Патент на изобретение №2383014
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) ЭКСТРАКЦИОННО-ВОЛЬТАМПЕРОМЕТРИЧЕСКИЙ СПОСОБ ОПРЕДЕЛЕНИЯ ЦИНКА, КАДМИЯ, СВИНЦА И МЕДИ В ПРИРОДНЫХ ВОДАХ
(57) Реферат:
Способ заключается в том, что металлы совместно с ртутью извлекают в расслаивающуюся систему вода-антипирин-сульфосалициловая кислота – тиоцианат калия, формируют нижнюю фазу ионной органической жидкости, аликвоту которой наносят на поверхность графитового электрода, который помещают в трехэлектродную ячейку с 0,1 моль/л KSCN, без задержки подают потенциал накопления ртути -1,4 В в течение 30 секунд и затем в режиме анодной развертки регистрируют вольтамперограмму в квадратно-волновом режиме в пределах -1,20 до +0,20 В в виде пиков анодного окисления цинка, кадмия, свинца, меди. Изобретение позволяет определять малые концентрации цинка, кадмия, свинца и меди, что позволяет проводить мониторинг в природных водах. 1 ил., 2 табл.
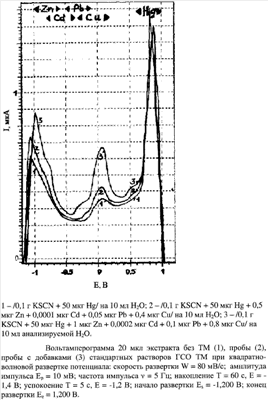
Изобретение относится к области аналитической химии объектов окружающей среды и направлено на разработку средств аналитического контроля параметров экосистем и полиэлементного фонового мониторинга природных вод и водных экосистем. Известен способ электрохимического определения цинка, кадмия, свинца и меди в воде природной, питьевой, очищенной сточной [Э.А.Захарова, Г.Б.Слепченко. Методика количественного химического анализа вод на содержание цинка, кадмия, свинца и меди методом инверсионной вольтамперометрии. МВИ 08-47/008. Томск. ТПУ. 1995]. Методика (аналог) основана на проведении инверсионно-вольтамперометрических измерений с накоплением цинка, кадмия, свинца и меди на индикаторном электроде (ртутно-пленочном, ртутно-графитовом) при заданном отрицательном потенциале электролиза – 1,4 В относительно хлоридсеребряного электрода сравнения. Процесс электрорастворения элементов с поверхности электрода и регистрация аналитических сигналов (анодных пиков) на вольтамперограмме проводятся при ступенчатой развертке потенциала 100 мВ/с в анодном направлении от -1,2 В до +0,15 В относительно электрода. Потенциалы максимумов регистрируемых анодных пиков (аналитических сигналов) цинка, кадмия, свинца и меди на фоне муравьиной кислоты соответственно равны (-0,9±0,1) В; (-0,6±0,1) В; (-0,4±0,1) В; (-0,05±0,10) В. Растворенный кислород удаляют инертным газом. Подготовка индикаторного ртутно-пленочного или ртутно-графитового электрода перед аналитической процедурой включает стадию формирования ртутной пленки на поверхности соответствующего рабочего (серебряного, графитового) электрода. В случае природных вод и других жидких проб аналог требует длительной, около одного часа, трудоемкой подготовки аналитического образца, например ультрафиолетового облучения (УФО) с фотокатализатором – двуокисью титана. УФО может занимать до двух часов в зависимости от наличия в пробе органического вещества. Наличие в водах органического вещества в миллиграммовых количествах отрицательно влияет на аналитические сигналы элементов вследствие сорбции его на поверхности ртутной пленки или графите. Сорбция органического вещества уменьшает величины предельных диффузионных токов элементов, с одной стороны, и смещает максимум токов пиков элементов из-за комплексующего действия растворенного органического вещества на определяемые элементы. Для разложения избыточного органического вещества в воде используют физическое воздействие: температуру, ультразвук и другие, либо химическое, например кислотную обработку сухого остатка воды после выпаривания. Подобного рода предварительная обработка водных образцов удлиняет аналитическую процедуру. Решающим недостатком аналога является сложность определения цинка и низких концентраций менее 0,01 мкг/л свинца и кадмия, например, в фильтрате снеговой воды (таблица 1). Недостаток аналога, заключающийся в сорбции органического вещества из природных вод на поверхность индикаторного электрода, заявляемый экстракционно-вольтамперометрический способ определения цинка, кадмия, свинца и меди в природных водах превращает в преимущество. Из известных технических решений наиболее близким по назначению и технической сущности к заявляемому способу является: электрохимический способ определения ртути модификацией графитового электрода ионной жидкостью (Патент РФ К недостаткам прототипа следует отнести: – недостаточную степень извлечения, цинка, кадмия, свинца и меди в нижнюю органическую фазу системы при расслаивании; степень извлечения составляет только 10-15%, причем для меди она наибольшая; способом, изложенным в прототипе, можно регистрировать только сигнал меди в области -0,1 – такая степень извлечения 10-15% металлов не позволяет регистрировать анодные сигналы цинка, кадмия, свинца и меди в анализируемой снеговой воде. Предлагаемое изобретение Экстракционно-вольтамперометрический способ определения цинка, кадмия, свинца и меди в природных водах заключается в извлечении металлов из жидкой водной фазы в органическую компоненту расслаивающей системы вода – антипирин – сульфосалициловая кислота – тиоцианат калия с помощью ртути, которую вводят в систему в микроколичествах. Добавка ртути вводится в систему вместе с анионами тиоцианата калия и навесками реагентов. Концентрирование ртути происходит из жидкой пробы, например фильтрата снеговой воды, в режиме «in situ», то есть во время формирования нижней фазы расслаивающейся системы (таблица 2). Ртуть присутствует в воде в виде тиоцианатного комплекса и выступает в качестве коллектора, соосаждающего тиоцианаты цинка, кадмия, свинца и меди. Отличие от прототипа состоит в дополнительном введении анионов тиоцианата в количестве 1-2 ммоль на 10 мл анализируемой воды. Увеличение концентрации более 200 моль/л ведет к выпадению прозрачных кристаллов переменного состава вследствие ограниченной растворимости роданидов в водной фазе. Заявляемый способ отличается от прототипа: 1) введением в систему тиоцианата в виде соли калия; 2) введением аликвоты стандартного раствора ртути; 3) квадратно-волновой регистрацией вольтамперограммы; 4) объемом 20 мкл (0,02 мл) аликвоты концентрата нижней фазы, используемой для модификации электрода из графита; 5) использование 0,1 моль/л раствора KSCN в качестве рабочего электролита. Осуществление изобретения Для предварительного формирования жидкой водной фазы (роданидных комплексов ртути и определяемых металлов) к фильтрату снеговой воды объемом 10 мл (объект анализа) добавляют 1,0-2,0 ммоль (0,1 г) тиоцианата калия KSCN марки ХЧ, 0,25 мкмоль (50 мкг) ртути Hg, порошкообразные химические реагенты: 16-20 ммоль (3 г) фармакопейного антипирина (брутто формула C11H12N2O, температура плавления 113°С, молекулярная масса 188,23 г/моль) и 6-10 ммоль (1,5 г) двухводной сульфосалициловой кислоты (ГОСТ 4478-78, брутто формула C7H6O6S×2H2O, молекулярная масса 254,21 г/моль). Затем стеклянную пробирку плотно закрывают пробкой, интенсивно встряхивают 5 мин, отстаивают 20-30 минут при 25°С, где и формируется плотная нижняя фаза органической компоненты. При растворении реагента – антипирина и органической кислоты (твердого вещества) в объекте анализа (водном растворе, содержащем микроколичества ртути и металлов) происходит кислотно-основное взаимодействие между протонированным антипирином и анионами органической сульфокислоты. Образующаяся органическая компонента, состоящая из ионного ассоциата органической соли, сульфосалицилата антипириния и антипирина, имеет кислую среду, обладает большой буферной емкостью и извлекает из кислого тиоцианатного раствора ртуть и металлы вследствие образования сложного комплекса металла с ионной по природе и органической по составу жидкости. Органическую компоненту расслаивающейся системы, формирующуюся в нижней фазе при расслаивании в виде ионной жидкости желтого цвета, используют для модификации графитового рабочего электрода. Объем органической компоненты (нижняя фаза) составляет около 3 мл при 25°С. Далее «ex situ» отбирают микродозатором 20 мкл (0,02 мл) нижней органической фазы и помещают на подготовленную поверхность графитового электрода. Перед каждым определением поверхность графитового электрода обновляют, полируют фильтровальной бумагой, обезжиривают в 10 мл 0,1 моль/л HCl с добавкой 0,2 мл 3%-ного щелочного раствора борогидрида натрия и промывают в кипящей дистиллированной воде для удаления избытка кислорода с его поверхности. На сухую графитовую поверхность наносят 20 мкл (0,02 мл) органической фазы с помощью микродозатора, равномерно распределяя по торцевой поверхности электрода (геометрическая площадь 0,13 см2). Такой пленочный электрод помещают в трехэлектродную ячейку с 10 мл 0,1 моль/л KSCN в качестве фонового раствора. Фоновый электролит не должен содержать растворенного кислорода. Растворенный в электролите кислород удаляют путем барботирования аргона в течение 3-5 минут. Далее без задержки подают потенциал накопления ртути -1,4 В в течение 30 сек. Затем в режиме анодной развертки регистрируют вольтамперограмму в квадратно-волновом режиме регистрации со скоростью анодной развертки потенциала 80 мВ/с (ТА-2, Томск, ТПУ) с 0,1 моль/л раствором KSCN, не содержащим растворенного кислорода, и накладывают перенапряжение – 1,4 В на 30 секунд относительно хлоридсеребряного электрода сравнения и затем регистрируют анодную вольтамперограмму в квадратно-волновом режиме в области потенциалов от – 1,2 В до +0,2 В. Типичные вольтамперограммы представлены на чертеже. Максимумы предельных диффузионных токов наблюдают примерно при тех же потенциалах, характерных для водных растворов (-0,800 Заявляемый способ обладает рядом преимуществ: 1) предварительно формирует тиоцианатные комплексы катионов тяжелых металлов с анионами тиоцианата; 2) концентрирует тиоцианатные комплексы катионов металлов из анализируемой воды в нижнюю органическую фазу при ее формировании путем соосаждения с ртутью; 3) введение тиоцианата в расслаивающуюся систему увеличивает степени извлечения металлов до 75-85%. Предлагаемый способ удешевляет процедуру анализа из-за применения значительно более простого и недорогого оборудования, не имеющего источников повышенной опасности для аналитика, исключает стадии высокотемпературного разложения аналита с целью его атомизации. Электрохимические анализаторы легче автоматизируются, значительно меньше по размерам и массе, а электрохимические методы позволяют дополнительно концентрировать аналит на поверхности рабочего электрода в вольтамперометрическом цикле. Вольтамперометрические анализаторы в полевом варианте позволяют контролировать элементы непосредственно на месте отбора проб воды. Сравнительный анализ таблиц 1 и 2 демонстрирует эффективность экстракционной вольтамперометрии в отношении металлов, особенно в отношении цинка. Цинк с применением аналога достоверно в снеговом фильтрате не определялся. Увеличивая степень извлечения анализируемых металлов до 85%, заявляемое изобретение обеспечивает достижение технического результата в сравнении с прототипом, т.е. достоверно позволяет определять малые концентрации цинка, кадмия, свинца и меди в фильтрованной снеговой воде и тем самым позволяет проводить мониторинг в природных водах (таблица 2).
Формула изобретения
Экстракционно-вольтамперометрический способ определения цинка, кадмия, свинца и меди в природных водах, заключающийся в извлечении металлов из жидкой водной фазы в органическую компоненту расслаивающей системы: антипирин – сульфосалициловая кислота и определении концентрата металлов на графитовом электроде, отличающийся тем, что для количественного определения цинка, кадмия, свинца и меди к 10 мл водного раствора анализируемой жидкости добавляют 16-20 ммоль антипирина, 6-10 ммоль сульфосалициловой кислоты, 1-2 ммоль тиоцианата калия KSCN и 0,25 мкмоль ртути, формируют нижнюю фазу ионной органической жидкости, затем отбирают аликвоту в количестве 20 мкл и наносят для модификации на поверхность графитового электрода, который помещают в трехэлектродную ячейку с 0,1 моль/л KSCN, и затем в режиме анодной развертки регистрируют вольтамперограмму в квадратно-волновом режиме в пределах -1,20 до +0,20 В с пиками анодного окисления цинка, кадмия, свинца, меди.
РИСУНКИ
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 0,0 В;
0,0 В; 0,95
0,95