Патент на изобретение №2381221
|
||||||||||||||||||||||||||
(54) СПОСОБ ПОЛУЧЕНИЯ (+)- И (-)-3-ОКСАБИЦИКЛО[3.3.0]ОКТ-6-ЕН-2-ОНОВ
(57) Реферат:
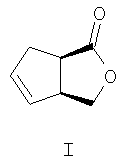
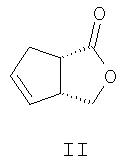
Изобретение относится к органической химии, конкретно к способу получения (-)- и (+)-3-оксабицикло[3.3.0]окт-6-ен-2-онов формул (I) и (II):
который включает взаимодействие рацемического 7,7-дихлорбицикло[3.2.0]-гепт-2-ен-6-она с (+)-
Изобретение относится к органической химии, конкретно к способу получения (-)- и (+)-3-оксабицикло[3.3.0]окт-6-ен-2-онов формул (I) и (II),
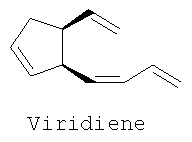
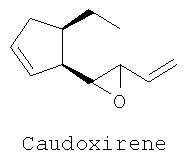
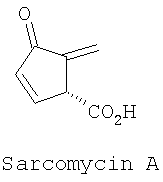
из которых лактон (I) был использован в хемоэнзиматических синтезах феромонов морских коричневых водорослей (Multifidenes, Viridiene, Caudoxirene) [1, 2]; энантиомерные лактоны (I) и (II) могут найти применение в качестве хиральных блок-синтонов в синтезе циклопентеновых антибиотиков [3], простагландинов [4], карбануклеозидов [5] и др. [6]. Данные о синтезе (±)-Sarcomycine А исходя из рацемической смеси (I+II) приведены в [7]. Получение рацемического 3-оксабицикло[3.3.0]окт-6-ен-2-она, а также использование его Ме3Si-аналога в синтезе Xanthocidin описаны в [8]. Сведения о синтезе и свойствах хирального лактона (II) в литературе отсутствуют. Синтезу лактона (I), основанному на использовании ферментативных реакций для оптического разрешения ахиральных субстратов, посвящены работы [1, 2]. Согласно [1] энантиодивергентным микробиологическим окислением по Байеру-Виллигеру (±)-бицикло[3.2.0]гепт-6-ен-2-она (III) получены хиральные лактоны (IV) и (I) с выходами 37 и 40% соответственно; энантиомерная чистота продуктов
Исходное соединение (III) получают в две стадии [2+2]-циклоприсоединением дихлоркетена к циклопентадиену с последующим восстановительным дехлорированием аддукта (V). Основным недостатком данной схемы является низкая региоселективность на стадии микробиологического окисления (образование примерно равных количеств целевого соединения (I) и побочного лактона (IV)), а также невозможность получения энантиомерного соединению (I) лактона (II). Кроме того, работа по данной схеме также неперспективна из-за необходимости приложения крайне трудоемкого хроматографического разделения однотипных региоизомерных лактонов (IV) и (I). Позднее был разработан улучшенный вариант синтеза (I) [2]. В этом случае с использованием на стадии окисления ((±)-III) бактерий гриба Cunninghamella echinulata NRRL 3665 удалось улучшить регио- и стереоселективность процесса и получить (I) с выходами 30-35% и оптической чистотой Недостатки приведенного метода заключаются в исключительной трудоемкости и специфичности подготовки и осуществления микробиологического процесса хирализации субстрата, что наглядно видно в ходе рассмотрения самой методики. Вначале клетки Cunninghamella echinulata NRRL 3665 (2*107 клеток в растворе Tween 80, 0.5%) в 5 л среды, содержащей 100 г зернового ликера, 20 г глюкозы, 5 г КН2РO4, 10 г К2НРO4, 10 г NaNO3, 2.5 г КСl, 2.5 г MgSO4, 0.1 г FeSO4, 1 мл Pluronic PE 8100 (BASF), 0.5 мл антивспенивателя ферментируют при 27°С в течение 6 ч, фильтруют, суспендируют в 5 л фосфатного буфера (рН 6.9) и добавляют раствор 5.2 г кетона (III) в 50 мл спирта. Инкубацию при встряхивании ферментера проводят при 27°С в течение 24-30 ч. После в массу добавляют НСl до рН 2 и экстрагируют СН2Сl2 в течение 48 ч. После осушки экстрактов над MgSO4 и упаривания остаток хроматографируют на колонке с SiO2. Получают ((+)-III) (1.7 г, 33%, 86% ее), смешанные фракции (9%) и целевое соединение ((-)-I) (2 г, 34%, Задача, на решение которой направлено заявляемое изобретение, заключается в упрощении процесса и «энантиодивергентном» получении энантиомерно чистых лактонов (I) и (II) с высоким выходом. Сущность изобретения заключается в следующем. Для одновременного получения обоих энантиомеров 3-оксабицикло[3.3.0]окт-6-ен-2-она из (V) предлагается следующий простой и практичный вариант. Вначале рацемический 7,7-дихлорбицикло[3.2.0]-гепт-2-ен-6-он (V), получаемый по [9], обрабатывают (+)- На завершающих этапах синтеза индивидуальные аминали (VIII) и (IX) восстанавливают борогидридом натрия до амидоспиртов (X) и (XI), которые в ходе кислотного гидролиза спонтанно лактонизуются с образованием энантиомерно чистых лактонов (I) и (II) с суммарным выходом более 72% в расчете на исходный 7,7-дихлорбицикло[3.2.0]-гепт-2-ен-6-он (V). Преимущества предложенного метода по сравнению с известным микробиологическим методом получения (I) заключаются в обеспечении возможности одновременного выхода к обеим целевым структурам (I) и (II) в энантиомерно чистом виде, в легкости их разделения простой колоночной хроматографией и в отсутствии необходимости в дорогостоящих реагентах и неизмеримо затруднительных условий микробиологических синтезов.
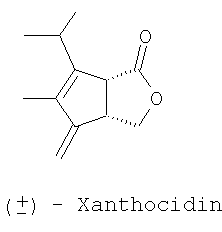
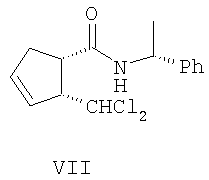
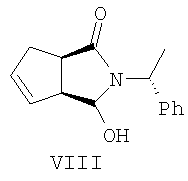
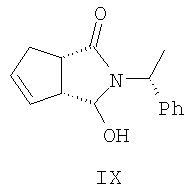
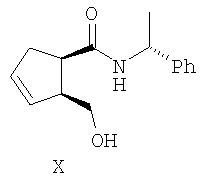
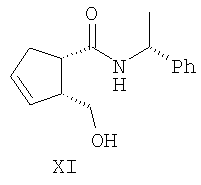
Сущность изобретения подтверждается следующими примерами. Пример 1. Синтез V. К непрерывно перемешиваемому раствору 53 г (0.8 м) свежеперегнанного циклопентадиена, 60 г (0.4 м) дихлорацетилхлорида в 400 мл гексана добавляют 59 мл сухого триэтиламина в 300 мл сухого гексана в период 1.5 ч. После чего реакционную массу перемешивают в течение 15 часов в атмосфере аргона. Полученную смесь фильтруют, осадок (гидрохлорид триэтиламина) промывают на фильтре 300 мл гексана. Растворитель упаривают в вакууме, остаток перегоняется в вакууме, отбирают фракцию, кипящую при температуре 49-50°С/ 0.3 мм рт.ст. Получают 55.23 г (84%) соединения (V) в виде желтоватой маслообразной жидкости. (±)-7,7-Дихлорбицикло[3.2.0]-гепт-2-ен-6-он (V). Желтоватая маслообразная жидкость с т. кип. 49-50°С/ 0.3 мм рт.ст. (Rf=0.8, петролейный эфир – этилацетат, 1:1). ИК-спектр, Пример 2. Синтез VI+VII. К раствору 1.5 г (8.5 ммоль) дихлоркетона (V) в 40 мл бензола прилили раствор 1.09 г (9 ммоль) (+)- 2-(Диxлopмeтил)-N-[(1R)-1-фенилэтил)]циклопент-3-ен-1-карбоксамиды (VI и VII). Желтые кристаллы (Rf=0.75, петролейный эфир – этилацетат, 1:1). ИК-спектр, Пример 3. Синтез VIII и IX. К раствору 1.0 г (3.33 ммоль) смеси 1:1 амидов (VI) и (VII) в 20 мл MeCN добавляют раствор 1.54 г (10 ммоль) ВаО в 5 мл Н2О и перемешивают полученную смесь при кипячении в течение 20 часов (контроль по ТСХ). После этого упаривают органический растворитель, водную фазу экстрагируют EtOAc (3×15 мл). Объединенные органические экстракты сушат над MgSO4, упаривают при пониженном давлении и сушат в вакууме. Остаток делят на SiO2 в системе петролейный эфир:этилацетат (7:3). Получают 385 мг (47%) желтых кристаллов соединения (VIII) и 393 мг (48%) белых кристаллов соединения (IX). (3aS,6aR)-3-Гидрокси-2-[(1R)-фенилэтил]-3,3а,6,6а-тетрагидроциклопента-[с]пиррол-1(2Н)-он (VIII). Желтые кристаллы с т. пл. 105-107°C (Rf=0.2, петролейный эфир – этилацетаг, 7:3). [ (3аR,6аS)-3-Гидрокси-2-[(1К)-фенилэтил]-3,3а,6,6а-тетрагидроциклопента-[с]пиррол-1(2Н)-он (IХ). Белые кристаллы с т. пл. 120-121°C (Rf=0.45, петролейный эфир – этилацетат, 7:3). [ Пример 4. Синтез Х и XI (1R,2S)-2-(Гидроксиметил)-N-[(1R)-1-фенилэтил]-пиклопент-3-ен-1-карбоксамид (X). В раствор 85 мг (0.35 ммоль) соединения (VIII) в 5 мл диоксана добавляют 1 мл воды и 130 мг (3.4 ммоль) NaBH4. Полученную смесь кипятят 5 часов (контроль по ТСХ). После чего упаривают органический растворитель, а водную фазу экстрагируют CH2Cl2 (3×10 мл). Объединенные органические экстракты сушат над MgSO4, упаривают при пониженном давлении и вакуумируют. Остаток делят на SiO2 в системе петролейный эфир:этилацетат (1:1). Получают 77 мг (90%) белых кристаллов соединения (X) с т.пл. 115-117°C (Rf=0.4, петролейный эфир – этилацетат, 1:1). [ (1S,2R)-2-(Гидроксиметил)-N-[(1R)-1-фенилэтил]-циклопент-3-ен-1-карбоксамид (XI). В раствор 130 мг (0.54 ммоль) соединения (IX) (в 5 мл диоксана добавляют 1 мл воды и 200 мг (5.3 ммоль) NaBH4. Полученную смесь кипятят 5 часов (контроль по ТСХ). После чего упаривают органический растворитель, а водную фазу экстрагируют СН2С2 (3×10 мл). Объединенные органические экстракты сушат над MgSO4, упаривают при пониженном давлении и вакуумируют. Остаток делят на SiO2 в системе петролейный эфир:этилацетат (1:1). Получают 120 мг белых кристаллов (91%) соединения (XI) с т.пл. 132-134°C (Rf=0.4, петролейный эфир – этилацетат, 1:1). [ Пример 5. Синтез I и II (-)-(1R,5S)-3-оксабицикло[3.3.0]окт-6-ен-2-он (I). В раствор 98 мг (0.4 ммоль) амидоспирта (X) в 2 мл диоксана добавляют 1 мл 9N H2SO4. Реакционную массу выдерживают 4 часа при кипячении (контроль по ТСХ). После чего упаривают органический растворитель, а водную фазу экстрагируют Et2O (3×10 мл). Объединенные органические экстракты сушат над MgSO4, упаривают при пониженном давлении и вакуумируют. Остаток делят на SiO2 в системе петролейный эфир:этилацетат (1:1). Получают 43 мг (88%) маслообразного светло-желтого соединения (I) (Rf=0.7, петролейный эфир – этилацетат, 1:1). [ (+)-(1S,5R)-3-оксабицикло[3.3.0]окт-6-ен-2-он (II). В раствор 70 мг (0.31 ммоль) амидоспирта (XI) в 2 мл диоксана добавляют 1 мл 9N H2SO4. Реакционную массу выдерживают 4 часа при кипячении (контроль по ТСХ). После чего упаривают органический растворитель, а водную фазу экстрагируют Et2O (3×10 мл). Объединенные органические экстракты сушат над MgSO4, упаривают при пониженном давлении и вакуумируют. Остаток делят на SiO2 в системе петролейный эфир:этилацетат (1:1). Получают 31 мг (89%) маслообразного светло-желтого жидкого соединения (II) (Rf=0.7, петролейный эфир – этилацетат, 1:1). [ С7Н8O2. Вычислено, %: С 67.74; Н 6.45. Литература 1. Alphand V., Archelas A. Furstoss R. Microbial Transformations 16. One-step synthesis of a pivotal prostaglandin chiral synthon via a highly enantioselective microbiological Baeyer-Villiger type reaction. Tetrahedron Lett. 1989, 30, 3663; Alphand V., Furstoss R. Microbial Transformations 22. Microbiologically Mediated Bayer-Villiger Reactions: A Unique Route to Several Bicyclic 2. Lebreton, J.; Alphand, V.; Furstoss, R. A short chemoenzymatic synthesis of (+)-multifidene and (+)-viridiene. Tetrahedron Lett. 1996, 37, 1011; Lebreton, J.; Alphand, V.; Furstoss, R. Chemoenzymatic Synthesis of Marine Brown Pheromones. Tetrahedron. 1997, 53, 145. 3. Scarboround, R.M., Jr.; Toder, B.H.; Smith, A.B. A stereospecific total synthesis of (±)-methylenomycin A and its epimer, (±)-epimethylenomycin A. J.Am.Chem.Soc. 1980, 102, 3904. 4. Roberts, S.M.; Santoro, M.; Sickle, E. The Emergence of the Cyclopentenone Prostaglandins as Important, Biologically Active Compounds. J. Chem. Soc. Perkin Trans. I, 2002, 1735. 5. Crimmins, M.T. New developments in the enantioselective synthesis of cyclopentyl carbocyclic nucleosides. Tetrahedron. 1998, 54, 9229. 6. Li, S.W.; Batey, R.A. Mild lanthamde(III) catalyzed formation of 4,5-diaminocyclopent-2-enones from 2-furaldehyde and secondary amines: a domino condensation/ring-opening/electrocyclization process. Chem.Comm. 2007, 3759; Toueg, J.; Prunet, J. Dramatic Solvent Effect on the Diastereoselectivity of Michael Addition: Study toward the Synthesis of the ABC Ring System of Hexacyclinic Acid. Org. Lett. 2008, 10, 45. 7. Govindan, S.V.; Hudlicky, Т.; Koszyk, F.J. Two topologically distinct total syntheses of (±)-sarkomycin. J. Org. Chem. 1983, 48, 3581. 8. Au-Yeung, B.-W.; Wang, Y. Allylsilane in synthesis: new syntheses of (±)-desepoxy-4,5-didehydromethylenomycin a and (±)-xanthocidin. J. Chem. Soc., Chem. Commun. 1985, 825. 9. Ghosez L., Montaigne R., Roussel A., Vanlierde H., Mollet P. Cycloadditions with dichloroketene. Tetrahedron Lett. 1966, 7, 135.
Формула изобретения
1. Способ получения (-)- и (+)-3-оксабицикло[3.3.0]окт-6-ен-2-онов формул (I) и (II), 2. Способ по п.1, отличающийся тем, что в качестве восстановителя используют борогидрид натрия.
|
||||||||||||||||||||||||||
 -метилбензиламином с образованием смеси диастереомерных амидов, гидратация которой приводит к разделяющимся колоночной хроматографией на SiO2 продуктам гидролиза – бициклическим аминалям. Последующее борогидридное восстановление индивидуальных соединений приводит к соответствующим амидоспиртам, при кислотном гидролизе которых образуются энантиомерно чистые целевые соединения – лактоны (I) и (II) с суммарным выходом более 72% в расчете на исходное соединение. Технический результат – одновременное получение лактонов I и II с высоким суммарным выходом в энантиомерно чистом виде. 1 з.п. ф-лы.
-метилбензиламином с образованием смеси диастереомерных амидов, гидратация которой приводит к разделяющимся колоночной хроматографией на SiO2 продуктам гидролиза – бициклическим аминалям. Последующее борогидридное восстановление индивидуальных соединений приводит к соответствующим амидоспиртам, при кислотном гидролизе которых образуются энантиомерно чистые целевые соединения – лактоны (I) и (II) с суммарным выходом более 72% в расчете на исходное соединение. Технический результат – одновременное получение лактонов I и II с высоким суммарным выходом в энантиомерно чистом виде. 1 з.п. ф-лы.
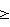 95%.
95%.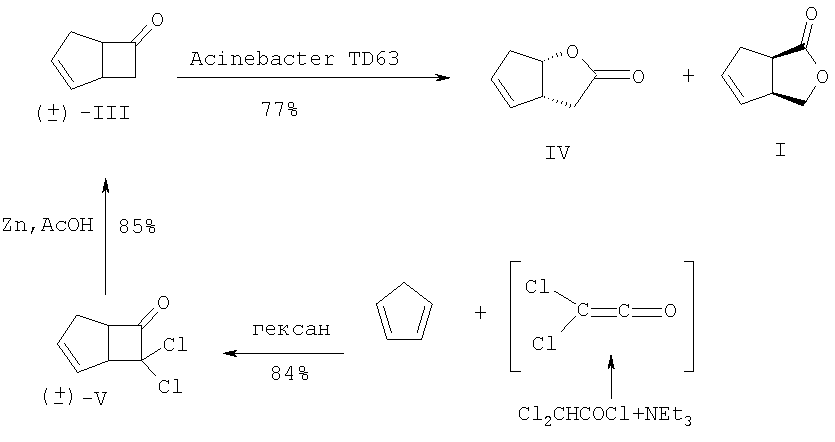
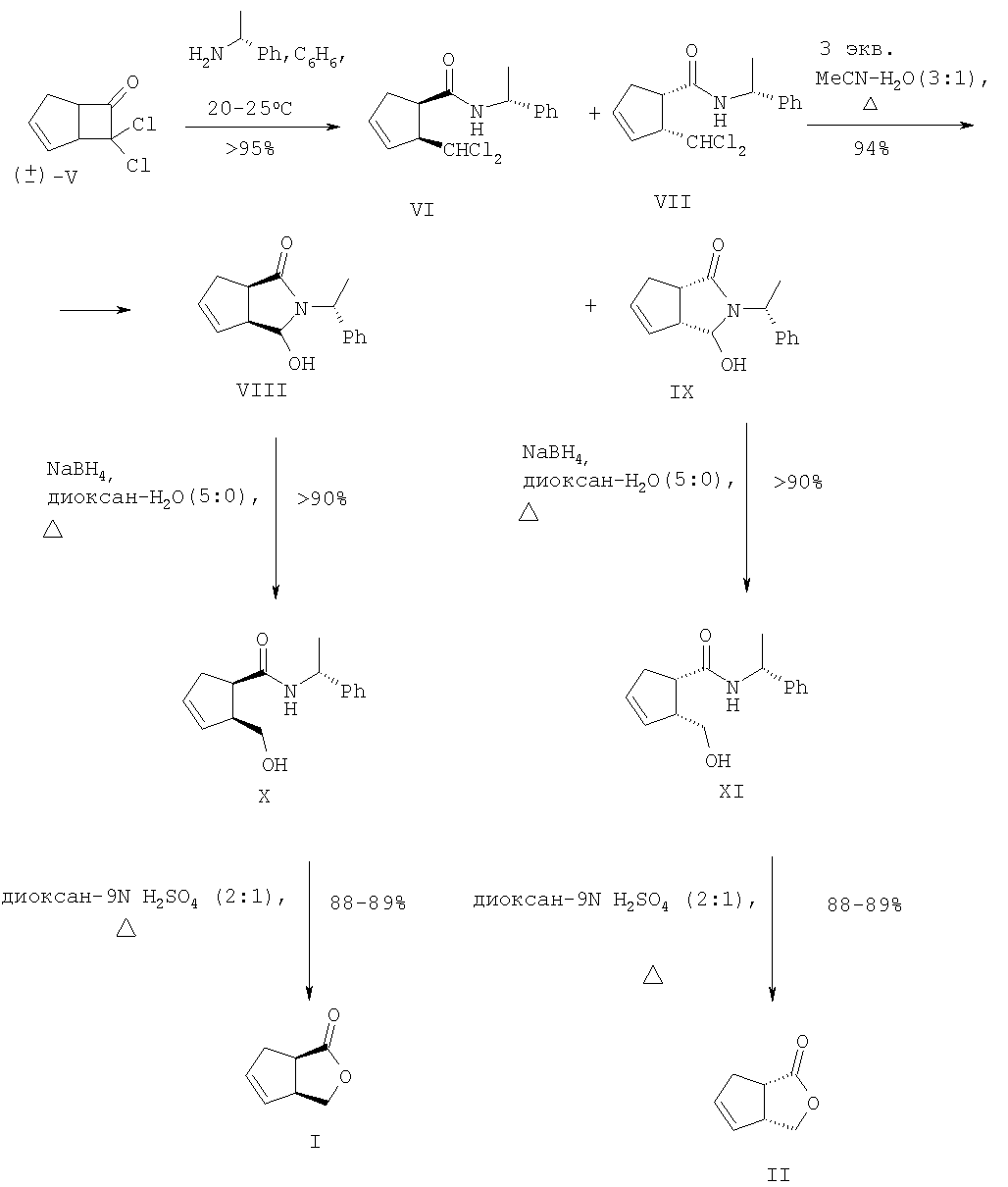
 , см-1: 1805, 1028, 887, 814, 797, 754, 731, 631. Спектр ЯМР 1Н,
, см-1: 1805, 1028, 887, 814, 797, 754, 731, 631. Спектр ЯМР 1Н,  , м.д.: 2.48-2.88 м (2Н, С4H, 4.04-4.22 м (1Н, С5H, 4.32-4.42 м (1Н, С1H), 5.78-5.90 м (1Н, С3H), 6.03-6.17 м (1Н, С2H). Спектр ЯМР 13С,
, м.д.: 2.48-2.88 м (2Н, С4H, 4.04-4.22 м (1Н, С5H, 4.32-4.42 м (1Н, С1H), 5.78-5.90 м (1Н, С3H), 6.03-6.17 м (1Н, С2H). Спектр ЯМР 13С,  20+142.4° (с 0.65, МеОН). ИК-спектр,
20+142.4° (с 0.65, МеОН). ИК-спектр, 
 -Lactones in High Enantiomeric Purity. J. Org. Chem. 1992, 57, 1306.
-Lactones in High Enantiomeric Purity. J. Org. Chem. 1992, 57, 1306.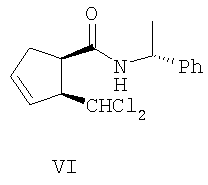
 ,
,
 ,
,
 ,
,