Патент на изобретение №2166982
|
||||||||||||||||||||||||||
(54) СПОСОБ РАЗДЕЛЕНИЯ ИЗОТОПОВ УГЛЕРОДА
(57) Реферат: Изобретение может быть использовано при извлечении изотопов углерода из технологических отходов, мониторинге окружающей среды, выделении радиоактивного изотопа 14С из радиоактивных отходов. Смесь изотопов углерода с содержанием 13С 1,104 ат.% очищают криогенной ректификацией, разделяют в каскаде ректификационных насадочных колонн. Полученный полупродукт гидрируют водородом до метана. Для гидрирования используют отвальный газ производства дейтерия ректификацией жидкого водорода с содержанием дейтерия 0,0015 ат.%. Продукт гидрирования подвергают глубокой сушке и подают в непрерывно работающую противоточную адсорбционную колонну, заполненную цеолитом NaX, NaA, СаА с обращением потоков фаз на концах: внизу – десорбция метана из цеолита при нагреве до 470 К, вверху – адсорбция метана цеолитом при охлаждении до 200 К. Давление в колонне поддерживают на уровне 105 Па. Концентрация изотопа 13С – не менее 99,95 ат.%, коэффициент разделения – не менее 1,02, коэффициент обогащения превышает аналогичный показатель для известного способа в 6 раз. 3 з.п. ф-лы, 3 табл. Изобретение относится к области изотопной химии и может быть использовано для разделения изотопных смесей углерода: при производстве его изотопов из природных изотопных смесей; при изотопной очистке препаратов углерода-14, производимых с использованием ядерной реакции 14N(n,p)14C; при извлечении (регенерации) изотопов углерода из технологических отходов, сопровождающих производство меченых этими изотопами химических соединений; при изотопной очистке от радиоактивного углерода-14 циркулирующих в производстве, а также сбрасываемых в окружающую среду технологических потоков предприятий ядерно-технологического цикла и, кроме того, при изотопном концентрировании (с целью повышения чувствительности и точности анализа) проб углерода, отбираемых на изотопный анализ в ходе проведения различных технологических процессов, мониторинга окружающей среды, а также разнообразных научных исследований. Известен способ разделения изотопов углерода химическим изотопным обменом с использованием двухфазных углеродсодержащих рабочих систем в непрерывноработающих противоточных колоннах с обращением потоков на их концах химическим либо термическим путем (см. Боресков Г.К., Катальников С.Г. Технология процессов химического изотопного обмена. М.: Изд-во МХТИ им. Д.И. Менделеева. 1974 г. С. 95-102). Недостатком этого способа являются: низкая скорость межфазного изотопного обмена углерода в рабочих системах, обусловленная протеканием в фазах наряду с диффузионными процессами углеродсодержащих веществ также реакции изотопного обмена между ними (следствием этого являются большие габариты массообменных колонн, а также большая длительность вывода их в стационарный режим работы); большой расход высокочистых химических реактивов на обращение потоков в колоннах и связанный с этим значительный объем химически агрессивных отходов; высокая токсичность, а также низкая термохимическая устойчивость рабочих веществ, требующая принятия специальных мер по обеспечению безопасности обслуживающего персонала, а также организации, наряду с основным процессом разделения изотопов углерода, вспомогательных процессов глубокой очистки циркулирующих в аппаратуре веществ от продуктов их разложения, и, наконец, низкая радиационная устойчивость веществ, входящих в состав рабочих систем, не позволяющая использовать этот способ для концентрирования радиоактивного изотопа углерода – 14C. Недостатком этого способа являются: большая энергоемкость, сложность накопления продуктов разделения, а также многократного умножения элементарного эффекта разделения. К недостаткам этого способа относятся: сложность многократного умножения элементарного эффекта разделения; необходимость использования в качестве исходных, сложных по построению, специально синтезируемых для целей разделения изотопов углерода, дорогостоящих химических соединений; высокая стоимость и сложность разделительной аппаратуры, а также химическая инертность получаемых изотопнообогащенных соединений, осложняющая их дальнейшую переработку в широко используемые меченые изотопами углерода химические вещества. Недостатком этого способа являются: сложность и высокая стоимость аппаратурного оформления; высокая энергоемкость, связанная с перекачкой газовых потоков, а также с испарением и перекачкой вспомогательных веществ; необходимость, как правило, работать с высокотоксичными веществами (пары ртути); низкая производительность. Например, при разделении изотопной смеси 12CH4 – 13CH4 масс-диффузией метана с исходным содержанием 13C 2,7% ат. через поток пара ртути при давлении 1333 Па в каскаде, состоящем из 70 масс-диффузионных разделительных насосов удается при концентрации 13C в продукте 88% ат. обеспечить производительность (отбор) всего 30 см3 газа в сутки. Известен способ разделения изотопов углерода в газовых термодиффузионных колоннах с использованием в качестве рабочего вещества метана (CH4 Недостатками этого способа являются: необходимость предварительной чрезвычайно глубокой очистки исходных газов от химических примесей, а также непрерывного вывода последних из термодиффузионных колонн в ходе разделения изотопов углерода; низкая производительность; большая энергоемкость и, как следствие, высокая себестоимость целевых продуктов. Например, для обогащения метана с исходным содержанием 13C 1,104% ат. методом термодиффузии до 20% ат. необходимо затратить мощность 6700 кВт  ч/г 13C. Использование термодиффузионного способа разделения изотопов углерода для концентрирования 13C из природных изотопных смесей экономически оправдано лишь при объемах производства не выше ~ (100-200) г 13CH4 в год с концентрацией 13C до 60% ат. Термодиффузия природного метана или CO без принятия специальных мер не позволяет обеспечить получение в колоннах высококонцентрированных изотопов углерода, в частности для 13C выше 94,8% ат. Последнее обусловлено концентрированием в колоннах, наряду с целевым продуктом, например 13CH4, разбавляющих его и равных ему по массе, например, 12CH3D, изотопнозамещенных по водороду форм.
14C, достигаются при использовании в нем в качестве углеродсодержащего соединения оксида углерода (II) (CO). При использовании этого соединения процесс ректификации осуществляется в области криогенных температур (вблизи 80 К) при давлении, близком к атмосферному (оптимальное давление 86,6 кПа). В этих условиях он характеризуется относительно большой величиной однократного коэффициента разделения ( ч/г 13C. Использование термодиффузионного способа разделения изотопов углерода для концентрирования 13C из природных изотопных смесей экономически оправдано лишь при объемах производства не выше ~ (100-200) г 13CH4 в год с концентрацией 13C до 60% ат. Термодиффузия природного метана или CO без принятия специальных мер не позволяет обеспечить получение в колоннах высококонцентрированных изотопов углерода, в частности для 13C выше 94,8% ат. Последнее обусловлено концентрированием в колоннах, наряду с целевым продуктом, например 13CH4, разбавляющих его и равных ему по массе, например, 12CH3D, изотопнозамещенных по водороду форм.
14C, достигаются при использовании в нем в качестве углеродсодержащего соединения оксида углерода (II) (CO). При использовании этого соединения процесс ректификации осуществляется в области криогенных температур (вблизи 80 К) при давлении, близком к атмосферному (оптимальное давление 86,6 кПа). В этих условиях он характеризуется относительно большой величиной однократного коэффициента разделения ( (13CO-12CO) (13CO-12CO)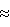 1,007), высокой скоростью межфазного изотопного обмена (высота эквивалентной теоретической ступени разделения в насадочных колоннах ВЭТС (2,2-2,6) см), а также большой плотностью орошения массообменных колонн углеродом.
Недостатком этого способа является трудность достижения в нем высоких концентраций тяжелого изотопа углерода из-за наличия в составе оксида углерода (II) тяжелого изотопа кислорода 18O. Например, природный оксид углерода (II) представляет собой в основном смесь 12C16O+13C16O+12C18O. Парциальные упругости паров 12C18O и 13C16O отличаются настолько мало ( = 1,002 при 74,4 К), что при ректификации в колонне происходит практически отделение только 12C16O от бинарной смеси 12C18O + 13C16O. Это ограничивает возможный уровень концентрации 13C в ректификационной колонне пределом < 95% ат. В реально работающих колоннах этот уровень не превышает 93% ат.
Наиболее близким решением к предлагаемому изобретению является способ разделения изотопов углерода, включающий: предварительное концентрирование целевого изотопа криогенной дистилляцией оксида углерода (II) в непрерывноработающих ректификационных колоннах; последующее гидрирование предварительно обогащенного продукта до метана водородом, полученным при электролизе воды природного изотопного состава, в котором содержание дейтерия составляет 5105 (0,005% ат. ); конечное концентрирование целевого изотопа в противоточных колоннах термодиффузией продукта гидрирования (метана) (см. Рабинович Г.Д. Разделение изотопов и других смесей термодиффузией. М.: Атомиздат. 1981 г. 98 с.) (прототип). Указанный способ обеспечивает концентрирование из изотопных смесей углерода как стабильных (12C; 13C), так и радиоактивных (14C) изотопов. Благодаря использованию в нем на стадии гидрирования CO водорода с пониженным содержанием дейтерия, на стадию конечного термодиффузионного концентрирования целевого изотопа, например 13C, в этом способе подается метан с пониженным содержанием дейтерозамещенных форм (в частности, CH3D). Это обеспечивает возможность достижения высоких концентраций целевого изотопа в продукте. Например, при концентрировании указанным способом 13C из природной изотопной смеси удается достичь содержания этого изотопа в продукте на уровне 99-99,8% ат.
К недостаткам этого способа следует отнести использование в нем на стадии гидрирования CO водорода, полученного при электролизе воды природного изотопного состава, а также проведение конечного концентрирования целевого изотопа термодиффузией метана в колоннах. Получаемый электролизом воды водород содержит до 0,5% об. примесей, в том числе до 25 г/см3 влаги. Кроме влаги в нем в качестве примесей присутствуют компоненты воздуха (O2, до 0,02% N2, Ar), а также CO2. Указанные примеси, поступая вместе с водородом на стадию гидрирования CO, способны не только загрязнять получаемый при этом метан и, как следствие, оказывать в последующем вредное воздействие на работу термодиффузионных колонн (снижая их производительность, а также вызывая в условиях высоких температур коррозию конструкционных материалов), но и приводить к потере обогащенного продукта за счет химического взаимодействия некоторых из них (O2, H2O) с CO и CH4, а также за счет его изотопного разбавления путем изотопного обмена и смешения с углеродом примесного CO2 природного изотопного состава. Наличие в используемом водороде указанных примесей приводит к необходимости либо его глубокой химической очистки перед стадией гидрирования, либо к последующей глубокой химической очистке получаемого с его использованием метана, используемого затем для питания термодиффузионных колонн. Оба процесса сложны в аппаратурном оформлении, их реализация ведет к повышению себестоимости целевого изотопного продукта. Получаемый при электролизе воды природного изотопного состава водород содержит пониженное относительно природного уровня (0,015% ат.) количество дейтерия (~ 0,005% ат.). Это, снижая на стадии гидрирования CO в подвергаемом термодиффузии метане содержание изотопнозамещенных форм (например, CH3D), способствует повышению достигаемого предела концентрации целевых изотопов (в частности, 13C) в продукте. Например, для 13C от 94,7 до 99 – 99,8% ат. Однако количество дейтерия в электролитическом водороде остается на высоком уровне и, как следствие, на высоком уровне остается содержание дейтерозамещенных форм метана в газе, подаваемом на термодиффузию. Это ограничивает возможности конечного термодиффузионного концентрирования изотопов углерода в рассматриваемом способе уровнем 99 – 99,8% ат. Использование в рассматриваемом способе на стадии конечного концентрирования термодиффузии метана ведет к резкому усложнению аппаратурного оформления процесса конечного концентрирования, к необходимости использования для его аппаратурного оформления драгоценных металлов, а также к низкой эффективности самого процесса конечного концентрирования, которая определяется коэффициентом разделения изотопов (например, для смеси 12CH4–13CH4 = 1,009) и высотой эквивалентной теоретической ступени (ВЭТС 4 см) разделения в термодиффузионной колонне. В связи с этим можно отметить, что из-за высоких температур в термодиффузионных колоннах (~ 695 К) горячие проволоки внутри них изготавливаются из платино-иридиевого сплава (80% Pt и 20% Ir). В прототипе для получения нескольких литров метана с концентрацией 13C 99 – 99,8% ат. из предварительно обогащенного дистилляцией CO до ~ 70% ат. 13C метана потребовался каскад из восьми последовательно соединенных колонн с внутренним диаметром 12,1 мм, эффективной высотой 2,85 м с горячей проволокой диаметром 0,4 мм. Низкое для термодиффузии значение и высокое ВЭТС=h ведут к большому объему (V ~ h/( – 1)2) и длине (H ~ h/ ( – 1)) массообменных колонн, к большой величине циркулирующих внутри них флегмовых потоков (L ~ 1/( -1)), а также к высокому уровню энергозатрат (E ~ 1/( -1)) на осуществление процесса разделения.
Задачей создания изобретения является увеличение предела концентрирования целевых изотопов углерода, упрощение аппаратурного оформления процесса их разделения, снижение капитальных и эксплуатационных затрат на его осуществление, а также увеличение коэффициента разделения изотопов углерода на стадии их конечного концентрирования.
Поставленная задача решается тем, что в способе разделения изотопов углерода, включающем предварительное концентрирование целевого изотопа криогенной ректификацией оксида углерода (II), гидрирование концентрата водородом с пониженным относительно природного уровня содержанием дейтерия до метана и конечное концентрирование целевого изотопа в противоточных колоннах в форме метана, в качестве водорода используют отвальный газ производства дейтерия криогенной ректификацией водорода природного изотопного состава, а конечное концентрирование осуществляют криогенной адсорбцией метана на цеолите. При этом в качестве цеолита преимущественно используют цеолиты NaX (13X), NaA (4A) или CaA (5A).
Предлагаемое изобретение иллюстрируется следующими примерами.
Пример 1. Природная изотопная смесь углерода с содержанием 13C 1,104% ат. в форме предварительно очищенного криогенной ректификацией в насадочной колонне от химических примесей оксида углерода (II) подвергается 81,6 К ( 1,007) разделению в каскаде ректификационных насадочных колонн до содержания 13C в отбираемом из этого каскада полупродукте 92% ат. Полученный таким образом полупродукт гидрируется водородом на промышленно выпускаемом катализаторе метанирования (ТО-2) до метана. При этом в качестве водорода используется отвальный газ производства дейтерия ректификацией жидкого водорода. Указанный газ содержит 0,0015% ат. дейтерия, что в 10 раз меньше содержания дейтерия в природном водороде и более чем в 3 раза меньше аналогичного содержания в водороде электролитическом, используемом в прототипе. Общее содержание химических примесей в этом газе не превышает 108% ат., что в 5109 раз ниже, чем в электролитическом водороде, используемом в прототипе. Продукт гидрирования (метан) после глубокой осушки подается в непрерывноработающую противоточную заполненную цеолитом NaX адсорбционную колонну с обращением потоков на ее концах: внизу – десорбцией метана из цеолита при нагреве до ~ 470 К, а вверху – адсорбцией метана цеолитом при охлаждении до ~ 200 К. В адсорбционной колонне поддерживаются давление ~ 105 Па и температура ~ 200 К (коэффициент разделения смеси 13CH4–12CH4 в этих условиях равен = 1,02, соответствующий ему коэффициент обогащения 1,007), высокой скоростью межфазного изотопного обмена (высота эквивалентной теоретической ступени разделения в насадочных колоннах ВЭТС (2,2-2,6) см), а также большой плотностью орошения массообменных колонн углеродом.
Недостатком этого способа является трудность достижения в нем высоких концентраций тяжелого изотопа углерода из-за наличия в составе оксида углерода (II) тяжелого изотопа кислорода 18O. Например, природный оксид углерода (II) представляет собой в основном смесь 12C16O+13C16O+12C18O. Парциальные упругости паров 12C18O и 13C16O отличаются настолько мало ( = 1,002 при 74,4 К), что при ректификации в колонне происходит практически отделение только 12C16O от бинарной смеси 12C18O + 13C16O. Это ограничивает возможный уровень концентрации 13C в ректификационной колонне пределом < 95% ат. В реально работающих колоннах этот уровень не превышает 93% ат.
Наиболее близким решением к предлагаемому изобретению является способ разделения изотопов углерода, включающий: предварительное концентрирование целевого изотопа криогенной дистилляцией оксида углерода (II) в непрерывноработающих ректификационных колоннах; последующее гидрирование предварительно обогащенного продукта до метана водородом, полученным при электролизе воды природного изотопного состава, в котором содержание дейтерия составляет 5105 (0,005% ат. ); конечное концентрирование целевого изотопа в противоточных колоннах термодиффузией продукта гидрирования (метана) (см. Рабинович Г.Д. Разделение изотопов и других смесей термодиффузией. М.: Атомиздат. 1981 г. 98 с.) (прототип). Указанный способ обеспечивает концентрирование из изотопных смесей углерода как стабильных (12C; 13C), так и радиоактивных (14C) изотопов. Благодаря использованию в нем на стадии гидрирования CO водорода с пониженным содержанием дейтерия, на стадию конечного термодиффузионного концентрирования целевого изотопа, например 13C, в этом способе подается метан с пониженным содержанием дейтерозамещенных форм (в частности, CH3D). Это обеспечивает возможность достижения высоких концентраций целевого изотопа в продукте. Например, при концентрировании указанным способом 13C из природной изотопной смеси удается достичь содержания этого изотопа в продукте на уровне 99-99,8% ат.
К недостаткам этого способа следует отнести использование в нем на стадии гидрирования CO водорода, полученного при электролизе воды природного изотопного состава, а также проведение конечного концентрирования целевого изотопа термодиффузией метана в колоннах. Получаемый электролизом воды водород содержит до 0,5% об. примесей, в том числе до 25 г/см3 влаги. Кроме влаги в нем в качестве примесей присутствуют компоненты воздуха (O2, до 0,02% N2, Ar), а также CO2. Указанные примеси, поступая вместе с водородом на стадию гидрирования CO, способны не только загрязнять получаемый при этом метан и, как следствие, оказывать в последующем вредное воздействие на работу термодиффузионных колонн (снижая их производительность, а также вызывая в условиях высоких температур коррозию конструкционных материалов), но и приводить к потере обогащенного продукта за счет химического взаимодействия некоторых из них (O2, H2O) с CO и CH4, а также за счет его изотопного разбавления путем изотопного обмена и смешения с углеродом примесного CO2 природного изотопного состава. Наличие в используемом водороде указанных примесей приводит к необходимости либо его глубокой химической очистки перед стадией гидрирования, либо к последующей глубокой химической очистке получаемого с его использованием метана, используемого затем для питания термодиффузионных колонн. Оба процесса сложны в аппаратурном оформлении, их реализация ведет к повышению себестоимости целевого изотопного продукта. Получаемый при электролизе воды природного изотопного состава водород содержит пониженное относительно природного уровня (0,015% ат.) количество дейтерия (~ 0,005% ат.). Это, снижая на стадии гидрирования CO в подвергаемом термодиффузии метане содержание изотопнозамещенных форм (например, CH3D), способствует повышению достигаемого предела концентрации целевых изотопов (в частности, 13C) в продукте. Например, для 13C от 94,7 до 99 – 99,8% ат. Однако количество дейтерия в электролитическом водороде остается на высоком уровне и, как следствие, на высоком уровне остается содержание дейтерозамещенных форм метана в газе, подаваемом на термодиффузию. Это ограничивает возможности конечного термодиффузионного концентрирования изотопов углерода в рассматриваемом способе уровнем 99 – 99,8% ат. Использование в рассматриваемом способе на стадии конечного концентрирования термодиффузии метана ведет к резкому усложнению аппаратурного оформления процесса конечного концентрирования, к необходимости использования для его аппаратурного оформления драгоценных металлов, а также к низкой эффективности самого процесса конечного концентрирования, которая определяется коэффициентом разделения изотопов (например, для смеси 12CH4–13CH4 = 1,009) и высотой эквивалентной теоретической ступени (ВЭТС 4 см) разделения в термодиффузионной колонне. В связи с этим можно отметить, что из-за высоких температур в термодиффузионных колоннах (~ 695 К) горячие проволоки внутри них изготавливаются из платино-иридиевого сплава (80% Pt и 20% Ir). В прототипе для получения нескольких литров метана с концентрацией 13C 99 – 99,8% ат. из предварительно обогащенного дистилляцией CO до ~ 70% ат. 13C метана потребовался каскад из восьми последовательно соединенных колонн с внутренним диаметром 12,1 мм, эффективной высотой 2,85 м с горячей проволокой диаметром 0,4 мм. Низкое для термодиффузии значение и высокое ВЭТС=h ведут к большому объему (V ~ h/( – 1)2) и длине (H ~ h/ ( – 1)) массообменных колонн, к большой величине циркулирующих внутри них флегмовых потоков (L ~ 1/( -1)), а также к высокому уровню энергозатрат (E ~ 1/( -1)) на осуществление процесса разделения.
Задачей создания изобретения является увеличение предела концентрирования целевых изотопов углерода, упрощение аппаратурного оформления процесса их разделения, снижение капитальных и эксплуатационных затрат на его осуществление, а также увеличение коэффициента разделения изотопов углерода на стадии их конечного концентрирования.
Поставленная задача решается тем, что в способе разделения изотопов углерода, включающем предварительное концентрирование целевого изотопа криогенной ректификацией оксида углерода (II), гидрирование концентрата водородом с пониженным относительно природного уровня содержанием дейтерия до метана и конечное концентрирование целевого изотопа в противоточных колоннах в форме метана, в качестве водорода используют отвальный газ производства дейтерия криогенной ректификацией водорода природного изотопного состава, а конечное концентрирование осуществляют криогенной адсорбцией метана на цеолите. При этом в качестве цеолита преимущественно используют цеолиты NaX (13X), NaA (4A) или CaA (5A).
Предлагаемое изобретение иллюстрируется следующими примерами.
Пример 1. Природная изотопная смесь углерода с содержанием 13C 1,104% ат. в форме предварительно очищенного криогенной ректификацией в насадочной колонне от химических примесей оксида углерода (II) подвергается 81,6 К ( 1,007) разделению в каскаде ректификационных насадочных колонн до содержания 13C в отбираемом из этого каскада полупродукте 92% ат. Полученный таким образом полупродукт гидрируется водородом на промышленно выпускаемом катализаторе метанирования (ТО-2) до метана. При этом в качестве водорода используется отвальный газ производства дейтерия ректификацией жидкого водорода. Указанный газ содержит 0,0015% ат. дейтерия, что в 10 раз меньше содержания дейтерия в природном водороде и более чем в 3 раза меньше аналогичного содержания в водороде электролитическом, используемом в прототипе. Общее содержание химических примесей в этом газе не превышает 108% ат., что в 5109 раз ниже, чем в электролитическом водороде, используемом в прототипе. Продукт гидрирования (метан) после глубокой осушки подается в непрерывноработающую противоточную заполненную цеолитом NaX адсорбционную колонну с обращением потоков на ее концах: внизу – десорбцией метана из цеолита при нагреве до ~ 470 К, а вверху – адсорбцией метана цеолитом при охлаждении до ~ 200 К. В адсорбционной колонне поддерживаются давление ~ 105 Па и температура ~ 200 К (коэффициент разделения смеси 13CH4–12CH4 в этих условиях равен = 1,02, соответствующий ему коэффициент обогащения  = – 1 превышает аналогичный коэффициент обогащения в прототипе в ~ 2,22 раза). Колонна заполняется гранулированным цеолитом NaX (13X) с размером гранул 1 мм. Теплота адсорбции (десорбции) метана на цеолите NaX(13X) в этих условиях равна ~ 4,3 кДж/моль. При удельной величине флегмового потока метана внутри колонны ~ 7,64 моль/(м2с), длине концентрирующей части колонны 2,7 м в ней на богатом конце достигнута концентрация 13C 99,95% ат., что превышает аналогичную концентрацию, достигнутую в прототипе (99-99,8 %). Высота эквивалентной теоретической ступени разделения в указанных условиях работы адсорбционной колонны составила h 1,33 см, что ~ в 3 раза ниже аналогичной величины в прототипе.
Пример 2. Используя метан, полученный в результате каталитического гидрирования (водородом, произведенным в качестве отвального газа при производстве дейтерия криогенной ректификацией жидкого водорода природного изотопного состава) предварительно обогащенного криогенной дистилляцией по 13C оксида углерода (II), методами однократного и двухкратного изотопного уравновешивания углерода в газовой и сорбированной на цеолите NaX (13X) фазах, путем масс-спектрометрического замера концентраций 13C в уравновешенных фазах определяют коэффициент разделения изотопной смеси 12CH4 – 13CH4 на стадии ее конечного концентрирования сорбцией на цеолите NaX (13X). Результаты определения отражены в таблице 1.
Из данных таблицы 1 видно, что коэффициент обогащения изотопной смеси 13CH4 – 12CH4 на стадии конечного концентрирования 13C в предлагаемом способе при (145 – 273) К в ~ 1,2-5,9 раз больше чем в прототипе.
Пример 3. В условиях, аналогичных примеру 2, определяют коэффициент разделения смеси 13CH4 – 12CH4 на стадии ее конечного концентрирования сорбцией на цеолите NaA (4A). Результаты определения отражены в таблице 2.
Из данных таблицы 2 видно, что коэффициент обогащения изотопной смеси 13CH4–12CH4 на стадии конечного концентрирования 13C в предлагаемом способе при (143-188) К в ~ 2,9-5,8 раз больше чем в прототипе.
Пример 4. В условиях, аналогичных примеру 2, определяют коэффициент разделения смеси 13CH4–12CH4 на стадии ее конечного концентрирования сорбцией на цеолите CaA (5A). Результаты определения отражены в таблице 3.
Из данных таблицы 3 видно, что коэффициент обращения изотопной смеси 13CH4–12CH4 на стадии конечного концентрирования 13C в предлагаемом способе при (142-210) К в ~ 1,9-5,7 раз больше чем в прототипе.
Приведенные примеры свидетельствуют, что предлагаемый способ по сравнению с прототипом обеспечивает достижение в конечном продукте более высоких концентраций целевого изотопа, характеризуется на стадии конечного концентрирования более высокими (до 6 раз) значениями коэффициента обогащения, а также более низкими ( ~ в 3 раза) значениями высоты эквивалентной теоретической ступени разделения, что свидетельствует о его большей эффективности. Кроме того, предлагаемый способ позволяет обеспечить конечное концентрирование целевых изотопов в более простой аппаратуре (одиночная колонна) существенно меньших размеров, выполненной из стекла или дешевых металлических сплавов. Его использование исключает применение драгоценных металлов (платины, иридия) для аппаратурного оформления процесса конечного концентрирования изотопов углерода. Указанные выше достоинства в совокупности обеспечивают снижение капитальных, а также эксплуатационных затрат на реализацию процесса разделения изотопов углерода и, как следствие, себестоимость целевых изотопов. Предлагаемый способ, также как и прототип, ввиду высокой радиационной устойчивости используемых в нем сорбентов и газов, обеспечивает возможность производства не только высококонцентрированных стабильных изотопов углерода, но и его радиоактивного изотопа углерода-14. = – 1 превышает аналогичный коэффициент обогащения в прототипе в ~ 2,22 раза). Колонна заполняется гранулированным цеолитом NaX (13X) с размером гранул 1 мм. Теплота адсорбции (десорбции) метана на цеолите NaX(13X) в этих условиях равна ~ 4,3 кДж/моль. При удельной величине флегмового потока метана внутри колонны ~ 7,64 моль/(м2с), длине концентрирующей части колонны 2,7 м в ней на богатом конце достигнута концентрация 13C 99,95% ат., что превышает аналогичную концентрацию, достигнутую в прототипе (99-99,8 %). Высота эквивалентной теоретической ступени разделения в указанных условиях работы адсорбционной колонны составила h 1,33 см, что ~ в 3 раза ниже аналогичной величины в прототипе.
Пример 2. Используя метан, полученный в результате каталитического гидрирования (водородом, произведенным в качестве отвального газа при производстве дейтерия криогенной ректификацией жидкого водорода природного изотопного состава) предварительно обогащенного криогенной дистилляцией по 13C оксида углерода (II), методами однократного и двухкратного изотопного уравновешивания углерода в газовой и сорбированной на цеолите NaX (13X) фазах, путем масс-спектрометрического замера концентраций 13C в уравновешенных фазах определяют коэффициент разделения изотопной смеси 12CH4 – 13CH4 на стадии ее конечного концентрирования сорбцией на цеолите NaX (13X). Результаты определения отражены в таблице 1.
Из данных таблицы 1 видно, что коэффициент обогащения изотопной смеси 13CH4 – 12CH4 на стадии конечного концентрирования 13C в предлагаемом способе при (145 – 273) К в ~ 1,2-5,9 раз больше чем в прототипе.
Пример 3. В условиях, аналогичных примеру 2, определяют коэффициент разделения смеси 13CH4 – 12CH4 на стадии ее конечного концентрирования сорбцией на цеолите NaA (4A). Результаты определения отражены в таблице 2.
Из данных таблицы 2 видно, что коэффициент обогащения изотопной смеси 13CH4–12CH4 на стадии конечного концентрирования 13C в предлагаемом способе при (143-188) К в ~ 2,9-5,8 раз больше чем в прототипе.
Пример 4. В условиях, аналогичных примеру 2, определяют коэффициент разделения смеси 13CH4–12CH4 на стадии ее конечного концентрирования сорбцией на цеолите CaA (5A). Результаты определения отражены в таблице 3.
Из данных таблицы 3 видно, что коэффициент обращения изотопной смеси 13CH4–12CH4 на стадии конечного концентрирования 13C в предлагаемом способе при (142-210) К в ~ 1,9-5,7 раз больше чем в прототипе.
Приведенные примеры свидетельствуют, что предлагаемый способ по сравнению с прототипом обеспечивает достижение в конечном продукте более высоких концентраций целевого изотопа, характеризуется на стадии конечного концентрирования более высокими (до 6 раз) значениями коэффициента обогащения, а также более низкими ( ~ в 3 раза) значениями высоты эквивалентной теоретической ступени разделения, что свидетельствует о его большей эффективности. Кроме того, предлагаемый способ позволяет обеспечить конечное концентрирование целевых изотопов в более простой аппаратуре (одиночная колонна) существенно меньших размеров, выполненной из стекла или дешевых металлических сплавов. Его использование исключает применение драгоценных металлов (платины, иридия) для аппаратурного оформления процесса конечного концентрирования изотопов углерода. Указанные выше достоинства в совокупности обеспечивают снижение капитальных, а также эксплуатационных затрат на реализацию процесса разделения изотопов углерода и, как следствие, себестоимость целевых изотопов. Предлагаемый способ, также как и прототип, ввиду высокой радиационной устойчивости используемых в нем сорбентов и газов, обеспечивает возможность производства не только высококонцентрированных стабильных изотопов углерода, но и его радиоактивного изотопа углерода-14.
Формула изобретения
РИСУНКИ
|
||||||||||||||||||||||||||