Патент на изобретение №2379312
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) СОЕДИНЕНИЯ НА ОСНОВЕ ЛИЗИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЭТИ СОЕДИНЕНИЯ, ПРИМЕНЕНИЕ УКАЗАННЫХ СОЕДИНЕНИЙ ДЛЯ ЛЕЧЕНИЯ ИЛИ ПРОФИЛАКТИКИ ВИЧ ИНФЕКЦИИ
(57) Реферат:
Настоящее изобретение относится к соединениям на основе лизина формулы (I) или его фармацевтически приемлемым солям, фармацевтическим композициям на их основе и применению для лечения или профилактики ВИЧ-инфекции. Соединения формулы (I), где n равно 3 или 4, где Х и Y, одинаковые или различные, выбраны из группы, состоящей из Н, F, Cl, Br, I и -NR4R5, где R6 выбран из группы, состоящей из неразветвленной алкильной группы, содержащей от 1 до 6 атомов углерода, и разветвленной алкильной группы, содержащей от 3 до 6 атомов углерода, где R3 выбран из группы, состоящей из группы формулы R3A-CO-, причем R3A выбран из группы, состоящей из неразветвленной или разветвленной алкильной группы, содержащей от 1 до 6 атомов углерода, алкилоксигруппы, содержащей от 1 до 6 атомов углерода, и 4-морфолинила, где R4 и R5, одинаковые, представляют собой Н, где R2 выбран из группы, состоящей из дифенилметильной группы, нафтил-1-СН2-группы, и нафтил-2-СН2-группы, где X’ и У’ одинаковые, представляют собой Н, и где R1 выбран из группы, состоящей из (НО)2Р(O) и (МО)2Р(O), где М представляет собой щелочной металл. Технический результат – соединения на основе лизина, представляющие собой эффективные ингибиторы аспартил-протеазы. 4 н. и 11 з.п. ф-лы, 3 табл.
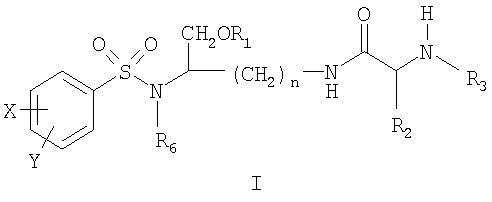
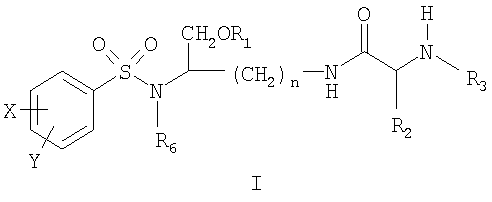
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ Данное изобретение относится к соединениям на основе лизина, которые демонстрируют хорошую растворимость и биологическую доступность. Более конкретно настоящее изобретение относится к соединениям на основе лизина, содержащим физиологически отщепляемое звено, посредством чего при отщеплении данного звена соединение способно высвобождать ингибитор протеазы ВИЧ. Соединения и фармацевтические композиции по настоящему изобретению особенно хорошо подходят для снижения лекарственной нагрузки и улучшения соблюдения больным режима и схемы лечения. УРОВЕНЬ ТЕХНИКИ Ингибиторы вирусной протеазы ВИЧ были разработаны относительно недавно, и их использование началось только в 1996 году. В настоящее время они рассматриваются как наиболее эффективные лекарственные препараты против ВИЧ-инфекции. К сожалению, большинство имеющихся в настоящее время ингибиторов протеаз представляют собой относительно крупные гидрофобные молекулы, которые обладают довольно низкой биологической доступностью. Поэтому для достижения терапевтической дозы у пациента требуется высокая лекарственная нагрузка. Это является сдерживающим фактором, который слишком часто приводит к несоблюдению больным режима и схемы лечения и к недостаточным результатам лечения. Данная ситуация ведет к субоптимальной терапевтической концентрации лекарственного препарата, что в свою очередь ведет к развитию устойчивых штаммов ВИЧ. Следовательно, существует неотложная потребность улучшения растворимости и биологической доступности ингибиторов протеазы. Примеры улучшенных соединений были разработаны в форме пролекарств ингибиторов аспартил-протеазы, таких как соединения, описанные, например, в патенте США Уникальный класс ароматических производных, которые являются ингибиторами аспартил-протеаз, описан в патенте США СУЩНОСТЬ ИЗОБРЕТЕНИЯ Настоящее изобретение предлагает новые соединения на основе лизина, источником которых является класс производных, являющихся эффективными ингибиторами аспартил-протеазы, и их фармацевтически приемлемые производные. Данные соединения могут легко расщепляться in vivo, высвобождая активный ингредиент. Активный ингредиент имеет сродство к аспартил-протеазам, в частности к аспартил-протеазе ВИЧ-1 (патент США Основная цель данного изобретения состоит в предложении улучшенного класса соединений на основе лизина, которые способны высвобождать ингибитор аспартил-протеазы и, особенно, ингибиторы ВИЧ аспартил-протеазы. Соединения на основе лизина по настоящему изобретению могут содержать отщепляемое звено, посредством чего при отщеплении данного звена соединение способно высвобождать ингибитор ВИЧ-протеазы. Настоящее изобретение также предлагает фармацевтические композиции, включающие описанные здесь соединения на основе лизина. Следовательно, настоящее изобретение в одном аспекте предлагает соединения на основе лизина, которые при in vivo физиологических условиях (например, метаболических, кишечных, желудочно-кишечных и т.д.) позволяют высвобождаться ингибитору протеазы (например, ингибитору аспартил-протеазы). Соединения по настоящему изобретению могут служить в качестве средства для улучшения растворимости и/или биологической доступности ингибиторов протеазы, и, поэтому, могут снижать лекарственную нагрузку, и могут благоприятствовать соблюдению больным режима и схемы лечения. Соединения по настоящему изобретению могут содержать, например, (например, физиологически) расщепляющуюся (например, гидролизующуюся) связь или звено, которые при отщеплении расщепляющейся связи или звена дают ингибитор протеазы (например, активный ингибитор протеазы). Ингибитор протеазы может действовать на аспартил-протеазу ВИЧ-1, включая мутантные или немутантные вирусные штаммы ВИЧ-1 (например, NL4.3), или на протеазу ВИЧ-2 (мутантного или немутантного) или даже на протеазу родственных вирусов (SIV и т.д.). Соединения по настоящему изобретению можно использовать в одиночку или в комбинации с другими терапевтическими или профилактическими средствами для лечения или профилактики, например, ВИЧ-инфекции. Соединения и фармацевтические композиции по настоящему изобретению могут высвобождать ингибитор протеазы (активный ингредиент) in vivo и, посредством этого, могут подавлять (например, in vivo) активность ВИЧ аспартил-протеазы, фермента, существенного для развития и инфективности вируса. Соединения и фармацевтические композиции по настоящему изобретению могут обладать более высокой биологической доступностью, а также могут подходить для снижения необходимых для подавления дозировок и, следовательно, могут улучшать лечение ВИЧ-инфицированных пациентов. Настоящее изобретение согласно своему одному аспекту предлагает соединение (например, соединение, способное генерировать ингибитор ВИЧ-протеазы) формулы I:
и его фармацевтически приемлемые соли и производные (например, когда соединение по настоящему изобретению включает аминогруппу, фармацевтически приемлемая соль может представлять собой соль аммония), где n может быть равно, например, 3 или 4, где X и Y, одинаковые или различные, могут быть выбраны, например, из группы, состоящей из H, неразветвленной алкильной группы, содержащей от 1 до 6 атомов углерода, разветвленной алкильной группы, содержащей от 3 до 6 атомов углерода, циклоалкильной группы, содержащей от 3 до 6 атомов углерода, F, Cl, Br, I, -CF3, -OCF3, -CN, -NO2, -NR4R5, -NHCOR4, -OR4, -SR4, -COOR4, -COR4 и -CH2OH, или X и Y вместе определяют алкилендиоксигруппу, выбранную из группы, состоящей из метилендиоксигруппы формулы -OCH2O- и этилендиоксигруппы формулы -OCH2CH2O-, где R6 может быть выбран, например, из группы, состоящей из неразветвленной алкильной группы, содержащей от 1 до 6 атомов углерода, разветвленной алкильной группы, содержащей от 3 до 6 атомов углерода, циклоалкилалкильной группы, содержащей от 3 до 6 атомов углерода в своей циклоалкильной части и от 1 до 3 атомов углерода в своей алкильной части, где R3 может быть выбран, например, из группы, состоящей из H, неразветвленной алкильной группы, содержащей от 1 до 6 атомов углерода, разветвленной алкильной группы, содержащей от 3 до 6 атомов углерода, циклоалкильной группы, содержащей от 3 до 6 атомов углерода, и группы формулы R3A-CO-, где R3A может быть выбран, например, из группы, состоящей из неразветвленной или разветвленной алкильной группы, содержащей от 1 до 6 атомов углерода (например, метильной, этильной, пропильной, изопропильной, бутильной, изобутильной, трет-бутильной, трет-бутил-CH2 и т.д.), циклоалкильной группы, содержащей от 3 до 6 атомов углерода (например, циклопропильной, циклогексильной и т.д.), циклоалкилалкильной группы, содержащей от 3 до 6 атомов углерода в своей циклоалкильной части и от 1 до 3 атомов углерода в своей алкильной части (например, циклопропил-CH2-, циклогексил-CH2– и т.д.), алкилоксигруппы, содержащей от 1 до 6 атомов углерода (например, CH3O-, CH3CH2O-, изобутил-O-, трет-бутил-O- (Boc) и т.д.), тетрагидро-3-фуранилокси, -CH2OH, -CF3, -CH2CF3, -CH2CH2CF3, пирролидинила, пиперидинила, 4-морфолинила, CH3O2C-, CH3O2CCH2-, ацетил-OCH2CH2-, HO2CCH2-, 3-гидроксифенила, 4-гидроксифенила, 4-CH3OC6H4CH2-, CH3NH-, (CH3)2N-, (CH3CH2)2N-, (CH3CH2CH2)2N-, HOCH2CH2NH-, CH3OCH2O-, CH3OCH2CH2O-, C6H5CH2O-, 2-пирролила, 2-пиридила, 3-пиридила, 4-пиридила, 2-пиразинила, 2-хинолила, 3-хинолила, 4-хинолила, 1-изохинолила, 3-изохинолила, 2-хиноксалинила, фенильной группы формулы
пиколильной группы, выбранной из группы, состоящей из
пиколилоксигруппы, выбранной из группы, состоящей из
замещенной пиридильной группы, выбранной из группы, состоящей из
и группы формулы
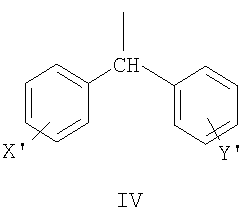
где X’ и Y’, одинаковые или различные, могут быть выбраны, например, из группы, состоящей из H, неразветвленной алкильной группы, содержащей от 1 до 6 атомов углерода, разветвленной алкильной группы, содержащей от 3 до 6 атомов углерода, циклоалкильной группы, содержащей от 3 до 6 атомов углерода, F, Cl, Br, I, -CF3, -NO2, -NR4R5, -NHCOR4, -OR4, -SR4, -COOR4, -COR4 и -CH2OH, где R4 и R5, одинаковые или различные, могут быть выбраны, например, из группы, состоящей из H, неразветвленной алкильной группы, содержащей от 1 до 6 атомов углерода, разветвленной алкильной группы, содержащей от 3 до 6 атомов углерода, циклоалкильной группы, содержащей от 3 до 6 атомов углерода, где R2 может быть выбран, например, из группы, состоящей из дифенилметильной группы, формулы IV
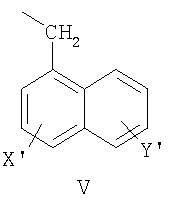
нафтил-1-CH2-группы формулы V
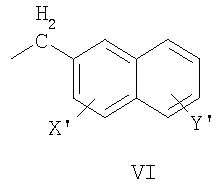
нафтил-2-CH2-группы формулы VI
бифенилметильной группы формулы VII
и антрил-9-CH2-группы формулы VIII
и где R1 может представлять собой отщепляемое звено (например, физиологически отщепляемое звено), посредством чего при отщеплении данного звена соединение высвобождает ингибитор протеазы (ингибитор протеазы ВИЧ), при условии, что R1 не является H. Например, R1 может представлять собой ферментно или метаболически отщепляемое звено или гидролизующуюся связь, которые могут быть отщеплены в условиях, существующих в кишечнике и/или желудочно-кишечном тракте (рН) или других физиологических условиях. По настоящему изобретению R1 можно выбрать, например, из группы, состоящей из (HO)2P(O) и (MO)2P(O), где M представляет собой щелочной металл (например, Na, K, Cs и т.д.) или щелочноземельный металл (Ca, Mg и т.д.). Далее по настоящему изобретению R1 может представлять собой группу формулы R1A-CO-, где R1A может быть выбран, например, из группы, состоящей из неразветвленной или разветвленной алкильной группы, содержащей от 1 до 6 атомов углерода (например, метильной, этильной, пропильной, изопропильной, бутильной, изобутильной, трет-бутильной, трет-бутил-CH2– и т.д.), циклоалкильной группы, содержащей от 3 до 6 атомов углерода (например, циклопропильной, циклогексильной и т.д.), циклоалкилалкильной группы, содержащей от 3 до 6 атомов углерода в своей циклоалкильной части и от 1 до 3 атомов углерода в своей алкильной части (например, циклопропил-CH2-, циклогексил-CH2– и т.д.), алкилоксигруппы, содержащей от 1 до 6 атомов углерода (например, CH3O-, CH3CH2O-, изобутил-O-, трет-бутил-O- (Boc) и т.д.), -CH2OH, CH3O2C-, CH3O2CCH2-, ацетил-OCH2CH2-, HO2CCH2-, 2-гидроксифенила, 3-гидроксифенила, 4-гидроксифенила, (CH3)2NCH2-, (CH3)2CHCH(NH2)-, HOCH2CH2NH-, CH3OCH2O-, CH3OCH2CH2O-, 2-пирролила, 2-пиридила, 3-пиридила, 4-пиридила, 1-метил-1,4-дигидро-3-пиридила, 2-пиразинила, 2-хинолила, 3-хинолила, 4-хинолила, 1-изохинолила, 3-изохинолила, 2-хиноксалинила, фенильной группы формулы
пиколильной группы, выбранной из группы, состоящей из
пиколилоксигруппы, выбранной из группы, состоящей из
замещенной пиридильной группы, выбранной из группы, состоящей из
и группы формулы
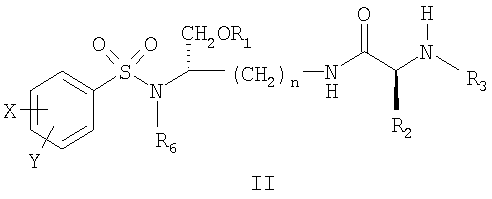
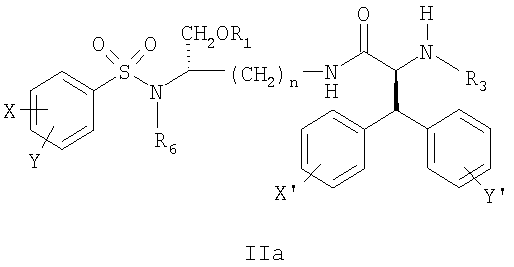
где X’, Y’, R4 и R5 являются такими, как определено в настоящем описании. В другом аспекте настоящее изобретение далее предлагает соединение формулы II,
и его фармацевтически приемлемые соли и производные (например, когда соединение по настоящему изобретению включает аминогруппу, фармацевтически приемлемая соль может представлять собой соль аммония), где n может быть равно 3 или 4, где X и Y, одинаковые или различные, могут быть выбраны, например, из группы, состоящей из H, неразветвленной алкильной группы, содержащей от 1 до 6 атомов углерода, разветвленной алкильной группы, содержащей от 3 до 6 атомов углерода, циклоалкильной группы, содержащей от 3 до 6 атомов углерода, F, Cl, Br, I, -CF3, -OCF3, -CN, -NO2, -NR4R5, -NHCOR4, -OR4, -SR4, -COOR4, -COR4 и -CH2OH, или X и Y вместе определяют алкилендиоксигруппу, выбранную из группы, состоящей из метилендиоксигруппы формулы -OCH2O- и этилендиоксигруппы формулы -OCH2CH2O-, где R6 может быть выбран, например, из группы, состоящей из неразветвленной алкильной группы, содержащей от 1 до 6 атомов углерода, разветвленной алкильной группы, содержащей от 3 до 6 атомов углерода, циклоалкилалкильной группы, содержащей от 3 до 6 атомов углерода в своей циклоалкильной части и от 1 до 3 атомов углерода в своей алкильной части, где R3 может быть выбран, например, из группы, состоящей из H, неразветвленной алкильной группы, содержащей от 1 до 6 атомов углерода, разветвленной алкильной группы, содержащей от 3 до 6 атомов углерода, циклоалкильной группы, содержащей от 3 до 6 атомов углерода, и группы формулы R3A-CO-, где R3A может быть выбран, например, из группы, состоящей из неразветвленной или разветвленной алкильной группы, содержащей от 1 до 6 атомов углерода (например, метильной, этильной, пропильной, изопропильной, бутильной, изобутильной, трет-бутильной, трет-бутил-CH2– и т.д.), циклоалкильной группы, содержащей от 3 до 6 атомов углерода (например, циклопропильной, циклогексильной и т.д.), циклоалкилалкильной группы, содержащей от 3 до 6 атомов углерода в своей циклоалкильной части и от 1 до 3 атомов углерода в своей алкильной части (например, циклопропил-CH2-, циклогексил-CH2– и т.д.), алкилоксигруппы, содержащей от 1 до 6 атомов углерода (например, CH3O-, CH3CH2O-, изобутил-O-, трет-бутил-O- (Boc) и т.д.), тетрагидро-3-фуранилокси, -CH2OH, -CF3, -CH2CF3, -CH2CH2CF3, пирролидинила, пиперидинила, 4-морфолинила, CH3O2C-, CH3O2CCH2-, ацетил-OCH2CH2-, HO2CCH2-, 3-гидроксифенила, 4-гидроксифенила, 4-CH3OC6H4CH2-, CH3NH-, (CH3)2N-, (CH3CH2)2N-, (CH3CH2CH2)2N-, HOCH2CH2NH-, CH3OCH2O-, CH3OCH2CH2O-, C6H5CH2O-, 2-пирролила, 2-пиридила, 3-пиридила, 4-пиридила, 2-пиразинила, 2-хинолила, 3-хинолила, 4-хинолила, 1-изохинолила, 3-изохинолила, 2-хиноксалинила, фенильной группы формулы
пиколильной группы, выбранной из группы, состоящей из
пиколилоксигруппы, выбранной из группы, состоящей из
замещенной пиридильной группы, выбранной из группы, состоящей из
и группы формулы
где X’ и Y’, одинаковые или различные, могут быть выбраны, например, из группы, состоящей из H, неразветвленной алкильной группы, содержащей от 1 до 6 атомов углерода, разветвленной алкильной группы, содержащей от 3 до 6 атомов углерода, циклоалкильной группы, содержащей от 3 до 6 атомов углерода, F, Cl, Br, I, -CF3, -NO2, -NR4R5, -NHCOR4, -OR4, -SR4, -COOR4, -COR4 и -CH2OH, где R4 и R5, одинаковые или различные, могут быть выбраны, например, из группы, состоящей из H, неразветвленной алкильной группы, содержащей от 1 до 6 атомов углерода, разветвленной алкильной группы, содержащей от 3 до 6 атомов углерода, и циклоалкильной группы, содержащей от 3 до 6 атомов углерода, где R2 может быть выбран, например, из группы, состоящей из дифенилметильной группы, формулы IV
нафтил-1-CH2-группы формулы V
нафтил-2-CH2-группы формулы VI
бифенилметильной группы формулы VII
и антрил-9-CH2-группы формулы VIII
и где R1 может представлять собой физиологически отщепляемое звено, посредством чего при отщеплении данного звена соединение может быть способно высвобождать ингибитор протеазы при условии, что R1 не является H. По настоящему изобретению R1 можно выбрать, например, из группы, состоящей из (HO)2P(O) и (MO)2P(O), где M представляет собой щелочной металл (например, Na, K, Cs и т.д.) или щелочноземельный металл (Ca, Mg и т.д.). Далее по настоящему изобретению R1 может представлять собой группу формулы R1A-CO-, где R1A может быть выбран из группы, состоящей из неразветвленной или разветвленной алкильной группы, содержащей от 1 до 6 атомов углерода (например, метильной, этильной, пропильной, изопропильной, бутильной, изобутильной, трет-бутильной, трет-бутил-CH2– и т.д.), циклоалкильной группы, содержащей от 3 до 6 атомов углерода (например, циклопропильной, циклогексильной и т.д.), циклоалкилалкильной группы, содержащей от 3 до 6 атомов углерода в своей циклоалкильной части и от 1 до 3 атомов углерода в своей алкильной части (например, циклопропил-CH2-, циклогексил-CH2– и т.д.), алкилоксигруппы, содержащей от 1 до 6 атомов углерода (например, CH3O-, CH3CH2O-, изобутил-O-, трет-бутил-O- (Boc) и т.д.), -CH2OH, CH3O2C-, CH3O2CCH2-, ацетил-OCH2CH2-, HO2CCH2-, 2-гидроксифенила, 3-гидроксифенила, 4-гидроксифенила, (CH3)2NCH2-, (CH3)2CHCH(NH2)-, HOCH2CH2NH-, CH3OCH2O-, CH3OCH2CH2O-, 2-пирролила, 2-пиридила, 3-пиридила, 4-пиридила, 1-метил-1,4-дигидро-3-пиридила, 2-пиразинила, 2-хинолила, 3-хинолила, 4-хинолила, 1-изохинолила, 3-изохинолила, 2-хиноксалинила, фенильной группы формулы
пиколильной группы, выбранной из группы, состоящей из
пиколилоксигруппы, выбранной из группы, состоящей из
замещенной пиридильной группы, выбранной из группы, состоящей из
и группы формулы
где X’, Y’, R4 и R5 являются такими, как определено в настоящем описании. В дальнейшем аспекте настоящее изобретение предлагает соединение формулы IIa:
его фармацевтически приемлемые соли и производные (например, когда соединение по настоящему изобретению включает аминогруппу, фармацевтически приемлемая соль может представлять собой соль аммония), где X и Y, одинаковые или различные, могут быть выбраны, например, из группы, состоящей из H, неразветвленной алкильной группы, содержащей от 1 до 6 атомов углерода, разветвленной алкильной группы, содержащей от 3 до 6 атомов углерода, циклоалкильной группы, содержащей от 3 до 6 атомов углерода, F, Cl, Br, I, -CF3, -OCF3, -CN, -NO2, -NR4R5, -NHCOR4, -OR4, -SR4, -COOR4, -COR4 и -CH2OH, или X и Y вместе определяют алкилендиоксигруппу, выбранную из группы, состоящей из метилендиоксигруппы формулы -OCH2O- и этилендиоксигруппы формулы -OCH2CH2O-, где X’ и Y’, одинаковые или различные, могут быть выбраны, например, из группы, состоящей из H, неразветвленной алкильной группы, содержащей от 1 до 6 атомов углерода, разветвленной алкильной группы, содержащей от 3 до 6 атомов углерода, циклоалкильной группы, содержащей от 3 до 6 атомов углерода, F, Cl, Br, I, -CF3, -NO2, -NR4R5, -NHCOR4, -OR4, -SR4, -COOR4, -COR4 и -CH2OH, и где n, R1, R3, R4, R5 и R6 являются такими, как определено в настоящем описании. В дополнительном аспекте настоящее изобретение предлагает соединение формулы IIb:
его фармацевтически приемлемые соли и производные (например, когда соединение по настоящему изобретению включает аминогруппу, фармацевтически приемлемая соль может представлять собой соль аммония), где X и Y, одинаковые или различные, могут быть выбраны, например, из группы, состоящей из H, неразветвленной алкильной группы, содержащей от 1 до 6 атомов углерода, разветвленной алкильной группы, содержащей от 3 до 6 атомов углерода, циклоалкильной группы, содержащей от 3 до 6 атомов углерода, F, Cl, Br, I, -CF3, -OCF3, -CN, -NO2, -NR4R5, -NHCOR4, -OR4, -SR4, -COOR4, -COR4 и -CH2OH, или X и Y вместе определяют алкилендиоксигруппу, выбранную из группы, состоящей из метилендиоксигруппы формулы -OCH2O- и этилендиоксигруппы формулы -OCH2CH2O-, где X’ и Y’, одинаковые или различные, могут быть выбраны, например, из группы, состоящей из H, неразветвленной алкильной группы, содержащей от 1 до 6 атомов углерода, разветвленной алкильной группы, содержащей от 3 до 6 атомов углерода, циклоалкильной группы, содержащей от 3 до 6 атомов углерода, F, Cl, Br, I, -CF3, -NO2, -NR4R5, -NHCOR4, -OR4, -SR4, -COOR4, -COR4 и -CH2OH, и где n, R1, R3, R4, R5 и R6 являются такими, как определено в настоящем описании. В еще одном дополнительном аспекте настоящее изобретение предлагает соединение формулы IIc:
его фармацевтически приемлемые соли и производные (например, когда соединение по настоящему изобретению включает аминогруппу, фармацевтически приемлемая соль может представлять собой соль аммония), и где n, X, Y, X’, Y’, R1, R3, R4, R5 и R6 являются такими, как определено в настоящем описании. В другом аспекте настоящее изобретение относится к соединению формулы IIA
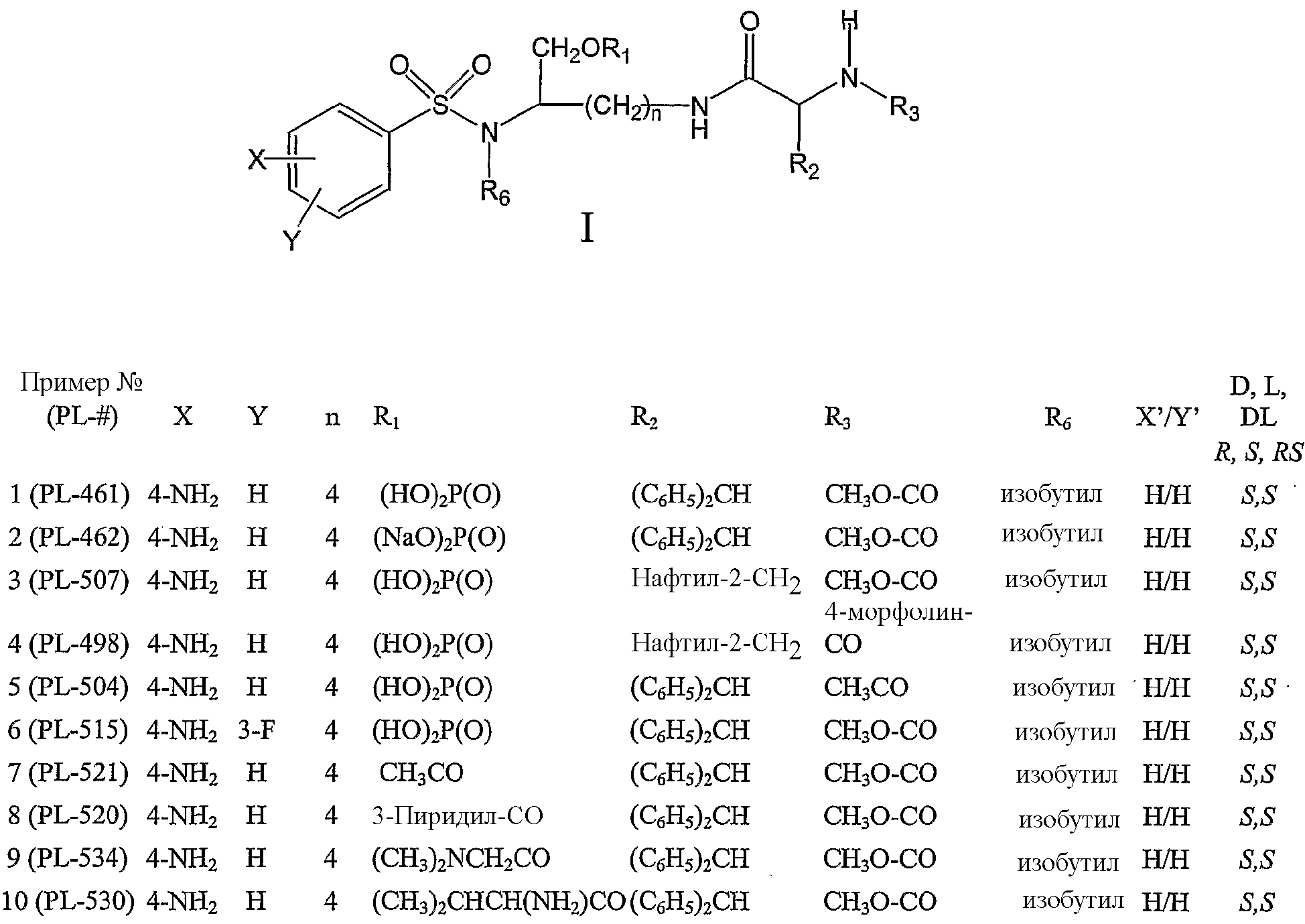
где Y, n, R1, R2, R3, X’ и Y’ являются такими, как определено в настоящем описании. По настоящему изобретению R1 может представлять собой, например, (HO)2P(O) или (NaO)2P(O). Далее по настоящему изобретению n может быть равно 4. Y может, например, представлять собой H. R3 может, например, представлять собой CH3O-CO. R2 может, например, представлять собой дифенилметильную группу формулы IV, где X’ и Y’ могут представлять собой, например, H,
Следовательно, соединения формулы IIA’, а также их фармацевтически приемлемые соли и производные охватываются настоящим изобретением,
такие как, например, соединение формулы IIA’, где R1 представляет собой (HO)2P(O), или соединение формулы IIA’, где R1 представляет собой (NaO)2P(O). В еще одном отличающемся аспекте настоящее изобретение относится к фармацевтической композиции, включающей, по меньшей мере, одно соединение формулы I, II, IIa, IIb, IIc, IIA, IIA’ или комбинацию соединений формулы I, II, IIa, IIb, IIc, IIA и/или IIA’. Фармацевтическая композиция может включать фармацевтически приемлемый носитель. Фармацевтическая композиция может включать, например, фармацевтически эффективное количество такого одного или нескольких соединений или, в соответствующих случаях, их фармацевтически приемлемых солей аммония. Например, фармацевтическая композиция по настоящему изобретению может включать одно или несколько из следующих ниже соединений: – соединение формулы IIa, где n равно 4, R1 представляет собой (HO)2P(O), X представляет собой 4-NH2, Y представляет собой H, X’ представляет собой H, Y’ представляет собой H, R6 представляет собой изобутил и R3 представляет собой CH3O-CO, – соединение формулы IIa, где n равно 4, R1 представляет собой (NaO)2P(O), X представляет собой 4-NH2, Y представляет собой H, X’ представляет собой H, Y’ представляет собой H, R6 представляет собой изобутил и R3 представляет собой CH3O-CO, – соединение формулы IIa, где n равно 4, R1 представляет собой (HO)2P(O), X представляет собой 4-NH2, Y представляет собой H, X’ представляет собой H, Y’ представляет собой H, R6 представляет собой изобутил и R3 представляет собой CH3CO, – соединение формулы IIa, где n равно 4, R1 представляет собой (HO)2P(O), X представляет собой 4-NH2, Y представляет собой 3-F, X’ представляет собой H, Y’ представляет собой H, R6 представляет собой изобутил и R3 представляет собой CH3O-CO, – соединение формулы IIa, где n равно 4, R1 представляет собой CH3CO, X представляет собой 4-NH2, Y представляет собой H, X’ представляет собой H, Y’ представляет собой H, R6 представляет собой изобутил и R3 представляет собой CH3O-CO, – соединение формулы IIa, где n равно 4, R1 представляет собой 3-пиридил-CO, X представляет собой 4-NH2, Y представляет собой H, X’ представляет собой H, Y’ представляет собой H, R6 представляет собой изобутил и R3 представляет собой CH3O-CO, – соединение формулы IIa, где n равно 4, R1 представляет собой (CH3)2NCH2CO, X представляет собой 4-NH2, Y представляет собой H, X’ представляет собой H, Y’ представляет собой H, R6 представляет собой изобутил и R3 представляет собой CH3O-CO, – соединение формулы IIa, где n равно 4, R1 представляет собой (CH3)2CHCH(NH2)CO, X представляет собой 4-NH2, Y представляет собой H, X’ представляет собой H, Y’ представляет собой H, R6 представляет собой изобутил и R3 представляет собой CH3O-CO, – соединение формулы IIb, где n равно 4, R1 представляет собой (HO)2P(O), X представляет собой 4-NH2, Y представляет собой H, X’ представляет собой H, Y’ представляет собой H, R6 представляет собой изобутил и R3 представляет собой CH3O-CO, и где нафтильная группа представляет собой нафтил-2-CH2 группу, – соединение формулы IIb, где n равно 4, R1 представляет собой (HO)2P(O), X представляет собой 4-NH2, Y представляет собой H, X’ представляет собой H, Y’ представляет собой H, R6 представляет собой изобутил и R3 представляет собой 4-морфолин-CO, и где нафтильная группа представляет собой нафтил-1-CH2 группу, или – комбинацию любого из вышеуказанных соединений. В дополнительном аспекте настоящее изобретение относится к использованию, по меньшей мере, одного соединения формулы I, II, IIa, IIb, IIc, IIA, IIA’ или комбинации соединений формулы I, II, IIa, IIb, IIc, IIA и/или IIA’ или его фармацевтически приемлемых солей или производных (а также их комбинаций) при изготовлении лекарственного препарата (или фармацевтической композиции) для лечения или профилактики ВИЧ-инфекции. В дальнейшем аспекте настоящее изобретение относится к использованию, по меньшей мере, одного соединения формулы I, II, IIa, IIb, IIc, IIA, IIA’ или комбинации соединений формулы I, II, IIa, IIb, IIc, IIA и/или IIA’ или его фармацевтически приемлемых солей или производных при лечении или профилактике ВИЧ-инфекции у млекопитающих, нуждающихся в этом, или для задержки возникновения СПИДа. В еще одном дополнительном аспекте настоящее изобретение относится к способу лечения или профилактики ВИЧ-инфекции (или для задержки возникновения СПИДа), включающему введение, по меньшей мере, одного соединения формулы I, II, IIa, IIb, IIc, IIA, IIA’ или комбинации соединений формулы I, II, IIa, IIb, IIc, IIA и/или IIA’ или его фармацевтически приемлемых солей или производных млекопитающему, нуждающемуся в этом. В другом аспекте настоящее изобретение относится к соединению формулы I, II, IIa, IIb, IIc, IIA или IIA’, его фармацевтически приемлемым солям или производным, для использования при лечении или профилактике ВИЧ-инфекции. В еще одном дополнительном аспекте настоящее изобретение относится к способу изготовления соединения на основе лизина, использующему любое из соединений, описанных в патенте США Соединения, перечисленные в настоящем описании, являются иллюстративными вариантами осуществления настоящего изобретения на практике и необходимо понимать, что настоящее изобретение не ограничивается только данными соединениями. Термин Термины Термин Термины Настоящее изобретение предлагает фармацевтически приемлемые производные соединений формулы I (такие как соединения формулы II, IIa, IIb, IIc, IIA и IIA’) и, где применимо, их фармацевтически приемлемые соли, такие как, например, соли аммония. Необходимо понимать, что Необходимо понимать, что Необходимо понимать, что Соли, полученные из соответствующих оснований, включают соли щелочных металлов (например, натрия), щелочноземельных металлов (например, магния), аммония и N-(C1-4алкил)4 Соединения по данному изобретению содержат один или более асимметричных атомов углерода и, таким образом, могут находиться в виде рацематов и рацемических смесей, отдельного энантиомера, диастереомерных смесей и индивидуальных диастереоизомеров. Все такие изомерные формы данных соединений определенно включены в настоящее изобретение. Каждый стереогенный атом углерода может иметь R или S конфигурацию. Фармацевтически приемлемые соли соединений по данному изобретению включают соли, полученные из фармацевтически приемлемых неорганических и органических кислот и оснований. Примеры таких солей кислот включают: ацетат, адипат, альгинат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, цитрат, камфорат, камфорсульфонат, циклопентанпропионат, диглюконат, додецилгидросульфат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептаноат, глицерофосфат, гликолят, гемисульфат, гептаноат, гексаноат, гидрохлорид, гидробромид, гидройодид, 2-гидроксиэтансульфонат, лактат, малеат, малонат, метансульфонат, 2-нафтилсульфонат, никотинат, нитрат, оксалат, памоат, пектинат, перхлорат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, салицилат, сукцинат, сульфат, тартрат, тиоцианат, тозилат и ундеканоат. Данное изобретение также охватывает кватернизацию любых содержащих основной азот групп соединений, раскрытых в настоящем описании. Основной азот можно кватернизировать любыми реагентами, известными специалисту в данной области, включая, например, низшие алкилгалогениды, такие как метил-, этил-, пропил- и бутил- хлориды, бромиды и йодиды; диалкилсульфаты, включая диметил-, диэтил-, дибутил- и диамилсульфаты; длинноцепочечные галогениды, такие как, децил-, лаурил-, миристил- и стеарил- хлориды, бромиды и йодиды, и аралкилгалогениды, включая бензил- и фенэтилбромиды. Такой кватернизацией можно получить водо- или маслорастворимые или дисперсные продукты. Здесь необходимо понимать, что если указывается – относительно числа атомов углерода, указание диапазона от 1 до 6 атомов углерода в настоящем описании необходимо понимать как включающее все до единого индивидуальные количества атомов углерода, а также поддиапазоны, такие как, например, 1 атом углерода, 3 атома углерода, от 4 до 6 атомов углерода и т.д.; – относительно времени реакции, время, равное 1 минуте или более, необходимо понимать, как конкретно включающее здесь все до единого индивидуальные времена, а также поддиапазон, больше 1 минуты, такие как, например, 1 минуту, от 3 до 15 минут, от 1 минуты до 20 часов, от 1 до 3 часов, 16 часов, от 3 часов до 20 часов и т.д.; – и аналогично относительно других параметров, таких как концентрации, элементы и т.д. В частности здесь необходимо понимать, что каждая формула соединения включает все до единого индивидуальные соединения, описанные таким образом, а также все до единого возможные классы, или подгруппы, или подклассы соединений, независимо от того, определен ли такой класс или подкласс, как определенно включающий конкретные соединения, как исключающий конкретные соединения или как комбинация этого; например, исключающее определение для формулы (например, I) можно читать следующим образом: Также необходимо понимать, что Соединения по настоящему изобретению можно легко получить, используя обычные методы из легкодоступных исходных веществ. Детальные описания данных подходов представлены, например, в схемах 1-5, обсужденных ниже. Схема 1 иллюстрирует характерный пример получения фосфатного сложного моноэфира III, полученного из первичного спирта (смотри I), соединения ингибиторов ВИЧ-протеазы (смотри пример 1 (стадии G и H) в экспериментальной части данного документа в качестве конкретного примера данного синтеза). Следует отметить: a) R2 и R3 являются такими, как определено в настоящем описании. При синтезе фосфатного сложного моноэфира III в качестве исходного вещества можно использовать ингибитор ВИЧ аспартил-протеазы (I, смотри патент США
Схема 1A представляет другой характерный пример получения фосфатного сложного моноэфира IIIA, полученного из первичного спирта (смотри IA), соединения ингибиторов ВИЧ-протеазы. Следует отметить: a) n, X, Y, R2, R3 и R6 являются такими, как определено в настоящем описании.
Синтез фосфатного сложного моноэфира IIIA осуществляют, как описано для получения III (схема 1). Схема 2 иллюстрирует характерный пример получения фосфатного сложного моноэфира III, соединения ингибиторов ВИЧ-протеазы, с другим подходом, используя в качестве исходного вещества (3S)-3-изобутиламиноазепан-2-он (IV). Следует отметить: a) R2 и R3 являются такими, как определено в настоящем описании. Как показано на схеме 2, производное, представляющее собой фосфатный сложный моноэфир III, получали из (3S)-3-изобутиламиноазепан-2-она (IV) в семистадийной последовательности реакций. В начале, (2S)-3-изобутиламиноазепан-2-он (IV) сульфонировали 4-ацетамидобензолсульфонилхлоридом в присутствии триэтиламина в дихлорметане, получая соединение V с превосходными выходами. Производное VI получали количественно при обработке V ди-трет-бутилпирокарбонатом и ДМАП в ацетонитриле. Восстановительное раскрытие цикла боргидридом натрия в этаноле приводит к ключевым промежуточным продуктам VII с хорошим выходом. Эфир диэтилфосфорной кислоты VIII получали с хорошим выходом при обработке диэтилхлорфосфатом и гидридом натрия в смеси тетрагидрофурана и триэтилфосфата. Boc защитные группы удаляли обработкой HCl в этаноле, получая соединение IX количественно (T.W. Greene and P.G.M. Wuts, Protective groups in Organic Synthesis, 3rd Edition, John Wiley & Sons, Inc. 1999). Затем сочетание свободной аминогруппы, присутствующей на промежуточном соединении IX, с рядом синтетических аминокислот в присутствии 1-гидроксибензотриазола (HOBt) и 1-[3-(диметиламино)пропил]-3-этилкарбодиимид гидрохлорида (EDAC) привело к производному II с выходами от хорошего до превосходного. Наконец, добавление триметилсилилбромида в дихлорметане (ДХМ) дало соединение III с выходами от хорошего до превосходного.
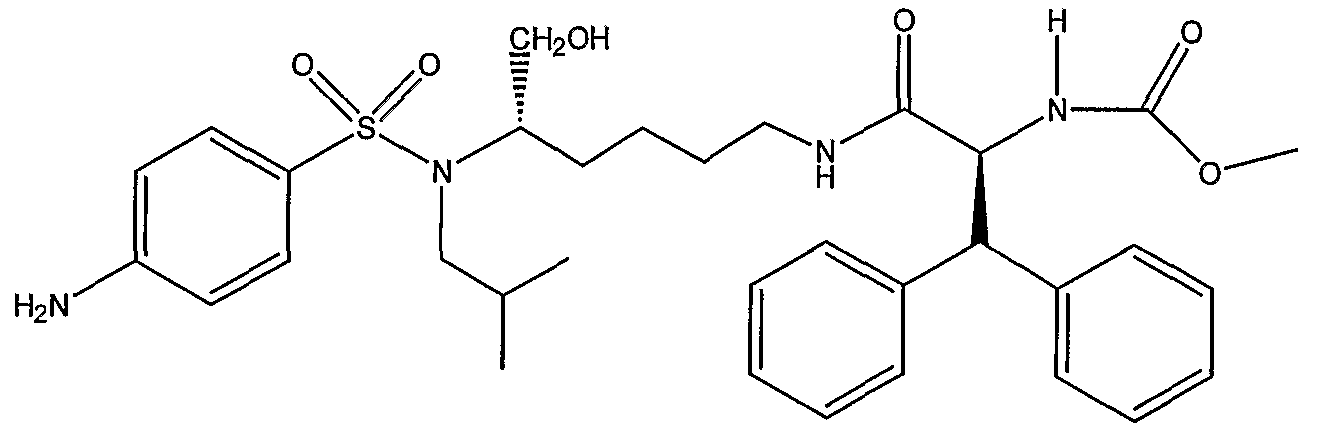
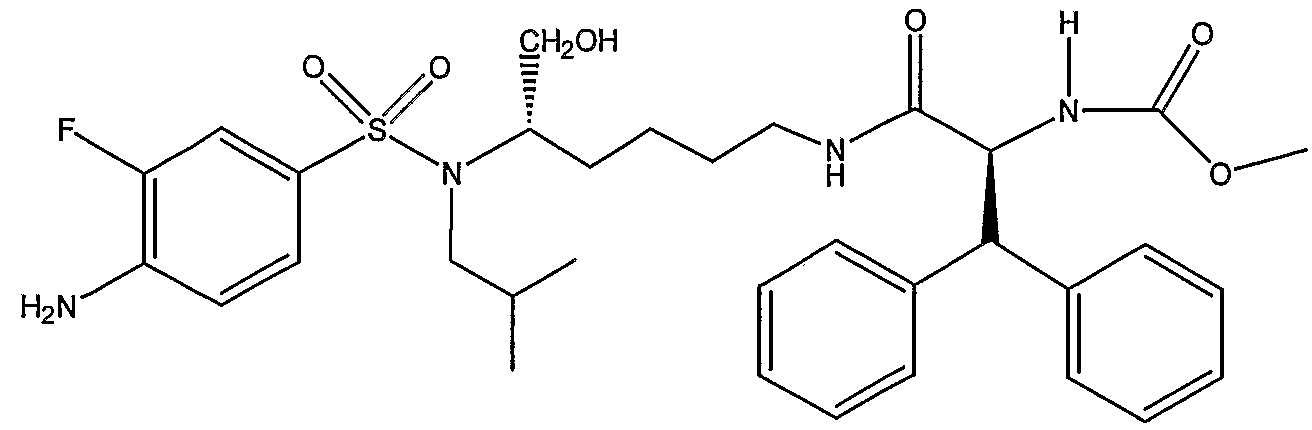
Схема 3 представляет превращение дифенилметильного производного; метилового эфира (1S,5S)-(1-{5-[(4-аминобензолсульфонил)изобутиламино]-6-гидроксигексилкарбамоил}-2,2-дифенилэтил)карбаминовой кислоты (PL-100) в его аналог XI, представляющий собой натриевую соль фторированного фосфатного сложного моноэфира. Данную последовательность реакций можно использовать для получения любых других аналогичных соединений, полученных из незамещенных (или замещенных) дифенилметильных, 1-нафтильных, 2-нафтильных, бифенильных и 9-антрильных групп, описанных в данном изобретении. Так, обработка PL-100 с помощью Selectfluor
Схема 4 иллюстрирует типичный пример превращения фосфотриэфира II в его фторированный аналог XIII в двухстадийной последовательности реакций. Данный типичный пример представляет второй подход к синтезу фторированных соединений по данному изобретению. В данном случае атом фтора добавляют к фосфотриэфиру II, а не к производному общей формулы I, представляющему собой первичный спирт, или более конкретно PL-100, как показано на схеме 3. Данную альтернативную последовательность реакций можно использовать для синтеза любых других аналогичных соединений, полученных из незамещенных (или замещенных) дифенилметильных, 1-нафтильных, 2-нафтильных, бифенильных и 9-антрильных групп, описанных в данном изобретении. Следует отметить: a) R2 и R3 являются такими, как определено в настоящем описании. Кратко, обработка производного II с помощью Selectfluor
Схема 5 иллюстрирует синтез различных соединений сложных эфиров XVIпо изобретению. Известно, что соединения сложных эфиров легко расщепляются in vivo ферментами эстеразы и в результате могут высвобождать активный ингредиент. В данной схеме R2 определен как дифенилметильная группа. Однако данную последовательность реакций можно использовать для синтеза любых других аналогичных соединений, полученных из незамещенных (или замещенных) дифенилметильных, 1-нафтильных, 2-нафтильных, бифенильных и 9-антрильных групп, описанных в данном изобретении. Следует отметить: a) R1A представляет Соединения XVI обычно получают в трехстадийной последовательности реакций с высокими выходами. Этерификация трет-бутилового эфира (1S)-{4-[(5-трет-бутоксикарбониламино-1-гидроксиметилпентил)изобутилсульфамоил]фенил}карбаминовой кислоты (VII) различной кислотой в присутствии 1-гидроксибензотриазола (HOBt) и 1-[3-(диметиламино)пропил]-3-этилкарбодиимид гидрохлорида (EDAC) привела к желаемым сложным эфирам XIV с превосходными выходами. Сложный уксуснокислый эфир получали количественно, используя уксусный ангидрид в присутствии N,N-диметиламинопиридина (ДМАП) в дихлорметане (ДХМ). Отрыв Boc защитной группы достигали количественно при обработке трифторуксусной кислотой (ТФУ) в ДХМ. Второе сочетание с (2S)-2-метоксикарбониламино-3,3-дифенилпропионовой кислотой осуществляют на первичной аминогруппе промежуточного соединения XV с HOBt и EDAC, получая желаемые соединения XVI с выходами от хорошего до превосходного. Если необходимо, осуществляют каталитическое гидрирование бензилоксикарбонильной группы, используя 10% палладий на угле, получая конечное соединение XVII.
Специалист в данной области поймет, что вышеуказанные синтетические схемы не имеют намерения представлять собой полный список всех средств, которыми можно синтезировать соединение, описанное и заявленное в данной заявке на изобретение, а представляют собой лишь иллюстрацию методов синтеза среди прочих. Дальнейшие методы будут очевидны специалистам в данной области. Соединения по данному изобретению можно модифицировать, добавляя соответствующие функциональности для усиления селективных биологических свойств. Такие модификации известны из уровня техники и включают модификации, которые увеличивают биологическую проницаемость в данную биологическую систему (например, кровь, лимфатическую систему, центральную нервную систему), увеличивают пероральную доступность, увеличивают растворимость, чтобы дать возможность введения инъекцией, изменяют метаболизм и изменяют скорость выделения. Как обсуждено выше, новые соединения могут высвобождать активные ингредиенты, которые являются превосходными лигандами по отношению к аспартил-протеазам, например, ВИЧ-1 протеазе. Соответственно, данные соединения, посредством высвобождения активного ингредиента, способны нацеливаться и подавлять события поздней фазы при репликации, т.е. процессинг вирусных полипротеинов ВИЧ-кодированной протеазой. Соединения по данному изобретению преимущественно подавляют способность ВИЧ-1 вируса инфицировать иммортализованные Т-клетки человека в течение периода, составляющего дни, как определено анализом, измеряющим количество внеклеточного p24 антигена; специфичного маркера репликации вируса (смотри, Meek et al., Nature, 343, pp.90-92 (1990)). Кроме их использования при профилактике или лечении ВИЧ или HTLV-инфекции, соединения по данному изобретению можно также использовать в качестве ингибирующих или прерывающих средств для других вирусов, которые используют аспартил-протеазы, аналогичные ВИЧ или HTLV аспартил-протеазам, в своем жизненном цикле. Такие соединения подавляют протеолитический процессинг предшественников вирусных полипротеинов, подавляя аспартил-протеазу. Поскольку аспартил-протеаза является существенной для продукции зрелых вирионов, подавление данного процессинга эффективно блокирует распространение вируса, подавляя продукцию и репродукцию инфекционных вирионов, в частности из остро и хронически инфицированных клеток. Соединения по данному изобретению преимущественно подавляют аспартил-протеазы, таким образом блокируя способность аспартил-протеаз катализировать гидролиз пептидных связей. Соединения по данному изобретению можно применять обычным способом для лечения или профилактики ВИЧ, HTLV или других вирусных инфекций, которые включают в свой жизненный цикл (репликацию) аспартил-протеазы. Такие методы лечения, их уровни дозировок и требования могут быть выбраны специалистом в данной области из имеющихся методов и методик. Например, соединение по данному изобретению можно объединить с фармацевтически приемлемым вспомогательным веществом для введения инфицированному вирусом пациенту в фармацевтически приемлемой манере и в количестве, эффективном для снижения тяжести вирусной инфекции. Альтернативно, соединения по данному изобретению можно использовать в вакцинах и способах защиты особей от вирусной инфекции в течение продолжительного периода времени. Соединения можно применять в таких вакцинах либо в одиночку, либо вместе с другими соединениями по данному изобретению в манере, согласующейся с традиционным использованием ингибиторов протеазы или производных ингибиторов протеазы в вакцинах. Например, соединение по данному изобретению можно объединить с фармацевтически приемлемыми вспомогательными веществами, или системами доставки, обычно используемыми в вакцинах, и ввести в профилактически эффективных количествах для защиты особей в течение продолжительного периода времени от вирусных инфекций, таких как ВИЧ-инфекция. По существу, новые соединения по настоящему изобретению (при отщеплении физиологически расщепляющегося звена) можно вводить в качестве агентов для лечения или профилактики вирусных инфекций, включая ВИЧ-инфекцию, у млекопитающих. Соединения по данному изобретению можно вводить здоровому или ВИЧ-инфицированному пациенту (до или после появления симптомов СПИД) в виде одиночного агента или в комбинации с другими противовирусными средствами, которые мешают циклу репликации ВИЧ. Вводя соединения по данному изобретению с другими противовирусными средствами, которые нацелены на другие события в жизненном цикле вируса, терапевтический эффект данных соединений усиливается. Например, совместно вводимое противовирусное средство может представлять собой средство, которое нацелено на ранние события жизненного цикла вируса, такие как присоединение к клеточному рецептору или входу клетки, обратную транскрипцию и интеграцию вирусной ДНК в клеточную ДНК. Противовирусные агенты, нацеленные на такие ранние события жизненного цикла, включают, среди прочего, полисульфированные полисахариды, sT4 (растворимый CD4)и другие соединения, которые блокируют связывание вируса с CD4 рецептором на несущих CD4 Т-лимфоцитах и других CD4(+) клетках, или подавляют слияние оболочки вируса с цитоплазматической мембраной, и диданозин (ddI), зальцитабин (ddC), ставудин (d4T), зидовудин (AZT) и ламивудин (3TC), которые ингибируют обратную транскрипцию. Например, с соединениями по настоящему изобретению можно использовать другой ингибитор протеазы. Другие антриретровирусные и противовирусные лекарственные средства также можно совместно вводить с соединениями по данному изобретению, чтобы обеспечить терапевтическое лечение для существенного снижения или исключения вирусной инфекционности и связанных с ней симптомов. Примеры других противовирусных средств включают ганцикловир, дидеоксицитидин, тринатрия фосфоноформат, эфлорнитин, рибавирин, ацикловир, альфа-интерферон и трименотрексат. Кроме того, для усиления действия соединений по данному изобретению можно использовать другие типы лекарственных средств, такие как ингибиторы утраты вирусом оболочки, ингибиторы Tat или Rev трансактивирующих белков, антисмысловые молекулы или ингибиторы интегразы вируса. Данные соединения также можно совместно вводить с другими ингибиторами ВИЧ аспартил-протеазы. Кроме того, может оказаться полезным вводить соединения по настоящему изобретению с другим лекарственным средством (другими противовирусными соединениями, антибиотиками, аналгизирующим средством и т.д.). Комбинированные терапии по данному изобретению оказывают синергическое действие при подавлении репликации ВИЧ, поскольку каждый компонентный агент комбинации действует на различные участки ВИЧ-репликации. Использование таких комбинаций также преимущественно снижает дозировки заданного традиционного антриретровирусного средства, которые потребовались бы для желаемого терапевтического или профилактического действия, по сравнению со случаем, когда такой агент вводят в виде монотерапии. Данные комбинации могут уменьшить или исключить побочные эффекты традиционных терапий одиночным антиретровирусным средством, в то же время не мешая антиретровирусной активности данных средств. Данные комбинации снижают потенциал резистентности к терапиям одиночным средством, в то же время сводя к минимуму сопутствующую токсичность. Данные комбинации также могут увеличить эффективность традиционного средства, не увеличивая сопутствующую токсичность. Комбинированные терапии, охватываемые настоящим изобретением, включают, например, введение соединения по изобретению с AZT, 3TC, ddI, ddC, d4T или другими ингибиторами обратной транскриптазы. Альтернативно, соединения по данному изобретению можно также совместно вводить с другими ингибиторами ВИЧ-протеазы, такими как Ro 31-8959 (Cаквинавир; Roche), L-735,524 (Индинавир; Merck), AG-1343 (Нелфинавир; Agouron), A-84538 (Ритонавир; Abbott), ABT-378/r (Лопинавир; Abbott) и VX-478 (Ампренавир; Glaxo), для увеличения эффекта терапии или профилактики против различных вирусных мутантов или членов других ВИЧ-квазивидов. Ведение соединений по настоящему изобретению можно осуществить, например, в виде одиночных средств или в комбинации с ингибиторами ретровирусной обратной транскриптазы или другими ингибиторами ВИЧ аспартил-протеазы. Совместное введение соединений по данному изобретению с ингибиторами ретровирусной обратной транскриптазы или ингибиторами ВИЧ аспартил-протеазы может оказать значительное синергическое действие, таким образом предотвращая, существенно уменьшая или полностью исключая инфекционность вируса и сопутствующие симптомы. Соединения по настоящему изобретению можно вводить таким образом или в форме, которая может допускать отщепление звена R1 для высвобождения ингибитора протеазы. Соединения по данному изобретению также можно вводить, например, в комбинации с иммуномодуляторами (например, бропиримином, античеловеческими антителами к альфа-интерферону, IL-2, GM-CSF, метионин-энцефалином, интерфероном альфа, диэтилдитиокарбаматом натрия, фактором некроза опухоли, налтрексоном и rEPO), антибиотиками (например, пентамидина изотионатом) или вакцинами для профилактики или борьбы с инфекцией или болезнью, связанными с ВИЧ-инфекцией, такими как СПИД и СПИД-ассоциированный комплекс. Когда соединения по данному изобретению вводят в комбинированных терапиях с другими средствами, их можно вводить пациенту последовательно или параллельно. Альтернативно, фармацевтические или профилактические композиции по данному изобретению могут содержать комбинацию одного или нескольких соединений по данному изобретению и другого терапевтического или профилактического средства. Хотя данное изобретение фокусируется на использовании соединений, раскрытых в настоящем описании, для профилактики и лечения ВИЧ-инфекции соединения по данному изобретению также можно использовать в качестве ингибирующих средств для других вирусов, которые зависят от аналогичных аспартил-протеаз для обязательных событий в их жизненном цикле. Данные вирусы включают, но не ограничиваются этим, ретровирусы, вызывающие подобные СПИД заболевания, такие как обезьяньи вирусы иммунодефицита, ВИЧ-2, HTLV-I и HTLV-II. Кроме того, соединения по данному изобретению также можно использовать для подавления других аспартил-протеаз и, в частности, других аспартил-протеаз человека, включая ренин и аспартил-протеазы, которые участвуют в процессинге предшественников эндотелина. Фармацевтические композиции по данному изобретению включают любое соединение по настоящему изобретению и его фармацевтически приемлемые соли с любым фармацевтически приемлемым носителем, вспомогательным веществом или растворителем. Фармацевтически приемлемые носители, вспомогательные вещества и растворители, которые можно использовать в фармацевтических композициях по данному изобретению, включают, но не ограничиваются этим, ионообменники, оксид алюминия, стеарат алюминия, лецитин, сывороточные белки, такие как человеческий альбумин, буферные вещества, такие как фосфаты, глицин, сорбиновую кислоту, сорбат калия, смеси неполных глицеридов насыщенных растительных жирных кислот, воду, соли или электролиты, такие как протамин сульфат, кислый фосфат динатрия, кислый фосфат дикалия, хлорид натрия, соли цинка, коллоидный кремнезем, трисиликат магния, поливинилпирролидон, вещества на основе целлюлозы, полиэтиленгликоль, натрий карбоксиметилцеллюлозу, полиакрилаты, воски, блок-сополимеры полиэтилен-полиоксипропилен, полиэтиленгликоль и ланолин. Фармацевтические композиции по данному изобретению можно вводить перорально, парентерально аэрозолем для ингаляции, местно, ректально, назально, трансбукально, вагинально или посредством имплантированного резервуара. Следовательно, здесь понятно, что пероральное введение или введение инъекцией охватывается настоящим изобретением. Например, соединения по настоящему изобретению можно вводить, например, перорально в водном растворе. Фармацевтические композиции по данному изобретению могут содержать любые обычные нетоксичные фармацевтически приемлемые носители, вспомогательные вещества или растворители. Используемый здесь термин Фармацевтические композиции могут быть в форме стерильного препарата для инъекций, например, в виде стерильной впрыскиваемой водной и масляной суспензии. Рецептуру данной суспензии можно составить согласно методикам, известным из уровня техники, используя подходящие диспергирующие или увлажняющие средства (такие как, например, Tween 80) и суспендирующие вещества. Стерильный впрыскиваемый препарат также может представлять собой стерильный впрыскиваемый раствор или суспензию в нетоксичном парентерально приемлемом разбавителе или растворителе, например, в виде раствора в 1,3-бутандиоле. Среди приемлемых носителей и растворителей, которые можно использовать, находятся аминокислоты, вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды обычно используют стерильные, нелетучие масла. Для данной цели можно использовать любое легкое нелетучее масло, включая синтетические моно- и диглицериды. Жирные кислоты, такие как олеиновая кислота и ее глицеридные производные применимы при получении препаратов для инъекций, как, например, природные фармацевтически приемлемые масла, такие как оливковое масло или касторовое масло, особенно в своих полиоксиэтилированных вариантах. Данные масляные растворы или суспензии также могут содержать разбавители или диспергирующие вещества, представляющие собой спирты с длинной цепью, такие как Ph. Helv. или аналогичный спирт. Фармацевтические композиции по данному изобретению можно вводить перорально в любой перорально приемлемой лекарственной форме, включая, но, не ограничиваясь этим, капсулы, таблетки и водные суспензии и растворы. В случае таблеток для перорального использования носители, которые обычно используют, включают лактозу и кукурузный крахмал. Также типично добавляют смазывающие вещества, такие как стеарат магния. Для перорального введения в форме капсулы, применимые разбавители включают лактозу и сухой кукурузный крахмал. Когда водные суспензии вводят перорально, активный ингредиент объединяют с эмульгирующими и суспендирующими веществами. Если желательно, можно добавить определенные подслащивающие вещества, и/или корригенты, и/или красители. Фармацевтические композиции по данному изобретению также можно вводить в форме суппозиториев для ректального введения. Данные композиции можно получить, смешивая соединение по изобретению с подходящим нераздражающим наполнителем, который является твердым при комнатной температуре, но жидким при ректальной температуре и, следовательно, будет плавиться в прямой кишке, высвобождая активные компоненты. Такие материалы включают, но не ограничиваются этим, масло какао, пчелиный воск и полиэтиленгликоли. Местное введение фармацевтических композиций по данному изобретению особенно применимо, когда желаемая терапия включает области или органы, легко доступные местным нанесением. Для нанесения местно на кожу фармацевтическая композиция должна быть составлена в виде подходящей мази, содержащей активные компоненты, суспендированные или растворенные в носителе. Носители для местного введения соединений по данному изобретению включают, но не ограничиваются этим, минеральное масло, жидкую нефть, белый вазелин, пропиленгликоль, соединение полиоксиэтилена или полиоксипропилена, эмульгирующий воск и воду. Альтернативно, фармацевтическую композицию можно составить в виде подходящего лосьона или крема, содержащего активное соединение, суспендированное или растворенное в носителе. Подходящие носители включают, но не ограничиваются этим, минеральное масло, сорбит моностеарат, полисорбат 60, цетиловые эфиры, воск, цетеариловый спирт, 2-октилдодеканол, бензиловый спирт и воду. Фармацевтические композиции по данному изобретению можно также наносить местно в нижнюю часть кишечника рецептурой ректального суппозитория или в подходящей чистой рецептуре. Местные трансдермальные пластыри также включаются в данное изобретение. Фармацевтические композиции по данному изобретению можно вводить назальным аэрозолем или ингаляцией. Такие композиции получают методиками, хорошо известными из уровня техники фармацевтических процедур, и их можно приготовить в виде растворов в насыщенных растворах соли, используя бензиловый спирт или другие подходящие консерванты, усилители абсорбции для увеличения биологической доступности, фторуглероды и/или другие солюбилизаторы или диспергирующие агенты, известные из уровня техники. Для профилактики или лечения вирусной инфекции, включая ВИЧ-инфекцию, пригодны уровни дозировок примерно от 0,01 до 25 мг/кг массы тела в день, например, приблизительно от 0,5 до 25 мг/кг массы тела в день соединения активного ингредиента. Типично фармацевтические композиции по данному изобретению будут вводить примерно от 1 до 5 раз в сутки или, альтернативно, в виде непрерывного вливания. Такое введение можно использовать в виде хронической или острой терапии. Количество активного ингредиента, которое можно объединить с веществами носителя для получения монолитной лекарственной формы, будет различаться в зависимости от пациента, которого лечат, и конкретного режима введения. Типичный препарат будет содержать примерно от 5% до 95% активного ингредиента (мас./мас.). Например, такие препараты могут содержать примерно от 20% до 80% активного ингредиента. При улучшении состояния пациента, можно вводить поддерживающую дозу соединения, композиции или комбинации по данному изобретению, если это необходимо. Затем дозировку или частоту введения, или то и другое, можно понизить в виде функции симптомов до уровня, при котором сохраняется улучшенное состояние. Когда симптомы облегчили до желаемого уровня, лечение приостанавливают. Однако пациенты могут требовать периодического лечения на долговременной основе при рецидиве симптомов заболевания. Специалисту в данной области понятно, что могут быть желательны более низкие или более высокие дозы, чем изложенные выше. Конкретные дозировки и режимы лечения для конкретного пациента могут зависеть от разнообразных факторов, включая активность конкретного применяемого соединения, возраст, массу тела, общее состояние здоровья, пол, режим питания, время введения, скорость выведения, комбинацию лекарственных препаратов, тяжесть и течение инфекции, склонность пациента к инфекции и мнение лечащего врача. В данном описании используют следующие сокращения:
ПРИМЕРЫ Данный раздел описывает синтез соединений на основе лизина, способных высвобождать ингибиторы ВИЧ аспартил-протеазы, как описано в настоящей заявке. Данные примеры даны только для иллюстративных целей и их не следует рассматривать как ограничивающие каким-либо образом объем изобретения. Данный раздел предоставляет детальный синтез соединений Вещества и методы Аналитическую тонкослойную хроматографию (ТСХ) проводили на 0,25 мм силикагелевых пластинах E. Merck 60 F254 и элюировали показанными системами растворителей. Препаративную хроматографию осуществляли флэш-хроматографией, используя силикагель 60 (EM Science) с показанными системами растворителей и избыточным давлением воздуха, чтобы обеспечить надлежащую скорость элюирования. Определение соединений осуществляли, воздействуя на подвергнутые элюированию пластины (аналитические или препаративные) йодом, УФ-излучением и/или обрабатывая аналитические пластины 2% раствором п-анизальдегида в этаноле, содержащем 3% серной кислоты и 1% уксусной кислоты, после чего следовало нагревание. Альтернативно, аналитические пластины можно обработать 0,3% раствором нингидрина в этаноле, содержащем 3% уксусной кислоты, и/или САМ раствором, изготовленным из 20 г (NH4)6Mo7O24 и 8,3 г Ce(SO4)2 полигидрата в воде (750 мл), содержащей концентрированную серную кислоту (90 мл). Препаративную ВЭЖХ проводили на приборе Gilson, оборудованном С18 колонкой, модулем подачи жидкости 215 и напорными насосами производительностью 25 мл/мин. ВЭЖХ работает с программным обеспечением Gilson UniPoint System. Условия полупрепаративной ВЭЖХ для очистки тестируемых соединений Система ВЭЖХ: 2 насоса Gilson #305-25 мл, устройство Gilson #215 для ввода и сбора жидкости и детектор УФ-видимого поглощения Gilson #155, все устройства контролируются программным обеспечением Gilson UniPoint V1.91. Колонка: Alltech (#96053) Hyperprep PEP, C-18, 100 Е Расход: 15 мл/мин Растворители: A: H2O, B: CH3CN Градиент: от 25% до 80% B в течение 40 мин Детектор: поглощение; Неочищенное вещество, растворенное в ацетонитриле до концентрации примерно от 50 до 80 мг/2 мл, инжектировали в каждом цикле. Фракции собирали в количестве 9 мл, имеющее отношение поглощение определяли УФ-детектором. Если не указано иным образом, все исходные вещества получали из коммерческих источников, таких как Aldrich Co. или Sigma Co. Температуры плавления (Тпл.) определяли на приборе для измерения температуры плавления B Масс-спектры записывали на системе Hewlett Packard LC/MSD 1100 с использованием источника APCI или электрораспылительного источника в отрицательном режиме или положительном режиме. Спектры ядерного магнитного резонанса (ЯМР) записывали на Bruker AMX-II-500, оборудованном реверсивным или QNP зондом. Образцы растворяли в дейтерированном хлороформе (CDCl3), дейтерированном ацетоне (ацетон-d6), дейтерированном метаноле (CD3OD) или дейтерированном диметилсульфоксиде (ДМСО-d6), используя тетраметилсилан в качестве внутреннего стандарта для получения данных. Химические сдвиги (*) выражены в миллионных долях (м.д.), константы взаимодействия (J) выражены в Герцах (Гц), тогда как мультиплетности обозначают как с для синглета, д для дублета, 2д для двух дублетов, дд для дублета дублетов, т для триплета, кв для квартета, квинт для квинтета, м для мультиплета и ушир. с для уширенного синглета. ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ ПРИМЕРЫ: Конкретные примеры получения производных общей формулы I Следующие ниже соединения получают из производных L-лизина, используя методики, суммированные в схемах 1, 1A, 2, 3, 4 и 5 данного изобретения. Пример 1. Получение метилового эфира (1S,5S)-(1-{5-[(4-аминобензолсульфонил)изобутиламино]-6-фосфоноксигексилкарбамоил}-2,2-дифенилэтил)карбаминовой кислоты (PL-461) Получение указанного в заголовке соединения основано на схемах 1 и 2 данного изобретения. Стадия A. Получение (3S)-3-изобутиламиноазепан-2-она (IV) L- 1Н ЯМР (CDCl3): 1Н ЯМР (CDCl3): Небольшой образец превращают в S-метилбензилмочевину, добавляя твердое вещество к раствору S-метилбензилизоцианата в MeCN. ЯМР дает 98% эи. Стадия B. Получение N N 1Н ЯМР (ДМСО-d6): * 0,93 (д, J=6,0, 3H), 0,96 (д, J=6,0, 3H), 1,39 (т, J=12,0, 1H), 1,85-1,65 (м, 3H), 2,08-2,18 (м и с, 6H), 2,90-2,97 (м, 1Н), 3,00-3,06 (м, 2H), 3,35 (дд, J=14,2, 8,5, 1H), 4,65 (д, J=8,7, 1H), 6,3 (с, 1H), 7,42 (д, J=8,8, 2H), 7,6 (д, J=8,8, 2H). Тпл. 230-233°C (EtOH). Стадия C. Получение трет-бутилового эфира (3S)-3-{[4-ацетил-трет-бутоксикарбониламино)бензолсульфонил]изобутиламино}-2-оксоазепан-1-карбоновой кислоты (Boc активация) (VI) 4,2 г N 1Н ЯМР (ДМСО-d6): * 0,68 (д, J=6,0, 3H), 0,85 (д, J=6,0, 3H), 1,39 (с, 10H), 1,47 (с, 9H), 1,85-1,65 (м, 3H), 2,15 (с, 3H), 2,80 (кв, J=4, 1Н), 3,10-3,36 (м, 2H), 4,01 (д, J=8,0, 1Н), 4,85 (д, J=8,7, 1H), 7,32 (д, J=8,8, 2H), 7,87 (д, J=8,8, 2H) Тпл. 123-124°C. Стадия D. Получение (1S)-4-амино-N-(5-амино-1-гидроксиметилпентил)-N-изобутилбензолсульфонамида (VII-без защиты) (восстановительное раскрытие цикла и снятие защиты) 3,0 г порцию трет-бутилового эфира (3S)-3-{[4-ацетил-трет-бутоксикарбониламино)бензолсульфонил]изобутиламино}-2-оксоазепан-1-карбоновой кислоты (VI, стадия C) растворяют в 40 мл EtOH, после чего добавляют 750 мг NaBH4. Кратковременное нагревание феном дает прозрачный раствор. ТСХ обнаруживает только полосчатое пятно через 20 мин (EtOAc). Раствор концентрируют до пасты, выливают в 40 мл 1 н. NaOH и экстрагируют этилацетатом, органическую фазу сушат над Na2SO4 и выпаривают, получая 2,8 г промежуточного продукта (VII); трет-бутилового эфира (1S)-{4-[(5-трет-бутоксикарбониламино-1-гидроксиметилпентил)изобутилсульфамоил]фенил}карбаминовой кислоты (VII) Вышеуказанный промежуточный продукт растворяют в 5 мл EtOH и добавляют 5 мл 12 н. HCl. В течение нескольких минут наблюдается энергичное выделение газа. Через 2 ч раствор выпаривают, делают щелочным концентрированным KOH и экстрагируют EtOAc, получая 1,75 г белого порошка. 1Н ЯМР (ДМСО-d6): * 0,82 (м, 6H), 0,97-1,12 (м, 2H), 1,15-1,30 (м, 3H), 1,57 (м, 1Н), 1,84 (м, 1H), 2,40 (т, J=7,8, 2H), 2,75 (м, 1H), 2,85 (м, 1H), 3,21 (м, 1H), 3,44 (д, J=6,4, 2H), 5,92 (ушир.с, 2H), 6,59 (д, J=8,0, 2H), 7,39 (д, J=8,0, 2H). Стадия E. Получение (2S)-2-метоксикарбониламино-3,3-дифенилпропионовой кислоты К раствору L-дифенилаланина (241 мг, 1,0 ммоль) (Peptech Corp.) в 5 мл 1 н. NaOH и 0,5 мл насыщенного Na2CO3 (полученный в результате раствор при рН 10) добавляют метоксикарбонилоксисукцинимид ((2,5-диоксопирролидин-1-ил)метиловый эфир углекислоты) (180 мг, 1,1 ммоль), растворенный в 5 мл. После этого реакционную смесь перемешивают при комнатной температуре в течение 2 час. Щелочной раствор один раз экстрагируют эфиром (10 мл) и водную фазу подкисляют 1 н. HCl. Данную фазу дважды экстрагируют 20 мл EtOAc и объединенные органические фазы промывают 50 мл 1 н. HCl. Органическую фазу сушат над Na2SO4, фильтруют и выпаривают до маслянистого вещества, которое отверждают, получая 250 мг (83%) желаемого вещества. Данное производное используют без дополнительной обработки в следующей стадии. Стадия F. Получение метилового эфира (1S,5S)-(1-{5-[(4-аминобензолсульфонил)изобутиламино]-6-гидроксигексилкарбамоил}-2,2-дифенилэтил)карбаминовой кислоты (PL-100) Указанное в заголовке соединение получают из (1S)-4-амино-N-(5-амино-1-гидроксиметилпентил)-N-изобутилбензолсульфонамида (VII-без защиты) (стадия D) и (2S)-2-метоксикарбониламино-3,3-дифенилпропионовой кислоты (стадия E), используя процедуру сочетания с HOBt и EDAC, описанную в примере 3 (стадия D). Конечный продукт получают с выходом 67% (121 мг). ЖХ-МС: 625,3 (M+H)+, 95% чист. 1Н ЯМР (CD3OD): 13С ЯМР (CD3OD): Стадия G. Получение метилового эфира (1S,5S)-{1-[5-[(4-аминобензолсульфонил)изобутиламино]-6-(диэтоксифосфорилокси)гексилкарбамоил]-2,2-дифенилэтил}карбаминовой кислоты Соединение PL-100 (продукт стадии F, 203 мг, 0,325 ммоль) растворяют в смеси сухого тетрагидрофурана (3 мл) и 0,2 мл триэтилфосфата под атмосферой N2. Смесь перемешивают при данной температуре в течение 15 мин, после чего добавляют диэтилхлорфосфат (0,061 мл, 0,423 ммоль). При 0°C добавляют гидрид натрия (60% в минеральном масле) (17 мг, 0,423 ммоль). Раствор перемешивают в течение 1 часа при 0°C и 12 ч при комнатной температуре. К данному раствору добавляют 20 мл Amberlite XAD-2 и гранулы тщательно перемешивают с растворителем. К смеси добавляют воду со льдом 2 мл и ТГФ выпаривают. Затем гранулы 6 раз промывают дистиллированной водой 100 мл, затем три раза экстрагируют этилацетатом (30 мл). Объединенную фазу выпаривают и остаток сушат под высоким вакуумом. Неочищенный продукт очищают флэш-хроматографией, используя смесь этилацетат/гексан (8/2), затем EtOAc 100% в качестве элюента. Выход данной реакции составляет 152 мг 61%. ЖХ-МС: 761,2 (M+H)+, 90% чист. 1Н ЯМР (CD3OD): 31P ЯМР (CD3OD): Стадия H. Получение метилового эфира (1S,5S)-(1-{5-[(4-аминобензолсульфонил)изобутиламино]-6-фосфоноксигексилкарбамоил}-2,2-дифенилэтил)карбаминовой кислоты (PL-461) Продукт из стадии G, приготовленный выше, (152 мг) растворяют в безводном дихлорметане (3,0 мл). При 0°C добавляют триметилсилилбромид (0,5 мл). Смесь перемешивают в течение 1 ч при данной температуре и в течение ночи при комнатной температуре. Растворитель выпаривают и к остатку добавляют 0,2 мл воды. Добавляют 3 мл EtOH, перемешивают и выпаривают. Данную стадию повторяют три раза и остаток сушат под вакуумом. Выход 98 мг 70% указанных в заголовке производных из данного первого примера. ЖХ-МС: 705,2 (M+H)+, 95% чист. 1Н ЯМР (CD3OD): 31P ЯМР (CD3OD): Пример 2. Получение натриевой соли метилового эфира (1S,5S)-(1-{5-[(4-аминобензолсульфонил)изобутиламино]-6-фосфоноксигексилкарбамоил}-2,2-дифенилэтил)карбаминовой кислоты (PL-462) 70,7 мг конечного продукта из примера 1 добавляют к 1 мл 0,1 н. NaOH и разбавляют 1 мл дистиллированной воды. Данный раствор затем замораживают и подвергают лиофилизации. Выход 67,2 мг (92%) желаемого вещества с чистотой 95%. 1Н ЯМР (CD3OD): 31P ЯМР (CD3OD): Пример 3. Получениеметилового эфира (1S,5S)-(1-{5-[(4-аминобензолсульфонил)изобутиламино]-6-фосфоноксигексилкарбамоил}-2-нафталин-2-илэтил)карбаминовой кислоты (PL-507) Получение указанного в заголовке соединения основано на схеме 2 данного изобретения. Схема A. Получение трет-бутилового эфира (1S)-(4-{[5-трет-бутоксикарбониламино-1-(диэтоксифосфорилоксиметил)пентил]изобутилсульфамоил}фенил)-карбаминовой кислоты (VIII) 2,00 г (3,7 ммоль) трет-бутилового эфира (1S)-{4-[(5-трет-бутоксикарбониламино-1-гидроксиметилпентил)изобутилсульфамоил]фенил}карбаминовой кислоты (VII) (пример 1, стадия D) растворяют в смеси 0,63 мл триэтилфосфата и 10 мл ТГФ при 0°C в инертной атмосфере аргона. Добавляют 0,63 мл (4,44 ммоль) диэтилхлорфосфата и затем порциями добавляют 0,25 г (6,2 ммоль) NaH 60% в масле. Смеси дают нагреться до комнатной температуры и оставляют перемешиваться в течение 2 ч (ЖХ-МС показывает завершение после 1 ч). К данному раствору добавляют 20 мл смолы Amberlite XAD-2, суспензию тщательно перемешивают и добавляют к 200 мл воды со льдом. После перемешивания в течение 15 мин суспензию смолы отфильтровывают и смолу несколько раз промывают дистиллированной водой (500 мл). Желаемый продукт десорбируют из смолы ацетоном (5 Х 50 мл), EtOAc (5 Х 50 мл), затем органическую фазу сушат над Na2SO4. После выпаривания растворителя получают 2,66 г (89%) прозрачного маслянистого вещества. Неочищенный продукт содержит фракцию с двумя диэтилфосфатами и используют без дополнительной обработки в следующей стадии. 1Н ЯМР (CD3OD): 31P ЯМР (CD3OD): Стадия B. Получение (диэтил)(6-амино-2-[(4-аминобензолсульфонил)изобутиламино]гексилового) эфира (2S)-фосфорной кислоты (IX) Неочищенный продукт, полученный в предыдущей стадии (VIII, 2,66 г) растворяют в 12 мл EtOH. Добавляют 4 мл HClконц. и кратковременно нагревают до 70°C, затем оставляют при комнатной температуре в течение 3 ч. Растворитель выпаривают и остаток растирают в 50 мл эфира. Затем вязкий остаток растворяют в 3 мл воды со льдом и рН корректируют до 12 50% NaOH. Полученную вязкую суспензию экстрагируют EtOAc (3 Х 50 мл) и органическую фазу сушат над Na2SO4. После отфильтровывания осушающего агента органическую фазу откачивают, получая 1,84 г (98%) желаемого продукта (IX). ЖХ-МС: 480,2 (M+H)+, 95% чист. 1H ЯМР (CD3OD): 31P ЯМР (CD3OD): Стадия C. Получение (2S)-2-метоксикарбониламино-3-нафталин-2-илпропионовой кислоты (или L-Moc-2-нафтилаланина) К раствору L-2-нафтилаланина (215 мг, 1 ммоль) (Peptech Corp.) в 5 мл 1 н. NaOH и 0,5 мл насыщенного Na2CO3 (pH полученного в результате раствора 10) добавляют метоксикарбонилоксисукцинимид (187 мг, 1,1 ммоль), растворенный в 5 мл. После этого реакционную смесь перемешивают при комнатной температуре в течение 2 ч. Щелочной раствор один раз экстрагируют эфиром (10 мл) и водную фазу подкисляют 1 н. HCl. Данную фазу дважды экстрагируют 20 мл EtOAc и объединенные органические фазы промывают 50 мл 1 н. HCl. Органическую фазу сушат над Na2SO4, фильтруют и выпаривают до маслянистого вещества, которое отверждают, получая 200 мг (73%) желаемого вещества. Данное промежуточное вещество (называемое N-замещенной аминокислотой) используют без дополнительной обработки в следующей стадии. Стадия D. Получение метилового эфира (1S,5S)-(1-{5-[(4-аминобензолсульфонил)изобутиламино]-6-фосфоноксигексилкарбамоил}-2-нафталин-2-илэтил)карбаминовой кислоты (PL-507) 100 мг L-Moc-2-нафтилаланина (стадия C) активируют 100 мг EDAC и 57 мг HOBt в 1,5 мл ДМФА в течение 30 минут. Затем добавляют 100 мг (6-амино-2-[(4-аминобензолсульфонил)изобутиламино]гексил)(диэтилового) эфира фосфорной кислоты (стадия B) и оставляют перемешиваться при комнатной температуре в течение 1 ч. К раствору ДМФА добавляют 40 мл 1М K2CO3 и оставляют в течение 10 мин. Затем добавляют 50 мл EtOAc и затем смесь энергично перемешивают. Осуществляют отделение фазы EtOAc, после чего следует экстракция один раз 5% лимонной кислотой (50 мл), затем 3 раза водой (50 мл) и, наконец, насыщенным раствором соли. Органическую фазу отделяют и выпаривают. Остаток отбирают в 50 мл ДХМ и повторно выпаривают. Остаток снова отбирают в 50 мл ДХМ и добавляют 0,5 мл TMSBr. Раствор оставляют на ночь (16 ч). ДХМ откачивают и добавляют охлажденный на ледяной бане раствор MeOH:Вода 1:1, кратковременно перемешивают и откачивают. Остаток растирают в эфире, затем растворяют в 1 н. NaOH. Прозрачный раствор экстрагируют эфиром и водную фазу подкисляют 6 н. HCl. Затем фильтрованием собирают белый осадок и сушат под вакуумом в течение ночи. Выход 88 мг указанного в заголовке соединения. ЖХ-МС: 679,8 (M+H)+, 95% чист. 1H ЯМР (CD3OD): 31P ЯМР (CD3OD): Пример 4. Получение моно-(2-[(4-аминобензолсульфонил)изобутиламино]-6-{2-[(морфолин-4-карбонил)амино]-3-нафталин-1-илпропиониламино}гексилового эфира (2S,2S)фосфорной кислоты (PL-498) Стадия A. Получение (2S)-2-[(морфолин-4-карбонил)амино]-3-нафталин-1-илпропионовой кислоты К раствору L-1-нафтилаланина (215 мг, 1 ммоль) (Peptech Corp.) в 5 мл 1 н. NaOH и 0,5 мл насыщенного Na2CO3 (pH полученного в результате раствора 10) добавляют морфолин-4-карбонилхлорид (150 мг, 1,0 ммоль), растворенный в 5 мл. После этого реакционную смесь перемешивают при комнатной температуре в течение 2 ч. Щелочной раствор один раз экстрагируют эфиром (10 мл) и водную фазу подкисляют 1 н. HCl. Данную фазу дважды экстрагируют 20 мл EtOAc и объединенные органические фазы промывают 50 мл 1 н. HCl. Органическую фазу сушат над Na2SO4, фильтруют и выпаривают до маслянистого вещества, которое отверждают, получая 125 мг (38%) желаемого вещества. Данное промежуточное соединение используют без дополнительной обработки в следующей стадии. Стадия B. Получение моно-(2-[(4-аминобензолсульфонил)изобутиламино]-6-{2-[(морфолин-4-карбонил)амино]-3-нафталин-1-илпропиониламино}гексилового эфира (2S,2S)фосфорной кислоты (PL-498) Данное соединение синтезируют, так же как в методе получения продукта из примера 3 (стадия D) с использованием 100 мг (2S)-2-[(морфолин-4-карбонил)амино]-3-нафталин-1-илпропионовой кислоты (стадия A данного примера). Полученный в результате осажденный остаток дополнительно очищают препаративной ВЭЖХ с обращенной фазой. Выход 41 мг конечного соединения. ЖХ-МС: 734,8 (M+H)+, 95% чист. 1H ЯМР (CD3OD): 31P ЯМР (CD3OD): Пример 5. Получение моно-{6-(2-ацетиламино-3,3-дифенилпропиониламино)-2-[(4-аминобензолсульфонил)изобутиламино]гексилового}эфира (2S,2S)-фосфорной кислоты (PL-504) Стадия A. Получение (2S)-2-ацетиламино-3,3-дифенилпропионовой кислоты К раствору L-дифенилаланина (100 мг, 0,4 ммоль) (Peptech Corp.) в 5 мл 1 н. NaOH и 0,5 мл насыщенного Na2CO3 (pH полученного в результате раствора 10) добавляют ацетилхлорид (0,5 ммоль), растворенный в 5 мл. После этого реакционную смесь перемешивают при комнатной температуре в течение 2 ч. Щелочной раствор один раз экстрагируют эфиром (10 мл) и водную фазу подкисляют 1 н. HCl. Данную фазу дважды экстрагируют 20 мл EtOAc и объединенные органические фазы промывают 50 мл 1 н. HCl. Органическую фазу сушат над Na2SO4, фильтруют и выпаривают до маслянистого вещества, которое отверждают, получая 70 мг (60%) желаемого вещества. Данное неочищенное промежуточное соединение используют без дополнительной обработки в следующей стадии. Стадия B. Получение моно-{6-(2-ацетиламино-3,3-дифенилпропиониламино)-2-[(4-аминобензолсульфонил)изобутиламино]гексилового} эфира (2S,2S)-фосфорной кислоты (PL-504) Данное соединение синтезируют, так же как в методе получения продукта из примера 3 (стадия D) с использованием 100 мг (2S)-2-ацетиламино-3,3-дифенилпропионовой кислоты (стадия A данного примера). Конечный продукт получают с выходом 30% (30 мг). ЖХ-МС: 689,3 (M+H)+, 95% чист. 1H ЯМР (CD3OD): 31P ЯМР (CD3OD): Пример 6. Получение метилового эфира (1S,5S)-(1-{5-[(4-амино-3-фторбензолсульфонил)изобутиламино]-6-фосфоноксигексилкарбамоил}-2,2-дифенилэтил)карбаминовой кислоты (PL-515) Первая методика: Получение указанного в заголовке соединения основано на схеме 3 данного изобретения. Стадия A. Получение метилового эфира (1-{5-[(4-амино-3-фторбензолсульфонил)изобутиламино]-6-гидроксигексилкарбамоил}-2,2-дифенилэтил)карбаминовой кислоты (X)(PL-337) Продукт из примера 1, стадия F (0,624 г, 1 ммоль) растворяют в 5 мл MeCN при 24°C. Одной порцией добавляют SelectFluor 0,35 г (1 ммоль) и перемешивают в течение 1 ч. Добавляют 1 мл воды и раствор впрыскивают непосредственно в препаративную ВЭЖХ с обращенной фазой. Продукт собирают и подвергают лиофилизации, получая выход белого твердого вещества 250 мг (38%). ЖХ-МС: 643,3 (M+H)+, 99% чист. 1H ЯМР (MeOD): Стадия B. Получение метилового эфира (1S,5S)-{1-[5-[(4-амино-3-фторбензолсульфонил)изобутиламино]-6-(диэтоксифосфорилокси)гексилкарбамоил]-2,2-дифенилэтил}карбаминовой кислоты Продукт из стадии A фосфорилируют хлордиэтилфосфатом, следуя процедуре, описанной в примере 1, стадия G. Выходы 157 мг, 68%. ЖХ-МС: 779,3 (M+H)+, 95% чист. 1H ЯМР (CD3OD): 31P ЯМР (CD3OD): Стадия C. Получение метилового эфира (1S,5S)-(1-{5-[(4-амино-3-фторбензолсульфонил)изобутиламино]-6-фосфоноксигексилкарбамоил}-2,2-дифенилэтил)карбаминовой кислоты (XI) (PL-515) Снятие защиты осуществляют, используя процедуру, описанную в примере 1, стадия G. Выходы 101 мг. ЖХ-МС: 723,2 (M+H)+, 95% чист. 1H ЯМР (CD3OD): 0,65-0,77 (м, 1H), 0,77-0,85 (м, 1H), 0,85-1,05 (м, 9H), 1,25-1,39 (м, 1H), 1,40-1,52 (м, 1H), 1,82-1,98 (м, 1Н), 2,58-2,72 (м, 1H), 2,82-2,92 (м, 1H), 2,92-3,05 (м, 2H), 3,54 (с, 3H), 3,64-3,75 (м, 1H), 3,80-3,92 (м, 1H), 3,91-4,04 (м, 1Н), 4,29 (д, J=11,4, 1H), 7,19 (т, J=6,6, 1H), 7,13-7,21 (м, 2H), 7,22-7,33 (м, 6H), 7,34-7,38 (м, 2H), 7,39-7,48 (м, 2H) 31P ЯМР (CD3OD): Вторая методика: Получение указанного в заголовке соединения основано на схеме 4 данного изобретения. Стадия A. Получение метилового эфира (1S,5S)-(1-{5-[(4-аминобензолсульфонил)изобутиламино]-6-фосфоноксигексилкарбамоил}-2,2-дифенилэтил)карбаминовой кислоты (PL-461). (2S)-2-метоксикарбониламино-3,3-дифенилпропионовую кислоту ((пример 1, стадия E) 0,9 г, 3 ммоль) активируют в ДМФА (5 мл) посредством EDAC (1,7 г, 9 ммоль) и HOBt (1,2 г, 9 ммоль). К данному раствору добавляют 1,17 г (диэтил)(6-амино-2-[(4-аминобензолсульфонил)изобутиламино]гексилового) эфира (2S)-фосфорной кислоты (IX) (пример 3, стадия B) и смесь перемешивают в течение 3 ч. Затем добавляют 20 г Amberlite XAD-2 и гранулы оставляют намокать в течение 10 мин. Смолу переносят на стеклянный фильтр и тщательно промывают дистиллированной водой (400 мл) и 200 мл 1М NaHCO3. Затем гранулы промывают 4 х 50 мл порциями MeOH, затем EtOAc 200 мл. Органическую фазу выпаривают. Остаток адсорбируют на силикагеле и пропускают через короткую силикагелевую колонку (EtOAc), получая 2,4 г (83%) белого твердого вещества после выпаривания. 1H ЯМР идентичен примеру 1, стадия H. Стадия B. Получение метилового эфира (1S,5S)-{1-[5-[(4-амино-3-фторбензолсульфонил)изобутиламино]-6-(диэтоксифосфорилокси)гексилкарбамоил]-2,2-дифенилэтил}карбаминовой кислоты (XII) Продукт из приведенной выше стадии A, метиловый эфир (1S,5S)-(1-{5-[(4-аминобензолсульфонил)изобутиламино]-6-фосфоноксигексилкарбамоил}-2,2-дифенилэтил)карбаминовой кислоты (0,555 г, 0,73 ммоль) растворяют в 5 мл MeCN. Добавляют Selectfluor (0,26 г, 0,7 ммоль) и смесь перемешивают в течение 30 мин. Смесь очищают препаративной ВЭЖХ с обращенной фазой и подвергают лиофилизации, получая 278 мг (выход 48%) белого твердого вещества. 1H ЯМР идентичен предшествующему описанию, смотри первую методику выше. Стадия C. Получение метилового эфира (1S,5S)-(1-{5-[(4-амино-3-фторбензолсульфонил)изобутиламино]-6-фосфоноксигексилкарбамоил}-2,2-дифенилэтил)карбаминовой кислоты (XIII, в данном конкретном случае представляет собой соединение XI)(PL-515) Процедура, дающая данное производное, является такой, как описано в стадии снятия защиты для описанной выше методики. Выходы 139 мг 70% после ВЭЖХ с обращенной фазой. 1H ЯМР идентичен предшествующему описанию, смотри первую методику выше. Пример 7. Получение 2-[(4-аминобензолсульфонил)изобутиламино]-6-(2-метоксикарбониламино-3,3-дифенилпропиониламино)гексилового эфира (2S,2S)уксусной кислоты (PL-521) Получение указанного в заголовке производного основано на схеме 5 данного изобретения. Стадия A. Получение 6-трет-бутоксикарбониламино-2-[(4-трет-бутоксикарбониламинобензолсульфонил)изобутиламино]гексилового эфира (2S)уксусной кислоты (XIV, R1A=CH3) К перемешиваемому раствору трет-бутилового эфира (1S)-{4-[(5-трет-бутоксикарбониламино-1-гидроксиметилпентил)изобутилсульфамоил]фенил}карбаминовой кислоты (промежуточный продукт (VII) из примера 1, стадия D, 97 мг, 0,18 ммоль) в безводном CH2Cl2 (3 мл) добавляют N,N-диметиламинопиридин (22 мг, 0,18 ммоль) и уксусный ангидрид (0,014 мл, 0,18 ммоль). Смесь перемешивают при комнатной температуре в течение 1 часа. Растворитель выпаривают. Добавляют этилацетат (50 мл) и органический слой промывают водой (30 мл), затем сушат над Na2SO4 и концентрируют. Остаток очищают флэш-хроматографией, элюируя этилацетатом. Полученный выход является количественным (100 мг). ЖХ-МС: 586,2 (M+H)+, 95% чист. Стадия B. Получение 6-амино-2-[(4-аминобензолсульфонил)изобутиламино]гексилового эфира (2S)-уксусной кислоты (XV, R1A=CH3) Данное производное получают из 6-трет-бутоксикарбониламино-2-[(4-трет-бутоксикарбониламинобензолсульфонил)изобутиламино]гексилового эфира (2S)-уксусной кислоты, как описано в примере 15, стадия B. Твердое вещество желтого цвета (66 мг) используют в следующей реакции без очистки. ЖХ-МС: 386,2 (M+H)+, 95% чист. Стадия C. Получение 2-[(4-аминобензолсульфонил)изобутиламино]-6-(2-метоксикарбониламино-3,3-дифенилпропиониламино)гексилового эфира (2S,2S)-уксусной кислоты (XVI, R1A=CH3) (PL-521) Данное производное получают из 6-амино-2-[(4-аминобензолсульфонил)изобутиламино]гексилового эфира (2S)-уксусной кислоты (продукт из стадии B), как описано в примере 15, стадия B. Конечный продукт очищают флэш-хроматографией смесью элюентов гексан/этилацетат (2/8). Получают желтое твердое вещество с выходом 70% (70 мг). ЖХ-МС: 667,3 (M+H)+, 95% чист. 1H ЯМР (ацетон-d6): Пример 8. Получение 2-[(4-аминобензолсульфонил)изобутиламино]-6-(2-метоксикарбониламино-3,3-дифенилпропиониламино)гексилового эфира (2S,2S)-никотиновой кислоты (PL-520) Стадия A. Получение 6-трет-бутоксикарбониламино-2-[(4-трет-бутоксикарбониламинобензолсульфонил)изобутиламино]гексилового эфира (2S)-никотиновой кислоты (XIV, R1A=3-пиридил) Трет-бутиловый эфир (1S)-{4-[(5-трет-бутоксикарбониламино-1-гидроксиметилпентил)изобутилсульфамоил]фенил}карбаминовой кислоты (промежуточный продукт (VII) из примера 1, стадия D, 130 мг, 0,24 ммоль) растворяют в безводном ДМФА (1 мл) и обрабатывают 0,066 мл (0,48 ммоль) триэтиламина, поле чего EDC (120 мг, 0,65 ммоль), HOBt (88 мг, 0,65 ммоль) и никотиновой кислотой (27 мг, 0,22 ммоль). Смесь перемешивают в течение ночи при комнатной температуре. Продукт экстрагируют смесью этилацетата (40 мл) и воды (40 мл). Органическую фазу отделяют и сушат над Na2SO4, затем выпаривают, получая 200 мг неочищенного продукта. Данное соединение очищают флэш-хроматографией с этилацетатом в качестве элюента. Получают прозрачное маслянистое вещество с выходом 100% (150 мг). ЖХ-МС: 649,3 (M+H)+, 95% чист. 1H ЯМР (ацетон-d6): Стадия B. Получение 6-амино-2-[(4-аминобензолсульфонил)изобутиламино]гексилового эфира (2S)-никотиновой кислоты (XV, R1A=3-пиридил) Продукт из стадии A, 6-трет-бутоксикарбониламино-2-[(4-трет-бутоксикарбониламинобензолсульфонил)изобутиламино]гексиловый эфир (2S)-никотиновой кислоты (150 мг, 0,23 ммоль) растворяют в CH2Cl2 (5 мл) и добавляют трифторуксусную кислоту (1 мл). Смесь перемешивают в течение 2 часов при комнатной температуре. Растворитель выпаривают и остаток экстрагируют смесью этилацетата (40 мл) и NaOH 1М (40 мл) (pH 10). Органическую часть отделяют, сушат над Na2SO4 и затем выпаривают. Остаток (100 мг) используют для следующей реакции без дополнительной очистки. Выход количественный. ЖХ-МС: 449,3 (M+H)+, 95% чист. Стадия C. Получение 2-[(4-аминобензолсульфонил)изобутиламино]-6-(2-метоксикарбониламино-3,3-дифенилпропиониламино)гексилового эфира (2S,2S)-никотиновой кислоты (PL-520) Продукт из стадии B, 6-амино-2-[(4-аминобензолсульфонил)изобутиламино]гексиловый эфир (2S)-никотиновой кислоты (100 мг, 0,22 ммоль) растворяют в безводном ДМФА (2 мл) и обрабатывают 0,062 мл (0,45 ммоль) триэтиламина, а затем EDC (100 мг, 0,56 ммоль), HOBt (75 мг, 0,56 ммоль) и (2S)-2-метоксикарбониламино-3,3-дифенилпропионовой кислотой (56 мг, 0,19 ммоль). Данную смесь перемешивают в течение ночи при комнатной температуре. Продукт экстрагируют смесью этилацетата (40 мл) и воды (40 мл). Органический слой отделяют, сушат над Na2SO4 и затем выпаривают, получая 160 мг неочищенного маслянистого вещества. Остаток очищают флэш-хроматографией со смесью элюентов гексан/этилацетат (2/8). Указанное в заголовке соединение получают в виде прозрачного маслянистого вещества с выходом 20% (25 мг). ЖХ-МС: 730,2 (M+H)+, 95% чист. 1H ЯМР (ацетон-d6): Пример 9. Получение 2-[(4-аминобензолсульфонил)изобутиламино]-6-(2-метоксикарбониламино-3,3-дифенилпропиониламино)гексилового эфира (2S,2S)-диметиламиноуксусной кислоты (PL-534) Стадия A. Получение 6-трет-бутоксикарбониламино-2-[(4-трет-бутоксикарбониламинобензолсульфонил)изобутиламино]гексилового эфира (2S)-диметиламиноуксусной кислоты (XIV, R1A=(CH3)2NCH2-) Данное указанное в заголовке соединение получают из трет-бутилового эфира (1S)-{4-[(5-трет-бутоксикарбониламино-1-гидроксиметилпентил)изобутилсульфамоил]фенил}карбаминовой кислоты (промежуточный продукт (VII) из примера 1, стадия D), как описано в примере 15, стадия A, с использованием N,N-диметилглицина. Получают прозрачное масло с выходом 100% (150 мг). ЖХ-МС: 629,3 (M+H)+, 95% чист. 1H ЯМР (ацетон-d6): Стадия B. Получение 6-амино-2-[(4-аминобензолсульфонил)изобутиламино]гексилового эфира (2S)-диметиламиноуксусной кислоты (XV, R1A=(CH3)2NCH2-) Указанное в заголовке производное получают из 6-трет-бутоксикарбониламино-2-[(4-трет-бутоксикарбониламинобензолсульфонил)изобутиламино]гексилового эфира (2S)-диметиламиноуксусной кислоты, как описано в примере 15, стадия B. Конечный продукт (100 мг) используют без дополнительной обработки в следующей стадии. ЖХ-МС: 429,3 (M+H)+, 90% чист. Стадия C. Получение 2-[(4-аминобензолсульфонил)изобутиламино]-6-(2-метоксикарбониламино-3,3-дифенилпропиониламино)гексилового эфира (2S,2S)-диметиламиноуксусной кислоты (PL-534) Данное указанное в заголовке соединение получают из 6-амино-2-[(4-аминобензолсульфонил)изобутиламино]гексилового эфира (2S)-диметиламиноуксусной кислоты, как описано в примере 15, стадия C. Неочищенный продукт очищают препаративной ЖХ. Конечное соединение получают с выходом 10% (10 мг). ЖХ-МС: 710,3 (M+H)+, 92% чист. 1H ЯМР (ацетон-d6): Пример 10. Получение 2-[(4-аминобензолсульфонил)изобутиламино]-6-(2-метоксикарбониламино-3,3-дифенилпропиониламино)гексилового эфира (2S,2S)-2-амино-3-метилмасляной кислоты (PL-530) Стадия A. Получение 6-трет-бутоксикарбониламино-2-[(4-трет-бутоксикарбониламинобензолсульфонил)изобутиламино]гексилового эфира (2S)-2-бензилоксикарбониламино-3-метилмасляной кислоты (XIV, R1A=(CH3)2CHCH(NH2)-) Данное указанное в заголовке соединение получают из трет-бутилового эфира (1S)-{4-[(5-трет-бутоксикарбониламино-1-гидроксиметилпентил)изобутилсульфамоил]фенил}карбаминовой кислоты (промежуточный продукт (VII) из примера 1, стадия D), как описано в примере 15, стадия A с использованием (2S)-2-бензилоксикарбониламино-3-метилмасляной кислоты. Неочищенный продукт очищают флэш-хроматографией, элюируя смесью гексан/этилацетат (1/1). Полученный выход равен 100% (150 мг). ЖХ-МС: 777,3 (M+H)+, 95% чист. 1H ЯМР (ацетон-d6): Стадия B. Получение 6-амино-2-[(4-аминобензолсульфонил)изобутиламино]гексилового эфира (2S)-бензилоксикарбониламино-3-метилмасляной кислоты (XV, R1A=(CH3)2CHCH(NH2)-) Данное производное получают из 6-трет-бутоксикарбониламино-2-[(4-трет-бутоксикарбониламинобензолсульфонил)изобутиламино]гексилового эфира (2S)-2-бензилоксикарбониламино-3-метилмасляной кислоты (продукт из стадии A), как описано в примере 15, стадия B. Конечное соединение получают с количественным выходом (110 мг) и используют в следующей стадии без очистки. ЖХ-МС: 577,3 (M+H)+, 90% чист. Стадия C. Получение 2-[(4-аминобензолсульфонил)изобутиламино]-6-(2-метоксикарбониламино-3,3-дифенилпропиониламино)гексилового эфира (2S,2S)-2-бензилоксикарбониламино-3-метилмасляной кислоты Указанное в заголовке соединение получают из 6-амино-2-[(4-аминобензолсульфонил)изобутиламино]гексилового эфира (2S)-бензилоксикарбониламино-3-метилмасляной кислоты (продукт из стадии B), как описано в примере 15, стадия C. Прозрачное маслянистое вещество получают с выходом 86% (120 мг). ЖХ-МС: 858,3 (M+H)+, 95% чист. Стадия D. Получение 2-[(4-аминобензолсульфонил)изобутиламино]-6-(2-метоксикарбониламино-3,3-дифенилпропиониламино)гексилового эфира (2S,2S)-2-амино-3-метилмасляной кислоты (PL-530) К перемешиваемому раствору 2-[(4-аминобензолсульфонил)изобутиламино]-6-(2-метоксикарбониламино-3,3-дифенилпропиониламино)гексилового эфира (2S,2S)-2-бензилоксикарбониламино-3-метилмасляной кислоты (стадия C, 120 мг, 0,14 ммоль) в безводном ТГФ (8 мл) в атмосфере азота добавляют палладий 10% мас. на активированном угле (160 мг). Смесь подвергают взаимодействию в атмосфере водорода в течение ночи при комнатной температуре. Раствор фильтруют и палладий на угле промывают ТГФ (50 мг). Растворитель выпаривают и остаток (110 мг) очищают флэш-хроматографией, используя этилацетат в качестве элюента. Получают прозрачное маслянистое вещество с выходом 47% (47 мг). ЖХ-МС: 796,4 (M+H)+, 95% чист. 1H ЯМР (ацетон-d6): 0,84-0,97 (м, 12H), 0,97-1,08 (м, 2H), 1,27-1,43 (м, 3H), 1,49-1,62 (м, 4H), 1,80-1,93 (м, 1H), 1,94-2,00 (м, 1Н), 2,36-2,46 (м, 1Н), 2,58-2,74 (м, 2H), 2,86-2,96 (м, 3H), 2,99-3,10 (м, 2H), 3,46-3,52 (с, 3H), 3,52-3,60 (м, 2H), 3,75-3,87 (м, 2H), 3,95-4,04 (м, 1Н), 4,10-4,18 (м, 1H), 4,37-4,44 (м, 1H), 4,89-4,97 (м, 1Н), 5,40-5,48 (м, 1H), 6,30-6,40 (м, 1Н), 6,76-6,83 (д, J=8,2, 1H), 6,87-7,03 (м, 2H), 7,14-7,22 (м, 1H), 7,23-7,34 (м, 3H), 7,35-7,45 (м, 4H), 7,50-7,56 (м, 1H), 7,57-7,65 (м, 1H). Биологическая доступность соединений Чтобы оценить степень отщепления фосфатной группы in vivo от предполагаемых соединений, соединения PL-100, PL-462 (на основе PL-100), PL-337 и PL-515 (на основе PL-337) вводят перорально (50 мг/кг) самцам крыс Спрег-Доули и их концентрации в плазме измеряют при различных временных интервалах после введения. PL-100 представляет собой активный ингредиент (ингибитор протеазы) следующей формулы:
PL-337 представляет собой активный ингредиент (ингибитор протеазы) следующей формулы:
Как было показано, данный активный ингредиент является эффективным по отношению к ВИЧ-1 аспартил-протеазе (патент США Все тестируемые продукты (PL-100, PL-462, PL-337 и PL-515) готовят в различных растворителях с конечной концентрацией 25 мг/мл. Композиция растворителя является следующей: (1) 20% этанола, 50% пропиленгликоля, 0,05% мас./об. Tween 20 и вода (Смесь); (2) забуференный фосфатом физиологический раствор (PBS). Тестируемые продукты вводят самцам крыс Спрег-Доули при однократной пероральной дозе 50 мг/кг. Каждый продукт тестируют для трех крыс. Образцы крови (0,2-0,3 мл) собирают через 10, 20, 40, 60, 120, 180 и 360 мин после введения дозы. Собранную кровь центрифугируют, чтобы выделить плазму. Полученную в результате плазму отделяют и хранят при -70°C. Образцы плазмы вместе со стандартами и образцами контроля качества обрабатывают, чтобы осадить белки, затем анализируют ВЭЖХ-МС на присутствие PL-462, PL-100, PL-515 и PL-337.
Результаты демонстрируют, что соединения PL-462 и PL-515 можно доставить перорально в водных растворах. Ни одно из соединений PL-462 и PL-515, доставленное в виде водного раствора, не обнаруживается в образцах крови, что говорит о быстром метаболизме исходных лекарственных веществ в PL-100 и PL-337. Установлено, что водные дозировки растворов PL-462 и PL-515 слегка превосходят доставку PL-100 и PL-337 по сравнению с неводными рецептурами PL-100 и PL-337. Основываясь на данных результатах все фосфорилированные соединения, описанные в настоящем изобретении, будут демонстрировать аналогичные фармакокинетические свойства. Коэффициенты распределения (LogP) выбранных соединений и соответствующих ингибиторов ВИЧ-протеазы (лекарственных средств) являются следующими:
LogP измеряют стандартно, растворяя 1 мг соединения в 0,8 мл октанола или фосфатного буфера рН 7,4 (0,04 М KHPO4). Концентрацию соединений в фазах определяют ЖХ-МС. Данный тест демонстрирует растворимость соединений при физиологическом значении рН. Полученные LogP показывают, что соединения хорошо растворимы по сравнению с соответствующими лекарственными средствами. Соединения, перечисленные в таблице 3, готовят по следующим схемам 1, 1A, 2, 3, 4 или 5; и более конкретно, как описано в каждом примере, перечисленном выше. Номера соединений, перечисленных в таблице 3 (Прим. Таблица 3 Структуры соединений на основе лизина по настоящему изобретению
Формула изобретения
1. Соединение формулы I 2. Соединение по п.1, которое представляет собой соединение формулы II, 3. Соединение по п.2, где R6 представляет собой изобутил и n равно 4. 4. Соединение по п.3, где R1 представляет собой (НО)2Р(О) или (NaO)2P(O). 5. Соединение по п.4, где R3 выбран из группы, состоящей из СН3СО, СН3О-СО и 4-морфолин-СО. 6. Соединение по п.5, где Х представляет собой 4-NH2, a Y представляет собой Н или F. 7. Соединение по п.6, где R2 выбран из группы, состоящей из дифенилметильной группы формулы IV, нафтил-1-CH2– группы формулы V и нафтил-2-СН2– группы формулы VI. 8. Соединение по п.2, которое представляет собой соединение формулы IIa 9. Соединение по п.8, где R6 представляет собой изобутил; n равно 4; R1 представляет собой (НО)2Р(О) или (NaO)2P(O); R3 выбран из группы, состоящей из СН3СО, СН3О-СО и 4-морфолин-СО; Х представляет собой NH2, а Y представляет собой Н или F. 10. Соединение по п.1, которое выбрано из группы, состоящей из следующих соединений и солей:
11. Соединение по п.10, которое представляет собой натриевую соль метилового эфира (1S,5S)-(1-{5-[(4-аминобензолсульфонил)изобутиламино]-6-фосфоноксигексакарбамоил}-2,2-дифенилэтил)карбаминовой кислоты. 12. Соединение по п.10, которое представляет собой метиловый эфир (1S,5S)-(1-{5-[(4-амино-3-фторбензолсульфонил)изобутиламино]-6-фосфоноксигексакарбамоил}-2,2-дифенилэтил)карбаминовой кислоты. 13. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении ВИЧ аспартилпротеазы, включающая, по меньшей мере, одно соединение, определенное в любом из пп.1-12, в фармацевтически эффективном количестве или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель. 14. Соединения, определенные в любом из пп.1-12, или их фармацевтически приемлемая соль, являющиеся эффективными ингибиторами аспартилпротеазы. 15. Способ лечения или профилактики ВИЧ инфекции, включающий введение, по меньшей мере, одного соединения, определенного в любом из пп.1-12, или его фармацевтически приемлемой соли млекопитающему, нуждающемуся в этом.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
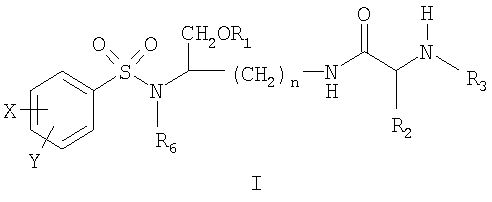
 6436989 Hale et al., полное содержание которого включено в описание в виде ссылки. Данный патент показывает новый класс молекул, характеризующихся подходящей растворимостью в воде, высокой пероральной биологической доступностью и легким генерированием in vivo активного ингредиента. Однако хорошо известно, что ВИЧ обладает способностью развивать устойчивость к имеющимся в настоящее время лекарственным препаратам. Таким образом, существует необходимость в альтернативных ингибиторах ВИЧ-протеазы, активных по отношению к диким типам вирусных штаммов и устойчивым вирусным штаммам. Так, для борьбы с устойчивыми вирусными штаммами желательными являются молекулы, полученные из имеющихся в настоящее время ингибиторов ВИЧ-протеазы, показывающих улучшенную растворимость и биологическую доступность.
6436989 Hale et al., полное содержание которого включено в описание в виде ссылки. Данный патент показывает новый класс молекул, характеризующихся подходящей растворимостью в воде, высокой пероральной биологической доступностью и легким генерированием in vivo активного ингредиента. Однако хорошо известно, что ВИЧ обладает способностью развивать устойчивость к имеющимся в настоящее время лекарственным препаратам. Таким образом, существует необходимость в альтернативных ингибиторах ВИЧ-протеазы, активных по отношению к диким типам вирусных штаммов и устойчивым вирусным штаммам. Так, для борьбы с устойчивыми вирусными штаммами желательными являются молекулы, полученные из имеющихся в настоящее время ингибиторов ВИЧ-протеазы, показывающих улучшенную растворимость и биологическую доступность.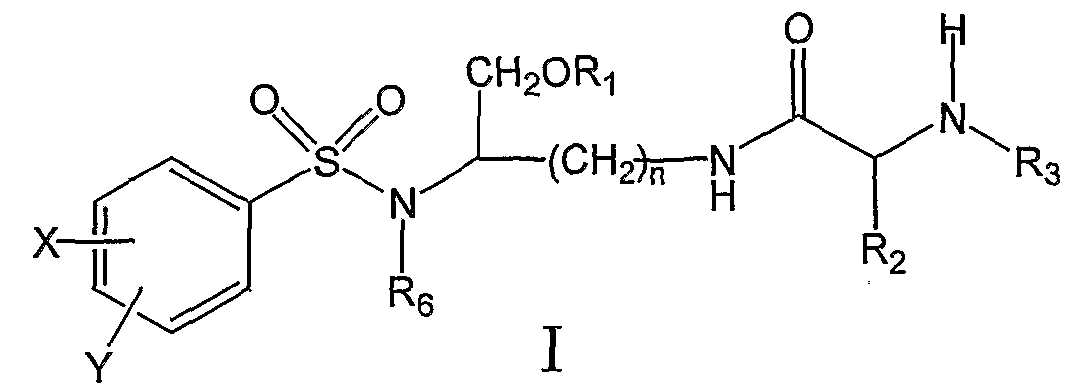
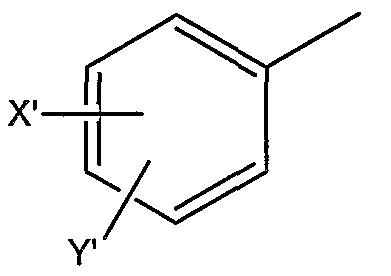
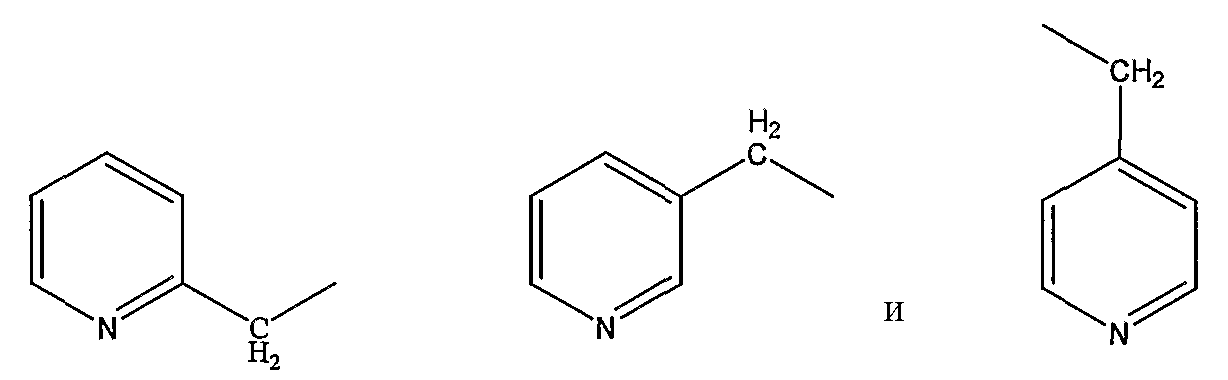
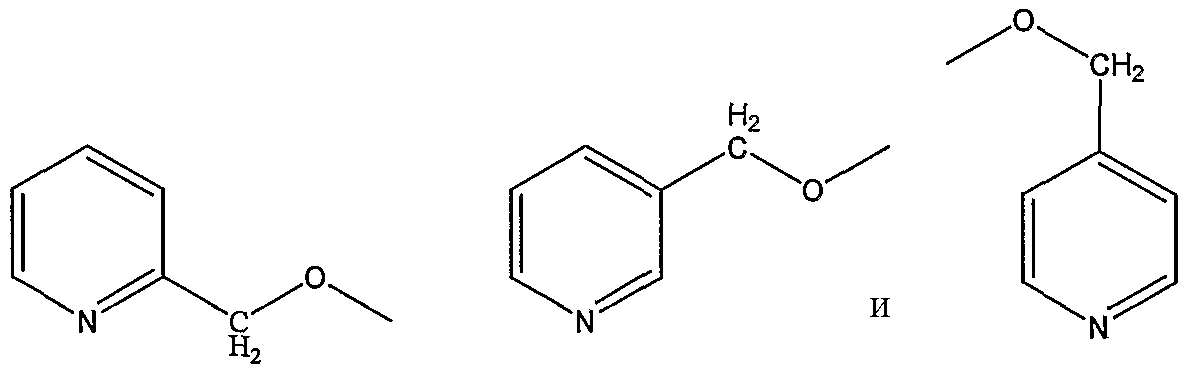
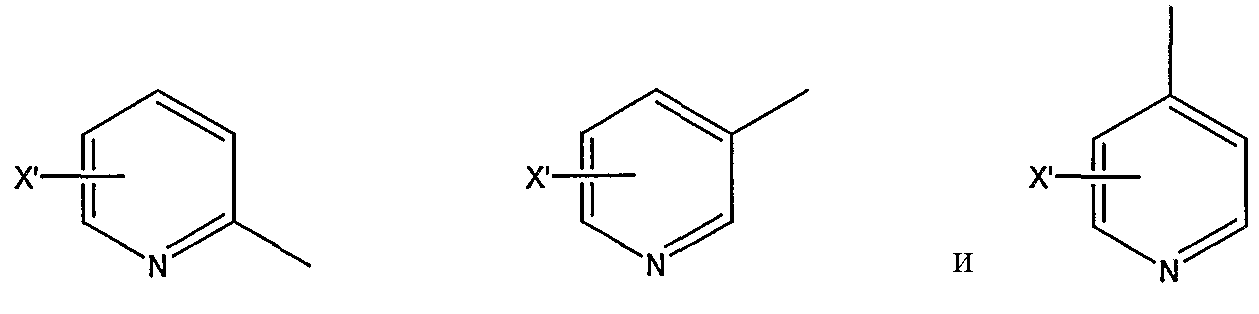
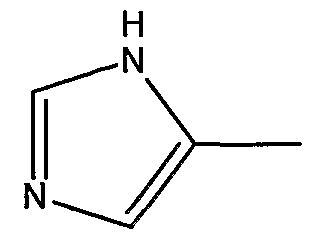
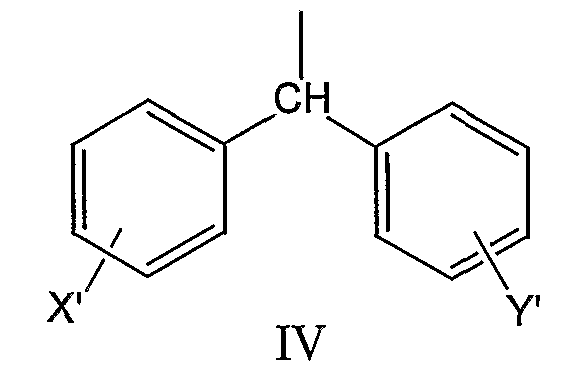
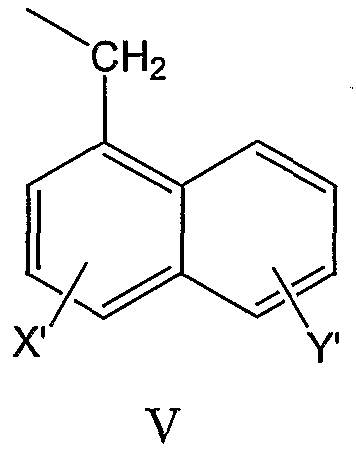
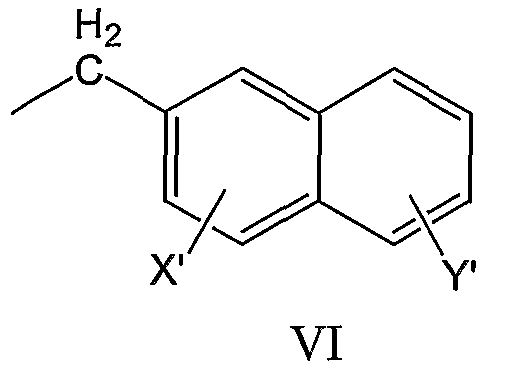
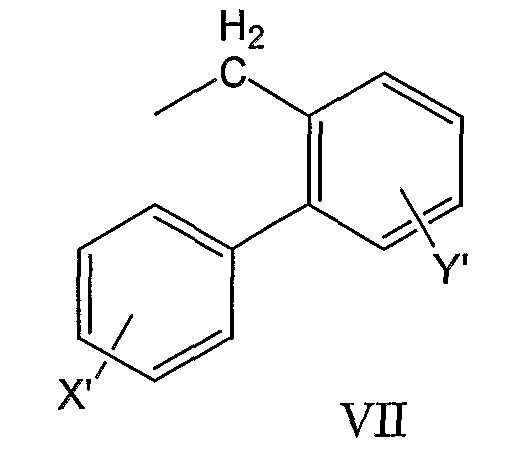
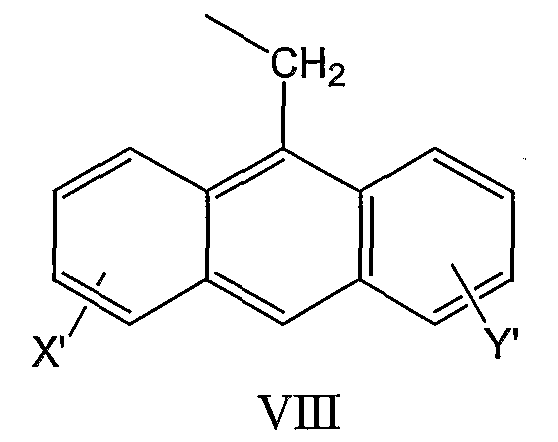
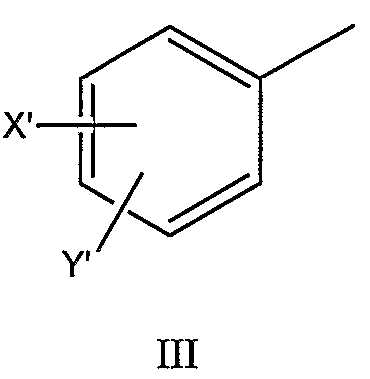
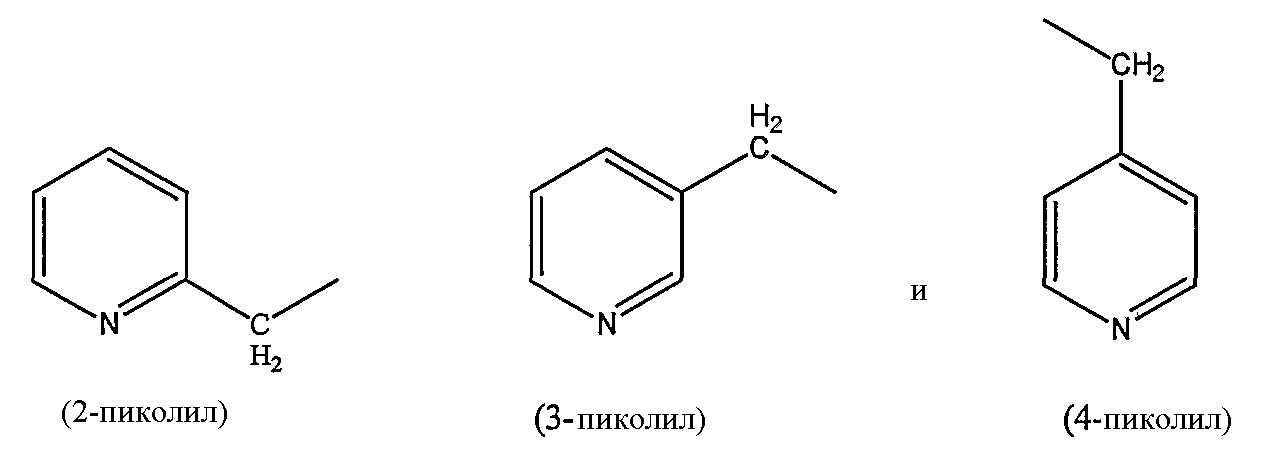
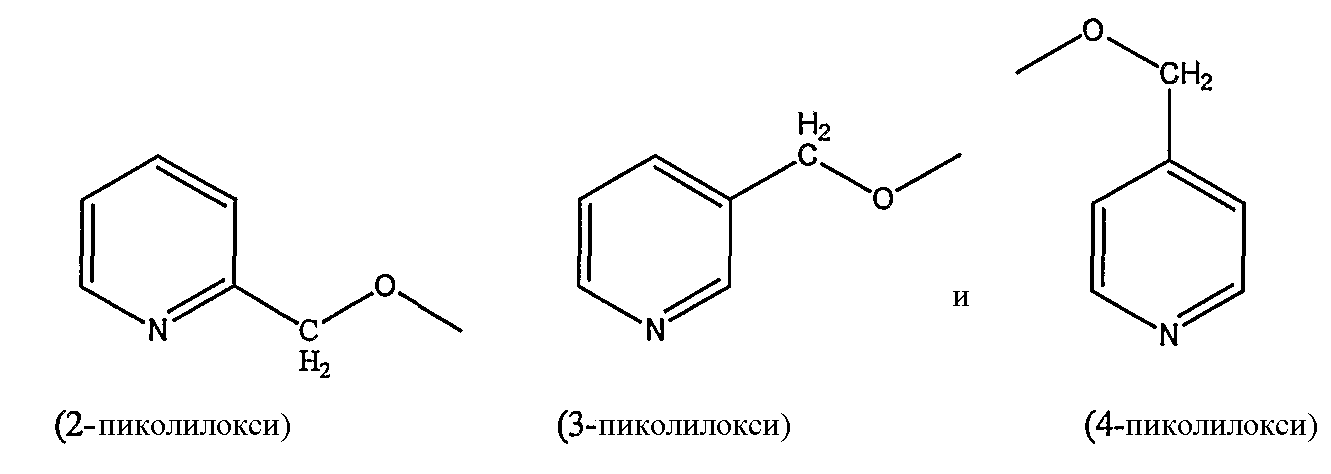
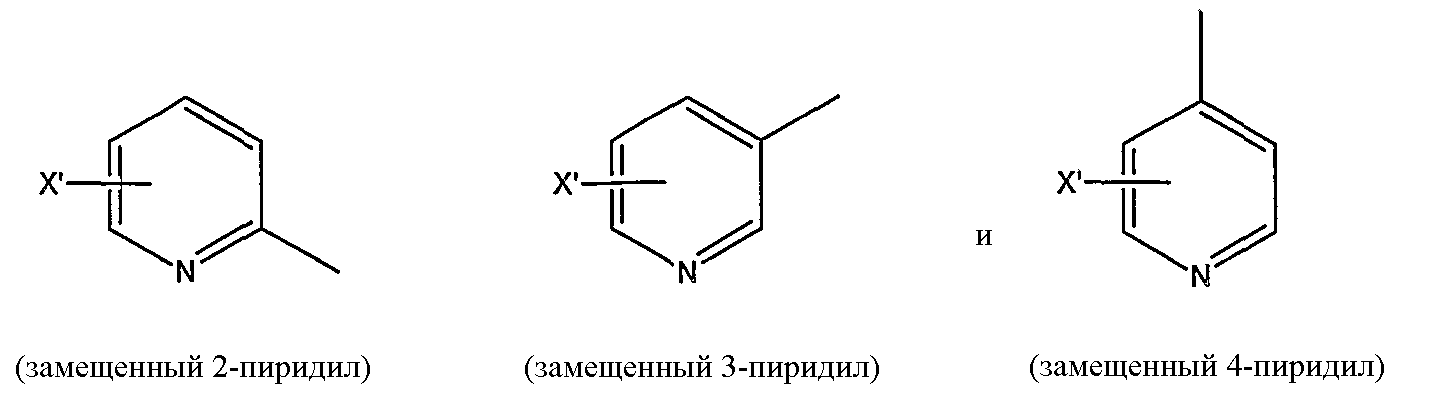
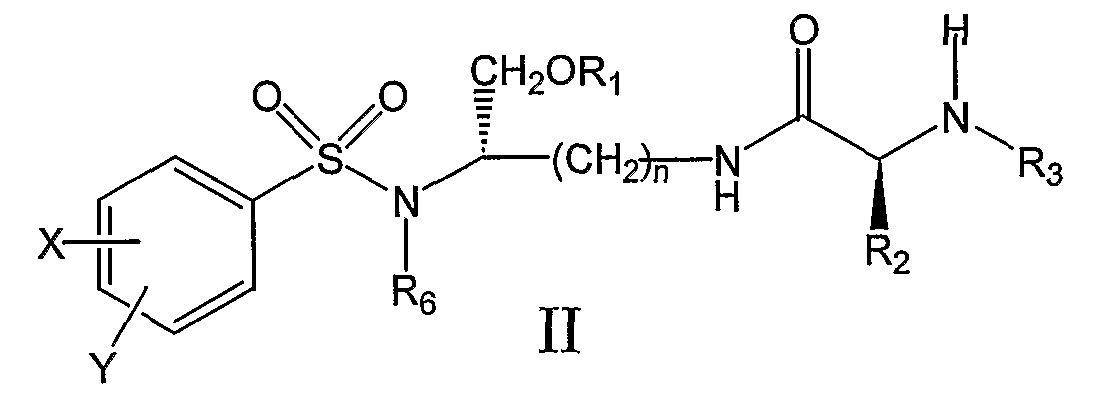
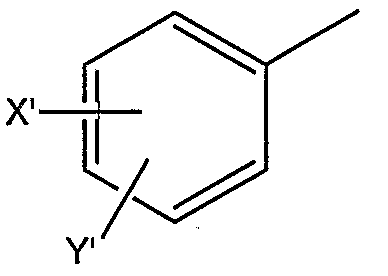
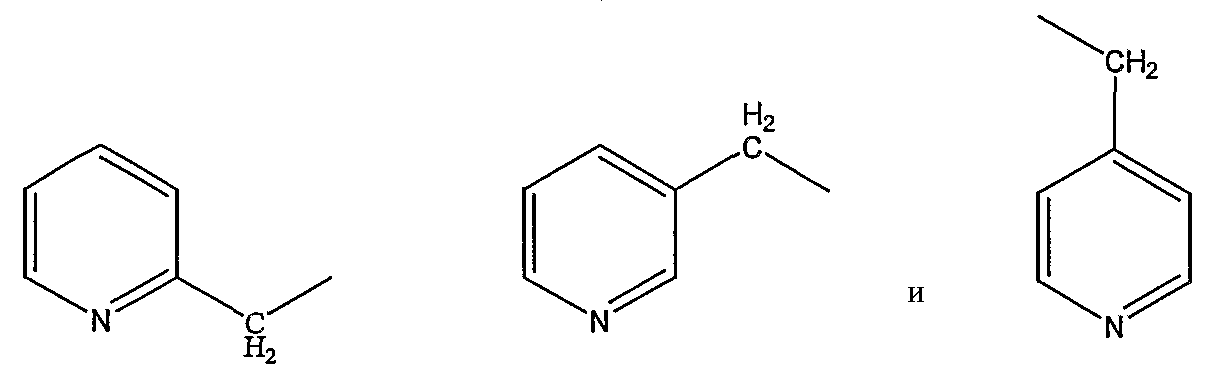
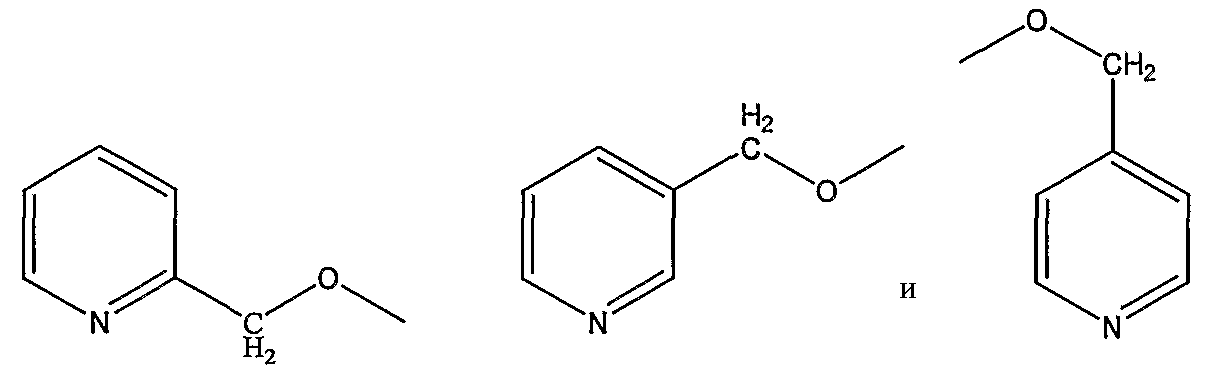
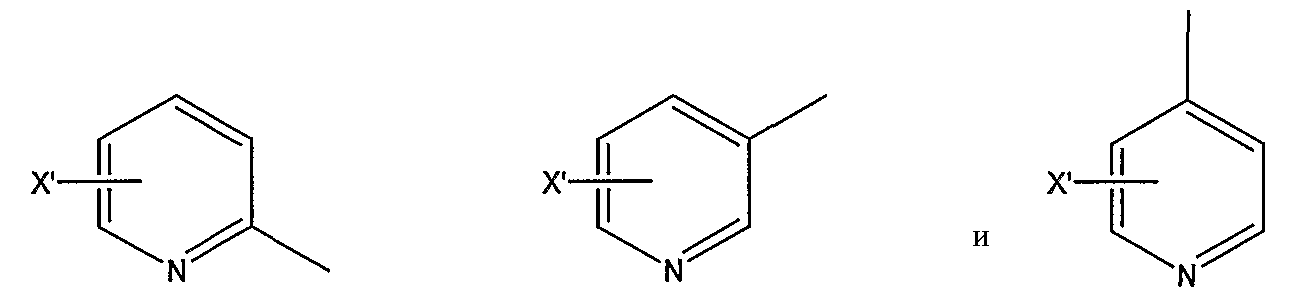
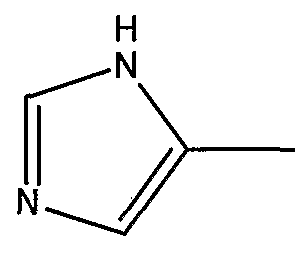
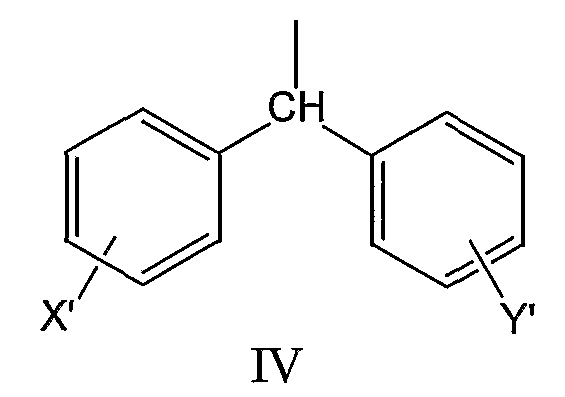
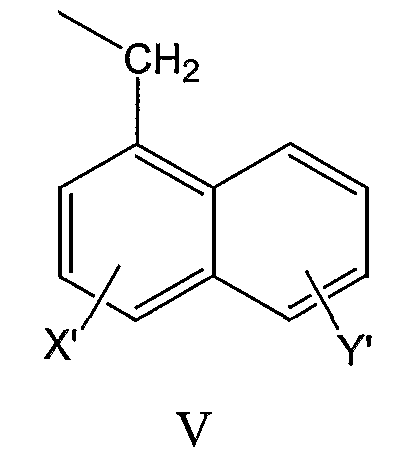
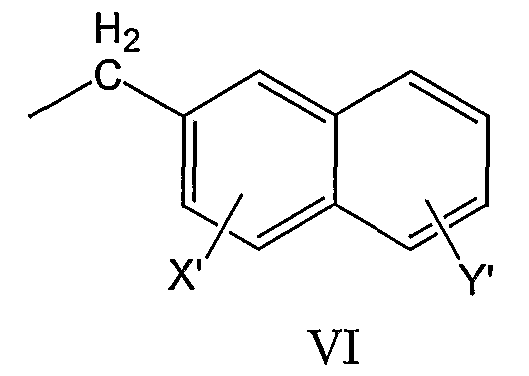
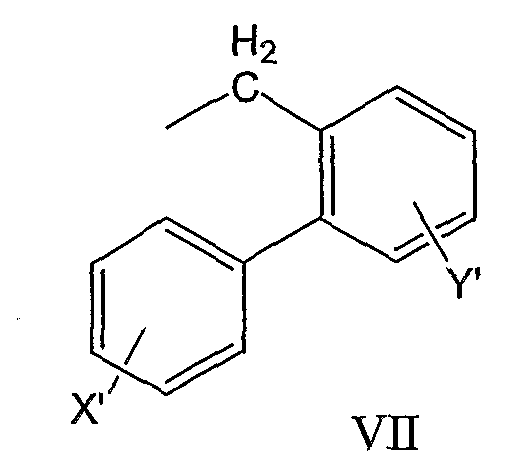
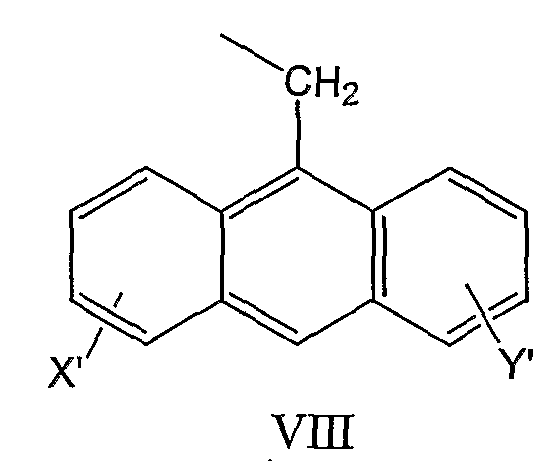
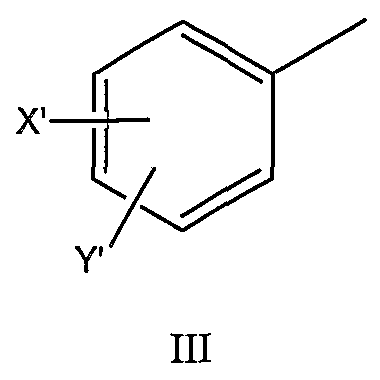
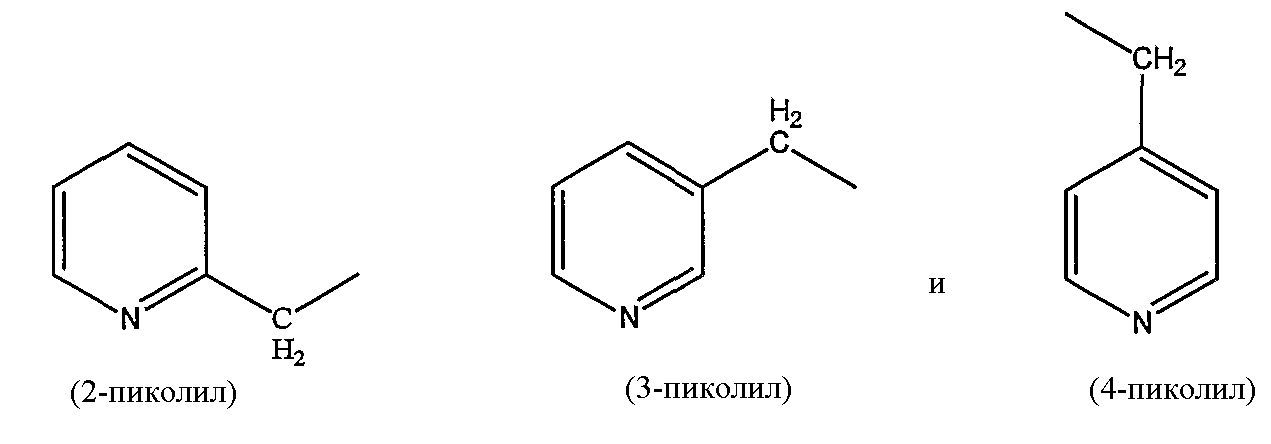
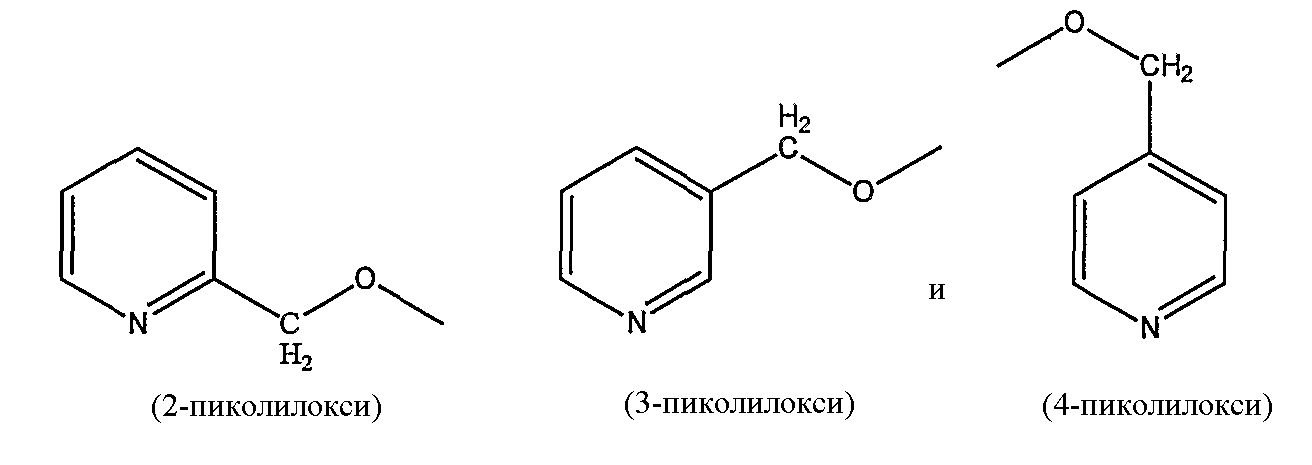
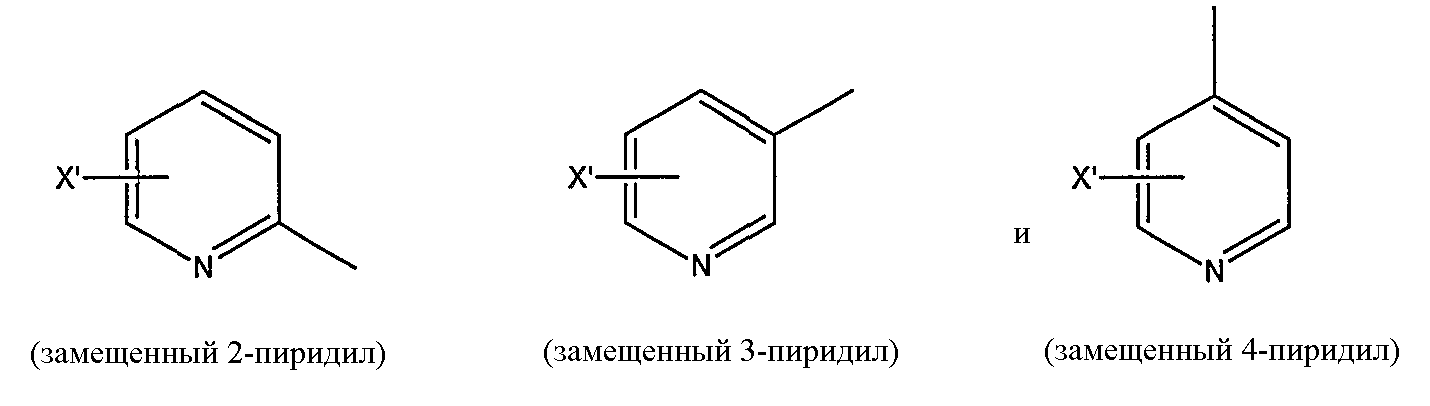
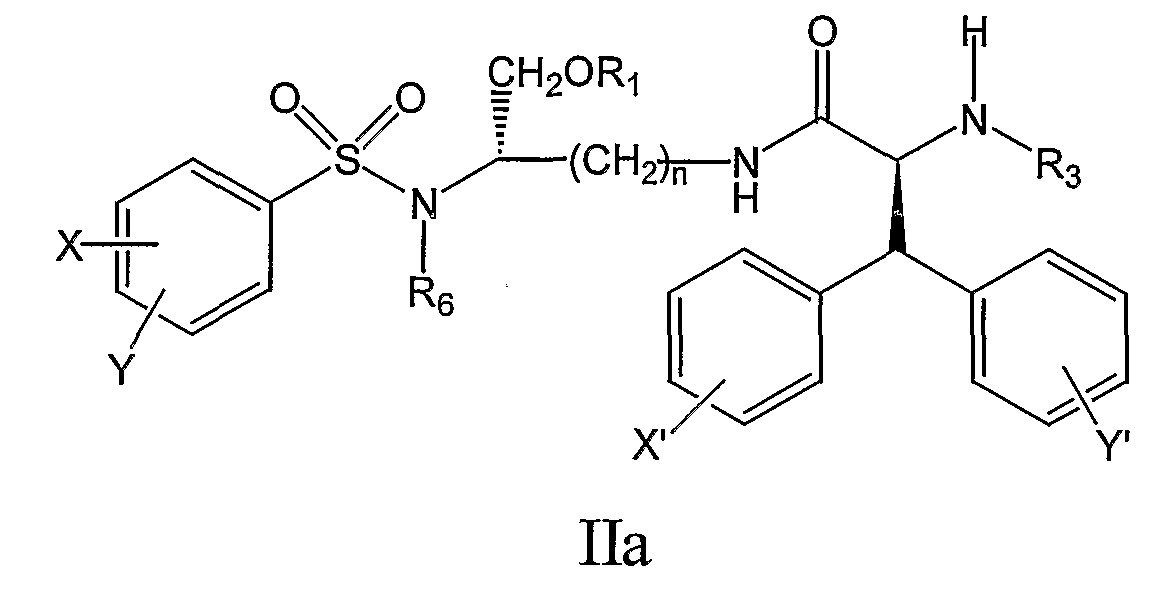
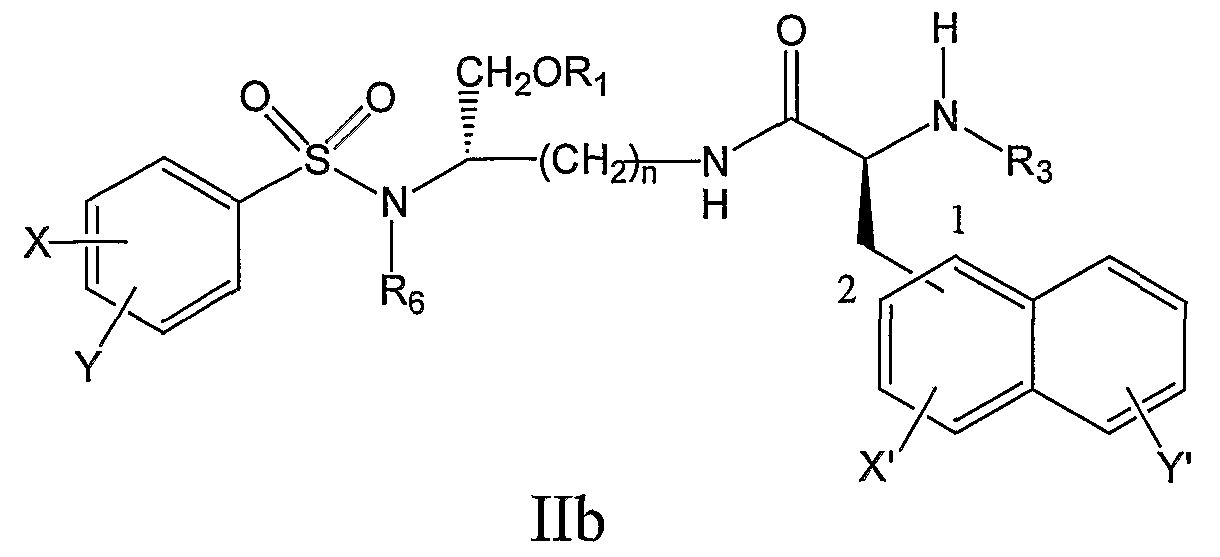
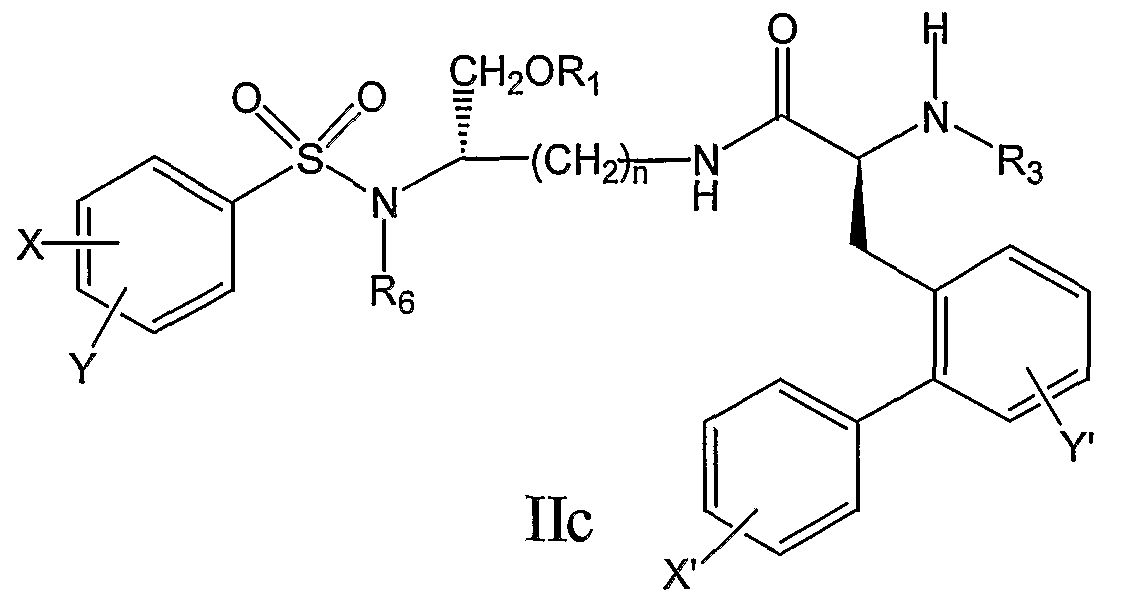
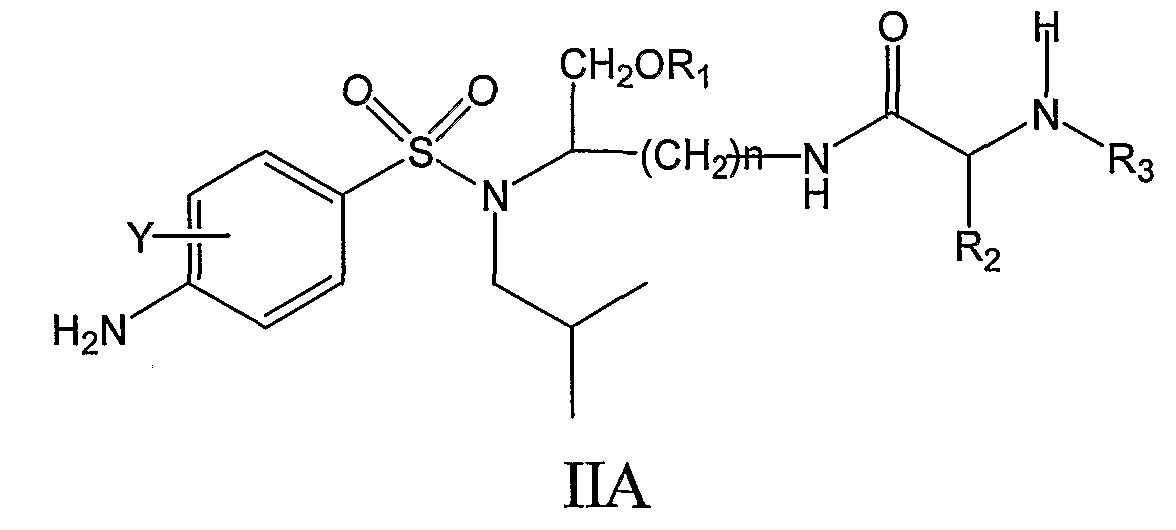
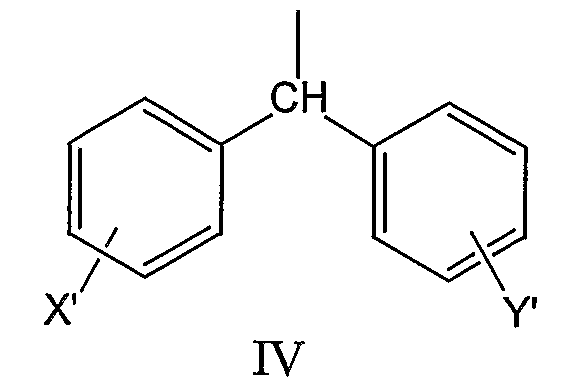
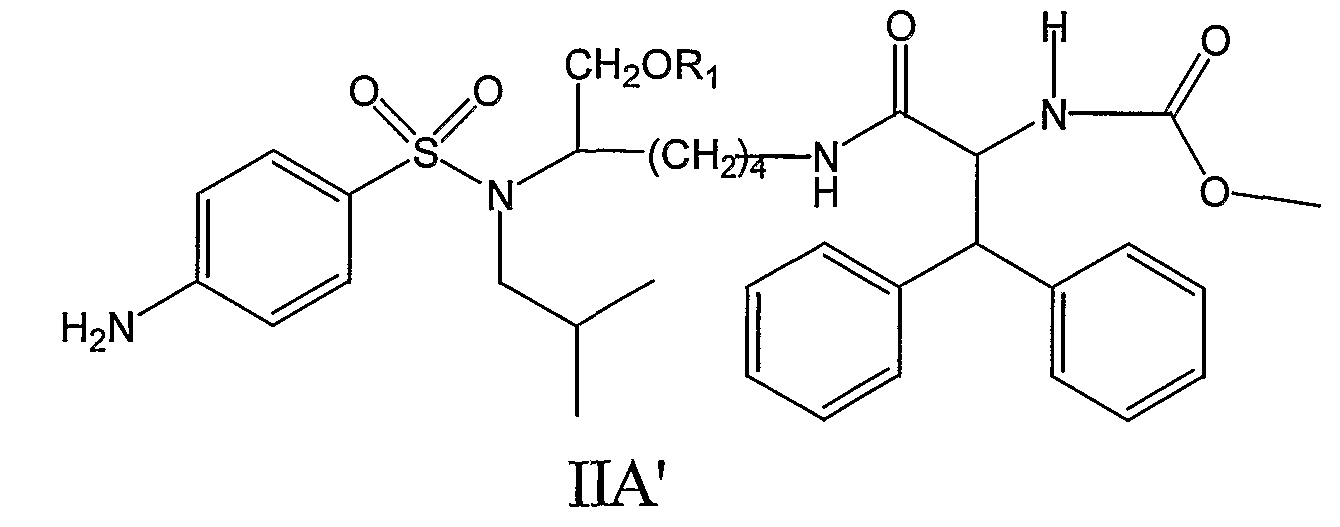
 фармацевтически эффективное количество
фармацевтически эффективное количество +.
+.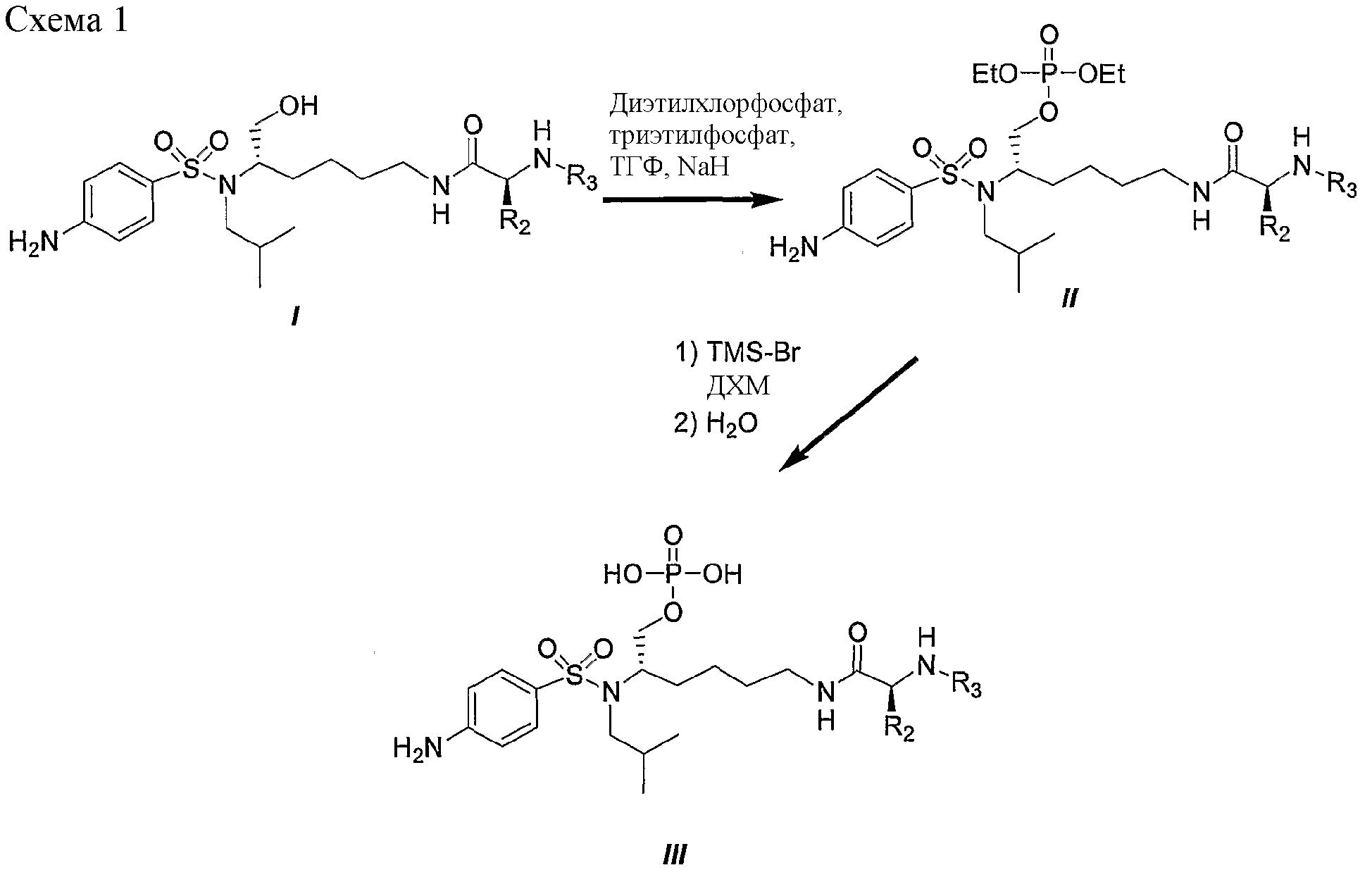
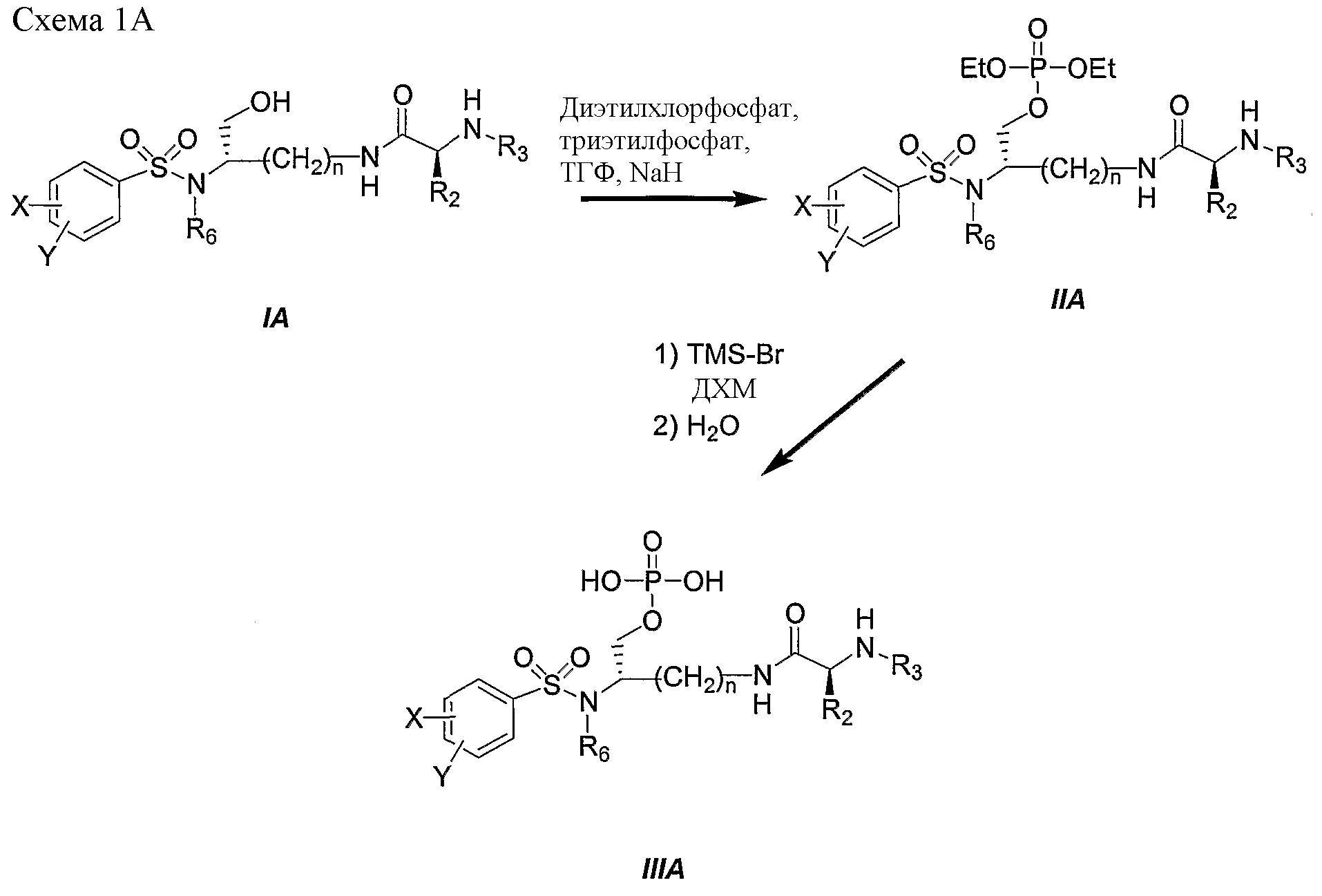
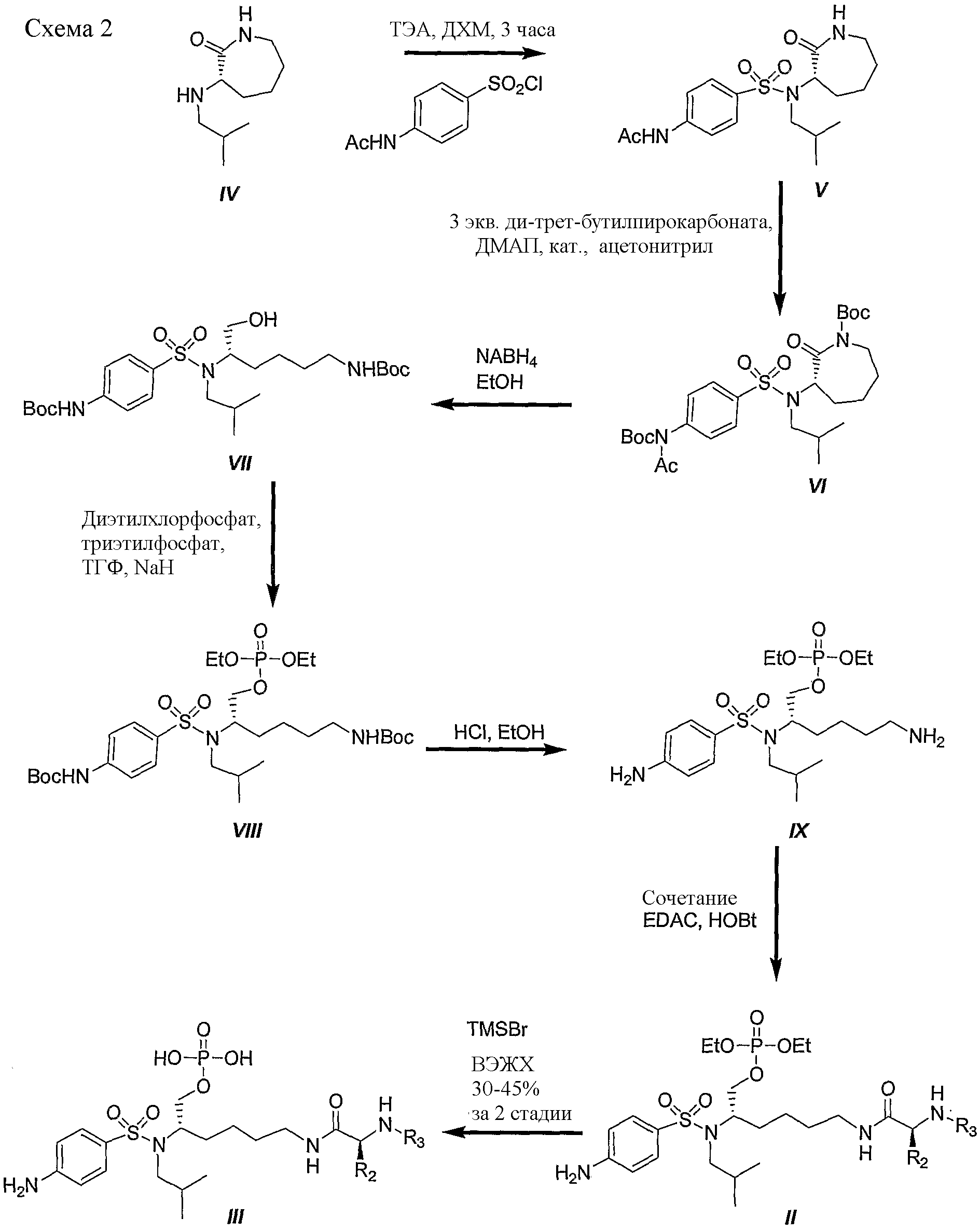
 в ацетонитриле дала производное X с выходом 38%. Введение группы фосфатного сложного моноэфира осуществляли, как описано ранее в схемах 1 и 2. Сначала промежуточное соединение, представляющее собой эфир диэтилтрифосфорной кислоты, получали с хорошим выходом при обработке диэтилхлорфосфатом и гидридом натрия в смеси тетрагидрофурана и триэтилфосфата. Затем, добавление триметилсилилбромида в дихлорметане (ДХМ) дало соединение фосфатного сложного моноэфира с выходами от хорошего до превосходного. Конечный продукт XI легко получали при обработке фосфатного сложного моноэфира раствором гидроксида натрия с хорошими выходами.
в ацетонитриле дала производное X с выходом 38%. Введение группы фосфатного сложного моноэфира осуществляли, как описано ранее в схемах 1 и 2. Сначала промежуточное соединение, представляющее собой эфир диэтилтрифосфорной кислоты, получали с хорошим выходом при обработке диэтилхлорфосфатом и гидридом натрия в смеси тетрагидрофурана и триэтилфосфата. Затем, добавление триметилсилилбромида в дихлорметане (ДХМ) дало соединение фосфатного сложного моноэфира с выходами от хорошего до превосходного. Конечный продукт XI легко получали при обработке фосфатного сложного моноэфира раствором гидроксида натрия с хорошими выходами.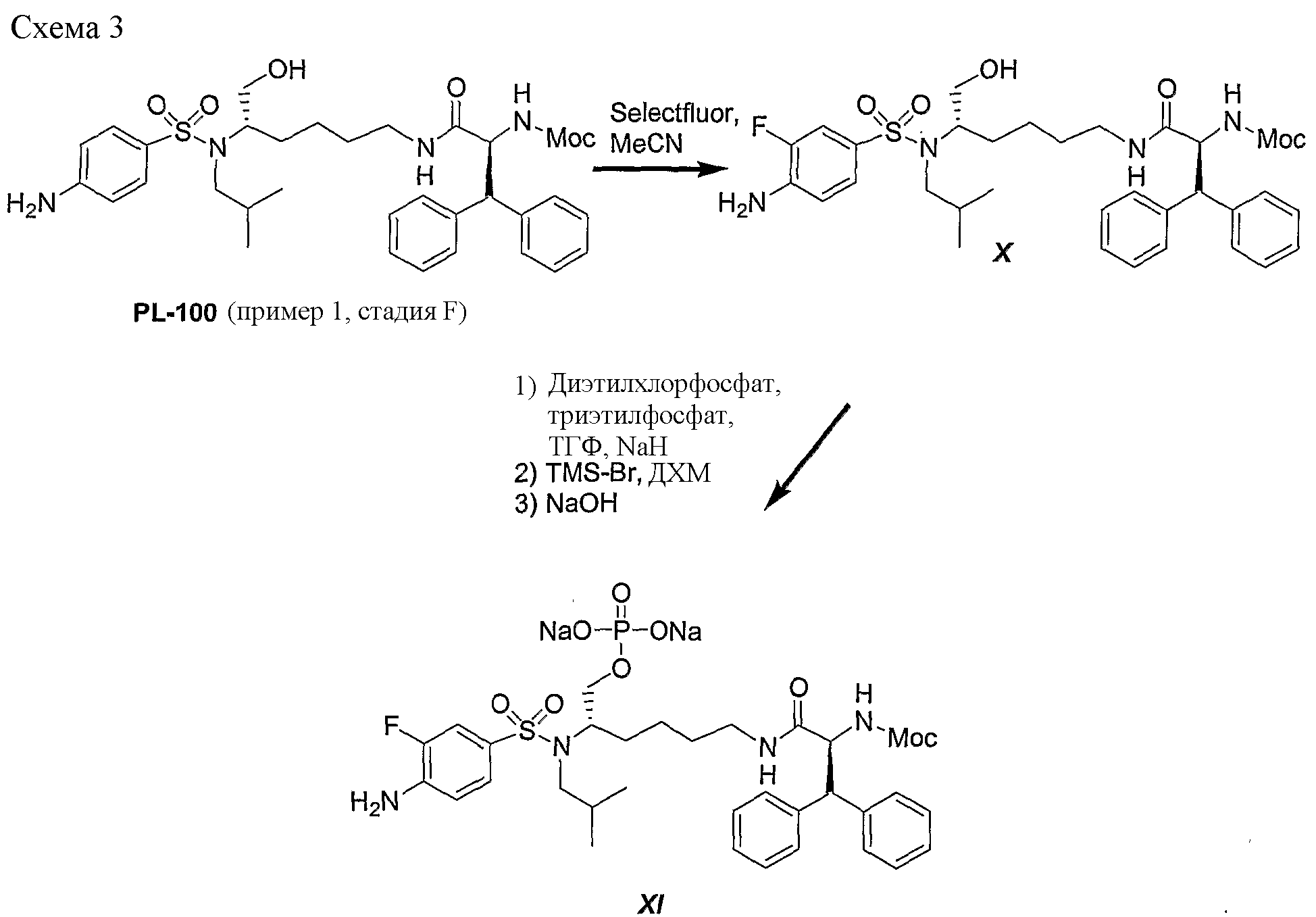
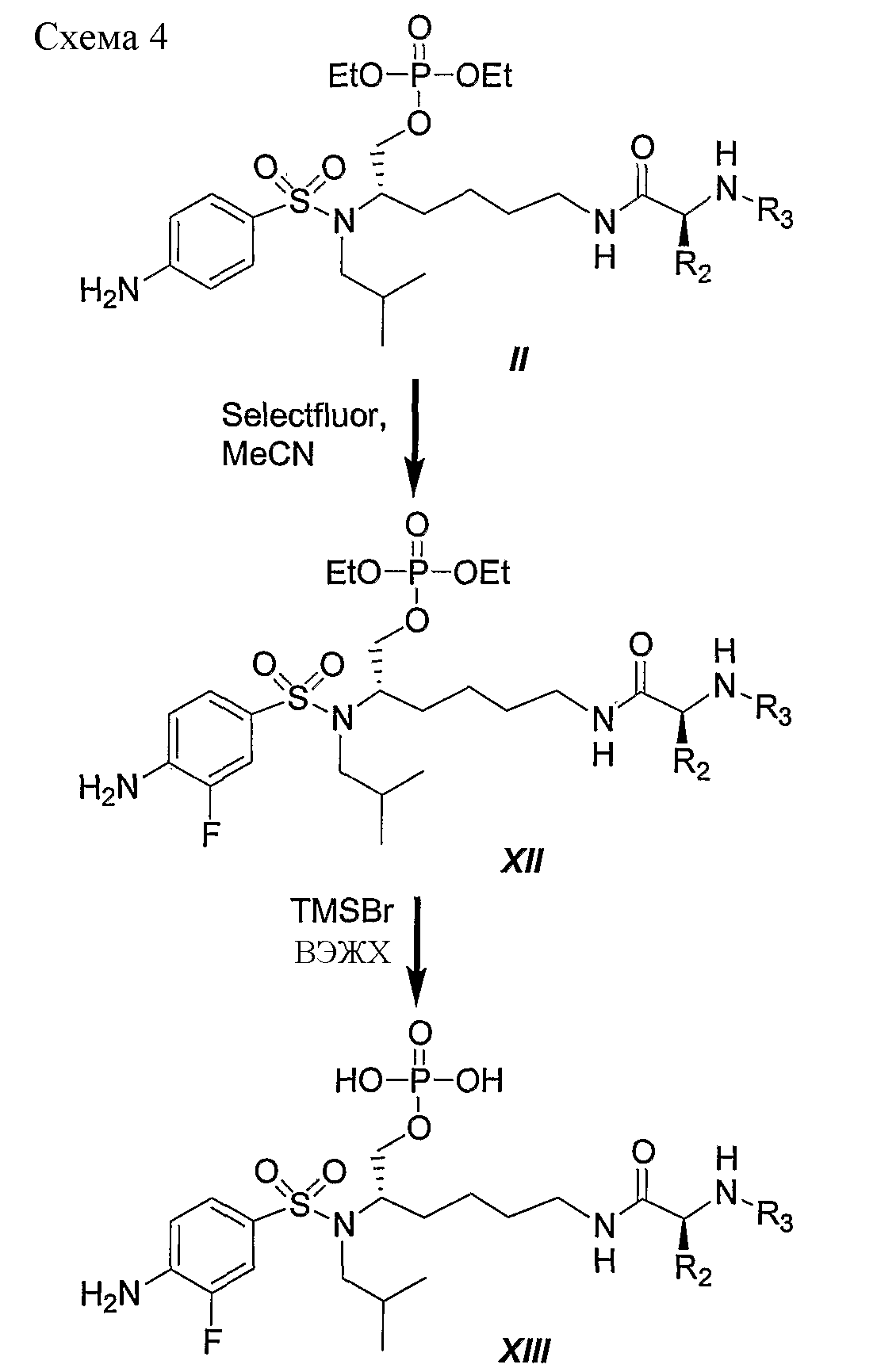
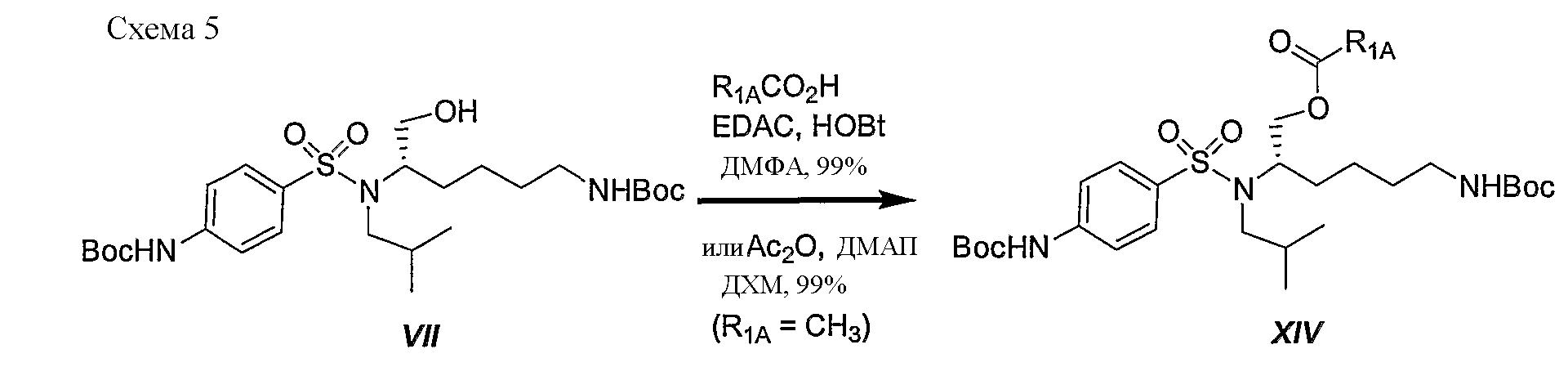
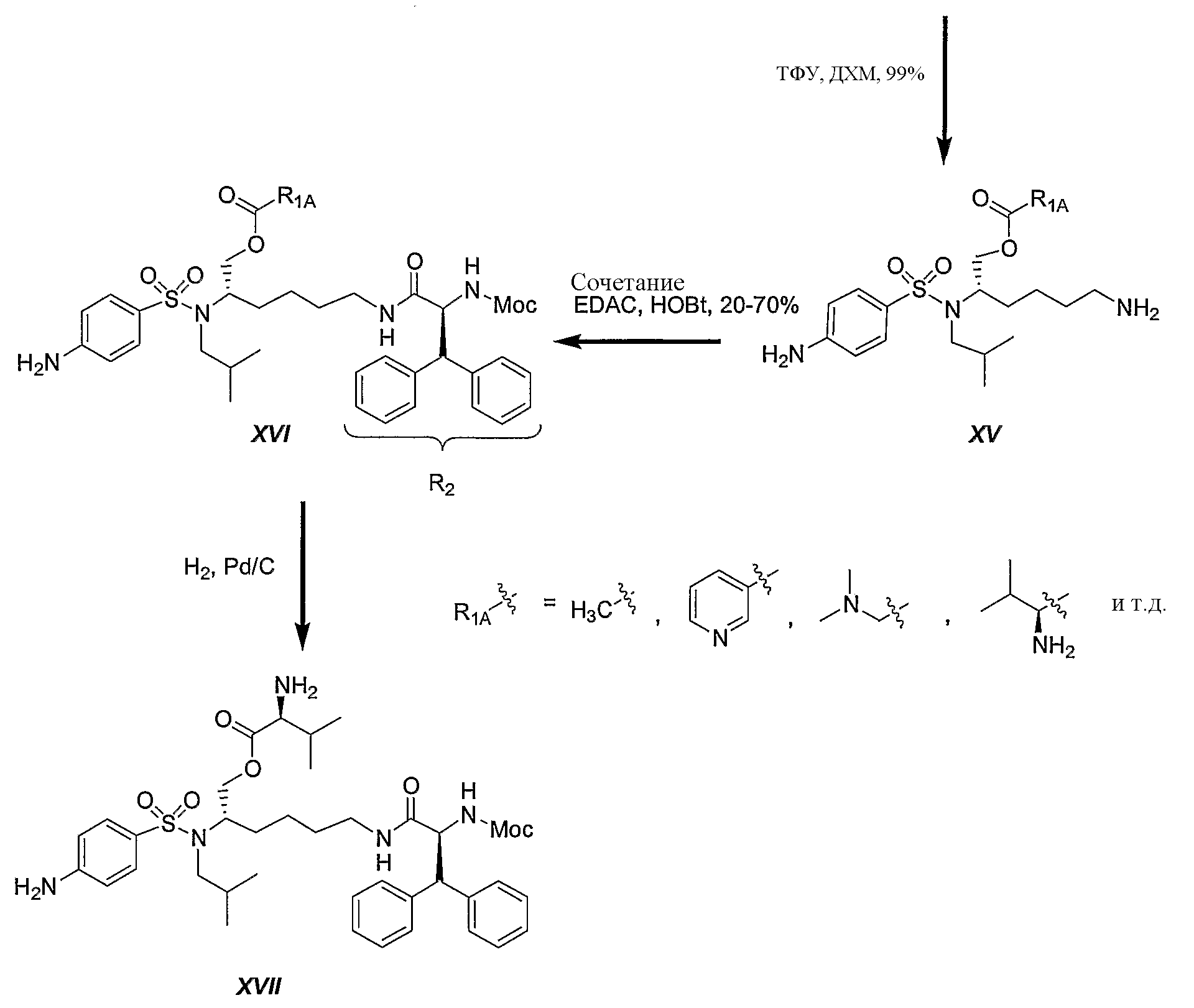
 , 8 мкм, 22х250 мм
, 8 мкм, 22х250 мм :210 & 265 нм
:210 & 265 нм chi 530 в капиллярных трубках, и они не корректировались.
chi 530 в капиллярных трубках, и они не корректировались. 0,93 (д, J=6,5, 3H), 0,97 (д, J=6,5, 3H), 1,39 (т, J=9,8, 1H), 1,47 (м, 1H), 1,78-1,65 (м, 2H), 2,00-1,93 (м, 2H), 2,32-2,2 (м, 2H), 2,38 (т, J=9,7, 1H), 3,16 (м, 3H), 6,62 (с, 1H (NH)). Тпл. 52-54°C (гексан).
0,93 (д, J=6,5, 3H), 0,97 (д, J=6,5, 3H), 1,39 (т, J=9,8, 1H), 1,47 (м, 1H), 1,78-1,65 (м, 2H), 2,00-1,93 (м, 2H), 2,32-2,2 (м, 2H), 2,38 (т, J=9,7, 1H), 3,16 (м, 3H), 6,62 (с, 1H (NH)). Тпл. 52-54°C (гексан).