Патент на изобретение №2376299
|
||||||||||||||||||||||||||
(54) ПИРРОЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ERK ПРОТЕИНКИНАЗ, ИХ СИНТЕЗ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ
(57) Реферат:
Настоящее изобретение относится к соединениям формулы I
С1-4-алифатическую группу; R2 обозначает R, фтор или хлор; m обозначает 0, 1 или 2 и R3 обозначает водород, C1-3-алифатическую группу, фтор или хлор, к композиции для ингибирования активности протеинкиназы ERK1 или ERK2 на основе этих соединений, к способу ингибирования активности протеинкиназы ERK1 или ERK2, а также к применению соединений формулы I или композиции на их основе для лечения или уменьшения серьезности заболевания. Технический результат: получены и описаны новые соединения, которые могут быть использованы в качестве ингибиторов протеинкиназ. 5 н.и 6 з.п. ф-лы, 3 табл.
ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ По настоящей заявке испрашивается приоритет на основании Предварительной Заявки на патент Соединенных Штатов 60/571309, поданной 14 мая 2004, все содержание которой тем самым включено в настоящую заявку путем ссылки. ТЕХНИЧЕСКАЯ ОБЛАСТЬ ИЗОБРЕТЕНИЯ Настоящее изобретение относится к соединениям, которые могут быть использованы как ингибиторы протеинкиназ. Изобретение также относится к фармацевтически приемлемым композициям, включающим соединения по изобретению, и к способам применения композиций в лечении различных нарушений. УРОВЕНЬ ТЕХНИКИ Поиску новых терапевтических средств значительно помогли в последние годы лучшее понимание структуры ферментов и других биомолекул, связанных с заболеваниями, к лечению которых относится изобретение. Один важный класс ферментов, который был объектом обширных исследований, составляют протеинкиназы. Протеинкиназы составляют большое семейство структурно родственных ферментов, которые являются ответственными за контроль разнообразных процессов проведения сигналов в пределах клетки (см. Hardie, G. and Hanks, S. (1995) The Protein Kinase Facts Book, I and II, Academic Press, San Diego, CA). Считается, что протеинкиназы эволюционировали от общего генетического предшественника, о чем свидетельствует консерватизм их структуры и каталитической функции. Почти все киназы содержат сходный каталитический домен из 250-300 аминокислот. Киназы могут быть классифицированы по семействам на основании субстратов, которые они фосфорилируют (например, белок-тирозин, белок-серин/треонин, липиды и т.д.). Были идентифицированы звенья последовательности, которые в целом соответствуют каждому из этих семейств киназ (см., например, Hiles et al., Cell, 70:419-429 (1992) Knighton et al., Science, 253:407-414 (1991); Hanks, S.K., Hunter, T., FASEB J., 9:576-596 (1995); Garcia-Bustos et al., EMBO J., 13:2352-2361 (1994 Kunz et al., Cell,73:585-596 (1993)). В целом, протеинкиназы опосредуют внутриклеточное проведение сигналов, осуществляя перенос фосфорила от нуклеозидтрифосфата к белковому акцептору, который участвует в осуществлении сигнального пути. Эти события фосфорилирования действуют как молекулярный включатель, который может модулировать или регулировать биологическую функцию целевого белка. Эти события фосфорилирования в конечном счете запускаются в ответ на различные внеклеточные и другие раздражители. Примеры таких раздражителей включают сигналы экологического и химического стресса (например, осмотический шок, тепловой шок, ультрафиолетовое излучение, бактериальный эндотоксин и H2O2), цитокины (например, интерлейкин-1 (IL-I) и фактор некроза опухоли (TNF- Многие заболевания связаны с патологическими клеточными реакциями, вызываемыми опосредуемыми протеинкиназой событиями. Эти заболевания включают аутоиммунные заболевания, воспалительные заболевания, заболевания костей, нарушения обмена веществ, неврологические и нейродегенеративные заболевания, рак, сердечно-сосудистые заболевания, аллергии и астму, болезнь Альцгеймера и связанные с гормонами заболевания. Соответственно, в области фармхимии предпринимались усилия для того, чтобы найти ингибиторы протеинкиназ, которые были бы эффективными как терапевтические средства. Однако, учитывая отсутствие в настоящее время доступных вариантов лечения для большинства состояний, связанных с протеинкиназами, все еще существует большая потребность в новых терапевтических средствах, которые ингибируют эти белковые мишени. Клетки млекопитающих отвечают на внеклеточные раздражители, активируя сигнальные каскады, которые опосредуются семейством митоген-активируемых протеинкиназ (МАР), которые включают регулируемые внеклеточным сигналом киназы (ERK), p38 МАР-киназы и c-Jun N-концевые киназы (JNK). МАР-киназы (MAPK) активируются различными сигналами, включая факторы роста, цитокины, УФ-излучение и стресс-индуцирующие агенты. MAPK представляют собой серин/треонин-киназы, и их активация происходит за счет двойного фосфорилирования треонина и тирозина на сегменте Thr-X-Tyr в активирующей петле. MAPK фосфорилируют различные субстраты, включая факторы транскрипции, которые в свою очередь регулируют экспрессию специфических наборов генов и, таким образом, опосредуют специфический ответ на раздражитель. ERK2 представляет собой широко распространенную протеинкиназу, которая достигает максимальной активности, когда одновременно Tyr183 и Tyr185 фосфорилируются МАР-киназ-киназой, MEK1 (Anderson et al., 1990, Nature 343, 651; Crews et al., 1992, Science 258,478). После активации ERK2 фосфорилирует многие регуляторные белки, включая протеинкиназы Rsk90 (Bjorbaek et al., 1995, J. Biol. Chem. 270, 18848) и MAPKAP2 (Rouse et al., 1994, Cell 78, 1027), и факторы транскрипции, такие как ATF2 (Raingeaud et al., 1996, Mol. Cell Biol. 16, 1247), Elk-I (Raingeaud et al. 1996), c-Fos (Chen et al., 1993 Proc. Natl. Acad. Sei. USA 90, 10952), и c-Myc (Oliver et al., 1995, Proc. Soc. Exp. Biol. Med. 210, 162). ERK2 также является мишенью на более поздних стадиях Ras/Raf-зависимых путей (Moodie et al., 1993, Science 260, 1658) и передает сигналы от этих потенциально онкогенных белков. Показано, что ERK2 играет роль в отрицательном контроле роста клеток рака молочной железы (Frey and Mulder, 1997, Cancer Res. 57, 628), и сообщалось о гиперэкспрессии ERK2 при раке молочной железы у человека (Sivaraman et al., 1997, J Clin. Invest. 99, 1478). Активированная ERK2 также участвует в пролиферации стимулируемых эндотелином гладких мышечных клеток дыхательных путей, что говорит о роли этой киназы в механизме астмы (Whelchel et al., 1997, Am. J. Respir. Cell MoI. Biol. 16, 589). Повышенная экспрессия рецептора тирозинкиназ, такого как EGFR и ErbB2 (Arteaga CL, 2002, Semin Oncol. 29, 3-9; Eccles SA, 2001, J. Mammary Gland Biol Neoplasia 6:393-406; Mendelsohn J & Baselga J, 2000, Oncogene 19, 6550-65), а также активирующие мутации в Ras ГТФ-азных белках (Nottage M & Siu LL, 2002, Curr Pharm Des 8, 2231-42; Adjei AA, 2001, J Natl Cancer Inst 93, 1062-74) или мутанты B-Raf (Davies H. et al., 2002, Nature 417, 949-54; Brose et al., 2002, Cancer Res 62, 6997-7000) являются основными причинами рака у человека. Эти генетические альтерации коррелируют с плохим клиническим прогнозом и приводят к активации Raf-1/2/3-MEK1/2-ERK1/2 каскада трансдукции сигнала в обширной группе опухолей у человека. Активированная ERK (то есть ERK1 и/или ERK2) является центральной сигнальной молекулой, которая связана с контролем пролиферации, дифференцировки, независимого от заякоревания выживания клетки и ангиогенеза, внося свой вклад в множество процессов, которые являются важными для формирования и прогрессии злокачественных опухолей. Эти данные позволяют предположить, что ингибитор ERK1/2 будет проявлять плейотропную активность, включая проапоптотические, антипролиферативные, антиметастатические и антиангиогенные эффекты, и предлагают терапевтический подход в отношении очень широкой группы опухолей у человека. Существует растущий набор очевидных данных о том, что конститутивная активация пути ERK МАРК вовлечена в осуществление онкогенного режима обсуждаемых видов рака. Активирующие мутации Ras обнаруживаются в ~30% всех видов рака, в некоторых, таких как рак поджелудочной железы (90%) и толстого кишечника (50%), достигая особенно высокой частоты мутации. Мутации Ras были также идентифицированы в 9-15% меланом, однако соматические миссенс-мутации B-Raf, придающие конститутивную активацию, являются более частыми и обнаруживаются в 60-66% злокачественных меланом. Активирующие мутации Ras, Raf и MEK способны онкогенно трансформировать фибробласты in vitro, и Ras или Raf мутации, в сочетании с потерей гена супрессора опухоли (например, p16INK4A), могут вызывать спонтанное развитие опухоли in vivo. Увеличенная активность ERK была показана в этих моделях, и о ней также широко сообщалось в отношении соответствующих опухолей у человека. При меланоме зарегистрирована высокая базовая активность ERK, происходящая из B-Raf или N-Ras мутации или аутокринной активации фактора роста, и ее связывают с быстрым ростом опухоли, увеличенной выживаемостью клеток и устойчивостью к апоптозу. Дополнительно, активацию ERK считают главной движущей силой высоко метастатического режима меланомы, связанной с увеличенной экспрессией как протеаз, разлагающих внеклеточную матрицу, так и промотирующих инвазию интегринов, наряду с ингибированием молекул адгезии E-кадгерина, которые в норме опосредуют взаимодействие кератиноцитов, контролируя рост меланоцитов. Эти данные, взятые вместе, показывают, что ERK являются многообещающей терапевтической мишенью в отношении лечения меланомы, в настоящее время не поддающегося лечению заболевания. КРАТКОЕ СОДЕРЖАНИЕ ИЗОБРЕТЕНИЯ В настоящее время обнаружено, что соединения по изобретению и их фармацевтически приемлемые композиции являются эффективными в качестве ингибиторов ERK протеинкиназы. Эти соединения имеют общую формулу I:
или их фармацевтически приемлемую соль, в которой m, R1, R2 и R3 имеют значения, определенные ниже. Эти соединения и их фармацевтически приемлемые композиции могут быть использованы для лечения или снижения серьезности различных нарушений, особенно пролиферативных нарушений, таких как рак. Соединения согласно настоящему изобретению также могут быть использованы для исследования киназ в биологических и патологических явлениях и в исследовании путей внутриклеточной трансдукции сигнала, опосредуемых такими киназами, и для сравнительной оценки новых ингибиторов киназ. ПОДРОБНОЕ ОПИСАНИЕ НЕКОТОРЫХ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ 1. Общее описание соединений по изобретению Настоящее изобретение относится к соединению формулы I:
или его фармацевтически приемлемой соли, в которой: R1 обозначает C1-6 алифатическую группу, причем R1 может быть замещен заместителями в числе до 2 групп, независимо от выбранных из -OR или -C1-3 галогеналкила; каждый R независимо обозначает водород или C1-4 алифатическую группу; каждый R2 независимо обозначает R, фтор или хлор; m обозначает 0, l или 2 и R3 обозначает водород, C1-3 алифатическую группу, фтор или хлор. 2. Соединения и определения Соединения по изобретению включают описываемые в целом выше и далее иллюстрируются классами, подклассами и разновидностями, раскрытыми в настоящем описании. В рамках изобретения должны использоваться следующие определения, если не указано иное. В целях этого изобретения, химические элементы идентифицированы в соответствии с Периодической таблицей элементов, версия CAS, Handbook of Chemistry and Physics, 75th Ed. Дополнительно, общие принципы органической химии описаны в “Organic Chemistry”, Thomas Sorrell, University Science Books, Sausalito: 1999, и “March’s Advanced Organic Chemistry”, 5th Ed., Ed.: Smith, M.B. and March, J., John Wiley & Sons, New York: 2001, все содержание которых тем самым включено в настоящее описание путем ссылки. В рамках изобретения, термин “пролекарство” относится к производному материнской молекулы лекарственного средства, которое требует трансформации в организме для высвобождения активного лекарственного средства и которое обладает улучшенными физическими свойствами и/или свойствами доставки по сравнению с материнской молекулой лекарственного средства. Пролекарства предназначены для того, чтобы усилить фармацевтические и/или фармакокинетические свойства, связанные с материнской молекулой лекарственного средства. Преимущество пролекарства заключается в его физических свойствах, таких как увеличенная водорастворимость для парентерального введения при физиологическом рН по сравнению с исходным лекарственным средством, или оно усиливает всасывание из пищеварительного тракта, или оно может увеличить стабильность лекарственного средства для длительного хранения. В последние годы несколько типов биообратимых производных были использованы для проектирования пролекарств. Использование сложных эфиров в качестве одного типа пролекарства для препаратов, содержащих карбоксильную или гидроксильную группу, известно в уровне техники и описано, например, в “The Organic Chemistry of Drug Design and Drug Interaction” Richard Silverman, published by Academic Press (1992). Как описано здесь, соединения по изобретению могут быть в случае необходимости замещены одним или более заместителями, такими как проиллюстрированные выше, или как иллюстрируется особыми классами, подклассами и разновидностями по изобретению. Следует понимать, что фраза «в случае необходимости замещенный» используется взаимозаменяемо с фразой «замещенный или незамещенный». В общем, термин «замещенный», предваряемый или нет термином «в случае необходимости», относится к замене водородных радикалов в данной структуре радикалом указанного заместителя. Если не указано иное, в случае необходимости замещенная группа может иметь заместитель в каждом пригодном для замещения положении группы, и в случае, если более одного положения в любой данной структуре может быть замещено более чем одним заместителем, выбранным из определенной группы, заместитель в каждом положении может быть тем же самым или отличным. Комбинации заместителей, которые могут быть использованы в соответствии с этим изобретением, предпочтительно являются такими, которые приводят к формированию стабильных или химически возможных соединений. Термин “стабильный” в рамках изобретения относится к соединениям, которые в основном не подвергаются изменениям под действием условий, необходимых для их получения, детекции и, предпочтительно, их рекуперации, очистки и использования для одной или более целей, раскрытых в настоящем изобретении. В некоторых вариантах осуществления стабильное соединение или химически возможное соединение представляет собой соединение, которое в основном не подвергается изменению при хранении при температуре 40°C или ниже, в отсутствие влажности или в других химически реактивных условиях, в течение по меньшей мере недели. Термин “алифатический радикал” или “алифатическая группа” в рамках изобретения обозначает прямую (то есть неразветвленную) или разветвленную, замещенную или незамещенную углеводородную цепь, которая является полностью насыщенной или содержит одно или более ненасыщенных звеньев, или моноциклический углеводород, который является полностью насыщенным или содержит одно или более ненасыщенных звеньев, но который не является ароматическим (также упомянутый здесь как “карбоцикл” “циклоалифатический радикал” или “циклоалкил”), который присоединен к остальной части молекулы в одной точке. В некоторых вариантах осуществления алифатические группы содержат 1-6 алифатических атомов углерода, а в других вариантах осуществления алифатические группы содержат 1-4 алифатических атома углерода. В некоторых вариантах осуществления “циклоалифатический радикал” (или “карбоцикл”, или “циклоалкил”) относится к моноциклическому С3-С6-углеводороду, который является полностью насыщенным или содержит одно или более ненасыщенных звеньев, но который не является ароматическим, который присоединен к остальной части молекулы в одной точке. Подходящие алифатические группы включают, но не ограничены ими, прямой или разветвленный, замещенный или незамещенный алкил, алкенил, алкинил и их гибриды, такие как (циклоалкил)алкил, (циклоалкенил)алкил или (циклоалкил)алкенил. Термин “ненасыщенный” в рамках изобретения означает, что часть имеет одно или более ненасыщенных звеньев. Термины “галогеналкил”, “галогеналкенил” и “галогеналкокси” означают алкил, алкенил или алкокси, в зависимости от обстоятельств замещенный одним или более атомами галогена. Термин “галоген” означает F, Cl, Br или I. Термин “арил”, использованный отдельно или как часть большего определения, такого как “аралкил”, “аралкокси” или “арилоксиалкил”, относится к моноциклической, бициклической и трициклической кольцевым системам, имеющим в общей сложности от пяти до четырнадцати кольцевых членов, причем по меньшей мере одно кольцо в системе является ароматическим, и причем каждое кольцо в системе содержит 3-7 кольцевых членов. Термин “арил” может использоваться взаимозаменяемо с термином “арильное кольцо”. Арил (включая аралкил, аралкокси, арилоксиалкил и т.п.) или гетероарил (включая гетероаралкил и гетероарилалкокси и т.п.) может содержать один или более заместителей. Подходящие заместители на ненасыщенном атоме углерода арила или гетероарила выбирают из галогена; R°; OR°; SR°; 1,2-метилен-диокси; 1,2-этилендиокси; фенила (Ph), в случае необходимости замещенного R°; -О(Ph), в случае необходимости замещенного R°; (CH2)1-2(Ph), в случае необходимости замещенного R°; CH=CH(Ph), в случае необходимости замещенного R°; NO2; CN; N(R°)2; NR°C(O)R°; NR°C(О)N(R°)2; NR°CO2R°; -NR°NR°C(O)R°; NR°NR°C(O)N(R°)2; NR°NR°CO2R°; C(O)C(O)R°; C(O)CH2C(O)R°; CO2R°; C(O)R°; C(О)N(R°)2; ОС(O)N(R°)2; С(O)2R°; SO2N(R°)2; С(O)R°; NR°SО2N(R°)2; NR°SO2R°; C(=S)N(R°)2; C(=NH)-N(R°)2; или (CH2)0-2NHC(О)R°, причем каждый R° независимо выбирают из водорода, в случае необходимости замещенного C1-6 алифатического радикала, незамещенных 5-6 членных гетероарила или гетероциклического ядра, фенила, O(Ph) или CH2(Ph), или, несмотря на определение, данное выше, два независимых R°, в том же самом заместителе или в разных заместителях, вместе с атомом(ами), с которым каждая группа R° связана, образуют 3-8-членный циклоалкил, гетероциклил, арил или гетероарил, имеющее 0-4 гетероатома, независимо выбранные из азота, кислорода или серы. Возможные заместители на алифатической группе R° выбраны из NH2, NH(C1-4-алифатической группы), N(C1-4-алифатической группы)2, галогена, C1-4-алифатической группы, ОН, О(C1-4-алифатической группы), NO2, CN, CO2H, CO2(C1-4-алифатической группы), О(галоген-C1-4-алифатической группы) или галоген-C1-4-алифатической группы, причем каждая из указанных C1-4-алифатической группы групп в R° является незамещенной. Алифатическая или гетероалифатическая группа или неароматическое гетероциклическое ядро могут содержать один или более заместителей. Подходящие заместители на насыщенном углероде алифатической или гетероалифатической группы или неароматического гетероциклического ядра выбраны из упомянутых выше для ненасыщенного углерода арила или гетероарила и дополнительно включают следующее: =О, =S, =NNHR*, =NN(R*)2, =NNHC(О)R*, =NNHCО2(алкил), =NNHSО2(алкил) или =NR*, где каждый R* независимо выбран из водорода или в случае необходимости замещенной C1-6-алифатической группы. Возможные заместители на алифатической группе R* выбраны из NH2, NH(C1-4-алифатической группы), N(C1-4-алифатической группы)2, галогена, C1-4-алифатической группы, ОН, О(C1-4-алифатической группы), NO2, CN, CO2H, CO2(C1-4-алифатической группы), О(галоген- C1-4-алифатической группы) или галоген(C1-4-алифатической группы), причем каждая из указанных C1-4-алифатических групп в R* является незамещенной. Возможные заместители на азоте неароматического гетероциклического ядра выбраны из R+, N(R+)2, C(O)R+, CO2R+, C(O)C(O)R+, C(O)CH2C(O)R+, SO2R+, SО2N(R+)2, C(=S)N(R+)2, C(=NH)-N(R+)2 или NR+SO2R+; причем R+ обозначает водород, в случае необходимости замещенную C1-6-алифатическую группу, в случае необходимости замещенный фенил, в случае необходимости замещенный O(Ph), в случае необходимости замещенный CH2(Ph), в случае необходимости замещенный (CH2)1-2(Ph); в случае необходимости замещенный CH=CH(Ph); или незамещенный 5-6-членный гетероарил или гетероциклическое кольцо, имеющие один-четыре гетероатома, независимо выбранные из кислорода, азота или серы, или, несмотря на определение, данное выше, два независимых R+, на том же самом заместителе или на разных заместителях, вместе с атомом(ами), с которым каждая группа R+ связана, образуют 3-8-членный циклоалкил, гетероциклил, арил или гетероарил, имеющее 0-4 гетероатома, независимо выбранные из азота, кислорода или серы. Возможные заместители на алифатической группе или фениле R+ выбраны из NH2, NH(C1-4-алифатической группы), N(C1-4-алифатической группы)2, галогена, C1-4-алифатической группы, ОН, О(C1-4-алифатической группы), NO2, CN, CO2H, CO2(C1-4-алифатической группы), О(галоген-C1-4-алифатической группы) или галоген(C1-4-алифатической группы), причем каждая из указанных C1-4-алифатических групп в R+ является незамещенной. Если не указано иное, структуры, изображенные здесь, также включают все изомерные (например, энантиомерные, диастереомерные и геометрические (или конформационные)) формы структуры; например, конфигурации R и С для каждого центра асимметрии, (Z) и (E) изомеров двойной связи и (Z) и (E) конформационных изомеров. Поэтому отдельные стереохимические изомеры, а также энантиомерные, диастереомерные и геометрические (или конформационные) смеси настоящих соединений находятся в рамках изобретения. Если не указано иное, все таутомерные формы соединений по изобретению также входят в рамки изобретения. Дополнительно, если не указано иное, структуры, изображенные здесь, также включают соединения, которые отличаются только присутствием одного или более изотопически обогащенных атомов. Например, соединения, имеющие структуры по изобретению, за исключением замены водорода дейтерием или тритием или замены углерода 13C- или l4C-обогащенным углеродом, также входят в рамки этого изобретения. Такие соединения могут быть использованы, например, как аналитические инструменты или зонды в биологических анализах. 3. Описание примеров соединений Согласно одному варианту осуществления настоящее изобретение относится к соединению формулы I, причем указанное соединение имеет формулу Ia или Ib:
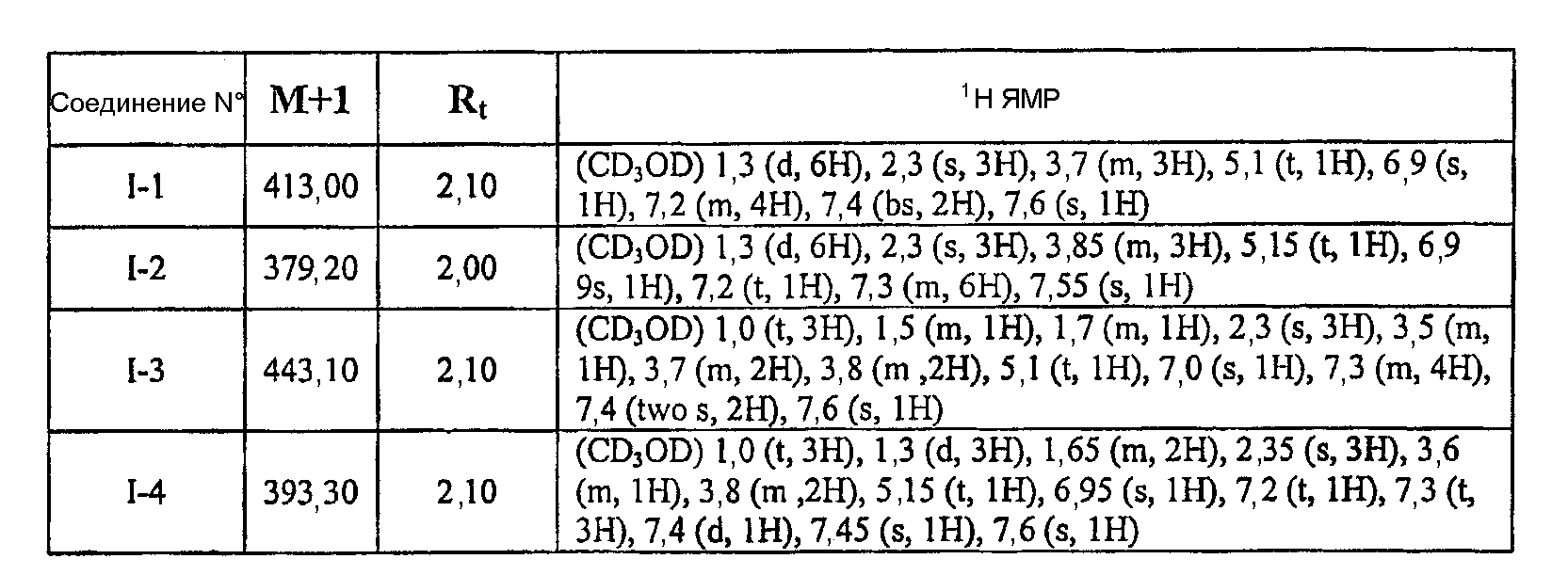
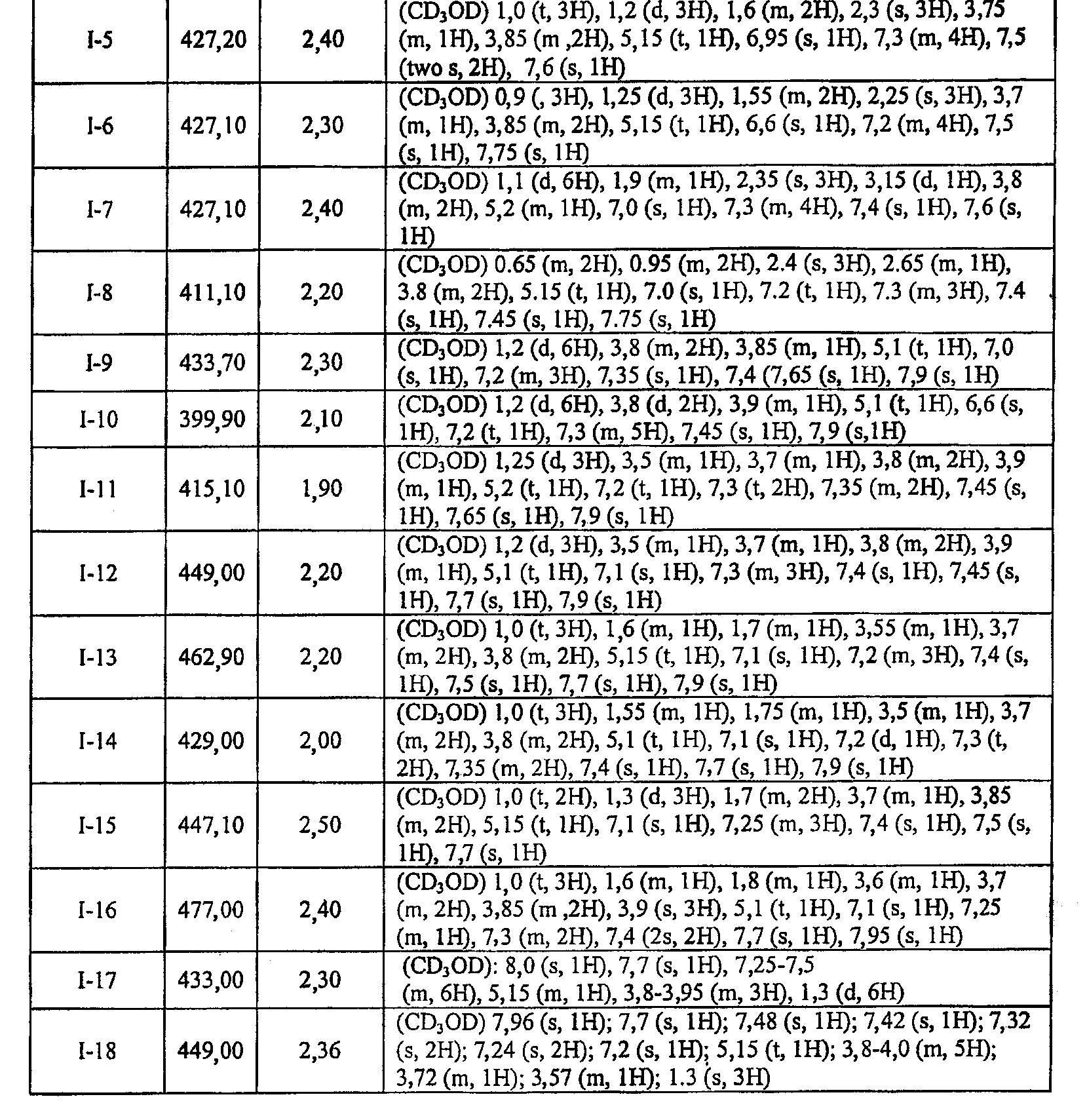
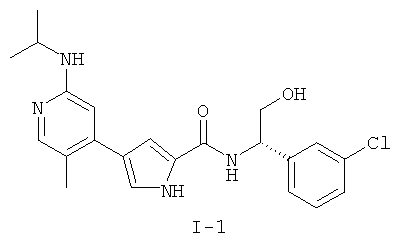
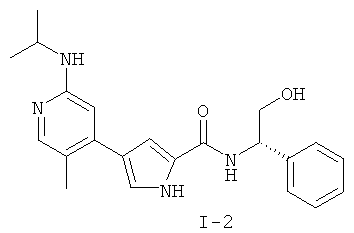
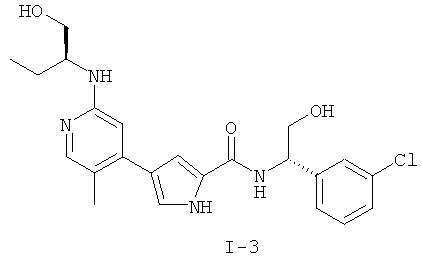
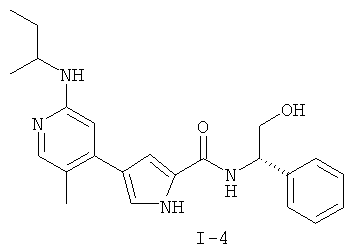
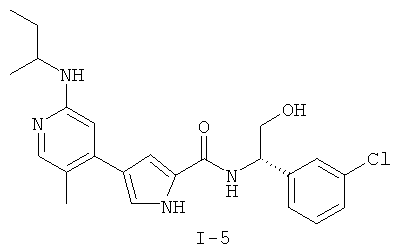
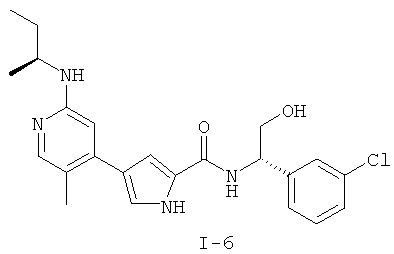
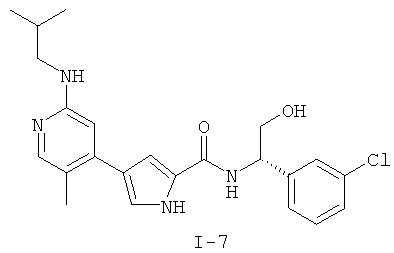
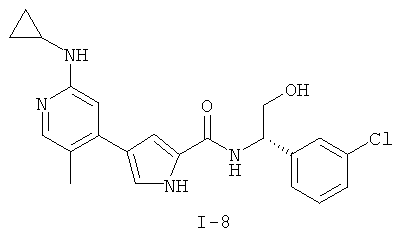
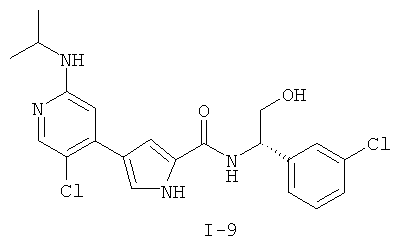
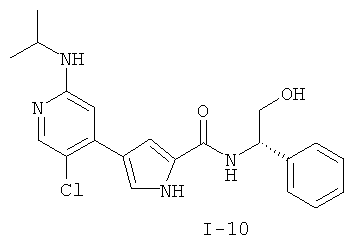
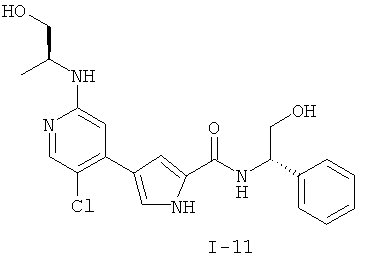
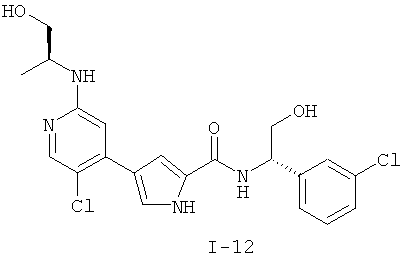
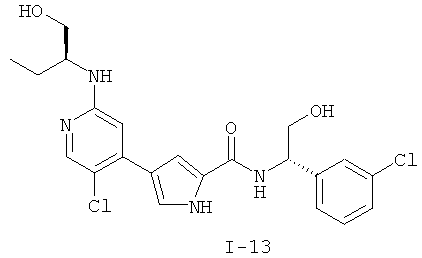
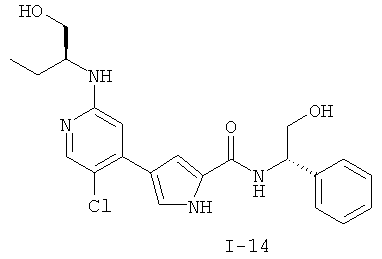
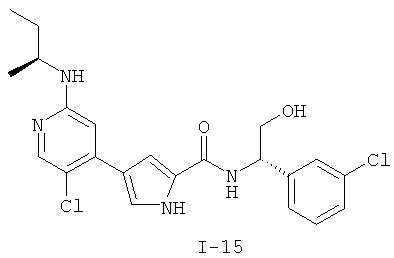
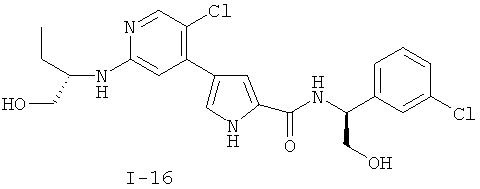
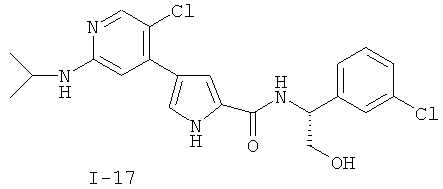
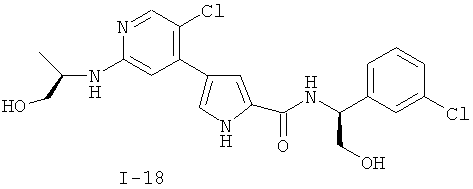
или к его фармацевтически приемлемой соли, в которой каждый m, R1, R2 и R3 имеет значение, определенное выше. Согласно некоторым вариантам осуществления R1 в любой из формул I, Ia и Ib обозначает C1-4-алифатическую группу, в случае необходимости замещенную -OR или -C1-3 галогеналкилом. В некоторых вариантах осуществления R1 в любой из формул I, Ia и Ib обозначает C1-4-алифатическую группу, в случае необходимости замещенную -ОН, -CH2F, -CHF2 или -CF3. В других вариантах осуществления, R1 в любой из формул I, Ia и Ib обозначает C1-4-алифатическую группу, в случае необходимости замещенную -ОН. В других вариантах осуществления R1 является незамещенным. Согласно другому варианту осуществления R1 в любой из формул I, Ia и Ib обозначает изопропил, 2-бутил, циклопропил или этил, причем каждый из указанных радикалов может быть замещен -ОН, -CHF2, -CH2F или -СF3. В определенных вариантах осуществления в любой из формул I, Ia и Ib может быть замещен -ОН или -CF3. Другой аспект настоящего изобретения относится к соединению любой из формул I, Ia и Ib, в которой R2 обозначает водород, C1-3-алифатическую группу или хлор. Согласно еще одному аспекту настоящее изобретение относится к соединению любой из формул I, Ia и Ib, в которой R2 обозначает хлор. В некоторых вариантах осуществления m=1. В других вариантах осуществления R3 в любой из формул I, Ia и Ib обозначает водород, метил или хлор. Примеры соединений формулы I сформулированы ниже в Таблице 1. Таблица 1. Примеры соединений формулы I:
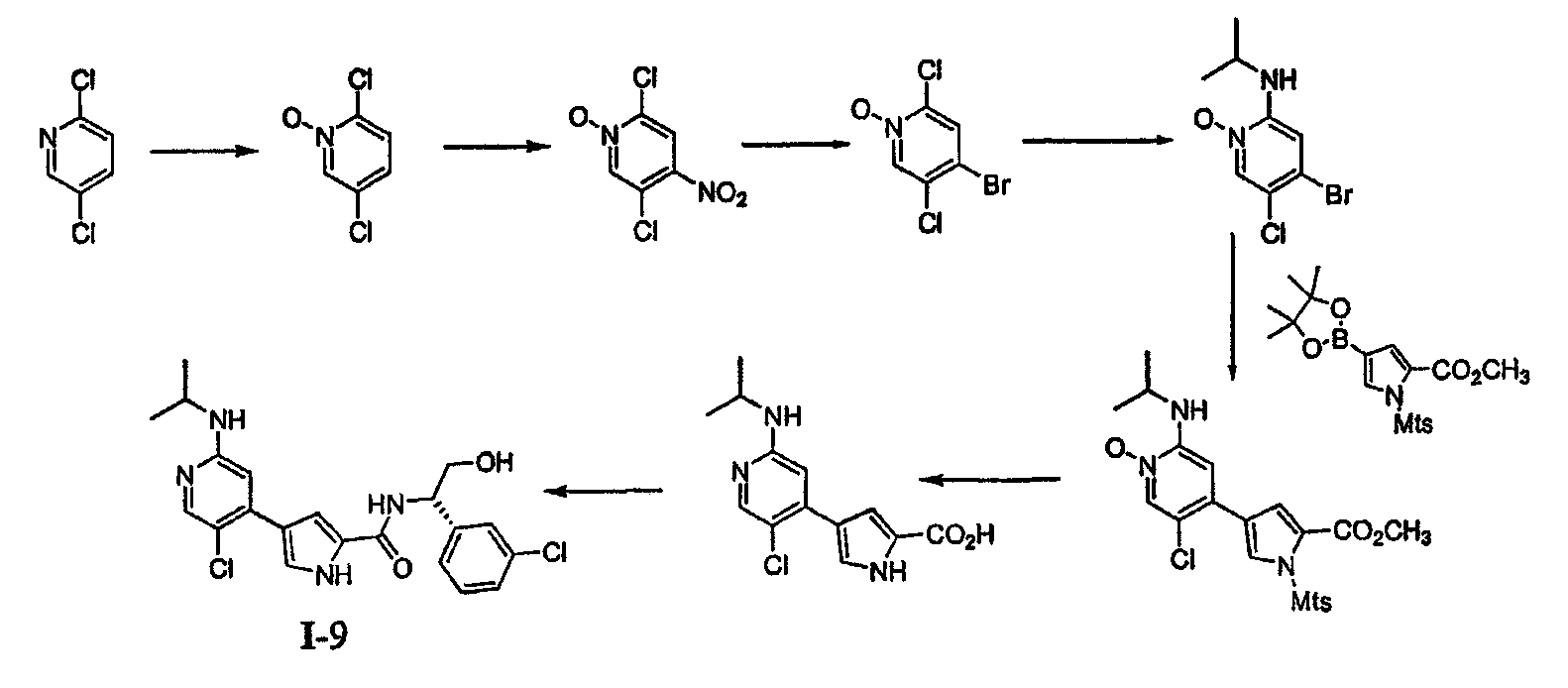
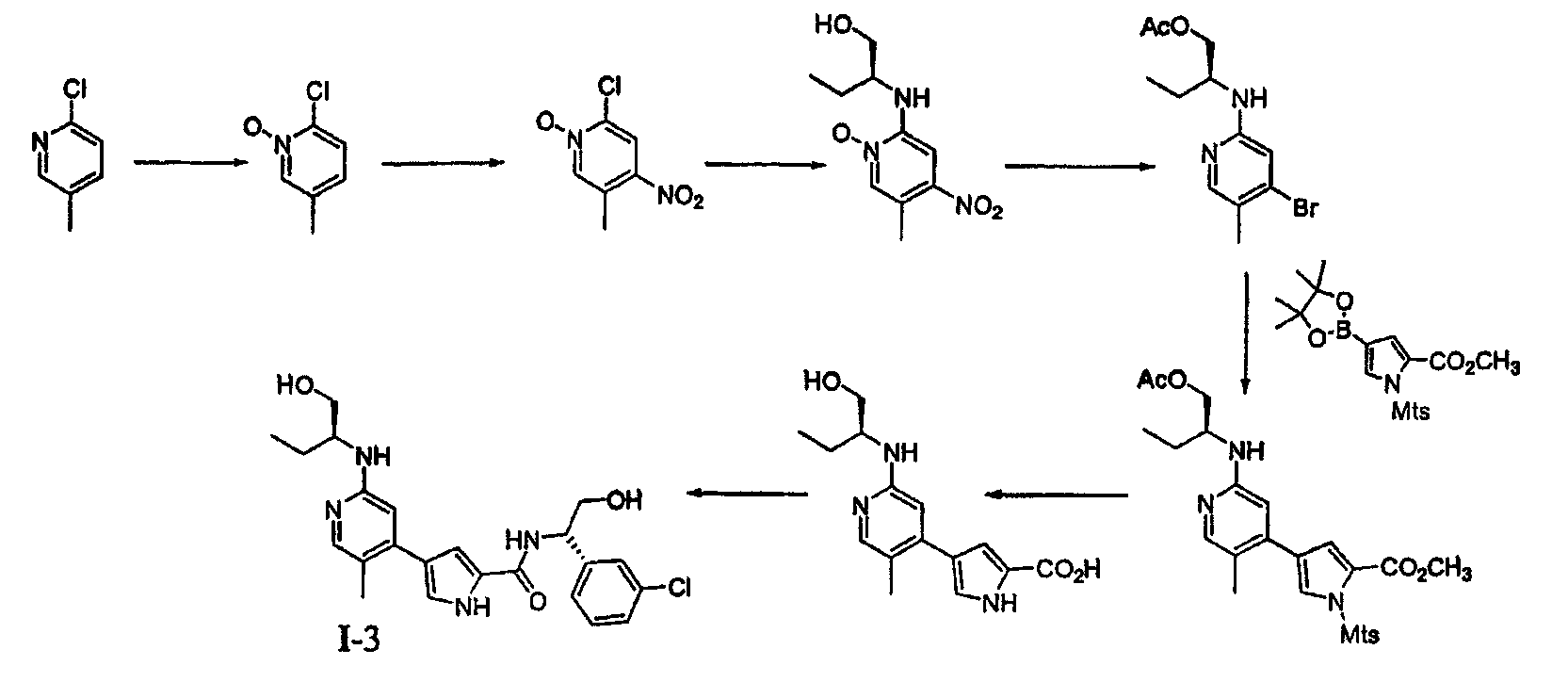
4. Общие способы получения соединений по изобретению Соединения по изобретению могут быть получены или выделены в общем синтетическими и/или псевдосинтетическими способами, известными специалисту в отношении аналогичных соединений и такими, как иллюстрируется ниже общими Схемами I, II и III и примерами получения, приведенными далее. Схема I
Реагенты и условия: (a) iIC1, CH2CL2, iiNaOMe, MeOH; (b) PG-CL, DMAP, триэтиламин; (c) Pd(dppf); (d)R1-NH2; (e) Pd(PPh3)4,4; (f) удаление защитной группы/омыление; (g) условия сочетания. Общая Схема I, приведенная выше, иллюстрирует общий способ получения соединений согласно настоящему изобретению. В стадии (a) пиррольное соединение 1 йодируют и этерифицируют, получая соединение 2. В стадии (b) пиррольное звено в случае необходимости защищают по -NH- с помощью подходящей аминозащитной группы, получая соединение 3. Аминозащитные группы известны в уровне техники и описаны подробно в Protecting Groups in Organic Synthesis, Theodora W. Greene and Peter G. M. Wuts, 1991, опубликованном John Wiley and Sons, который тем самым в полном объеме включен в настоящее описание путем ссылки. Йод из соединения 3 вытесняют подходящей бороновой кислотой или сложным эфиром. Как изображено выше, используют бис(пинаколато)диборан, получая соединение 4, хотя в этой реакции могут быть использованы другие бороновые кислоты или их эфиры, выбор которых очевиден для специалиста. Поскольку соединения по изобретению касаются полизамещенного пиридина, рассматривают такой порядок реакции и используют такие способы активации положений в пиридине, чтобы направлять региохимию. В стадии (d) первая удаляемая группа L1 может быть вытеснена по желанию спиртом, амином или тиолом. Для специалиста является очевидным, что различные удаляемые группы L1 могут быть подвергнуты этой реакции. Примеры таких групп включают, но не ограничены ими, галоген и активированные простые эфиры. Эта реакция может сопровождаться, в стадии (e), заменой второй удаляемой группы L2 или с использованием катализируемой металлом реакции сочетания или через нуклеофильное замещение, с получением соединения 7. Для специалиста в данной области очевидно, что различные удаляемые группы L2 могут быть подвергнуты этой реакции. Примеры таких групп включают, но не ограничены ими, галоген, активированные простые эфиры, бороновую кислоту или эфир бороновой кислоты. В стадии (f) защитную группу на пиррольном звене удаляют способами, подходящими для удаления используемой аминозащитной группы. В зависимости от используемой аминозащитной группы условия, подходящие для этого удаления, могут одновременно приводить к омылению или иначе обеспечить получение карбоксилатного звена, как изображено выше для соединения 8. Если условия, подходящие для удаления аминозащитной группы, не являются подходящими для того, чтобы обеспечить получение карбоксилатного соединения 8, может использоваться другая стадия. Соединения формулы I получают из 8 путем сочетания полученного карбоксилата с желаемым амином, как изображено в стадии (g). Для специалиста в данной области очевидно, что для указанной реакции сочетания пригодны различные условия, и они могут включать стадию образования карбоксилатного звена соединения 8 до или одновременно с обработкой желаемым амином. Такие состояния включают, но не ограничены ими, соединения, описанные подробно в разделе Примеров, приведенном ниже. Схема II
Реагенты и условия: (a) Ac2О/H2О2; (b) HNО3/H2SО4; (c) R1-NH2; (d) L2. Схема, приведенная выше, изображает альтернативный путь получения промежуточного соединения 6, полезного для получения соединений согласно настоящему изобретению. В стадии (a) N-оксид соединения 9 получают обработкой перекисью. N-оксид соединения 10 затем обрабатывают азотной кислотой, чтобы получить нитросоединение 11. Группу L1 в соединении 11 заменяют желаемым амином R1-NH2, получая соединение 12, и затем группу L2 вводят на стадии (d), чтобы получить промежуточное соединение 6. Соединение 6 может затем использоваться для получения соединений по изобретению согласно общей Схеме I, приведенной выше, и Примерам, представленным ниже. Схема III
Схема III, представленная выше, демонстрирует альтернативный способ получения соединения 7 из соединения 6. В этом способе группу L2 пирридинила 6 заменяют подходящей бороновой кислотой или ее эфирным производным, получая соединение 13, причем Rx и Ry имеют значения, как определено ниже для соединений формулы А. Это боронатное звено затем заменяют удаляемой группой L3 пиррола, изображенного выше, причем L3 является подходящей удаляемой группой с получением соединения 7. Соединение 7 затем используют, чтобы получить соединения согласно настоящему изобретению способами, сформулированными выше в Схемах I и II, описанными в разделе Примеров, и способами, известными специалисту в данной области техники. Для специалиста очевидно, что различные соединения по изобретению могут быть получены согласно общему способу Схем I, II и III и Примерам синтеза, сформулированным ниже. Согласно другому варианту осуществления настоящее изобретение относится к соединению формулы A:
или его соли, в которой: PG обозначает подходящую аминозащитную группу; Rz обозначает подходящую защитную группу карбоксилата и Rх и Ry независимо обозначают водород или в случае необходимости замещенную С1-6 алифатическую группу или Rх и Ry вместе образуют в случае необходимости замещенное 5-7-членное кольцо. Подходящие аминозащитные группы известны в уровне техники и включают описанные подробно в Protecting Groups in Organic Synthesis, Theodora W. Greene and Peter G. M. Wuts, 1991, опубликованном John Wiley and Sons. В некоторых вариантах осуществления группа PG соединения A обозначает алкил или арилсульфонил. Примеры таких групп включают метилсульфонил, тозил, нозил, брозил и 2,4,6-триметилбензолсульфонил (“Мts”). Другие такие группы включают Bn, PMB, Ms, Тs, SiR3, MOM, BOM, Tr, Ac, CO2R, CH2OCH2CH2Si(CH3)3. Подходящие защитные группы карбоксилата известны в уровне техники и описаны подробно в Protecting Groups in Organic Synthesis, Theodora W. Greene and Peter G. M. Wuts, 3rd Edition, 1999, опубликованном John Wiley and Sons. В некоторых вариантах осуществления группа Rz соединения A обозначает в случае необходимости замещенную С1-6 алифатическую группу или в случае необходимости замещенный арил. Примеры подходящих групп Rz включают метил, этил, пропил, изопропил, бутил, изобутил, бензил и фенил, причем каждая группа может замещена. В некоторых вариантах осуществления одна или обе группы Rх и Ry обозначают водород. В других вариантах осуществления Rх и Ry вместе образуют в случае необходимости замещенное 5-6-членное кольцо. В других вариантах осуществления Rх и Ry вместе образуют 4,4,5,5-тетраметилдиоксаборолановое звено. Другие подходящие боронатные производные, рассматриваемые в соответствии с настоящим изобретением, включают бороновую кислоту, B(О-С1-10-алифатическую группу)2 и B(О-арил)2. Согласно еще одному варианту осуществления настоящее изобретение относится к соединению формулы B:
или его соли, в которой: R1 обозначает С1-6-алифатическую группу, причем R1 может быть замещен заместителями в числе до 2 групп, независимо выбранными из -OR или -C1-3 галогеналкила; каждый R независимо обозначает водород или С1-4-алифатическую группу; R3 обозначает водород, С1-3-алифатическую группу, фтор или хлор и L2 обозначает подходящую удаляемую группу. В некоторых вариантах осуществления настоящее изобретение относится к соединению формулы B, как определено в целом и в классах и подклассах, описанных выше и здесь, в которой L2 не обозначает йод, когда R3 обозначает хлор и R1 обозначает изопропил. Подходящая удаляемая группа представляет собой радикал, который легко заменяется желаемым химическим звеном. Таким образом, выбор определенной подходящей удаляемой группы основывается на ее способности к легкой замене на химическое звено формулы A. Подходящие удаляемые группы известны в уровне техники, например, см., “Advanced Organic Chemistry,” Jerry March, 5th Ed., pp. 351-357, John Wiley and Sons, N.Y. Такие удаляемые группы включают, но не ограничены ими, галоген, алкокси, сульфонилокси, в случае необходимости замещенный алкилсульфонил, в случае необходимости замещенный алкенилсульфонил, в случае необходимости замещенный арилсульфонил и диазоний. Примеры подходящих удаляемых групп включают хлор, йод, бром, фтор, метансульфонил (метилсульфонил), тозил, трифлат, нитро-фенилсульфонил (нозил) и бром-фенилсульфонил (брозил). В некоторых вариантах осуществления группа L2 в формуле B обозначает йод. Согласно альтернативному варианту осуществления подходящая удаляемая группа может быть получена in situ в реакционной среде. Например, L2 в соединении формулы B может быть получена in situ от предшественника этого соединения формулы B, причем указанный предшественник содержит группу, которая легко заменяется группой L2 in situ. В частном случае такой замены указанный предшественник соединения формулы B содержит группу (например, хлор или гидроксильную группу), которая in situ заменяется группой L2, такую как йод. Источник йода может быть, например, йодидом натрия. Такая генерация in situ подходящей удаляемой группы известна в уровне техники, например, см., “Advanced Organic Chemistry,” Jerry March, pp. 430-431,5th Ed., John Wiley and Sons, N. Y. Согласно некоторым вариантам осуществления звено R1 формулы B представляет собой С1-4-алифатическую группу, в случае необходимости замещенную -OR или -C1-3 галогеналкилом. В некоторых вариантах осуществления звено R1 формулы B обозначает С1-4-алифатическую группу, в случае необходимости замещенную -ОН, -CH2F, -CHF2 или -CF3. В других вариантах осуществления звено R1 формулы B обозначает С1-4-алифатическую группу, в случае необходимости замещенную -ОН. В других вариантах осуществления R1 является незамещенным. Согласно другому варианту осуществления звено R1 формулы B обозначает изопропил, 2-бутил, циклопропил или этил, причем каждое звено в случае необходимости замещено -ОН или -CF3. В других вариантах осуществления звено R3 формулы B обозначает водород, метил или хлор. Соединение формулы B может быть получено из соединения формулы B’:
или его соли, в которой: R3 обозначает водород, С1-3-алифатическую группу, фтор или хлор и L1 и L2 каждый независимо обозначают подходящую удаляемую группу. В некоторых вариантах осуществления настоящее изобретение относится к соединению формулы B’, как определено в целом и в классах и подклассах, описанных выше и здесь, в которой L2 не обозначает боронатное звено, когда R3 обозначает хлор и R1 обозначает фтор. В некоторых вариантах осуществления изобретение относится к соединению B’, в котором L2 обозначает -B(ORx)(ORy). В других вариантах осуществления одна или обе группы Rx и Ry обозначают водород. В других вариантах осуществления Rx и Ry вместе образуют в случае необходимости замещенное 5-6-членное кольцо. В других вариантах осуществления Rx и Ry вместе образуют 4,4,5,5-тетраметилдиоксаборолановое звено. Как описано выше, подходящая удаляемая группа представляет собой радикал, который легко заменяется желаемым химическим звеном. Подходящие удаляемые группы известны в уровне техники, например, см., “Advanced Organic Chemistry,” Jerry March, 5th Ed., pp. 351-357, John Wiley and Sons, N.Y. Такие удаляемые группы включают, но не ограничены ими, галоген, алкокси, сульфонилокси, в случае необходимости замещенный алкилсульфонил, в случае необходимости замещенный алкенилсульфонил, в случае необходимости замещенный арилсульфонил и диазоний. Примеры подходящих удаляемых групп включают хлор, йод, бром, фтор, метансульфонил (метилсульфонил), тозил, трифлат, нитро-фенилсульфонил (нозил) и бром-фенилсульфонил (брозил). В некоторых вариантах осуществления группа L1 в формуле B’ обозначает галоген. В другом варианте осуществления группа L1 в формуле B’ обозначает в случае необходимости замещенный алкилсульфонил, в случае необходимости замещенный алкенилсульфонил или в случае необходимости замещенный арилсульфонил. В других вариантах осуществления группа L1 в формуле B’ обозначает фтор. Согласно альтернативному варианту осуществления подходящая удаляемая группа может быть получена in situ в реакционной среде. Например, L1 или L2 в соединении формулы B’ может быть получена in situ от предшественника этого соединения формулы B’, причем указанный предшественник содержит группу, которая легко заменяется группами L1 или L2 in situ. Такая генерация in situ подходящей удаляемой группы известна в уровне техники, например, см., “Advanced Organic Chemistry,” Jerry March, pp. 430-431,5th Ed., John Wiley and Sons, N.Y. Согласно другому варианту осуществления настоящее изобретение относится к способу получения соединения формулы B:
или его соли, включающему стадию введения в реакцию соединения формулы B’:
или его соли, с соединением формулы R1-NH2, причем указанную реакцию осуществляют в подходящей среде, и причем: R1 обозначает С1-6-алифатическую группу, причем R1 может быть замещен 2 группами, независимо выбранными из -OR или -C1-3 галогеналкила; каждый R независимо обозначает водород или С1-4-алифатическую группу; R3 обозначает водород, С1-3-алифатическую группу, фтор или хлор; и L1 и L2 независимо обозначают подходящую удаляемую группу. В некоторых вариантах осуществления указанную реакцию в случае необходимости осуществляют в присутствии подходящего основания. Для специалиста очевидно, что замена удаляемой группы аминогруппой может быть осуществлена в присутствии или в отсутствие подходящего основания. Такие подходящие основания известны в уровне техники и включают органические и неорганические основания. Подходящая среда представляет собой растворитель или смесь растворителей, которые, в комбинации с объединенными соединениями, могут облегчить развитие реакции между ними. Подходящий растворитель может солюбилизировать один или более компонентов реакции, или, альтернативно, подходящий растворитель может облегчить перемешивание суспензии один или более компонентов реакции. Примерами подходящих растворителей, пригодных для использования в настоящем изобретении, являются протонный растворитель, галогенированный углеводород, простой эфир, ароматический углеводород, полярный или неполярный апротонный растворитель или любые их смеси. Такие смеси включают, например, смеси протонного и непротонного растворителей, такие как бензол/метанол/вода; бензол/вода; DME/вода и т.п. Эти и другие подходящие растворители известны в уровне техники, например, см., “Advanced Organic Chemistry”, Jerry March, 5th edition, John Wiley and Sons, N.Y. Согласно еще одному варианту осуществления один или более реагентов могут действовать как подходящий растворитель. Например, органическое основание, такое как триэтиламин или диизопропилэтиламин, если оно используется в указанной реакции, может служить растворителем в дополнение к его роли как реагента повышения основности. В некоторых вариантах осуществления настоящее изобретение относится к соединению формулы B’, в которой R1 и R3 являются такими, как определено в целом и в классах и подклассах, описанных выше и здесь. Согласно другому аспекту настоящее изобретение относится к соединению формулы C:
или его соли, в которой: PG обозначает подходящую аминозащитную группу; Rz обозначает подходящую защитную группу карбоксилата; R1 обозначает С1-6-алифатическую группу, причем R1 может быть замещен заместителями в числе до 2 групп, независимо выбранными из -OR или -C1-3 галогеналкила; каждый R независимо обозначает водород или С1-4-алифатическую группу и R3 обозначает водород, С1-3-алифатическую группу, фтор или хлор. Как отмечено выше, подходящие аминозащитные группы известны в уровне техники и включают описанные подробно в Protecting Groups in Organic Synthesis, Theodora W. Greene and Peter G. M. Wuts, 1991, опубликованном John Wiley and Sons. В некоторых вариантах осуществления группа PG в соединении C обозначает алкил или арилсульфонил. Примеры таких групп включают метилсульфонил, тозил, нозил, брозил и 2,4,6-триметилбензолсульфонил (“Мts”). Подходящие защитные группы карбоксилата известны в уровне техники и описаны подробно в Protecting Groups in Organic Synthesis, Theodora W. Greene and Peter G. M. Wuts, 1991, опубликованном John Wiley and Sons. В некоторых вариантах осуществления группа Rz в соединении C обозначает в случае необходимости замещенную С1-6-алифатическую группу или в случае необходимости замещенный арил. Примеры подходящих групп Rz включают метил, этил, пропил, изопропил, бутил, изобутил, бензил и фенил, причем каждая группа может быть замещена. Согласно некоторым вариантам осуществления группа R1 в формуле C обозначает С1-4-алифатическую группу, в случае необходимости замещенную группой -OR или -C1-3 галогеналкилом. В некоторых вариантах осуществления группа R1 в формуле C обозначает С1-4-алифатическую группу, в случае необходимости замещенную -ОН, -CH2F, -CHF2 или -CF3. В других вариантах осуществления группа R1 в формуле C обозначает С1-4-алифатическую группу, в случае необходимости замещенную -ОН. В других вариантах осуществления R1 является незамещенной. Согласно другому варианту осуществления группа R1 в формуле C обозначает изопропил, 2-бутил, циклопропил или этил, причем каждая группа может быть замещена -ОН или -CF3. В других вариантах осуществления группа R3 в формуле C обозначает водород, метил или хлор. Еще один аспект настоящего изобретения относится к способу получения соединения формулы C:
или его соли, включающему стадию введения в реакцию соединения формулы A:
или его соли, с соединением формулы B:
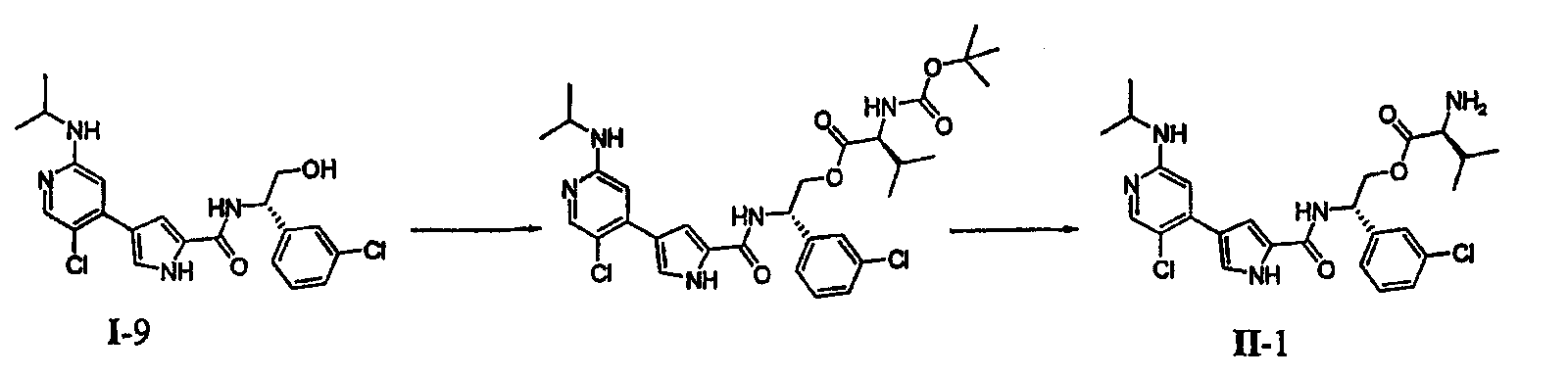
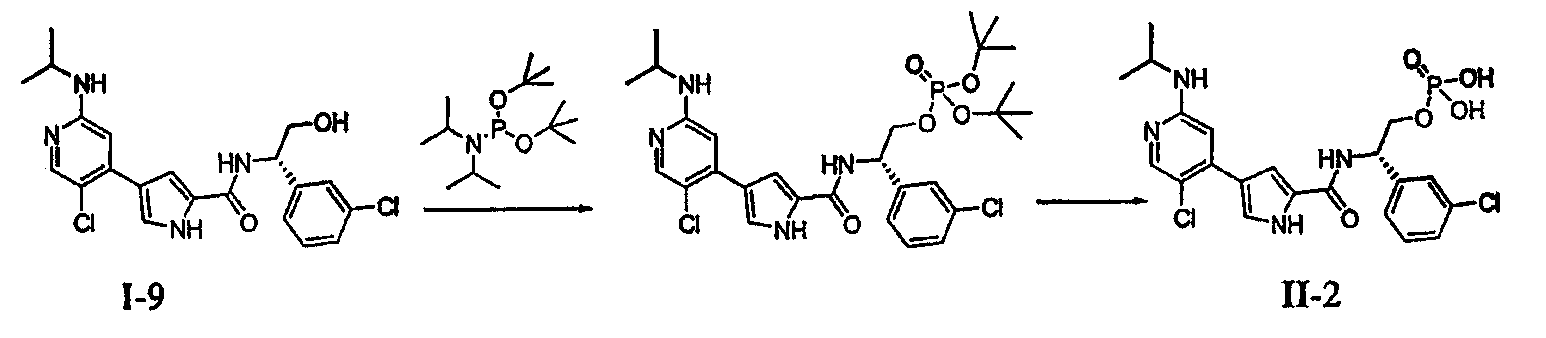
или его солью, причем указанную реакцию осуществляют в подходящей среде и причем: PG обозначает подходящую аминозащитную группу; L2 обозначает подходящую удаляемую группу; Rz обозначает подходящую защитную группу карбоксилата; Rх и Ry независимо обозначают водород или в случае необходимости замещенную С1-6-алифатическую группу, или: Rх и Ry вместе образуют в случае необходимости замещенное 5-7-членное кольцо; R1 обозначает С1-6-алифатическую группу, причем R1 может быть замещен заместителями в числе до 2 групп, независимо выбранными из -OR или -C1-3 галогеналкила; каждый R независимо обозначает водород или С1-4-алифатическую группу; и R3 обозначает водород, С1-3-алифатическую группу, фтор или хлор. В некоторых вариантах осуществления указанную реакцию осуществляют в присутствии Ni (II), Pd (0) или Pd (II), где каждый катализатор может быть связан с лигандом, таким как лиганды на основе ферроцена или фосфина. В других вариантах осуществления указанную реакцию осуществляют в присутствии Pd(PPh3)4. Подходящая среда представляет собой растворитель или смесь растворителей, которая, в комбинации с объединенными соединениями, может облегчить развитие реакции между ними. Подходящий растворитель может солюбилизировать один или более компонентов реакции, или, альтернативно, подходящий растворитель может облегчить перемешивание суспензии одного или более компонентов реакции. Примерами подходящих растворителей, которые могут быть использованы в настоящем изобретении, являются протонный растворитель, галогенированный углеводород, простой эфир, ароматический углеводород, полярный или неполярный апротонный растворитель или любые их смеси. Такие смеси включают, например, смеси протонного и непротонного растворителей, такие как бензол/метанол/вода; бензол/вода; DME/вода и т.п. Эти и другие такие подходящие растворители известны в уровне техники, например, см., “Advanced Organic Chemistry”, Jerry March, 5th edition, John Wiley and Sons, N.Y. В некоторых вариантах осуществления реакцию между соединениями A и B с получением соединения C осуществляют в смеси DME и воды. В некоторых вариантах осуществления реакцию между соединениями A и B с получением соединения C осуществляют при температуре в пределах от приблизительно 20°C до 150°C. В других вариантах осуществления реакцию между соединениями A и B с получением соединения C осуществляют при микроволновом облучении при температуре в пределах от приблизительно 100°C до 250°C. В других вариантах осуществления реакцию между соединениями A и B с получением соединения C осуществляют при несколько основном рН. Согласно другому варианту осуществления настоящее изобретение относится к пролекарству соединения формулы I, причем указанное пролекарство имеет формулу II:
или к его фармацевтически приемлемой соли, в которой: R1 обозначает С1-6-алифатическую группу, причем R1 может быть замещен 2 группами, независимо выбранными из -OR, -OR4 или -C1-3 галогеналкила; каждый R независимо обозначает водород или С1-6-алифатическую группу; каждый R2 независимо обозначает R, фтор или хлор; m=0, 1 или 2; R3 обозначает водород, С1-3-алифатическую группу, фтор или хлор; каждый R4 обозначает независимо водород, -C(R)2O-R5 или R5, при условии, что по меньшей мере одна из групп R4 или R8 отлична от водорода; каждый R5 независимо обозначает -C(O)R6, -C(O)OR6, -C(O)-Q-R6, -C(O)-(CH2)n-C(O)OR6, -C(О)-(CH2)n-C(О)N(R7)2, -C(O)-(CH2)n-CH(R6)N(R7)2, -P(O)(OR6)2; каждый R6 независимо обозначает водород, в случае необходимости замещенную С1-6-алифатическую группу или в случае необходимости замещенное 5-8-членное насыщенное, частично ненасыщенное или полностью ненасыщенное кольцо, имеющее 0-4 гетероатома, независимо выбранные из азота, кислорода или серы; каждый R7 независимо обозначает водород, -C(O)R6, -C(O)OR6, -S(O)2R6, -OR6, в случае необходимости замещенную С1-6-алифатическую группу или в случае необходимости замещенное 5-8-членное насыщенное, частично ненасыщенное или полностью ненасыщенное кольцо, имеющее 0-4 гетероатома, независимо выбранные из азота, кислорода или серы, или: два R6 на одном и том же атоме азота вместе с атомом азота, связанным с ними, образуют 4-7-членное насыщенное, частично ненасыщенное или полностью ненасыщенное кольцо, имеющее 1-3 гетероатома помимо атома азота, независимо выбранные из азота, кислорода или серы; каждый n=0-6; Q обозначает в случае необходимости замещенную C1-10 алкилиденовую цепь, причем от нуля до четырех метиленовых звеньев в Q независимо заменены -О-, -N(R)-, -S-, -S(O)-, -S(O)2– или -C(O)-; и R8 обозначает водород или -C(R)2O-R5. Согласно некоторым вариантам осуществления группа R1 в формуле II обозначает С1-4-алифатическую группу, в случае необходимости замещенную -OR или -C1-3 галогеналкилом. В некоторых вариантах осуществления группа R1 в формуле II обозначает С1-4-алифатическую группу, в случае необходимости замещенную -ОН, -CH2F, -CHF2 или -CF3. В других вариантах осуществления группа R1 в формуле II обозначает С1-4-алифатическую группу, в случае необходимости замещенную -ОН. В других вариантах осуществления R1 является незамещенной. Согласно другому варианту осуществления группа R1 в формуле II обозначает изопропил, 2-бутил, циклопропил или этил, причем каждая группа может быть замещена -О, -CHF2, -CH2F или -CF3. Согласно еще одному варианту осуществления группа R1 в формуле II обозначает изопропил, 2-бутил, циклопропил или этил, причем каждая группа может быть замещена -ОН или -CF3. Другой аспект настоящего изобретения относится к соединению формулы II, в которой каждый R2 независимо обозначает водород, С1-3-алифатическую группу или хлор. Согласно еще одному аспекту настоящее изобретение относится к соединению формулы II, в которой R2 обозначает хлор и m=1. В других вариантах осуществления группа R3 в формуле II обозначает водород, метил или хлор. В некоторых вариантах осуществления R4 обозначает C(O)-Q-R6. Другие варианты осуществления относятся к соединению формулы II, в которой R4 обозначает C(O)-Q-R6 и Q обозначает в случае необходимости замещенную C1-8 алкилиденовую цепь, причем от нуля до четырех метиленовых звеньев в Q независимо замещены -O-, -N(R)-, -С-, -S(O)-, -S(O)2- или -C(O)-, и R6 имеет значения, как определено в общем и в классах и подклассах, описанных выше и здесь. Согласно другому варианту осуществления, Q обозначает в случае необходимости замещенную C1-8 алкилиденовую цепь, причем от двух до четырех метиленовых звеньев в Q независимо замещены -О-. Такие группы Q включают -CH2OCH2CH2O-, -CH2OCH2CH2OCН2CH2O- и т.п. Еще один аспект настоящего изобретения относится к соединению формулы II, в которой R4 обозначает C(О)-(CH2)n-CH(R6)N(R7)2. В некоторых вариантах осуществления n=0-2. В других вариантах осуществления R6 обозначает в случае необходимости замещенную С1-6-алифатическую группу. Примеры таких групп R6 включают метил, бензил, этил, изопропил, трет-бутил и т.п. Группы R7 в C(O)-(CH2)n-CH(R6)N(R7)2 в формуле II включают водород и в случае необходимости замещенную С1-6-алифатическую группу. Примеры таких групп включают метил, бензил, этил, изопропил, трет-бутил и т.п. Согласно одному аспекту настоящее изобретение относится к соединению формулы II, в которой R8 обозначает водород. Согласно одному варианту осуществления R4 обозначает сложный эфир L-валина. В некоторых вариантах осуществления настоящего изобретения группа R4 в формуле II обозначает -P(О)(ОR6)2. В других вариантах осуществления каждый R6 независимо обозначает водород или в случае необходимости замещенную С1-6-алифатическую группу. Примеры таких групп R6 включают метил, бензил, этил, изопропил, трет-бутил и т.п. В других вариантах осуществления группа R4 в формуле II обозначает -P(O)(ОН)2. В некоторых вариантах осуществления соединение формулы II обеспечивает улучшение в отношении одной или более физических или физиологических характеристик. В других вариантах осуществления соединение формулы II обеспечивает улучшение в отношении одной или более физических и физиологических характеристик. Примеры соединений формулы II сформулированы ниже в Таблице 2. Таблица 2.
Способы получения таких пролекарств включают сформулированные подробно ниже в разделе секцию Примеров и способы, известные специалисту в данной области техники. 5. Применения, состав и введение ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ КОМПОЗИЦИИ Как обсуждалось выше, настоящее изобретение относится к соединениям, которые являются ингибиторами протеинкиназ, и таким образом соединения по изобретению могут быть использованы для лечения заболеваний, нарушений и состояний, включая, но не ограничиваясь ими, рак, аутоиммунные нарушения, нейродегенеративные и неврологические нарушения, шизофрению, нарушения, связанные с костями, заболевание печени и кардиологические нарушения. Соответственно, в другом аспекте настоящее изобретение относится к фармацевтически приемлемым композициям, причем эти композиции включают любое из соединений, описанных в настоящей заявке, и в случае необходимости включают фармацевтически приемлемую основу, адъювант или носитель. В некоторых вариантах осуществления эти композиции в случае необходимости дополнительно содержат одно или более дополнительных терапевтических средств. Следует также отметить, что некоторые из соединений согласно настоящему изобретению могут существовать в свободной форме для лечения или, в случае необходимости, в виде фармацевтически приемлемого производного. Согласно настоящему изобретению фармацевтически приемлемое производное включает, но не ограничено ими, фармацевтически приемлемые соли, сложные эфиры, соли таких сложных эфиров или любой другой аддукт или производное, которое при введении пациенту способно обеспечить, прямо или косвенно, соединение, как описано здесь, или его метаболит или остаток. В рамках изобретения термин “фармацевтически приемлемая соль” относится к таким солям, которые, с точки зрения медицины, пригодны для использования в контакте с тканями человека и животных без нежелательной токсичности, раздражения, аллергической реакции и т.п. и имеют приемлемое отношение выгоды/риска. “Фармацевтически приемлемая соль” означает любую нетоксичную соль или соль сложного эфира соединения по изобретению, которая, при введении реципиенту, способна обеспечить, прямо или косвенно, соединение по изобретению или его ингибиторно активный метаболит или остаток. В рамках изобретения термин Фармацевтически приемлемые соли известны в уровне техники. Например, S.M. Berge et al. подробно описывают фармацевтически приемлемые соли в J. Pharmaceutical Sciences, 1977, 66, 1-19, включенном в настоящее описание путем ссылки. Фармацевтически приемлемые соли соединений по изобретению включают полученные с подходящими неорганическими и органическими кислотами и основаниями. Примерами фармацевтически приемлемых нетоксичных солей присоединения с кислотой являются соли аминогруппы, образованные с минеральными кислотами, такими как соляная кислота, бромистоводородная кислота, фосфорная кислота, серная кислота и перхлорная кислота, или с органическими кислотами, такими как уксусная кислота, щавелевая кислота, малеиновая кислота, винная кислота, лимонная кислота, янтарная кислота или малоновая кислота, или при использовании других способов, используемых в данной области, таких как ионный обмен. Другие фармацевтически приемлемые соли включают адипат, альгинат, аскорбат, аспартат, бензолсульфонат, бензоат, бисульфат, борат, бутират, камфорат, камфорсульфонат, цитрат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептаноат, глицерофосфат, глюконат, гемисульфат, гептаноат, гексаноат, гидройодид, 2-гидрокси-этансульфонат, лактобионат, лактат, лаурат, лаурилсульфат, малат, малеат, малонат, метансульфонат, 2-нафталинсульфонат, никотинат, нитрат, олеат, оксалат, пальмитат, памоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикринат, пивалат, пропионат, стеарат, сукцинат, сульфат, тартрат, тиоцианат, п-толуолсульфонат, ундеканоат, валерат и т.п. Соли, полученные с подходящими основаниями, включают соли щелочного металла, щёлочноземельного металла, аммония и N+(С1-4-алкил)4. Это изобретение также предусматривает кватернизацию любых содержащих основной атом азота групп в соединениях, раскрытых здесь. Водорастворимые, или жирорастворимые, или диспергируемые продукты могут быть получены такой кватернизацией. Примеры солей щелочного или щёлочноземельного металла включают соли натрия, лития, калия, кальция, магния и т.п. Другие фармацевтически приемлемые соли включают, в случае необходимости, катионы нетоксичного аммония, четверичного аммония и амина, образованные с использованием противоиона, такого как галид, гидроксид, карбоксилат, сульфат, фосфат, нитрат, (низший алкил)сульфонат и арилсульфонат. Как описано выше, фармацевтически приемлемые композиции согласно настоящему изобретению дополнительно включают фармацевтически приемлемую основу, адъювант или носитель, которые, в рамках изобретения, включают любые и все растворители, разбавители или другие жидкие носители, диспергирующие или суспендирующие вспомогательные вещества, поверхностно-активные вещества, изотонические средства, загустители или эмульгаторы, консерванты, твердые связующие, лубриканты и т.п., подходящие для конкретной желаемой лекарственной формы. В Remington’s Pharmaceutical Sciences, Sixteenth Edition, E. W. Martin (Mack Publishing Co., Easton, Pa., 1980) раскрыты различные основы, используемые в составлении фармацевтически приемлемых композиций, и известные способы их получения. За исключением случаев, когда обычная среда носителя является несовместимой с соединениями по изобретению, например, производя любой нежелательный биологический эффект или иначе взаимодействуя нежелательным образом с любым другим компонентом(ами) фармацевтически приемлемой композиции, ее использование возможно в рамках этого изобретения. Некоторые примеры материалов, которые могут служить фармацевтически приемлемыми носителями, включают, но не ограничены ими, иониты, оксид алюминия, стеарат алюминия, лецитин, сывороточные белки, такие как человеческий сывороточный альбумин, буферные вещества, такие как фосфаты, аминоуксусная кислота, сорбиновая кислота или сорбат калия, смеси частичных глицеридов насыщенных растительных жирных кислот, вода, соли или электролиты, такие как протамин сульфат, динатрийгидрофосфат, дикалийгидрофосфат, хлорид натрия, соли цинка, коллоидный оксид кремния, трисиликат магния, поливинил пирролидон, полиакрилаты, воски, полиэтилен-полиоксипропилен-блокполимеры, ланолин, сахара, такие как лактоза, глюкоза и сахароза; крахмал, такой как кукурузный крахмал и картофельный крахмал; целлюлоза и ее производные, такие как натриевая соль карбоксиметилцеллюлозы, этилцеллюлоза и ацетилцеллюлоза; порошкованный трагакант; солод; желатин; тальк; эксципиенты, такие как масло какао и воски для суппозиториев; масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; гликоли, такие как пропиленгликоль или полиэтиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные средства, такие как гидроксид магния и гидроксид алюминия; альгиновая кислота; апирогенная вода; изотонический солевой раствор; раствор Рингера; этиловый спирт и фосфатные буферные растворы, а также другие нетоксичные совместимые лубриканты, такие как лаурилсульфат натрия и стеарат магния, а также красители, агенты высвобождения, агенты покрытия, подсластители, ароматизаторы и отдушки, консерванты и антиоксиданты могут также присутствовать в композиции по усмотрению составителя. ПРИМЕНЕНИЯ СОЕДИНЕНИЙ И ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ КОМПОЗИЦИИ В еще одном аспекте изобретение относится к способу лечения или уменьшения серьезности рака, аутоиммунного нарушения, нейродегенеративного или неврологического нарушения, заболевания печени или кардиологического нарушения, включающему введение пациенту эффективного количества соединения согласно настоящему изобретению, или фармацевтически приемлемой композиции, включающей соединение согласно настоящему изобретению. В некоторых вариантах осуществления настоящего изобретения “эффективное количество” соединения или фармацевтически приемлемой композиции представляет собой количество, эффективное для лечения или уменьшения серьезности заболевания, состояния или нарушения, выбранного из рака, аутоиммунного нарушения, нейродегенеративного или неврологического нарушения, шизофрении, нарушения, связанного с костями, заболевания печени или кардиологического нарушения. Соединения и композиции, согласно способу по изобретению, можно вводить, используя любое количество и любой путь введения, эффективные для лечения или уменьшения серьезности рака, аутоиммунного нарушения, нейродегенеративного или неврологического нарушения, шизофрении, нарушения, связанного с костями, заболевания печени или кардиологического нарушения. Точное требуемое количество варьируют от пациента к пациенту в зависимости от вида, возраста и общего состояния пациента, серьезности инфекции, конкретного средства, способа введения и т.п. Соединения по изобретению предпочтительно составляют в форме разовой лекарственной формы для обеспечения простоты введения и однородности дозировки. Выражение “разовая лекарственная форма” в рамках изобретения относится к физически отдельной единице средства, адаптированной к пациенту. Следует, однако, понимать, что решение о полном суточном применении соединений и композиций согласно настоящему изобретению будет принято лечащим врачом по его усмотрению. Определенный эффективный уровень дозы для любого конкретного пациента или организма будет зависеть от различных факторов, включая излечиваемое нарушение и серьезность нарушения; активность конкретного используемого соединения; конкретную используемую композицию; возраст, массу тела, общее состояние здоровья, пол и диету пациента; время введения, путь введения и скорость выведения конкретного используемого соединения; продолжительность лечения; препараты, используемые в комбинации или одновременно с конкретным используемым соединением, и т.п. факторы, известные в области медицины. Термин “пациент” в рамках изобретения означает животное, предпочтительно млекопитающее и наиболее предпочтительно человека. Фармацевтически приемлемые композиции по изобретению могут вводиться человеку и животным перорально, ректально, парентерально, интрацистернально, внутривлагалищно, интраперитонеально, топически (например, в виде порошков, мазей или капель), буккально, например, в виде перорального или носового спрея, и т.п. в зависимости от серьезности излечиваемой инфекции. В некоторых вариантах осуществления получения желаемого терапевтического эффекта соединения по изобретению могут вводиться перорально или парентерально в количестве от приблизительно 0,01 мг/кг до приблизительно 50 мг/кг и предпочтительно от приблизительно 1 мг/кг до приблизительно 25 мг/кг массы тела пациента в сутки один или более раз в день. Жидкие лекарственные формы для перорального введения включают, но не ограничены ими, фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. В дополнение к активным соединениям, жидкие лекарственные формы могут содержать инертные разбавители, обычно используемые в уровне техники, например, воду или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (в частности, масло семян хлопчатника, арахисовое, кукурузное, зародышевое, оливковое, касторовое и кунжутное масла), глицерин, тетрагидрофуриловый спирт, полиэтиленгликоли и эфиры жирной кислоты и сорбитана и их смеси. Помимо инертных разбавителей пероральные композиции могут также включать адъюванты, такие как смачивающие вещества, эмульгаторы и суспендирующие агенты, подсластители, ароматизаторы и отдушки. Инъецируемые препараты, например, стерильные водные или масляные суспензии для инъекций могут быть составлены согласно известному уровню техники с использованием подходящих диспергаторов или смачивающих веществ и суспендирующих агентов. Стерильный инъецируемый препарат может также быть стерильным инъецируемым раствором, суспензией или эмульсией в нетоксичном парентерально приемлемом разбавителе или растворителе, например, раствором в 1,3-бутандиоле. Среди приемлемых носителей и растворителей, которые могут использоваться, можно назвать воду, раствор Рингера, U.S.P. и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды традиционно используют стерильные нелетучие масла. С этой целью может использоваться любое мягкое нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, в препарате для инъекций используют жирные кислоты, такие как олеиновая кислота. Составы для инъекций можно стерилизовать, например, фильтрацией через бактериальный-сдерживающий фильтр или путем включения стерилизующих средств в форме стерильных твердых композиций, которые могут быть растворены или диспергированы в стерилизованной воде или другой стерильной инъецируемой среде перед использованием. Для пролонгирования эффекта соединения по изобретению часто бывает желательно замедлить всасывание соединения, вводимого подкожной или внутримышечной инъекцией. Это может быть достигнуто при помощи жидкой суспензии кристаллического или аморфного материала с недостаточной водорастворимостью. Скорость всасывания соединения будет тогда зависеть от его скорости растворения, которая, в свою очередь, может зависеть от размера кристаллов и кристаллической формы. Альтернативно, отсроченное всасывание парентерально вводимой формы соединения может быть достигнуто за счет растворения или суспендирования соединения в масляном носителе. Инъецируемые формы депо получают, формируя микроинкапсулируемый матрикс соединения в биоразлагаемых полимерах, таких как полилактид-полигликолид. В зависимости от отношения соединения к полимеру и природе конкретного используемого полимера можно контролировать скорость высвобождения соединения. Примеры других биоразлагаемых полимеров включают сложные поли(орто-эфиры) и поли(ангидриды). Инъецируемые составы депо также получают, захватывая соединение в липосомы или микроэмульсии, которые являются совместимыми с тканями организма. Композиции для ректального или влагалищного введения предпочтительно представляют собой суппозитории, которые можно получить, смешивая соединения по изобретению с подходящими нераздражающими эксципиентами или носителями, такими как масло какао, полиэтиленгликоль или воск для суппозиториев, которые являются твердыми при температуре окружающей среды, но жидкими при температуре тела и поэтому плавятся в полости прямой кишки или влагалища и высвобождают активное соединение. Твердые лекарственные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых лекарственных формах активное соединение смешано с по меньшей мере одним инертным, фармацевтически приемлемым эксципиентом или носителем, таким как цитрат натрия или фосфат дикальция, и/или a) наполнителями, такими как крахмалы, лактоза, сахароза, глюкоза, маннит и кремневая кислота, b) связующими, такими как, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидинон, сахароза и гуммиарабик, c) увлажнителями, такими как глицерин, d) дезинтеграторами, такими как агар-агар, карбонат кальция, картофельный крахмал или крахмал тапиоки, альгиновая кислота, некоторые силикаты и карбонат натрия, e) агентами, замедляющими растворение, такими как парафин, f) ускорителями всасывания, такими как четвертичные аммониевые соединения, g) смачивающими веществами, такими как, например, цетиловый спирт и глицерин моностеарат, h) абсорбентами, такими как каолин и бентонит, и i) лубрикантами, такими как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия, и их смесями. В случае капсул, таблеток и пилюль лекарственная форма может также включать буферные агенты. Твердые композиции подобного типа могут также использоваться как наполнители в желатиновых капсулах с мягким или твердым наполнением, в которых используют такие эксципиенты, как лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли и т.п. Твердые лекарственные формы в виде таблеток, драже, капсул, пилюль и гранул могут быть получены с покрытиями и оболочками, такими как энтеросолюбильные покрытия и другие покрытия, известные в области фармацевтических составов. Они могут, в случае необходимости, содержать рентгеноконтрастные средства и могут также представлять собой такие композиции, которые высвобождают активные ингредиенты, только, или предпочтительно, в определенной части кишечника, в случае необходимости, отсроченным образом. Примеры композиций, которые могут использоваться, включают полимерные вещества и воски. Твердые композиции подобного типа могут также использоваться как наполнители в желатиновых капсулах с мягким или твердым наполнением, в которых используют такие эксципиенты, как лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли и т.п. Активные соединения могут также быть в микроинкапсулированной форме с одним или более эксципиентами, как отмечено выше. Твердые лекарственные формы в виде таблеток, драже, капсул, пилюль и гранул могут быть получены с покрытиями и оболочками, такими как энтеросолюбильные покрытия, покрытия для контроля высвобождения и другие покрытия, известные в области фармацевтических составов. В таких твердых лекарственных формах активное соединение может быть смешано по меньшей мере с одним инертным разбавителем, таким как сахароза, лактоза или крахмал. Такие лекарственные формы могут также включать, в соответствии с обычной практикой, дополнительные вещества, отличные от инертных разбавителей, например, лубриканты для таблетирования и другие добавки для таблетирования, такие как стеарат магния и микрокристаллическая целлюлоза. В случае капсул, таблеток и пилюль лекарственные формы могут также включать буферные агенты. Они могут, в случае необходимости, содержать рентгеноконтрастные средства и могут также представлять собой такие композиции, которые высвобождают активные ингредиенты, только, или предпочтительно, в определенной части кишечника, в случае необходимости, отсроченным образом. Примеры композиций, которые могут использоваться, включают полимерные вещества и воски. Лекарственные формы для топического или чрескожного введения соединения по изобретению включают мази, пасты, кремы, лосьоны, гели, порошки, растворы, спреи, средства для ингаляции или пластыри. Активный компонент смешивают в стерильных условиях с фармацевтически приемлемым носителем и любыми необходимыми консервантами или буферами, которые могут потребоваться. Офтальмологический состав, ушные капли и капли для глаз также входят в рамки этого изобретения. Дополнительно в настоящем изобретении рассматривается использование чрескожных пластырей, которые имеют дополнительное преимущество в том, что обеспечивают контролируемую доставку соединения в организм. Такие лекарственные формы можно получить, растворяя или диспергируя соединение в соответствующей среде. Усилители всасывания могут также использоваться, чтобы усилить прохождение соединения через кожу. Скорость всасывания можно контролировать, либо используя контролирующую скорость мембрану, либо диспергируя соединение в полимерной матрице или геле. Как описано выше, соединения по изобретению могут быть использованы как ингибиторы протеинкиназ ERK. В одном варианте осуществления соединения и композиции по изобретению представляют собой ингибиторы одной или обеих из протеинкиназ ERK1 и ERK2, и, таким образом, вне связи с какой бы то ни было конкретной теорией, соединения и композиции особенно пригодны для лечения или уменьшения серьезности заболевания, состояния или нарушения, где активация одной или обеих из протеинкиназ ERK1 и ERK2 участвует в механизме заболевания, состояния или нарушения. Когда активация протеинкиназ ERK1 и/или ERK2 участвует в механизме конкретного заболевания, состояния или нарушения, это заболевание, состояние или нарушение может также быть указано как “ERK1- или ERK2-опосредованное заболевание”, состояние или болезненный симптом. Соответственно, в другом аспекте настоящее изобретение относится к способу лечения или уменьшения серьезности заболевания, состояния или нарушения, где активация одной или обеих из протеинкиназ ERK1 и ERK2 участвует в механизме указанного заболевания, состояния или нарушения. Активность соединения, используемого в этом изобретении в качестве ингибитора протеинкиназ ERK1 и/или ERK2, может быть испытана in vitro, in vivo или на линии клеток. Тесты in vitro включают тесты, которые определяют ингибирование фосфорилирующей активности или АТФ-азной активности активированной протеинкиназы ERK1 или ERK2. Другие тесты in vitro количественно определяют способность ингибитора связываться с протеинкиназами ERK1 или ERK2. Связывание ингибитора может быть измерено путем радиомечения ингибитора до связывания, выделения комплекса ингибитор/ERK1 или ингибитор/ERK2 и определения количества связанной радиометки. Альтернативно, связывание ингибитора может быть определено путем конкурентного эксперимента, где новые ингибиторы инкубируют с протеинкиназами ERK1 или ERK2, связанными с известными мечеными лигандами. Термин “измеримо ингибирует” в рамках изобретения означает измеримое изменение активности протеинкиназы ERK1 или ERK2 между образцом, включающим указанную композицию и протеинкиназу ERK1 или ERK2, и эквивалентным образцом, включающим протеинкиназу ERK1 или ERK2 в отсутствие указанной композиции. Такие способы измерения активности протеинкиназы известны специалисту и включают способы, сформулированные ниже. Согласно другому варианту осуществления изобретение относится к способу ингибирования активности протеинкиназы ERK1 или ERK2 у пациента, включающему стадию введения указанному пациенту соединения по изобретению или композиции, содержащей указанное соединение. Термин “ERK-опосредованное состояние” или “заболевание” в рамках изобретения означает любое заболевание или другое аномальное состояние, в отношении которого известно, что в его механизме играет роль ERK. Термин “ERK-опосредованное состояние” или “заболевание” также означает заболевания или состояния, которые облегчаются за счет лечения с использованием ингибитора ERK. Такие состояния включают, без ограничения, рак, инсульт, диабет, гепетомегалию, сердечно-сосудистое заболевание, включая кардиомегалию, болезнь Альцгеймера, муковисцидоз, вирусное заболевание, аутоиммунные заболевания, атеросклероз, рестеноз, псориаз, аллергические нарушения, включая астму, воспаление, неврологические нарушения и связанные с гормонами заболевания. Термин “рак” включает, но не ограничен ими, следующие виды рака: рак груди, яичника, шейки, предстательной железы, яичка, мочеполовых путей, пищевода, гортани, глиобластому, нейробластому, желудка, кожи, кератоакантому легкого, плоскоклеточный рак, крупноклеточный рак, мелкоклеточный рак, аденокарциному легкого, кости, толстого кишечника, аденому поджелудочной железы, аденокарциному щитовидной железы, фолликулярную карциному, недифференцированный рак, папиллярную карциному, семиному, меланому, саркому, рак мочевого пузыря, рак печени и желчных путей, рак почек, нарушения спинного мозга, лимфоидные нарушения, болезнь Ходжкина, волосатоклеточный лейкоз, рак полости рта и глотки (оральный), губы, языка, рта, глотки, тонкого кишечника, колоректальный, толстого кишечника, прямой кишки, мозга и центральной нервной системы и лейкоз. Соответственно, другой вариант осуществления настоящего изобретения относится к лечению или уменьшению серьезности одного или более заболеваний, о которых известно, что в их механизме играет роль ERK. В частности, настоящее изобретение относится к способу лечения или уменьшения серьезности заболевания или состояния, выбранного из рака, инсульта, диабета, гепатомегалии, сердечно-сосудистого заболевания, включая кардиомегалию, болезни Альцгеймера, муковисцидоза, вирусного заболевания, аутоиммунных заболеваний, атеросклероза, рестеноза, псориаза, аллергических нарушений, включая астму, воспаления, неврологических нарушений и связанных с гормонами заболеваний, причем указанный способ включает введение пациенту композиции согласно настоящему изобретению. Согласно другому варианту осуществления настоящее изобретение относится к способу лечения рака, выбранного из рака груди, яичника, шейки, предстательной железы, яичка, мочеполовых путей, пищевода, гортани, глиобластомы, нейробластомы, желудка, кожи, кератоакантомы, легкого, плоскоклеточного рака, крупноклеточного рака, мелкоклеточного рака, аденокарциномы легкого, кости, толстого кишечника, аденомы, поджелудочной железы, аденокарциномы, щитовидной железы, фолликулярной карциномы, недифференцированного рака, папиллярной карциномы, семиномы, меланомы, саркомы, рака мочевого пузыря, рака печени и желчных путей, рака почек, нарушений спинного мозга, лимфоидных нарушений, болезни Ходжкина, волосатоклеточного лейкоза, рака полости рта и глотки (орального), губы, языка, рта, глотки, тонкого кишечника, колоректального, толстого кишечника, прямой кишки, мозга и центральной нервной системы и лейкоза. Другой вариант осуществления относится к способу лечения меланомы, рака молочной железы, рака толстого кишечника или рака поджелудочной железы у пациента. Также следует отметить, что соединения и фармацевтически приемлемые композиции согласно настоящему изобретению могут использоваться в комбинированных терапиях, то есть соединения и фармацевтически приемлемые композиции могут вводиться одновременно с, до или после одной или более других желаемых терапий или медицинских процедур. В конкретной комбинации терапий (терапевтических средств или процедур) для использования в комбинированном режиме следует учитывать совместимость желаемых терапевтических средств и/или процедур и желаемого терапевтического эффекта. Также следует отметить, что используемые терапии могут обеспечивать желаемый эффект в отношении того же самого нарушения (например, соединение по изобретению может вводиться одновременно с другим средством, используемым для лечения того же самого нарушения), или они могут обеспечивать другие эффекты (например, контроль побочных эффектов). В рамках изобретения дополнительные терапевтические средства, которые обычно вводят для лечения или профилактики конкретного заболевания или состояния, известны как “соответствующие излечиваемому заболеванию или состоянию”. Например, химиотерапевтические средства или другие антипролиферативные средства могут быть скомбинированы с соединениями по изобретению для лечения пролиферативных заболеваний и рака. Примеры известных химиотерапевтических средств включают, но не ограничены ими, например, другие противораковые терапии или средства, которые могут использоваться в комбинации с противораковыми средствами по изобретению, включают хирургию, лучевую терапию (например, гамма-облучение, нейтронная лучевая радиотерапия, электронная лучевая радиотерапия, протонная терапия, брахитерапия и системные радиоактивные изотопы), эндокринную терапию, модификаторы биологического ответа (например, интерфероны, интерлейкины и фактор некроза опухоли (TNF)), гипертермию и лечение холодом, средства для смягчения побочных эффектов (например, противорвотные средства) и другие апробированные химиотерапевтические препараты, включая, но не ограничиваясь ими, алкилирующие препараты (меклоретамин, хлорамбуцил, циклофосфамид, мельфалан, ифосфамид), антиметаболиты (Метотрексат), антагонисты пурина и антагонисты пиримидина (6-меркаптопурин, 5-Фторурацил, цитарабил, гемцитабин), нервно-мышечные яды (Винбластин, Винкристин, Винорелбин, Паклитаксел), подофиллотоксины (Этопозид, Иринотекан, Топотекан), антибиотики (Доксорубицин, Блеомицин, Митомицин), нитрозомочевины (Кармустин, Ломустин), неорганические ионы (Цисплатин, Карбоплатин), ферменты (Аспарагиназа) и гормоны (Тамоксифен, Лейпролид, Флутамид и Мегестрол), Gleevec Другие примеры средств, с которыми ингибиторы по изобретению могут также быть скомбинированы, включают, без ограничения: средства для лечения болезни Альцгеймера, такие как Aricept® и Excelon®; для лечения болезни Паркинсона, такие как L-ДОФА/карбидофа, энтакапон, ропинрол, прамипексол, бромокриптин, перголид, тригексефендил и амантадин; средства для лечения рассеянного склероза (MS), такие как бета-интерферон (например, Avonex® и Rebif®), Copaxone® и митоксантрон; для лечения астмы, такие как альбутерол и Singulair®; средства для лечения шизофрении, такие как зипрекса, риспердал, сероквел и галоперидол; противовоспалительные средства, такие как кортикостероиды, блокаторы TNF, IL-1 RA, имуран, циклофосфамид и сульфасалазин; иммуномодуляторы и иммуносупрессивные средства, такие как циклоспорин, такролимус, рапамицин, мофетил микофенолат, интерфероны, кортикостероиды, циклофосфамид, имуран и сульфасалазин; нейротрофные факторы, такие как ингибиторы ацетилхолинэстеразы, ингибиторы МАО, интерфероны, противосудорожные средства, блокаторы ионных каналов, рилузол и антипаркинсонические средства; средства для лечения сердечно-сосудистых заболеваний, такие как бета-блокаторы, АСЕ ингибиторы, диуретики, нитраты, блокаторы кальциевых каналов и статины; средства для лечения заболеваний печени, такие как кортикостероиды, холестирамин, интерфероны и противовирусные средства; средства для лечения нарушений крови, такие как кортикостероиды, противолейкозные средства и факторы роста; и средства для лечения нарушений, связанных с иммунодефицитом, такие как гамма-глобулин. Количество дополнительного терапевтического средства в композициях по изобретению не должно превышать количество, которое обычно используется в композиции, включающей это терапевтическое средство в качестве единственного активного агента. Предпочтительно, количество дополнительного терапевтического средства в описываемых композициях составляет приблизительно от 50% до 100% от количества, обычно представленного в композиции, включающей это средство в качестве единственного активного агента. В другом варианте осуществления способы по изобретению, в которых используют композиции, которые не содержат дополнительное терапевтическое средство, включают дополнительную стадию отдельного введения указанному пациенту дополнительного терапевтического средства. Когда эти дополнительные терапевтические средства вводят отдельно, они могут вводиться пациенту до, одновременно с или после введения композиций по изобретению. Соединения по изобретению или содержащие их фармацевтически приемлемые композиции могут также быть включены в композиции для покрытия имплантируемых медицинских устройств, таких как протезы, искусственные клапаны, сосудистые трансплантаты, стенты и катетеры. Соответственно, настоящее изобретение, в другом аспекте, включает композицию для покрытия имплантируемого устройства, содержащую соединение согласно настоящему изобретению, как описано выше в целом, и в классах, и подклассах, и носитель, подходящий для нанесения покрытия на указанное имплантируемое устройство. В другом аспекте настоящее изобретение включает имплантируемое устройство, покрытое композицией, содержащей соединение по изобретению, как описано выше в целом, и в классах и подклассах, и носитель, подходящий для нанесения покрытия на указанное имплантируемое устройство. Сосудистые стенты, например, используют, чтобы преодолеть рестеноз (сужение стенок сосудов после повреждения). Однако пациенты, использующие стенты или другие имплантируемые устройства, подвергаются риску формирования сгустка или активации тромбоцитов. Эти нежелательные эффекты могут быть предотвращены или смягчены путем предварительного нанесения покрытия на устройство с использованием фармацевтически приемлемой композиции, включающей ингибитор киназы. Подходящие покрытия и общее получение покрытых имплантируемых устройств описаны в Патентах США 6099562; 5886026 и 5304121. Покрытия обычно представляют собой биологически совместимые полимерные материалы, такие как гидрогелевые полимеры, полиметилдисилоксан, поликапролактон, полиэтиленгликоль, полимолочную кислоту, этиленвинилацетат и их смеси. Покрытия могут в случае необходимости быть далее покрыты подходящим поверхностным покрытием из фторсиликона, полисахаридов, полиэтиленгликоля, фосфолипидов или их комбинаций, чтобы придать композиции характеристики контролируемого высвобождения. Другой аспект изобретения относится к ингибированию активности протеинкиназы ERK1 или ERK2 в биологическом образце или в организме пациента, включающему введение пациенту соединения согласно настоящему изобретению или композиции, содержащей указанное соединение, или контактирование указанного биологического образца с соединением согласно настоящему изобретению или композицией, содержащей указанное соединение. Термин “биологический образец” в рамках изобретения включает, без ограничения, клеточные культуры или их экстракты; материал биопсии, полученный от млекопитающего, или его экстракты; и кровь, слюну, мочу, кал, сперму, слезы или другие жидкости организма или их экстракты. Ингибирование активности протеинкиназы ERK1 или ERK2 в биологическом образце может быть использовано в различных целях, которые известны специалисту. Примеры таких целей включают, но не ограничены ими, переливание крови, трансплантацию органа, хранение биологического образца и биологические исследования. ПРИМЕРЫ СИНТЕЗА В рамках изобретения термин «Rt» относится ко времени удерживания в ВЭЖХ, в минутах, в отношении соединения. Если не указано иное, способ ВЭЖХ, используемый, чтобы получить время удерживания, осуществляют следующим образом: Колонка: Agilent / ZORBAX SB-CL8 / 5 мкм / 3,0×150 мм / PN 883975-302 / SN USBM001410 Градиент: 10-90% MeCN в течение 8 минут Поток: 1,0 мл/мин Детекция: 214 и 254 нм Если не указано иное, каждый 1H ЯМР был получен при 500 МГц в CDCl3, и номера соединений соответствуют номерам соединений, указанным в Таблице 1. Пример 1 Соединение I-9 было получено следующим образом:
2,2,2-Трихлор-1-(4-йод-1Н-пиррол-2-ил)этанон К перемешиваемому раствору 50 г (235 ммоль, 1,0 экв.) 2,2,2-трихлор-1-(1Н-пиррол-2-ил)этанона в сухом дихлорметане (400 мл) в атмосфере азота добавляют по каплям раствор монохлорида йода (39 г, 240 ммоль, 1,02 эквивалента) в дихлорметане (200 мл). Полученную смесь перемешивали при комнатной температуре в течение 2 часов. Раствор промывали 10% карбоната калия, водой, 1,0 М тиосульфата натрия, насыщенным хлоридом натрия, высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Твердое вещество перекристаллизовывали из смеси гексан/метилацетат, получая целевое соединение (68,5 г, 86%) в виде бесцветного твердого вещества (86%). MS FIA: 335,8, 337,8 ES-. Метиловый эфир 4-йод-1Н-пиррол-2-карбоновой кислоты К перемешиваемому раствору 2,2,2-трихлор-1-(4-йод-1Н-пиррол-2-ил)этанона (68 г, 201 ммоль, 1,0 эквивалент) в сухом метаноле (400 мл) в атмосфере азота за 10 минут был добавлен раствор метилата натрия в метаноле (4,37 М, 54 мл, 235 ммоль, 1,2 эквивалента). Полученную смесь перемешивали при комнатной температуре в течение 1 часа. Летучие компоненты удаляли при пониженном давлении и сырой продукт разделяли между водой и трет-бутилметиловым эфиром. Органическую фазу отделяли, промывали два раза водой, насыщенным хлоридом натрия, высушивали над сульфатом натрия, фильтровали и концентрировали под вакуумом, получая целевое соединение (48 г, 96%) в виде бесцветного твердого вещества, которое использовали непосредственно без дальнейшей очистки. Метиловый эфир 4-йод-1-(толуол-4-сульфонил)-1Н-пиррол-2-карбоновой кислоты: метиловый эфир 4-йод-1Н-пиррол-2-карбоновой кислоты (24,6 г, 98 ммоль, 1,0 эквивалент) растворяли в дихлорметане (150 мл) и триэтиламине (30 мл, 215,6 ммоль, 2,2 эквивалента). Добавляли 4-(диметиламино)пиридин (1,2 г, 9,8 ммоль, 0,1 эквивалента) и п-толуолсульфонилхлорид (20,6 г, 107,8 ммоль, 1,1 эквивалента) и реакционную смесь перемешивали в течение 16 часов при комнатной температуре. Реакцию останавливали 1 М HCl и органический слой промывали водным бикарбонатом натрия и рассолом. После высушивания над сульфатом магния растворитель удаляли при пониженном давлении и остаток кристаллизовали из трет-бутилметилового эфира, получая целевое соединение в виде твердого вещества светло-желтого цвета (30 г, 75%). Rt(мин) 8,259 минут. Метиловый эфир 4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1-(толуол-4-сульфонил)-1Н-пиррол-2-карбоновой кислоты: К дегазированному раствору метилового эфира 4-йод-1-(толуол-4-сульфонил)-1Н-пиррол-2-карбоновой кислоты (20 г, 49,4 ммоль, 1,0 эквивалент) и бис(пинаколято)диборана (15 г, 65 ммоль, 1,3 эквивалента) в ДМФ (200 мл) в атмосфере азота добавляли аддукт дихлорметана и дихлор[1,1′-бис(дифенилфосфин)ферроцен]палладия (II) (3,6 г, 4,9 ммоль, 0,1 эквивалент). Реакционную смесь перемешивали при 80°C в течение 18 часов. После удаления ДМФ при пониженном давлении полученное густое масло суспендировали в диэтиловом эфире (500 мл), и твердое вещество сразу осаждалось. Это твердое вещество удаляли фильтрацией и фильтрат промывали с 1M HCl, водой, рассолом и высушивали над MgSO4. Концентрацией получали целевое соединение как белое твердое вещество, которое использовали без дальнейшей очистки (10 г, 50%). LC/MS: Rt (мин) 4,6; 406,4 ES+. MS FIA: 406,2 ES +. 1H ЯМР N,N’-2-(5-хлор-4-йодопиридил)-изопропиламин: Способ A. (Микроволновой) В микроволновой пробирке на 10 мл 5-хлор-2-фтор-4-йодопиридин (1,0 г, 3,9 ммоль, 1,0 эквивалент) растворяли в ДМСО (4,0 мл) и затем добавляли изопропиламин (0,99 мл, 11,7 ммоль, 3,0 эквивалента). Пробирку герметизировали и помещали под микроволновое излучение на 600 секунд в 150°C. Эту реакцию повторяли шесть раз. Реакционные смеси объединяли, затем разбавляли в этилацетате и промывали водой. После высушивания над сульфатом натрия растворитель выпаривали, получая целевое соединение в виде густого коричневого масла (5,6 г, 80%), которое использовали непосредственно без дальнейшей очистки. Rt (мин) 4,614; MS FIA: 296,9 ES+. 1H ЯМР Способ B: (Тепловой) 5-хлор-2-фтор-4-йодопиридин (400 мг, 1,55 ммоль, 1,0 эквивалент) растворяли в этаноле (5,0 мл) и затем добавляли изопропиламин (0,66 мл, 7,8 ммоль, 5,0 эквивалентов). Полученный раствор перемешивали при 80°C в течение 48 часов. Реакционную смесь разбавляли в этилацетате и промывали водой. После высушивания над сульфатом натрия выпаривали растворитель и получали густое коричневое масло, которое очищали флэш-хроматографией на силикагеле с элюированием смесями гексан/этилацетат (от 99:1 до 80:20), получая целевое соединение в виде твердого вещества светло-желтого цвета (96 мг, 21%). Метиловый эфир 4-(5-хлор-2-изопропиламинопиридин-4-ил)-1-(толуол-4-сульфонил)-1Н-пиррол-2-карбоновой кислоты: К раствору N,N’-2-(5-хлор-4-йодопиридил)-изопропиламина (0,53 г, 1,8 ммоль, 1,0 эквивалент) и метилового эфира 4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1-(толуол-4-сульфонил)-1Н-пиррол-2-карбоновой кислоты (0,78 г, 1,8 ммоль, 1,0 эквивалент) в DME (4,0 мл) был добавлен раствор водного 2М карбоната натрия (1,0 мл), затем Pd(PPh3)4 (0,21 мг, 0,18 ммоль, 0,1 эквивалента). Микроволновую пробирку герметизировали и реакционную смесь облучали микроволновым излучением в течение 1800 секунд при 170°C. Сырой продукт шести реакций объединяли и разбавляли в этилацетате и промывали водой. После высушивания органического слоя над сульфатом натрия удаляли растворитель и полученное густое масло адсорбировали на силикагеле. Сырой продукт очищали флэш-хроматографией на силикагеле, элюировали смесями гексан/этилацетат (от 99:1 до 70:30), получая целевое соединение как желтое твердое вещество (3,1 г, 61% за две стадии). Rt(мин) 6,556. MS FIA: 448,1 ES+. 1H ЯМР Метиловый эфир 4-(5-хлор-2-изопропиламинoпиридин-4-ил)-1-(2,4,6-триметилбензолсульфонил)-1Н-пиррол-2-карбоновой кислоты: К раствору N,N’-2-(5-хлор-4-йодопиридил)-изопропиламина (96 мг, 0,32 ммоль, 1,0 эквивалент) и метилового эфира 4-(4,4,5,5-тетраметил-[1, 3,2]диоксаборолан-2-ил)-1-(2,4,6-триметилбензолсульфонил)-1Н-пиррол-2-карбоновой кислоты (152 мг, 0,35 ммоль, 1,1 эквивалента) в DME (2 мл) был добавлен раствор 2М водного карбоната натрия (0,2 мл), затем Pd(PPh3)4 (37 мг, 0,032 ммоль, 0,1 эквивалента). Реакционную смесь перемешивали при 80°C в течение 16 часов. Сырой продукт разбавляли в этилацетате и промывали водой. После высушивания органического слоя над сульфатом натрия удаляли растворитель, и полученное густое масло было адсорбировано на силикагеле. Сырой продукт очищали флэш-хроматографией на силикагеле, элюировали смесями гексан/этилацетат (от 99:1 до 80:20), получая целевое соединение в виде желтого твердого вещества (65 мг, 43%). Rt (мин) 7,290. MS FIA: 476,1 ES+. 4-(5-хлор-2-изопропиламинопиридин-4-ил)-1Н-пиррол-2-карбоновая кислота: Способ А (Микроволновой). Раствор метилового эфира 4-(5-хлор-2-изопропиламинопиридин-4-ил)-1-(толуол-4-сульфонил)-1Н-пиррол-2-карбоновой кислоты (3,1 г, 6,9 ммоль, 1,0 эквивалент) в ТГФ (2,0 мл) был добавлен к раствору моногидратированного гидроксида лития (710 мг, 17,3 ммоль, 2,5 эквивалента) в воде (3,0 мл). Микроволновую пробирку герметизировали и реакционную смесь подвергали микроволновому облучению в течение 1200 секунд при 150°C. Сырой раствор подкисляли водной 6н. HCl. Растворитель выпаривали, получая целевое соединение, которое использовали непосредственно без дальнейшей очистки. Rt (мин):3,574. MS FIA: 279,9 ES+; 278,2 ES-. Способ B: (Тепловой) Раствор метилового эфира 4-(5-хлор-2-изопропиламинопиридин-4-ил)-1-(2,4,6-триметилбензолсульфонил)-1Н-пиррол-2-карбоновой кислоты (0,69 г, 1,4 ммоль, 1,0 эквивалента) в ТГФ (3,0 мл) был добавлен к раствору моногидратированного гидроксида лития (1,19 г, 29 ммоль, 20,0 эквивалентов) в воде (3,0 мл). Смесь нагревали с обратным холодильником в течение 8 часов. Сырой раствор подкисляли водной 6н. HCl до помутнения, органический растворитель частично удаляли и продукт осаждали. Целевое соединение выделяли фильтрацией и промывали водой и диэтиловым эфиром, получая твердое вещество белого цвета (0,38 г, 96 %). [1-(3-хлорфенил)-2-гидроксиэтил]амид 4-(5-хлор-2-изопропиламинопиридин-4-ил)-1H-пиррол-2-карбоновой кислоты: К суспензии 4-(5-хлор-2-изопропиламинопиридин-4-ил)-1Н-пиррол-2-карбоновой кислоты (1,93 г, 6,9 ммоль, 1,0 эквивалент) в ДМФ (5,0 мл) добавляли EDCI (1,45 г, 7,6 ммоль, 1,1 эквивалента), HOBt (0,94 г, 6,9 ммоль, 1,0 эквивалент) и (S)-3-хлорфенилглицинол (1,58 г, 7,6 ммоль, 1,1 эквивалента). Затем добавляли диизопропилэтиламин (2,7 мл) и полученную смесь перемешивали при комнатной температуре в течение ночи. Смесь вливали в воду и экстрагировали этилацетатом. После высушивания над сульфатом натрия растворитель удаляли и сырой продукт адсорбировали на силикагеле. Очистка была произведена флэш-хроматографией на силикагеле при элюировании со смесями гексан/ацетон (от 80:20 до 60:40), с получением целевого соединения в виде белого твердого вещества (1,9 г, 64%). Rt (мин) 4,981s. FIA MS: 433,1 ES+; 431,2 ES-. 1H ЯМР (CD3OD) Пример 2 Соединение I-9 было также получено согласно следующему альтернативному способу:
2,5-дихлор-4-нитропиридин-N-оксид: К суспензии 2-хлор-5-хлорпиридина (10 г, 0,067 моль) в уксусном ангидриде (25 мл) малыми порциями добавляли перекись водорода 30% (25 мл). Эту смесь перемешивали при комнатной температуре в течение 24 часов и затем нагревали при 60°C в течение 30 часов. После удаления избытка уксусной кислоты при пониженном давлении остаток малыми порциями добавляли к концентрированной серной кислоте (15 мл). Полученный раствор добавляли к смеси концентрированной серной кислоты (15 мл) и дымящей азотной кислоты (25 мл) и затем нагревали при 100°C в течение 90 минут. Реакционную смесь лили на лед, нейтрализовывали с твердым карбонатом аммония и, наконец, водным раствором аммиака до достижения основного рН и образования осадка. Осадок собирали фильтрацией, получая целевое соединение в виде твердого вещества светло-желтого цвета (3,1 г), Rt (мин) 3,75. MS FIA не показывает пиков. 1H ЯМР (ДМСО-d6) 4-бром-2-хлор-5-N-изопропилпиридин-2-амин N-оксид: К 2,5-дихлор-4-нитропиридин N-оксиду (400 мг, 1,9 ммоль) очень медленно добавляли ацетил бромид (2 мл). Реакционную смесь затем нагревали при 80°C в течение 10 минут. Растворитель удаляли под током азота и сырой продукт высушивали под сильным вакуумом. Сырой материал (165 мг, 0,62 ммоль) растворяли в этаноле (2 мл), добавляли изо-пропиламин (0,53 мл) и полученную смесь нагревали при 80°C в течение 2 часов. Сырой раствор очищали полностью ВЭЖХ с обратной фазой (ацетонитрил/вода/ТФК 1%), получая целевое соединение в виде твердого вещества светло-желтого цвета (60 мг, 36,6%). Rt (мин) 5,275. MS FIA 264,8, 266,9 ES+. [l-(3-хлорфенил)-2-гидроксиэтил] амид 4-(5-хлор-2-изопропиламинопиридин-4-ил)-1Н-пиррол-2-карбоновой кислоты (I-9): 4-бром-2-хлор-5-N-изопропилпиридин-2-амин N-оксид (25 мг, 0,094 ммоль, 1,0 эквивалент) и метиловый эфир 4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1-(2,4,6-триметилбензолсульфонил)-1Н-пиррол-2-карбоновой кислоты (39 мг, 0,094 ммоль, 1,0 эквивалент) растворяли в бензоле (5 мл), затем добавляли водный раствор 2M Na2CO3 (1 мл) и Pd(PPh3)4 (115,6 мг, 0,1 ммоль, 0,2 эквивалента) и полученную суспензию нагревали с обратным холодильником при 80°C в течение 16 часов. Реакционную смесь разбавляли в этилацетате, промывали водой и высушивали над безводным сульфатом натрия, получая N-оксид метилового эфира 4-(5-хлор-2-изопропиламино-пиридин-4-ил)-1-(2,4,6-триметил-бензолсульфонил)-1Н-пиррол-2-карбоновой кислоты (Rt (мин) 6,859. MS FIA: 492,0 ES+), который затем обрабатывают 2М раствором PCl3 в дихлорметане (1 мл) при комнатной температуре. Через 10 минут растворитель удаляли под током азота и сырое масло растворяли в метаноле (1 мл) и водном 1М растворе NaOH (1 мл). Полученную смесь нагревали с обратным холодильником в течение 16 часов, сырой раствор подкисляли, используя водный 1М раствор HCl, и растворитель удаляли. Полученную 4-(5-хлор-2-изопропиламино-пиридин-4-ил)-1H-пиррол-2-карбоновую кислоту (Rt (мин) 3,527. MS FIA: 279,4 ES+; 278,2 Es-) суспендировали в ДМФ (3 мл) вместе с EDCI (36 мг, 0,19 ммоль, 2 эквивалента), HOBt (26 мг, 0,19 ммоль, 2 эквивалента), соли HCl (S)-3-хлорфенилглицинола (59 мг, 0,28 ммоль, 3 эквивалента) и DIEA (0,12 мл, 0,75 ммоль, 8 эквивалентов). Полученную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь разбавляли в этилацетате, промывали водой и высушивали над сульфатом натрия. После удаления растворителя при пониженном давлении сырой продукт очищали ВЭЖХ с обратной фазой (ацетонитрил/вода/ТФК 1%), получая целевое соединение в виде белого твердого вещества (4,8 мг, 8,1%). Пример 3 Соединение I-3 получали следующим образом:
2-хлор-5-метил-4-нитропиридин N-оксид: целевое соединение получают способом, в основном аналогичным описанному Z. Talik, A. Puszko, Roczniki Chemii Ann. Soc. Chim. Polonorum 1976, 50, 2209, следующим образом. К суспензии 2-хлор-5-метилпиридина (10 г, 0,078 моль) в уксусном ангидриде (25 мл) малыми порциями добавляют перекись водорода 30% (25 мл). Эту смесь перемешивали при комнатной температуре в течение 24 часов и затем нагревали при 60°C в течение 30 часов. После удаления избытка уксусной кислоты при пониженном давлении остаток добавляют малыми порциями к концентрированной серной кислоте (15 мл). Полученный раствор добавляют к смеси концентрированной серной кислоты (15 мл) и дымящей азотной кислоты (25 мл) и затем нагревают при 100°C в течение 90 минут. Реакционную смесь выливают на лед, нейтрализуют твердым карбонатом аммония и, наконец, с водным раствором аммиака до достижения основного рН и формирования осадка. Этот осадок собирают фильтрацией, получая целевое соединение в виде твердого вещества светло-желтого цвета (9,4 г, 0,050 моль, Rt (мин) 3,272, FIA ES+ 188,9, ES- 188,0). 2-((S)-1-гидроксиметилпропиламино)-5-метил-4-нитро-пиридин N-оксид: 2-Хлор-5-метил-4-нитропиридин N-оксид (200 мг, 1,06 ммоль, 1,0 эквивалент) растворяли в этаноле (1,5 мл). Добавляли (S)-2-Аминобутанол (284 мг, 3,2 ммоль, 3,0 эквивалента) и полученную смесь нагревали с обратным холодильником в течение 16 часов. Сырой раствор очищали ВЭЖХ с обратной фазой (ацетонитрил/вода/ТФК), получая целевое соединение в виде коричневого твердого вещества (146 мг, 0,61 ммоль, Rt (мин) 3,787; FIA ES+ 241,8, ES- не наблюдается). 2-(4-бром-5-метилпиридин-2-иламино)-бутиловый эфир уксусной кислоты: 2-((S)-1-гидроксиметилпропиламино)-5-метил-4-нитро-пиридин N-оксид (146 мг, 0,61 ммоль) растворяли в ацетилбромиде (1,5 мл). Смесь нагревали при 90°C в течение 3 часов. Ацетилбромид выпаривали под током азота и сырой материал растворяли в растворе 2M PCl3 в дихлорметане (2 мл). Полученную смесь перемешивали при комнатной температуре в течение 1 часа и реакционную смесь вливали в водный раствор насыщенного NaHCO3 и экстрагировали этилацетатом. Органический слой промывали водой, растворитель высушивали над Na2SO4 и удаляли при пониженном давлении, получая целевое соединение в виде коричневого масла (149 мг, Rt (мин) 4,146; FIA ES+ 300,9, ES- не наблюдается). Метиловый эфир 4-[2-(S)-(1-ацетоксиметилпропиламино)-5-метилпиридин-4-ил]-1-(2,4,6-триметилбензолсульфонил)-1Н-пиррол-2-карбоновой кислоты: К раствору 2-(4-бром-5-метилпиридин-2-иламино)-бутилового эфира уксусной кислоты (149 мг, 0,5 ммоль, 1,0 эквивалент) и метилового эфира 4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1-(2,4,6-триметилбензолсульфонил)-1H-пиррол-2-карбоновой кислоты (215 мг, 0,50 ммоль, 1,0 эквивалент) в бензоле (5 мл) добавляли водный раствор 2M Na2CO3 (1 мл) и Pd(PPh3)4 (115,6 мг, 0,1 ммоль, 0,2 эквивалента). После нагревания с обратным холодильником в течение 16 часов смесь вливали в воду и экстрагировали этилацетатом. Органический экстракт высушивали над Na2SO4 и растворитель удаляли при пониженном давлении, получая целевое соединение (Rt (мин) 6,684; FIA ES+ 528,3, ES- не наблюдается), которое переносили на следующую стадию. [1-(S)-(3-хлорфенилглицинол]амид 4-[2-(S)-гидроксиметилпропиламино)-5-метилпиридин-4-ил]-1Н-пиррол-2-карбоновой кислоты (I-3): Сырой метиловый эфир 4-[2-(S)-(1-ацетоксиметилпропиламино)-5-метилпиридин-4-ил]-1-(2,4,6-триметилбензолсульфонил)-1Н-пиррол-2-карбоновой кислоты растворяли в метаноле (1,5 мл) и водном растворе 1M NaOH (2 мл) и смесь нагревали с обратным холодильником в течение 16 часов. После подкисления водным раствором 1M HCl (2,2 мл) растворитель удаляли при пониженном давлении и сырой продукт суспендировали в ДМФ (5 мл). После добавления EDCI (192 мг, 1,0 ммоль), HOBt (135 мг, 1,0 ммоль) и DIEA (0,48 мл, 3,0 ммоль) смесь перемешивали в течение 30 минут при комнатной температуре, после чего добавляли соль HCl (S)-3-хлорфенилглицинола (312 мг, 1,5 ммоль) и реакционную смесь перемешивали в течение 16 часов при комнатной температуре. Реакционную смесь растворяли в этилацетате, промывали водой, органический слой высушивали над Na2SO4 и растворитель удаляли при пониженном давлении. Сырой остаток очищали ВЭЖХ с обратной фазой (ацетонитрил/вода/ТФК), получая целевое соединение в виде бесцветного твердого вещества (29,3 мг, 0,07 ммоль, Rt (мин) 4,563; FIA ES + 443,1, ES- 441,5, ES+ 443,1, ES- 441,5; 1H ЯМР (CD3OD) Пример 4 Данные oхарактеризовывания Соединения согласно настоящему изобретению были получены способами, в основном подобными описанным в приведенных выше Примерах 1-3, и способами, известными специалисту. Данные охарактеризовывания для этих соединений приведены ниже в Таблице 3 и включают данные ВЭЖХ, MS и 1H ЯМР. Если не указано иное, данные 1H ЯМР были получены при 500 МГц, и все указанные химические сдвиги даны в ppm. Номера соединений соответствуют номерам соединений, перечисленным в Таблицах 1, 2 и 3. В рамках изобретения термин «Rt» относится ко времени удерживания, в минутах, полученному для определяемого соединения с использованием способа ВЭЖХ, описанного выше. В случае, если для данного соединения, как описано здесь, было получено более одного результата аналитического измерения, приводится только один результат. Таблица 3. Данные охарактеризовывания для выбранных соединений формулы I
Пример 5 Синтез пролекарств Пролекарства формулы II получают из гидроксильных соединений формулы I различными способами, известными специалисту. Эти способы включают, но не ограничены ими, ацилирование желаемой карбоновой кислотой или образование фосфата. Когда гидроксильная группа в формуле I ацилируется желаемой аминокислотой, аминогруппа аминокислоты может быть в случае необходимости защищена подходящей аминозащитной группой, как описано выше. Получение L-валинового пролекарства соединения I-9 описано подробно ниже.
(2S)-(S)-2-(4-(5-хлор-2-(изопропиламино)пиридин-4-ил)-1H-пиррол-2-карбоксамидо)-2-(3-хлорфенил)этил-2-амино-2-метилбутаноат (II-1): К раствору соединения I-9 (1 г, 2,3 ммоль, 1,0 эквивалент) в дихлорметане (50 мл) добавляли DIEA (1,1 мл, 6,9 ммоль, 3,0 эквивалента) и N-BOC-L-валин (1,2 г, 5,52 ммоль, 2,4 эквивалента). Затем медленно добавляли PyBOP (2,9 г, 5,75 ммоль, 2,5 эквивалента) и полученную смесь перемешивали при комнатной температуре в течение 48 часов. Смесь затем промывали водой и высушивали над сульфатом натрия. Сырое твердое вещество адсорбировали на силикагеле и затем очищали флэш-хроматографией при элюировании со смесями гексан/этилацетат (от 90:10 до 50:50), получая Вос-защищенное соединение в виде белого твердого вещества (786 мг). Это промежуточное соединение (761 мг, 1,2 ммоль) растворяли в диоксане (1 мл) и обрабатывали 4М раствором HCl в диоксане. Полученную смесь перемешивали в течение 16 часов при комнатной температуре. Растворитель удаляли и получали 2xHCl соль целевого соединения в виде белого твердого вещества (571 мг). Rt ВЭЖХ: 4,56 минуты. MS FIA: 531,9 ES+; 529,8 ES-. LC/MS: Rt: 2,07 минуты; 532,0 ES+; 530,1 ES-. 1H ЯМР (CD3OD) Получение фосфатного пролекарства соединения I-9 описано подробно ниже.
Ди-трет-бутил-4-(5-хлор-2-(изопропиламино)пиридин-4-ил)-N-((S)-1-(3-хлорфенил)- 2-гидроксиэтил)-1Н-пиррол-2-карбоксамидфосфат: Соединение I-9 (1 г, 2,3 ммоль, 1,0 эквивалент) и тетразол (241 мг, 3,45 ммоль, 1,5 эквивалента) растворяли в дихлорметане (5 мл) и ацетонитриле (5 мл) в атмосфере азота при комнатной температуре. Ди-трет-бутилдиизопропилфосфоамидит (1,1 мл, 3,45 ммоль, 1,5 эквивалента) добавляли по каплям и полученную смесь перемешивали в течение 16 часов. Реакционную смесь затем охлаждали на ванне со льдом, обрабатывали раствором 6 М трет-бутилгидроксипероксида (3 мл) и перемешивали в течение 20 минут. Прозрачный раствор разбавляли в дихлорметане и малом количестве метанола, промывали Na2S2O3, водой и высушивали над сульфатом натрия. Сырое масло адсорбировали на силикагеле и сначала очищали флэш-хроматографией с элюированием со смесями гексан/ацетон (от 90:10 до 60:40) и затем ВЭЖХ с обратной фазой (ацетонитрил/вода/1% ТФК), получая промежуточный ди-трет-бутиловый эфир в виде белого твердого вещества (336 мг). ВЭЖХ Rt: 6,53 минут, MS FIA: 625,0 ES+; 623,1 ES-. 4-(5-хлор-2-(изопропиламино)пиридин-4-ил)-N-((S)-1-(3-хлорфенил)-2-гидроксиэтил)-1Н-пиррол-2-карбоксамид фосфат (II-2): Трет-бутилфосфатное промежуточное соединение (336 мг, 0,54 ммоль) суспендировали в диоксане (5 мл) и добавляли раствор 4М HCl в диоксане. Полученную смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель удаляли, свободный фосфат растворяли в растворе диметилсульфоксида (15 мл), метаноле (50 мл) и воде (25 мл) и обрабатывали 2М раствором Na2CO3 (0,25 мл). Метанол удаляли при пониженном давлении и смесь вода/ДМСО удаляли с использованием лиофилизатора, получая целевое соединение в виде твердого вещества белого цвета (187 мг). ВЭЖХ: Rt: 4,24 минуты. MS FIA: 512,9 ES+; 510,9 ES-. LC/MS: Rt 2,39 минуты; 512,9 ES+; 510,9 ES-. 1H ЯМР (CD3OD) Пример 6 Исследование ингибирования ERK2 Соединения были протестированы в отношении ингибирования ERK2 спектрофотометрическим сдвоенным ферментным анализом (Fox et al (1998) Protein Sсi 7,2249). В этом тесте фиксированную концентрацию активированной ERK2 (10 нМ) инкубировали с различными концентрациями соединения в ДМСО (2,5%) в течение 10 минут при 30°C в буфере HEPES (0,1 М), рН 7,5, содержащем 10 мМ MgCl2, 2,5 мМ фосфоенолпирувата, 200 мкМ НАДН, 150 мкг/мл пируваткиназы, 50 мкг/мл лактатдегидрогеназы и 200 мкМ эрктида пептида. Реакция была инициирована добавлением 65 мкМ АТФ. Отслеживали степень уменьшения поглощения при 340 нм. IC50 рассчитывали на основании этих данных в зависимости от концентрации ингибитора. Было обнаружено, что соединения согласно настоящему изобретению являются ингибиторами протеинкиназы ERK2. В некоторых вариантах осуществления было обнаружено, что соединения ингибируют ERK2-киназу в концентрации <0,1 мкM. В других вариантах осуществления было обнаружено, что соединения ингибируют ERK2-киназу в концентрации <0,01 мкМ. Пример 7 Исследование ингибирования ERK1 Соединения были протестированы в отношении ингибирования ERK1 спектрофотометрическим сдвоенным-ферментным анализом (Fox et al (1998) Protein Sci 7,2249). В этом тесте фиксированную концентрацию активированной ERK1 (20 нМ) инкубировали с различными концентрациями соединения в ДМСО (2,0%) в течение 10 минут при 30°C в буфере HEPES (0,1 М), рН 7,6, содержащем 10 мМ MgCl2, 2,5 мМ фосфоенолпирувата, 200 мкМ НАДН, 30 мкг/мл пируваткиназы, 10 мкг/мл лактатдегидрогеназы и 150 мкМ эрктида пептида. Реакция была инициирована добавлением 140 мкМ АТФ. Отслеживали степень уменьшения поглощения при 340 нм. Ki рассчитывали на основании этих данных в зависимости от концентрации ингибитора. Несмотря на то, что было описано множество вариантов осуществления этого изобретения, очевидно, что приведенные основные примеры могут быть изменены, чтобы обеспечить другие варианты осуществления, в которых можно использовать соединения и способы согласно изобретению. Поэтому следует отметить, что объем этого изобретения должен быть определен в соответствии с приложенной формулой изобретения, а не в соответствии с определенными вариантам осуществления, которые были представлены посредством примеров.
Формула изобретения
1. Соединение формулы I: 2. Соединение по п.1, в котором R обозначает С1-4-алифатическую группу, в случае необходимости замещенную -OR или -C1-3галогеналкилом. 3. Соединение по п.2, в котором R1 обозначает С1-4-алифатическую группу, в случае необходимости замещенную -ОН, -CHF2, -CH2F или -CF3. 4. Соединение по п.3, в котором R1 обозначает изопропил, 2-бутил, циклопропил или этил, причем каждая группа может быть замещена -ОН или -CF3. 5. Соединение по п.1, в котором R2 обозначает водород, С1-3-алифатическую группу или хлор. 6. Соединение по п.1, в котором R3 обозначает водород, метил или хлор. 7. Соединение, выбранное из группы, состоящей из: 8. Соединение по п.1, в котором: 9. Композиция для ингибирования активности протеинкиназы ERK1 или ERK2, содержащая соединение по п.1 и фармацевтически приемлемую основу, адъювант или носитель. 10. Способ ингибирования активности протеинкиназы ERK1 или ERK2 в биологическом образце, включающий введение в контакт указанного биологического образца с: 11. Применение соединения по п.1 или композиции по п.9 для лечения или уменьшения серьезности заболевания, состояния или нарушения у пациента, причем указанное заболевание, нарушение или состояние выбрано из меланомы, рака толстого кишечника, рака поджелудочной железы, рака почки, рака легкого, рака яичника или рака предстательной железы.
|
||||||||||||||||||||||||||
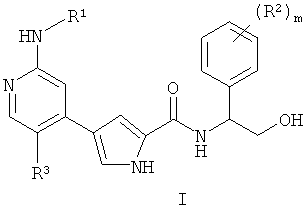 или его фармацевтически приемлемой соли, в которой R1 обозначает С1-6-алифатическую группу, причем R1 может быть замещен заместителями в числе до 2 групп, независимо выбранных из -OR или -С1-3галогеналкила; каждый R независимо обозначает водород или
или его фармацевтически приемлемой соли, в которой R1 обозначает С1-6-алифатическую группу, причем R1 может быть замещен заместителями в числе до 2 групп, независимо выбранных из -OR или -С1-3галогеналкила; каждый R независимо обозначает водород или )), и факторы роста (например, колониестимулирующий фактор гранулоцита-макрофага (GM-CSF), и фактор роста фибробласта (FGF)). Внеклеточный раздражитель может воздействовать на одну или более клеточных реакций, относящихся к росту, миграции, дифференцировки клеток, секреции гормонов, активации факторов транскрипции, сокращения мышцы, метаболизма глюкозы, контролю синтеза белка и регуляции клеточного цикла.
)), и факторы роста (например, колониестимулирующий фактор гранулоцита-макрофага (GM-CSF), и фактор роста фибробласта (FGF)). Внеклеточный раздражитель может воздействовать на одну или более клеточных реакций, относящихся к росту, миграции, дифференцировки клеток, секреции гормонов, активации факторов транскрипции, сокращения мышцы, метаболизма глюкозы, контролю синтеза белка и регуляции клеточного цикла.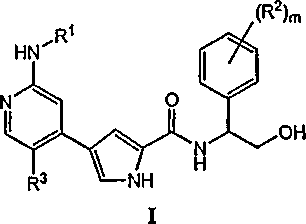
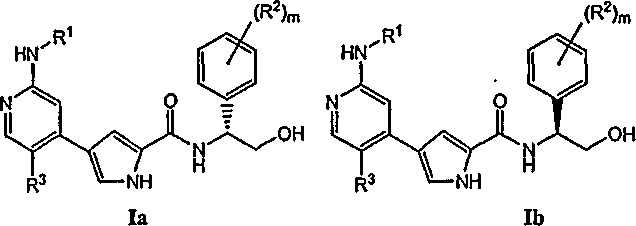
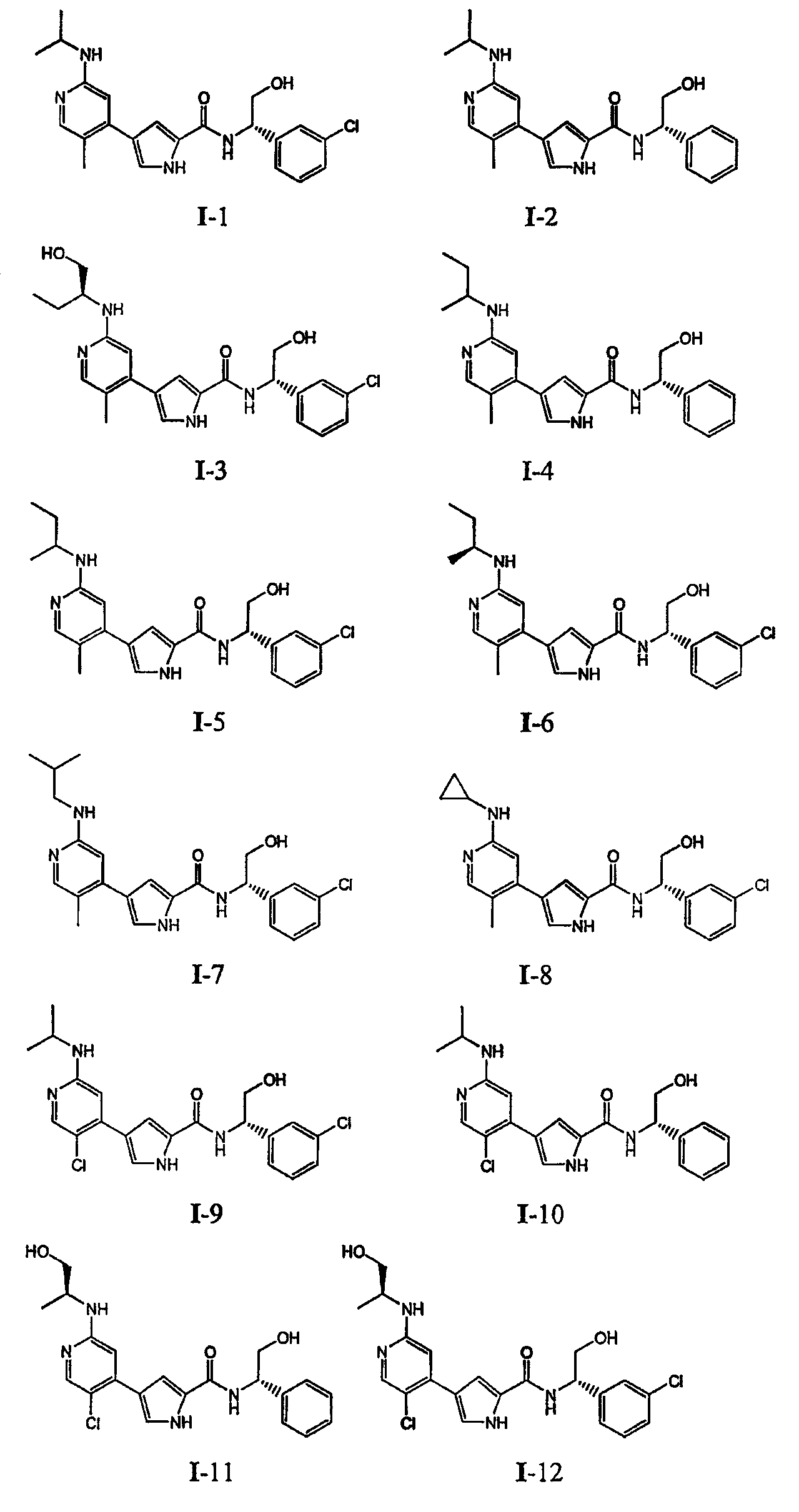
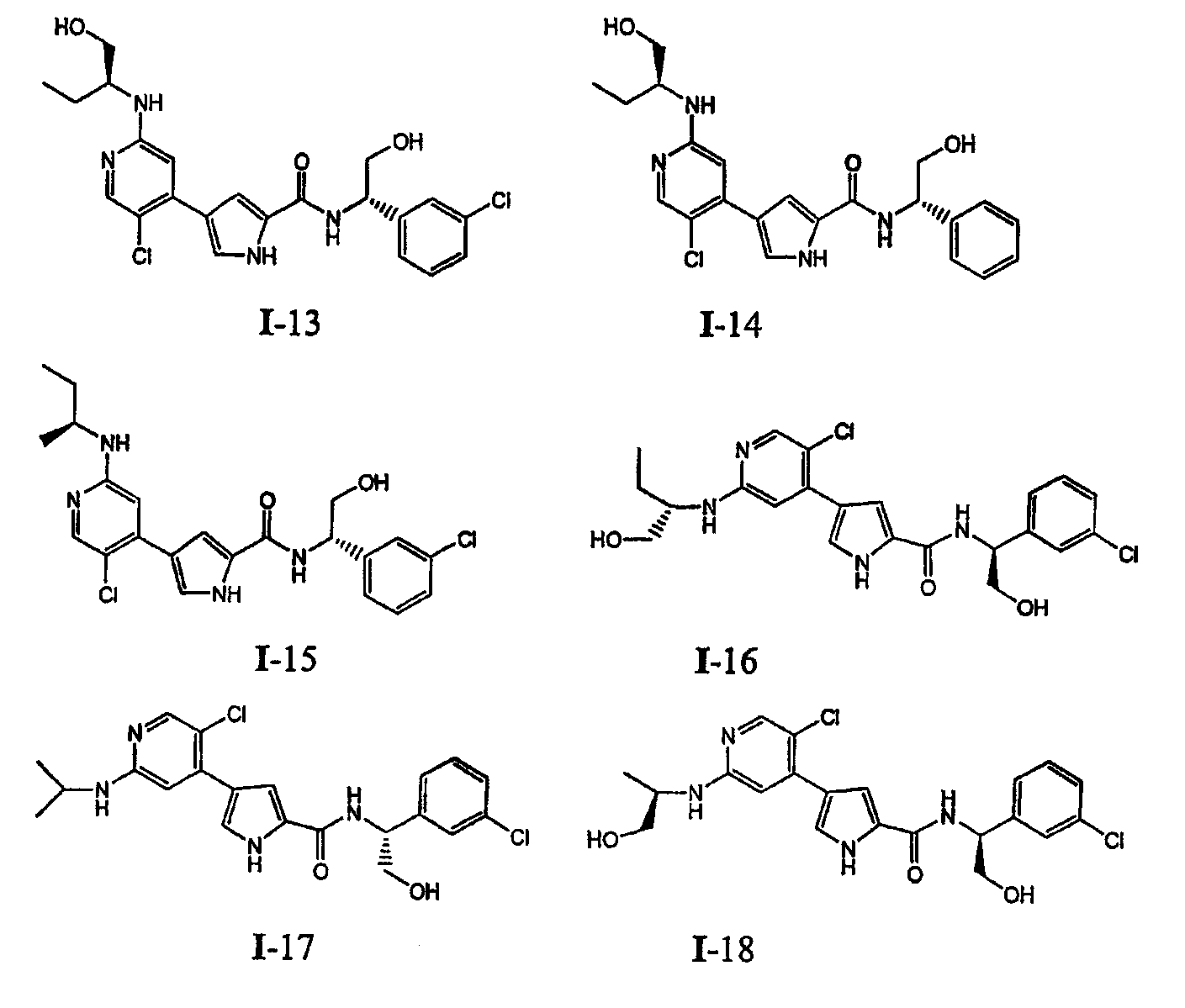
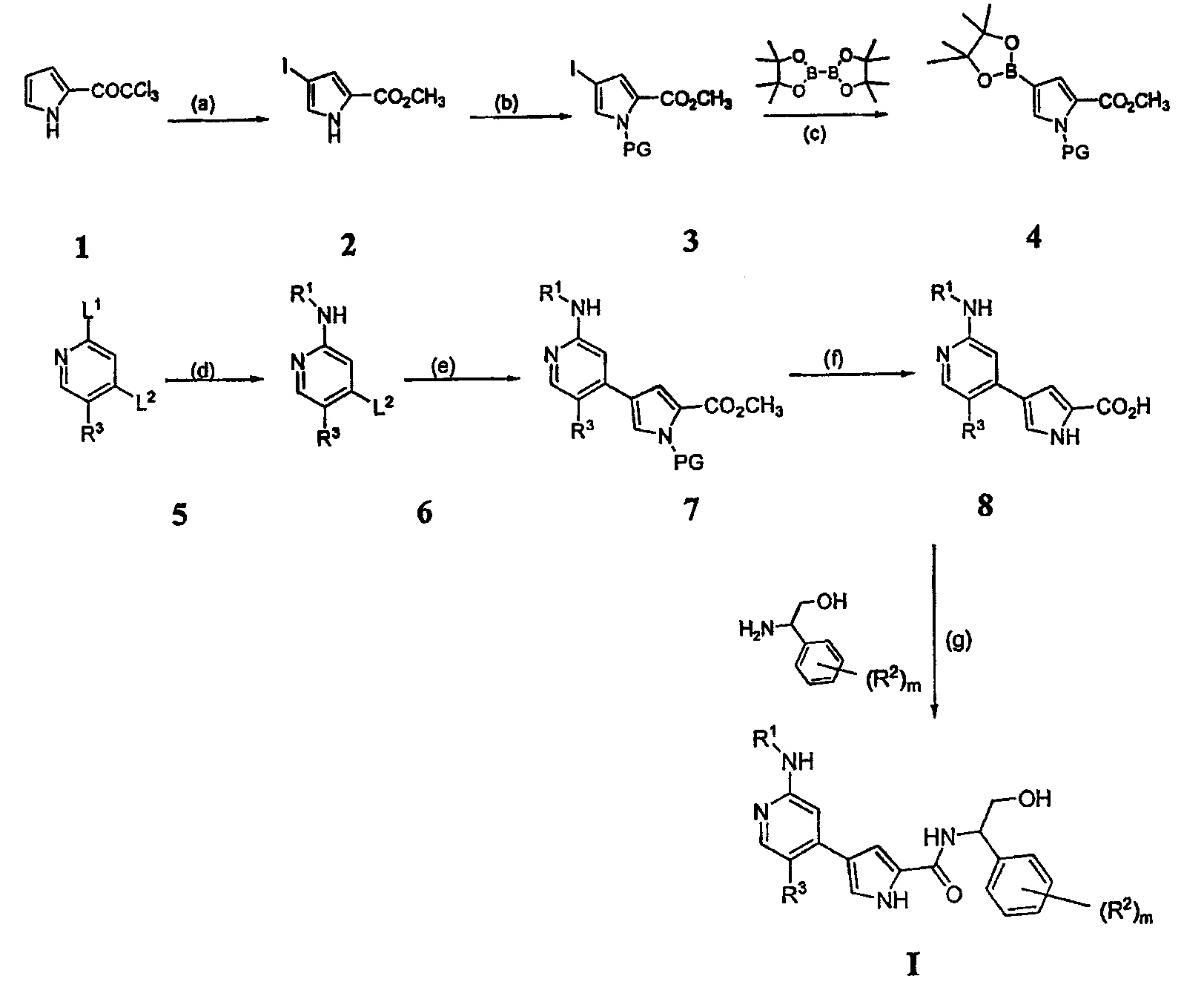
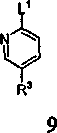
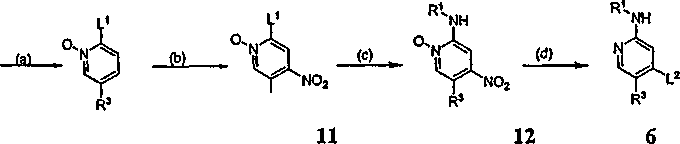
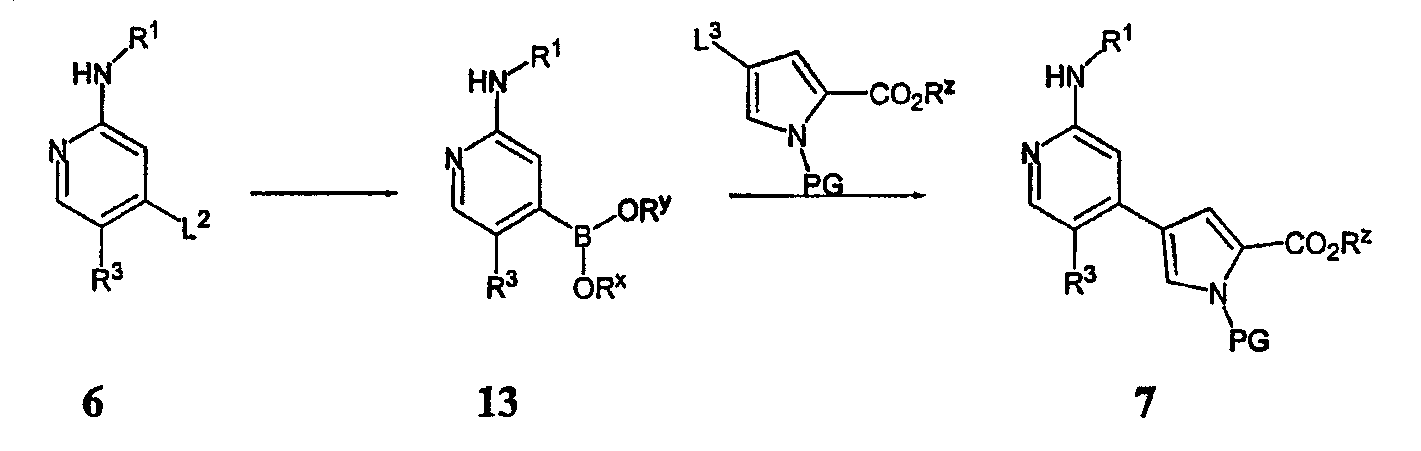
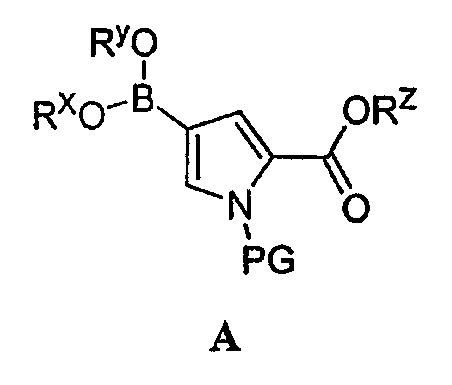
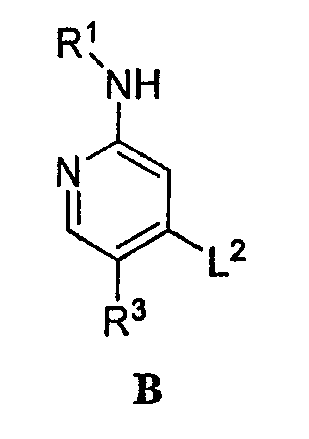
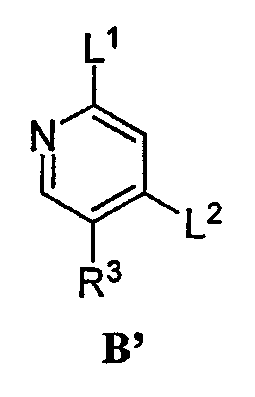
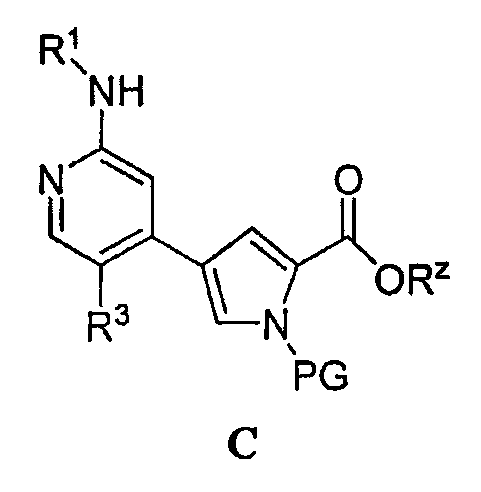
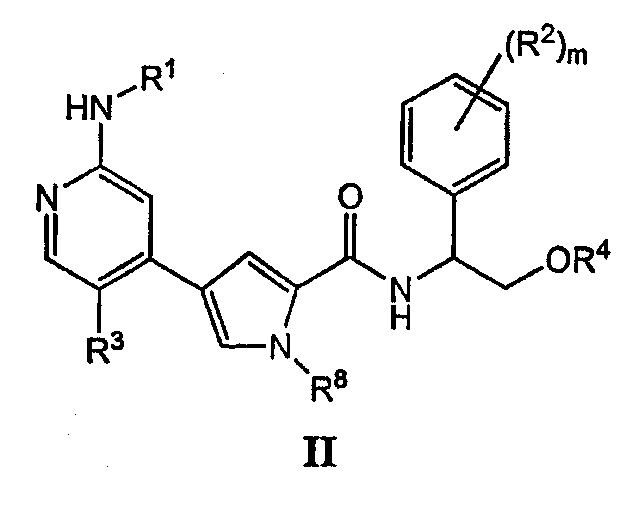
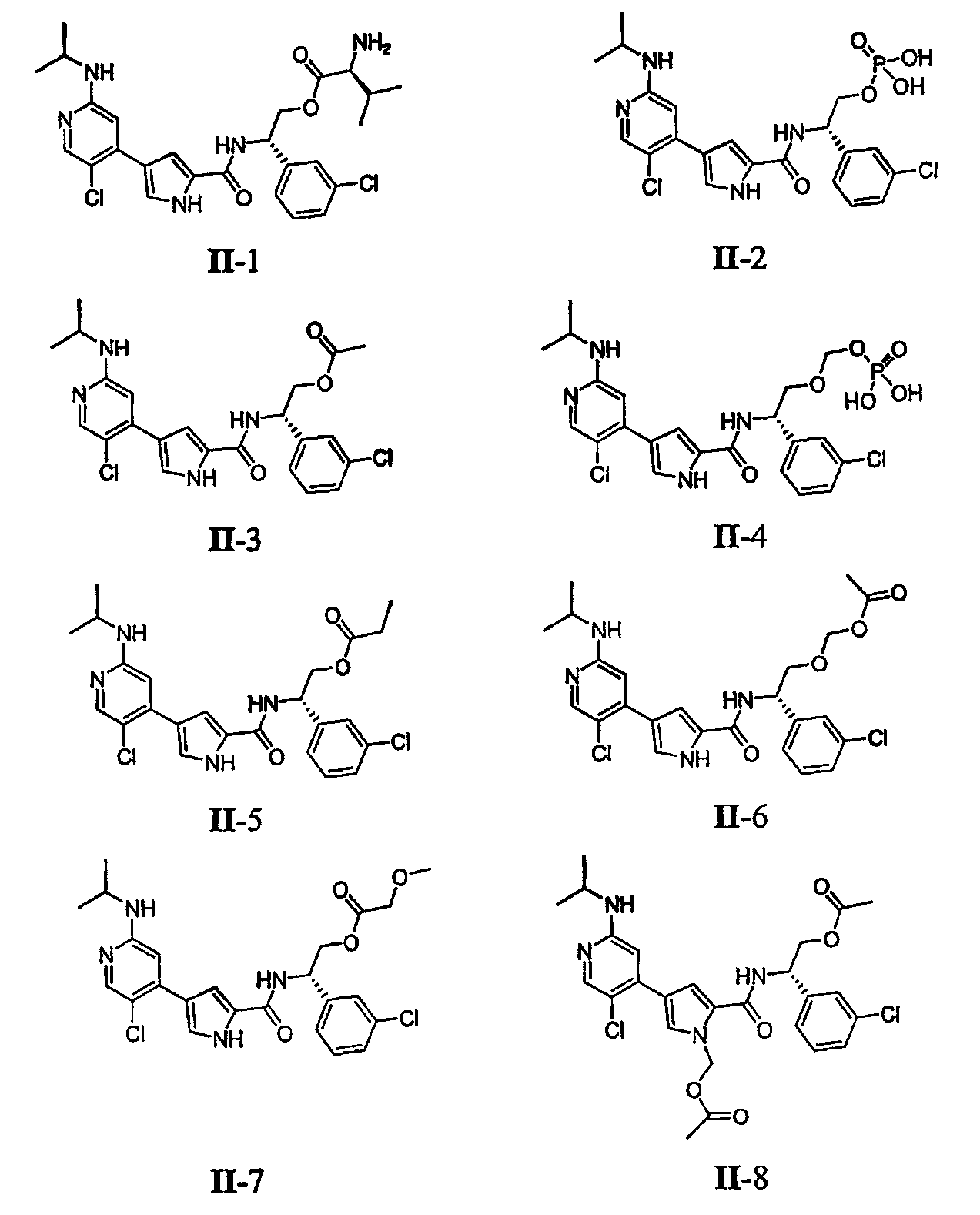
 ингибиторно активный метаболит или остаток
ингибиторно активный метаболит или остаток означает, что метаболит или остаток также является ингибитором протеинкиназы ERK2.
означает, что метаболит или остаток также является ингибитором протеинкиназы ERK2.
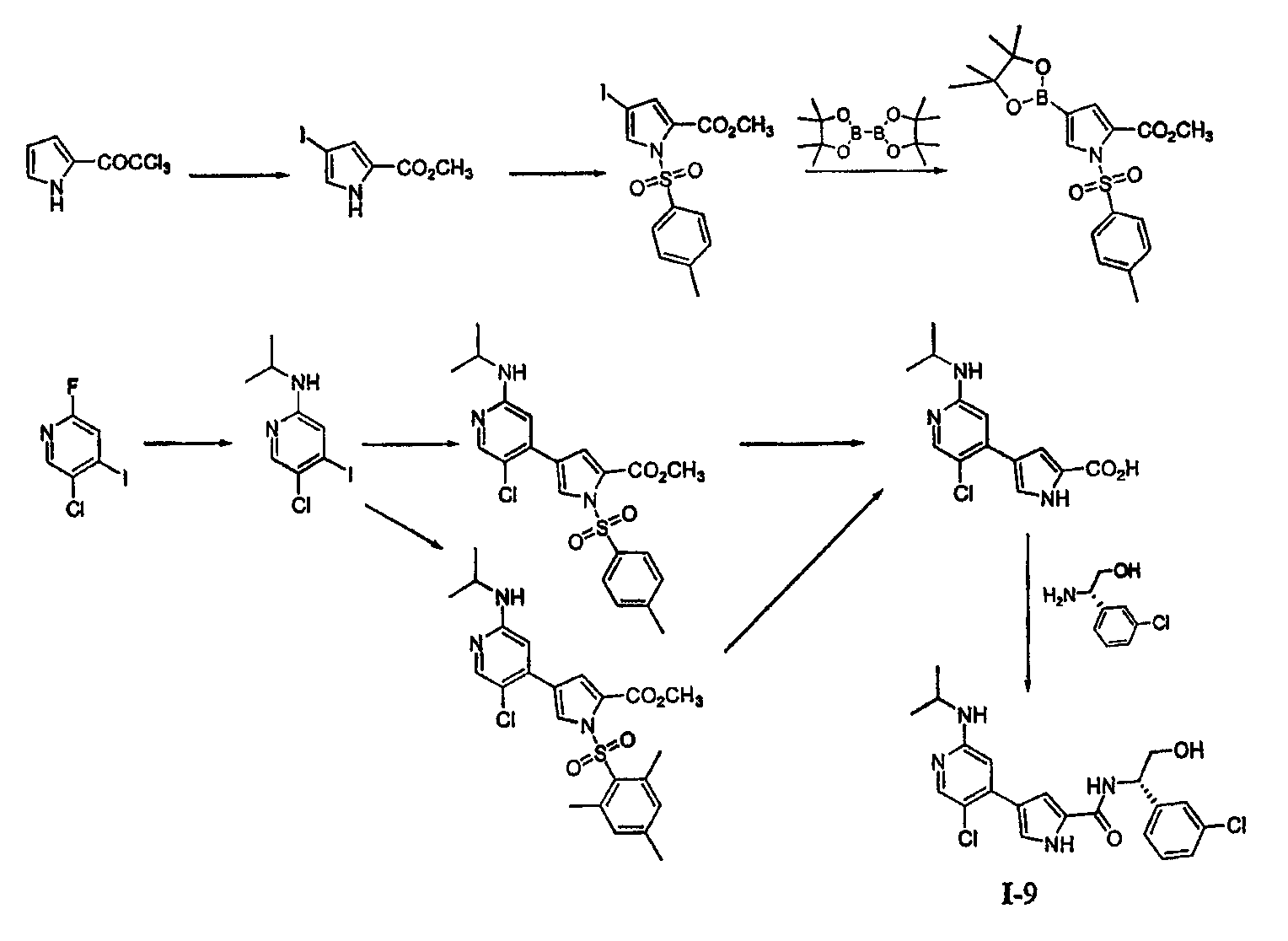
 1,2 (с, 12Н), 2,35 (с, 3H), 3,8 (с, 3H), 7,2 (м, 3H), 7,8 (д, 2H), 8,0 (с, 1H).
1,2 (с, 12Н), 2,35 (с, 3H), 3,8 (с, 3H), 7,2 (м, 3H), 7,8 (д, 2H), 8,0 (с, 1H).