 8-16 клеток и переносили в те же самые микрокапли, содержащие бластомеры. Указанные эмбрионы с перенесенными ядрами включали как предкомпактные эмбрионы (например, эмбрионы на стадии 8 клеток), так и компактные эмбрионы (например, эмбрионы на стадии 16 клеток). Затем 4-6 бластомеров переносили в эмбрионы с перенесенными ядрами конической микропипеткой (диаметром 35 мкм), используя стандартные способы микроманипуляции. После переноса бластомеров эмбрионы культивировали, как описано для эмбрионов с перенесенными ядрами. На седьмой и восьмой день оценивали развитие химерных эмбрионов до бластоцисты. Бластоцисты также анализировали в присутствии красителя мембран DiI, который добавляли к клеткам из оплодотворенного in vitro эмбриона перед тем, как их инъецировали в эмбрион с перенесенным ядром. Клетки метили на четвертый день и наблюдали на седьмой день. Этот краситель сохранялся в течение нескольких клеточных делений в потомстве исходно окрашенной клетки, позволяя анализировать химерный эмбрион после нескольких делений клеток. На основании данного анализа клетки из оплодотворенного in vitro эмбриона включались в химерный эмбрион. При необходимости можно осуществить флуоресцентный анализ гибридизации in situ (FISH) с зондом, специфичным по отношению к нуклеиновой кислоте либо в оплодотворенном in vitro эмбрионе, либо в эмбрионе с перенесенным ядром, используя стандартные способы (смотри, например, Ausubel et al., Current Protocols in Molecular Biology, John Wiley and Sons, New York, pp. 14.7.1-14.7.12, 1995). Данный FISH-анализ можно использовать для определения распределения клеток, полученных из каждого эмбриона, в химерном эмбрионе (например, чтобы определить, какой процент клеток включен во внутреннюю клеточную массу и какой процент включен в трофэктодерму) при его культивировании in vitro и в плоде или потомстве, полученном из эмбриона. Альтернативно можно добавить репортерный ген, такой как ген зеленого флуоресцирующего белка, к клеткам из одного эмбриона и использовать для наблюдения за включением клеток в плаценту и различные эмбриональные ткани химерного эмбриона. Перенос эмбрионов На 7 и 8 день бластоцисты с перенесенными ядрами 1 и 2 сорта, полученные из эмбрионов с перенесенными ядрами и химерных эмбрионов с перенесенными ядрами, переносили синхронизированным на 6 и 7 день реципиентным телочкам. Реципиентов синхронизировали, используя однократную инъекцию лутализа (Pharmacia and Upjohn, Kalamazoo, MI) с последующей регистрацией эструса. Реципиентов обследовали на 30 день и 60 день после переноса эмбрионов ультрасонографией в отношении присутствия оплодотворенного яйца, а после этого каждый 30 дней ректальной пальпацией вплоть до 240 дней. Результаты относительно беременности на 40 день для химерных эмбрионов и для контрольных эмбрионов, полученных слиянием трансгенного фибробласта коров с ооцитом, сравниваются в таблице 13. Полученные результаты свидетельствуют, что вплоть до 40 дня выживало большее количество химерных эмбрионов.
Альтернативные способы получения химерных эмбрионов Можно использовать стандартные способы, чтобы модифицировать описанный выше способ получения химерных эмбрионов. Например, природный эмбрион можно хирургически выделить из млекопитающего (например, коровы), или ооцит можно партеногенетически активировать, используя стандартные способы, и использовать вместо оплодотворенного in vitro эмбриона. При желании можно инъецировать меньше клеток из оплодотворенного in vitro эмбриона, природного эмбриона или партеногенетически активированного эмбриона (например, 1, 2, 3, 4 или 5 клеток) в эмбрион с перенесенным ядром, чтобы уменьшить процент инъецированных клеток и их потомства, которые будут включены в эмбриональную ткань. Альтернативно можно инъецировать больше клеток (например, 6, 7, 8, 9, 10, 11 или более клеток), чтобы увеличить процент инъецированных клеток и их потомства, которые включаются в плацентарную ткань. Кроме того, можно использовать клетки из эмбрионов на других клеточных стадиях. Например, оплодотворенные in vitro, природные или партеногенетически активированные эмбрионы на стадии 4, 8, 16, 32, 64, 128, 256, 512 клеток или более поздней клеточной стадии можно инъецировать в эмбрионы с перенесенным ядром на стадии 4, 8, 16, 32, 64, 128, 256, 512 клеток или более поздней клеточной стадии. Инъецированные клетки и эмбрионы с переносом ядра могут быть на одной и той же клеточной стадии или на разных клеточных стадиях. В одном варианте осуществления оплодотворенный in vitro, природный или партеногенетически активированный эмбрион имеет увеличенную плоидность (например, содержание ДНК 4n) по сравнению с эмбрионом с перенесенным ядром, что далее смещает инъецированные клетки к трофэктодерме (т.е. самому крайнему слою клеток эмбриона, который главным образом образует плацентарную ткань). При необходимости может быть сохранена вся или часть zona pellucida, окружающая инъецированные клетки, а не удалена перед инъекцией. В других альтернативных способах клетки из предкомпактного или компактного оплодотворенного in vitro, природного или партеногенетического эмбриона инкубируют с клетками из предкомпактного эмбриона с перенесенным ядром в условиях, при которых возможна реорганизация клеток из каждого эмбриона с образованием одного химерного эмбриона (Wells and Powell, Cloning 2: 9-22, 2000). Предполагается, что клетки из оплодотворенного in vitro, природного или партеногенетического эмбриона главным образом вносят вклад в трофэктодерму и, в конечном итоге, в плацентарную ткань, а клетки из эмбриона с перенесенным ядром, предположительно, главным образом вносят вклад во внутреннюю клеточную массу и, в конечном итоге, в ткань плода. Клетки из обоих эмбрионов могут быть на одной и той же клеточной стадии или на разных клеточных стадиях и можно объединять одинаковые или разные количества клеток из каждого эмбриона, чтобы образовать агрегированный эмбрион. При необходимости клетку из полученного клонированного плода или клонированного потомства можно использовать для второго раунда переноса ядер, чтобы дополнительно создать клонированное потомство. Клетки из исходного клонированного плода или клонированного потомства также могут быть заморожены для образования клеточной линии, используемой в качестве источника донорных клеток для создания дополнительных клонированных копытных животных. ПРИМЕР 19: Тестирование в отношении экспрессии Ig человека Тестирование телят, у которых сохраняется HAC или другая нуклеиновая кислота, кодирующая ксеногенное антитело, может начинаться сразу после рождения и включает оценку (1) экспрессии ксеногенного Ig, (2) ответ на иммунизацию, (3) созревание аффинности и (4) передачу HAC потомству. Экспрессию Ig человека можно контролировать посредством взятия крови у животных и оценки наличия экспрессии тяжелой и легкой цепи человека с помощью анализов ELISA, ОТ-ПЦР или FACS (смотри, например, публикацию PCT No. WO01/35735). После того как определено, что животные продуцируют Ig человека, животных иммунизируют столбнячным токсоидом в адъюванте. После иммунизации у животных берут кровь один раз в неделю и определяют ответы на антиген посредством ELISA или FACS и сравнивают с предварительно взятыми перед иммунизацией образцами крови. Через месяц после начальной иммунизации животным проводят бустер-иммунизацию водной формой антигена. Через неделю после бустер-иммунизации у животных берут кровь и измеряют ответ на антиген посредством ELISA или FACS и сравнивают с предварительно взятыми образцами крови. Анализ ELISA или FACS позволяет измерять наибольший титр ответа, а также продуцируемые изотипы тяжелой цепи. Указанные данные позволяют определить увеличение титра антител, а также наличие переключения классов. Также измеряют оценки средней аффинности, чтобы определить, происходит ли созревание аффинности во время ответа на антиген. После получения трансгенных коров, как описано выше, их используют для получения трансгенных Ig, предпочтительно человеческих, но возможно и Ig других видов, например собаки, кошки, примата, отличного от человека, других копытных животных, таких как овца, свинья, коза, грызунов, таких как мышь, крыса, морская свинка, кролик и т.д. Как указано, известно, что гены Ig являются консервативными среди видов. Трансгенные антисыворотки и молоко, содержащее ксеногенные антитела Корова (или другое копытное животное) дает трансгенную антисыворотку, направленную к любому антигену(нам), которым на нее воздействуют эндогенно, или к экзогенно введенному антигену(нам). Например, копытному животному могут быть введены антигены, чтобы получить желаемые антитела, реагирующие с антигенами, включая такие антигены, как патогены (например, бактерии, вирусы, простейшие, дрожжи или грибы), опухолевые антигены, рецепторы, ферменты, цитокины и т.д. Примеры патогенов для продукции антител включают, без ограничения этим, вирус гепатита (например, гепатита C), вирус иммунодефицита (например, ВИЧ), вирус герпеса, парвовирус, энтеровирус, вирус Эбола, вирус бешенства, вирус кори, вирус вакцинии, Streptococcus (например, Streptococcus pneumoniae), Haemophilus (например, Haemophilus influenza), Neisseria (например, Neisseria meningitis), Coryunebacterium diptheriae, Haemophilus (например, Haemophilus pertussis), Clostridium (например, Clostridium botulinium), Staphylococcus, Pseudomonas (например, Pseudomonas aeruginosa) и респираторно-синцитиальный вирус (RSV). Трансгенному копытному животному могут быть введены один или несколько патогенов, чтобы создать гипериммунную сыворотку, применимую для профилактики, стабилизации или лечения конкретного заболевания. Например, трансгенному копытному животному могут быть введены патогены, связанные с респираторной инфекций у детей, чтобы создать антисыворотку, реагирующую с патогенами (например, Streptococcus pneumoniae, Haemophilus influenza и/или Neisseria meningitis). Эти патогены, необязательно, могут быть обработаны, чтобы уменьшить их токсичность (например, воздействием тепла или химических веществ, таких как формальдегид), перед введением копытному животному. Для создания Ig широкого спектра трансгенному копытному животному может быть введено множество патогенов (например, множественные бактериальные и/или вирусные патогены). Такую гипериммунную сыворотку можно использовать для профилактики, стабилизации или лечения инфекции у млекопитающих (например, человека) и особенно она применима для лечения млекопитающих с генетическими или приобретенными иммунодефицитами. Кроме того, антитела, полученные способами по изобретению, можно использовать для подавления иммунной системы, например чтобы лечить невропатии, а также, чтобы удалить конкретные клетки человека и модулировать специфичные молекулы. Например, антиидиотипические антитела (т.е. антитела, которые ингибируют другие антитела) и антитела, реактивные по отношению к T-клеткам, B-клеткам или цитокинам, могут использоваться для лечения аутоиммунного заболевания или невропатии (например, невропатии вследствие воспаления). Такие антитела могут быть получены из трансгенных копытных животных, которым не вводили антиген, или они могут быть получены из трансгенных копытных животных, которым ввели антиген, такой как B-клетка, T-клетка или цитокин (например, TNF Трансгенные антисыворотки, созданные с использованием трансгенных копытных животных, которым не вводили антиген, можно использовать для производства фармацевтических препаратов, содержащих поликлональные антитела человека, предпочтительно молекулы IgG человека. Указанные человеческие антитела можно использовать вместо антител, полученных от людей, в виде терапевтических внутривенных иммуноглобулинов (IVIG). Трансгенная антисыворотка, необязательно, может быть обогащена антителами, реагирующими на один или несколько интересующих антигенов. Например, антисыворотку можно очистить с использованием стандартных способов, таких как способы, описанные Ausubel et al. (Current Protocols in Molecular Biology, Volume 2, p. 11.13.1-11.13.3, John Wiley and Sons, 1995). Предпочтительные способы очистки включают преципитацию с использованием шариков, покрытых антигеном или антителом, колоночную хроматографию, такую как аффинная хроматография, аффинную очистку на магнитных шариках и пэннинг с использованием связанного с планшетом антигена. Кроме того, трансгенную антисыворотку можно подвергать контакту с одним или несколькими представляющими интерес антигенами, и антитела, которые связываются с антигеном, можно отделить от несвязанного антитела на основе увеличенного размера комплекса антитело/антиген. Для очистки молекул IgG также можно использовать белок A и/или белок G. Если экспрессия эндогенных антител не исключена, то белок A и/или антитело против легкой цепи лямбда Ig человека (Pharmingen) можно использовать для отделения желаемых антител человека от эндогенных антител копытного животного или химерных антител копытное животное/человек. Белок A имеет более высокую аффинность по отношению к тяжелой цепи Ig человека, чем к тяжелой цепи Ig коровы, и может быть использован для отделения желаемых молекул Ig, содержащих две тяжелые цепи человека, от других антител, содержащих одну или две тяжелых цепи копытного животного. Антитело против легкой цепи лямбда Ig человека можно использовать для отделения желаемых молекул Ig, имеющих две цепи лямбда Ig человека, от молекул, имеющих одну или две легких цепи Ig копытного животного. Дополнительно или альтернативно одно или несколько антител, которые являются специфичными по отношению к тяжелой или легкой цепи Ig копытного животного, можно использовать на стадии негативной селекции, чтобы удалить молекулы Ig, содержащие одну или две тяжелые и/или легкие цепи копытного животного. Полученные антисыворотки как таковые можно использовать для пассивной иммунизации против антигена. Альтернативно антисыворотка имеет диагностическое, профилактическое применение или применяется для очистки, например для достижения очистки антигенов. Альтернативно после введения антисыворотки можно выделить B-клетки из трансгенной коровы и использовать для получения гибридом. Например, можно использовать стандартные способы, чтобы слить B-клетку от трансгенного копытного животного с миеломой для получения гибридомы, секретирующей представляющее интерес моноклональное антитело (Mocikat, J. Immunol. Methods 225: 185-189, 1999; Jonak et al., Hum. Antibodies Hybridomas 3: 177-185, 1992; Srikumaran et al., Science 220: 522, 1983). Предпочтительные гибридомы включают гибридомы, созданные слиянием B-клетки с миеломой от млекопитающего того же самого рода или вида, что и трансгенное копытное животное. Другие предпочтительные миеломы получают от мышей Balb/C или человека. В этом случае получают гибридомы, которые производят ксеногенные моноклональные антитела против конкретного антигена. Например, данный способ можно использовать для получения моноклональных антител человека, собаки, кошки и т.д. (в зависимости от конкретной искусственной хромосомы), которые специфичны по отношению к патогенам. Способы селекции гибридом, которые продуцируют антитела, обладающие желаемыми свойствами, т.е. повышенной аффинностью связывания, авидностью, хорошо известны. Альтернативно B-клетку от трансгенного копытного животного можно генетически модифицировать, чтобы экспрессировать онкоген, такой как ras, myc, abl, bcl2 или neu, или инфицировать трансформирующим ДНК- или РНК-вирусом, таким как вирус Эпштейн-Барра или вирус SV40 (Kumar et al., Immunol. Lett. 65: 153-159, 1999; Knight et al., Proc. Nat. Acad. Sci. USA 85: 3130-3134, 1988; Shammah et al., J. Immunol. Methods 160-19-25, 1993; Gustafsson and Hinkula, Hum. Antibodies Hybridomas 5: 98-104, 1994; Kataoka et al., Differentiation 62: 201-211, 1997; Chatelut et al., Scand. J. Immunol. 48: 659-666, 1998). Полученные иммортализованные B-клетки также можно использовать для получения теоретически неограниченного количества антител. Так как Ig также секретируется в молоко копытных животных, то молоко копытных животных также можно использовать в качестве источника ксеногенных антител. Другие варианты осуществления изобретения Из приведенного выше описания будет очевидно, что могут быть осуществлены изменения и модификации изобретения, приведенного в данном описании, чтобы его приспособить к различным применениям и условиям. Например, можно использовать другое время или температуры инкубирования (а именно 25, 30, 35, 37, 38,5 или 40°C или более высокие или более низкие температуры) для пермеабилизации мембран, перепрограммирования донорного генетического материала, возвращения мембран к непроницаемому состоянию и/или переноса генетического материала в ооциты. Такие варианты также входят в объем следующей формулы изобретения. Все публикации, упоминаемые в этом описании, приведены здесь в качестве ссылки в такой же мере, как в случае, когда для каждой независимой публикации или заявки на выдачу патента конкретно и отдельно указано, что они приведены здесь в качестве ссылки.
Формула изобретения
1. Трансгенная корова для продукции сельскохозяйственных и фармацевтических продуктов, содержащая неприродную мутацию в одном или обоих аллелях эндогенной нуклеиновой кислоты, кодирующей прион, где указанная мутация включает вставку последовательности терминации транскрипции и/или стоп-кодона в указанную нуклеиновую кислоту, кодирующую прион, где у указанной коровы снижена функциональная продукция прионов. 2. Трансгенная корова по п.1, где указанная мутация является гемизиготной. 3. Трансгенная корова по п.1, где указанная мутация является гомозиготной и где у указанной коровы отсутствует функциональная продукция прионов. 4. Трансгенная корова по п.1, где указанная мутация включает инсерцию маркера позитивной селекции в нуклеиновую кислоту, кодирующую прион. 5. Трансгенная корова по п.3, где указанная мутация включает инсерцию стоп-кодона в нуклеиновую кислоту, кодирующую прион. 6. Трансгенная корова по п.5, где указанный стоп-кодон встроен ниже начального кодона ATG в экзоне 3 указанной нуклеиновой кислоты, кодирующей прион. 7. Трансгенная корова по п.6, где указанный стоп-кодон встроен в пределах 10 нуклеотидов указанного ATG-кодона. 8. Трансгенная корова по п.1, содержащая одну или несколько нуклеиновых кислот, включающих в себя весь ген или часть гена ксеногенного иммуноглобулина (Ig), который подвергается перестройке, и экспрессирующая более одной молекулы ксеногенного Ig. 9. Фибробласт трансгенной коровы, использующийся для создания коровы, обладающей пониженной функциональной экспрессией прионов, содержащий неприродную мутацию в одном или обоих аллелях эндогенной нуклеиновой кислоты, где указанная мутация включает вставку последовательности терминации транскрипции и/или стоп-кодона в указанную нуклеиновую кислоту, кодирующую прион, и причем у указанного фибробласта снижена функциональная продукция прионов. 10. Способ получения трансгенной клетки коровы, имеющей пониженную экспрессию функционального прионного белка, заключающийся во введении в клетку коровы первого вектора, направленного на ген, кодирующий прион, в условиях, при которых возможна гомологичная рекомбинация между указанным первым вектором и первым аллелем эндогенной нуклеиновой кислоты, кодирующей прион указанной клетки, и, таким образом, введение гемизиготной мутации в указанную клетку, где указанная мутация включает вставку последовательности терминации транскрипции и/или стоп-кодона в указанную нуклеиновую кислоту, кодирующую прион. 11. Способ получения трансгенной коровы, имеющей пониженную экспрессию функционального прионного белка, при этом указанный способ включает в себя стадии: 12. Способ получения трансгенной коровы, имеющей пониженную экспрессию функционального прионного белка, при этом указанный способ включает в себя стадии:
РИСУНКИ
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 517
517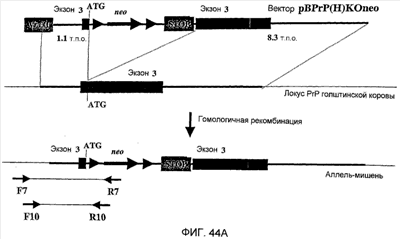
 HAC или
HAC или  ,
,  ,
,  ,
,  или
или  , и легкая цепь является легкой цепью лямбда или каппа. В других предпочтительных вариантах осуществления нуклеиновая кислота, кодирующая ксеногенную цепь иммуноглобулина или антитело, находится в своей не подвергнутой перестройке форме. В других предпочтительных вариантах осуществления копытное животное продуцирует более одного класса ксеногенного антитела. В различных вариантах осуществления копытное животное продуцирует более чем один различный ксеногенный Ig или антитело. Ксеногенное антитело может быть поликлональным или моноклональным антителом.
, и легкая цепь является легкой цепью лямбда или каппа. В других предпочтительных вариантах осуществления нуклеиновая кислота, кодирующая ксеногенную цепь иммуноглобулина или антитело, находится в своей не подвергнутой перестройке форме. В других предпочтительных вариантах осуществления копытное животное продуцирует более одного класса ксеногенного антитела. В различных вариантах осуществления копытное животное продуцирует более чем один различный ксеногенный Ig или антитело. Ксеногенное антитело может быть поликлональным или моноклональным антителом. и IgH, и положение маркера для селекции neo. Из CHO-клона HAC переносили в эмбриональные фибробласты коровы с помощью методики MMCT. Tc-фибробласты сливали с лишенным ядра ооцитом для переноса ядер. Реконструированные Tc-эмбрионы культивировали in vitro до стадии бластоцисты, затем имплантировали коровам-реципиентам. Примерно на 60 день беременности Tc-плоды извлекали; линию фибробластов восстанавливали, оценивали и использовали для дальнейшего переноса ядер. Повторно клонированные Tc-эмбрионы переносили реципиентам, чтобы получить Tc-телят.
и IgH, и положение маркера для селекции neo. Из CHO-клона HAC переносили в эмбриональные фибробласты коровы с помощью методики MMCT. Tc-фибробласты сливали с лишенным ядра ооцитом для переноса ядер. Реконструированные Tc-эмбрионы культивировали in vitro до стадии бластоцисты, затем имплантировали коровам-реципиентам. Примерно на 60 день беременности Tc-плоды извлекали; линию фибробластов восстанавливали, оценивали и использовали для дальнейшего переноса ядер. Повторно клонированные Tc-эмбрионы переносили реципиентам, чтобы получить Tc-телят. C, и супернатант (митотический экстракт) делили на аликвоты, замораживали в жидком азоте и хранили при -80
C, и супернатант (митотический экстракт) делили на аликвоты, замораживали в жидком азоте и хранили при -80
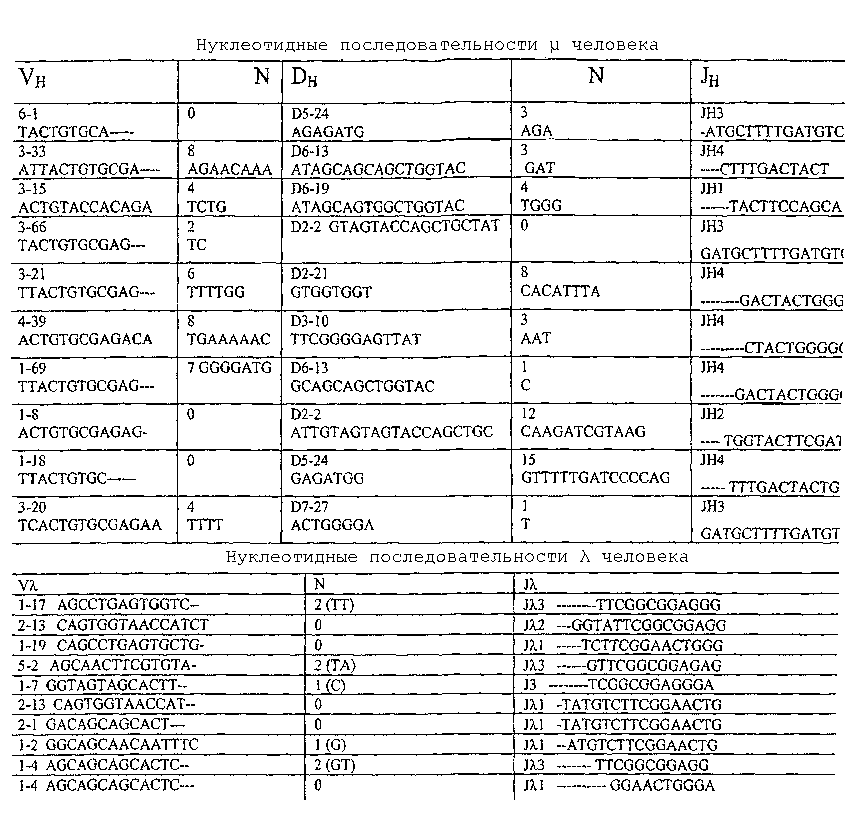
 -меркаптоэтанола (Gibco) и антибиотик-противогрибкового средства (Gibco). Через три дня после посева клетки собирали с помощью трипсина-EDTA и замораживали в
-меркаптоэтанола (Gibco) и антибиотик-противогрибкового средства (Gibco). Через три дня после посева клетки собирали с помощью трипсина-EDTA и замораживали в  ‘ на фиг. 32), свидетельствует, что часть сборки NuMA в пронуклеусах ядерных трансплантатов является результатом транскрипции de novo гена NuMA на стадии пронуклеусов.
‘ на фиг. 32), свидетельствует, что часть сборки NuMA в пронуклеусах ядерных трансплантатов является результатом транскрипции de novo гена NuMA на стадии пронуклеусов.