Патент на изобретение №2378047
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) РОДИЙСОДЕРЖАЩИЕ КАТАЛИЗАТОРЫ
(57) Реферат:
Настоящее изобретение относится к способу получения катализатора или прокатализатора, к катализаторам для получения алкенилалканоатов и способу получения алкенилалканоатов. Описан способ получения катализатора или прокатализатора, подходящих для содействия получению алкенилалканоатов, включающий введение палладий-, золото- и родийсодержащих предшественников в контакт с материалом носителя, прокаливание в невосстанавливающей атмосфере, восстановление палладий-, золото- и родийсодержащих предшественников в результате введения восстанавливающей среды в контакт с материалом носителя и введение в контакт ацетата щелочного металла с восстановленным материлом носителя. Описана каталитическая композиция для получения алкенилалканоатов, содержащая: материал носителя, содержащий введенные с ним в контакт, по меньше мере, палладий, родий золото и ацетат щелочного металла, с получением катализатора или прокатализатора получения алкенилалканоатов, где, по меньшей мере, палладий и родий подвергают прокаливанию в невосстанавливающей атмосфере. Описан также способ получения алкенилалканоатов, включающий введение исходного сырья, содержащего алкен, алкановую кислоту и окислитель, в контакт с описанным выше катализатором или прокатализатором. Технический эффект – повышение активности и селективности катализатора получения алкенилалканоатов. 3 н. и 38 з.п. ф-лы, 6 табл.
Данная заявка заявляет преимущества предварительной заявки США Область техники Настоящее изобретение относится к катализаторам, способам получения катализаторов и способам получения алкенилалканоатов. Говоря более конкретно, изобретение относится к способам получения винилацетата. Уровень техники Определенные алкенилалканоаты, такие как винилацетат (VA), представляют собой товарные химические продукты, пользующиеся высоким спросом в их мономерной форме. Например, VA используют для получения поливинилацетата (PVAc), который обычно используется для изготовления клеев и составляет большую долю использования VA. Другие варианты использования VA включали поливиниловый спирт (PVOH), сополимеры этилена-винилацетата (EVA), винилацетата-этилена (VAE), поливинилбутираль (PVB), сополимер этилена-винилового спирта (EVOH), поливинилформаль (PVF) и сополимер винилхлорида-винилацетата. PVOH обычно используют при изготовлении текстилей, пленок, клеев и фоточувствительных покрытий. При изготовлении пленок и изоляции для проводов и кабелей в определенном количестве зачастую используют EVA. Основные сферы применения для сополимера винилхлорида-винилацетата включают покрытия, краски, а в клеях зачастую используют VAE, в определенном количестве содержащий VA. VAE, который содержит более 50 процентов VA, главным образом используют в качестве добавок к цементу, красок и клеев. PVB в основном используют при изготовлении нижнего слоя в многослойных ветровых стеклах, покрытий и типографских красок. EVOH используют при изготовлении непроницаемых пленок и конструкционных полимеров. PVF используют при изготовлении эмали для проводов и магнитной ленты. Поскольку VA является базисом для изготовления такого большого количества коммерчески значимых материалов и продуктов, потребность в VA велика, и производство VA зачастую осуществляют в относительно больших масштабах, например, в размере 50000 метрических тонн или более в год. Данное крупномасштабное производство означает то, что возможной является значительная экономия, обусловленная увеличением масштаба производства, и относительно тонкие изменения в способе, условиях реализации способа или характеристиках катализатора могут оказывать значительное с точки зрения экономии влияние на затраты при производстве VA. Сообщалось о большом количестве методик получения алкенилалканоатов. Например, при получении VA широко используемая методика включает катализируемую газофазную реакцию между этиленом, уксусной кислотой и кислородом, как это видно из следующей далее реакции: С2Н4+СН3СООН+0,5О2 Возможно прохождение нескольких побочных реакций, в том числе таких, как образование СО2. Результаты для данной реакции обсуждаются при выражении через выход в граммах в час на единицу объема катализатора (STY), где STY представляет собой количество граммов VA, полученного на один литр катализатора за один час времени реакции (г/л·час). Состав материала исходного сырья может варьироваться в широких пределах. Обычно материал исходного сырья включает 30-70% этилена, 10-30% уксусной кислоты и 4-16% кислорода. Исходное сырье также может включать и инертные материалы, такие как CO2, азот, метан, этан, пропан, аргон и/или гелий. Основное ограничение для состава исходного сырья заключается в том, что уровень содержания кислорода в потоке продуктов, покидающем реактор, должен быть достаточно низким, так чтобы состав потока не попадал бы в пределы области воспламеняемости. На уровень содержания кислорода в продуктах оказывают влияние уровень содержания кислорода в потоке материалов исходного сырья, степень превращения О2 в реакции и количество любого инертного материала в продуктах. Газофазную реакцию проводят тогда, когда поток материалов исходного сырья пропускают в реакторы с неподвижным слоем катализатора либо поверх слоя, либо сквозь него. Благоприятные результаты получали при использовании температур реакции в диапазоне от 125°С до 200°С, в то время как обычными давлениями при проведении реакции являются 1-15 атмосфер. Несмотря на то, что данные системы обеспечивают получение надлежащих выходов, продолжает сохраняться потребность в пониженном получении побочных продуктов, повышенных объемах получения VA и пониженном потреблении энергии во время получения. Один подход заключается в улучшении характеристик катализатора, в особенности в том, что касается селективности в отношении получения СО2 и/или активности катализатора. Еще один подход заключается в модифицировании условий реакции, таких как соотношение между компонентами материалов исходного сырья, степень превращения О2 в реакции, объемная скорость (SV) для потока материалов исходного сырья и рабочие температуры и давления. Образование СО2 представляет собой один аспект, действие которого можно ослабить благодаря использованию улучшенных катализаторов. Селективность в отношении получения СО2 представляет собой процентную долю подвергшегося конверсии этилена, который превратился в СО2. Уменьшение селективности в отношении получения СО2 делает возможным получение большего количества VA при расчете на единицу объема и в единицу времени в существующих установках даже при сохранении всех других условий реакции на прежнем уровне. На объем получения VA в конкретной реакционной системе оказывают влияние несколько других факторов, в том числе активность катализатора, соотношение между компонентами материалов исходного сырья, степень превращения O2 в реакции, объемная скорость (SV) для потока материалов исходного сырья и рабочие температуры и давления. Все данные факторы в результате их совокупного действия определяют выход в граммах в час на единицу объема катализатора (STY) в реакционной системе, где STY обсуждается при выражении через количество граммов VA, полученного на один литр катализатора за один час времени реакции, или г/л·час. В общем случае при определении STY существенным фактором является активность, но на STY значительное влияние могут оказывать еще и другие факторы. Обычно, чем выше активность катализатора, тем выше будет значение STY, которое катализатор будет способен обеспечить. Степень превращения O2 представляет собой меру того, как много кислорода вступает в реакцию в присутствии катализатора. Степень превращения O2 является температурозависимым параметром, так что в общем случае степень превращения увеличивается по мере роста температуры реакции. Однако параллельно увеличению степени превращения O2 увеличивается также и количество полученного CO2. Поэтому степень превращения O2 выбирают таким образом, чтобы добиться достижения баланса между желательным объемом получения VA и количеством полученного CO2. Катализатор, обладающий более высокой активностью, обозначает то, что общую температуру реакции можно будет уменьшить при одновременном сохранении той же самой степени превращения O2. В альтернативном варианте катализатор, обладающий более высокой активностью, будет обеспечивать достижение более высокой степени превращения O2 при данных температуре и объемной скорости. Обычной практикой является то, что в катализаторе используют один или несколько каталитических компонентов, нанесенных на относительно инертный материал носителя. В случае катализаторов VA каталитические компоненты обычно представляют собой смесь металлов, которые могут быть однородно распределены по всему материалу носителя («катализаторы с активной зоной по всему материалу»), только на поверхности материала носителя («корковые катализаторы»), только под поверхностью материала носителя («катализаторы со структурой активной зоны типа «белок в яйце»») или в сердцевине материала носителя («катализаторы со структурой активной зоны типа «желток в яйце»»). Для использования в катализаторе получения VA были предложены многочисленные различные типы материалов носителей, в том числе диоксид кремния, диоксид кремния, допированный церием, оксид алюминия, диоксид титана, диоксид циркония и смеси оксидов. Но исследования в отношении различий между материалами носителей были проведены в очень малой степени. Фактически в качестве материалов носителей в коммерческих масштабах главным образом использовались только диоксид кремния и оксид алюминия. Одна комбинация металлов, подходящая для катализа при получении VA, представляет собой палладий и золото. Катализаторы Pd/Au обеспечивают достижение надлежащих селективности в отношении получения СО2 и активности, но продолжает сохраняться потребность в улучшенных катализаторах, позволяющих добиться экономии, обусловленной увеличением масштаба производства, которая является возможной при производстве VA. Один способ получения катализаторов Pd/Au обычно включает стадии импрегнирования носителя водными растворами растворимых в воде солей палладия и золота; проведения реакции между импрегнированными растворимыми в воде солями и подходящим щелочным соединением, например гидроксидом натрия, для осаждения (зачастую называемого фиксацией) металлических элементов в виде соединений, нерастворимых в воде, например гидроксидов; промывания зафиксированного материала носителя для удаления незафиксированных соединений и для очищения катализатора другим способом от любых потенциальных ядов, например хлорида; восстановления нерастворимых в воде соединений при использовании обычного восстановителя, такого как водород, этилен или гидразин, и добавления соединения щелочного металла, такого как ацетат калия или натрия. Были предложены различные модификации данного базового способа. Например, в патенте США В патенте США В патенте США Обобщая вышеизложенное, можно сказать, что изобретатели исходили из осознания и обращения к потребности в непрерывном внедрении усовершенствований в области катализаторов получения VA для обеспечения улучшенного производства VA при пониженных затратах. Краткое изложение изобретения Настоящее изобретение обращается, по меньшей мере, к четырем различным аспектам, относящимся к структуре катализаторов, способам получения данных катализаторов и способам использования данных катализаторов для получения алкенилалканоатов. По отдельности или совместно в комбинации различные аспекты изобретения направлены на усовершенствование производства алкенилалканоатов и VA, в частности, в том числе на уменьшение количества побочных продуктов и улучшенную эффективность производства. Первый аспект настоящего изобретения относится к уникальным палладий/золотосодержащим катализатору или прокатализатору (необязательно прокаленным), которые содержат родий или другой металл. Второй аспект относится к палладий/золотосодержащим катализатору или прокатализатору, основой которых является слоистый материал носителя, где один слой материала носителя по существу не содержит каталитических компонентов. Третий аспект относится к палладий/золотосодержащим катализатору или прокатализатору, нанесенным на материал носителя, содержащий диоксид циркония. Четвертый аспект относится к палладий/золотосодержащим катализатору или прокатализатору, которые получают из каталитических компонентов, по существу не содержащих хлоридов. Подробное описание Катализаторы Для целей настоящего изобретения катализатор представляет собой любой материал носителя, который содержит, по меньшей мере, один каталитический компонент и который способен катализировать реакцию, в то время как прокатализатор представляет собой любой материал, который в результате получают на любой из обсуждающихся в настоящем документе стадий получения катализатора. Катализаторы и прокатализаторы настоящего изобретения могут включать те материалы, которые характеризуются, по меньшей мере, одним из следующих далее признаков: 1) катализатор будет являться палладий- и золотосодержащим катализатором, который содержит, по меньшей мере, еще один каталитический компонент, например родий, где один или несколько каталитических компонентов подвергают прокаливанию; 2) катализатор будет нанесен на слоистый носитель; 3) катализатор будет нанесен на материал носителя, содержащий диоксид циркония; 4) катализатор будет получен при использовании не содержащих хлоридов предшественников, или любой комбинацией из приведенных выше признаков. В соответствии с этим, эффективное использование катализатора должно способствовать улучшению селективности в отношении получения СО2, активности или обеих характеристик одновременно, в особенности в том, что касается получения VA. Необходимо понимать, что настоящее изобретение описывается в контексте определенных иллюстративных вариантов реализации, но оно может варьироваться в любом из нескольких аспектов в зависимости от потребностей в конкретной сфере применения. В порядке примера и не в порядке ограничения катализаторы могут содержать каталитические компоненты, однородно распределенные по всему материалу носителя, или же они могут быть корковыми катализаторами, где каталитические компоненты находятся в относительно тонкой поверхностной оболочке, охватывающей сердцевину материала носителя. Подходящими также могут быть и катализаторы со структурой активной зоны типа «белок в яйце», где каталитические компоненты находятся по существу на удалении от центра материала носителя. Подходящими также могут оказаться и катализаторы со структурой активной зоны типа «желток в яйце». Каталитические компоненты В общем случае катализаторы и прокатализаторы настоящего изобретения содержат металлы и, в частности, содержат комбинацию, по меньшей мере, двух металлов. В частности, комбинация металлов содержит, по меньшей мере, один из группы VIIIB и, по меньшей мере, один из группы IB. Необходимо понимать, что «каталитический компонент» используют для обозначения металла, который, в конечном счете, придает катализатору каталитическую функциональность, но данный термин также содержит и металл в разнообразных состояниях, таких как соль, раствор, золь-гель, суспензии, коллоидальные суспензии, свободный металл, сплав или их комбинации. Предпочтительные катализаторы в качестве каталитических компонентов содержат палладий и золото. Один вариант реализации катализатора включает комбинацию каталитических компонентов, включающую палладий и золото в комбинации с третьим каталитическим компонентом. Третий каталитический компонент, предпочтительно, выбирают из группы VIIIB, при этом Rh является наиболее предпочтительным. Другие предпочтительные катализаторы включают те из них, в которых третий каталитический компонент выбирают из W, Ni, Nb, Та, Ti, Zr, Y, Re, Os, Fe, Cu, Co, Zn, In, Sn, Ce, Ge, Ga и их комбинаций. Еще один вариант реализации катализатора включает комбинацию каталитических компонентов, в том числе частей палладия, золота и родия. Вместо Rh в данный вариант реализации необязательно также можно включить третий каталитический компонент (согласно приведенному выше перечню). В еще одном варианте реализации возможно использование двух или более каталитических компонентов из приведенного выше перечня. В одном примере палладий и золото можно комбинировать с Rh с получением катализатора, который демонстрирует улучшенную селективность в отношении получения СО2 (то есть пониженное образование СО2) в сопоставлении с катализаторами на основе Pd/Au, в которых отсутствует Rh. Кроме того, добавление Rh, по-видимому, не оказывает негативного влияния на активность катализатора. Селективность в отношении получения СО2 у палладий-, золото-, родийсодержащего катализатора также можно улучшить в результате прокаливания во время получения катализатора и/или в результате использования растворимых в воде предшественников, не содержащих галогенидов (оба варианта обсуждаются далее) несмотря на то, что оба варианта не являются обязательными для получения эффекта от Rh. Атомное отношение между третьим каталитическим компонентом и палладием может находиться в диапазоне от приблизительно 0,005 до приблизительно 1,0, более предпочтительно, от приблизительно 0,01 до приблизительно 1,0. В одном варианте реализации катализатор содержит от приблизительно 0,01 до приблизительно 5,0 г третьего каталитического компонента на один литр катализатора. Еще один предпочтительный вариант реализации катализатора содержит от приблизительно 1 до приблизительно 10 г палладия и от приблизительно 0,5 до приблизительно 10 г золота на один л катализатора. Количество золота, предпочтительно, находится в диапазоне от приблизительно 10 до приблизительно 125% (мас.) при расчете на массу палладия. В одном варианте реализации измельченных катализаторов, для измельченных катализаторов предпочтительными могут оказаться атомные отношения между Au и Pd в диапазоне от приблизительно 0,5 до приблизительно 1,00. Атомное отношение можно регулировать для установления баланса между активностью и селективностью в отношении получения СО2. Использование более высоких массовых или атомных отношений Au/Pd имеет тенденцию к благоприятствованию получения более активных, более селективных катализаторов. Выражаясь по-другому, можно сказать, что катализатор, характеризующийся атомным отношением, равным приблизительно 0,6, является менее селективным в отношении получения СО2, но также отличается и меньшей активностью в сопоставлении с катализатором, характеризующимся отношением, приблизительно равным 0,8. Влияние высокого атомного отношения Au/Pd на измельченный материал носителя также можно. увеличить в результате использования относительно высокого избытка гидроксид-иона, как это обсуждается далее в отношении стадии фиксации. Измельченный катализатор может представлять собой катализатор, в котором каталитические компоненты вводят в контакт с материалом носителя с последующим уменьшением размера частиц (например, в результате истирания или измельчения в шаровой мельнице), или катализатор, в котором каталитические компоненты вводят в контакт с материалом носителя после того, как размер частиц материала носителя был уменьшен. В случае корковых катализаторов толщина поверхностной оболочки, образованной каталитическими компонентами на материале носителя, находится в диапазоне от приблизительно 5 мкм до приблизительно 500 мкм. Более предпочтительные диапазоны включают интервал от приблизительно 5 мкм до приблизительно 300 мкм. Материалы носителя Как было указано, в одном аспекте изобретения каталитические компоненты настоящего изобретения в общем случае будут наносить на материал носителя. Подходящие материалы носителя обычно включают материалы, которые являются по существу однородными по природе, или смесь материалов. В целом материалы носителя обычно являются инертными в проводимой реакции. Материалы носителя могут состоять из любого подходящего вещества, предпочтительно выбираемого таким образом, чтобы материалы носителя характеризовались бы относительно высоким значением площади поверхности на единицу массы или объема, таким как у пористой структуры, структуры молекулярных сит, сотовой структуры или другой подходящей структуры. Например, материал носителя может содержать диоксид кремния, оксид алюминия, диоксид кремния-оксид алюминия, диоксид титана, диоксид циркония, диоксид ниобия, силикаты, алюмосиликаты, титанаты, шпинель, карбид кремния, нитрид кремния, углерод, кордиерит, стеатит, бентонит, глины, металлы, стекла, кварц, пемзу, цеолиты, нецеолитные молекулярные сита, их комбинации и тому подобное. Подходящей также может быть и любая из различных кристаллических форм материалов, например альфа- или гамма-оксид алюминия. Наиболее предпочтительными являются материалы носителей, содержащие диоксид кремния и диоксид циркония. В дополнение к этому для использования в настоящем изобретении подходящими также являются многослойные материалы носителей. Материал носителя в катализаторе данного изобретения может состоять из частиц, имеющих любую из различных правильных или неправильных форм, такую как форма сфер, таблеток, цилиндров, дисков, колец, звезд или другие формы. Материал носителя может иметь размеры, такие как диаметр, длина или ширина, в диапазоне от приблизительно 1 до приблизительно 10 мм, предпочтительно, от приблизительно 3 до приблизительно 9 мм. В частности, в случае правильной формы (например, сферической) в качестве предпочтительного его наибольшего размера будет иметь место величина в диапазоне от приблизительно 4 мм до приблизительно 8 мм. В дополнение к этому, подходящим может оказаться измельченный или порошкообразный материал носителя, такой, чтобы материал носителя имел бы правильную или неправильную форму при диаметре в диапазоне от приблизительно 10 микронов до приблизительно 1000 микронов, при этом предпочтительные размеры находятся в диапазоне от приблизительно 10 до приблизительно 700 микронов, а наиболее предпочтительные размеры находятся в диапазоне от приблизительно 180 микронов до приблизительно 450 микронов. Возможно использование более крупных или более мелких размеров, а также полидисперсных наборов размеров частиц. Например, в случае псевдоожиженного слоя катализатора предпочтительный диапазон размеров будет включать интервал от 10 до 150 микронов. В случае предшественников, используемых в слоистых катализаторах, предпочтительным является диапазон размеров от 10 до 250 микронов. Площади удельной поверхности, возможные для носителя компонентов катализатора, согласно измерениям по методу БЭТ (Брунауэра-Эмметта-Теллера) в общем случае могут находиться в диапазоне от приблизительно 1 м2/г до приблизительно 500 м2/г, предпочтительно, от приблизительно 100 м2/г до приблизительно 200 м2/г. Например, в случае пористого носителя объем пор материала носителя в общем случае может находиться в диапазоне от приблизительно 0,1 до приблизительно 2 мл/г, а предпочтительно, от приблизительно 0,4 до приблизительно 1,2 мл/г. Желательным, но не обязательным, является средний размер пор в диапазоне, например, от приблизительно 50 до приблизительно 2000 ангстрем. Примеры подходящих материалов носителей, содержащих диоксид кремния, включают КА160 от компании Sud Chemie, Aerolyst 350 от компании Degussa и другие марки пирогенного или не содержащего микропор диоксида кремния, характеризующиеся размером частиц в диапазоне от приблизительно 1 мм до приблизительно 10 мм. Примеры подходящих материалов носителей, содержащих диоксид циркония, включают материалы от компании NorPro, Zirconia Sales (America), Inc., Daichi Kigenso Kagaku Kogyo и Magnesium Elektron Inc (MEI). Подходящие материалы носителей, содержащие диоксид циркония, характеризуются широким диапазоном площадей удельной поверхности от менее, чем приблизительно 5 м2/г до более, чем 300 м2/г. Предпочтительные материалы носителей, содержащие диоксид циркония, характеризуются площадями удельной поверхности в диапазоне от приблизительно 10 м2/г до приблизительно 135 м2/г. Материалы носителей могут иметь поверхности, подвергнутые обработке на стадии прокаливания, на которой исходный материал носителя подвергают нагреванию. Нагревание (например, прокаливание) приводит к уменьшению площади удельной поверхности материала носителя. Это обеспечивает получение способа создания материалов носителей, характеризующихся площадями удельной поверхности, которые в других случаях было бы невозможно легко получить от поставщиков. В еще одном варианте реализации предполагается использовать, по меньшей мере, множественную комбинацию материалов носителей, каждый из которых обладает своей собственной характеристикой. Например, по меньшей мере, два материала носителей (например, диоксид циркония), обладающие различными характеристиками, могут демонстрировать различные активности и селективности в отношении получения СО2, таким образом, делая возможным получение катализаторов, обладающих желательным набором характеристик, то есть может быть найден баланс между активностью катализатора и селективностью катализатора в отношении получения СО2. В одном варианте реализации несколько различных носителей используют в слоистой конфигурации. При использовании любого из нескольких различных подходов можно добиться получения слоистой структуры, такой как множества ламелей, которые в общем случае являются плоскими, волнистыми или комбинированными. Один конкретный подход заключается в использовании последовательно охватывающих слоев по отношению к первоначальному сердцевинному слою. В общем случае в настоящем изобретении слоистые материалы носителей обычно включают, по меньшей мере, внутренний слой и наружный слой, по меньшей мере, частично окружающий внутренний слой. Наружный слой, предпочтительно, содержит существенно больше каталитических компонентов в сопоставлении с внутренним слоем. В одном варианте реализации внутренний и наружный слои получают из различных материалов; но материалы могут быть и одинаковыми. В то время как внутренний слой может быть непористым, другие варианты реализации включают внутренний слой, который является пористым. Слоистый материал носителя, предпочтительно, дает в результате форму коркового катализатора. Но слоистый материал носителя устанавливает хорошо определенную границу между поверхностями материала носителя, которые содержат каталитические компоненты, и поверхностями, которые их не содержат. Кроме того, наружный слой может быть согласованно сформирован имеющим желательную толщину. Граница и однородная толщина наружного слоя совместно в результате приводят к получению коркового катализатора, то есть поверхностной оболочки, образованной каталитическими компонентами, которая характеризуется однородной и известной толщиной. Известно несколько методик создания слоистых материалов носителей, и они включают те, что описываются в патентах США Данные материалы, которые составляют внутренний слой, могут иметься в разнообразных формах, таких как частицы правильной формы, частицы неправильной формы, гранулы, диски, кольца, звезды, «вагонные колеса», сотовые структуры или другие формованные тела. Предпочтительным является внутренний слой в виде сферической частицы. Внутренний слой, будучи или не будучи сферическим, характеризуется эффективным диаметром в диапазоне от приблизительно 0,02 мм до приблизительно 10,0 мм, а предпочтительно, от приблизительно 0,04 мм до приблизительно 8,0 мм. Наиболее удаленный от центра слой в любой многослойной структуре представляет собой слой, который является пористым, характеризуется площадью удельной поверхности в диапазоне от приблизительно 5 м2/г до приблизительно 300 м2/г. Материалом наружного слоя являются металл, керамика или их комбинация, а в одном варианте реализации его выбирают из оксида алюминия, диоксида кремния, диоксида кремния-оксида алюминия, диоксида титана, диоксида циркония, диоксида ниобия, силикатов, алюмосиликатов, титанатов, шпинели, карбида кремния, нитрида кремния, углерода, кордиерита, стеатита, бентонита, глин, металлов, стекол, кварца, пемзы, цеолитов, нецеолитных молекулярных сит и их комбинаций, а предпочтительно, он содержит оксид алюминия, диоксид кремния, диоксид кремния/оксид алюминия, цеолиты, нецеолитные молекулярные сита (NZMS), диоксид титана, диоксид циркония и их смеси. Конкретные примеры включают диоксид циркония, диоксид кремния и оксид алюминия или их комбинации. Несмотря на то, что наружный слой обычно окружает по существу весь внутренний слой, это не является обязательным, и возможно использование селективного покрытия внутреннего слоя наружным слоем. Наружный слой можно подходящим образом наносить в виде покрытия на слой, расположенный под ним. В одном варианте реализации используют суспензию материала наружного слоя. Нанесение покрытия на внутренний слой при использовании суспензии можно осуществлять в соответствии со способами, такими как прокатка, погружение, распыление, протравное нанесение покрытия, другие методики нанесения суспензионного покрытия, их комбинации и тому подобное. Одна предпочтительная методика включает использование неподвижного или псевдоожиженного слоя частиц внутреннего слоя и распыление суспензии в слое для нанесения на частицы равномерного покрытия. Суспензию можно наносить неоднократно в небольших количествах с промежуточным высушиванием, получая наружный слой, который является высокооднородным по толщине. Суспензия, используемая для нанесения покрытия на внутренний слой, может также содержать любую из нескольких добавок, таких как поверхностно-активное вещество, органическое или неорганическое связующее, которое способствует адгезии наружного слоя к слою, расположенному под ним, или их комбинации. Примеры данного органического связующего включают нижеследующее, но не ограничиваются только им: PVA, гидроксипропилцеллюлоза, метилцеллюлоза и карбоксиметилцеллюлоза. Количество органического связующего, которое добавляют к суспензии, может варьироваться так, как в диапазоне от приблизительно 1% (мас.) до приблизительно 15% (мас.) при расчете на массу комбинации наружного слоя и связующего. Примеры неорганических связующих выбирают из связующего на основе оксида алюминия (например, Bohmite), связующего на основе диоксида кремния (например, Ludox, Teos), связующего на основе диоксида циркония (например, ацетата диоксида циркония или коллоидального диоксида циркония) или их комбинаций. Примеры связующих на основе диоксида кремния включают золь кремниевой кислоты и гель кремниевой кислоты, в то время как связующие на основе оксида алюминия включают золь оксида алюминия, бентонит, бомит и нитрат алюминия. Количество неорганического связующего может находиться в диапазоне от приблизительно 2% (мас.) до приблизительно 15% (мас.) при расчете на массу комбинации наружного слоя и связующего. Толщина наружного слоя может находиться в диапазоне от приблизительно 5 микронов до приблизительно 500 микронов, а предпочтительно, от приблизительно 20 микронов до приблизительно 250 микронов. Как только на внутренний слой будет нанесено покрытие в виде наружного слоя, получающийся в результате слоистый носитель будут высушивать таким образом, как в результате нагревания при температуре в диапазоне от приблизительно 100°С до приблизительно 320°С (например, в течение промежутка времени продолжительностью в диапазоне от приблизительно 1 до приблизительно 24 часов), а после этого его необязательно можно подвергнуть прокаливанию при температуре в диапазоне от приблизительно 300°С до приблизительно 900°С (например, в течение промежутка времени продолжительностью в диапазоне от приблизительно 0,5 до приблизительно 10 часов) для улучшения сцепления между наружным слоем и слоем, расположенным под ним, по меньшей мере, на части их поверхности и получения слоистого носителя катализатора. Стадии высушивания и прокаливания можно скомбинировать в одну стадию. Получающийся в результате слоистый материал носителя можно вводить в контакт с каталитическими компонентами точно так же, как и любой другой материал носителя при получении катализаторов, как это описывается далее. В альтернативном варианте материал носителя наружного слоя вводят в контакт с каталитическими компонентами до того, как его нанесут в виде покрытия на слой, расположенным под ним. В еще одном варианте реализации слоистого носителя добавляют второй наружный слой для охватывания первоначального наружного слоя и получения, по меньшей мере, трех слоев. Материал второго наружного слоя может быть тем же самым, что и материал первого наружного слоя, или может отличаться от него. Подходящие материалы включают те, что обсуждались в отношении первого наружного слоя. Способ нанесения второго наружного слоя может быть тем же самым, что и способ, используемый при нанесении среднего слоя, или может отличаться от него, и подходящие способы включают те, что обсуждались в отношении первого наружного слоя. При получении второго наружного слоя в подходящем случае возможно использование описанных органических или неорганических связующих. Первоначальный наружный слой может содержать, а может и не содержать каталитические компоненты. Подобным же образом каталитические компоненты может содержать, а может и не содержать второй наружный слой. Если оба наружных слоя содержат каталитический компонент, то тогда в каждом слое, предпочтительно, используют различные каталитические компоненты, хотя это и не является обязательным. В одном предпочтительном варианте реализации первоначальный наружный слой не содержит каталитического компонента. Введение каталитического компонента в контакт с наружными слоями можно осуществить в результате импрегнирования или нанесения покрытия распылением, как это описывается далее. В вариантах реализации, в которых первоначальный наружный слой содержит каталитический компонент, один способ получения этого заключается во введении каталитического компонента в контакт с материалом первоначального наружного слоя до того, как материал нанесут на внутренний слой. Второй наружный слой можно наносить на первоначальный наружный слой, не содержащий примесей или содержащий каталитический компонент. Для получения трехслойного материала носителя, в котором один или несколько наружных слоев содержат каталитические компоненты, возможно использование и других подходящих методик. И действительно, на слоистый материал носителя ограничения в виде наличия трех слоев не накладываются, и он может включать четыре, пять или более слоев, из которых все или некоторые могут содержать каталитические компоненты. В дополнение к этому, от слоя к слою могут варьироваться количество и тип каталитических компонентов, которые меняются при переходе от слоя к слою в слоистом материале носителя, другие характеристики (например, пористость, размер частиц, площадь удельной поверхности, объем пор и тому подобное) материала носителя. Способы получения катализаторов В общем случае способ включает введение каталитических компонентов в контакт с материалом носителя и восстановление каталитических компонентов. Предпочтительные способы настоящего изобретения включают импрегнирование каталитическими компонентами материала носителя, прокаливание материала носителя, содержащего каталитические компоненты, восстановление каталитических компонентов и модифицирование восстановленных каталитических компонентов на материале носителя. В способ получения катализатора или прокатализатора также можно включить и дополнительные стадии, такие как фиксация каталитических компонентов на материале носителя и промывание зафиксированных каталитических компонентов. Некоторые из стадий, перечисленных выше, являются необязательными, а другие можно исключить (например, стадии промывания и фиксации). В дополнение к этому, некоторые стадии можно повторять (например, несколько стадий импрегнирования или фиксации), и порядок стадий может отличаться от того, что приведен выше (например, стадия восстановления может предшествовать стадии прокаливания). В определенной степени стадия введения в контакт будет определять то, какие последующие стадии потребуются для получения катализатора. Стадия введения в контакт Один конкретный подход к введению в контакт представляет собой подход, относящийся к тому, будут ли получать катализатор или прокатализатор со структурой активной зоны типа «желток в яйце», будут ли получать катализатор или прокатализатор со структурой активной зоны типа «белок в яйце», будут ли получать катализатор или прокатализатор с активной зоной по всему материалу, или будут получать корковый катализатор или прокатализатор или их комбинацию. В одном варианте реализации предпочтительными являются методики, которые позволяют получить корковые катализаторы. Стадию введения в контакт можно проводить при использовании любого из описанных выше материалов носителей, при этом наиболее предпочтительными являются диоксид кремния, диоксид циркония и слоистые материалы носителей, содержащие диоксид циркония. Стадию введения в контакт, предпочтительно, проводят в условиях температуры и давления окружающей среды; однако, возможно использование и пониженных или повышенных температур или давлений. В одной предпочтительной стадии введения в контакт материал носителя импрегнируют одним или несколькими водными растворами каталитических компонентов (называемыми растворами предшественников). Физическое состояние материала носителя во время стадии введения в контакт может представлять собой сухое твердое тело, суспензию, золь-гель, коллоидальную суспензию и тому подобное. В одном варианте реализации каталитические компоненты, содержащиеся в растворе предшественника, представляют собой растворимые в воде соли, полученные из каталитических компонентов, в том числе нижеследующее, но не ограничиваясь только им: хлориды, другие галогениды, нитраты, нитриты, гидроксиды, оксиды, оксалаты, ацетаты (ОАс) и амины, при этом предпочтительными являются соли, не содержащие галогенидов, а более предпочтительными являются соли, не содержащие хлоридов. Примеры паллалиевых солей, подходящих для использования в растворах предшественников, включают PdCl2, Na2PdCl4, Pd(NH3)2(NO2)2, Pd(NH3)4(OH)2, Pd(NH3)4(NO3)2, Pd(NO3)2, Pd(NH3)4(OAc)2, Pd(NH3)2(OAc)2, Pd(OAc)2 в КОН и/или NMe4OH и/или NaOH, Pd(NH3)4(НСО3)2 и оксалат палладия. В числе содержащих хлориды палладийсодержащих предшественников наиболее предпочтительным является Na2PdCl4. В числе не содержащих хлоридов солей палладийсодержащих предшественников наиболее предпочтительными являются следующие четыре: Pd(NH3)4(NO3)2, Pd(NO3)2, Pd(NH3)2(NO2)2, Pd(NH3)4(OH)2. Примеры солей золота, подходящих для использования в растворах предшественников, включают AuCl3, HAuCl4, NaAuCl4, KAuO2, NaAuO2, NMe4AuO2, Au(OAc)3 в КОН и/или NMe4OH, а также HAu(NO3)4 в азотной кислоте, при этом в числе не содержащих хлоридов золотосодержащих предшественников наиболее предпочтительным является KAuО2. Примеры родийсодержащих солей, подходящих для использования в растворах предшественников, включают RhCl3, Rh(OAc)3 и Rh(NO3)2. Также можно выбрать и подобные соли для описанных выше третьих каталитических компонентов. Кроме того, в конкретном растворе предшественника возможно использование более чем одной соли. Например, в одном растворе предшественника палладиевую соль можно объединить с солью золота, или можно объединить друг с другом две различные палладиевые соли. Растворы предшественников обычно можно получать в результате растворения выбранных соли или солей в воде, в присутствии или в отсутствие модификаторов растворимости, таких как кислоты, основания или другие растворители. Подходящими также могут быть и другие неводные растворители. Растворами предшественников можно импрегнировать материал носителя одновременно (например, совместное импрегнирование) или последовательно, и импрегнирование можно проводить при использовании одного или нескольких растворов предшественников. В случае трех или более каталитических компонентов возможно использование комбинации одновременного и последовательного импрегнирования. Например, палладием и родием можно импрегнировать при использовании одного раствора предшественника (что называется совместным импрегнированием), после чего проводят импрегнирование раствором золотосодержащего предшественника. В дополнение к этому, каталитическим компонентом можно импрегнировать материал носителя в ходе нескольких стадий таким образом, чтобы каждый раз вводить в контакт часть каталитического компонента. Например, один подходящий протокол может включать импрегнирование с использованием Pd с последующим импрегнированием с использованием Au, с последующим импрегнированием еще раз с использованием Au. Порядок импрегнирования материала носителя растворами предшественников не является критичным моментом; хотя могут существовать и некоторые преимущества при определенных порядках, как это обсуждается далее в отношении стадии прокаливания. Предпочтительно первым материал носителя импрегнируют палладийсодержащим каталитическим компонентом, при этом золотом импрегнируют после палладия или последним. Родием или другим третьим каталитическим компонентом, в случае его использования, можно импрегнировать совместно с палладием, совместно с золотом или только им одним. Кроме того, материал носителя можно импрегнировать несколько раз одним и тем же каталитическим компонентом. Например, сначала можно ввести в контакт часть всего золота, содержащегося в катализаторе, после чего ввести в контакт вторую часть золота. В промежутке между стадиями, на которых золото вводят в контакт с материалом носителя, можно провести одну или несколько других стадий, например прокаливание, восстановление и/или фиксацию. Влияние может оказать кислотно-основной профиль растворов предшественников вне зависимости от того, будут ли использовать совместное импрегнирование или последовательное импрегнирование. Таким образом, на стадии совместного импрегнирования должны совместно использоваться только растворы предшественников, характеризующиеся подобным кислотно-основным профилем; это исключит протекание любых кислотно-основных реакций, которые могут отравлять растворы предшественников. В случае стадии импрегнирования объем раствора предшественника выбирают таким образом, чтобы он соответствовал бы величине в диапазоне от приблизительно 85% до приблизительно 110% от объема пор материала носителя. Предпочтительными являются объемы в диапазоне от приблизительно 95% до приблизительно 100% от объема пор материала носителя, а более предпочтительно, от приблизительно 98% до приблизительно 99% от объема пор. Обычно раствор предшественника добавляют к материалу носителя, и материалу носителя дают возможность впитывать раствор предшественника. Это можно осуществить при покапельном введении до тех пор, пока по существу не будет достигнута начальная влажность материала носителя. В альтернативном варианте материал носителя можно помещать в аликвоты раствора предшественника или помещать в раствор предшественника периодически. Для обеспечения надежного контакта между материалом носителя и раствором предшественника возможно использование аппарата для погружения с вращением или другого вспомогательного аппарата. Кроме того, возможно использование распылительного устройства таким образом, чтобы раствор предшественника распылялся бы через сопло на материал носителя, где он бы впитывался. Для удаления любой избыточной жидкости, не впитавшейся в материал носителя, или для высушивания материала носителя после импрегнирования необязательно можно использовать декантирование, нагревание или действие пониженного давления. В случае стадии импрегнирования объем раствора предшественника выбирают таким, чтобы он соответствовал бы величине в диапазоне от приблизительно 85% до приблизительно 110% от объема пор материала носителя. Предпочтительными являются объемы от приблизительно 95% до приблизительно 100% от объема пор материала носителя, а более предпочтительно, от приблизительно 98% до приблизительно 99% от объема пор. Обычно раствор предшественника добавляют к материалу носителя, и материалу носителя дают возможность впитывать раствор предшественника. Это можно осуществить при покапельном введении до тех пор, пока по существу не будет достигнута начальная влажность материала носителя. В альтернативном варианте материал носителя можно помещать в аликвоты раствора предшественника или помещать в раствор предшественника периодически. Для обеспечения надежного контакта между материалом носителя и раствором предшественника возможно использование аппарата для погружения с вращением или другого вспомогательного аппарата. Кроме того, возможно использование распылительного устройства таким образом, чтобы раствор предшественника распылялся бы через сопло на материал носителя, где он бы впитывался. Для удаления любой избыточной жидкости, не впитавшейся в материал носителя, или для высушивания материала носителя после импрегнирования необязательно можно использовать декантирование, нагревание или действие пониженного давления. Во избежание стадии фиксации, тем не менее, при одновременном получении коркового катализатора можно воспользоваться и другими методиками введения в контакт. Например, каталитические компоненты можно вводить в контакт с материалом носителя при использовании способа химического осаждения из паровой фазы, такого как описанный в документе US 2001/0048970, который включается в настоящий документ для справки. Кроме того, нанесение покрытия по способу распыления или наслаивание другим образом предварительно однородно импрегнированного материала носителя в качестве наружного слоя на внутренний слой эффективно приводит к получению коркового катализатора, который можно описать как слоистый материал носителя. В еще одной методике при получении корковых катализаторов возможно использование металлоорганических предшественников каталитических компонентов, в особенности, в том, что касается золота, как это описывается в патенте США Для получения корковых катализаторов подходящей также может быть методика физического формирования поверхностной оболочки. В данном случае раствор предшественника можно распылять на нагретый материал носителя или на слоистый материал носителя, где растворитель раствора предшественника испаряется при контакте с нагретым материалом носителя, таким образом обеспечивается осаждение каталитических компонентов в поверхностной оболочке на материале носителя. Предпочтительно, могут быть использованы температуры в диапазоне приблизительно от 40 до 140°С. Толщину поверхностной оболочки можно регулировать в результате выбора температуры материала носителя и расхода раствора через распылительное сопло. Например, при температурах, превышающих приблизительно 100°С, образуется относительно тонкая поверхностная оболочка. В случае использования не содержащих хлоридов предшественников данный вариант реализации может оказаться в особенности подходящим для содействия улучшению формирования поверхностной оболочки на материале носителя. Специалист в соответствующей области должен понимать, что подходящим способом получения введенного в контакт материала носителя может оказаться комбинирование стадий введения в контакт. Стадия фиксации Может оказаться желательным превращение, по меньшей мере, части каталитических компонентов на введенном в контакт материале носителя из формы, растворимой в воде, в форму, нерастворимую в воде. Такая стадия может называться стадией фиксации. Ее можно провести в результате нанесения фиксирующего вещества (например, дисперсии в жидкости, такой как раствор) на импрегнированный материал носителя, что приведет к осаждению, по меньшей мере, части каталитических компонентов. Данная стадия фиксации способствует получению коркового катализатора, но она не является обязательной для получения корковых катализаторов. Возможно использование любого подходящего фиксирующего вещества, при этом предпочтительными являются гидроксиды (например, гидроксиды щелочных металлов), силикаты, бораты, карбонаты и бикарбонаты в водных растворах. Предпочтительным фиксирующим веществом является NaOH. Фиксацию можно осуществить в результате добавления фиксирующего вещества к материалу носителя до, во время или после того, как растворами предшественников будут импрегнировать материал носителя. Обычно фиксирующее вещество используют после стадии введения в контакт таким образом, чтобы введенный в контакт материал носителя имел бы возможность пропитаться раствором фиксирующего вещества в течение промежутка времени продолжительностью в диапазоне от приблизительно 1 до приблизительно 24 часов. Конкретное время зависит от комбинации раствора предшественника и фиксирующего вещества. Подобно стадии импрегнирования на стадии фиксации с выгодой можно использовать вспомогательное устройство, такое как аппарат для погружения с вращением, описанный в патенте США Стадию фиксации можно провести в виде одной или нескольких стадий, называемых совместной фиксацией или раздельной фиксацией. В случае совместной фиксации после того, как все соответствующие растворы предшественников будут введены в контакт с материалом носителя, на введенный в контакт материал носителя наносят один или несколько объемов раствора фиксирующего вещества вне зависимости от того, было ли введение в контакт осуществлено в результате использования одного или нескольких растворов предшественников. Например, фиксация после последовательного импрегнирования раствором палладийсодержащего предшественника, раствором золотосодержащего предшественника и раствором родийсодержащего предшественника будет представлять собой совместную фиксацию, как и фиксация после совместного импрегнирования раствором палладий/родийсодержащего предшественника с последующим импрегнированием раствором золотосодержащего предшественника. Пример совместной фиксации можно обнаружить в патенте США С другой стороны раздельная фиксация будет включать нанесение раствора фиксирующего вещества во время или после каждого импрегнирования раствором предшественника. Например, следующие далее протоколы будут представлять собой раздельную фиксацию: а) импрегнирование палладием с последующей фиксацией с дальнейшим импрегнированием золотом с последующей фиксацией; или b) совместное импрегнирование палладием и родием с последующей фиксацией с дальнейшим импрегнированием золотом с последующей фиксацией. В промежутке между фиксацией и последующим импрегнированием любую избыточную жидкость можно удалить, а материал носителя высушить, хотя это и не является обязательным. Пример раздельной фиксации можно обнаружить в патенте США В еще одном варианте реализации стадию фиксации и стадию введения в контакт проводят одновременно, один пример чего описывается в патенте США В еще одном варианте реализации, в особенности подходящем для использования совместно с предшественниками, не содержащими хлоридов, материал носителя подвергают предварительной обработке фиксирующим веществом для регулирования свойств материала носителя. В данном варианте реализации материал носителя сначала импрегнируют раствором либо кислоты, либо основания, обычно не содержащим металлов. После высушивания материал носителя импрегнируют раствором предшественника, который имеет противоположную кислотность/щелочность в сопоставлении с высушенным материалом носителя. Последующая кислотно-основная реакция приводит к образованию на материале носителя поверхностной оболочки из каталитических компонентов. Например, можно воспользоваться азотной кислотой для предварительной обработки материала носителя, который, в свою очередь, импрегнируют раствором основного предшественника, такого как Pd(OH)2 или Au(ОН)3. Данная методика получения может рассматриваться в качестве использования стадии фиксации с последующей стадией введения в контакт. Концентрация фиксирующего вещества в растворе обычно соответствует молярному избытку в сопоставлении с количеством каталитических компонентов, импрегнированных на материал носителя. Количество фиксирующего вещества должно находиться в диапазоне от приблизительно 1,0- до приблизительно 2,0-, предпочтительно, от приблизительно 1,1- до приблизительно 1,8-кратного в сопоставлении с количеством, необходимым для вступления в реакцию с каталитически активными катионами, присутствующими в растворимой в воде соли. В одном варианте реализации использование высокого значения атомного или массового отношения Au/Pd, увеличенного молярного избытка гидроксид-иона приводит к улучшению селективности в отношении получения СО2 и активности у получающегося в результате катализатора. Объем поданного раствора фиксирующего вещества в общем случае должен представлять собой количество, достаточное для покрытия всех доступных свободных поверхностей импрегнированного материала носителя. Это можно осуществить, например, в результате введения объема, который превышает объем пор у введенного в контакт материала носителя. Комбинирование стадий импрегнирования и фиксации может привести к получению катализатора, относящегося к типу коркового катализатора. Но использование растворов предшественников, не содержащих галогенидов, также делает возможным получение коркового катализатора при необязательном исключении стадии фиксации. В отсутствие хлоридсодержащего предшественника без стадии промывания, обсуждающейся далее, можно и обойтись. Кроме того, способ может не включать стадию фиксации каталитических компонентов, что в других случаях было бы необходимо для успешного прохождения через стадию промывания. Поскольку никакой стадии промывания не требуется, каталитические компоненты не требуется фиксировать для успешного прохождения через стадию промывания. Последующие стадии в способе получения катализатора не требуют фиксации каталитических компонентов, и, таким образом, оставшуюся часть стадии можно провести без дополнительных подготовительных стадий. В целом использование не содержащих хлоридов предшественников делает возможным создание способа получения катализатора или прокатализатора, который не включает стадии промывания, что, таким образом, приводит к уменьшению количества стадий, необходимых для получения катализатора, и устранению потребности в утилизации хлоридсодержащих отходов. Стадия промывания В частности, если используют растворы предшественников, содержащих галогениды, а при желании и при других вариантах нанесения, после стадии фиксации для подвергнутого фиксации материала носителя можно провести промывание для удаления любого остаточного количества галогенидов на носителе или обработку другим образом для устранения потенциального негативного влияния загрязнителя на материал носителя. Стадия промывания включала прополаскивание подвергнутого фиксации материала носителя в воде, предпочтительно, деионизованной воде. Промывание можно проводить в периодическом или непрерывном режиме. Промывание при комнатной температуре должно продолжаться до тех пор, пока отходящие промывные воды не будут характеризоваться уровнем содержания галогенид-иона, меньшим приблизительно 1000 ч/млн, а более предпочтительно, до тех пор, пока конечный продукт не даст отрицательного результата в испытании с использованием нитрата серебра. Стадию промывания можно проводить после или во время стадии восстановления, обсуждающейся далее, но, предпочтительно, ее проводят до нее. Как уже обсуждалось выше, использование растворов предшественников, не содержащих галогенидов, делает возможным исключение стадии промывания. Стадия прокаливания После того, как, по меньшей мере, один каталитический компонент будет введен в контакт с материалом носителя, можно провести стадию прокаливания. Стадию прокаливания обычно проводят до стадии восстановления и после стадии фиксации (если такую стадию используют), но она может присутствовать и на других участках процесса. В еще одном варианте реализации стадию прокаливания проводят после стадии восстановления. Стадия прокаливания включает нагревание материала носителя в невосстанавливающей атмосфере (то есть окисляющей или инертной). Во время прокаливания каталитические компоненты на материале носителя, по меньшей мере, частично разлагаются, превращаясь из их солей в смесь их оксидной формы и формы свободного металла. Например, стадию прокаливания проводят при температуре в диапазоне от приблизительно 100°С до приблизительно 700°С, предпочтительно, от приблизительно 200°С до приблизительно 500°С. Невосстанавливающие газы, используемые для прокаливания, могут включать один или несколько инертных или окисляющих газов, таких как гелий, азот, аргон, неон, оксиды азота, кислород, воздух, диоксид углерода, их комбинации и тому подобное. В одном варианте реализации стадию прокаливания проводят в атмосфере по существу чистых азота, кислорода, воздуха или их комбинаций. Времена прокаливания могут варьироваться, но, предпочтительно, они находятся в диапазоне приблизительно от 1 до 5 часов. Степень разложения солей каталитических компонентов зависит от использованной температуры и продолжительности периода времени, в течение которого импрегнированный катализатор будут прокаливать, и ее можно отслеживать, проводя мониторинг летучих продуктов разложения. Можно проводить одну или несколько стадий прокаливания таким образом, что в любой точке после того, как, по меньшей мере, один каталитический компонент будет введен в контакт с материалом носителя, его можно будет подвергнуть прокаливанию. Предпочтительно, последнюю стадию прокаливания проводят перед введением золотосодержащего каталитического компонента в контакт с материалом носителя на основе диоксида циркония. В альтернативном варианте прокаливание золотосодержащего материала носителя на основе диоксида циркония проводят при температурах, меньших приблизительно 300°С. Благодаря тому, что прокаливание золотосодержащего материала носителя на основе диоксида циркония не проводят при температурах, превышающих приблизительно 300°С, уменьшается опасность возникновения негативного влияния на селективность в отношении получения СО2 у получающегося в результате катализатора. Примерные протоколы, включающие стадию прокаливания, включают: а) импрегнирование палладием с последующим прокаливанием с дальнейшим импрегнированием золотом; b) совместное импрегнирование палладием и родием с последующим прокаливанием с дальнейшим импрегнированием с использованием Au; с) импрегнирование палладием с последующим прокаливанием с дальнейшим импрегнированием родием с последующим прокаливанием с дальнейшим импрегнированием золотом; или d) импрегнирование палладием и родием с последующим импрегнированием золотом с дальнейшим прокаливанием. Стадия восстановления Еще одна стадия, в общем случае используемая в настоящем изобретении, по меньшей мере, для частичного превращения любых остаточных каталитических компонентов из формы соли или оксида в каталитически активное состояние, такого как в результате проведения стадии восстановления. Обычно ее проводят в результате воздействия на соли или оксиды восстановителем, примеры которого включают аммиак, монооксид углерода, водород, углеводороды, олефины, альдегиды, спирты, гидразин, первичные амины, карбоновые кислоты, соли карбоновых кислот, сложные эфиры карбоновых кислот и их комбинации. Предпочтительными восстановителями считаются водород, этилен, пропилен, гидразин в щелочной среде и формальдегид в щелочной среде и их комбинации, при этом в особенности предпочтительными являются этилен и водород, смешанные с инертными газами. Несмотря на то, что предпочтительным является восстановление с использованием газообразной среды, также возможно использование и стадии восстановления, проведенной с применением жидкой среды (например, с применением восстанавливающего раствора). Температура, выбранная для восстановления, может находиться в диапазоне от температуры окружающей среды вплоть до приблизительно 550°С. Времена восстановления обычно будут варьироваться в диапазоне от приблизительно 1 до приблизительно 5 часов. Поскольку способ, используемый для восстановления каталитических компонентов, может оказывать влияние на характеристики конечного катализатора, условия, используемые для восстановления, могут варьироваться в зависимости от того, будут ли желательными высокая активность, высокая селективность или определенный баланс данных свойств. В одном варианте реализации палладий вводят в контакт с материалом носителя, фиксируют и восстанавливают до того, как будет введено в контакт и восстановлено золото, что описывается в патентах США Примерные протоколы, включающие стадию восстановления, включают: а) импрегнирование палладием с последующим необязательным прокаливанием с дальнейшим импрегнированием золотом с последующим восстановлением; b) совместное импрегнирование палладием и золотом с последующим необязательным прокаливанием с дальнейшим восстановлением; или с) импрегнирование палладием с последующим необязательным прокаливанием с дальнейшим восстановлением с последующим импрегнированием золотом. Стадия модифицирования Обычно после стадии восстановления и перед использованием катализатора желательной является стадия модифицирования. В то время, как катализатор можно использовать после проведения стадии модифицирования, данная стадия приводит к получению некоторых благоприятных результатов, в том числе к удлинению эксплуатационного срока службы катализатора. Стадию модифицирования иногда называют стадией активации, и ее можно проводить в соответствии с обычной практикой. А именно, восстановленный материал носителя перед использованием вводят в контакт с модификатором, таким как карбоксилат щелочного металла и/или гидроксид щелочного металла. Для данной цели используют обычные карбоксилаты щелочных металлов, такие как натриевые, калиевые, литиевые и цезиевые соли С2-4алифатических карбоновых кислот. Предпочтительным активатором при производстве VA является ацетат щелочного металла, при этом наиболее предпочтительным является ацетат калия (КОАс). Материал носителя необязательно можно импрегнировать раствором модификатора. После высушивания катализатор может содержать, например, от приблизительно 10 до приблизительно 70, предпочтительно, от приблизительно 20 до приблизительно 60 г модификатора на один литр катализатора. Способы получения алкенилалканоатов Настоящее изобретение можно использовать для получения алкенилалканоатов из алкена, алкановой кислоты и кислородсодержащего газа в присутствии катализатора. Предпочтительные алкеновые материалы исходного сырья содержат от двух до четырех атомов углерода (например, этилен, пропилен и н-бутен). Предпочтительные материалы исходного сырья в виде алкановых кислот, используемые в способе данного изобретения для получения алкенилалканоатов, содержат от двух до четырех атомов углерода (например, уксусная, пропионовая и масляная кислоты). Предпочтительными продуктами в способе являются VA, винилпропионат, винилбутират и аллилацетат. Наиболее предпочтительными материалами исходного сырья являются этилен и уксусная кислота, при этом VA является наиболее предпочтительным продуктом. Таким образом, настоящее изобретение является подходящим для использования при получении сложных эфиров карбоновых кислот с ненасыщенностью олефинового типа из соединения с ненасыщенностью олефинового типа, карбоновой кислоты и кислорода в присутствии катализатора. Несмотря на то, что в остальной части описания изобретения обсуждается исключительно VA, необходимо понимать, что катализаторы, способ получения катализаторов и способы производства являются в равной степени применимыми и к другим алкенилалканоатам, и описание не предполагает ограничения заявки изобретения вариантом VA. Если VA получают при использовании катализатора настоящего изобретения, то тогда поток газа, который содержит этилен, кислород или воздух, и уксусную кислоту пропускают через катализатор. Состав газового потока может варьироваться в широких пределах при учете наличия области воспламеняемости для потока продуктов. Например, молярное отношение между этиленом и кислородом может находиться в диапазоне от приблизительно 80:20 до приблизительно 98:2, молярное отношение между уксусной кислотой и этиленом может находиться в диапазоне от приблизительно 100:1 до приблизительно 1:100, предпочтительно, приблизительно от 10:1 до 1:10, а наиболее предпочтительно, от приблизительно 1:1 до приблизительно 1:8. Газовый поток также может содержать газообразный ацетат щелочного металла и/или инертные газы, такие как азот, диоксид углерода и/или насыщенные углеводороды. Температурами реакции, которые можно использовать, являются повышенные температуры, предпочтительно, температуры в диапазоне приблизительно 125-220°С. Используемым давлением может являться несколько пониженное давление, обычное давление или повышенное давление, предпочтительно, давление вплоть до приблизительно 20 атмосфер избыточного давления. В дополнение к реакторам с неподвижным слоем катализатора способы получения алкенилалканоатов и катализатор настоящего изобретения подходящим образом также могут быть использованы и в других типах реакции, например в реакторах с псевдоожиженным слоем катализатора. ПРИМЕРЫ Следующие далее примеры предлагаются только для иллюстрации и не предполагаются в роли ограничения. Количества растворителей и реагентов являются приблизительными. Атомное отношение Au/Pd можно пересчитать в массовое отношение Au/Pd и наоборот при использовании нижеследующих уравнений: атомное отношение Au/Pd=0,54* (массовое отношение Au/Pd) и массовое отношение Au/Pd=1,85 (атомное отношение Au/Pd). Восстановление можно сокращенно обозначить как “R” с последующим указанием температуры в °С, при которой проводят восстановление. Подобным же образом, прокаливание можно сокращенно обозначить как “С” с последующим указанием температуры в °С, при которой проводят прокаливание, в то время как стадию высушивания можно сокращенно обозначить как “dry”. Катализатор из примеров 1-11 можно получить так, как это описывается в примере, и подвергнуть испытанию в соответствии со следующей далее методикой, где друг с другом можно сопоставлять катализатор из примеров 1-7, и друг с другом можно сопоставлять катализатор из примеров 8-11. Результаты приведены, если они были доступны. Катализаторы из примеров подвергали испытаниям для определения их активности и селективности в отношении получения различных побочных продуктов при получении винилацетата в результате проведения реакции между этиленом, кислородом и уксусной кислотой. Для достижения этого приблизительно 60 мл катализатора, полученного так, как описано, помещали в корзину из нержавеющей стали при температуре, которую можно было измерять при помощи термопары как в верхней части, так и в нижней части корзины. Корзину помещали в емкостной реактор с постоянным перемешиванием Berty, относящийся к типу с рециркуляцией, и при помощи электрически обогреваемого кожуха выдерживали при температуре, которая обеспечивала достижение степени превращения кислорода, равной приблизительно 45%. Газовую смесь, образованную из приблизительно 50 нормальных литров (измеренных при нормальных температуре и давлении) этилена, приблизительно 10 нормальных литров кислорода, приблизительно 49 нормальных литров азота, приблизительно 50 г уксусной кислоты и приблизительно 4 мг ацетата калия, под давлением, равным приблизительно 12 атмосферам, пропускали через корзину, и катализатор выдерживали при данных условиях реакции в течение, по меньшей мере, 16 часов перед проведением двухчасового прогона работы системы, после которого реакцию завершали. Анализ продуктов проводили с использованием анализа по методу газовой хроматографии в реальном режиме времени в комбинации с анализом жидких продуктов в автономном режиме, проводимым благодаря конденсации потока продуктов приблизительно при 10°С с обеспечением проведения оптимального анализа конечных продуктов в виде диоксида углерода (СО2), тяжелых фракций (НЕ) и этилацетата (EtOAc), где данные результаты можно использовать для вычисления процентных селективностей (селективность в отношении получения CO2) в отношении получения данных материалов для каждого примера. Относительную активность в реакции, выраженную в виде коэффициента активности (активности), можно рассчитать на компьютере при использовании серии уравнений, которые устанавливают корреляцию между коэффициентом активности и температурой катализатора (во время реакции), степенью превращения кислорода и серией кинетических параметров для реакций, которые имеют место во время синтеза VA. В более общем случае коэффициент активности обычно является обратно пропорциональным температуре, необходимой для достижения постоянной степени превращения кислорода. Примеры родиевого катализатора Пример 1 Материал носителя, содержащего металлические палладий и родий, получали следующим образом: материал носителя в количестве 250 мл, состоящий из сфер диоксида кремния Sud Chemie КА-160, характеризующихся номинальным диаметром 7 мм, плотностью, приблизительно равной 0,569 г/мл, абсорбционной способностью, приблизительно равной 0,568 г Н2О/г носителя, площадью удельной поверхности в диапазоне приблизительно от 160 до 175 м2/г и объемом пор, равным приблизительно 0,68 мл/г, сначала импрегнировали до достижения начальной влажности при использовании 82,5 мл водного раствора натрий-тетрахлорпалладия (II) (Na2PdCl4) и тригидрита хлорида родия (RhCl3·3H2O), что было достаточным для получения приблизительно 7 г элементарного палладия и приблизительно 0,29 г элементарного родия на один литр катализатора, при атомном отношении между родием и палладием в диапазоне от приблизительно 0,01 до приблизительно 0,5. Носитель встряхивали в растворе в течение 5 минут для обеспечения полного впитывания раствора. После этого палладий и родий фиксировали на носителе в виде гидроксидов палладия (II) и родия (III) при использовании погружения с вращением в течение 2,5 часов приблизительно при 5 об/мин в ходе операции введения подвергнутого обработке носителя в контакт с 283 мл водного раствора гидроксида натрия, полученного из раствора NaOH/Н2О с концентрацией 50% (мас./мас.) в количестве 120% от того, количества, которое необходимо для превращения палладия и родия в их гидроксиды. Раствор с подвергнутого обработке носителя сливали, а носитель после этого промывали деионизованной водой и высушивали при 100°С в сушилке с псевдоожиженным слоем в течение 1,2 часа. Затем материал носителя, содержащий гидроксиды палладия и родия, импрегнировали водным раствором (81 мл), содержащим 1,24 г Au от NaAuCl4 и 2,71 г 50%-ного раствора NaOH (1,8 эквивалента по отношению к Au), при использовании способа с достижением начальной влажности, причем атомное отношение между золотом и палладием находится в диапазоне от приблизительно 0,50 до приблизительно 1,00. Гранулы, подвергнутые обработке с использованием NaOH, оставляли стоять в течение ночи для обеспечения осаждения соли Au с получением нерастворимого гидроксида. Гранулы тщательно промывали деионизованной водой (~5 часов) для удаления хлорид-ионов и после этого высушивали при 100°С в сушилке с псевдоожиженным слоем в течение 1,2 часа. После этого палладий-, родий- и золотосодержащий носитель прокаливали при 400°С в течение 2 часов на воздухе, а затем оставляли для естественного охлаждения до комнатной температуры. Палладий, родий и золото восстанавливали в результате приведения носителя в контакт с С2Н4 (1% в азоте) в паровой фазе при 150°С в течение 5 часов. В заключение, катализатор импрегнировали до достижения начальной влажности при использовании водного раствора 10 г ацетата калия в 81 мл Н2О и высушивали в сушилке с псевдоожиженным слоем при 100°С в течение 1,2 часа. Пример 2 Материал носителя, использующий гидроксиды палладия и родия, получали так, как это описывается в примере 1. После этого палладий- и родийсодержащий носитель прокаливали при 400°С в течение 2 часов на воздухе, а затем оставляли для естественного охлаждения до комнатной температуры. Затем подвергнутый прокаливанию материал носителя, содержащий гидроксиды палладия и родия, импрегнировали водным раствором (81 мл), содержащим 1,24 г Au от NaAuCl4 и 2,71 г 50%-ного раствора NaOH (1,8 эквивалента по отношению к Au), при использовании способа с достижением начальной влажности, при этом атомное отношение между родием и палладием находится в диапазоне от приблизительно 0,01 до приблизительно 0,5, а атомное отношение между золотом и палладием находится в диапазоне от приблизительно 0,50 до приблизительно 1,00. Гранулы, подвергнутые обработке с использованием NaOH, оставляли стоять в течение ночи для обеспечения осаждения соли Au с получением нерастворимого гидроксида. Гранулы тщательно промывали деионизованной водой (~5 часов) для удаления хлорид-ионов и после этого высушивали при 100°С в сушилке с псевдоожиженным слоем в течение 1,2 часа. Затем палладий, родий и золото восстанавливали в результате приведения носителя в контакт с С2Н4 (1% в азоте) в паровой фазе при 150°С в течение 5 часов. В заключение, катализатор импрегнировали до достижения начальной влажности при использовании водного раствора 10 г ацетата калия в 81 мл Н2О и высушивали в сушилке с псевдоожиженным слоем при 100°С в течение 1,2 часа. Пример 3 Материал носителя, содержащий гидроксиды палладия и родия, получали так, как это описывается в примере 1. После этого палладий- и родийсодержащий носитель прокаливали при 400°С в течение 2 часов на воздухе, а затем оставляли для естественного охлаждения до комнатной температуры. Затем подвергнутый прокаливанию материал носителя, содержащий гидроксиды палладия и родия, восстанавливали в результате приведения носителя в контакт с С2Н4 (1% в азоте) в паровой фазе при 150°С в течение 5 часов. После этого носитель, содержащий металлические палладий и родий, импрегнировали водным раствором (81 мл), содержащим 1,24 г Au от NaAuCl4 и 2,71 г 50%-ного раствора NaOH (1,8 эквивалента по отношению к Au), при использовании способа с достижением начальной влажности, так что атомное отношение между родием и палладием находится в диапазоне от приблизительно 0,01 до приблизительно 0,5, а атомное отношение между золотом и палладием находится в диапазоне от приблизительно 0,50 до приблизительно 1,00. Гранулы, подвергнутые обработке с использованием NaOH, оставляли стоять в течение ночи для обеспечения осаждения соли Au с получением нерастворимого гидроксида. Гранулы тщательно промывали деионизованной водой (~5 часов) для удаления хлорид-ионов и после этого высушивали при 100°С в сушилке с псевдоожиженным слоем в течение 1,2 часа. Затем палладий, родий и золото восстанавливали в результате приведения носителя в контакт с С2Н4 (1% в азоте) в паровой фазе при 150°С в течение 5 часов. В заключение, катализатор импрегнировали до достижения начальной влажности при использовании водного раствора 10 г ацетата калия в 81 мл Н2О и высушивали в сушилке с псевдоожиженным слоем при 100°С в течение 1,2 часа. Пример 4 Материал носителя, содержащий гидроксиды палладия и родия, получали так, как это описывается в примере 1. После этого палладий- и родийсодержащий носитель прокаливали при 400°С в течение 2 часов на воздухе, а затем оставляли для естественного охлаждения до комнатной температуры. Затем подвергнутый прокаливанию материал носителя, содержащий гидроксиды палладия и родия, восстанавливали в результате приведения носителя в контакт с С2Н4 (1% в азоте) в паровой фазе при 150°С в течение 5 часов. После этого носитель, содержащий металлические палладий и родий, импрегнировали водным раствором (81 мл), содержащим 1,1 г Au от KAuO2, при использовании способа с достижением начальной влажности, так что атомное отношение между родием и палладием находится в диапазоне от приблизительно 0,01 до приблизительно 0,5, а атомное отношение между золотом и палладием находится в диапазоне от приблизительно 0,50 до приблизительно 1,00. Гранулы затем высушивали при 100°С в сушилке с псевдоожиженным слоем в течение 1,2 часа. Затем палладий, родий и золото восстанавливали в результате приведения носителя в контакт с С2Н4 (1% в азоте) в паровой фазе при 150°С в течение 5 часов. В заключение, катализатор импрегнировали до достижения начальной влажности при использовании водного раствора 10 г ацетата калия в 81 мл Н2О и высушивали в сушилке с псевдоожиженным слоем при 100°С в течение 1,2 часа. Пример 5 Материал носителя, содержащий гидроксиды палладия и родия, получали так, как это описывается в примере 1. После этого палладий- и родийсодержащий носитель прокаливали при 400°С в течение 2 часов на воздухе, а затем оставляли для естественного охлаждения до комнатной температуры. Затем подвергнутый прокаливанию материал носителя, содержащий гидроксиды палладия и родия, импрегнировали водным раствором (81 мл), содержащим 1,1 г Au от KAuO2, при использовании способа с достижением начальной влажности, так что атомное отношение между родием и палладием находится в диапазоне от приблизительно 0,01 до приблизительно 0,5, а атомное отношение между золотом и палладием находится в диапазоне от приблизительно 0,50 до приблизительно 1,00. Гранулы затем высушивали при 100°С в сушилке с псевдоожиженным слоем в течение 1,2 часа. Затем палладий, родий и золото восстанавливали в результате приведения носителя в контакт с С2Н4 (1% в азоте) в паровой фазе при 150°С в течение 5 часов. В заключение, катализатор импрегнировали до достижения начальной влажности при использовании водного раствора 10 г ацетата калия в 81 мл Н2О и высушивали в сушилке с псевдоожиженным слоем при 100°С в течение 1,2 часа. Пример 6 Материал носителя, содержащий гидроксиды палладия и родия, получали так, как это описывается в примере 1. После этого палладий- и родийсодержащий носитель прокаливали при 400°С в течение 2 часов на воздухе, а затем оставляли для естественного охлаждения до комнатной температуры. Затем подвергнутый прокаливанию материал носителя, содержащий гидроксиды палладия и родия, восстанавливали в результате приведения носителя в контакт с С2Н4 (1% в азоте) в паровой фазе при 150°С в течение 5 часов. После этого носитель, содержащий металлические палладий и родий, импрегнировали водным раствором (81 мл), содержащим 1,1 г Au от KAuO2 и 10 г ацетата калия, при использовании способа с достижением начальной влажности, так что атомное отношение между родием и палладием находится в диапазоне от приблизительно 0,01 до приблизительно 0,5, а атомное отношение между золотом и палладием находится в диапазоне от приблизительно 0,50 до приблизительно 1,00. Гранулы затем высушивали при 100°С в сушилке с псевдоожиженным слоем в течение 1,2 часа. Пример 7 (сравнительный катализатор) Материал носителя, содержащего металлический палладий, получали следующим образом: материал носителя в количестве 250 мл, состоящий из сфер диоксида кремния Sud Chemie KA-160, характеризующихся номинальным диаметром 7 мм, плотностью, приблизительно равной 0,569 г/мл, абсорбционной способностью, приблизительно равной 0,568 г Н2О/г носителя, площадью удельной поверхности в диапазоне приблизительно от 160 до 175 м2/г и объемом пор, равным приблизительно 0,68 мл/г, сначала импрегнировали до достижения начальной влажности при использовании 82,5 мл водного раствора натрий-тетрахлорпалладия (II) (Na2PdCl4), что было достаточным для получения приблизительно 7 г элементарного палладия на один литр катализатора. Носитель встряхивали в растворе в течение 5 минут для обеспечения полного впитывания раствора. После этого палладий фиксировали на носителе в виде гидроксидов палладия (II) при использовании погружения с вращением в течение 2,5 часов приблизительно при 5 об/мин в ходе операции введения подвергнутого обработке носителя в контакт с 283 мл водного раствора гидроксида натрия, полученного из раствора NaOH/Н2О с концентрацией 50% (мас./мас.) в количестве 110% от необходимого для превращения палладия в его гидроксид. Раствор с подвергнутого обработке носителя сливали, а носитель после этого промывали деионизованной водой и высушивали при 100°С в сушилке с псевдоожиженным слоем в течение 1,2 часа. Затем материал носителя, содержащий гидроксид палладия, импрегнировали водным раствором (81 мл), содержащим 1,24 г Au от NaAuCl4 и 2,71 г 50%-ного раствора NaOH (1,8 эквивалента по отношению к Au), при использовании способа с достижением начальной влажности. Гранулы, подвергнутые обработке с использованием NaOH, оставляли стоять в течение ночи для обеспечения осаждения соли Аи с получением нерастворимого гидроксида. Гранулы тщательно промывали деионизованной водой (~5 часов) для удаления хлорид-ионов и после этого высушивали при 100°С в сушилке с псевдоожиженным слоем в течение 1,2 часа. После этого палладий- и золотосодержащий носитель восстанавливали в результате приведения носителя в контакт с С2Н4 (1% в азоте) в паровой фазе при 150°С в течение 5 часов. В заключение, катализатор импрегнировали до достижения начальной влажности при использовании водного раствора 10 г ацетата калия в 81 мл H2O и высушивали в сушилке с псевдоожиженным слоем при 100°С в течение 1,2 часа. Таблица 1 демонстрирует сопоставление селективности в отношении получения СО2 и активности для катализатора из примеров 1 и 7.
Примеры слоистого носителя Пример 8 40 г ZrO2 (RC-100, поставляемого от компании DKK) прокаливали при 650°С в течение 3 часов. Получающийся в результате материал характеризовался площадью удельной поверхности, равной 38 м2/г согласно измерениям по методу БЭТ. Материал в течение 6 часов измельчали в шаровой мельнице в присутствии 120 мл деионизованной воды. Золь перемешивали с 22,5 г связующего в виде ацетата циркония, поставляемого от компании DKK (ZA-20), и распыляли на 55 г сфер из бентонита КА-160 с наружным диаметром ~7,5 мм. Гранулы с нанесенным покрытием подвергали прокаливанию в течение 3 часов при 600°С. Наблюдение через микроскоп свидетельствовало об образовании однородной поверхностной оболочки с толщиной 250 мкм. Пример 9 20 г ZrO2 (XZ16075, площадь удельной поверхности, равная 55 м2/г согласно измерениям по методу БЭТ), импрегнировали раствором Pd(NO3)2 (Aldrich) с получением величины Pd-наполнения, равной 39 мг/г ZrO2. Импрегнированный материал высушивали и прокаливали при 450°С в течение 4 часов. Материал в течение 4 часов измельчали в шаровой мельнице в присутствии 60 мл деионизованной воды, перемешивали с 11 г связующего (ZA-20) и распыляли на 30 г сфер из бентонита КА-160. Гранулы подвергали прокаливанию в течение 3 часов при 450°С. Данная методика в результате приводила к образованию прочной однородной поверхностной оболочки с толщиной 160 мкм. Пример 10 Гранулы из примера 8 импрегнировали раствором ацетата калия с получением величины наполнения 40 мг КОАс/мл КА-160, высушивали и прокаливали при 300°С в течение 4 часов. После этого на данные гранулы распыляли раствор, содержащий 9,4 мМ Pd (от Pd(NH3)4(ОН)2, поставляемого от компании Heraeus) и 4,7 мМ Au (от раствора с концентрацией 1М, полученного при растворении Au(OH)3 “Alfa” в 1,6М КОН). Материал восстанавливали при помощи смеси: 5% Н2, 95% N2 при 200°С в течение 4 часов. Гранулы измельчали и подвергали испытанию в микрореакторе с неподвижным слоем катализатора в условиях, описанных в экспериментальной секции. Достигали значений селективности в отношении получения СО2 ~6% и степени превращения кислорода 45%. Пример 11 (сравнительный катализатор) В данном случае в качестве сравнительного катализатора использовали тот же самый катализатор, что и полученный в примере 7. Таблица 2 демонстрирует сопоставление селективности в отношении получения СО2 и активности для катализатора из примеров 9-11.
Примеры материала носителя на основе диоксида циркония и предшественников, не содержащих хлоридов Для данного набора примеров использовали следующую далее общую методику. Катализаторы, содержащие материал носителя на основе диоксида циркония, получали следующим далее образом: различные формованные носители катализаторов измельчали и просеивали. Материалы носителей на основе диоксида циркония поставляли от компаний NorPro (XZ16052 и XZ16075), DKK и MEI. Материалы носителей на основе диоксида кремния поставляли от компаний Degussa и Sud Chemie. Ситовую фракцию 180-425 мкм импрегнировали (либо одновременно, либо последовательно с промежуточной стадией высушивания при 110°С и необязательно с промежуточной стадией прокаливания) до достижения начальной влажности при использовании раствора предшественника Pd и Au, необязательно прокаливали на воздухе, восстанавливали при использовании пластового газа 5% H2/N2, подвергали последующему импрегнированию раствором КОАс, высушивали при 100°С в атмосфере N2 и подвергали испытаниям в 8×6-многоканальном реакторе с неподвижным слоем катализатора. В качестве предшественника Au использовали раствор Au(ОН)3 в КОН. В качестве предшественников Pd использовали водные растворы Pd(NH3)4(ОН)2, Pd(NH3)2(NO2)2, Pd(NH3)4(NO3)2 и Pd(NO3)2. Сравнительный катализатор, содержащий материал носителя на основе диоксида кремния, получали следующим далее образом. Материал носителя, содержащего металлические палладий и родий, получали следующим образом: материал носителя в количестве 250 мл, состоящий из сфер диоксида кремния Sud Chemie KA-160, характеризующихся номинальным диаметром 7 мм, плотностью, приблизительно равной 0,569 г/мл, абсорбционной способностью, приблизительно равной 0,568 г Н2О/г носителя, площадью удельной поверхности в диапазоне приблизительно от 160 до 175 м2/г и объемом пор, равным приблизительно 0,68 мл/г, сначала импрегнировали до достижения начальной влажности при использовании 82,5 мл водного раствора натрий-тетрахлорпалладия (II) (Na2PdCl4), что было достаточным для получения приблизительно 7 г элементарного палладия на один литр катализатора. Носитель встряхивали в растворе в течение 5 минут для обеспечения полного впитывания раствора. После этого палладий фиксировали на носителе в виде гидроксидов палладия (II) при использовании погружения с вращением в течение 2,5 часов приблизительно при 5 об/мин в ходе операции введения подвергнутого обработке носителя в контакт с 283 мл водного раствора гидроксида натрия, полученного из раствора NaOH/Н2О с концентрацией 50% (мас./мас.) в количестве 110% от необходимого для превращения палладия в его гидроксид. Раствор с подвергнутого обработке носителя сливали, а носитель после этого промывали деионизованной водой и высушивали при 100°С в сушилке с псевдоожиженным слоем в течение 1,2 часа. Затем материал носителя, содержащий гидроксид палладия, импрегнировали водным раствором (81 мл), содержащим 1,24 г Au от NaAuCl4 и 2,71 г 50%-ного раствора NaOH (1,8 эквивалента по отношению к Au), при использовании способа с достижением начальной влажности. Гранулы, подвергнутые обработке с использованием NaOH, оставляли стоять в течение ночи для обеспечения осаждения соли Au с получением нерастворимого гидроксида. Гранулы тщательно промывали деионизованной водой (~5 часов) для удаления хлорид-ионов и после этого высушивали при 100°С в сушилке с псевдоожиженным слоем в течение 1,2 часа. После этого палладий- и золотосодержащий носитель восстанавливали в результате приведения носителя в контакт с С2Н4 (1% в азоте) в паровой фазе при 150°С в течение 5 часов. В заключение, катализатор импрегнировали до достижения начальной влажности при использовании водного раствора 10 г ацетата калия в 81 мл Н2О и высушивали в сушилке с псевдоожиженным слоем при 100°С в течение 1,2 часа. Перед проведением испытаний катализатор измельчали и просеивали. Использовали ситовую фракцию с диапазоном размеров 180-425 мкм. Были разработаны библиотеки катализаторов с массивами в виде 8 строк × 6 столбцов стеклянных емкостей, и стойку с 36 стеклянными емкостями устанавливали на прибор для встряхивания и перемешивания, и перемешивание проводили при одновременном дозировании растворов предшественников металлов при использовании роботов для дозирования жидкостей Cavro Степень введения КОАс приводили в виде граммов КОАс на один литр объема катализатора или в виде мкмоль КОАс на 0,4 мл носителя. Для описания степени введения Au относительное атомное отношение между Au и Pd приводили в виде Au/Pd. Степень введения Pd указывали в виде мг Pd на 0,4 мл объема носителя (то есть абсолютное количество Pd в реакционной емкости). Протокол испытаний использовал линейное увеличение температуры от 145°С до 165°С с приращением в 5°С в условиях фиксированной объемной скорости 175% (при 1,5 мг Pd на 0,4 мл носителя). 100%-ную объемную скорость определяли в виде следующих далее потоков: 5,75 стандартного кубического сантиметра азота в минуту, 0,94 стандартного кубического сантиметра кислорода в минуту, 5,94 стандартного кубического сантиметра этилена в минуту и 5,38 микролитров уксусной кислоты в минуту через каждую из 48 емкостей с катализаторами (из которых все характеризовались внутренним диаметром, приблизительно равным 4 мм). Строили графическую зависимость селективности в отношении получения СО2 от степени превращения кислорода, подбирали прямую, согласующуюся с экспериментальными данными, и рассчитывали (в большинстве случаев интерполировали) селективность в отношении получения СО2 при степени превращения кислорода 45%, представленную в приведенных далее свободных таблицах эксплуатационных характеристик. Также приводили температуру при степени превращения кислорода 45%, рассчитанную из линейного увеличения Т (прямые, описывающие зависимость селективности в отношении получения СО2 от степени превращения кислорода, как функция температуры реакции). Чем меньше будет данная рассчитанная температура, тем выше будет активность катализатора. Выход в граммах в час на единицу объема катализатора (STY; г VA, полученного на один мл объема катализатора за один час) при степени превращения кислорода 45% представляет собой меру производительности катализатора. Пример 12 400 мкл носителей на основе ZrO2 XZ16075 (55 м2/г, без дополнительной обработки) и XZ16052 (предварительное прокаливание при 650°С/2 часа для уменьшения площади удельной поверхности до 42 м2/г) импрегнировали, используя 3 различных раствора Pd, до достижения начальной влажности, высушивали при 110°С в течение 5 часов, импрегнировали, используя KAuO2 (маточный раствор, содержащий 0,97М Au), до достижения начальной влажности, высушивали при 110°С в течение 5 часов, восстанавливали при 350°С в течение 4 часов при использовании пластового газа 5% H2/N2, подвергали последующему импрегнированию, используя КОАс, и высушивали при 110°С в течение 5 часов. После этого образцы Pd/Au/ZrO2 (поверхностные оболочки) разбавляли в пропорции 1/9,3, используя разбавитель для КА160 (с предварительным введением 40 г/л КОАс), то есть в реакционные емкости загружали 43 мкл поверхностной оболочки на основе Pd/Au/ZrO2 и 357 мкл разбавителя (совокупный объем неподвижного слоя 400 мкл). Степень введения Pd составляла 14 мг Pd в 400 мкл поверхностной оболочки из ZrO2 (или 14·43/400=14/9,3=1,5 мг Pd в реакционной емкости для всех элементов библиотеки). Предшественники Pd представляли собой Pd(NH3)2(NO2)2 в столбцах 1 и 4, Pd(NH3)4(ОН)2 в столбцах 2 и 5, Pd(NH3)4(NO3)2 в столбцах 3 и 6. Au/Pd=0,3 в строке 2 и строке 5, Au/Pd=0,6 в строке 3, Au/Pd=0,9 в строке 4, строке 6 и строке 7. Степень введения КОАс составляла 114 мкмоль в строках 2, 3, 5 и 147 мкмоль в строках 4, 6, 7. Сравнительный катализатор на основе диоксида кремния загружали в строке 1. Библиотеку подвергали испытанию при использовании протокола проведения испытаний с линейным изменением Т при фиксированной SV. Результаты проведения испытаний обобщены в таблице 3.
Пример 13 400 мкл носителей на основе ZrO2 XZ16075 (55 м2/г, без дополнительной обработки) и XZ16052 (предварительное прокаливание при 650°С/2 часа для уменьшения площади удельной поверхности до 42 м2/г) импрегнировали, используя Pd(NH3)4(ОН)2 (маточный раствор, содержащий 1,117М Pd), до достижения начальной влажности, прокаливали при 350°С в течение 4 часов на воздухе, импрегнировали, используя KAuO2 (маточный раствор, содержащий 0,97М Au), до достижения начальной влажности, высушивали при 110°С в течение 5 часов, восстанавливали при 350°С в течение 4 часов при использовании пластового газа 5% H2/N2, подвергали последующему импрегнированию, используя КОАс, и высушивали при 110°С в течение 5 часов. После этого образцы Pd/Au/ZrO2 (поверхностные оболочки) разбавляли в пропорции 1/12, используя разбавитель для КА160 (с предварительным введением 40 г/л КОАс), то есть, в реакционные емкости загружали 33,3 мкл катализатора на основе Pd/Au/ZrO2 и 366,7 мкл разбавителя (совокупный объем неподвижного слоя 400 мкл). Структура библиотеки и составы элементов библиотеки представляли собой нижеследующее: ZrO2 XZ16075 в столбцах 1-3 (левая половина библиотеки) и ZrO2 XZ16052 (650°С) в столбцах 4-6 (правая половина библиотеки). Степень введения Pd составляла 18 мг Pd в 400 мкл поверхностной оболочки из ZrO2 (или 18·33/400=18/12 мг Pd в реакционной емкости) в ячейке G2, столбце 3 (ячейки B3-G3), ячейке G5, столбце 6 (ячейки B6-G6); 10 мг Pd в 400 мкл поверхностной оболочки из ZrO2 (или 10·33/400=10/12 мг Pd в реакционной емкости) в столбце 1 (ячейки A1-G1) и столбце 4 (ячейки A4-G4); 14 мг Pd в 400 мкл поверхностной оболочки из ZrO2 (или 14·33/400=14/12 мг Pd в реакционной емкости) в столбце 2 (ячейки B2-F2) и столбце 5 (ячейки B5-F5). Au/Pd=0,3 в строке 2 и строке 5, Au/Pd=0,5 в строке 3 и строке 6, Au/Pd=0,7 в строке 4 и строке 7 (за исключением ячеек А1, А4, G2, G5, где Au/Pd составляло 0,3). Степень введения КОАс составляла 114 мкмоль (за исключением ячеек D3, G3, D6, G6, где степень введения КОАс составляла 147 мкмоль). Сравнительный катализатор на основе диоксида кремния загружали в строке 1. Библиотеку подвергали испытанию при использовании протокола проведения испытаний с линейным изменением Т при фиксированной SV. Результаты проведения испытаний обобщены в таблице 4.
Пример 14 Носитель на основе ZrO2 (поставляемый от компании NorPro, XZ16075, ситовая фракция 180-425 мкм, плотность 1,15 г/мл, объем пор 475 мкл/г, площадь удельной поверхности 55 м2/г согласно измерению по методу БЭТ) импрегнировали раствором предшественника Pd(NO3)2 до достижения начальной влажности, высушивали при 110°С, прокаливали при 250°С (столбцы 1-2), 350°С (столбцы 3-4), 450°С (столбцы 5-6) на воздухе, импрегнировали раствором KAuO2 (полученным в результате растворения Au(ОН)3 в КОН), высушивали при 110°С, восстанавливали при 350°С в течение 4 часов при использовании пластового газа 5% H2/N2 и подвергали последующему импрегнированию раствором КОАс. Библиотека имела градиент КОАс от 25 до 50 г/л на участке от строки 2 до строки 7. Степень введения Pd составляла 1,5 мг Pd на 0,4 мл носителя. Выбирали две различных степени введения Au (Au/Pd=0,5 в столбцах 1, 3, 5 и Au/Pd=0,7 в столбцах 2, 4, 6). Сравнительный катализатор на основе диоксида кремния загружали в строке 1. Библиотеку подвергали испытанию при использовании протокола проведения испытаний с линейным изменением Т при фиксированной SV в реакторе получения VA MCFB48. Результаты проведения испытаний обобщены в таблице 5.
Пример 15 Носитель на основе ZrO2 (поставляемый от компании NorPro, XZ16075, ситовая фракция 180-425 мкм, плотность 1,15 г/мл, объем пор 575 мкл/г, площадь удельной поверхности 55 м2/г согласно измерению по методу БЭТ) импрегнировали раствором предшественника Pd(NO3)2 до достижения начальной влажности, высушивали при 110°С, прокаливали при 450°С на воздухе, импрегнировали раствором KAuO2 (полученным в результате растворения Au(ОН)3 в КОН), высушивали при 110°С, восстанавливали при 200°С (столбцы 1-2), 300°С (столбцы 3-4) или 400°С (столбцы 5-6) при использовании пластового газа 5% H2/N2 и подвергали последующему импрегнированию раствором КОАс. Библиотека имела градиент КОАс от 15 до 40 г/л на участке от строки 2 до строки 7. Степень введения Pd составляла 1,5 мг Pd на 0,4 мл носителя. Выбирали две различных степени введения Au (Au/Pd=0,5 в столбцах 1, 3, 5 и Au/Pd=0,7 в столбцах 2, 4, 6). Сравнительный катализатор на основе диоксида кремния загружали в строке 1. Библиотеку подвергали испытанию при использовании протокола проведения испытаний с линейным изменением Т при фиксированной SV в реакторе получения VA MCFB48. Результаты проведения испытаний обобщены в таблице 6.
Кроме того, необходимо понимать, что функции или структуры нескольких компонентов или стадий можно комбинировать с получением одного компонента или одной стадии, или функции или структуры одной стадии или одного компонента можно разделить между несколькими стадиями или компонентами. Настоящее изобретение предусматривает все данные комбинации. Если только не будет указано другого, то размеры и геометрии различных структур, описанных в настоящем документе, не предполагают ограничивать собой изобретение, и возможными являются и другие размеры или геометрии. Несколько структурных компонентов или стадий может представлять одна интегрированная структура или стадия. В альтернативном варианте одну интегрированную структуру или стадию можно разделить на несколько отдельных компонентов или стадий. В дополнение к этому, несмотря на то, что признак настоящего изобретения может описываться в контексте только одного из проиллюстрированных вариантов реализации, такой признак можно комбинировать с одним или несколькими другими признаками других вариантов реализации для любой данной сферы применения. Кроме этого, исходя из вышеизложенного, необходимо понимать, что получение уникальных структур в настоящем изобретении и их использование также составляют способы, соответствующие настоящему изобретению. Объяснения и иллюстрации, представленные в настоящем документе, предназначены для ознакомления других специалистов в соответствующей области с изобретением, его принципами и его практическим применением. Специалисты в соответствующей области могут адаптировать и использовать изобретение во множестве его форм, которые могут наилучшим образом соответствовать требованиям конкретного варианта использования. В соответствии с этим, конкретные предложенные варианты реализации настоящего изобретения не предполагаются как исчерпывающие или ограничивающие изобретение. Поэтому объем изобретения необходимо определять не ссылкой на приведенное выше описание, но вместо этого его необходимо определять ссылкой на прилагаемую формулу изобретения с одновременным включением полного объема эквивалентов, на которые такая формула изобретения будет давать право. Описания всех статей и ссылок, в том числе патентных заявок и публикаций, для всех целей включаются в настоящий документ для справки.
Формула изобретения
1. Способ получения катализатора или прокатализатора, подходящих для содействия получению алкенилалканоатов, включающий: 2. Способ по п.1, где стадия введения в контакт включает совместное импрегнирование палладий- и родийсодержащими предшественниками с последующим импрегнированием золотосодержащим предшественником. 3. Способ по любому из пп.1 или 2, где стадия введения в контакт включает совместное импрегнирование палладий-, родий- и золотосодержащими предшественниками. 4. Способ по п.1, где стадию прокаливания проводят до импрегнирования золотосодержащим предшественником. 5. Способ по п.1, дополнительно включающий, по меньшей мере, одну стадию фиксации для фиксации, по меньшей мере, одного из предшественников при помощи фиксирующего вещества. 6. Способ по п.1, где порядок стадий в способе включает: 7. Способ по п.1, где порядок стадий в способе включает: 8. Способ по п.1, где порядок стадий в способе включает: 9. Способ по п.1, дополнительно включающий фиксацию золотосодержащего предшественника при помощи фиксирующего вещества. 10. Способ по п.1, где растворимый в воде золотосодержащий предшественник является основным раствором, так что стадия импрегнирования в результате приводит к получению зафиксированного золота. 11. Способ по п.9, где стадия фиксации включает одновременную фиксацию палладия, родия и золота. 12. Способ по любому из пп.7 и 8, где стадия фиксации включает фиксацию палладия и родия отдельно от фиксации золота. 13. Способ по п.1, где стадия прокаливания включает нагревание введенного в контакт материала носителя при температуре в диапазоне от приблизительно 200°С до приблизительно 700°С. 14. Способ по п.3, где стадия импрегнирования включает импрегнирование материала носителя, по меньшей мере, одним, по существу, не содержащим хлоридов растворимым в воде палладий-, родий- или золотосодержащим предшественником. 15. Способ по п.14, где стадия импрегнирования включает импрегнирование до достижения начальной влажности. 16. Способ по п.15, где за стадией импрегнирования следует подвод тепла для удаления избыточной жидкости. 17. Способ по п.1, где материал носителя содержит диоксид кремния. 18. Способ по п.1, где ацетатом щелочного металла является ацетат калия. 19. Способ по п.1, где ацетат щелочного металла присутствует в количестве в диапазоне приблизительно от 10 до 70 г на один литр катализатора. 20. Способ по п.1, где стадии способа в результате приводят к получению коркового катализатора или прокатализатора, катализатора или прокатализатора со структурой активной зоны типа «желток в яйце», катализатора или прокатализатора со структурой активной зоны типа «белок в яйце» или катализатора или прокатализатора с активной зоной по всему материалу. 21. Способ по п.1, где стадия введения в контакт включает введение в контакт родия и палладия при атомном отношении между родием и палладием в диапазоне от приблизительно 0,01 до приблизительно 0,5. 22. Способ по п.1, где стадия введения в контакт включает введение в контакт от приблизительно 1 до приблизительно 10 г палладия и от приблизительно 0,5 до приблизительно 10 г золота на один литр катализатора, при этом количество золота находится в диапазоне от приблизительно 10 до приблизительно 125 мас.% при расчете на массу палладия. 23. Способ по п.22, где атомное отношение между золотом и палладием находится в диапазоне от приблизительно 0,50 до приблизительно 1,00. 24. Каталитическая композиция для получения алкенилалканоатов, содержащая: 25. Композиция по п.24, где материал носителя содержит диоксид кремния. 26. Композиция по п.24, где палладий, родий и золото подвергают восстановлению. 27. Композиция по п.24, где перед восстановлением золото не прокаливают. 28. Композиция по п.24, где золото не восстанавливают. 29. Композиция по п.24, где катализатор или прокатализатор содержат родий и палладий при атомном отношении между родием и палладием в диапазоне от приблизительно 0,01 до приблизительно 0,5. 30. Композиция по п.24, где катализатор или прокатализатор содержат от приблизительно 1 до приблизительно 10 г палладия и от приблизительно 0,5 до приблизительно 10 г золота на один литр катализатора, при этом количество золота находится в диапазоне от приблизительно 10 до приблизительно 125 мас.% при расчете на массу палладия. 31. Композиция по п.30, где атомное отношение между золотом и палладием находится в диапазоне от приблизительно 0,50 до приблизительно 1,00. 32. Композиция по п.24, где ацетатом щелочного металла является ацетат калия. 33. Композиция по п.24, где ацетат щелочного металла присутствует в количестве в диапазоне приблизительно от 10 до 70 г на один литр катализатора. 34. Композиция по п.24, где катализатор или прокатализатор включают корковый катализатор или прокатализатор, катализатор или прокатализатор со структурой активной зоны типа «желток в яйце», катализатор или прокатализатор со структурой активной зоны типа «белок в яйце» или катализатор или прокатализатор с активной зоной по всему материалу. 35. Способ получения алкенилалканоатов, включающий: 36. Способ по п.35, где алкеном является этилен, алкановой кислотой является уксусная кислота, а окислителем является кислородсодержащий газ. 37. Способ либо по п.35, либо по п.36, где катализатор или прокатализатор содержат родий и палладий при атомном отношении между родием и палладием в диапазоне от приблизительно 0,01 до приблизительно 0,5. 38. Способ по п.35, где катализатор или прокатализатор содержат от приблизительно 1 до приблизительно 10 г палладия и от приблизительно 0,5 до приблизительно 10 г золота на один литр катализатора, при этом количество золота находится в диапазоне от приблизительно 10 до приблизительно 125 мас.% при расчете на массу палладия. 39. Способ по п.38, где атомное отношение между золотом и палладием находится в диапазоне от приблизительно 0,50 до приблизительно 1,00. 40. Способ по п.35, где материал носителя содержит диоксид кремния. 41. Способ по п.35, где катализатор или прокатализатор представляют собой корковый катализатор или прокатализатор.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
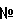 60/530937, поданной 19 декабря 2003 года, которая, таким образом, включается в настоящий документ для справки.
60/530937, поданной 19 декабря 2003 года, которая, таким образом, включается в настоящий документ для справки. СН3СООСН=СН2+Н2О.
СН3СООСН=СН2+Н2О.
 . 0,4 мл носителя использовали для каждого элемента библиотеки, для синтеза в стеклянной емкости, а также загружали в каждую реакционную емкость.
. 0,4 мл носителя использовали для каждого элемента библиотеки, для синтеза в стеклянной емкости, а также загружали в каждую реакционную емкость.