Патент на изобретение №2378257
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 1,2,3,4-ТЕТРАГИДРОИЗОХИНОЛИНА
(57) Реферат:
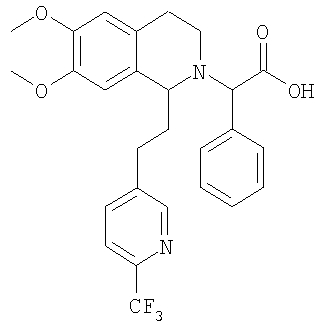
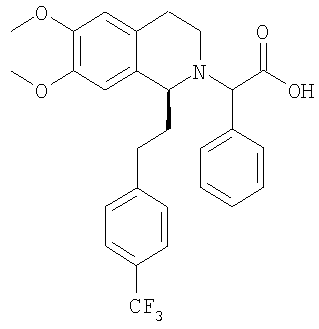
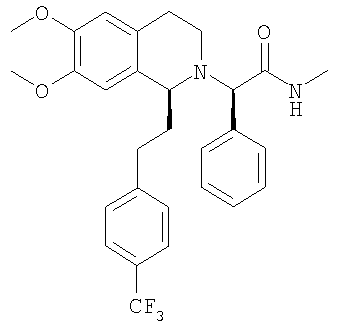
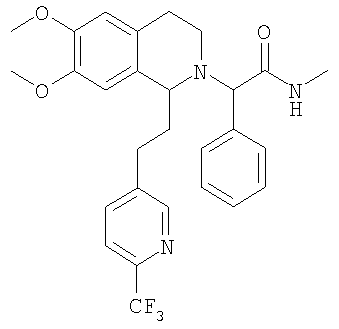
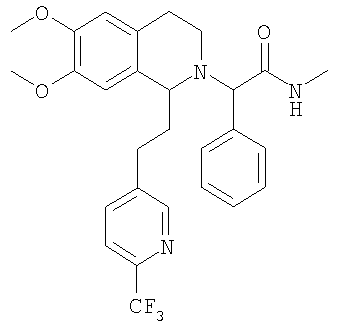
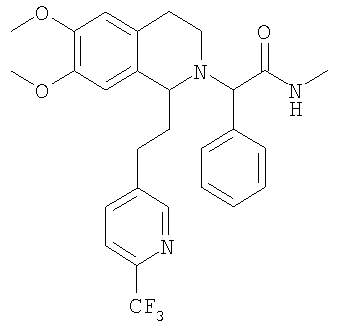
Изобретение относится к области органической химии, а именно к (2R)-2-{(1S)-6,7-диметокси-1-[2-(4-трифторметилфенил)-этил]-3,4-дигидро-1Н-изохинолин-2-ил}-N-метил-2-фенилацетамиду или его фармацевтически приемлемой соли. Изобретение также относится к применению и фармацевтической композиции на основе указанного соединения, обладающей активностью не пептидного антагониста орексиновых рецепторов человека. Технический результат – получение нового соединения и его соли, которое может найти применение в медицине в качестве лекарственного средства для предупреждения или лечения заболеваний, таких как нарушение питания и нарушение сна. 3 н. и 8 з.п. ф-лы, 3 табл.
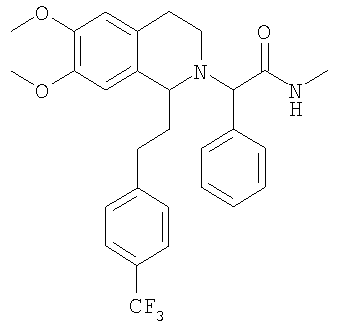
Настоящее изобретение относится к новым замещенным производным 1,2,3,4-тетрагидроизохинолина общей формулы (I) и их применению в качестве лекарственных средств. Настоящее изобретение также включает родственные объекты, в том числе способы получения соединений, фармацевтические композиции, содержащие одно или большее количество соединений общей формулы (I), и предпочтительно их применение в качестве антагонистов орексинового рецептора. Орексины (орексин А, или ОХ-А, и орексин В, или ОХ-В) являются новыми нейропептидами, обнаруженными в 1998 двумя группами исследователей, орексин А является содержащим 33 аминокислоты пептидом и орексин В является содержащим 28 аминокислот пептидом (Sakurai Т. et al., Cell, 1998, 92, 573-585). Орексины продуцируются в отдельных нейронах боковой части гипоталамуса и связываются со связанными с G-белком рецепторами (рецепторами OX1 и ОХ2). Орексиновый рецептор 1 (OX1) селективен по отношению к ОХ-А, а орексиновый рецептор 2 (ОХ2) способен связывать и ОХ-А, и ОХ-В. Обнаружено, что орексины стимулируют потребление корма крысами, что указывает на физиологическую функцию этих пептидов, как медиаторов механизма центральной обратной связи, который регулирует кормовое поведение (Sakurai Т. et al., Cell, 1998, 92, 573-585). С другой стороны, также обнаружено, что орексины регулируют состояние сна и бодрствования и тем самым создают возможность потенциально новых терапевтических способов лечения нарколепсии, а также инсомнии и других нарушений сна (Chemelli R.M. et al., Cell, 1999, 98, 437-451). Орексиновые рецепторы обнаружены в головном мозге млекопитающих, и они могут принимать участие в патологиях, включая депрессию; тревогу; привыкания; обсессивно-компульсивное нарушение; аффективный невроз; депрессивный невроз; невроз тревоги; дистимическое нарушение; нарушение настроения; половую дисфункцию; психополовую дисфункцию; половое нарушение; шизофрению; маниакальную депрессию; белую горячку; слабоумие; тяжелую умственную отсталость и дискинезии, такие как болезнь Гентингтона и болезнь Туретта; нарушения питания; нарушения сна; сердечно-сосудистые заболевания, диабет; нарушения аппетита/чувства вкуса; тошноту/рвоту; астму; болезнь Паркинсона; синдром/болезнь Кушинга; базофильную аденому; пролактиному; гиперпролактинемию; гипопитуитаризм; опухоль/аденому гипофиза; заболевания гипоталамуса; воспалительную болезнь кишечника; желудочную дискинезию; язву желудка; синдром Фрелиха; заболевания гипофиза, гипоталамический гипогонадизм; синдром Кальмана (аносмия, гипосмия); функциональную или психогенную аменорею; гипоталамический гипотиреоз; гипоталамически-адреналиновую дисфункцию; идиопатическую гиперпролактинемию; нарушения гипоталамуса, приводящие к дефициту гормона роста; идиопатический недостаточный рост; карликовость; гигантизм; акромегалию; нарушения биологического и суточного ритма; нарушения сна, связанные с заболеваниями, такими как неврологические нарушения, невропатическая боль и синдром усталых ног; заболевания сердца и легких, острую и застойную сердечную недостаточность; гипотензию; гипертензию; задержку мочи; остеопороз; стенокардию; инфаркт миокарда; ишемический или геморрагический удар; субарахноидальное кровотечение; язвенные заболевания; аллергии; доброкачественную гиперплазию предстательной железы; хроническую почечную недостаточность; заболевание почек; нарушение переносимости глюкозы; мигрень; гиперальгезию; боль; повышенную или чрезмерную чувствительность к боли, такую как гиперальгезия, каузалгия и аллодиния; острую боль; жгучую боль; атипичную лицевую боль; невропатическую боль; боль в пояснице; комплексный регионарный болевой синдром I и II; артритную боль; боль вследствие спортивной травмы; боль, связанную с инфекцией, например HIV, боль после химиотерапии; боль после удара; послеоперационную боль; невралгию; патологические состояния, при которых проявляется висцеральная боль, такие как синдром раздраженной толстой кишки, мигрень и ангина; недержание мочи, например неотложное недержание мочи; переносимость наркотиков и отмену наркотиков; апноэ во сне; нарколепсию; инсомнию; парасомнию; и нейродегенеративные нарушения, включая нозологические проявления, такие как комплекс растормаживание-слабоумие-паркинсонизм-амиотрофия; паллидо-понто-нигральную дегенеративную эпилепсию; припадки и другие заболевания, связанные с общим нарушением функции орексиновой системы. Настоящее изобретение относится к замещенным производным 1,2,3,4-тетрагидроизохинолина, которые являются непептидными антагонистами орексиновых рецепторов человека. Эти соединения перспективны для применения при лечении, например, нарушения питания или нарушения сна. К настоящему времени известны некоторые низкомолекулярные соединения, возможно являющиеся антагонистами специфически по отношению к ОХ1 или ОХ2, или одновременно по отношению к обоим рецепторам. В некоторых заявках на патенты, например фирмы SmithKline Beecham, описаны фенилмочевиновые, фенилтиомочевиновые и циннамидные производные, как селективные антагонисты OX1 (WO 99/09024, WO 00/47576 и WO 00/47580). Недавно в своих заявках на патенты фирма SmithKline Beecham предложила производные 2-аминометилпиперидина (WO 01/96302), 3-производные аминометилморфолина (WO 02/44172) и N-ароил-циклические амины (WO 02/090355, WO 03/002559 м WO 03/002561) в качестве антагонистов орексинового рецептора. В WO 01/85693 фирма Banyu Pharmaceuticals описала в формуле изобретения производные N-ацилтетрагилроизохинолина. Другие антагонисты орексинового рецептора, такие как новые производные бензазепина, раскрыты в WO 02/051838. Фирма Actelion Pharmaceuticals Ltd. описала в формуле изобретения производные 1,2,3,4-тетрагидроизохинолина и их применение в качестве активных ингредиентов для приготовления фармацевтической композиции (WO 01/68609). Кроме того, описано применение химических реакций в жидкой фазе для предварительной оптимизации производных 1,2,3,4-тетрагидроизохинолина, как возможных антагонистов орексинового рецептора (Chimia, 2003, 57, 5, 270-275). Также хорошо известно, что надлежащее регулирование концентраций лекарственных средств в плазме в период лечения является одним из критически важных аспектов лечения. Одним очень важным механизмом этого регулирования является окисление лекарственного вещества ферментами цитохрома Р450 (CYP). Окисление лекарственного вещества ферментами CYP должно соответствовать необходимому терапевтическому показанию и следует избегать сильного ингибирования ферментов CYP. Это обусловлено проблемой лекарственных взаимодействий, т.е. повышением концентрации лекарственного средства в плазме вследствие ингибирования фермента CYP другим лекарственным средством. Основными представителями группы CYP 450, осуществляющими метаболизм лекарственны средств, являются CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1 и CYP3A4, которые составляют примерно 30% всех ферментов CYP. Многие лекарственные вещества преобразуются с помощью CYP3A4 и у многих лекарственных веществ не имеется другого пути метаболизма, кроме включающего этот конкретный цитохром. Поэтому слабое ингибирование CYP3A4 является абсолютно необходимым для того, чтобы стало возможным применение химического вещества в качестве лекарственного средства. Согласно изобретению установлено, что соединения, предлагаемые в настоящем изобретении, обладают низким сродством к CYP3A4. Кроме того, также установлено, что эти соединения являются очень активными при пероральном введении. Поэтому соединения, предлагаемые в настоящем изобретении, применимы для лечения, например, таких заболеваний, как нарушения питания или нарушения сна. Ниже приведены определения различных химических фрагментов соединений, предлагаемых в настоящем изобретении, и они будут использоваться во всем описании и формуле изобретения, если явно не приведенное утверждение не приводит к более широкому определению. Термин “алкил”, по отдельности или в комбинации с другими группами, означает обладающую линейной или разветвленной цепью алкильную группу, содержащую от 1 до 6 атомов углерода, предпочтительно – обладающую линейной или разветвленной цепью алкильную группу, содержащую от 1 до 4 атомов углерода. Примерами обладающих линейной или разветвленной цепью C1-С6алкильных групп являются метил, этил, пропил, изопропил, бутил, вторбутил, изобутил, третбутил, пентил, гексил, изомерные пентилы, изомерные гексилы, предпочтительно – метил, этил, пропил, изопропил, бутил, вторбутил, изобутил или третбутил. Термин “алкоксигруппа”, по отдельности или в комбинации с другими группами, означает группу R-O-, в которой R обозначает алкильную группу, определенную выше, такую как метокси-, этокси-, н-пропокси-, изопропокси-, н-бутокси-, изобутокси-, вторбутокси- и третбутоксигруппу, предпочтительно – метокси- и этоксигруппу. Выражение “фармацевтически приемлемые соли” включает соли с неорганическими кислотами или с органическими кислотами, такими как хлористо-водородная кислота, бромисто-водородная кислота, йодисто-водородная кислота, серная кислота, фосфорная кислота, азотная кислота, лимонная кислота, муравьиная кислота, уксусная кислота, малеиновая кислота, винно-каменная кислота, фумаровая кислота, бензойной кислота, памоевая кислота, стеариновая кислота, метансульфоновая кислота, п-толуолсульфоновая кислота, салициловая кислота, янтарная кислота, трифторуксусная кислота и т.п., которые нетоксичны для живых организмов или в случае соединений формулы (I) являются кислотными по природе, соли с неорганическим основанием, таким как основание щелочного или щелочноземельного металла, например гидроксид натрия, гидроксид калия, гидроксид кальция и т.п. Другие примеры фармацевтически приемлемых солей приведены в публикации “Salt selection for basic drugs”, Int. J. Pharm. (1986), 33, 201-217. Солеобразующими группами являются группы или радикалы, обладающие основными или кислыми свойствами. Соединения, содержащие по меньшей мере одну основную группу или по меньшей мере один основной радикал, например аминогруппу, вторичную аминогруппу, не образующую пептидную связь, или пиридильный радикал, могут образовать молекулярные соли с кислотами, например с неорганическими кислотами. Если имеется несколько основных групп, то могут образовываться молекулярные соли с одной или несколькими кислотами. Соединения, содержащие кислотные группы, такие как карбоксигруппа или фенольная гидроксигруппа, могут образовывать соли с металлами или аммониевые соли, такие как соли со щелочными металлами или щелочноземельными металлами, например натриевые, калиевые, магниевые или кальциевые соли, или аммониевые соли с аммиаком или подходящими органическими аминами, такими как третичные моноамины, например триэтиламин или три(2-гидроксиэтил)-амин, или гетероциклические основания, например N-этилпиперидин или N,N’-диметилпиперазин. Возможные смеси солей. Соединения, содержащие и кислотные, и основные группы, могут образовывать внутренние соли. Для выделения или очистки, а также в случае соединений, которые затем будут использоваться в качестве промежуточных продуктов, можно использовать фармацевтически неприемлемые соли, например пикраты. Однако для терапевтических целей можно использовать только фармацевтически приемлемые нетоксичные соли, и поэтому такие соли являются предпочтительными. Первым объектом настоящего изобретения являются новые замещенные производные 1,2,3,4-тетрагидроизохинолина следующей общей формулы (I):
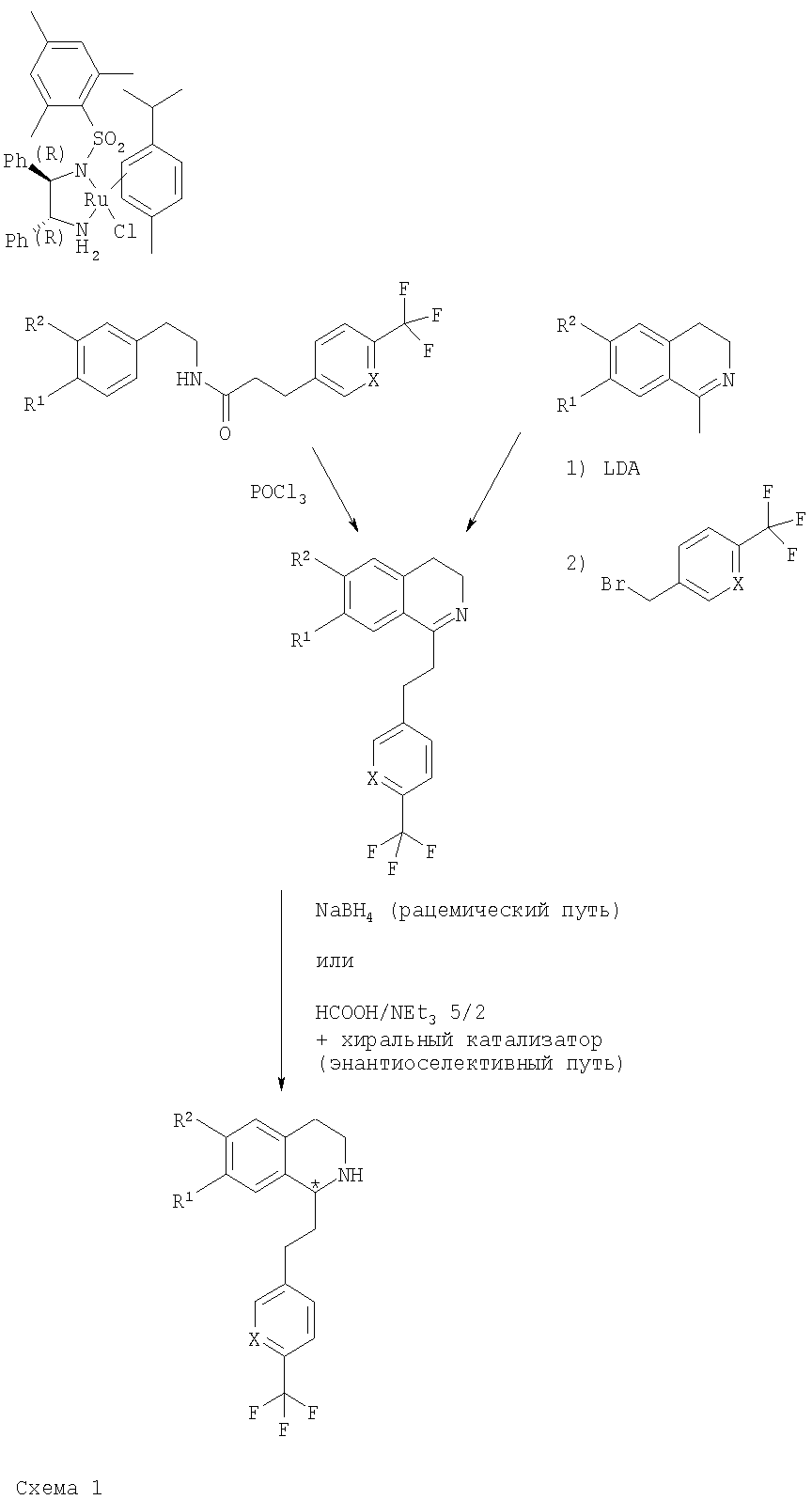
в которой R1 и R2 независимо обозначают водород или С1-С4-алкоксигруппу; R3 обозначает С1-С6-алкил; Х обозначает -СН- или атом азота. Настоящее изобретение также включает соединения формулы I и их оптически чистые энантиомеры, смеси энантиомеров, рацематы, оптически чистые диастереоизомеры, смеси диастереоизомеров, рацематы диастереоизомеров, смесь рацематов диастереоизомеров, мезоформы и фармацевтически приемлемые соли, комплексы с растворителями и морфологические формы. Любое указание на соединение общей формулы (I) следует понимать как указание и на конфигурационные изомеры, смеси энантиомеров, такие как рацематы, диастереоизомеры, смеси диастереоизомеров, рацематы диастереоизомеров и смеси рацематов диастереоизомеров, а также соли, предпочтительно – фармацевтически приемлемые соли, комплексы с растворителями и морфологические формы в соответствии с тем, что является целесообразным и подходящим. Как отмечено выше, настоящее изобретение также включает сольватированные комплексы соединений общей формулы (I). Сольватация может происходить во время приготовления или может происходить отдельно, например вследствие гигроскопичности исходного безводного соединения общей формулы (I). Настоящее изобретение также включает различные морфологические формы, например кристаллические формы соединений общей формулы (I) и их соли и сольватированные комплексы. В частности, гетероморфные формы могут обладать другими характеристиками растворения, характеристиками стабильности и т.п., и все они включены в объем настоящего изобретения. Предпочтительными замещенными производными 1,2,3,4-тетрагидроизохинолина являются такие, в которых R1 и R2 оба обозначают С1-С4-алкоксигруппу, предпочтительно – метоксигруппу. В предпочтительном варианте осуществления настоящего изобретения Х обозначает -СН-. В другом предпочтительном варианте осуществления Х обозначает атом азота. В другом предпочтительном варианте осуществления настоящего изобретения R3 обозначает метильную группу. В особенно предпочтительном варианте осуществления настоящего изобретения R1 и R2 обозначают метоксигруппу, Х обозначает -СН- и R3 обозначает С1-С6-алкил. Примеры предпочтительных соединений выбираются из группы, включающей: 2-{6,7-диметокси-1-[2-(4-трифторметилфенил)-этил]-3,4-дигидро-1H-изохинолин-2-ил}-N-метил-2-фенилацетамид; 2-{6,7-диметокси-1-[2-(6-трифторметилпиридин-3-ил)-этил]-3,4-дигидро-1H-изохинолин-2-ил}-N-метил-2-фенилацетамид. Соединения общей формулы (I) применимы для приготовления лекарственного средства, предназначенного для предупреждения или лечения заболеваний, выбранных из группы, включающей депрессию; тревогу; привыкания; обсессивно-компульсивное нарушение; аффективный невроз; депрессивный невроз; невроз тревоги; дистимическое нарушение; нарушение настроения; половую дисфункцию; психополовую дисфункцию; шизофрению; маниакальную депрессию; белую горячку; слабоумие; тяжелую умственную отсталость и дискинезии, такие как болезнь Гентингтона и болезнь Туретта; диабет; нарушения аппетита/чувства вкуса; тошноту/рвоту; астму; болезнь Паркинсона; синдром/болезнь Кушинга; базофильную аденому; пролактиному; гиперпролактинемию; гипопитуитаризм; опухоль/аденому гипофиза; заболевания гипоталамуса; воспалительную болезнь кишечника; желудочную дискинезию; язву желудка; синдром Фрелиха; заболевания гипофиза, гипоталамический гипогонадизм; синдром Кальмана (аносмия, гипосмия); функциональную или психогенную аменорею; гипоталамический гипотиреоз; гипоталамически-адреналиновую дисфункцию; идиопатическую гиперпролактинемию; нарушения гмпоталамуса, приводящие к дефициту гормона роста; идиопатический недостаточный рост; карликовость; гигантизм; акромегалию; нарушения биологического и суточного ритма; нарушения сна, связанные с заболеваниями, такими как неврологические нарушения, невропатическая боль и синдром усталых ног; заболевания сердца и легких, острую и застойную сердечную недостаточность; гипотензию; гипертензию; задержку мочи; остеопороз; стенокардию; инфаркт миокарда; ишемический или геморрагический удар; субарахноидальное кровотечение; язвенные заболевания; аллергии; доброкачественную гиперплазию предстательной железы; хроническую почечную недостаточность; заболевание почек; нарушение переносимости глюкозы; мигрень; боль; повышенную или чрезмерную чувствительность к боли, такую как гипералгезия, каузалгия и аллодиния; острую боль; жгучую боль; атипичную лицевую боль; невропатическую боль; боль в пояснице; комплексный регионарный болевой синдром I и II; артритную боль; боль вследствие спортивной травмы; боль, связанную с инфекцией, например с ВИЧ; боль после химиотерапии; боль после удара; послеоперационную боль; невралгию; патологические состояния, при которых проявляется висцеральная боль, такие как синдром раздраженной толстой кишки; мигрень и ангина; недержание мочи, например неотложное недержание мочи; переносимость наркотиков и отмену наркотиков; нарушения сна; нарушения питания; сердечно-сосудистые нарушения; нейродегенеративные нарушения; апноэ во сне; нарколепсию; инсомнию; парасомнию; и нейродегенеративные нарушения, включая нозологические проявления, такие как комплекс растормаживание-слабоумие-паркинсонизм-амиотрофия; паллидо-понто-нигральную дегенеративную эпилепсию; припадки и другие заболевания, связанные с общими нарушениями функции орексиновой системы. Соединения общей формулы (I) являются особенно подходящими для применения при лечении заболеваний или нарушений, выбранных из группы, включающей нарушения питания и нарушения сна. Нарушения питания можно определить, как включающие нарушения метаболизма; нарушение регуляции аппетита; компульсивные ожирения; эметобулимию или нервную анорексию. Это патологически измененное потребление пищи может быть обусловлено нарушением аппетита (тягу к пище или отвращение к пище); изменением энергетического баланса (потребления по сравнению с расходом); нарушением восприятия качества пищи (высокого содержания жиров или углеводов, высокой вкусовой привлекательности); нарушением доступности пищи (неограниченный рацион или голодание) или нарушением водного баланса. Нарушения сна включают инсомнии, нарколепсию и другие нарушения, такие как чрезмерная сонливость, связанные со сном дистонии; синдром усталых ног; апноэ во сне; синдром десинхронизации физиологических циклов после трансмеридиональных перелетов; синдром посменной работы, синдром задержки или опережения фазы сна. Инсомнии определяются, как включающие нарушения сна, связанные с возрастом; периодическое лечение хронической инсомнии; ситуационную преходящую инсомнию (новое окружение, шум) или кратковременную инсомнию, обусловленную стрессом; чувство горя; боль или болезненное состояние. Другим объектом настоящего изобретения является фармацевтическая композиция, содержащая по меньшей мере одно соединение общей формулы (I) и фармацевтически приемлемый носитель. Еще одним объектом настоящего изобретения является способ лечения или профилактики заболеваний, которые связаны с орексиновыми рецепторами, таких как нарушения питания или нарушения сна, включающее введение пациенту терапевтически эффективного количества производного 1,2,3,4-тетрагидроизохинолина общей формулы (I). В предпочтительном варианте осуществления настоящего изобретения это количество составляет от 1 до 1000 мг в сутки, предпочтительно – от 2 до 500 мг в сутки, более предпочтительно – от 5 до 200 мг в сутки. Настоящее изобретение также относится к способу получения фармацевтической композиции, включающей производное 1,2,3,4-тетрагидроизохинолина общей формулы (I), путем смешивания одного или большего количества ингредиентов общей формулы (I) с носителем, выполняемого способом, который сам по себе известен, энтерально, например перорально (например, в виде таблеток, таблеток с покрытием, драже, капсул из твердого или мягкого желатина, растворов, эмульсий или суспензий), назально (например, в виде назальных аэрозолей) или ректально (например, в виде суппозиториев). Однако введение можно проводить и парентерально, например внутримышечно или внутривенно (например, в виде растворов для инъекций) или местно, например в виде мазей, кремов или масел. Соединения общей формулы (I) и их фармацевтически приемлемые соли можно приготовить совместно с фармацевтически инертными неорганическими или органическими вспомогательными веществами, предназначенными для изготовления таблеток, таблеток с покрытием, драже и капсул из твердого или мягкого желатина. В качестве таких вспомогательных веществ для изготовления таблеток, драже и капсул из твердого желатина можно использовать, например, лактозу, кукурузный крахмал или его производные, тальк, стеариновую кислоту или ее соли и т.п. Для капсул из мягкого желатина подходящими вспомогательными веществами являются, например, растительные масла, воски, жиры, полужидкие вещества и жидкие полиолы и т.п. Для приготовления растворов и сиропов подходящими вспомогательными веществами являются, например, вода, спирт, полиолы, сахароза, инвертный сахар, глюкоза и т.п. Для растворов для инъекций подходящими вспомогательными веществами являются, например, вода, спирты, полиолы, глицерин, растительные масла и т.п. Для суппозиториев подходящими вспомогательными веществами являются, например, натуральные или отвержденные масла, воски, жиры, полужидкие или жидкие полиолы и т.п. Описанные выше компоненты для вводимых перорально или предназначенных для инъекций композиций являются просто типичными примерами. Другие материалы, а также методики обработки и т.п. приведены в публикации Remington’s Pharmaceutical Sciences, 20th Edition, 2001, Marck Publishing Company, Easton, Pennsylvania, которая включена в настоящее изобретение в качестве ссылки. Соединения, предлагаемые в настоящем изобретении, также можно вводить в виде форм замедленного выделения путем использования известных систем доставки лекарственных препаратов замедленного выделения. Другим объектом настоящего изобретения является способ получения производных 1,2,3,4-тетрагидроизохинолина общей формулы (I). Соединения общей формулы (I), предлагаемые в настоящем изобретении, получают по общей последовательности реакций, представленной на приведенных ниже схемах, на которых X, R1, R2 и R3 являются такими, как определено для общей формулы (I). Полученные соединения также можно превратить в их фармацевтически приемлемые соли способом, который сам по себе известен. Как показано ниже на схеме 1, основными промежуточными продуктами при синтезе соединений общей формулы (I) являются 1-замещенные производные 3,4-дигидроизохинолина. Эти соединения получают или путем циклизации N-фенетилпропионамидов с помощью POCl3, или путем алкилирования 1-метил-3,4-дигидроизохинолинов алкилбромидами. Полученные 3,4-дигидроизохинолины восстанавливают в 1,2,3,4-тетрагидроизохинолины с помощью борогидрида натрия и получают продукты в виде рацемических смесей. Энантиомерно высокообогащенные 1,2,3,4-тетрагидроизохинолины получают путем гидрирования с переносом соответствующего 3,4-дигидроизохинолина в присутствии хирального комплекса Ru(II) (хирального катализатора), который впервые был описан в публикациях R.Noyori et al. (J. Am. Chem. Soc. 1996, 118, 4916-4917 и WO 97/20789). Используемый хиральный катализатор (комплекс Ru(II)) имеет вид:
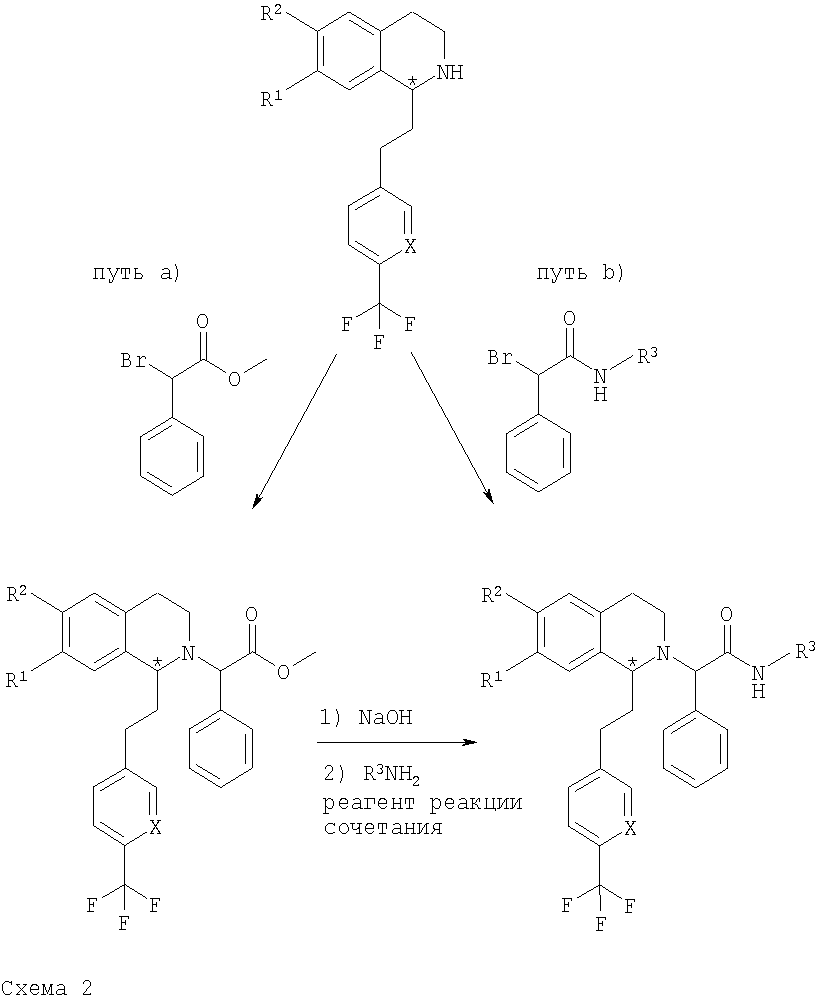
Как показано ниже на схеме 2 и схеме 3, промежуточные 1,2,3,4-тетрагидроизохинолины, предлагаемые в настоящем изобретении, можно превратить в соединения общей формулы (I) с помощью одного из трех разных путей синтеза, а), b) или с). На пути а) 1,2,3,4-тетрагидроизохинолин алкилируют замещенным метиловым эфиром 2-бромуксусной кислоты. Полученный сложный эфир гидролизуют в соответствующую кислоту и в заключение превращают в амид по реакции сочетания, предназначенной для получения амида, с необходимым амином в присутствии реагента реакции сочетания. На пути b) боковую цепь вводят путем прямого алкилирования соответствующего 1,2,3,4-тетрагидроизохинолина с помощью производного 2-бромацетамида:
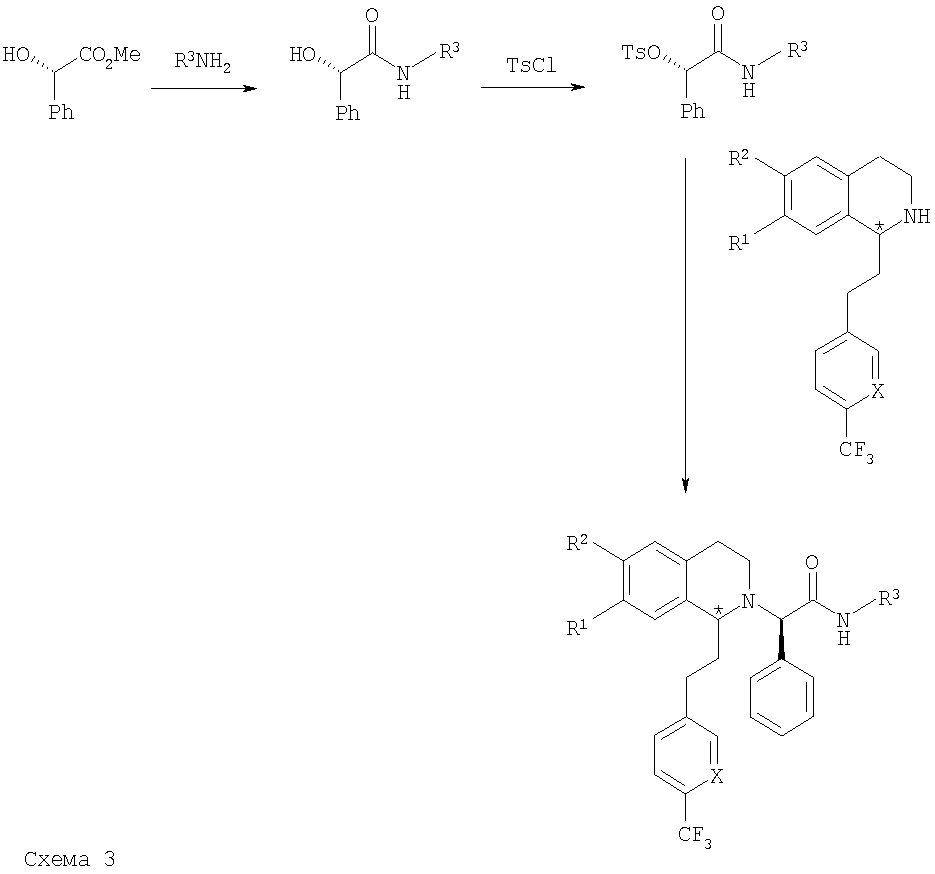
Производные 1,2,3,4-тетрагидроизохинолина общей формулы (I) также можно получить по стереоселективной схеме, используя в качестве исходного вещества энантиомерно чистый метил-(S)-(+)-манделат, по пути с) (см. ниже схему 3). Путем обработки сложного эфира спиртовым раствором амина получают соответствующий амид, который можно тозилировать п-толуолсульфонилхлоридом. На последней стадии тозилат вводят в реакцию сочетания с производным 1,2,3,4-тетрагидроизохинолина и получают соответствующее соединение общей формулы (I).
Производные 1,2,3,4-тетрагидроизохинолина, представляемые в качестве примера в настоящем изобретении, можно получить из легко доступных исходных веществ с помощью приведенных ниже общих методик и процедур. Следует понимать, что если указаны типичные или предпочтительные условия проведения эксперимента (т.е. температуры, длительность проведения реакций, молярные количества реагентов, растворителей и т.п.), то, если не указано иное, можно использовать другие условия проведения эксперимента. Оптимальные условия проведения реакции меняются в зависимости от конкретных использующихся реагентов или растворителей, но специалист в данной области техники может определить такие условия с помощью стандартных методик оптимизации. Экспериментальный раздел Аббревиатуры:
Химический раздел Приведенные ниже примеры иллюстрируют получение фармакологически активных соединений, предлагаемых в настоящем изобретении, но не ограничивают его объем. Все температуры приведены в °С. Все исследования с помощью аналитической и препаративной ВЭЖХ на нехиральных фазах проведены с использованием колонок на основе RP-C18. Исследования с помощью аналитической ВЭЖХ выполнены с помощью 2 разных приборов с длительностью цикла, равной ~2,5 мин и ~3,5 мин соответственно. Для разделения с помощью ВЭЖХ на хиральных фазах использовали колонку Chiralcel OD, выпускающуюся фирмой Daicel Chemical Industries. Соединения охарактеризованы с помощью 1Н-ЯМР (300 МГц) или 13С-ЯМР (75 МГц) (Varian Oxford; химические сдвиги приведены в мас.ч./млн относительно использованного растворителя; мультиплетности: s = синглет, d = дублет, t = триплет; q = квартет, m = мультиплет, b = широкий, константы спин-спинового взаимодействия приведены в Гц); с помощью ЖХ-МС, значения ВУ приведены в минутах; с помощью ТСХ (пластины для ТСХ, выпускающиеся фирмой Merck, силикагель 60 F254); или с помощью температуры плавления. А. Синтез производных пропионовой кислоты: 1. Синтез 3-(6-трифторметилпиридин-3-ил)-пропионовой кислоты: 1.1 Синтез метилового эфира 3-(6-трифторметилпиридин-3-ил)-акриловой кислоты:
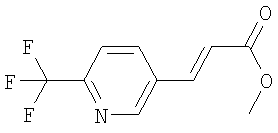
Раствор 6-трифторметилпиридин-3-карбальдегида (570 мг) в ДХМ (1,0 мл) прибавляют к раствору метилового эфира (трифенил- 1Н-ЯМР (300 МГц, CDCl3): 1.2 Синтез метилового эфира 3-(6-трифторметилпиридин-3-ил)-пропионовой кислоты:
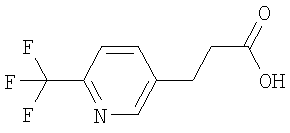
Раствор метилового эфира 3-(6-трифторметилпиридин-3-ил)-акриловой кислоты (720 мг) в метаноле (5,0 мл) обрабатывают с помощью Pd/C (10%, 240 мг) и перемешивают в атмосфере азота (~1 бар) при КТ в течение 20 ч. Суспензию фильтруют через целит и концентрируют в вакууме и получают эфир пропионовой кислоты в виде бесцветного масла. 1Н-ЯМР (300 МГц, CDCl3): 1.3. Синтез 3-(6-трифторметилпиридин-3-ил)-пропионовой кислоты:
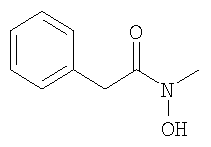
Моногидрат гидроксида лития (330 мг) одной порцией прибавляют к раствору метилового эфира 3-(6-трифторметилпиридин-3-ил)-пропионовой кислоты (610 мг) в смеси ТГФ (15 мл) с водой (5 мл). Смесь перемешивают в течение 20 ч при КТ. Прибавляют ДХМ и водный раствор HCl (1,0 М), слои разделяют, и водный слой дважды экстрагируют с помощью ДХМ. Объединенные органические экстракты сушат над MgSO4 и концентрируют в вакууме и получают искомую пропионовую кислоту в виде бежевого твердого вещества. 1Н-ЯМР (300 МГц, CDCl3): В. Синтез производных 2-бромацетамида: 1. Синтез 2-бром-N-метил-2-фенилацетамида: 1.1. Синтез N-гидрокси-N-метил-2-фенилацетамида:
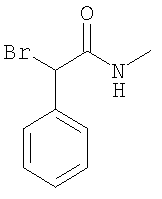
При 0°С фенилацетилхлорид (11,2 мл) по каплям прибавляют к раствору N-метилгидроксиламингидрохлорида (7,07 г) и триэтиламина (59 мл) в ДХМ (300 мл). После перемешивания в течение 90 мин прибавляют насыщенный водный раствор NaHCO3, слои разделяют, и водный слой дважды экстрагируют с помощью ДХМ (2×200 мл). Растворители удаляют в вакууме и остаток очищают с помощью флэш-хроматографии (ЭА/гептан 1/1) и получают искомый N-гидрокси-ацетамид в виде бесцветной жидкости. ЖХ-МС: ВУ = 0,63 мин, 166 (М+1, ЭР+). 1.2. Синтез 2-бром-N-метил-2-фенилацетамида:
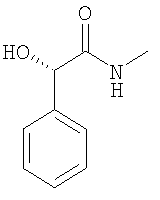
При 0°С триэтиламин (5,49 мл) прибавляют к раствору N-гидрокси-N-метил-2-фенилацетамида (6,5 г) в ДХМ (200 мл). Смесь по каплям обрабатывают раствором метансульфонилхлорида (3,21 мл) в ДХМ (60 мл). Через 2 ч прибавляют воду (150 мл), слои разделяют и водный слой дважды экстрагируют с помощью ЭА (2×100 мл). Органические экстракты объединяют и концентрируют в вакууме и получают неочищенный мезилат в виде бледно-желтого масла. Мезилат растворяют в ацетонитриле (200 мл). Прибавляют бромид лития (15,3 г) и реакционную смесь обрабатывают ультразвуком в течение 5 мин. После прибавления диизопропилэтиламина (6,78 мл) смесь повторно обрабатывают ультразвуком в течение 5 мин и перемешивают в течение еще 60 мин при комнатной температуре. Прибавляют воду (150 мл) и этилацетат (200 мл), слои разделяют и водный слой дважды экстрагируют этилацетатом (2×200 мл). Объединенные органические экстракты концентрируют в вакууме и очищают с помощью флэш-хроматографии (этилацетат/гептан 2:3) и получают искомый бромид в виде белого твердого вещества. ЖХ-МС: ВУ = 0,75 мин, 228 (М+1, ЭР+). С. Синтез (S)-метилкарбамоил-фенил-метилового эфира толуол-4-сульфоновой кислоты: 1. Синтез (S)-2-гидрокси-N-метил-2-фенилацетамида:
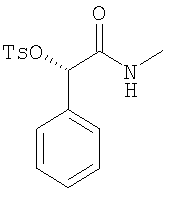
Метил-(S)-(+)-манделат (17 г) растворяют в раствор метиламина в метаноле (230 мл, 2,0 М) и выдерживают при КТ в течение 1 дня. Прибавляют еще одну порцию метиламина в метаноле (10 мл, 2,0 М). Еще через день прибавляют третью порцию метиламина в метаноле (10 мл, 2,0 М). Еще через 24 ч растворители удаляют в вакууме и получают искомый манделамид в виде бледно-желтых кристаллов, которые используют без дополнительной очистки. ЖХ-МС: ВУ = 0,52 мин, 166 (М+1, ЭР+). 2. Синтез (S)-метилкарбамоил-фенил-метилового эфира толуол-4-сульфоновой кислоты:
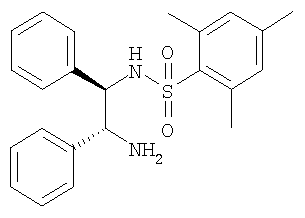
При КТ ДИПЭА (2,74 мл) и ДМАП (145 мг) прибавляют к раствору (S)-2-гидрокси-N-метил-2-фенилацетамида (2,4 г) в ДХМ (50 мл). Смесь обрабатывают порциями п-толуолсульфонилхлорида (2,75 г) и выдерживают в течение 2 ч при КТ. Растворитель удаляют в вакууме и остаток растворяют в этилацетате. Раствор дважды промывают насыщенным раствором NaHCO3 и один раз рассолом, растворители удаляют в вакууме и остаток перекристаллизовывают из смеси этилацетат/третбутилметиловый эфир и получают тозилат в виде белых кристаллов. ЖХ-МС: ВУ = 0,93 мин, 320 (М+1, ЭР+). D. Синтез N-((1R,2R)-2-амино-1,2-дифенилэтил)-2,4,6-триметилбензолсульфонамида (предшественник катализатора):
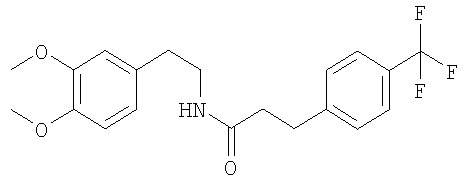
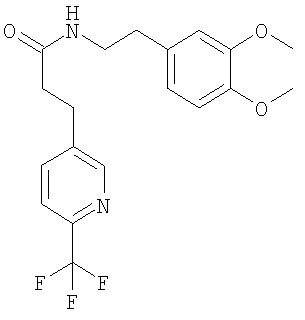
При 0°С раствор мезитиленсульфонилхлорида (3,09 г) в ТГФ (150 мл) по каплям прибавляют к суспензии (1R, 2R)-1,2-дифенилэтан-1,2-диамина (3,00 г), диизопропилэтиламина (3,87 мл) и карбоната калия (3,12 г) в смеси ТГФ (120 мл) и ДМФ (30 мл). Через 3 ч прибавляют воду (300 мл) и этилацетат (300 мл), слои разделяют и водный слой трижды экстрагируют этилацетатом (3×300 мл). Растворители удаляют в вакууме и остаток очищают с помощью препаративной ВЭЖХ. Для удаления муравьиной кислоты полученный продукт экстрагируют смесью насыщенный раствор NaHCO3/этилацетат и получают моносульфонамид в виде белого твердого вещества. ЖХ-МС: ВУ = 0,82 мин, 395 (М+1, ЭР+). Е. Синтез фенилэтиламинов (общая методика): Раствор соответствующего фенилэтиламина (110 ммоль) в толуоле (350 мл) обрабатывают с помощью соответствующего производного пропионовой кислоты (110 ммоль), кипятят с обратным холодильником в течение 90 ч с использованием ловушки Дина-Штарка и медленно охлаждают до КТ. Осадок отфильтровывают и сушат в вакууме и получают искомый амид. 1. Синтез N-[2-(3,4-диметоксифенил)-этил]-3-(4-трифторметилфенил)-пропионамида:
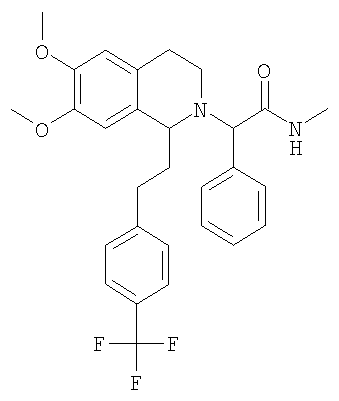
Это соединение получают по реакции 3,4-диметоксифенилэтиламина и 4-(трифторметил)-гидрокоричной кислоты. ЖХ-МС: ВУ = 0,97 мин, 382 (М+1, ЭР+). 2. Синтез N-[2-(3,4-диметоксифенил)-этил]-3-(6-трифторметилпиридин-3-ил)-пропионамида:
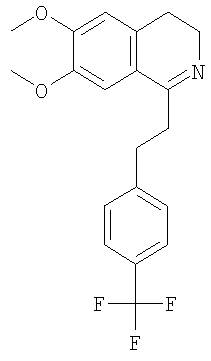
Это соединение получают по реакции 3,4-диметоксифенилэтиламина и 3-(6-трифторметилпиридин-3-ил)-пропионовой кислоты. ЖХ-МС: ВУ = 0,88 мин, 383 (М+1, ЭР+). F. Синтез 3,4-дигидроизохинолин производных путем циклизации амида (общая методика): Оксихлорид фосфора (123 ммоль) прибавляют к суспензии соответствующего амида (55,3 ммоль) в ацетонитриле (300 мл). Смесь кипятят с обратным холодильником в течение 90 мин и растворители удаляют в вакууме. Прибавляют метанол (100 мл) и повторно выпаривают. Полученный продукт перекристаллизовывают из диоксана или смеси диоксан/этанол. После фильтрования полученный гидрохлорид превращают в свободное основание путем прибавления насыщенного водного раствора NaHCO3 и экстракции дихлорметаном. Растворители удаляют в вакууме и получают соответствующий дигидроизохинолин. 1. Синтез 6,7-диметокси-1-[2-(4-трифторметилфенил)-этил]-3,4-дигидроизохинолина:
Это соединение получают с помощью циклизации N-[2-(3,4-диметоксифенил)-этил]-3-(4-трифторметилфенил)-пропионамида. ЖХ-МС: ВУ = 0,81 мин, 364 (М+1, ЭР+). 2. Синтез 6,7-диметокси-1-[2-(6-трифторметилпиридин-3-ил)-этил]-3,4-дигидроизохинолина:
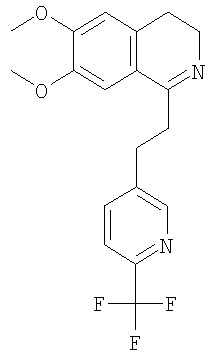
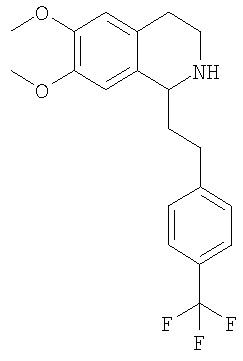
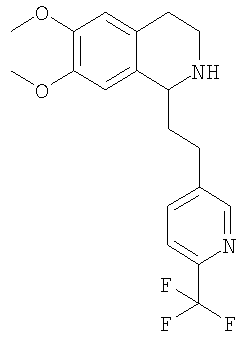
Это соединение получают с помощью циклизации N-[2-(3,4-диметоксифенил)-этил]-3-(6-трифторметилпиридин-3-ил)-пропионамида. ЖХ-МС: ВУ = 0,73 мин, 365 (М+1, ЭР+). G. Синтез 1,2,3,4-тетрагидроизохинолинов: 1. Синтез 1,2,3,4-тетрагидроизохинолинов по реакции Бишлера-Напиральского (общая методика): К суспензии соответствующего амида (44,8 ммоль) в ацетонитриле (500 мл) прибавляют оксихлорид фосфора (224 ммоль). Смесь кипятят с обратным холодильником в течение 2 ч и растворитель удаляют в вакууме. Полученное масло растворяют в толуоле или МеОН (20 мл), выпаривают досуха, растворяют в МеОН (200 мл) и охлаждают до 0°С. Небольшими порциями прибавляют NaBH4 (135 ммоль) и реакционную смесь перемешивают в течение 2 ч. Растворитель удаляют в вакууме, ЭА (400 мл) и прибавляют воду (400 мл), слои разделяют и водный слой трижды экстрагируют с помощью ЭА (3×200 мл). Объединенные органические экстракты концентрируют в вакууме и получают указанные ниже 1,2,3,4-тетрагидроизохинолины в виде рацемических смесей, которые очищают путем кристаллизации гидрохлорида из изопропанола. 1.1. Синтез рацемата 6,7-диметокси-1-[2-(4-трифторметилфенил)-этил]-1,2,3,4-тетрагидроизохинолина:
Это соединение получают по реакции N-[2-(3,4-диметоксифенил)-этил]-3-(4-трифторметилфенил)-пропионамида. ЖХ-МС: ВУ = 0,85 мин, 366 (М+1, ЭР+). 1.2. Синтез 6,7-диметокси-1-[2-(6-трифторметилпиридин-3-ил)-этил]-1,2,3,4-тетрагидроизохинолина:
Это соединение получают по реакции N-[2-(3,4-диметоксифенил)-этил]-3-(6-трифторметилпиридин-3-ил)-пропионамида. ЖХ-МС: ВУ = 0,73 мин, 367 (М+1, ЭР+). 2. Синтез 1,2,3,4-тетрагидроизохинолинов путем гидрирования с переносом (общая методика): Димер дихлор-(п-кумол)рутения(II) (0,20 ммоль) прибавляют к раствору N-((1R,2R)-2-амино-1,2-дифенилэтил)-2,4,6-триметилбензолсульфонамида (0,40 ммоль) и триэтиламина (0,80 ммоль) в ацетонитриле (3,0 мл). Смесь перемешивают в течение 1 ч при 80°С и прибавляют к раствору соответствующего дигидроизохинолина (28,0 ммоль) в дихлорметане (30 мл). Прибавляют азеотропную смесь муравьиной кислоты и триэтиламина (5:2, 14 мл) (происходит выделение газа). Через 90 мин к темно-красному раствору прибавляют насыщенный водный раствор NaHCO3 (200 мл). Слои разделяют, водный слой дважды экстрагируют с помощью ДХМ (2×200 мл) и объединенные органические экстракты концентрируют в вакууме. Остаток растворяют в изопропаноле (1600 мл) и обрабатывают раствором HCl в изопропаноле (5-6 М, 10 мл). Полученный гидрохлорид перекристаллизовывают и получают соответствующий 1,2,3,4-тетрагидроизохинолин с большим энантиомерным избытком, определенным с помощью хиральной ВЭЖХ. Гидрохлорид превращают в свободное основание путем экстракции смесью насыщенный раствор NaHCO3/дихлорметан. Абсолютную конфигурацию соответствующего продукта определяют по аналогии с литературной методикой (N.Uematsu, A.Fujii, S.Hashiguchi, Т.Ikariya, R.Noyori, J. Am. Chem. Soc. 1996, 118, 4916-4917). 2.1 Синтез (1S)-6,7-диметокси-1-[2-(4-трифторметилфенил)-этил]-1,2,3,4-тетрагидроизохинолина:
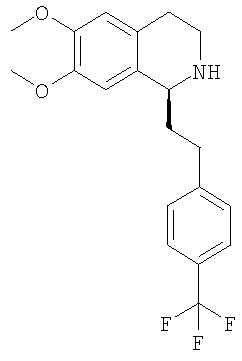
Это соединение получают с помощью гидрирования с переносом 6,7-диметокси-1-[2-(4-трифторметилфенил)-этил]-3,4-дигидроизохинолина. ЖХ-МС: ВУ = 0,80 мин, 366 (М+1, ЭР+). Хиральная ВЭЖХ: ВУ=12,0 мин (гексан/этанол 9/1; энантиомер: ВУ=17,1 мин). 2.2 Синтез (1S)-6,7-диметокси-1-[2-(6-трифторметилпиридин-3-ил)-этил]-1,2,3,4-тетрагидроизохинолина:
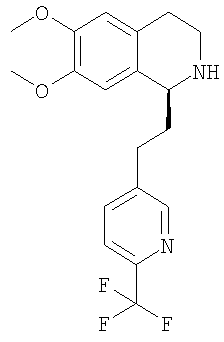
Это соединение получают с помощью гидрирования с переносом 6,7-диметокси-1-[2-(6-трифторметилпиридин-3-ил)-этил]-3,4-дигидроизохинолина. ЖХ-МС: ВУ = 0,73 мин, 367 (М+1, ЭР+). Хиральная ВЭЖХ: ВУ = 10,9 мин (гексан/этанол 4/1; энантиомер: ВУ = 24,4 мин). 3. Синтез 1,2,3,4-тетрагидроизохинолинов путем алкилирования 1-метил-3,4-дигидроизохинолинов (общая методика): При 0°С раствор n-BuLi в гексане (1,6 М, 0,63 ммоль) по каплям прибавляют к смеси 6,7-диметокси-1-метил-3,4-дигидроизохинолина (0,50 ммоль) и диизопропиламина (0,63 ммоль) в ТГФ (1,0 мл). Реакционную смесь перемешивают при КТ в течение 1 ч и при 0°С прибавляют к раствору соответствующего бензилбромида (0,50 ммоль) в ТГФ (1,0 мл). Раствор перемешивают в течение 1 ч, нагревают до КТ и разбавляют с помощью ДХМ (3,0 мл). Во второй колбе димер дихлор-(п-кумол)рутения(II) (0,15 ммоль) прибавляют к раствору N-((1R,2R)-2-амино-1,2-дифенилэтил)-2,4,6-триметилбензолсульфонамида (0,30 ммоль) и триэтиламина (0,60 ммоль) в ацетонитриле (3,3 мл). Смесь перемешивают в течение 1 ч при 80°С. Порцию этого раствора (0,10 мл) прибавляют к раствору соответствующего дигидроизохинолина (описанного выше). Прибавляют азеотропную смесь муравьиной кислоты и триэтиламина (5:2, 0,3 мл) (происходит выделение газа). Через 2 дня смесь концентрируют в вакууме и очищают с помощью препаративной ВЭЖХ и получают соответствующий 1,2,3,4-тетрагидроизохинолин. Энантиомерный избыток определяют с помощью хиральной ВЭЖХ. Абсолютную конфигурацию соответствующего продукта определяют по аналогии с литературной методикой (N.Uematsu, A.Fujii, S.Hashiguchi, T.Ikariya, R.Noyori, J. Am. Chem. Soc. 1996, 118, 4916-4917). 3.1. Синтез (1S)-6,7-диметокси-1-[2-(4-трифторметилфенил)-этил]-1,2,3,4-тетрагидроизохинолина:
Это соединение получают с помощью алкилирования 6,7-диметокси-1-метил-3,4-дигидроизохинолина 1-бромметил-4-трифторметилбензолом. ЖХ-МС: ВУ = 0,80 мин, 366 (М+1, ЭР+). Хиральная ВЭЖХ: ВУ = 12,0 мин (гексан/этанол 9/1; энантиомер: ВУ = 17,1 мин). Н. Синтез производных метилового эфира (3,4-дигидро-1Н-изохинолин-2-ил)-фенилуксусной кислоты (общая методика): ДИПЭА (43,0 ммоль) и метиловый эфир 1. Синтез метилового эфира {6,7-диметокси-1-[2-(4-трифторметилфенил)-этил]-3,4-дигидро-1Н-изохинолин-2-ил}-фенилуксусной кислоты:
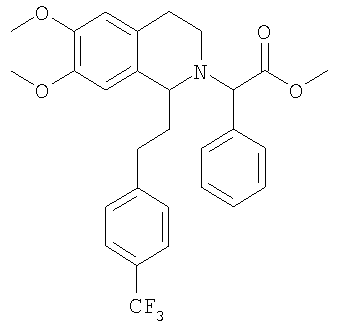
Это соединение получают по реакции 6,7-диметокси-1-[2-(4-трифторметилфенил)-этил]-1,2,3,4-тетрагидроизохинолина с метиловым эфиром ЖХ-МС: ВУ = 0,93 мин, 514 (М+1, ЭР+). 2. Синтез метилового эфира {6,7-диметокси-1-[2-(6-трифторметилпиридин-3-ил)-этил]-3,4-дигидро-1Н-изохинолин-2-ил}-фенилуксусной кислоты:
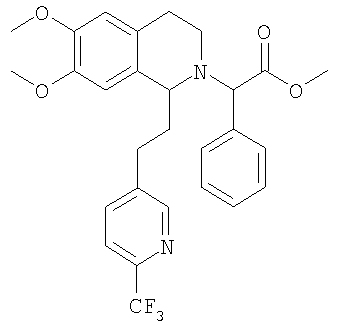
Это соединение получают по реакции 6,7-диметокси-1-[2-(6-трифторметилпиридин-3-ил)-этил]-1,2,3,4-тетрагидроизохинолина с метиловым эфиром ЖХ-МС: ВУ = 1,68 мин, 515 (М+1, ЭР+). 3. Синтез метилового эфира {(1S)-6,7-диметокси-1-[2-(4-трифторметилфенил)-этил]-3,4-дигидро-1Н-изохинолин-2-ил}-фенилуксусной кислоты:
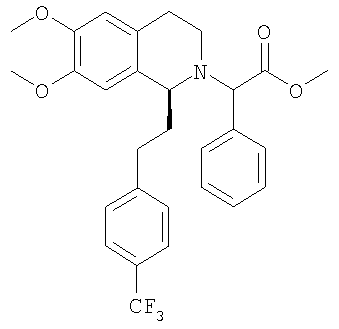
Это соединение получают по реакции (1S)-6,7-диметокси-1-[2-(4-трифторметилфенил)-этил]-1,2,3,4-тетрагидроизохинолина с метиловым эфиром ЖХ-МС: ВУ = 0,93 мин, 514 (М+1, ЭР+). I. Синтез производных (3,4-дигидро-1Н-изохинолин-2-ил)-фенилуксусной кислоты (общая методика): Водный раствор гидроксида натрия (2,0 М, 50 мл) прибавляют к раствору соответствующего сложного эфира (21,5 ммоль) в метаноле (400 мл). Смесь нагревают до 60°С и перемешивают в течение 20 ч. Большую часть метанола удаляют в вакууме и остаток растворяют в растворе гидроксида натрия (2,0 М, 20 мл), воде (100 мл) и ДХМ (100 мл). Слои разделяют и водный слой трижды экстрагируют с помощью ДХМ (3×100 мл). Объединенные органические экстракты концентрируют в вакууме и получают соответствующую карбоновую кислоту, которую используют без дополнительной очистки. 1. Синтез {6,7-диметокси-1-[2-(4-трифторметилфенил)-этил]-3,4-дигидро-1Н-изохинолин-2-ил}-фенилуксусной кислоты:
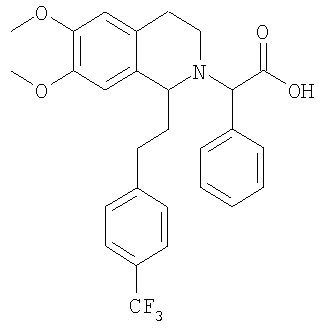
Это соединение получают с помощью омыления метилового эфира {6,7-диметокси-1-[2-(4-трифторметилфенил)-этил]-3,4-дигидро-1Н-изохинолин-2-ил}-фенилуксусной кислоты. ЖХ-МС: ВУ = 0,88 мин, 500 (М+1, ЭР+). 2. Синтез {6,7-диметокси-1-[2-(6-трифторметилпиридин-3-ил)-этил]-3,4-дигидро-1Н-изохинолин-2-ил}-фенилуксусной кислоты:
Это соединение получают с помощью омыления метилового эфира {6,7-диметокси-1-[2-(6-трифторметилпиридин-3-ил)-этил]-3,4-дигидро-1Н-изохинолин-2-ил}-фенилуксусной кислоты. ЖХ-МС: ВУ = 1,18 мин, 499 (М-1, ЭР-), 501 (М+1, ЭР+). 3. Синтез {(1S)-6,7-диметокси-1-[2-(4-трифторметилфенил)-этил]-3,4-дигидро-1Н-изохинолин-2-ил}-фенилуксусной кислоты:
Это соединение получают с помощью омыления метилового эфира {(1S)-6,7-диметокси-1-[2-(4-трифторметилфенил)-этил]-3,4-дигидро-1Н-изохинолин-2-ил}-фенилуксусной кислоты. ЖХ-МС: ВУ = 0,88 мин, 500 (М+1, ЭР+). Пример 1: Синтез 2-{6,7-диметокси-1-[2-(4-трифторметилфенил)-этил]-3,4-дигидро-1H-изохинолин-2-ил}-N-метил-2-фенилацетамида
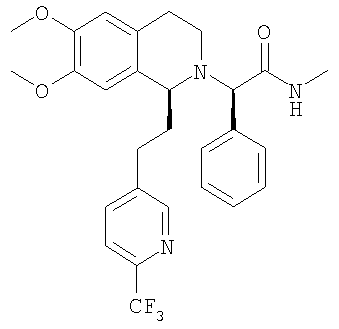
При 0°С метиламингидрохлорид (15,0 ммоль) и NaHCO3 (20,0 ммоль) прибавляют к раствору {6,7-диметокси-1-[2-(4-трифторметилфенил)-этил]-3,4-дигидро-1Н-изохинолин-2-ил}-фенилуксусной кислоты (10,0 ммоль) в ДМФ (200 мл). Через 15 мин прибавляют HOBt (12,0 ммоль) и EDC-гидрохлорид (22,0 ммоль). Смесь перемешивают в течение 10 мин и выдерживают в течение еще 14 ч при 0°С без перемешивания. Прибавляют воду (100 мл), ЭА (300 мл) и циклогексан (100 мл), слои разделяют и водный слой дважды экстрагируют смесью ЭА/циклогексан 3:1 (2×150 мл). Объединенные органические экстракты промывают насыщенным водным раствором NaHCO3 (100 мл) и рассолом (100 мл) и сушат над Na2SO4. Растворители удаляют в вакууме и остаток очищают с помощью флэш-хроматографии (градиентный режим: от ЭА/гептан 1/2 до ЭА/этанол/гептан 2/1/2) и получают искомые амиды в виде смесей всех 4 возможных стереоизомеров. ЖХ-МС: ВУ = 0,89 мин, 513 (М+1, ЭР+). Пример 2: Синтез (2R)-2-{(1S)-6,7-диметокси-1-[2-(4-трифторметилфенил)-этил]-3,4-дигидро-1Н-изохинолин-2-ил}-N-метил-2-фенилацетамида
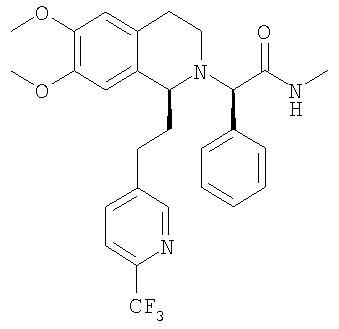
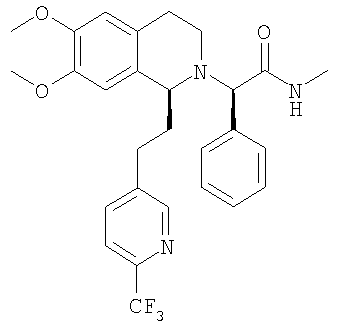
a) Методика I (посредством сочетания с амидом): При 0°С метиламингидрохлорид (23,7 ммоль) и NaHCO3 (2,01 г, 23,9 ммоль) прибавляют к раствору {(1S)-6,7-диметокси-1-[2-(4-трифторметилфенил)-этил]-3,4-дигидро-1Н-изохинолин-2-ил}-фенилуксусной кислоты (21,5 ммоль) в ДМФ (300 мл). Через 5 мин прибавляют HOBt (23,8 ммоль) и EDC-гидрохлорид (47,6 ммоль). Смесь перемешивают в течение 2 ч и выдерживают в течение еще 14 ч при 0°С без перемешивания. Прибавляют воду (300 мл) и ЭА (300 мл), слои разделяют и водный слой трижды экстрагируют с помощью ЭА (3×150 мл). Объединенные органические экстракты промывают водой (3×100 мл) и рассолом (100 мл). Растворители удаляют в вакууме и остаток очищают с помощью флэш-хроматографии (ЭА/гептан 3/2) и получают искомые амиды в виде разделенных диастереоизомеров. b) Методика II (посредством алкилирования бромпроизводным): ДИПЭА (119 ммоль) прибавляют к раствору 2-бром-N-метил-2-фенилацетамида (59,6 ммоль) в ТГФ (150 мл). Прибавляют раствор (1S)-6,7-диметокси-1-[2-(4-трифторметилфенил)-этил]-1,2,3,4-тетрагидроизохинолина (62,7 ммоль) в ТГФ (200 мл) и реакционную смесь перемешивают при 60°С в течение 7 дней. Прибавляют этилацетат (200 мл) и насыщенный водный раствор NaHCO3 (200 мл), слои разделяют и водный слой дважды экстрагируют этилацетатом (2×100 мл). Объединенные органические экстракты промывают водой (3×50 мл), сушат над MgSO4 и концентрируют в вакууме. Остаток очищают с помощью флэш-хроматографии (этилацетат/гептан 3/2) и получают искомые амиды в виде разделенных диастереоизомеров. c) Методика III (посредством алкилирования тозилатным производным): Раствор (1S)-6,7-диметокси-1-[2-(4-трифторметилфенил)-этил]-1,2,3,4-тетрагидроизохинолина (100 мг), (S)-метилкарбамоилфенилметилового эфира толуол-4-сульфоновой кислоты (100 мг) и ДИПЭА (0,065 мл) в бутаноне (5,0 мл) кипятят с обратным холодильником в течение 3 дней и охлаждают до КТ. Прибавляют этилацетат и смесь промывают насыщенным водным раствором NaHCO3 и рассолом. Органический слой сушат над Na2SO4 и растворители удаляют в вакууме. К неочищенному продукту прибавляют ТГФ (2,0 мл) и раствор HCl в изопропаноле (5-6 М, 0,10 мл) и растворители удаляют в вакууме. Полученное твердое вещество перекристаллизовывают из ТГФ (2,0 мл) и получают искомый амид в виде белых кристаллов. Данные приведены для более активного диастереоизомера свободного основания (IC50, СУПВФ). Rf = 0,21 (ЭА/гептан 2/1); ЖХ-МС: ВУ = 0,90 мин, 513 (М+1, ЭР+); Хиральная ВЭЖХ: ВУ = 18,9 мин (гексан/этанол 95/5; диастереоизомер: ВУ = 22,3 мин; два других возможных стереоизомера с противоположной конфигурацией 1,2,3,4-тетрагидроизохинолиновой кольцевой системы получают по аналогии с описанным выше синтезом путем использования N-((1S,2S)-2-амино-1,2-дифенилэтил)-2,4,6-триметилбензолсульфонамида (стадия G.2) для гидрирования с переносом: эти изомеры обладают следующими временами удерживания: ВУ = 26,2 мин, 33,8 мин); 1H-ЯМР (300 МГц, CDCl3): 13С-ЯМР (75 МГц, CDCl3): Пример 3: Синтез 2-{6,7-диметокси-1-[2-(6-трифторметилпиридин-3-ил)-этил]-3,4-дигидро-1H-изохинолин-2-ил}-N-метил-2-фенилацетамида
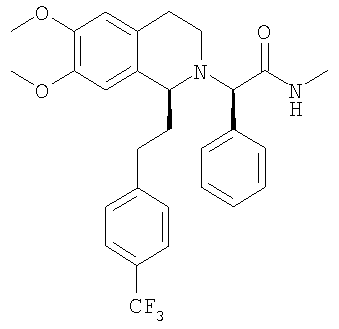
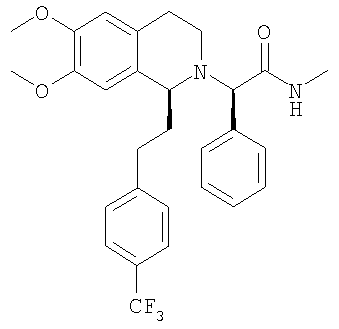
Смесь {6,7-диметокси-1-[2-(6-трифторметилпиридин-3-ил)-этил]-3,4-дигидро-1Н-изохинолин-2-ил}-фенилуксусной кислоты (0,20 ммоль), метиламингидрохлорида (0,20 ммоль), РуВОР (0,20 ммоль) и ДИПЭА (0,46 ммоль) в ДМФ (1,0 мл) перемешивают при КТ в течение 20 ч. Прибавляют воду и ЭА прибавляют, слои разделяют и водный слой экстрагируют с помощью ЭА. Объединенные органические экстракты сушат над MgSO4 и концентрируют в вакууме. Остаток очищают с помощью флэш-хроматографии (ЭА) и получают искомый продукт в виде вязкого масла. ЖХ-МС: ВУ = 1,17 мин, 514 (М+1, ЭР+). Пример 4: Синтез (2R)-2-{(1S)-6,7-диметокси-1-[2-(6-трифторметилпиридин-3-ил)-этил]-3,4-дигидро-1Н-изохинолин-2-ил}-N-метил-2-фенилацетамида
ДИПЭА (20,8 ммоль) прибавляют к раствору (1S)-6,7-диметокси-1-[2-(6-трифторметилпиридин-3-ил)-этил]-1,2,3,4-тетрагидроизохинолина (10,0 ммоль) в ТГФ (40 мл). Прибавляют 2-бром-N-метил-2-фенилацетамид (10,4 ммоль) и смесь перемешивают при 60°С в течение 5 дней. Прибавляют воду (100 мл) и этилацетат (200 мл), слои разделяют и водный слой дважды экстрагируют этилацетатом (2×100 мл). Объединенные органические экстракты концентрируют в вакууме и остаток очищают с помощью флэш-хроматографии (этилацетат/гептан 3/1) и получают искомые амиды в виде разделенных диастереоизомеров. Данные приведены для более активного диастереоизомера (IC50, СУПВФ). Rf = 0,15 (ЭА/гептан 3/1); ЖХ-МС: ВУ = 0,81 мин, 514 (М+1, ЭР+); 1Н-ЯМР (300 МГц, CDCl3): Биологические анализы Анализ in vitro Антагонистическую активность соединений общей формулы (I) по отношению к орексиновому рецептору определяют по следующей экспериментальной методике. Экспериментальная методика Исследования внутриклеточного кальция Клетки яичника китайского хомячка (ЯКХ), экспрессирующие орексиновый рецептор 1 человека и орексиновый рецептор 2 человека соответственно, выращивают в культуральной среде (Ham F-12 м L-глутамином), содержащей 300 мкг/мл G418, 100 Ед/мл пенициллина, 100 мкг/мл стрептомицина и 10%-ной инактивированной фетальной телячьей сыворотки (ФТС). Клетки высевают по 80000 клеток/лунка в черные с прозрачным дном стерильные 96-луночные планшеты (Costar), на которые предварительно нанесено покрытие с помощью 1%-ного раствора желатина в сбалансированном солевом растворе Хенкса (ССРХ). Все реагенты получены от фирмы Gibco BRL. Засеянные планшеты инкубируют в течение ночи при 37°С в атмосфере, содержащей 5% CO2. Орексин А человека, использующийся в качестве агониста, готовят в виде 1 мМ исходного раствора в смеси метанол:вода (1:1), разведенной в ССРХ, содержащем 0,1% бычьего сывороточного альбумина (БСА) и 2 мМ ГЭПЭС, и используют для анализа при конечной концентрации, равной 10 нМ. Антагонисты готовят в виде 10 мМ исходного раствора в ДМСО, затем разводят в 96-луночных планшетах, сначала в ДМСО, затем в ССРХ, содержащем 0,1% бычьего сывороточного альбумина (БСА) и 2 мМ ГЭПЭС. В день проведения анализа в каждую лунку прибавляют 100 мкл загрузочной среды (ССРХ, содержащей 1% ФТС, 2 мМ ГЭПЭС, 5 мМ пробенецида (Sigma) и 3 мкМ флуоресцентного индикатора кальция, fluo-3 AM (1 мМ исходный раствор в ДМСО с прибавлением 10%-ной плуроновой кислоты) (Molecular Probes). 96-Луночные планшеты инкубируют в течение 60 мин при 37°С в атмосфере, содержащей 5% СО2. Затем загрузочный раствор отсасывают и клетки 3 раза промывают с помощью 200 мкл ССРХ, содержащего 2,5 мМ пробенецида, 0,1% БСА, 2 мМ ГЭПЭС. В каждой лунке оставляют 100 мкл этого же буфера. Внутри считывающего устройства для планшетов с визуализацией флуоресценции (СУПВФ, Molecular Devices) в лунки прибавляют антагонисты по 50 мкл, инкубируют в течение 20 мин и в заключение прибавляют 100 мкл агониста. Для каждой лунки с 1-секундными интервалами измеряют флуоресценцию и высоту каждого пика флуоресценции сопоставляют с высотой пика флуоресценции, вызванного с помощью 10 нМ орексина-А с буфером, прибавленного вместо антагониста. Для каждого антагониста определяют значение IC50 (концентрацию соединения, необходимую для ингибирования агонистической реакции на 50%). Антагонистическая активность соединений находится в наномолярном диапазоне. Исследования ингибирующей активности по отношению к разным CYP Исследования ингибировния CYP проводили с использованием микросом печени человека (смесь образцов, взятых у 10 человек), описанных в литературе селективных по отношению к изоформам CYP субстратов, и путем количественного определения посредством или ЖХ-МС/МС (для CYP3A4 и CYP2C9) или обычной ВЭЖХ с флуоресцентным детектированием (для CYP2D6). Специфическими реакциями являлись 1′-гидроксилирование мидазоламом для CYP3A4, 3-гидроксилирование декстрометорфаном для CYP2D6 и 4′-гидроксилирование диклофенаком для CYP2C9. Эксперименты проводили дважды в 96-луночных планшетах при концентрациях субстратов, близких к соответствующим значениям Km (сводка условий проведения экспериментов приведена в таблице 1), и 7 концентрациях ингибитора вплоть до 50 мкМ. В каждом планшете параллельно проводили контрольные исследования (сульфафеназол для CYP2C9, флуоксетин для CYP2D6 и никардипин для CYP3A4).
Как показано ниже в таблице 2, соединения, описанные в примерах 1-4, обладают заметно меньшим сродством по отношению к CYP3A4.
Анализ in vivo Для лабораторных крыс с помощью радиотелеметрии в собственных клетках определяли самопроизвольную активность и температуру тела. Задачей настоящего исследования являлась регистрация суточной поведенческой активности крыс после перорального введения соединения общей формулы (I), предлагаемого в настоящем изобретении. Уменьшение активности в собственных клетках, зарегистрированное с помощью телеметрии у самцов крыс Wistar, рассматривали как указание на способность вызывать сонливость у ограниченного количества высокооптимизированных производных 1,2,3,4-тетрагидроизохинолина. Хорошо известно, что после перорального введения лабораторным животным психотропные лекарственные препараты, такие как антидепрессанты, нейролептики, снотворные или психостимуляторы, уменьшают или усиливают активность в собственных клетках и температуру тела. Терморегулирование является сложным процессом, который участвует в гомеостазе путем координации метаболизма, баланса энергии и поведения. Температура тела меняется с изменением суточной поведенческой активности и повышается при усилении локомоции. Эти два параметра определяют с помощью телеметрии у находящихся в сознании свободно перемещающихся крыс Wistar. При асептических условиях анестезированным животным в брюшную полость имплантируют телеметрическое устройство регистрации температуры тела/ активности. Более чем через 2 недели после имплантации телеметрической системы данные накапливают с 5-минутными интервалами в течение 96 ч. Для каждой крысы рассчитывают часовые средние значения. Данные для первых 48 ч используют в качестве внутреннего контроля и проводят сопоставление действия лекарственного препарата с действием плацебо. Эта методика прошла фармакологическую проверку путем определения амплитуды и зависимости от времени для гипоактивности и гипотермии, вызванных модуляторами рецептора GABA-A, такими как золпидем. Как показано ниже в таблице 3, антагонисты орексинового рецептора, предлагаемые в настоящем изобретении, такие как описанные в примерах 1-4, являются активными при пероральном введении.
Формула изобретения
1. Соединение (2R)-2-{(1S)-6,7-диметокси-1-[2-(4-трифторметилфенил)-этил]-3,4-дигидро-1Н-изохинолин-2-ил}-N-метил-2-фенилацетамид или его фармацевтически приемлемая соль. 2. Соединение по п.1, которое представляет собой (2R)-2-{(1S)-6,7-диметокси-1-[2-(4-трифторметилфенил)-этил]-3,4-дигидро-1Н-изохинолин-2-ил}-N-метил-2-фенилацетамид. 3. Соединение по п.1, которое представляет собой (2R)-2-{(1S)-6,7-диметокси-1-[2-(4-трифторметилфенил)-этил]-3,4-дигидро-1Н-изохинолин-2-ил}-N-метил-2-фенилацетамид или его фармацевтически приемлемую соль, где кислотный компонент указанной фармацевтически приемлемой соли выбирают из группы, включающей хлористоводородную кислоту, бромистоводородную кислоту, йодистоводородную кислоту, серную кислоту, фосфорную кислоту, азотную кислоту, лимонную кислоту, муравьиную кислоту, уксусную кислоту, малеиновую кислоту, виннокаменную кислоту, фумаровую кислоту, бензойную кислоту, памовую кислоту, стеариновую кислоту, метансульфоновую кислоту, п-толуолсульфоновую кислоту, салициловую кислоту, янтарную кислоту, трифторуксусную кислоту. 4. Соединение по п.1, которое представляет собой гидрохлорид (2R)-2-{(1S)-6,7-диметокси-1-[2-(4-трифторметилфенил)-этил]-3,4-дигидро-1Н-изохинолин-2-ил}-N-метил-2-фенилацетамида. 5. Соединение по п.1, которое представляет собой кристаллический гидрохлорид (2R)-2-{(1S)-6,7-диметокси-1-[2-(4-трифторметилфенил)-этил]-3,4-дигидро-1Н-изохинолин-2-ил}-N-метил-2-фенилацетамида, полученный посредством 6. Соединение по одному из пп.1-5, предназначенное для использования в качестве лекарственного средства, являющегося не пептидным антагонистом орексиновых рецепторов человека. 7. Фармацевтическая композиция, обладающая активностью не пептидного антагониста орексиновых рецепторов человека, содержащая соединение по любому из пп.1-5 и фармацевтически приемлемый носитель. 8. Применение соединения по одному из пп.1-5 для приготовления лекарственного средства для предупреждения или лечения заболеваний, выбранных из группы, включающей нарушения питания и нарушения сна. 9. Применение по п.8, в котором указанные нарушения питания включают нарушение метаболизма, нарушение регуляции аппетита, компульсивные ожирения, эметобулимию или нервную анорексию. 10. Применение по п.8, в котором указанные нарушения являются нарушениями сна. 11. Применение по п.8 или 10, в котором указанные нарушения сна включают инсомнии, нарколепсию и другие нарушения, такие как чрезмерная сонливость, связанные со сном дистонии, синдром усталых ног, апноэ во сне, синдром десинхронизации физиологических циклов после трансмеридиональных перелетов, синдром посменной работы, синдром задержки или опережения фазы сна.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 11
11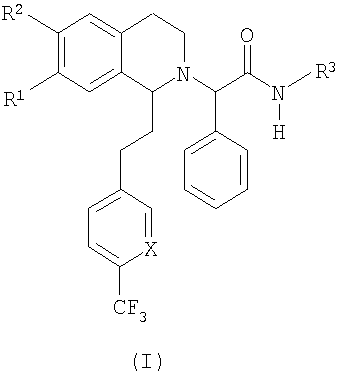
 5-фосфанилиден)-уксусной кислоты (990 мг) в ДХМ (2,5 мл). Смесь перемешивают в атмосфере азота при кипячении с обратным холодильником в течение 20 ч и концентрируют в вакууме. Остаток очищают с помощью флэш-хроматографии (ЭА/гептан 3/7) и получают искомый ненасыщенный сложный эфир в виде белого твердого вещества.
5-фосфанилиден)-уксусной кислоты (990 мг) в ДХМ (2,5 мл). Смесь перемешивают в атмосфере азота при кипячении с обратным холодильником в течение 20 ч и концентрируют в вакууме. Остаток очищают с помощью флэш-хроматографии (ЭА/гептан 3/7) и получают искомый ненасыщенный сложный эфир в виде белого твердого вещества. =3,85 (s, 3Н), 6,59 (d, J=16,2 Гц, 1Н), 7,70 (d, J=16,2 Гц, 1Н), 7,71 (d, J=8,1 Гц, 1Н), 7,98 (dd, J=8,1 Гц, J=2,1 Гц, 1Н), 8,84 (bs, 1Н).
=3,85 (s, 3Н), 6,59 (d, J=16,2 Гц, 1Н), 7,70 (d, J=16,2 Гц, 1Н), 7,71 (d, J=8,1 Гц, 1Н), 7,98 (dd, J=8,1 Гц, J=2,1 Гц, 1Н), 8,84 (bs, 1Н).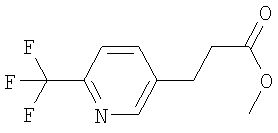
 -бромфенилуксусной кислоты (21,5 ммоль) последовательно прибавляют к раствору соответствующего 1,2,3,4-тетрагидроизохинолина (21,5 ммоль) в ТГФ, диоксане или толуоле (150 мл). Смесь кипятят с обратным холодильником в течение 20 ч, и ей дают достичь КТ. Прибавляют воду (250 мл) и ЭА (200 мл), слои разделяют и водный слой дважды экстрагируют с помощью ЭА (2×100 мл). Объединенные органические экстракты концентрируют в вакууме и/или очищают с помощью флэш-хроматографии, или используют без дополнительной очистки. Получают описанные ниже производные сложных эфиров.
-бромфенилуксусной кислоты (21,5 ммоль) последовательно прибавляют к раствору соответствующего 1,2,3,4-тетрагидроизохинолина (21,5 ммоль) в ТГФ, диоксане или толуоле (150 мл). Смесь кипятят с обратным холодильником в течение 20 ч, и ей дают достичь КТ. Прибавляют воду (250 мл) и ЭА (200 мл), слои разделяют и водный слой дважды экстрагируют с помощью ЭА (2×100 мл). Объединенные органические экстракты концентрируют в вакууме и/или очищают с помощью флэш-хроматографии, или используют без дополнительной очистки. Получают описанные ниже производные сложных эфиров.