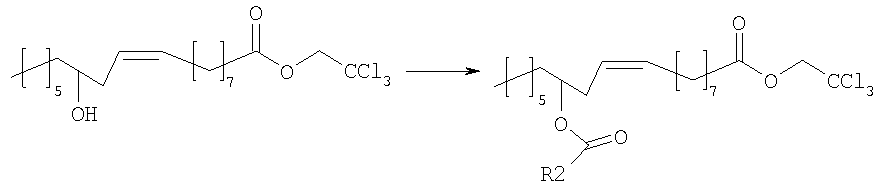Патент на изобретение №2379282
|
|||||||||||||||||||||||||||||||||||||||||||||||||||
(54) АГОНИСТЫ TRPV1, СОДЕРЖАЩИЕ ИХ ПРЕПАРАТЫ И ИХ ПРИМЕНЕНИЕ
(57) Реферат:
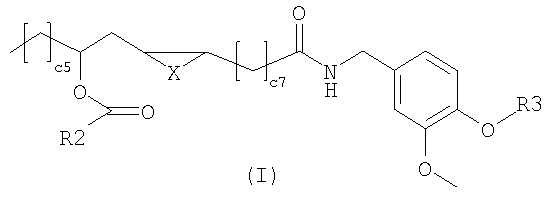
Настоящее изобретение относится к новым соединениям общей формулы (I),
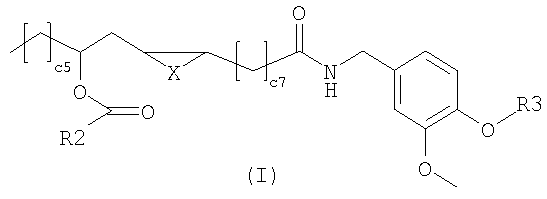
композиции, а также к применению данных соединений для получения лекарственных средств для лечения патологий, опосредованных рецепторами типа I ваниллоида (TRPV1). В общей формуле (I) X представляет собой два атома водорода,
Настоящее изобретение относится к производным рицинолевой кислоты с агонистической активностью к рецептору типа-1 ваниллоида. УРОВЕНЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ Рецептор типа 1 (VR1 или TRPV1) ваниллоида принадлежит к широкому семейству транзиторных рецепторов потенциальных катионных каналов (TRPV) с шестью транс-мембранными доменами. TRPV1 представляет собой единственный канал TRPV, который, как известно в настоящее время, активируется некоторыми природными продуктами, причем наиболее известными и изученными являются капсаицин и резинифератоксин (Sterner and Szallasi, Trends Pharmacol. Sci. 1999, 20, 459-465). В настоящее время обнаружено, что в то время как другие каналы TRPV, и такие как TRPV2, TRPV3 и TRPV4 (также известные как «VR1-подобные (VRL) рецепторы»), являются исключительно восприимчивыми к механическому, осмотическому или термическому стимулу и по существу равномерно распределяются в различных тканях млекопитающих, TRPV1 специфически действует как молекулярный интегратор стимуляции боли, индуцированной, например, нагреванием, протонами и растительными токсинами и, главным образом, экспрессируется в периферических сенсорных волокнах типа С и А Изучение дефицита VR1 на трансгенной мыши однозначно доказало роль TRPV1 в частичной перцепции и трансмиссии «термической» или «воспалительной» боли (Caterina et al., Science 2000, 288, 306-313; Davis et al., Nature 2000, 405, 183-187). Другие исследования позволяют предполагать, что TRPV1 также участвует в кишечных воспалительных нарушениях (Yiangou et al., Lancet 2001, 357, 1338-1339), невропатической боли (Walker et.al., J. Pharmacol. Exp. Ther. 2003. 304, 56-62), недержании стула (фекалий) и патологическом кашле (Chung and Chang, Pulm. Pharmacol. Ther. 2002, 15, 335-338). TRPV1 очевидно также играет фундаментальную роль в регулировании функции мочевого пузыря (Birder et al., Nat.Neurosci. 2002, 5, 856-860) и в регулировании нейрональной пластичности, температуры тела, всасывания пищи, расходования энергии и двигательной активности (Di Marzo et al., Eur. J. Pharmacol. 2001, 420, 123-131). Нейроны, экспрессирующие рецепторы TRPV1, могут быть десенсибилизированы немедленно после активации некоторыми агонистами, например капсаицином. На практике начальное ощущение жжения вследствие воздействия агониста преодолевается парадоксальным действием. Тахифилаксию VR1 можно также объяснить другими медицинскими воздействиями, описанными для капсаицина и чилийского перца такими, как хорошо известное противорвотное и противовоспалительное действия и нейрозащитное действие против эксцитотоксичности глутамата. Капсаицин и его аналог резиниферотоксин также применяют при лечении недержания мочи (при котором нервные окончания характеризуются присутствием VR, участвующем в передаче рефлекса опорожнения мочевого пузыря), в то время как синтетические производные капсаицина, причем наиболее известным является олванил, запатентованы как пероральные обезболивающие средства. Однако фармацевтическая промышленность по-прежнему заинтересована в разработке более сильнодействующих агонистов TRPV1. ОПИСАНИЕ ИЗОБРЕТЕНИЯ Соединения изобретения имеют следующую общую формулу (I)
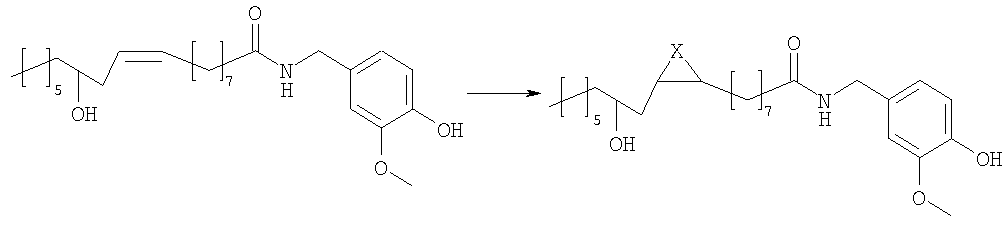
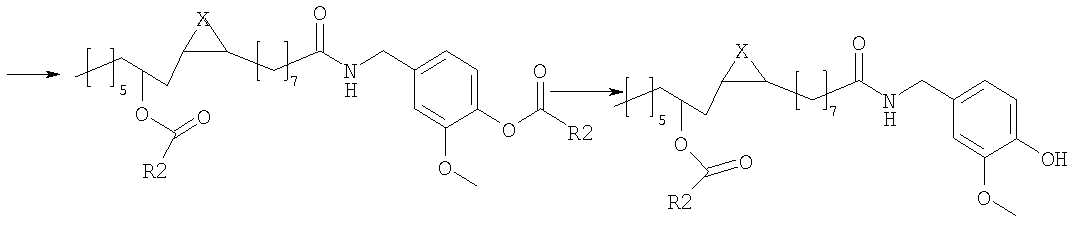
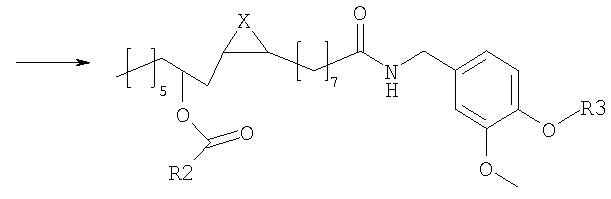
в которой Х представляет собой два атома водорода, R2 представляет собой С6-С12-арильный или арилалкильный остаток; R3 представляет собой водород, 2-гидроксиэтил или 2-аминоэтил. R2 представляет собой, предпочтительно, фенил, бензил или фенетил и R3 представляет собой, предпочтительно, водород. Эти соединения являются сильнодействующими агонистами TRPV1 и, следовательно, являются полезными для лечения боли или недержания мочи, или кишечных воспалительных нарушений. Настоящее изобретение также относится к фармацевтическим композициям, содержащим соединения формулы (I) в качестве активных ингредиентов, в эффективном количестве. Соединения изобретения могут быть в рацемической или энантиомерно чистой форме, более предпочтительно, в форме 12R. Конфигурация двойной связи может быть E и Z, более предпочтительно, Z. Настоящее изобретение также включает в себя фармакологически приемлемые соли соединений формулы (I). Соединения изобретения можно синтезировать, например, посредством способов, указываемых далее в тексте; можно выбрать другие реагенты и исходные вещества для того, чтобы получить другие соединения формулы (I). Согласно наиболее простому пути синтеза, соединения изобретения получают из ванилламида рицинолевой кислоты, согласно схеме 1. Схема 1
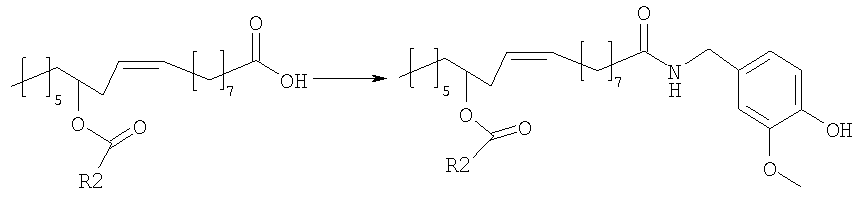
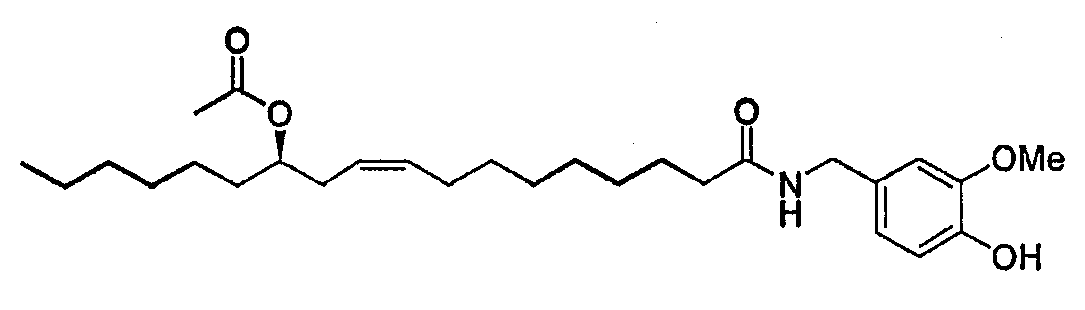
Ванилламид рицинолевой кислоты, полученный согласно методике, описанной в литературе, необязательно подвергают циклопропанированию или эпоксидированию и образующийся полупродукт синтеза этерифицируют подходящим ацилирующим средством. Активированные производные карбоновых кислот, подходящие для этерификации, представляют собой галогенангидриды кислот, в особенности хлорангидриды, а также смешанные ангидриды или аддукты с карбодиимидом согласно методам, известным в данной области. Затем сложный эфир пара-гидроксила остатка ванилламина селективно гидролизуют с образованием соответствующего гидроксипроизводного, которое представляет собой целевое соединение настоящего изобретения. Если требуется, фенольный гидроксил можно затем этерифицировать аминоэтильным или гидроксиэтильным остатком. В альтернативном варианте, полупродукты, применяемые для синтеза соединений настоящего изобретения, можно также получать исходя из рицинолеата трихлорэтанола согласно схеме 2. Схема 2
Рицинолеат трихлорэтанола, полученный согласно методике, описанной в литературе, ацилируют в положении 12 активированной карбоновой кислотой, аналогично описаному выше. Затем трихлорэтиловый эфир селективно гидролизуют с образованием 12-ацилрицинолевой кислоты, которую затем превращают в амид конденсацией с ванилламином. Некоторые способы можно успешно применять к транс и 12-S изомерам, или к насыщенным аналогам природной рицинолевой кислоты. Биологическую активность продуктов изобретения измеряли как in vitro, так и in vivo. In vivo действие соединений формулы (I) на TRPV1 изучали измерением концентрации внутриклеточного кальция в клетках НЕК-293 (которые сверхэкспрессируют TRPV1 человека); концентрацию кальция в присутствии 4 мкМ иономицина использовали как максимальную ссылочную (стандартную) величину (Hayes et al., Pain 2000, 88, 205-215). Результаты представлены в следующей таблице 1.
Активность соединения примера 2 также оценивали на крысах при помощи теста in vivo индуцированного недержания мочи. Крыс Sprague Dawley (250 ± 10 г) анестезировали хлоральгидратом. Мочевой пузырь вскрывали путем надрезания вдоль средней линии живота и удаляли жировую ткань, окружающую мочеиспускательный канал и мочеточники. Затем проксимальный мочеиспускательный канал перевязывали рассасывающейся хирургической нитью для создания частичной обструкции мочеиспускательного канала. Через 8 недель наблюдали изменения в функционировании мочевого пузыря и изменение цистометрической картины, причем, в частности, увеличилось число позывов к мочеиспусканию в час. На день лечения, соединение примера 2, соединение, полученное согласно Appendino et al., и резиниферотоксин в качестве ссылочного эталона солюбилизировали в этаноле и вводили каплями в мочевой пузырь при концентрации 50 нМ. Инстилляцию продолжали 30 мин: в течение этого периода насос для инфузии солевого раствора был выключен. После инкубации мочевой пузырь опорожняли каким-либо лекарственным раствором, затем насос для инфузии снова включали. Значительное увеличение количества позывов к мочеиспусканию в час наблюдали у неподвергаемых лечению оперированных животных. Напротив, у животных, подвергаемых лечению соединением изобретения и резиниферотоксином, количество позывов к мочеиспусканию оставалось аналогичным количеству позывов к мочеиспусканию у неоперированных животных. Очень низкую активность также наблюдали для продукта Appendino et al. Результаты эксперимента представлены в следующей таблице 2.
В качестве сильнодействующих агонистов рецептора TRPV1 соединения (I) можно применять при лечении недержания мочи, для ослабления невропатической боли и при терапии воспалительных кишечных нарушений. Изобретение также относится к фармацевтическим композициям, включающим соединения (I) в комбинации с подходящими носителями или разбавителями. Фармацевтические композиции изобретения можно вводить посредством различных путей введения, например, через пероральный, ректальный, внутривенный, внутримышечный, подкожный, внутритрахеальный, эпидуральный или интрацеребровентрикулярный путь. Подходящие носители для инъецируемых препаратов включают в себя масла, пропилен- или этиленгликоль, физиологический раствор, этанол, растительные масла и изопропилмиристат, или другие растворители, обычно используемые для получения растворов для инъекций. Для получения инъецируемых препаратов соединения изобретения можно растворять, суспендировать или эмульгировать в водных растворителях, таких как физиологический раствор, 5% декстроза, или в неводных растворителях, таких как растительное масло, насыщенные синтетические глицериды, эфиры алифатических кислот с длинной цепью или пропиленгликоль. Препараты могут также включать в себя общепринятые эксципиенты, такие как солюбилизирующие средства, изотонические средства, суспендирующие средства, эмульгаторы, стабилизирующие средства и консерванты. Для местного применения соединения по изобретению могут быть изготовлены как кремы или мази. Фармацевтическую композицию изобретения можно применять: – для ослабления боли, вызванной постгерпетической невралгией, диабетической невропатией, синдромом после мастэктомии, симпатической рефлекторной дистрофией, невралгией тройничного нерва, невропатической болью рта, остеоартритом, ревматоидным артритом, фибромиалгией, синдромом Гийена-Барре; – для ослабления неподдающейся лечению боли, вызванной билатеральной периферической невропатией; – для ослабления зуда, вызванного псориазом, гемодиализом, аквагенного зуда, вестибулита женских наружных половых органов, парестезической боли в спине, брахиорадиального зуда; – для лечения мигрени, вазомоторного ринита или аллергического ринита (в виде капель в нос); – для лечения повышенной чувствительности мочевого пузыря или гиперрефлексии спинальной мышцы-сжимателя (в виде раствора внутрь мочевого пузыря). Соединения настоящего изобретения обладают сильнодействующим обезболивающим действием и потенциальной противовоспалительной активностью, и содержащие их фармацевтические препараты можно применять для ослабления или лечения острой или хронической воспалительной боли, воспаления или недержания мочи. Соединения изобретения можно применять в форме фармакологически приемлемых солей, как таковых или в подходящей комбинации, необязательно в смеси с другими активными ингредиентами. Доза соединений изобретения изменяется в зависимости от состояния и массы тела пациента, тяжести заболевания, фармацевтической формы, пути и длительности введения и может устанавливаться специалистом-клиницистом. По существу, доза будет составлять в интервале от 0,1 мкг до 100 мг/кг, предпочтительно, от 1 мкг до 100 мг/кг/лекарственная форма. Препарат можно вводить в виде одной или многократной (повторяемой) дозы. Процентное содержание соединений в композициях может быть в интервале от 0,0001 до 10 мас.%, предпочтительно, от 0,0001 до 1% на массу композиции. Следующие примеры более подробно иллюстрируют изобретение. ПРИМЕР I – 12,4′-Дифенилацетилринванил К раствору 1,56 г ринванила (3,6 ммоль) в толуоле (20 мл) добавляли 2 мол. экв. фенилуксусной кислоты (1,0 г, 7,2 ммоль), 2 мол. экв. дициклогексилкарбодиимида (1,45 г, 7,2 ммоль) и 1 мол. экв. DMAP (440 мг, 3,6 ммоль). Реакционную смесь оставляли для перемешивания при комнатной температуре и контролировали методом ТСХ (смесь 6:4 петролейный эфир/этилацетат, Rfp=0,31; Rfa=0,60). Через 3 часа смесь отфильтровывали и растворитель выпаривали. Образовавшийся неочищенный продукт можно либо использовать как таковой для последующей стадии, либо очищать колоночной хроматографией. 1Н-ЯМР (300 МГц, CDCl3): 13C-ЯМР (75 МГц, CDCl3): 175,5 (с), 173,9 (с), 149,3 (с), 147,6 (с), 134,4 (с), 132,1 (д), 130,4 (с), 128,3 (д), 128,6 (д), 127,1 (д), 124,2 (д), 120,8 (д), 114,4 (д), 110,8 (д), 74,8 (д), 56,0 (кв), 43,5 (т), 41,8 (т), 36,9 (т), 33,5 (т), 31,8 (т), 29,5 (т), 29,3 (т), 28,18 (т), 27,4 (т), 25,8 (т), 25,2 (т), 22,6 (т), 14,2 (кв). CI-MS: 670(M+H)+. ПРИМЕР II – 12-Фенилацетилринванил Неочищенный 12,4′-дифенилацетилринванил (3,6 ммоль, вычисленное количество) примера I растворяли в дихлорметане (20 мл) и обрабатывали 5 мол. экв. пирролидина (1,52 мл, 1,30 г, 18,0 ммоль). Реакционную смесь перемешивали магнитной мешалкой при комнатной температуре и контролировали методом ТСХ (смесь 6:4 петролейный эфир/этилацетат, Rfp=0,8; Rfa=0,5). Через 3 часа реакционную смесь обрабатывали промыванием 2 н. H2SO4 и насыщенным раствором соли. Органическую фазу сушили (Na2SO4), фильтровали и выпаривали, а остаток очищали колоночной хроматографией (37 г силикагеля, заполненного смесью 7:3 петролейный эфир/этилацетат и элюированного смесью 6:4, 4:6; собирали фракции приблизительно 20 мл). Получали 1,6 г (80%) фенилацетилринванила. Соединение представляет собой масло при комнатной температуре, но при замораживании оно затвердевает, образуя белый порошок. 1Н-ЯМР (300 МГц, CDCl3): 13C-ЯМР (75 МГц, CDCl3): 177,0 (с), 173,5 (с), 171,7 (с), 156,6 (с), 147,2 (с), 145,4 (с), 141,6 (д), 136,8 (с), 132,7 (С), 130,2 (д), 129,5 (д), 129,3 (д), 128,5 (д), 128,3 (д), 127,5 (д), 127,0 (д), 126,2 (д), 124,1 (д), 120,6 (д), 114,9 (д), 110,9 (д), 106,1 (д), 74,6 (д), 56,3 (кв), 43,8 (т), 43,4 (т), 41,9 (т), 36,6 (т),4, 31,8 (т), 29,1 (т), 27,2 (т), 25,9 (т), 25,3 (т), 22,6 (т), 13,9 (кв). СI-MS: 552 (M+H)+. ПРИМЕР III – 2′,2′,2′-Трихлорэтилрицинолеат 3 г Рицинолевой кислоты (м.м.=298,47: 10,07 ммоль) растворяли в 30 мл толуола, добавляли 2 мол. экв. трихлорэтанола (М.м.=149,40; 20,14 ммоль; 3,0 г; d=1,55; 1,9 мл), 1 мол. экв. дициклогексилкарбодиимида (м.м.=202; 10,07 ммоль; 2,0 г) и 1 мол. экв. DMAP (м.м.=122; 10,07 ммоль; 1,23 г). Образовавшуюся смесь перемешивали магнитной мешалкой при комнатной температуре и реакцию контролировали методом ТСХ (смесь 8:2 гексан/этилацетат, Rfp=0,14; Rfa=0,53). Через 18 ч смесь отфильтровывали и растворитель выпаривали. Неочищенный продукт очищали фильтрованием через силикагель, используя в качестве элюента смесь 9:1 петролейный эфир/этилацетат. Получали 4,3 г продукта (количественный выход). 1Н-ЯМР (300 МГц): ПРИМЕР IV – 2′,2′,2′-Трихлорэтил-12-фенилацетилрицинолеат 4,3 г 2′,2′,2′-Трихлорэтилрицинолеата (10,7 ммоль) растворяли в 30 мл толуола и добавляли 2,5 мол. экв. фенилуксусной кислоты (3,4 г, 25,2 ммоль), 2,5 мол. экв. дициклогексилкарбодиимида (5,0 г, 25,2 ммоль) и 1,5 мол. экв. DMAP (1,8 г, 15,0 ммоль). Смесь перемешивали при комнатной температуре, реакцию контролировали методом ТСХ (оксид алюминия, смесь петролейный эфир/этилацетат 8:2, Rfp=0,50; Rfa=0,76). Через 30 мин дициклогексилкарбамид отделяли фильтрованием, а растворитель выпаривали с получением неочищенного продукта, который затем очищали колоночной хроматографией (35 г геля оксида алюминия, смесь петролейный эфир/этилацетат 95:5, фракции: приблизительно 20 мл). Получали 4,6 г продукта (84%). 1Н-ЯМР (300 МГц, CDCl3): ПРИМЕР V – 12-Фенилацетилрицинолевая кислота 4,6 г (8,4 ммоль) 2′,2′,2′-Трихлорэтил-12-фенилацетилрицинолеата растворяли в 40 мл раствора уксусная кислота/МеОН 1:1, затем добавляли 4,6 г активированного порошка цинка при энергичном перемешивании, и реакцию контролировали методом ТСХ (смесь петролейный эфир/этилацетат 8:2; Rfp=0,69; Rfa=0,36). Через 18 ч смесь профильтровали через целит, промывая этилацетатом. Фильтрат концентрировали, промывали водой и насыщенным раствором бикарбоната натрия, затем сушили над сульфатом натрия, фильтровали и выпаривали. Неочищенный продукт очищали колоночной хроматографией (60 г силикагеля, смесь петролейный эфир/этилацетат 95:5, фракции: приблизительно 20 мл). Получали 1,85 мг продукта (53%). 1Н-ЯМР (300 МГц): ПРИМЕР VI – 12-Фенилацетилринванил 1,85 г 12-Фенилацетилрицинолевой кислоты растворяли (4,4 ммоль) в 15 мл сухого дихлорметана и добавляли 2 мол. экв. гидрохлорида ванилламина (835 мг, 4,4 ммоль), 4 мол. экв. ТЕА (2,45 мл, 1,78 г, 17,6 ммоль) и 1,2 мол. экв. полифосфорной кислоты (50% раствор EtOH, 3,4 мл, 1,68 г, 5,28 ммоль). Реакционную смесь оставляли для перемешивания при комнатной температуре и контролировали методом ТСХ (смесь растворителей 6:4; Rfp=0,67; Rfa=0,37). Через 3 ч растворитель выпаривали и неочищенный продукт очищали колоночной хроматографией (50 г силикагеля, элюировали смесью 7:3 петролейный эфир/этилацетат, фракции: приблизительно 20 мл). Продукт дополнительно очищали фильтрованием через оксид алюминия (промывали смесью 6:4-4:6 петролейный эфир/этилацетат). Получали 512 мг фенилацетилринванила (23%). ПРИМЕР VII – 2′,2′,2′-Трихлорэтил-12-бензоилрицинолеат 200 мг 2′,2′,2′-Трихлорэтилрицинолеата (м.м.=429,85; 0,46 ммоль) растворяли в 2 мл толуола и добавляли 1 мол. экв. бензойной кислоты (м.м.=122,12; 0,46 ммоль, 56 мг), 1 мол. экв. дициклогексилкарбодиимида (м.м.=206,33; 0,46 ммоль, 95 мг) и 1 мол. экв. DMAP (м.м.=122,17; 0,46 ммоль, 56 мг). Смесь перемешивали при комнатной температуре и реакцию контролировали методом ТСХ (смесь 95:5 гексан/этилацетат, Rfp=0,05; Rfa=0,32). Через 30 мин добавляли еще один эквивалент бензойной кислоты, дициклогексилкарбодиимида и DMAP. Даже если реакция не заканчивалась, реакционную смесь обрабатывали спустя 18 ч дополнительного перемешивания: дициклогексилкарбамид отделяли фильтрованием и фильтрат выпаривали. Неочищенный продукт очищали колоночной хроматографией (5 г силикагеля, смесь 95:5 гексан/этилацетат, собирали фракции приблизительно 5 мл). Получали 195 мг продукта (79%). 1Н-ЯМР (300 МГц): 13C-ЯМР (75 МГц, CDCl3): 177,0 (с), 173,5 (с), 171,7 (с), 156,6 (с), 147,2 (с), 145,4 (с), 141,6 (д), 136,8 (с), 132,7 (С), 130,2 (д), 129,5 (д), 129,3 (д), 128,5 (д), 128,3 (д), 127,5 (д), 127,0 (д), 126,2 (д), 124,1 (д), 120,6 (д), 114,9 (д), 110,9 (д), 106,1 (д), 74,6 (д), 56,3 (кв), 43,8 (т), 43,4 (т), 41,9 (т), 36,6 (т),4, 31,8 (т), 29,1 (т), 27,2 (т), 25,9 (т), 25,3 (т), 22,6 (т), 13,9 (кв). CI-MS: 552 (M+H)+. ПРИМЕР VIII – 12-Бензоилрицинолевая кислота 185 мг 2′,2′,2′-Трихлорэтил-12-бензоилрицинолеата (м.м.=533,95; 0,35 ммоль) растворяли в 2 мл раствора 1:1 уксусная кислота/МеОН, затем добавляли 200 мг активированного порошка цинка при энергичном перемешивании и реакцию контролировали методом ТСХ (смесь 8:2 гексан/этилацетат; Rfp=0,42; Rfa=0,17). Через 3 ч смесь фильтровали через целит, промывая этилацетатом. Органическую фазу концентрировали, промывали водой и раствором бикарбоната натрия, затем сушили над сульфатом натрия, фильтровали и выпаривали. Остаток очищали колоночной хроматографией (2,5 г силикагеля, смесь 95:5 петролейный эфир/этилацетат, собирали фракции приблизительно 5 мл). Получали 68 мг продукта (48%). 1Н-ЯМР (300 МГц, CDCl3): ПРИМЕР IX – 12-Бензоилринванил 60 мг 12-Бензоилрицинолевой кислоты (м.м.=402,57; 0,15 ммоль) растворяли в 2 мл сухого дихлорметана и добавляли 2 мол. экв. гидрохлорида ванилламина (м.м.=189,64; 0,30 ммоль; 56,89 мг), 8 мол. экв. ТЕА (М.м.=101; 1,2 ммоль; 121 мг; d=0,726; 167 мкл) и 3 мол. экв. полифосфорной кислоты (м.м.=318,19; 0,45 ммоль; 34,2 мг; раствор 50% EtOH, 68 мкл). Смесь оставляли для перемешивания при комнатной температуре, реакцию контролировали методом ТСХ (смесь 8:2 петролейный эфир/этилацетат; Rfp=0,41; Rfa=0). Через 2 ч растворитель реакционной смеси выпаривали и неочищенный продукт очищали колоночной хроматографией (2,5 г силикагеля, смесь 8:2 петролейный эфир/этилацетат, фракции: приблизительно 5 мл). Получали 31 мг бензоилринванила (38%). 1Н-ЯМР (300 МГц, CDCl3): ПРИМЕР Х – 9,10-Метилринванил В двухгорлой круглодонной колбе в атмосфере азота растворяли 300 мг ринванила (м.м.=433,62; 0,69 ммоль) в 29 мл безводного толуола (29 мл) и обрабатывали 15 мол. экв. диэтилцинка (1,0 М в гексане; 10,35 ммоль; 10,35 мл) и 15 мол. экв. бис-иодметана (м.м.=268,84; 10,35 ммоль; 2,78 г; d=3,325 г/мл; 837 мкл). Раствор перемешивали при 65°С и раствор становился розовым, когда начинало осаждаться белое твердое вещество. Реакцию контролировали методом ТСХ на силикагеле с серебром (смесь 6:4 петролейный эфир/этилацетат; Rfp=0; Rfa=0,1). Через 7 ч смесь охлаждали до 0°С, добавляли 2 н. H2SO4 и экстрагировали этилацетатом, затем органическую фазу промывали NaHCO3 и насыщенным раствором соли. Затем сушили (Na2SO4) и выпаривали, неочищенный продукт очищали колоночной хроматографией (15 мл оксида кремния, наполненного смесью петролейный эфир/этилацетат 7:3 и элюированного смесью тех же растворителей 6:4; фракции: приблизительно 8 мл). Получали 119 мг продукта (40%). 1Н-ЯМР (300 МГц, CDCl3): CI-MS: 448 (M+H)+. ПРИМЕР XI – 9,10-Метилен-12,4′-дифенилацетилринванил 100 мг 9,10-Метилринванила (м.м.=447,65; 0,22 ммоль) растворяли в 2 мл толуола и добавляли 2 мол. экв. фенилуксусной кислоты (м.м.=136; 0,44 ммоль; 60 мг), 2 мол. экв. дициклогексилкарбодиимида (м.м.=202; 0,44 ммоль; 89 мг) и 1 мол. экв. DMAP (м.м.=122; 0,22 ммоль; 27 мг). Смесь оставляли для перемешивания при комнатной температуре и реакцию контролировали методом ТСХ (смесь 6:4 петролейный эфир/этилацетат, Rfp=0,23; Rfa=0,42). Через 3 ч дициклогексилкарбамид удаляли фильтрованием и растворитель выпаривали. Образовавшийся неочищенный продукт можно использовать как таковой для последующей реакции или очищать хроматографией (5 г силикагеля, в качестве элюента смесь 8:2 петролейный эфир/этилацетат). 1Н-ЯМР (300 МГц, CDCl3): 13C-ЯМР (75 МГц, CDCl3): 175,5 (с), 173,9 (с), 149,7 (с), 147,1 (с), 134,5 (с), 132,2 (д), 130,4 (с), 128,2 (д), 128,1 (д), 127,5 (д), 124,2 (д), 120,8 (д), 114,6 (д), 110,3 (д), 75,6 (д), 56,0 (кв), 43,6 (т), 41,9 (т), 33,2 (т),4, 31,8 (т), 30,1 (т), 29,5 (т), 39,4 (т), 25,9 (т), 25,1 (т), 22,6 (т), 14,2 (кв), 11,0 (т). CI-MS: 684 (M+H)+. ПРИМЕР XII – 9,10-Метилен-12-фенилацетилринванил I Неочищенный 9,10-метилен-12,4′-дифенилацетилринванил из примера XI (0,22 ммоль, теоретическое количество) растворяли в дихлорметане (20 мл) и добавляли 5 мол. экв. пирролидина (м.м.=71,12; 1,1 ммоль; 78 мг; d=0,86 г/мл; 90 мкл). Через 3 ч реакцию завершали (ТСХ на оксиде алюминия, элюент: смесь 6:4 петролейный эфир/этилацетат; Rfp=0,8; Rfa=0,5; Rfb=0,45; ТСХ на оксиде алюминия с таким же элюентом; Rfp=0,42; Rfa=0,39; присутствие только одного пятна является доказательством). Органическую фазу промывали 2 н. H2SO4 и насыщенным раствором соли, и сушили над сульфатом натрия. После выпаривания растворителя, неочищенный продукт очищали колоночной хроматографией (10 мл оксида кремния, смесь растворителей от 7:3 до 4:6 петролейный эфир/этилацетат). Получали 75 мг продукта (60%). 1Н-ЯМР (300 МГц): ПРИМЕР XIII – Анализ связывания TPRV1 Аффинность в отношении к TRPV1 человека измеряли замещением [3H]RTX (48 Ci/ммоль, NEN-Dupont) из мембран НЕК-клеток (50 мкг/пробирка) согласно способу, описанному Ross (Ross et al., Br. J. Pharmacol. 2001, 132, 631-640). В этих условиях Kd и Bmax для [3H]RTX составляло 0,5 нМ и 1,39 пикомоль/мг белка. Величину Ki замещения 1 нМ [3H]RTX вычисляли из величины IC50 (полученной с применением программного обеспечения GraphPad Software) и применением уравнения Cheng-Prusoff. Величину специфического связывания вычисляли с использованием 1 мкМ RTX (Alexis Biochemicals), она составила 48,1+5,6%. Величины для соединений изобретения приведены в таблице 1. ПРИМЕР XIV – 4′-(2-Аминоэтил)-12-фенилацетилринванил (гидрохлорид) В двухгорлой круглодонной колбе в атмосфере азота к суспензии NaH (60%, 57 мг, 1,4 ммоль, 2 молярных эквивалента) в ТГФ (10 мл) добавляли раствор 382 мг фенилацетилринванила (м.м.=552; 0,69 ммоль), растворенного в ТГФ (5 мл). После перемешивания при комнатной температуре в течение 10 мин, добавляли избыток 1,2-дибромэтана (0,7 мл). Раствор перемешивали при комнатной температуре в течение 16 ч, затем разбавляли насыщенным раствором NH4Cl и экстрагировали эфиром. Выпаривание растворителя позволяло получить масло, которое фильтровали через тонкий слой оксида кремния (15 г), используя петролейный эфир (100 мл) для удаления избытка 1,2-дибромэтана, затем смесь 1:1 петролейный эфир/этилацетат (100 мл) для элюирования продукта. После выпаривания растворителя получали клейкий осадок, который непосредственно растворяли в диметилформамиде (10 мл) и обрабатывали избытком азида натрия (NaN3) (300 мг). После перемешивания при комнатной температуре в течение 16 ч реакционную смесь разбавляли водой (приблизительно 50 мл) и экстрагировали смесью 3:1 петролейный эфир/эфир (2 х 30 мл). После промывания насыщенным раствором NaCl и сушки (Na2SO4) органическую фазу выпаривали и остаток растворяли в ТГФ (20 мл), затем добавляли трифенилфосфин (917 мг, 3,5 ммоль) и воду (0,62 мл, 3,5 ммоль). Вслед за перемешиванием в течение 5 ч при комнатной температуре, реакционную смесь разбавляли водой и экстрагировали этилацетатом. После сушки (Na2SO4) и выпаривания остаток очищали колоночной хроматографией на силикагеле (10 г) с применением этилацетата в качестве элюента. Получали 130 мг продукта (общий выход: 40%). 1Н-ЯМР (300 МГц, CDCl3): 13C-ЯМР (75 МГц, CDCl3) 177,1 (с), 173,7 (с), 171,1 (с), 158,9 (с), 147,3 (с), 145,1 (с), 143,6 (д), 138,3 (с), 132,7 (с), 130,8 (д), 129,0 (д), 129,8 (д), 128,5 (д), 128,8 (д), 128,0 (д), 127,2 (д), 126,5 (д), 123,9 (д), 121,1 (д), 115,0 (д), 112,2 (д), 109,2 (д), 74,9 (д), 71,4 (т), 56,1 (кв), 47,9 (т), 43,9 (т), 43,1 (т), 42,1 (т),36,6 (т),4, 31,0 (т), 29,2 (т), 27,2 (т), 26,1 (т), 25,2 (т), 22,6 (т), 13,2 (кв). CI-MS: 596 (М+Н)+. Хлористоводородную соль получали растворением продукта в минимальном количестве ТГФ и добавлением 1 эквивалента 1,0 М раствора соляной кислоты в диэтиловом эфире при 0°С. После выпаривания растворителя осадок собирали и сушили в вакууме. ПРИМЕР ХV – 9,10-Эпокси-12-фенилацетилринванил Раствор 12,4′-дифенилацетилринванила, полученного как в примере 1 (300 мг, 0,45 ммоль), в безводном СН2Cl2 (6 мл) смешивали с 2,5 молярными эквивалентами м-хлорпербензойной кислоты (МСРВА, 242 мг 80% кислоты, 1,12 ммоль), раствор перемешивали магнитной мешалкой в течение 3 ч, затем промывали Na2S2O3, сушили (Na2SO4), фильтровали и выпаривали. Остаток непосредственно растворяли в СН2Cl2 (5 мл) и добавляли пирролидин (155 мг, 0,180 мл, 5 молярных эквив.). После перемешивания при комнатной температуре в течение 16 ч, смесь промывали 2 н. H2SO4 и насыщенным раствором NaCl, затем сушили (Na2SO4). После выпаривания растворителя остаток очищали колоночной хроматографией на нейтральном оксиде алюминия (3 г, в качестве элюента использовали смесь 6:4 петролейный эфир/этилацетат) для получения 120 мг продукта (41%). 1Н-ЯМР (300 МГц, CDCl3): 13C-ЯМР (75 МГц, CDCl3): 173,0 (с), 171,4 (с), 146,8 (с), 145,2 (с), 134,2 (с), 130,4 (с), 129,3 (д), 128,6 (д), 128,3 (д), 127,3 (д), 127,2 (д), 120,9 (д), 114,5 (д), 110,8 (д), 73,0 (д), 57,0, 56,4 (д), 56,0 (т), 53,6, 53,0 (д), 43,6 (т), 41,8 (т), 36,8 (т),4, 31,7 (т), 29,4 (т), 29,2 (т), 27,5 (т), 25,8 (т), 25,3 (т), 22,6 (т), 14,2 (кв). CI-MS: 568 (М+Н)+. ПРИМЕР ХVI – Раствор для инъекций хлористоводородной соли 4′-(2-аминоэтил)-12-фенилацетилринванил Раствор для инъекций соли 4′-(2-аминоэтил)-12-фенилацетилринванил·HCl состоял из 4′-(2-аминоэтил)-12-фенилацетилринванил·HCl (1,0 мг), хлористого натрия (9,0 мг), бензилового спирта (15,0 мг) и воды для инъекций (до 1 мл). В стандартной методике хлористый натрий и бензиловый спирт растворяли в воде для инъекций, затем добавляли гидрохлорид 4′-(2-аминоэтил)-12-фенилацетилринванила. ПРИМЕР XVII – Раствор для инъекций 12-фенилацетилринванила Раствор для инъекций 12-фенилацетилринванила состоял из 12-фенилацетилринванила (1,0 мг), пропилгаллата (0,5 мг), оливкового масла для инъецируемых препаратов (до 1 мл). ПРИМЕР XVIII – Эмульсия для местного применения Эмульсии для местного применения, например эмульсии 12-фенилацетилринванила, состояли из 12-фенилацетилринванила (1,0 г), жидкого парафина (25,0 г), стеарилового спирта (12,0 г), цетилового спирта (5,0 г), метил-п-гидроксибензоата (0,028 г), пропил-п-гидроксибензоата (0,012 г), PEG-40-стеарата (1,0 г), глицерина (12,0 г), очищенной воды (до 100 г). В стандартной методике, жидкий парафин, стеариловый спирт и цетиловый спирт расплавляли при 70-75°С при перемешивании, затем в образовавшейся фазе растворяли 12-фенилацетилринванил, выдерживая раствор при 70-75°С. Затем при энергичном перемешивании при 70-75°С добавляли остальные компоненты, предварительно растворенные в очищенной воде при 70-75°С. Образовавшийся продукт медленно охлаждали при перемешивании.
Формула изобретения
1. Соединения общей формулы (I) 2. Соединения по п.1, в которых R3 представляет собой водород. 3. Применение соединений по пп.1 и 2 для получения лекарственных средств для лечения патологий, опосредованных рецепторами типа I ваниллоида (TRPV1), таких как недержание мочи, невропатическая боль и кишечные воспалительные нарушения. 4. Композиция, проявляющая свойства агониста TRVP1, включающая в себя эффективное количество одного или нескольких соединений по любому из пп.1 и 2 в смеси с подходящими носителями.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||
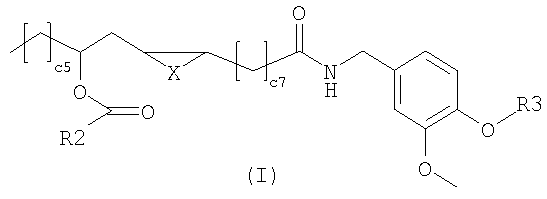
 -связь, атом кислорода или метилен; R2 представляет собой фенил, бензил и фенетил; R3 представляет собой водород, 2-гидроксиэтил или 2-аминоэтил. 3 н. и 1 з.п. ф-лы, 2 табл.
-связь, атом кислорода или метилен; R2 представляет собой фенил, бензил и фенетил; R3 представляет собой водород, 2-гидроксиэтил или 2-аминоэтил. 3 н. и 1 з.п. ф-лы, 2 табл. (Gunthorpe et al., Trends Pharmacol. Sci. 2002, 23, 183-191).
(Gunthorpe et al., Trends Pharmacol. Sci. 2002, 23, 183-191).