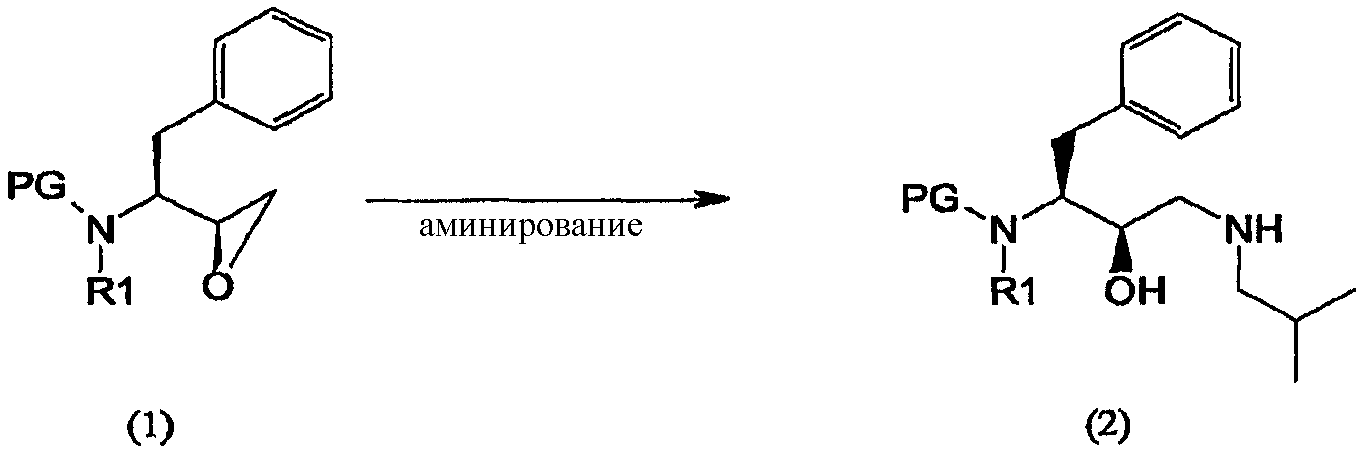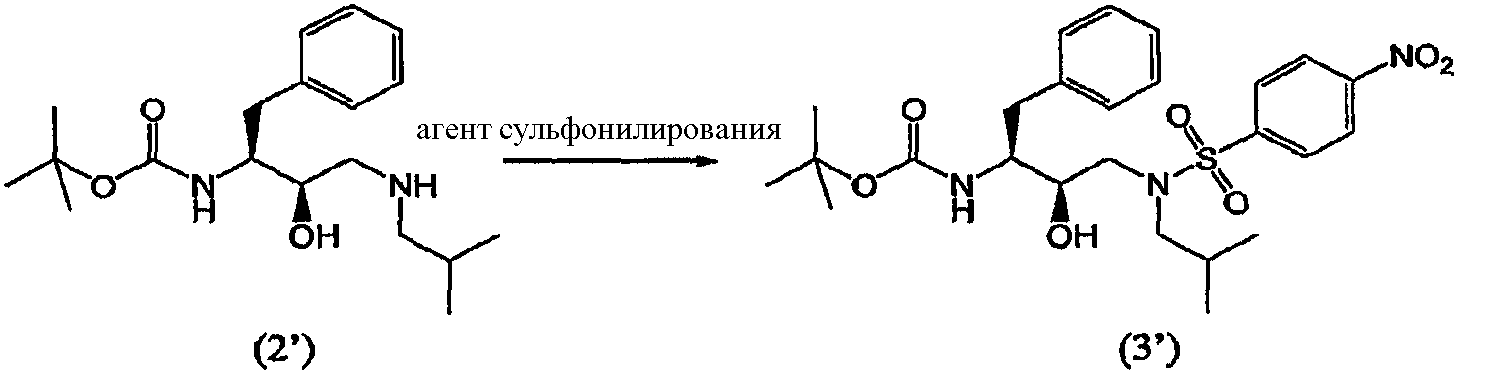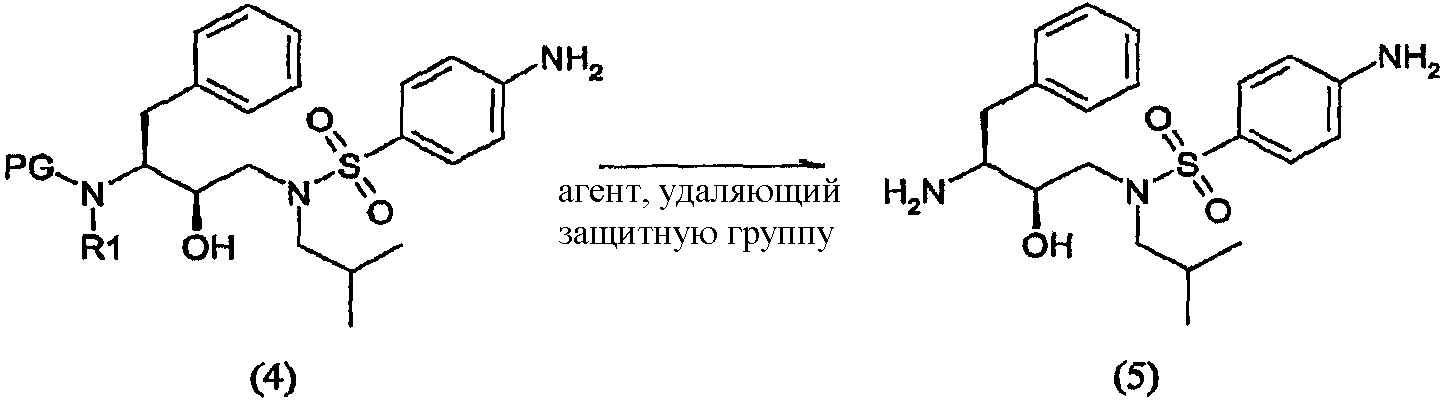Патент на изобретение №2376308
|
||||||||||||||||||||||||||
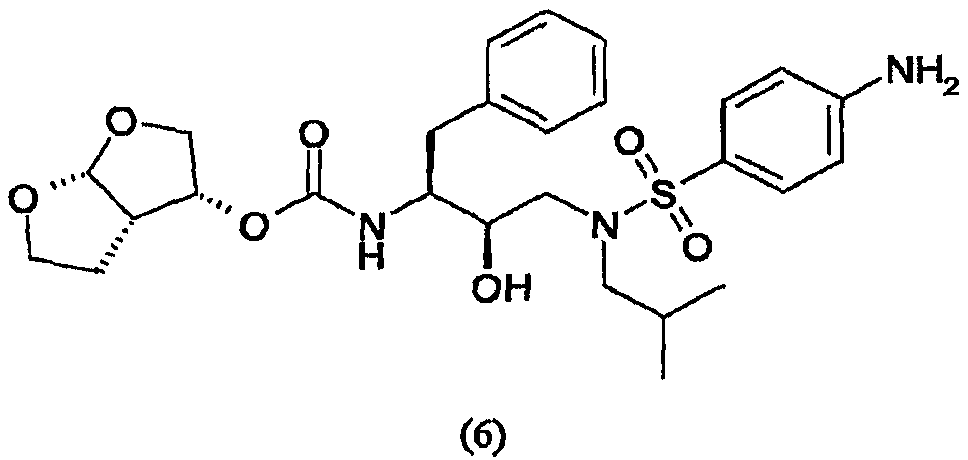
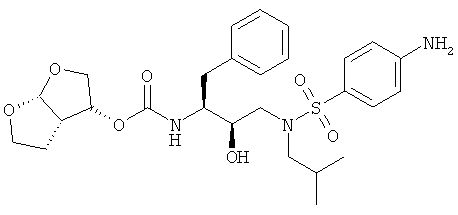
(54) СПОСОБ ПОЛУЧЕНИЯ (3R, 3aS,6aR)-ГЕКСАГИДРОФУРО[2,3-b]ФУРАН-3-ИЛ(1S,2R)-3-[[(4-АМИНОФЕНИЛ)СУЛЬФОНИЛ](ИЗОБУТИЛ)АМИНО]-1-БЕНЗИЛ-2-ГИДРОКСИПРОПИЛКАРБАМАТА
(57) Реферат:
Изобретение относится к способу получения (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ил(1S,2R)-3-[[(4-аминофенил)сульфонил](изобутил)амино]-1-бензил-2-гидроксипропилкарбамата, который осуществляют с применением промежуточного соединения 4-амино-N-[(2R,3S)-3-амино-2-гидрокси-4-фенилбутил]-N-(изобутил)бензолсульфонамида. (3R,3аS,6аR)-Гексагидрофуро[2,3-b]фуран-3-ил(1S,2R)-3-[[(4-аминофенил)сульфонил](изобутил)амино]-1-бензил-2-гидроксипропилкарбамат применим, в частности, как ингибитор протеазы ВИЧ. Технический результат – упрощение процесса за счет исключения необходимости выделения фенилэтинила. 13 з.п. ф-лы.
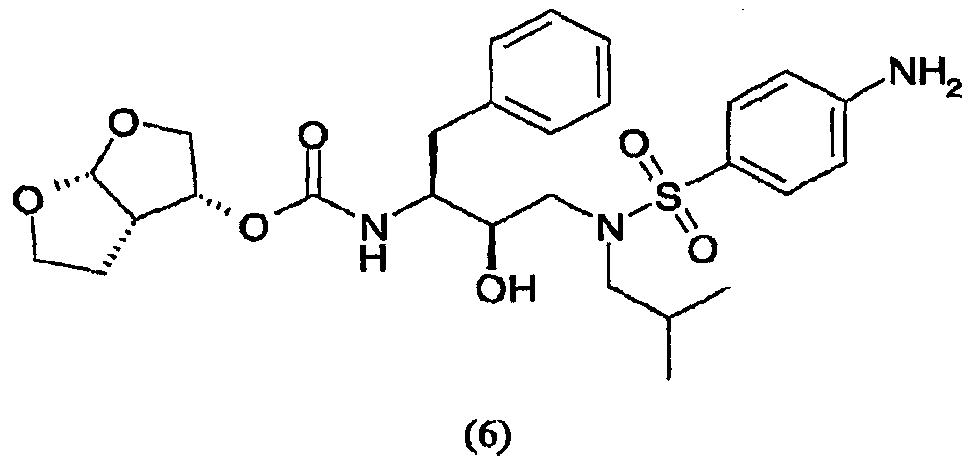
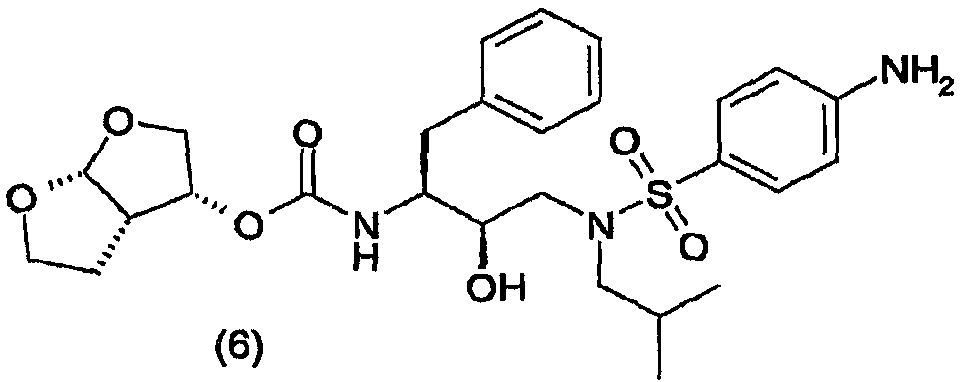
Область техники, к которой относится изобретение Настоящее изобретение относится к способу получения (3R,3аS,6аR)-гексагидрофуро[2,3-b]фуран-3-ил(1S,2R)-3-[[(4-аминофенил)сульфонил](изобутил)амино]-1-бензил-2-гидроксипропилкарбамата, а также промежуточных соединений для применения в указанном способе. В частности, изобретение относится к способам получения (3R,3аS,6аR)-гексагидрофуро[2,3-b]фуран-3-ил(1S,2R)-3-[[(4-аминофенил)сульфонил](изобутил)амино]-1-бензил-2-гидроксипропилкарбамата, которые осуществляют с применением промежуточного соединения 4-амино-N-[(2R,3S)-3-амино-2-гидрокси-4-фенилбутил]-N-изобутилбензолсульфонамида, которые можно применять в промышленном масштабе. Уровень техники Вирус, вызывающий синдром приобретенного иммунодефицита (СПИД), известен под разными названиями, в том числе как Т-лимфоцитарный вирус III (HTLV-III), или лимфаденопатии-ассоциированный вирус (LAV), или вирус СПИДа (ARV), или вирус иммунодефицита человека (ВИЧ). До настоящего времени идентифицированы два различных семейства, т.е. ВИЧ-1 и ВИЧ-2. Далее в описании аббревиатура ВИЧ будет использоваться для общего обозначения таких вирусов. Одним из определяющих этапов в жизненном цикле ретровирусов является процессинг белков-предшественников ретровирусной протеазой. Например, во время цикла репликации вируса ВИЧ продукты транскрипции генов gag и gag-pol транслируются как белки, которые затем процессируются кодированной вирусом протеазой с образованием ферментов и структурных белков сердцевины вируса. Чаще всего белки-предшественники gag процессируются в коровые белки, а белки-предшественники pol процессируются в вирусные ферменты, например обратную транскриптазу и ретровирусную протеазу. Правильный процессинг белков-предшественников ретровирусной протеазой необходим для сборки инфекционных вирионов, что таким образом делает ретровирусную протеазу привлекательной мишенью для противовирусной терапии. В частности, протеаза ВИЧ является привлекательной мишенью для лечения ВИЧ. На рынке и в разработке имеется несколько ингибиторов протеаз. Гидроксиэтиламиносульфонамидные ингибиторы протеазы ВИЧ, например 4-аминобензолгидроксиэтиламиносульфонамиды, описаны как обладающие благоприятными фармакологическими и фармакокинетическими свойствами против вируса ВИЧ дикого типа и мутантов. Типичным представителем 4-аминобензолгидроксиэтиламиносульфонамидного класса ингибиторов протеаз является ампренавир. Способ синтеза ампренавира описан в WO 99/48885 (Glaxo Group Ltd.). 4-Аминобензолгидроксиэтиламиносульфонамиды также можно получить согласно процедурам, описанным в ЕР 715618, WO 99/67417, US 6248775 и в Bioorganic and Chemistry Letters, Vol.8, pp. 687-690, “Potent HIV protease inhibitors incorporating high-affinity P2-ligands and (R)-(hydroxyethylamino) sulfonamide isostere”, и все указанные работы включены в данное описание в качестве ссылок. В частности, описание (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ил(1S,2R)-3-[[(4-аминофенил)сульфонил](изобутил)амино]-1-бензил-2-гидроксипропилкарбамата, называемого в данном описании соединением (6), и способы его получения можно найти в WO 99/67417 (USA, The Secretary, Dpt. of Health and Human Services) и в РСТ/ЕР 03/50176 (Tibotec N.V.). WO 03/057665 (Ajinomoto K.K.) относится к способу получения кристаллов бензолсульфонамидных производных. В частности, описывается кристаллизация (2R,3S)-N-(3-амино-2-гидрокси-4-фенилбутил)-N-изобутил-4-аминобензолсульфонамида, который является промежуточным соединением, представляющим интерес для получения (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ил(1S,2R)-3-[[(4-аминофенил)сульфонил](изобутил)амино]-1-бензил-2-гидроксипропилкарбамата. Указанное интересующее промежуточное соединение получают, как раскрывается, исходя из (2S,3S)-3-бензилоксикарбониламино-1,2-эпокси-4-фенилбутана, во взаимодействие с которым вводят изобутиламин, с последующим сочетанием с п-нитробензолсульфонилхлоридом с образованием (2R,3S)-N-(3-бензилоксикарбониламино-2-гидрокси-4-фенилбутил)-N-изобутил-4-нитробензолсульфонамида, который восстанавливают с одновременным удалением защитной группы и получают интересующее промежуточное соединение. В частности, в способе в качестве аминозащитной группы для коровой молекулы используют бензилоксикарбонил (Cbz или Z). Отмечается, что одновременное восстановление нитрогруппы и удаление защитной группы Cbz из (2R,3S)-N-(3-бензилоксикарбониламино-2-гидрокси-4-фенилбутил)-N-изобутил-4-нитробензолсульфонамида приводит к сильно экзотермической реакции. Экзотермических реакций, по возможности, следует избегать или сводить их к минимуму, так как в этом случае труднее регулировать температуру реакций, т.е. если температура реакции будет слишком низкой, скорость реакции мала и требуется длительное время; если температура реакции будет слишком высокой, скорость реакции слишком высокая и происходит недостаточное перемешивание, что способствует неравномерному протеканию реакции, быстрому окислению образовавшихся продуктов, или могут происходить нежелательные побочные реакции, в результате чего селективность по продукту снижается. С другой стороны, также отмечается, что каталитическая реакция, раскрываемая в WO 03/057665, не включает обработку в кислой среде. В отсутствие обработки кислотой катализатор, используемый во время восстановления и удаления защитной группы Cbz, будет отравляться серой п-нитробензолсульфонилхлорида. Отравленный катализатор неизбежно приведет в появлению побочных продуктов реакции, снижая селективность по продукту. Для того чтобы химический способ был приемлем для промышленного масштаба, он должен позволять получать соединения с приемлемым выходом и степенью чистоты, причем в то же время являться легким и простым в осуществлении, а также эффективным по затратам. Такой новый способ, приемлемый в промышленном масштабе, найден для синтеза соединения формулы (6).
В частности, настоящее изобретение относится к удобному способу получения соединения формулы (6) и промежуточных соединений для его получения, его солей присоединения, полиморфных и/или псевдополиморфных форм в промышленном масштабе. Конкретнее, настоящее изобретение относится к приемлемому способу синтеза соединения формулы (6), который также имеет преимущество улучшенной и эффективной по затратам кристаллизации (2R,3S)-N-(3-амино-2-гидрокси-4-фенилбутил)-N-изобутил-4-аминобензолсульфонамида с приемлемыми степенью чистоты и выходом. Даже конкретнее, настоящее изобретение касается раздельного способа проведения реакций восстановления и удаления защитной группы, включающему обработку кислотой, что в итоге приводит к более регулируемому, селективному и эффективному по затратам способу. В одном воплощении настоящее изобретение относится к усовершенствованному способу кристаллизации с регулированием рН и концентрации в определенном интервале, в то время как в WO 03/057665 в связи с кристаллизацией упоминается только нагревание раствора в полярном растворителе для того, чтобы улучшить выход, или нагревание раствора (30-80 оС) для того, чтобы растворить кристаллы, присутствующие в растворе в полярном растворителе, для того, чтобы улучшить очистку. Настоящее изобретение также имеет преимущество применения коммерчески доступного исходного вещества, такого как трет-бутиловый эфир 1-оксиранил-2-фенилэтилкарбаминовой кислоты. Кроме того, предшественника соединения формулы (6), т.е. (2R,3S)-N-(3-амино-2-гидрокси-4-фенилбутил)-N-изобутил-4-аминобензолсульфонамид или соединение формулы (5), можно получить процедурой в одном реакторе, что приводит к его эффективному использованию и исключению стадий очистки промежуточного соединения. Также реагенты, используемые в указанном способе, являются безопасными и доступными в большом количестве. Кроме того, каждую стадию указанного способа проводят в регулируемых условиях и получают требуемое соединение с оптимальным выходом. Кроме того, каждую стадию указанного способа проводят стереоселективно, что позволяет синтезировать чистые стереоизомерные формы требуемых соединений. Другие цели и преимущества настоящего изобретения станут очевидны из следующего далее подробного описания в сочетании с прилагаемыми примерами. В ЕР 0754669 (Kaneka Corporation) описываются способы получения альфа-галогенкетонов, альфа-галогенгидринов и эпоксидов; в ЕР 1029856 (Kaneka Corporation) раскрывается способ получения (2R,3S)-3-амино-1,2-оксирана; и ЕР 1067125, также Kaneka Corporation, относится к способу получения трео-1,2-эпокси-3-амино-4-фенилбутана. В ЕР 774453 (Ajinomoto Co., Inc.) описывается способ получения 3-амино-2-оксо-1-галогенпропановых производных. В WO 01/12599 (Samchully Pharm Co. Ltd.) описываются новые этилгидразиновые производные и способы их получения. В WO 01/46120 (Aerojet Fine Chemicals LLC) раскрывается улучшенный способ получения гидрохлорида 2S,3S-N-изобутил-N-(2-гидрокси-3-амино-4-фенилбутил)-п-нитробензолсульфонамида и других производных 2-гидрокси-1,3-диаминов. В WO 96/28418 (G.D. Searle & Co., Inc.) раскрываются сульфонилалканоиламиногидроксиэтиламиносульфонамидные ингибиторы ретровирусных протеаз. В WO 94/04492 (G.D. Searle & Co., Inc.) раскрываются гидроксиэтиламиносульфонамиды альфа- и бета-аминокислот, применяемые в качестве ингибиторов ретровирусных протеаз. В WO 97/21685 (Abbott) раскрывается получение пептидных аналогов как ингибиторов ретровирусных протеаз. В WO 94/05639 (Vertex Pharmaceuticals) описываются сульфонамидные ингибиторы аспартилпротеазы ВИЧ-1. Подробное описание изобретения Настоящее изобретение относится к способу получения соединения формулы (6)
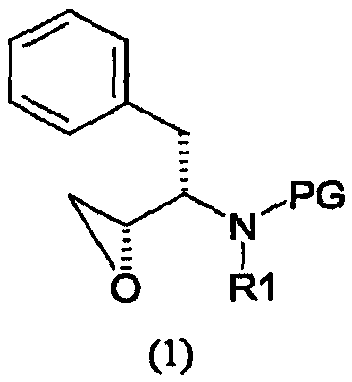
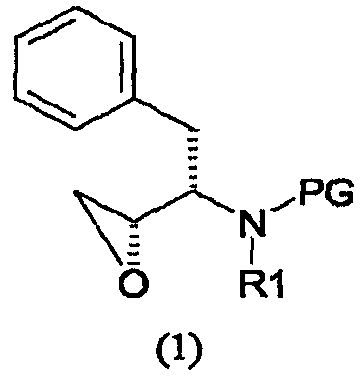
его солей присоединения, полиморфных и/или псевдополиморфных форм, включающему (i) введение изобутиламиногруппы в соединение формулы (1)
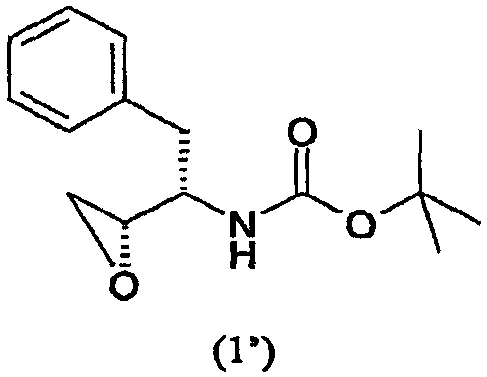
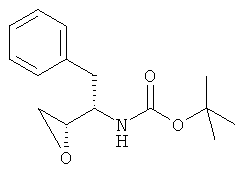
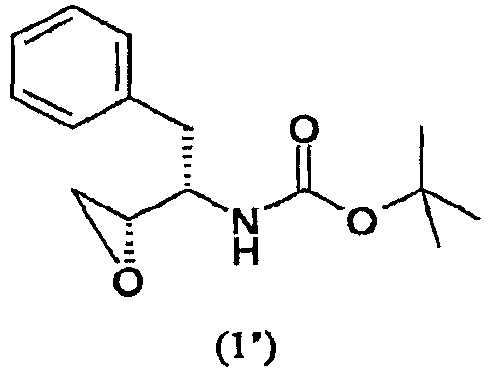
где PG представляет аминозащитную группу; R1 представляет собой водород или С1-6-алкил; (ii) введение п-нитрофенилсульфонильной группы в соединение, полученное на стадии (i); (iii) восстановление нитрогруппы соединения, полученного на стадии (ii); (iv) удаление защитной группы из соединения, полученного на стадии (iii); и (v) реакцию сочетания соединения, полученного на стадии (iv), с (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ильным производным с образованием соединения формулы (6). В одном воплощении настоящее изобретение относится к способу получения соединения формулы (6), отличающемуся тем, что указанный способ включает стадии введения изобутиламиногруппы в соединение формулы (1′)
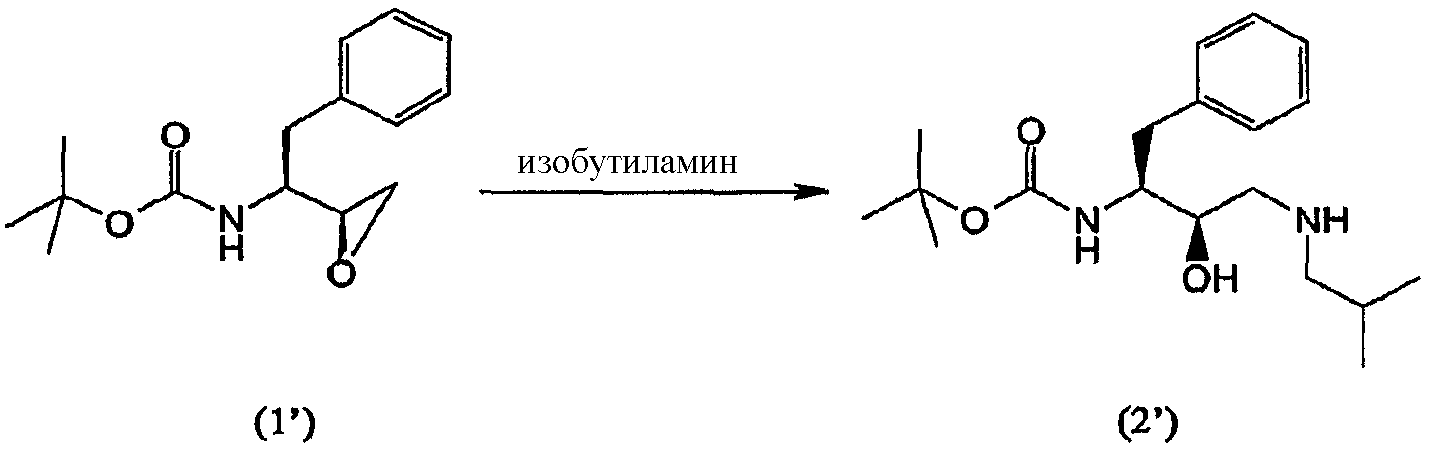
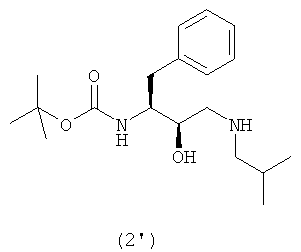
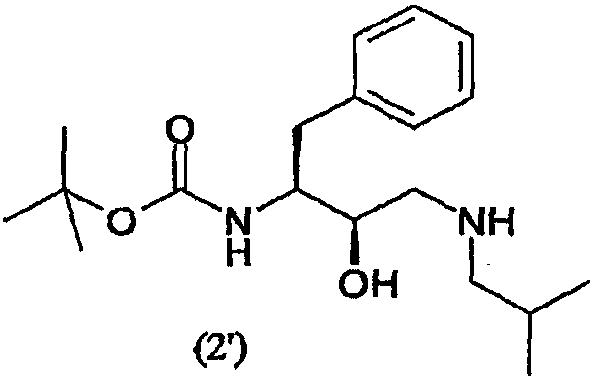
с образованием соединения формулы (2′)
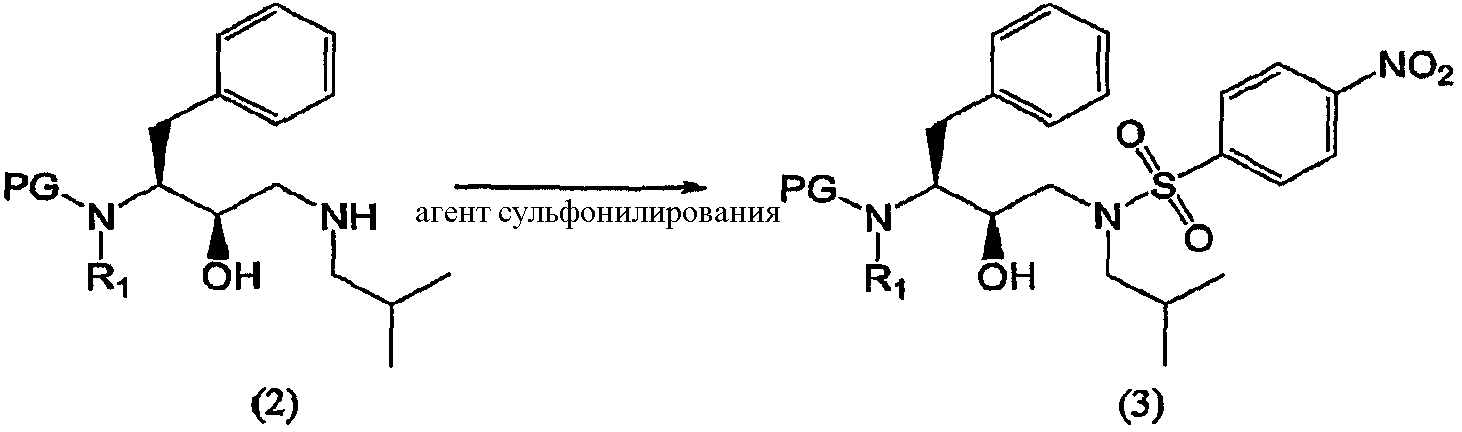
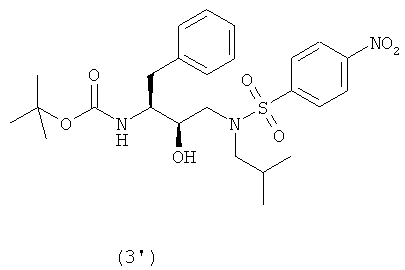
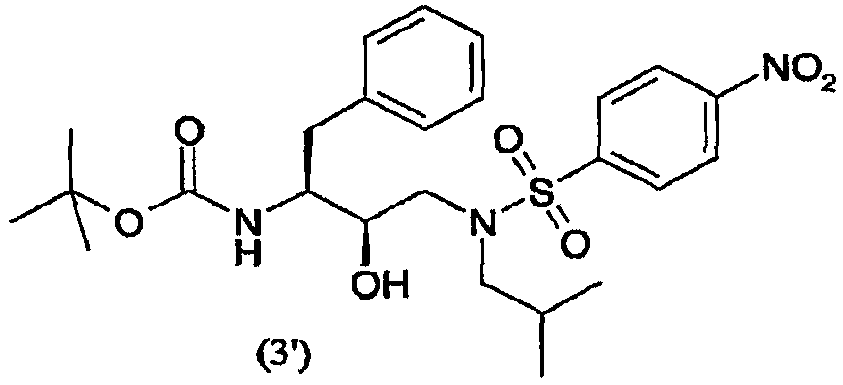
введения п-нитрофенилсульфонильной группы в соединение формулы (2′) с образованием соединения формулы (3′)
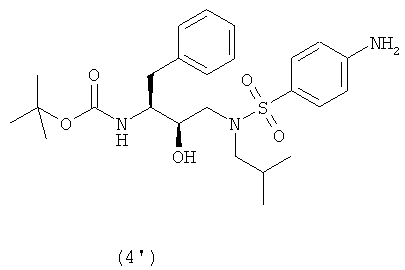
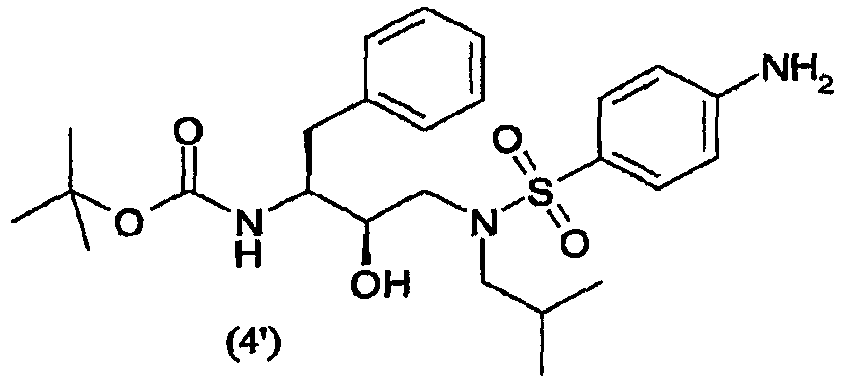
восстановления нитрогруппы соединения формулы (3′) с образованием соединения формулы (4′)
удаления защитной группы из соединения формулы (4′) с образованием соединения формулы (5)
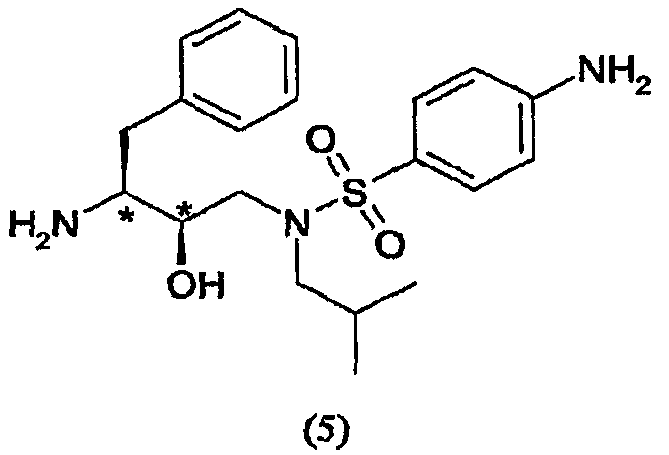
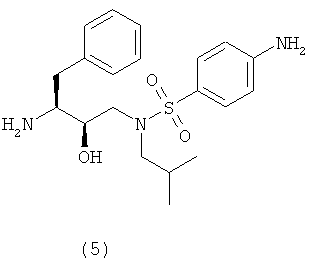
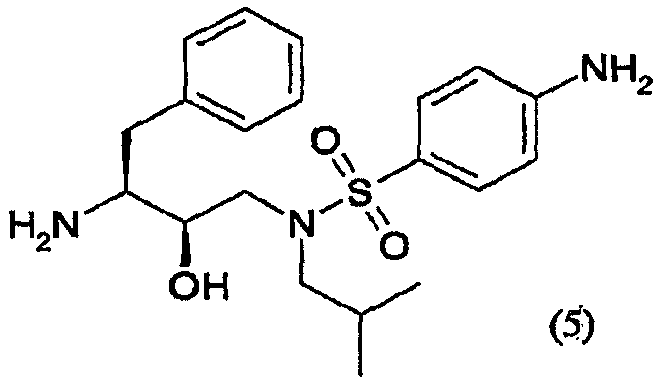
сочетания соединения формулы (5) с (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ильным производным с образованием соединения формулы (6). Таким образом, настоящее изобретение включает способы получения соединения формулы (6), его солей присоединения, полиморфных и/или псевдополиморфных форм через промежуточное соединение формулы (5)
Предпочтительно соединение формулы (5) кристаллизуют как свободное основание. С другой стороны, соединение формулы (5) кристаллизуют в виде солей с сильными кислотами, такими как соляная кислота, бромоводородная кислота, метансульфоновая кислота, серная кислота, щавелевая кислота, лимонная кислота и т.п. Кристаллизация соединения формулы (5) улучшает степень его чистоты и выход – оба фактора благоприятны для получения соединения формулы (6). С другой стороны, соединение формулы (5) можно кристаллизовать в виде его полиморфной и/или псевдополиморфной формы. Предпочтительно соединение формулы (6) кристаллизуют в виде псевдополиморфной формы, предпочтительно в виде алкоголята, предпочтительнее в виде этанолята. Соединение формулы (1) Соединение формулы (1) представляет собой
где PG представляет аминозащитную группу; R1 представляет собой водород или С1-6-алкил. Термин “аминозащитная группа”, используемый в данном описании, относится к одному или нескольким селективно удаляемым заместителям в аминогруппе, обычно используемым для блокировки или защиты функциональной аминогруппы от нежелательных побочных реакций во время синтетических процедур, и включает все обычные аминозащитные группы. Примерами аминозащитных групп являются уретановые блокирующие группы, такие как трет-бутоксикарбонил (“Вос”), 2-(4-бифенил)пропил(2)оксикарбонил (“Врос”), 2 фенилпропил(2)оксикарбонил (“Рос”), 2-(4-ксенил)изопроксикарбонил, изопропоксикарбонил, 1,1-дифенилэтил(1)оксикарбонил, 1,1-дифенилпропил(1)оксикарбонил, 2-(3,5-диметоксифенил)пропил(2)оксикарбонил (“Ddz”), 2-(п-5-толуил)пропил(2)оксикарбонил, 1-метилциклопентанилоксикарбонил, циклогексанилоксикарбонил, 1-метилциклогексанилоксикарбонил, 2-метилциклогексанилоксикарбонил, этоксикарбонил, 2-(4-толуилсульфонил)этоксикарбонил, 2-(метилсульфонил)этоксикарбонил, 2-(трифенилфосфино)этоксикарбонил, 9-флуороенилметоксикарбонил (“Fmoc”), 2-(триметилсилил)этоксикарбонил, аллилоксикарбонил, 1-(триметилсилилметил)проп-1-енилоксикарбонил, 5-бензизоксалилметоксикарбонил, 4-ацетоксибензилоксикарбонил, 2,2,2-трихлорэтоксикарбонил, трибромэтоксикарбонил, 2-этинил(2)пропоксикарбонил, циклопропилметоксикарбонил, изоборнилоксикарбонил, 1-пиперидилоксикарбонил, бензилоксикарбонил (“Z” или “Cbz”), 4-фенилбензилоксикарбонил, 2-метилбензилоксикарбонил, Другими примерами аминозащитных групп являются фенилацетил, формил (“For”), тритил (Trt), ацетил, трифторацетил (TFA), трихлорацетил, дихлорацетил, хлорацетил, бромацетил, иодацетил, бензоил, трет-амилоксикарбонил, трет-бутоксикарбонил, 3,4-диметоксибензилоксикарбонил, 4-(фенилазо)бензилоксикарбонил, 2-фурфурилоксикарбонил, дифенилметоксикарбонил, 1,1-диметилпропоксикарбонил, фталил или фталимидо, сукцинил, аланил, лейцил и 8-хинолилоксикарбонил, бензил, дифенилметил, 2-нитрофенилтио, 2,4-динитрофенилтио, метансульфонил, пара-толуолсульфонил, N,N-диметиламинометилен, бензилиден, 2-гидроксибензилиден, 2-гидрокси-5-хлорбензилиден, 2-гидрокси-1-нафтилметилен, 3-гидрокси-4-пиридилметилен, циклогексилиден, 2-этоксикарбонилциклогексилиден, 2-этоксикарбонилциклопентилиден, 2-ацетилциклогексилиден, 3,3-диметил-5-оксициклогексилиден, дифенилфосфорил, дибензилфосфорил, 5-метил-2-оксо-2Н-1,3-диоксол-4-илметил, триметилсилил, триэтилсилил, трифенилсилил, 2-(п-бифенил)-1-метилэтоксикарбонил, диизопропилметоксикарбонил, циклопентилоксикарбонил, адамантилоксикарбонил, трифенилметил, триметилсилан, фенилтиокарбонил, пара-нитробензилкарбонил. Другими аминозащитными группами являются 2,7-ди-трет-бутил[9-(10,10-диоксо-10,10,10,10-тетрагидротиоксантил)]метилоксикарбонил; 2-триметилсилилэтилоксикарбонил; 2-фенилэтилоксикарбонил; 1,1-диметил-2,2-дибромэтилоксикарбонил; 1-метил-1-(4-бифенил)этилоксикарбонил; п-нитробензилоксикарбонил; 2-(п-толуолсульфонил)этилоксикарбонил; м-хлор-п-ацилоксибензилоксикарбонил; 5-бензилизоксазолилметилоксикарбонил; п-(дигидроксиборил)бензилоксикарбонил; м-нитрофенилоксикарбонил; о-нитробензилоксикарбонил; 3,5-диметоксибензилоксикарбонил; 3,4-диметокси-6-нитробензилоксикарбонил; N’-п-толуолсульфониламинокарбонил; трет-амилоксикарбонил; п-децилоксибензилоксикарбонил; 2,2-диметоксикарбонилвинилоксикарбонил; ди(2-пиридил)метилоксикарбонил; 2-фуранилметилоксикарбонил; дитиасукцинимид; 2,5-диметилпиррол; 5-дибензилсуберил и метансульфонамидная группа. Предпочтительной аминозащитной группой является Вос. Другие примеры аминозащитных групп хорошо известны в технике органического синтеза и пептидов и описаны, например, в T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, 2nd ed., John Wiley and Sons, New York, Chapter 7, 1991; M. Bodanzsky, Principles of Peptide Synthesis, 1st and 2nd reviser ed., Springer-Verlag, New York, 1984 and 1993; Stewart and Young, Solid Phase Peptide Synthesis, 2nd ed., Pierce Chemical Co., Rockford, IL 1984; L. Fieser and M. Fieser, Fieser and Fieser’s Reagents for Organic Synthesis, John Wiley and Sons (1994); L. Paquette, ed., Encyclopedia of Reagents for Organic Synthesis, John Wiley and Sons (1995). Подходящие аминозащитные группы также приводятся в, например, WO 98/07685. Термин “С1-6-алкил” в отношении групппы или части группы обозначает линейные или разветвленные насыщенные углеводородные радикалы с 1-6 атомами углерода, такие как метил, этил, изопропил, бутил, пентил, гексил, 2-метилбутил, 3-метилпентил и подобные. Предпочтительно соединение формулы (1) представляет собой соединение формулы (1′), приведенной ниже, где PG представляет собой трет-бутоксикарбонил или “Вос” и R1 представляет собой водород. Соединения формулы (1) и (1′) коммерчески доступны и могут быть получены разными способами, описанными в литературе, например, так, как описано в WO 95/06030 (Searle & Co.), как описано в ЕР 0754669, ЕР 1029856 и ЕР 1067125, Kaneka Corporation, и так, как описано в EP 1081133 и ЕР 1215209, Ajinomoto KK.
Соединение формулы (2) Соединение формулы (1) подвергают аминированию по эпоксидной группе и получают соединение формулы (2)
Термин “аминирование”, используемый в данном описании, относится к процессу, в котором первичный амин, изобутиламин, вводят в органическую молекулу формулы (1). Аминирование соединения формулы (1) можно осуществить несколькими способами, описанными в литературе, например, так, как описано в WO 95/06030, включенной в данное описание в качестве ссылки. В предпочтительном воплощении соединение формулы (1′) вводят во взаимодействие с изобутиламином и получают соединение формулы (2′)
Аминирование эпоксидных соединений описывается, например, в March, Advanced Organic Chemistry, 368-69 (3rd Ed., 1985), и в McManus et al., 3 Synth. Comm. 177 (1973), включенных в данное описание в качестве ссылок. Соединения формулы (2) и (2′) можно получить подходящим образом согласно процедуре, описанной в WO 97/18205. Агент аминирования изобутиламин может также выполнять функцию растворителя, и в таком случае будет добавляться его избыток. В других воплощениях процесс аминирования осуществляют в присутствии одного или нескольких растворителей иных, чем изобутиламин. В предпочтительном воплощении указанные растворители используют при обработке соединений формулы (2) и (2′). Подходящие растворители включают протонные, апротонные и биполярные апротонные органические растворители, такие как, например, спирты, а именно метанол, этанол, изопропанол, н-бутанол, трет-бутанол и т.п.; кетоны, такие как ацетон; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан и т.п.; сложные эфиры, такие как этилацетат; амины, такие как триэтиламин; амиды, такие как N,N-диметилформамид или диметилацетамид; хлорсодержащие растворители, такие как дихлорметан, и другие растворители, такие как толуол, диметилсульфоксид, ацетонитрил, и их смеси. Предпочтительным растворителем является толуол. Взаимодействие обычно можно проводить в широком интервале температур, например, от примерно -20°С до примерно 200°С, но предпочтительно, хотя и не обязательно, взаимодействие проводят при температуре кипения растворителя, т.е., между 40°С и 100°С, предпочтительнее, между 60°С и 90°С. Подходящие эквивалентные соотношения между соединением формулы (1) и агентом аминирования могут колебаться от 1:1 до 1:99 соответственно. Предпочтительно соотношение эквивалентов между соединением формулы (2) и агентом аминирования составляет от 1:5 до 1:20, еще более предпочтительно соотношение составляет от 1:10 до 1:15. В воплощении изобретения реакцию аминирования проводят в присутствии примерно 15 эквивалентов изобутиламина с использованием в качестве растворителя толуола и нагревание при примерно 79°С. Соединения формулы (3) Соединение формулы (3) получают введением сульфонильной группы п-нитробензол-SO2 в промежуточное соединение формулы (2)
Так, в предпочтительном воплощении соединение формулы (3′) будут получать сульфонилированием соединения формулы (2′)
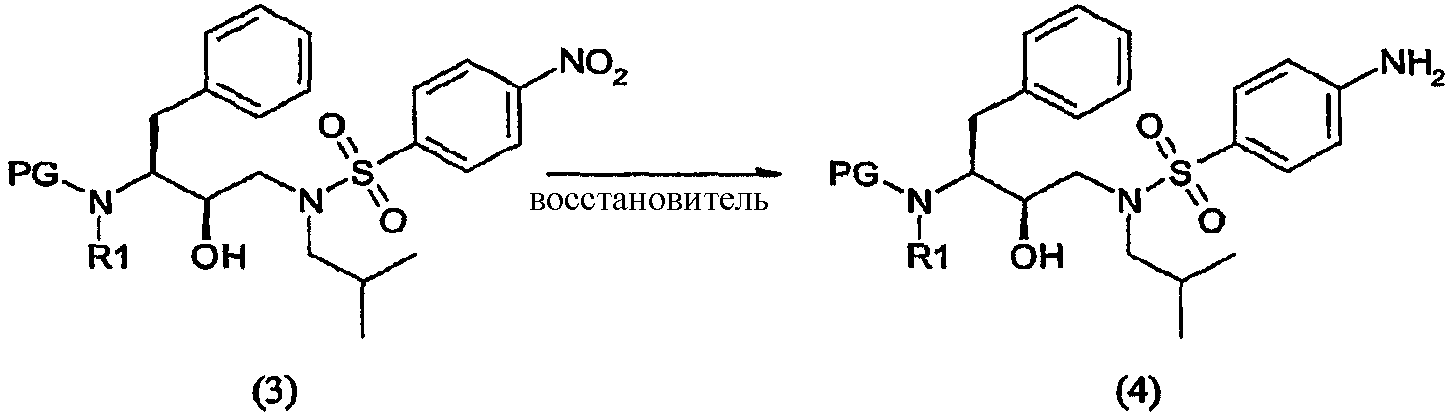
Как таковые, соединения формулы (2) и (2′) будут взаимодействовать с агентом сульфонилирования с превращением в соединения формулы (3) и (3′). Термин “сульфонилирование”, используемый в данном описании, относится к процессу, в котором п-нитробензолсульфонильную группу вводят в органическую молекулу формулы (2) и (2′). Термин “сульфирование”, используемый в данном описании, относится к процессу получения агента сульфонилирования. Термин “агент сульфонилирования” относится к п-нитробензолсульфонильным производным, таким как п-нитробензолсульфонильные галогенопроизводные. Агенты сульфонилирования, в частности п-нитробензолсульфонильные галогенопроизводные, можно получить окислением тиолов до сульфонилхлоридов с использованием хлора в присутствии воды в строго регулируемых условиях. Кроме того, можно превратить сульфоновые кислоты в сульфонилгалогениды с использованием таких реагентов, как PCl5, а также в ангидриды с использованием соответствующих дегидратирующих реагентов. Сульфоновые кислоты, в свою очередь, можно получить с использованием процедур, хорошо известных в технике. Также такие сульфоновые кислоты доступны коммерчески. Агенты сульфонилирования также можно получить процедурами сульфирования, описанными в работах “Sulfonation and Related Reactions”, E.E. Gilbert, R.E. Krieger Publishing Co., Hungtington, N.Y. (1977); “Mechanistic Aspects of Aromatic Sulfonation and Desulfonation”, H. Cerfontain, Interscience Publishers, NY (1968), и в US 6455738 “Process for sulfonation of an aromatic compound”, которые все включены в данное описание в качестве ссылок. Обработку соединений формулы (2) и (2′) агентом сульфонилирования можно осуществить в присутствии растворителя при нагревании приблизительно от 25°С до 250°С, предпочтительно от 70°С до 100°С, и перемешивании. После сульфонилирования оставшийся агент сульфонилирования или любые соли предпочтительно, хотя необязательно, удаляют из реакционной смеси. Удаление осуществляют повторяющимися промывками водой, изменением рН, разделением органической и водной фаз, ультрафильтрацией, обратным осмосом, центрифугированием и/или фильтрацией или подобными способами. Соединения формулы (3) и (3′) получают взаимодействием сульфонилирующего агента с промежуточными соединениями формулы (2) и (2′) в подходящих растворителях в щелочной среде. Подходящими щелочными средами являются обычные ненуклеофильные неорганические или органические основания и/или акцепторы кислоты. Обычные ненуклеофильные неорганические или органические основания включают, например, гидриды, гидроксиды, амиды, алкоголяты, ацетаты, карбонаты или гидрокарбонаты щелочноземельных металлов или гидриды щелочных металлов, такие как, например, гидрид натрия, гидрид калия или гидрид кальция, и амиды металлов, такие как амид натрия, амид калия, диизопропиламид лития или гексаметилдисилазид калия, и алканы металлов, такие как метилат натрия, этилат натрия, трет-бутилат калия, гидроксид натрия, гидроксид калия, гидроксид аммония, ацетат натрия, ацетат калия, ацетат кальция, ацетат аммония, карбонат натрия, бикарбонат натрия, карбонат калия, бикарбонат калия, карбонат цезия, гидрокарбонат калия, гидрокарбонат натрия или карбонат аммония, а также органические азотсодержащие соединения, такие как триалкиламины, такие как, например, триметиламин, триэтиламин, трибутиламин, N,N-диметиланилин, N,N-диметилбензиламин, N,N-диизопропилэтиламин, пиридин, 1,4-диазабицикло[2.2.2]октан (DABCO), 1,5-диазабицикло[4.3.0]нон-5-ен (DBN) или 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU), или можно использовать избыток подходящего пиперидина. Предпочтительно используют триэтиламин. Подходящие растворители указаны выше при получении соединений формул (2) и (2′), причем предпочтительны такие инертные растворители, как, например, толуол, этилацетат, метиленхлорид, дихлорметан и тетрагидрофуран. Обычно отношения эквивалентов, рассчитанные для соединений формулы (1) или (1′) и агента сульфонилирования, колеблются от 1:1 до 1:3 соответственно. Предпочтительно отношение эквивалентов соединений формулы (1) или (1′) и агента сульфонилирования составляет от 1:1 до 1:2, предпочтительнее отношение составляет примерно 1:1,15. Соединения формулы (4) Соединения формулы (4) и (4′) получают взаимодействием нитрогруппы промежуточных соединений формулы (3) и (3′) соответственно с восстановителем, необязательно, в атмосфере водорода
Восстановители, подходящие для восстановления нитрогруппы, представляют собой металлосодержащие восстановители, такие как комплексы бора, диборан, борогидрид натрия, борогидрид лития, борогидрид натрия-LiCl, алюмогидрид лития или диизобутилалюмогидрид; такие металлы, как железо, цинк, олово и т.п.; и содержащие переходные металлы, такие как палладий-на-угле, оксид платины, никель Ренея, родий, рутений и т.п. Когда применяют каталитическое восстановление, в качестве источника водорода можно использовать формиат аммония, дигидрофосфат натрия, гидразин. Подходящие растворители для восстановления нитрогруппы можно выбрать из воды, спиртов, таких как метанол, этанол, изопропанол, трет-бутиловый спирт, сложных эфиров, таких как этилацетат, амидов, таких как диметилформамид, уксусной кислоты, дихлорметана, толуола, ксилола, бензола, пентана, гексана, гептана, петролейного эфира, 1,4-тиоксана, диэтилового эфира, диизопропилового эфира, тетрагидрофурана, 1,4-диоксана, 1,2-диметоксиэтана, диметилсульфоксида или их смесей. Вообще, можно использовать любой растворитель, приемлемый для использования в процессе химического восстановления. Указанную стадию восстановления можно осуществлять при температурах от -78°С до 55°С, предпочтительно от -10°С до 50°С, причем предпочтительные температуры находятся в интервале от 0°С до 50°С, более предпочтительно от 5°С до 30°С. Время реакции может варьироваться от 30 минут до 2 суток, более предпочтительно от 1 часа до 24 часов. Согласно предпочтительному воплощению стадию восстановления осуществляют с использованием палладия-на-угле, суспендированного в метаноле. В другом предпочтительном воплощении можно использовать дополнительное количество угля. Отношения эквивалентов соединений формулы (3) или (3′) и водорода колеблются от 1:1 до 1:10 соответственно. Более предпочтительно, отношение эквивалентов соединений формулы (3) или (3′) и водорода составляет от 1:1 до 1:5, предпочтительнее отношение составляет примерно 1:3. Соединения формулы (5) Соединение формулы (5) получают удалением защитной группы из промежуточных соединений формулы (4) и (4′) в обычных условиях в кислой среде. С другой стороны, можно использовать щелочную среду
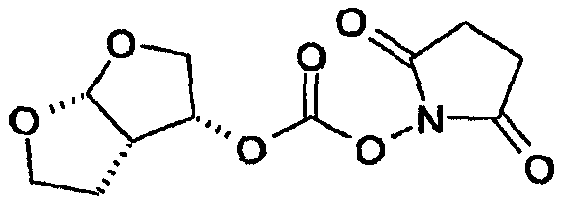
Удаления аминозащитной группы можно достичь в условиях, которые не влияют на остальную часть молекулы. Такие способы хорошо известны в технике и включают гидролиз в кислой среде, гидрогенолиз и т.п., причем также используют обычно известные кислоты в подходящих растворителях. Примеры кислот, используемых при удалении аминозащитной группы, включают такие неорганические кислоты, как соляная кислота, азотная кислота, хлороводород, серная кислота и фосфорная кислота: такие органические кислоты, как уксусная кислота, трифторуксусная кислота, метансульфоновая кислота и п-толуолсульфоновая кислота: кислоты Льюиса, такие как трифторид бора; катионообменные смолы кислотного характера, такие как Dowex 50Wтм. Из указанных кислот предпочтительными являются неорганические и органические кислоты. Более предпочтительными являются соляная кислота, серная кислота, фосфорная кислота трифторуксусная кислота и наиболее предпочтительна соляная кислота. Растворитель, используемый во время удаления защитной группы из промежуточных соединений формулы (4) и (4′), не оказывает вредного влияния на реакцию и, по меньшей мере, до некоторой степени растворяет исходные вещества. Подходящими растворителями являются такие алифатические углеводороды, как гексан, гептан и петролейный эфир; такие ароматические углеводороды, как бензол, толуол, ксилол и мезитилен; такие галогенсодержащие углеводороды, как метиленхлорид, хлороформ, четыреххлористый углерод и дихлорэтан; такие простые эфиры, как диэтиловый эфир, тетрагидрофуран, 1,4-диоксан и 1,2-диметоксиэтан; такие спирты, как метанол, этанол, пропанол, изопропанол и бутанол; такие сложные эфиры, как метилацетат, этилацетат, метилпропионат и этилпропионат; такие нитрилы, как ацетонитрил; такие амиды, как N,N-диметилформамид и N,N-диметилацетамид; такие сульфоксиды, как диметилсульфоксид и их смеси. Предпочтительными являются ароматические углеводороды, спирты и сложные эфиры. Спирты и вода являются более предпочтительными, и вода, изопропанол, этанол и метанол являются особенно предпочтительными. Также предпочтительными являются смеси метанола, воды и изопропанола или этанола и смеси этанола и воды. Температура реакции зависит от таких факторов, как природа исходных веществ, растворителей и кислот. Однако, как правило, она составляет от -20°С до 150°С и предпочтительно от 30°С до 100°С, даже предпочтительнее это температура образования флегмы. Используемое время реакции зависит от реагентов, температуры и подобных факторов. Типично оно составляет от 5 минут до 72 часов и предпочтительно от 15 минут до 4 часов. Примеры реагентов и способов удаления из аминов аминозащитных групп можно также найти в работе Protective Groups in Organic Synthesis, Theodora W. Greene, New York, John Wiley and Sons, Inc., 1981, включенной в данное описание в качестве ссылки. Как известно специалистам в данной области техники, выбор способа будет определять реагенты и методики, используемые при удалении аминозащитной группы. Соотношения эквивалентов соединений формулы (3) или (3′) и кислоты в растворителе могут варьироваться от 1:2 до 1:50 соответственно. Предпочтительно соотношение эквивалентов соединений формулы (3) или (3′) и кислоты составляет от 1:2 до 1:8, более предпочтительно соотношение составляет примерно 1:2. В предпочтительном воплощении настоящего изобретения соединение формулы (5) кристаллизуют. Кристаллизацию соединения формулы (5) проводят, растворяя соединение формулы (5) в смеси растворителей, устанавливая рН раствора и концентрацию соединения формулы (5). Можно добавлять кристаллы соединения формулы (5). Смесь растворителей, используемая при кристаллизации, может состоять из одного или нескольких смешивающихся с водой растворителей и воды, или система растворителей состоит из одного или нескольких не смешивающихся с водой растворителей и воды. Примеры растворителей, смешивающихся с водой, включают такие спирты С1-С4, как метанол, этанол, н-пропанол, изопропанол, н-бутанол, изобутанол; такие циклические простые эфиры, как тетрагидрофуран или диоксан; такие амиды, как диметилформамид, диметилацетамид, N-метилпирролидон; диметилсульфоксид, ацетонитрил, смесь вышеуказанных растворителей друг с другом или с водой или воду. Примерами растворителей, не смешивающихся с водой, являются такие углеводороды, как пентан, гексан, циклогексан, метилциклогексан, гептан, толуол, ксилол; такие сложные эфиры С4-С8, как метилформиат, этилформиат, метилацетат, этилацетат; такие простые эфиры С4-С8, как диэтиловый эфир, трет-бутилметиловый эфир, изопропиловый эфир; такие хлорсодержащие растворители, как метиленхлорид, дихлорметан, хлороформ, дихлорэтан, хлорбензол; или их двойные или многокомпонентные смеси. Когда используют такие не смешивающиеся с водой растворители, соединение формулы (5) будет извлекаться разделением органической и водной фаз. Регулирование концентрации соединения формулы (5) можно осуществлять добавлением воды или других подходящих растворителей или выпариванием, отгонкой растворителя или любыми другими методами концентрирования. В предпочтительном способе кристаллизации поддерживают концентрацию соединения формулы (5) от 0,1% до 40% (мас./мас.), предпочтительно от 1% до 30%, более предпочтительно от 2% до 20% и еще более предпочтительно от 4% до 15%, мас./мас. Контроль или непосредственный (текущий) контроль концентрации соединения формулы (5) в растворе можно осуществлять любым способом, известным специалистам в данной области техники, например хроматографией ВЭЖХ, измерением плотности, титрованием и т.п. Предпочтительно растворитель, используемый при кристаллизации соединения формулы (5), является тем же самым растворителем, который используют при удалении защитной группы из промежуточного соединения формулы (4) или (4′). С другой стороны, если используют несколько растворителей, один или несколько растворителей, используемых при кристаллизации соединения формулы (5), являются теми же самыми одним или несколькими растворителями, которые используют при удалении защитной группы из промежуточного соединения формулы (4) или (4′). Регулирование рН раствора, содержащего соединение формулы (5), можно осуществлять добавлением оснований, таких как гидроксид натрия, карбонат натрия, гидроксид калия, гидроксид лития, аммиак, гидразин, гидроксид кальция, метиламин, этиламин, анилин, этилендиамин, триэтиламин, гидроксид тетраэтиламмония, амины С2-С18, гидроксид аммония С4-С18, метоксид натрия, метоксид калия, органическое основание С1-С4, любое из оснований, перечисленных выше, и их смеси. рН раствора, содержащего соединение формулы (5), будет поддерживаться в щелочной области, предпочтительно более 7, более предпочтительно рН будет составлять более 8, еще более предпочтительно более 9. В одном воплощении после добавления основания суспензию перемешивают в течение 1-48 часов, предпочтительно 1-10 часов, еще более предпочтительно в течение 1-5 часов. Рабочая температура, используемая при осаждении соединения формулы (5), может варьироваться от -20°С до 50°С. Предпочтительно рабочая температура во время осаждения может варьироваться от -15°С до 10°С, еще более предпочтительно от -10°С до 10°С, наиболее предпочтительно около 5°С. В другом воплощении соединение формулы (5) собирают центрифугированием и сушат в вакууме при примерно 65°С. Предпочтительное кристаллическое соединение формулы (5) находится в форме свободного основания. С другой стороны, другими подходящими соединениями являются кристаллические соединения формулы (5) в форме соли, где соль выбирают из числа гидрохлорида, гидробромида, трифторацетата, фумарата, хлорацетата и метансульфоната и подобных солей. Промежуточные соединения формулы (5) также являются активными ингибиторами ретровирусных протеаз. (3R,3aS,6aR)-Гексагидрофуро[2,3-b]фуран-3-ильное производное (3R,3aS,6aR)-Гексагидрофуро[2,3-b]фуран-3-ол и его предшественников можно синтезировать так, как описано в WO 03/022853. (3R,3aS,6aR)-Гексагидрофуро[2,3-b]фуран-3-ол и его предшественники подходящим образом активируются агентами сочетания с образованием (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ильного производного, которое может подвергаться карбамоилированию соединением формулы (5). Активацию (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ола и его предшественников проводят перед их сочетанием с соединением формулы (5). Указанная активация (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ола и его предшественников и их сочетание с соединением формулы (5) проводят в одном реакторе, так как извлечение активированного промежуточного соединения не является неообходимым, что является дополнительным преимуществом способа. Предшественниками (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ола являются такие соединения, в которых кислород функциональной спиртовой группы защищен такими О-защитными группами, как трет-бутилэфирная группа (“Вос”), ацетатные, бензильные группы, бензилэфирные группы, аллильные, такие силилзащитные группы, как трет-бутилдиметилсилил (TBS), триметилсилилэтоксиметил (SEM), такие алкоксиалкильные группы, как метоксиэтоксиметил (МЕМ), метоксиметил (МОМ), тетрагидропиранил (ТНР), тетрагидропиранил (ТНЕ), и подобными группами. Когда используют предшественников 3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ола, удаление защитной группы можно осуществить до сочетания или непосредственно in situ. Удалить спиртозащитные группы можно в кислой или щелочной средах, причем предпочтительна кислая среда. Защитные группы хорошо известны в технике, см., например, Greene T.W., Protective Groups in Organic Synthesis, John Wiley and Sons, Inc., New York, 1991. С другой стороны, (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ол и его предшественников можно получить через динамическое диастереоселективное разделение рецемических смесей гексагидрофуро[2,3-b]фуран-3-ола. В таком случае рецемическую смесь подвергают действию определенных ферментов, таких как панкреатическая липаза свиньи, панкреатин candida cylindracca и т.п., в присутствии подходящих растворителей и реагентов, таких как уксусный ангидрид и винилацетат. Такой альтернативный способ позволяет получить in situ требуемый энантиомер (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ол, который можно удобно активировать по способу в одном реакторе; ненужный стереоизомер блокируется или становится инертным. Примерами агентов сочетания, используемых в реакциях карбамоилирования, являются такие карбонаты, как бис(4-нитрофенил)карбонат, дисукцинимидилкарбонат (DSC), карбонилдиимидазол (CDI). Другими агентами сочетания являются такие хлорформиаты, как п-нитрофенилхлорформиат, такие фосгены, как фосген и трифосген. В частности, когда (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ол обрабатывают дисукцинимидилкарбонатом, получают 1-([[(3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-илокси]карбонил]окси)-2,5-пирролидиндион. Указанное соединение является предпочтительным производным (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ила
В случае активации (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ола и его предшественников агентом сочетания рекомендуется присутствие спирта в концентрациях от 1% до 20% (мас./мас.), предпочтительно в концентрации от 2% до 15% (мас./мас.), еще более предпочтительно в концентрации от 4% до 10% (мас./мас.). Взаимодействие производного (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ила с соединением формулы (5) будет осуществляться в присутствии таких подходящих растворителей, как тетрагидрофуран, диметилформамид, ацетонитрил, диоксан, дихлорметан или хлороформ, и, необязательно, таких оснований, как триэтиламин, хотя также воплощаются сочетания растворителей и оснований, раскрытые в данном описании выше. Из числа растворителей предпочтительными являются апротонные, такие как тетрагидрофуран, ацетонитрил, диметилформамид, этилацетат и подобные. В одном воплощении во время сочетания производного (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ила с соединением формулы (5) указанное производное присутствует в концентрации от 1% до 15% (мас./мас.), предпочтительно в концентрации от 5% до 12% (мас./мас.), еще более предпочтительно в концентрации от 8% до 12% (мас./мас.). Реакцию карбомоилирования осуществляют при температуре от -70°С до 40°С, предпочтительно от -10°С до 20°С. Соединение, полученное сочетанием производного (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ила с соединением формулы (5), представляет собой соединение формулы (6). Соединение формулы (6) предпочтительно будет сольватироваться спиртами, такими как этанол, метанол, причем предпочтительной формой сольвата является этанолят. Сольватация соединения формулы (6) описана в РСТ/ЕР 03/50176 (Tibotec N.V.), включенной в данное описание в качестве ссылки
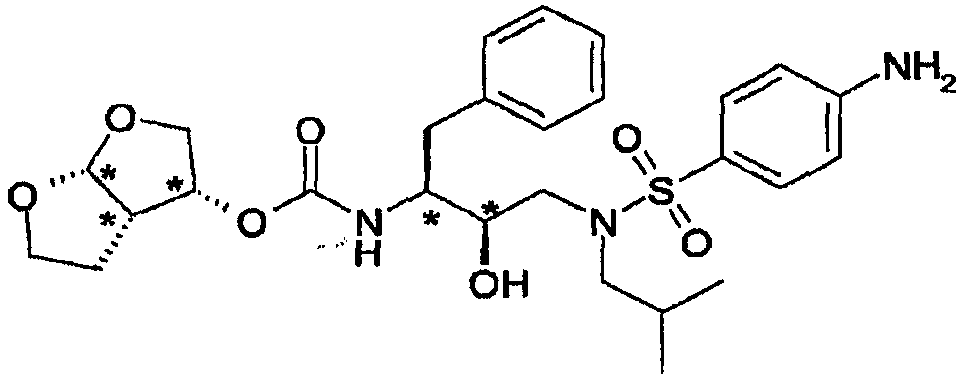
В каждом способе получения, представленном выше, продукты реакции, например соединения формулы (3), (3′), (4), (4′), (5) и конечный продукт формулы (6), можно выделить из реакционной смеси и, при необходимости, дополнительно очистить по методологиям, известным в технике, таким как, например, экстракция, кристаллизация, перегонка, растирание в порошок и хроматография. Для лечебного применения соли соединений по изобретению представляют собой соли, в которых противоион является фармацевтически или физиологически приемлемым. Однако соли с фармацевтически неприемлемыми противоионами также могут найти применение, например, при получении или очистке фармацевтически приемлемого соединения по настоящему изобретению. Все соли, являются ли они фармацевтически приемлемыми или нет, включены в объем настоящего изобретения. Фармацевтически приемлемые соли соединений по изобретению, т.е. в форме продуктов, растворимых или диспергируемых в воде или масле, включают обычные нетоксичные соли или соли четвертичного аммония, которые образуются, например, с неорганическими или органическими кислотами или основаниями. Примеры таких солей присоединения кислот включают ацетаты, адипаты, альгинаты, аспартаты, бензоаты, бензолсульфонаты, бисульфаты, бутираты, цитраты, камфораты, камфорсульфонаты, циклопентанпропионаты, диглюконаты, додецилсульфаты, этансульфонаты, фумараты, глюкогептаноаты, глицерофосфаты, гемисульфаты, гептаноаты, гексаноаты, гидрохлориды, гидробромиды, гидроиодиды, 2-гидроксиэтансульфонаты, лактаты, малеаты, метансульфонаты, 2-нафталинсульфонаты, никотинаты, оксалаты, фосфаты, памоаты, пектинаты, персульфаты, 3-фенилпропионаты, пикраты, пивалаты, пропионаты, сукцинаты, тартраты, тиоцианаты, тозилаты и ундеканоаты. Примеры солей присоединения оснований включают соли аммония, соли щелочных металлов, такие как натриевые и калиевые соли, соли щелочноземельных металлов, такие как кальциевые и магниевые соли, соли с органическими основаниями, такие как соли дициклогексиламина, соли N-метил-D-глюкамина и соли с аминокислотами, такими как аргинин, лизин и т.д. Также основные азотсодержащие группы могут быть кватернизованы такими агентами, как низшие алкилгалогениды, такие как метил-, этил-, пропил- и бутилхлорид, -бромид и -иодид, диалкилсульфаты, подобные диметил-, диэтил-, дибутил- и диамилсульфатам, длинноцепочечные галогениды, такие как децил-, лаурил-, миристил- и стеарилхлорид, -бромид и -иодид, аралкилгалогениды, такие как бензил- и фенилэтилбромид, и другими. Другие фармацевтически приемлемые соли включают этаноляты и сульфаты солей сульфатов. Термин “полиморфная форма” относится к свойству соединений формулы (5) и (6) существовать в аморфной форме, в полиморфной форме, в кристаллической форме с различными структурами, изменяющимися по твердости, форме и размерам кристаллов. Различные кристаллические формы можно обнаружить методами кристаллографии или косвенно, оценивая различия в физических и/или химических свойствах, связанных с определенным полиморфом. Полиморфы имеют различия в физических свойствах, таких как растворимость, растворение, стабильность в твердом состоянии, а также в поведении при переработке в сыпучести порошка и уплотнения во время таблетирования. Термины “псевдополимерная форма” или “сольваты” относятся к агрегатам, состоящим из молекул соединения формулы (6) и его солей с захваченными молекулами растворителями или образовавшими с ними комплекс в отношении моль/моль и с разной степенью сольватации. Промежуточные соединения по изобретению также могут существовать в таутомерных формах. Подразумевается, что такие формы, хотя и не указанны явно в соединениях, описанных в данном описании, входят в объем настоящего изобретения. Чистые стереоизомерные формы соединений и промежуточных соединений, упоминаемых в данном описании, определяются как изомеры, по существу, свободные от других энантиомерных или диастереомерных форм той же основной молекулярной структуры указанных соединений или промежуточных соединений. В частности, термин “стереоизомерно чистый” относится к соединениям или промежуточным соединениям со стереоизомерным избытком, по меньшей мере, 80% (т.е. имеющим минимум 90% одного изомера и максимум 10% других возможных изомеров) и до стереоизмерного избытка 100% (т.е. имеющим 100% одного изомера и никаких других изомеров), в особенности соединениям или промежуточным соединениям со стереоизомерным избытком от 90% до 100%, еще более предпочтительно со стереоизомерным избытком от 94% до 100% и наиболее предпочтительно со стереоизомерным избытком от 97% до 100%. Термины “энантиомерно чистый” и “диастереомерно чистый” следует понимать подобным образом, но в отношении энантиомерного избытка и, соответственно, диастереомерного избытка в смесях, представляющих интерес. Чистые стереоизомерные формы соединений и промежуточных соединений данного изобретения можно получить, применяя процедуры, известные в технике. Например, энантиомеры можно отделить друг от друга избирательной кристаллизацией их диастереомерных солей с оптически активными кислотами или основаниями. Их примерами являются винная кислота, дибензоилвинная кислота, дитолуоилвинная кислота и камфорсульфоновая кислота. С другой стороны, энантиомеры можно разделить хроматографическими методами с использованием оптически активных неподвижных фаз. Указанные чистые стереохимические изомерные формы также можно получить из соответствующих чистых стереохимических изомерных форм соответствующих исходных веществ, при условии, что реакция стереоспецифична. Предпочтительно в случае, если требуется определенный стереоизомер, указанное соединение можно синтезировать стереоспецифичными способами. В указанных способах будут преимущественно использоваться энантиомерно чистые исходные вещества. Рацематы диастереомеров соединений и промежуточных соединений данного изобретения можно разделить отдельно обычными способами. Наиболее выгодными физическими способами разделения являются, например, избирательная кристаллизация и хроматография, например, колоночная. Специалисту в данной области техники понятно, что соединения и промежуточные соединения данного изобретения содержат, по меньшей мере, два асимметрических центра и таким образом могут существовать в различных стереоизомерных формах. Такие асимметрические центры на фигурах, приведенных ниже, указаны звездочками (*)
Абсолютная конфигурация каждого асимметрического центра, который может присутствовать в соединениях и промежуточных соединениях данного изобретения, может быть указана стереохимическими дескрипторами R и S, причем такое обозначение R и S соответствует правилам, прописанным в Pure Appl. Chem., 1976, 45, 11-30. Также подразумевается, что настоящее изобретение включает все изотопы, встречающиеся в соединениях настоящего изобретения. Изотопы включают атомы с одинаковым атомным номером, но с разными массовыми числами. Например, как общий пример без ограничения, изотопы водорода включают тритий и дейтерий. Изотопы углерода включают С-13 и С-14. Реагенты и растворители, используемые в описании, можно заменять их функциональными альтернативами или функциональными производными, известными специалистам в данной области техники. Также такие условия реакций, как время перемешивания, очистку и температуру, можно оптимизировать для реакции. Подобным образом, продукты реакции можно извлекать из среды и, при необходимости, дополнительно очищать согласно методологиям, вообще известным в технике, таким как, например, экстракция, кристаллизация, растирание в порошок и хроматография. Ряд промежуточных соединений и исходных веществ, используемых в вышеописанных способах получения, являются известными соединениями, в то время как другие можно получить согласно способам, известным в технике получения указанных или подобных соединений. Соединения формулы (5) и все промежуточные соединения, ведущие к образованию стереоизомерно чистых соединений, представляют особый интерес при получении 4-аминобензолсульфонамидов как ингибиторов протеазы ВИЧ, как раскрывается в WO 95/06030, WO 96/22287, WO 96/28418, WO 96/28463, WO 96/28464, WO 96/28465, WO 97/18205 и WO 02/092595, включенных в данное описание в качестве ссылок, и в частности, ингибитора протеазы ВИЧ соединения формулы (6), как и любые их соли присоединения, полиморфные и/или псевдополиморфные формы. Таким образом, настоящее изобретение также относится к ингибиторам протеазы ВИЧ, таким как соединение формулы (6), и любым их солям присоединения, полиморфным и/или псевдополиморфным формам, полученным с использованием любого промежуточного соединения, описанного в данном описании, где и промежуточные соединения, и соединение формулы (6) получают, как описано в настоящем изобретении. Таким образом, настоящее изобретение также относится к ингибиторам протеазы ВИЧ, таким как соединение формулы (6), и любым их солям присоединения, полиморфным и/или псевдополиморфным формам, полученным с использованием соединения (5) в качестве промежуточного соединения, где как соединение формулы (5), так и соединение формулы (6) получают так, как описано в настоящем изобретении. Приведенные далее примеры предназначены для пояснения настоящего изобретения. Примеры приводятся как примеры изобретения и не должны рассматриваться как ограничение объема изобретения. ПРИМЕРЫ Пример 1. Получение трет-бутилового эфира (1-бензил-2-гидрокси-3-изобутиламинопропил)карбаминовой кислоты К 154,4 кг изобутиламина добавляют трет-бутиловый эфир (1-оксиранил-2-фенилэтил)карбаминовой кислоты (53,3 кг) и затем раствор нагревают с обратным холодильником. При пониженном давлении из реакционной смеси удаляют изобутиламин и затем заменяют его толуолом. Пример 2. Получение трет-бутилового эфира (1-бензил-2-гидрокси-3-[изобутил(4-нитробензолсульфонил)амино]пропил)карбаминовой кислоты К раствору, полученному в примере 1, добавляют 26,7 кг триэтиламина и полученный раствор нагревают до 82-88°С. К раствору постепенно добавляют раствор 4-нитробензолсульфонилхлорида (53 кг) в толуоле и перемешивают. Полученную реакционную смесь промывают водой. Промытый раствор трет-бутилового эфира (1-бензил-2-гидрокси-3-[изобутил(4-нитробензолсульфонил)амино]пропил)карбаминовой кислоты нагревают и затем добавляют толуол и н-гептан. Полученный раствор охлаждают и вносят затравку кристаллов трет-бутилового эфира (1-бензил-2-гидрокси-3-[изобутил(4-нитробензолсульфонил)амино]пропил)карбаминовой кислоты. После того как наблюдают выпадение кристаллического вещества в осадок, раствор перемешивают и затем постепенно охлаждают до 20-30°С. Полученное кристаллическое вещество отфильтровывают и промывают смешанным растворителем, состоящим из толуола и н-гептана, и получают влажные кристаллы трет-бутилового эфира (1-бензил-2-гидрокси-3-[изобутил(4-нитробензолсульфонил)амино]пропил) карбаминовой кислоты (выход 87-91% относительно трет-бутилового эфира (1-оксиранил-2-фенилэтил)карбаминовой кислоты). Пример 3. Получение трет-бутилового эфира (1-бензил-2-гидрокси-3-[изобутил(4-аминобензолсульфонил)амино]пропил) карбаминовой кислоты Влажные кристаллы трет-бутилового эфира (1-бензил-2-гидрокси-3-[изобутил(4-нитробензолсульфонил)амино]пропил) карбаминовой кислоты суспендируют в этаноле (примерно 950 л) и затем гидрируют в присутствии 10 мас.% палладия на угле при температуре примерно 5-30°С. Палладий на угле из реакционной смеси удаляют фильтрацией и затем фильтрат концентрируют при пониженном давлении с получением раствора трет-бутилового эфира (1-бензил-2-гидрокси-3-[изобутил(4-аминобензолсульфонил)амино]пропил)карбаминовой кислоты в этаноле. Пример 4. Получение 4-амино-N-(2R,3S)(3-амино-2-гидрокси-4-фенилбутил)-N-изобутилбензолсульфонамида Раствор трет-бутилового эфира (1-бензил-2-гидрокси-3-[изобутил(4-аминобензолсульфонил)амино]пропил) карбаминовой кислоты, полученный в примере 3, кипятят с обратным холодильником и затем добавляют концентрированную соляную кислоту (35-37 кг). Раствор перемешивают. Затем полученный раствор охлаждают до температуры 40±3°С и затем добавляют воду. рН раствора доводят до примерно 9,5 водным раствором гидроксида натрия, при этом образуются кристаллы 4-амино-N-(3-амино-2-гидрокси-4-фенилбутил)-N-изобутилбензолсульфонамида. К полученному раствору еще добавляют воду для доведения концентрации 4-амино-N-(3-амино-2-гидрокси-4-фенилбутил)-N-изобутилбензолсульфонамида до 5,5-5,8 мас.% и затем полученный раствор охлаждают до температуры 6±4°С. Полученное кристаллическое вещество отфильтровывают и промывают смешанным растворителем, состоящим из воды и этанола, и затем промывают водой. Полученное влажное кристаллическое вещество сушат в вакууме и получают продукт (2R,3S)-N-(3-амино-2-гидрокси-4-фенилбутил)-N-изобутил-4-аминобензолсульфонамид. Выход составляет 75-85% относительно трет-бутилового эфира (1-бензил-2-гидрокси-3-[изобутил(4-нитробензолсульфонил)амино]пропил)карбаминовой кислоты. Пример 5. Получение 4-амино-N-((2R,3S)-3-амино-2-гидрокси-4-фенилбутил)-N-(изобутил)бензолсульфонамида Суспендируют в метаноле 50,00 г трет-бутилового эфира (1-бензил-2-гидрокси-3-[изобутил(4-нитробензолсульфонил)амино]пропил) карбаминовой кислоты, полученного согласно процедурам, описанным в WO 99/48885, WO 01/12599 и WO 01/46120, 2 мол.% этаноламина и палладий на активированном угле, продувают инертным газом и создают вакуум. При температуре в реакторе 22-30°С подают примерно 3,0 экв. водорода при избыточном давлении. Затем катализатор удаляют фильтрацией. Бесцветный (до слегка желтоватого) раствор обрабатывают 21,70 г соляной кислоты, (37 мас.%), и смесь кипятят с обратным холодильником в течение 2 часов. По завершении превращения метанол удаляют перегонкой. Проводят осаждение из смеси растворителей МеОН/вода/IPA, 1:8:6,5. При температуре 0-7°С добавляют порциями 30% раствор гидроксида натрия до тех пор, пока величина рН не станет pH>12,5. Через 4-48 часов выпавшее в осадок вещество белого цвета отфильтровывают, промывают водой и изопропанолом. Влажный продукт реакции сушат в вакууме при 65 оС. Способ дает 36,94 г порошка от белого до желтоватого цвета. Пример 6. Получение 4-амино-N-((2R,3S)-3-амино-2-гидрокси-4-фенилбутил)-N-изобутил)бензолсульфонамида Суспендируют в этаноле 50,00 г трет-бутилового эфира (1-бензил-2-гидрокси-3-[изобутил(4-нитробензолсульфонил)амино]пропил) карбаминовой кислоты, полученного согласно процедурам, описанным в WO 99/48885, WO 01/12599 и WO 01/46120, и палладий на активированном угле продувают инертным газом, и создают вакуум. При температуре в реакторе 22-30°С подают примерно 3,0 экв. водорода при избыточном давлении. Затем катализатор удаляют фильтрацией. После отгонки спирта в колбе остается трет-бутиловый эфир (1-бензил-2-гидрокси-3-[изобутил(4-аминобензолсульфонил)амино]пропил) карбаминовой кислоты в виде бесцветной пены с выходом 97%. трет-Бутиловый эфир (1-бензил-2-гидрокси-3-[изобутил(4-аминобензолсульфонил)амино]пропил) карбаминовой кислоты растворяют в метаноле, обрабатывают 21,70 г соляной кислоты (37 мас.%) и кипятят с обратным холодильником в течение 2 часов. После завершения превращения большую часть спирта удаляют отгонкой. Солянокислую соль 4-амино-N-((2R,3S)-3-амино-2-гидрокси-4-фенилбутил)-N-изобутил)бензолсульфонамида осаждают, удаляя большую часть спирта отгонкой и добавляя дихлорметан к теплому (40°С) раствору. При перемешивании и охлаждении до комнатной температуры солянокислая соль сразу же выпадает в осадок. Осаждение 4-амино-N-((2R,3S)-3-амино-2-гидрокси-4-фенилбутил)-N-изобутил)бензолсульфонамида проводят, растворяя хлороводородную соль в смеси растворителей EtОН/вода, 1:1. При температуре 0-7 оС добавляют порциями 30% раствор гидроксида натрия до тех пор, пока величина рН не достигнет pH>12,5. Через 4-48 часов выпавшее в осадок вещество белого цвета отфильтровывают, промывают водой и сушат в вакууме. Способ дает 33,78 г порошка от белого до желтоватого цвета. Пример 7. Получение этанолята (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ил(1S,2R)-3-[[(4-аминофенил)сульфонил](изобутил)амино]-1-бензил-2-гидроксипропилкарбамата К 120 ммоль дисукцинимидилкарбоната (95%) в ацетонитриле добавляют 100 ммоль (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ола в этилацетате. Затем добавляют раствор 140 ммоль триэтиламина в этилацетате и перемешивают. Смесь охлаждают и обрабатывают суспензией 92 ммоль 4-амино-N-((2R,3S)-3-амино-2-гидрокси-4-фенилбутил)-N-(изобутил)бензолсульфонамида в этилацетате. Добавляют в этаноле 20 ммоль метиламина в виде 41% водного раствора и смесь греют. Реакционную смесь дважды промывают 10% раствором Na2CO3 и водой. Растворитель выпаривают и добавляют этанол. Отгоняют еще одну часть растворителя. Поддерживают температуру примерно 40-45°С и инициируют кристаллизацию затравкой. После перемешивания смесь охлаждают, перемешивают еще в течение 90 мин, охлаждают и снова перемешивают в течение 60 мин. Выпавшее в осадок вещество отфильтровывают и промывают этанолом. Влажный продукт реакции сушат в вакууме при 40°С. Суспендируют и растворяют в абсолютном этаноле 43,5 г (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ил(1S,2R)-3-[[(4-аминофенил)сульфонил](изобутил)амино]-1-бензил-2-гидроксипропилкарбамата. Прозрачный раствор охлаждают и вносят затравку. При охлаждении смеси происходит кристаллизация. Продолжают перемешивание еще в течение 60 мин, затем смесь охлаждают, перемешивают и отфильтровывают продукт, который промывают холодным абсолютным этанолом. Влажный продукт сушат в вакууме при 40°С. Выход 42,1 г = 71%. Пример 8. Получение этанолята (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ил(1S,2R)-3-[[(4-аминофенил)сульфонил](изобутил)амино]-1-бензил-2-гидроксипропилкарбамата К 105 ммоль бис(4-нитрофенил)карбоната в ацетонитриле добавляют 100 ммоль (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ола в этилацетате. Затем добавляют раствор 250 ммоль триэтиламина в этилацетате и перемешивают. Смесь обрабатывают суспензией 95 ммоль 4-амино-N-((2R,3S)-3-амино-2-гидрокси-4-фенилбутил)-N-(изобутил)бензолсульфонамида в этилацетате. Добавляют в этаноле 20 ммоль метиламина в виде 41% водного раствора. Реакционную смесь три раза промывают 10% раствором К2CO3 и водой. Растворитель выпаривают и добавляют этанол. Отгоняют еще одну часть растворителя. Поддерживают температуру примерно 40-45°С и инициируют кристаллизацию затравкой. После перемешивания смесь охлаждают, перемешивают еще в течение 90 мин, охлаждают и снова перемешивают в течение 60 мин. Выпавшее в осадок вещество отфильтровывают и промывают этанолом. Влажный продукт реакции сушат в вакууме при 40°С. Суспендируют и растворяют в абсолютном этаноле 43,5 г (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ил(1S,2R)-3-[[(4-аминофенил)сульфонил](изобутил)амино]-1-бензил-2-гидроксипропилкарбамата. Прозрачный раствор охлаждают и вносят затравку. При охлаждении смеси происходит кристаллизация. Продолжают перемешивание еще в течение 60 мин, затем смесь охлаждают, перемешивают и отфильтровывают продукт, который промывают холодным абсолютным этанолом. Влажный продукт сушат в вакууме при 40°С. Выход 47,9 г = 81%. Пример 9. Получение этанолята (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ил(1S,2R)-3-[[(4-аминофенил)сульфонил](изобутил)амино]-1-бензил-2-гидроксипропилкарбамата К 110 ммоль дисукцинимидилкарбоната (95%) в ацетонитриле добавляют 100 ммоль (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ола в ацетонитриле. Затем добавляют 300 ммоль пиридина и перемешивают. Смесь охлаждают и обрабатывают суспензией 95 ммоль 4-амино-N-((2R,3S)-3-амино-2-гидрокси-4-фенилбутил)-N-(изобутил)бензолсульфонамида в ацетонитриле и затем добавляют 100 ммоль триэтиламина. Затем добавляют 20 ммоль метиламина в виде 41% водного раствора и смесь греют. Отгоняют 80 г растворителя, добавляют МТВЕ и реакционную смесь промывают 10% раствором Na2CO3, смесью сульфата натрия и серной кислоты и снова 10% раствором Na2CO3. Растворитель выпаривают и добавляют этанол. Отгоняют еще одну часть растворителя. Поддерживают температуру примерно 40-45°С и инициируют кристаллизацию затравкой. После перемешивания смесь охлаждают, перемешивают еще в течение 90 мин, охлаждают и снова перемешивают в течение 60 мин. Выпавшее в осадок вещество отфильтровывают и промывают этанолом. Влажный продукт реакции сушат в вакууме при 40°С. Суспендируют и растворяют в абсолютном этаноле 43,5 г (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ил(1S,2R)-3-[[(4-аминофенил)сульфонил](изобутил)амино]-1-бензил-2-гидроксипропилкарбамата. Прозрачный раствор охлаждают и вносят затравку. При охлаждении смеси происходит кристаллизация. Продолжают перемешивание еще в течение 60 мин, затем смесь охлаждают, перемешивают и отфильтровывают продукт, который промывают холодным абсолютным этанолом. Влажный продукт сушат в вакууме при 40°С. Выход 48,1 г = 81%.
Формула изобретения
1. Способ получения соединения формулы (6) 2. Способ по п.1, где стадию (i) осуществляют в толуоле. 3. Способ по п.2, где стадию (ii) осуществляют в толуоле, этилацетате, метиленхлориде, дихлорметане или тетрагидрофуране. 4. Способ по п.3, где стадию (iii) осуществляют в присутствии до 10 мол.% первичного или вторичного амина с палладием-на-угле в атмосфере водорода. 5. Способ по п.4, где стадию (iv) осуществляют в кислой или щелочной среде. 6. Способ по п.5, где соединение формулы (5) кристаллизуют, растворяя его в смеси растворителей, доводя рН до величины более 9 и поддерживая концентрацию соединения формулы (5) в растворе на уровне от 4 до 15% (мас./мас.). 7. Способ по п.6, где соединение формулы (5) кристаллизуют при температуре от 0 до 10°С. 8. Способ по п.7, где во время кристаллизации добавляют затравочные кристаллы соединения формулы (5). 9. Способ по п.8, где смесь растворителей содержит один или несколько растворителей, смешивающихся с водой, и воду. 10. Способ по п.8, где смесь растворителей содержит один или несколько растворителей, несмешивающихся с водой, и воду. 11. Способ по п.9, где смесь растворителей представляет собой метанол, изопропанол и воду в соотношении 1:6,5:8 соответственно. 12. Способ по п.11, где (3R,3аS,6аR)-гексагидрофуро[2,3-b]фуран-3-ол или его предшественника перед сочетанием с соединением формулы (5) вводят во взаимодействие с бис (4-нитрофенил)карбонатом. 13. Способ по п.11, где (3R,3аS,6аR)-гексагидрофуро[2,3-b]фуран-3-ол или его предшественника перед сочетанием с соединением формулы (5) вводят во взаимодействие с дисукцинимидилкарбонатом. 14. Способ по п.12 или 13, где взаимодействие (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ола или его предшественника с производным карбоновой кислоты активируют основанием.
|
||||||||||||||||||||||||||
 -2,4,5,-тетраметилбензилоксикарбонил (“Tmz”), 4-метоксибензилоксикарбонил, 4-фторбензилоксикарбонил, 4-хлорбензилоксикарбонил, 3-хлорбензилоксикарбонил, 2-хлорбензилоксикарбонил, дихлорбензилоксикарбонил, 4-бромбензилоксикарбонил, орто-бромбензилоксикарбонил, 3-бромбензилоксикарбонил, 4-нитробензилоксикарбонил, 4-цианобензилоксикарбонил, 4-(децилокси)бензилоксикарбонил и т.п., бензоилметилсульфонильная группа, дитиосукциноильная (“Dts”) группа, 2-(нитро)фенилсульфенильная группа (“Nps”), дифенилфосфиноксидная группа и подобные группы. Виды используемых аминозащитных групп, как правило, не являются определяющим фактором до тех пор, пока дериватизированная аминогруппа устойчива в условиях последующих реакций и может быть удалена в подходящий момент без разрушения остальной части соединения.
-2,4,5,-тетраметилбензилоксикарбонил (“Tmz”), 4-метоксибензилоксикарбонил, 4-фторбензилоксикарбонил, 4-хлорбензилоксикарбонил, 3-хлорбензилоксикарбонил, 2-хлорбензилоксикарбонил, дихлорбензилоксикарбонил, 4-бромбензилоксикарбонил, орто-бромбензилоксикарбонил, 3-бромбензилоксикарбонил, 4-нитробензилоксикарбонил, 4-цианобензилоксикарбонил, 4-(децилокси)бензилоксикарбонил и т.п., бензоилметилсульфонильная группа, дитиосукциноильная (“Dts”) группа, 2-(нитро)фенилсульфенильная группа (“Nps”), дифенилфосфиноксидная группа и подобные группы. Виды используемых аминозащитных групп, как правило, не являются определяющим фактором до тех пор, пока дериватизированная аминогруппа устойчива в условиях последующих реакций и может быть удалена в подходящий момент без разрушения остальной части соединения.