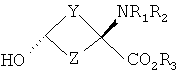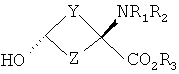Патент на изобретение №2376282
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) СТЕРЕОСЕЛЕКТИВНЫЙ СИНТЕЗ АМИНОКИСЛОТ ДЛЯ ПОЛУЧЕНИЯ ИЗОБРАЖЕНИЯ ОПУХОЛИ
(57) Реферат:
Изобретение относится к новым по существу чистым син-аминокислотам формул I и II, которые обладают способностью специфического связывания в биологической системе и могут быть использованы для получения изображения опухоли
В формулах I и II Y и Z независимо выбраны из группы, состоящей из СН2 и (CR4R5)n, n=1, 2; R1-R3 независимо выбраны из группы, состоящей из Н и алкила C1-C4; R4, R5=H и R7=18F. Изобретение относится также к способу синтеза син-аминокислот формулы II, который включает в себя стадии превращения кетона в транс-спирт формулы I и превращения полученного транс-спирта в син-аминокислоту формулы II, а также к фармацевтической композиции для получения изображения опухоли и способу получения изображения опухоли. 5 н. и 7 з.п. ф-лы, 1 табл., 3 ил.
Перекрестная ссылка на родственные заявки Данная заявка заявляет приоритет предварительной заявки Подтверждение о финансировании исследования Федеральными органами Данное изобретение выполнено с правительственной поддержкой по гранту Уровень техники Данное изобретение относится к способу синтеза аналогов син-аминокислот и соединениям, синтезированным согласно данному способу, в частности аналогам син-1-амино-3-циклобутан-1-карбоновой кислоты (АСВС). Аналоги аминокислот изобретения обладают способностью специфического связывания в биологической системе и их можно применять в способах получения изображения позитронно-эмиссионной томографией (ПЭТ) и однофотонной эмиссионной компьютерной томографией (ОЭКТ). Разработка меченых радиоактивными изотопами аминокислот для применения в качестве метаболических индикаторов для получения изображения опухолей с применением позитронно-эмиссионной томографии (ПЭТ) и однофотонной эмиссионной компьютерной томографии (ОЭКТ) проводилась в течение некоторого времени. Хотя меченые радиоактивными изотопами аминокислоты применяли для различных типов опухолей, их применению для внутричерепных опухолей уделяли значительное внимание вследствие потенциальных преимуществ относительно других модальностей получения изображений. После хирургической резекции и/или радиотерапии опухолей головного мозга общепринятые методы получения изображения, такие как СТ (компьютерная томография) и MRI (ЯМР-томография), надежно не отличают остаточную или рецидивирующую опухоль от повреждения ткани вследствие вмешательства и не являются оптимальными для мониторинга эффективности лечения или обнаружения рецидива опухоли [Buonocore, E (1992), Clinical Positron Emission Tomography. Mosby-Yaer Book, Inc. St. Louis, МО, p. 17-22; Langleben, DD et al. (2000), J. Nucl. Med. 41: 1861-1867]. Основной применяемый в ПЭТ агент для диагноза и получения изображения опухолей 2-[18F]фтopдeoкcиглюкoзa (FDG) имеет ограничения при получении изображения опухолей головного мозга. Нормальная кортикальная ткань головного мозга показывает высокое поглощение [18F]FDG, как и воспалительная ткань, которая может иметь место после лучевой терапии или хирургической терапии; эти факторы могут осложнять интерпретацию изображений, полученных с [18F]FDG [Griffeth, LK et al. (1993), Radiology. 186: 37-44; Conti, PS (1995)]. В ряде публикаций сообщается, что получение изображения методом ПЭТ и ОЭКТ с мечеными радиоактивными изотопами аминокислотами позволяет лучше определить границы опухоли в нормальном головном мозге, чем СТ или MRI, что позволяет лучше планировать лечение [Ogawa, Т et al. (1993), Radiology, 186: 45-53; Jager, PL et al. (2001), Nucl. Med., 42: 432-445]. Кроме того, некоторые исследования позволяют предположить, что степень поглощения аминокислоты коррелирует со стадией развития опухоли, которая может обеспечить важную прогностическую информацию [Jager, PL et al. (2001) J. Nucl. Med. 42: 432-445]. Аминокислоты являются питательными веществами, требуемыми для пролиферации клеток опухоли. Были получены различные аминокислоты, содержащие испускающие позитроны изотопы углерод-11 и фтор-18. Их оценивали для потенциального применения в клинической онкологии для получения изображения опухолей у пациентов с опухолями головного мозга и системных опухолей (опухолей во всем организме) и получили превосходные характеристики по сравнению с 2-[18F]FDG для некоторых опухолей. Эти аминокислотные кандидаты можно разделить на две основные категории. Первая категория представлена мечеными радиоактивными изотопами – существующими в природе аминокислотами, такими как [11С]валин, I-[11С]лейцин, L-[11С]метионин (MET) и L-[1-11С]тирозин и структурно подобными аналогами, такими как 2-[18F]фтop-L-тиpoзин и 4-[18F]фтop-L-фeнилaлaнин. Движение этих аминокислот через мембраны клеток опухолей преимущественно происходит посредством носителя, опосредуемого переносом натрий-независимой системой переноса лейцина, аминокислоты типа “L”. Повышенное поглощение и пролонгированное удерживание этих существующих в природе, меченых радиоактивным изотопом аминокислот в опухолях по сравнению с нормальной тканью является отчасти результатом значительного и быстрого регионального включения в белки. Из этих меченых радиоактивными изотопами аминокислот [11C]MET наиболее широко применяли клинически для обнаружения опухолей. Хотя обнаружено, что [11C]MET является пригодным при обнаружении опухолей головного мозга и системных поражений опухолями, он является восприимчивым in vivo к метаболизму посредством многочисленных путей, приводящему к образованию многочисленных меченых радиоактивными изотопами метаболитов. Поэтому графический анализ с необходимой точностью для надежного измерения метаболической активности опухоли является невозможным. Изучения кинетического анализа поглощения опухолью [11С]MET у людей убедительно позволяют предположить, что перенос аминокислот может обеспечить более чувствительное измерение пролиферации опухолевых клеток, чем синтез белка. Недостатки, связанные с [11С]МЕТ, могут быть преодолены второй категорией аминокислот. Они являются неприродными аминокислотами, такими как 1-аминоциклобутан-1-[11С]карбоновая кислота ([11C]ACBC). Преимуществом [11C]ACBC по сравнению с [11C]MET является то, что она не метаболизируется. Значительным ограничением в применении меченых углеродом-11 аминокислот для клинического применения является короткий 20-минутный период полураспада углерода-11. 20-Минутный период полураспада углерода-11 требует присутствия ускорителя частиц на участке для получения меченой углеродом-11 аминокислоты. Кроме этого, только одну или относительно очень мало доз можно получить из каждой партии получения меченой углеродом-11 аминокислоты. Следовательно, меченые углеродом-11 аминокислоты являются слабыми кандидатами с региональным распределением для широко распространенного клинического применения. Для преодоления ограничения применения углерода-11 из-за физического периода полураспада авторы изобретения в последнее время сосредоточили свои усилия на разработке нескольких новых меченых фтором-18 неприродных аминокислот, некоторые из которых описаны в патентах США 5808146 и 5817776, оба из которых включены здесь в качестве ссылки. Эти аминокислоты включают в себя анти-1-амино-3-[18F]фторциклобутил-1-карбоновую кислоту (анти-[18F]FACBC), син-1-амино-3-[18F]фторциклобутил-1-карбоновую кислоту (син-[18F]FACBC), син- и анти-1-амино-3-[18F]фторметилциклобутан-1-карбоновую кислоту (син- и aнти-[18F]FMACBC). Эти фтор-18-аминокислоты можно применять для получения изображения головного мозга и системных опухолей in vivo, основанного на переносе аминокислоты, способом получения изображения позитронно-эмиссионной томографией (ПЭТ). Разработка авторов включала меченые фтором-18 циклобутиламинокислоты, которые движутся через капилляры опухоли посредством опосредуемого носителем переноса, включающего в себя системы переноса в основном большой нейтральной аминокислоты типа “L” и в меньшей степени аминокислоты типа “А”. Предварительная оценка авторами циклобутиламинокислот, меченых излучателями позитронов, которые являются прежде всего субстратами для системы транспорта “L”, показала превосходный потенциал в клинической онкологии для получения изображения опухоли у пациентов с опухолью головного мозга и системными опухолями. Основными причинами для предложения 18F-меченых циклобутил/разветвленных аминокислот вместо 11С (t1/2=20 мин) являются существенные логистические и экономические преимущества, получаемые при применении 18F-меченых радиофармацевтических средств вместо 11С-меченых радиофармацевтических средств при клинических применениях. Преимущество получения изображений опухолей с применением 18F-меченых радиофармацевтических средств в отделении медицинской радиологии, в первую очередь, обусловлено более длительным периодом полураспада 18F (t1/2=110 мин). Более длительный период полураспада 18F делает возможным распределение вне участка и получения многих доз при одном получении партии радиоактивного индикатора. Кроме того, эти неметаболизированные аминокислоты могут также иметь широкое применение в качестве агентов для получения изображения некоторых системных солидных опухолей, хорошее изображение которых нельзя получить ПЭТ с применением 2-[18F]FDG. В WO 03/093412, который включен здесь в качестве ссылки, дополнительно описаны примеры фторированных аналогов Хотя преимущества аналогов аминокислот, содержащих изотопы, испускающие позитроны, для получения изображения опухолей у пациентов с опухолями головного мозга и системными опухолями достаточно одобрены в данной области, все же имеется потребность в надежном и эффективном синтетическом способе, который может обеспечить получение большого количества стереоспецифических изомеров этих соединений. Когда соединение-кандидат делает возможным переход от исследования валидации в моделях клеток и животных к применению для людей, применяемые синтетические способы должны быть адаптированы для общепринятого, надежного получения такого соединения. С этой целью авторы изобретения здесь разработали надежную стереоселективную синтетическую стратегию для получения аналогов син-1-амино-3-циклобутан-1-карбоновой кислоты (АСВС). В приведенном ниже описании будет очевидно, что стереоселективная синтетическая стратегия является подходящей для синтеза различных аналогов аминокислот, особенно аналогов, содержащих радиоактивные индикаторы, для получения изображения опухоли методами ПЭТ и ОЭКТ. Сущность изобретения Изобретение предлагает синтетическую стратегию, которая позволяет получать определенный стереоизомер ключевого предшественника для синтеза аналога аминокислоты в син(syn)-изомерной форме. Эта стратегия является особенно пригодной для синтеза аналогов син-1-амино-3-циклобутан-карбоновой кислоты (АСВС). Ключевая стадия в синтезе заключается в восстановлении синтонов-предшественников в транс-спирты, которые превращают в конечный продукт в син-изомерной форме. Синтетическая стратегия, описанная здесь, является надежной, эффективной и позволяет проводить получения в масштабе граммов ключевого предшественника для радиосинтеза аналогов син-АСВС. Кроме того, синтетическая стратегия, описанная здесь, включает введение подходящего изотопа в качестве последней стадии для максимизации пригодного периода полураспада изотопа. Настоящее изобретение предлагает транс-спирты, имеющие формулу:
Y и Z независимо представляют собой =СН2, N, О, S, Se, (CR4R5)n, n равно 1-4, R1-R3 независимо представляют собой Н, алкил, циклоалкил, ацил, арил, алкенил, алкинил, галогеналкил, галогенацил, гетероарил, галогенарил, галогенгетероарил, галогеналкенил, галогеналкинил, R4-R5 независимо представляют собой Н, алкил, циклоалкил, ацил, арил, галоген, галогеналкил, галогенацил, гетероарил, галогенарил, галогенгетероарил, алкинил, алкенил, галогеналкенил, галогеналкинил, где галогеном является нерадиоактивный F, CI, Вr, I. Изобретение предлагает также способы синтеза транс-спиртов, имеющих общую структуру формулы 1. Ключевой стадией в синтезе транс-спиртов указанной формулы является непосредственное восстановление гидридом металла с применением восстанавливающих агентов, связанных с полимером (например, продукта связывания борогидрид-полимер Aldrich 32864-2, осажденного на амберлит IRA 400; осажденного продукта связывания цианоборогидрид-полимер Aldrich 52630-4; продукта связывания борогидрид-полимер Aldrich 35994-7, осажденного на амберлит А-26; продукта связывания цинкборогидрид-полимер Aldrich 59603-5). На схеме 3 здесь приведен пример такой реакции с применением триизобутилборана лития и ZnCl2. Описанную синтетическую стратегию можно применять для получения син-изомеров различных аминокислотных соединений для применения при детектировании и оценке опухоли головного мозга и опухолей всего тела и других применений. Эти соединения объединяют благоприятные свойства 1-аминоциклоалкил-1-карбоновой кислоты, а именно ее быстрое поглощение и пролонгированное удерживание в опухолях, со свойствами галогенных заместителей, включающих в себя некоторые пригодные изотопы галогенов, в том числе фтор-18, иод-123, иод-125, иод-131, бром-75, бром-76, бром-77, бром-82, астат-210, астат-211 и другие изотопы астата. Кроме того, соединения можно метить изотопами технеция и рения с применением хелатированных комплексов. Подробное описание см. в WO 03/093412 и патенте США 5817776. Аналоги син-аминокислот можно получить с применением изобретательской синтетической стратегии, включающей в себя транс-спирты, которые включают в себя, но не ограничиваются ими, соединения, имеющие следующую формулу:
Y и Z независимо представляют собой =СН2, N, О, S, Se, (CR4R5)n, n равно 1-4, R1-R3 независимо представляют собой Н, алкил, циклоалкил, ацил, арил, алкенил, алкинил, галогеналкил, галогенацил, гетероарил, галогенарил, галогенгетероарил, галогеналкенил, галогеналкинил, R4-R5 независимо представляют собой Н, алкил, циклоалкил, ацил, арил, галоген, галогеналкил, галогенацил, гетероарил, галогенарил, галогенгетероарил, алкенил, алкинил, галогеналкенил, галогеналкинил, где галогеном является нерадиоактивный F, Cl, Вr, I, R7 представляет собой галоген, галогеналкил, галогеналкенил, галогеналкинил, галогенгетероалкил, галогенгетероалкенил, галогенгетероалкинил, галогенарил, галогенгетероарил, где галоген представляет собой F, Cl, Вr, I, причем включаются меченые соединения, такие как содержащие F-18, I-123, I-124, Тс-99m и Re хелаты. Конкретные, меченые радиоактивным изотопом аналоги аминокислот, которые можно получить с применением изобретательских способов, описанных здесь, включают в себя, но не ограничиваются перечисленным, фтор-, бром- или иодзамещенные циклопропил-, циклобутил-, циклопентил-, циклогексил-, циклогептил-, циклооктил-, циклононил-, циклодециламинокислоты, имеющие структуру, показанную выше, или алициклические соединения, содержащие гетероатом, т.е. N, О и S и Se. Производные аминокислот, полученные согласно изобретению, обладают высокой специфичностью для опухолевой ткани при введении субъекту in vivo. В соответствии с этим изобретение предлагает также фармацевтические и диагностические композиции, включающие в себя аналоги син-аминокислот, полученные согласно способу изобретения. Предпочтительные производные аминокислот, проявляющие отношение мишени к немишени, по меньшей мере, 2:1, являются стабильными in vivo и по существу локализуются в мишени через 1 час после введения. Примеры предпочтительных производных аминокислот включают в себя син-[18F]-1-амино-3-фторциклобутан-1-карбоновую кислоту (FACBC), син-[123I]-1-амино-3-иодциклобутан-1-карбоновую кислоту (IАСВС) и син-[18F]-1-амино-3-фторалкилциклобутан-1-карбоновую кислоту, например син-[18F]-1-амино-3-фторметилциклобутан-1-карбоновую кислоту (FMACBC). Аналоги аминокислот изобретения являются пригодными в качестве агента для получения изображения опухоли для детектирования и/или проведения мониторинга развития опухолей у субъекта. Аналог аминокислоты, применяемый в качестве агента для получения изображения опухоли, вводят in vivo и применяют для проведения мониторинга с применением способа, подходящего для метки. Предпочтительные способы детектирования и/или проведения мониторинга с применением аналога аминокислоты в качестве агента для получения изображения включают в себя позитронно-эмиссионную томографию (ПЭТ) и однофотонную эмиссионную компьютерную томографию (ОЭКТ). Краткое описание графического материала На фиг.1 показано поглощение in vivo соединения в опухолях 9L. Результаты выражены как процентное поглощение относительно контроля через 60 минут после инъекции. Подробности см. в примере 2. На фиг.2 показано поглощение in vivo соединений в контралатеральном нормальном головном мозге через 60 минут после инъекции. На фиг.3 показано отношение поглощения in vivo соединений в клетках опухоли к поглощению нормальными клетками через 60 минут после инъекции. Отношение получали из процентных величин, показанных на фиг.1 и 2. Подробное описание изобретения Данное изобретение относится к новым способам синтеза аналогов син-аминокислот, пригодных для получения изображения опухоли среди других применений. Авторы настоящего изобретения разработали синтетическую стратегию, которая позволяет проводить стереоселективный синтез ключевого предшественника в транс-изомерной форме для синтеза аналогов син-АСВС. Аналоги АСВС, полученные изобретательской синтетической стратегией, являются по существу чистой син-изомерной формой. Термин «по существу чистый», применяемый здесь, означает, что продукт в его изомерной форме имеет чистоту, по меньшей мере, 60%, предпочтительно чистоту 70%, более предпочтительно чистоту выше 90% в син-изомерной форме. Предполагается, что здесь включены все промежуточные величины от 60% до 100% и все промежуточные диапазоны в них, независимо от того, перечислены ли они по отдельности или не перечислены. В общем термины и фразы, применяемые здесь, имеют их принятое в данной области значение, которое можно найти ссылкой на стандартные книги, журнальные ссылки и контексты, известные специалисту в данной области. Нижеследующие определения предложены для прояснения их специфического применения в контексте изобретения. Термин «фармацевтически приемлемая соль», применяемый здесь, относится к тем карбоксилатным солям или кислотно-аддитивным солям соединений настоящего изобретения, которые являются подходящими для применения в контакте с тканями пациентов без излишней токсичности, раздражения, аллергической реакции и тому подобного, имеют приемлемое отношение польза/риск и являются эффективными для их предполагаемого применения, а также к цвиттерионным формам, когда возможно, соединений изобретения. Термин «фармацевтически приемлемая соль» относится к относительно нетоксичным, аддитивным солям соединений настоящего изобретения и неорганическим и органическим кислотам. Включены также такие соли, полученные из нетоксичных органических кислот, таких как алифатические моно- и дикарбоновые кислоты, например уксусная кислота, фенилзамещенные алкановые кислоты, гидроксиалкановые и алкандиовые кислоты, ароматические кислоты и алифатические и ароматические сульфоновые кислоты. Эти соли можно получить in situ во время последнего выделения и очистки соединений или отдельно реакцией очищенного соединения в его форме свободного основания с подходящей органической или неорганической кислотой и выделением таким образом образованной соли. Далее репрезентативные соли включают в себя гидробромид, гидрохлорид, сульфат, бисульфат, нитрат, ацетат, оксалат, валерат, олеат, пальмитат, стеарат, лаурат, борат, бензоат, лактат, фосфат, тозилат, цитрат, малеат, фумарат, сукцинат, тартрат, нафтилат, мезилат, глюкогептонат, лактиобионат и лаурилсульфонат, пропионат, пивалат, цикламат, изетионат и тому подобное. Они могут включать в себя катионы на основе щелочных и щелочноземельных металлов, таких как натрий, литий, калий, кальций, магний и тому подобное, а также нетоксичные катионы аммония, четвертичного аммония и амина, включающие в себя, но не ограничивающиеся перечисленным, катионы аммония, тетраметиламмония, тетраэтиламмония, метиламина, диметиламина, триметиламина, триэтиламина, этиламина и тому подобное. См., например, публикацию Berge S. M., et al., Pharmaceutical Salts, J. Pharm. Sci. 66; 1-19 (1977), которая включена здесь в качестве ссылки. Аналогично этому термин «фармацевтически приемлемый носитель», применяемый здесь, является органической или неорганической композицией, которая служит в качестве носителя/стабилизатора/разбавителя активного ингредиента настоящего изобретения в фармацевтической или диагностической композиции. В некоторых случаях фармацевтически приемлемыми носителями являются соли. Следующие примеры фармацевтически приемлемых носителей включают в себя, но не ограничиваются перечисленным, воду, забуференный фосфатом солевой раствор, солевой раствор, регулирующие рН агенты (например, кислоты, основания, буферы), стабилизаторы, такие как аскорбиновая кислота, изотонизирующие агенты (например, хлорид натрия), водные растворители, поверхностно-активное вещество (ионогенное и неионогенное), такое как полисорбат или твин 80(TWEEN 80). Термин «алкил», применяемый здесь сам по себе или как часть другой группы, относится к насыщенному углеводороду, который может быть неразветвленным, разветвленным или циклическим и содержит вплоть до 10 атомов углерода, предпочтительно 6 атомов углерода, более предпочтительно 4 атома углерода, такому как метил, этил, пропил, изопропил, бутил, трет-бутил и изобутил. Алкильные группы изобретения включают в себя такие группы, необязательно замещенные, где один или несколько атомов углерода в главной цепи могут быть заменены гетероатомом, один или несколько атомов водорода могут быть заменены на галоген или -ОН. Термин «арил», применяемый здесь сам по себе или как часть другой группы, относится к моноциклическим или бициклическим ароматическим группам, содержащим 5-12 атомов углерода в циклической части, предпочтительно 6-10 атомов углерода в циклической части, таким как фенил, нафтил или тетрагидронафтил. Одно или несколько колец арильной группы могут включать в себя конденсированные кольца. Арильные группы могут быть замещены одной или несколькими алкильными группами, которые могут быть неразветвленными, разветвленными или циклическими. Арильные группы могут также быть замещены в положениях кольца заместителями, которые не оказывают значительное вредное влияние на функцию соединения или части соединения, в котором он найден. Замещенные арильные группы включают в себя также группы, имеющие гетероциклические ароматические кольца, в которых один или несколько гетероатомов (например, N, О или S, необязательно с атомами водорода или заместителями для подходящей валентности) заменены одним или несколькими атомами углерода в кольце. Термин «алкокси» применяют здесь для обозначения алкильного радикала с неразветвленной или разветвленной цепью, как указано выше, если только, кроме того, длина цепи не ограничена, связанного с атомом кислорода, такой термин включает в себя, но не ограничивается перечисленным, метокси, этокси, н-пропокси, изопропокси и тому подобное. Цепь алкокси в длину предпочтительно содержит 1-6 атомов углерода, более предпочтительно 1-4 атома углерода в длину. «Ацильной» группой является группа, которая включает в себя группу – СО-. Термин «моноалкиламин», применяемый здесь сам по себе или как часть другой группы, относится к аминогруппе, которая замещена одной алкильной группой, указываемой выше. Термин «диалкиламин», применяемый здесь сам по себе или как часть другой группы, относится к аминогруппе, которая замещена двумя алкильными группами, указываемыми выше. Термин «галоген», применяемый здесь сам по себе или как часть другой группы, относится к атому хлора, брома, фтора или иода. Термин «гетероцикл» или «гетероциклическое кольцо», применяемый здесь, за исключением случаев, когда указано иначе, представляет собой систему стабильного 5-7-членного моногетероциклического кольца, которое может быть насыщенным или ненасыщенным и который состоит из атомов углерода и одного-трех гетероатомов, выбранных из группы, состоящей из N, О и S, где атом азота и серы может быть необязательно окислен. Особенно пригодными являются кольца, которые содержат один атом азота в комбинации с одним атомом кислорода или серы или два атома азота. Примеры таких гетероциклических групп включают в себя пиперидинил, пирролил, пирролидинил, имидазолил, имидазолинил, имидазолидинил, пиридил, пиразинил, пиримидинил, оксазолил, оксазолидинил, изоксазолил, изоксазолидинил, тиазолил, тиазолидинил, изотиазолил, гомопиперидинил, гомопиперазинил, пиридазинил, пиразолил и пиразолидинил, наиболее предпочтительно тиаморфолинил, пиперазинил и морфолинил. Термин «гетероатом» применяют здесь для обозначения атома кислорода (“О”), атома серы (“S”) или атома азота (“N”). Должно быть понятно, что когда гетероатомом является атом азота, он может образовывать часть NRaRb, где Ra и Rb представляют собой, независимо друг от друга, водород или С1-4алкил, С2-4аминоалкил, С1-4галогеналкил, галогенбензил или Ra и Rb взяты вместе с образованием 5-7-членного гетероциклического кольца, необязательно имеющего О, S или NRc в указанном кольце, где Rc представляет собой водород или С1-4алкил. Соединения изобретения являются пригодными в качестве связывающихся с опухолью агентами и в качестве лигандов, связывающих рецептор NMDA, и в форме с радиоактивным изотопом являются особенно пригодными в качестве соединений-индикаторов для способов получения изображений опухолей, в том числе получения изображения способами ПЭТ и ОЭКТ. Особенно пригодными в качестве агента для получения изображения являются такие соединения, меченые F-18, поскольку F-18 имеет период полураспада 110 минут, который обеспечивает достаточное время для получения меченого радиоактивным изотопом индикатора, для очистки и введения в организм человека или животного. Кроме того, F-18-меченые соединения можно применять в установках, удаленных от циклотрона в радиусе, более чем приблизительно до 200 миль. В приборе для получения изображения ОЭКТ применяют изотопные индикаторы, которые испускают фотоны высокой энергии ( Соответственно этому, соединения изобретения можно быстро и эффективно метить [123I] для применения в анализе ОЭКТ в качестве альтернативы получения изображения ПЭТ. Кроме того, из-за того факта, что одно и то же соединение можно метить любым изотопом, можно сравнивать результаты, полученные ПЭТ и ОЭКТ с применением одного и того же индикатора. Для получения изображения ПЭТ или ОЭКТ или для общепринятого мечения изотопным индикатором можно применять другие изотопы галогенов. Они включают в себя 75Br, 76Вr, 77Вr и 82Вr, как имеющие приемлемые периоды полураспада и характеристики эмиссии. В общем, существует химический способ для замены описанных изотопов любой галогенной частью. Следовательно, биохимические или физиологические активности любого галогенированного гомолога соединений изобретения являются теперь доступными для применения специалистами в данной области, включая гомологи со стабильным изотопом галогена. Другие изотопы галогена можно заменить на астат, [210At] излучает альфа-частицы с периодом полураспада 8,3 час. Следовательно, At-замещенные соединения являются пригодными для терапии опухолей, когда связывание является достаточно опухоль-специфическим. Изобретение предлагает способы получения изображения с применением ПЭТ и ОЭКТ. Эти способы включают в себя введение субъекту (которым может быть человек или животное для экспериментальных и/или диагностических целей) создающего изображение количества соединения изобретения, меченого подходящим изотопом, и затем измерение распределения соединения ПЭТ, если применяют [18F] или другой излучатель позитронов, или ОЭКТ, если применяют [123I] или другой гамма-излучатель. Создающим изображение количеством является количество, которое, по меньшей мере, способно создавать изображение в сканере ПЭТ или ОЭКТ, при принятии во внимание чувствительности детектирования и уровень помех сканера, срок службы изотопа, размер тела субъекта и путь введения, причем все такие параметры являются типовыми параметрами, известными и объясняемыми вычислениями и измерениями, известными специалисту в данной области без применения излишнего экспериментирования. Будет понятно, что соединения изобретения можно метить изотопом любого атома или комбинацией атомов в структуре. Хотя [18F], [123I] и [125] выделены здесь как особенно пригодные для ПЭТ, ОЭКТ и анализа методом меченых атомов, рассматриваются другие применения, включающие в себя применения, которые вытекают из физиологических и фармакологических свойств гомологов со стабильными изотопами и должны быть очевидными для специалистов в данной области. Соединения изобретения можно также метить технецием (ТС) с помощью аддуктов Тc. Изотопы Тc, а именно Tc99m, применяли для получения изображения опухолей. Настоящее изобретение предлагает комплексные аддукты Тc и соединений изобретения, которые являются пригодными для получения изображения опухоли. Такие аддукты являются координационными комплексами Tс, связанными с циклической аминокислотой цепью с 4-6 атомами углерода, которая может быть насыщенной или имеет двойную или тройную связь. Когда присутствует двойная связь, можно синтезировать либо Е (транс)-, либо Z (цис)-изомеры и можно применять любой изомер. Изобретательские соединения, меченые Тс, синтезируют включение изотопа 99mТс в качестве последней стадии для максимизации пригодного срока службы изотопа. В патенте США 5817776 описана десятистадийная последовательность реакций для синтеза (анти[18F]-1-амино-3-фторциклобутан-1-карбоновой кислоты (FACBC)), которая включает в себя разделение трудоемкой полупрепаративной жидкостной хроматографией при высоком давлении после стадии 4 смеси 75:25 ключевых промежуточных соединений, цис-1-амино-3-бензилоксициклобутан-1-карбоновой кислоты и транс-1-амино-3-бензилоксициклобутан-1-карбоновой кислоты соответственно. Очищенный основной изомер, цис-1-амино-3-бензилоксициклобутан-1-карбоновую кислоту, затем превращают в трифлатный предшественник в шестистадийной последовательности реакций. При попытке усовершенствовать синтетические методы авторы изобретения разработали стереоселективный синтез транс(анти)-1-амино-3-[18F]фторциклобутан-1-карбоновой кислоты (aнти-[18F]FACBC) для синтеза в больших масштабах как предшественника для радиомечения, метилового эфира цис-1-трет-бутилкабамат-3-трифторметансульфонокси-1-циклобутан-карбоновой кислоты (8), так и транс-1-амино-3-фторциклобутан-1-карбоновой кислоты (aнти-[18F]FACBC) (10). На схемах 1 и 2 показаны стадии синтеза анти-FACBC. С применением показанных синтетических стадий авторы смогли получить трифлатный предшественник (8) семистадийной последовательностью реакций. Ключевой стадией в синтезе является получение синтона, 3-бензилоксициклобутанона (2). Получение циклобутанона 3 включает в себя циклизацию обработкой 1-бром-2-бензилокси-3-бромпропана (1) метилэтилсульфоксидом и н-бутиллитием. Кетон 2 превращали непосредственно в гидантоины 3 и 4 в условиях реакции Bucherer-Strecker. Смесь 80:20 цис:транс-изомеров гидантоина легко очищали флэш-хроматографией, получая при этом требуемый цис-гидантоин 4. Превращение 4 в трифлатный предшественник, метиловый эфир цис-1-трет-бутилкарбамат-3-трифторметансульфонокси-1-циклобутан-карбоновой кислоты (8) проводили последовательностью реакций, описанных в патенте США 5817776. С применением этого метода авторы изобретения смогли получить граммовые количества соединения 9 [McConathy et al. (2003) Jour. Of Applied Radiation and Isotopes, 58: 657-666].
а) бензилбромид, Hg2Cl2, 150°C; b) nBuLi, CH3S(O)CH2SCH3, THF then 35% HClO4/Et2O; c) NH4(CO3)2, NH4Cl, KCN,1:1 EtOH: H2O, 60°C; d) 3N NaOH, 180°C then Boc2O, 9:1 CH3OH:Et3N; e) (CH3)3SiCHN2, 1:1 CH3OH:THF; f) 10% Pd/C, H2, CH3OH.
g) (CF3SO2)2O, пиридин, CH2Cl2; h) K18F, K222, K2CO3, 90°C; i) 4N HCl, 120°C. Для получения достаточных количеств аналогов аминокислот в син-изомерной форме для получения изображения опухолей, в частности цис(син)-1-амино-3-фторциклобутан-1-карбоновой кислоты (син-FACBC), разработали новый общий синтетический подход, как показано на схемах 3-5, для получения в больших масштабах метилового эфира транс-1-трет-бутилкарбамат-3-трифторметансульфонокси-1-циклобутан-1-карбоновой кислоты. Ключевая стадия в синтезах включает в себя восстановление синтонов, метилового эфира 1-трифторацетамидоциклобутан-3-он-1-карбоновой кислоты (11а), метилового эфира 1-фталамидоциклобутан-3-он-1-карбоновой кислоты (11b), метилового эфира 1-трет-бутилкарбаматциклобутан-3-он-1-карбоновой кислоты (11с) и метилового эфира 1-бензамидоциклобутан-3-он-1-карбоновой кислоты (11d). Кетоны 11a-d превращали непосредственно в транс(анти)-спирты с выходом 63-80% обработкой литийтриизобутилборана и ZnCl2. Этот метод дал смеси 95:5, 97:3, 70:30 и 90:10 транс:цис-спиртов 12а, 12b, 12с и 12d соответственно. Спирты 12a-12d легко очищали флэш-хроматографией с получением требуемых транс-спиртов 12a-d. Превращение 12a-d в трифлатные предшественники можно проводить последовательностью реакций, описанных в патенте США 5817776. Разработка этих синтетических подходов является существенной для обеспечения легкодоступной поставки предшественника для распределения в центрах ПЭТ для будущих клинических исследований во многих центрах для подтверждения пригодности син- и анти-FACBC в качестве ценного агента для получения изображения для диагностики и проведения лечения рака.
Указанную выше реакцию проводят следующим образом. К раствору кетона (11а, b, с или d) в ТГФ (безводн.) добавляют 2 эквивалента ZnCl2 (безводн., в ТГФ) при комнатной температуре (к.т.) в атмосфере аргона. Раствор перемешивают при комнатной температуре в течение 30 мин с последующим добавлением 1,5 эквивалента LiBR’3H при -78°C. Смесь перемешивают при -78°С в течение 2 час, затем при к.т. на протяжении ночи. Добавляют NH4Cl (1 н. водный раствор, 3 экв.) и смесь перемешивают при к.т. в течение 30 мин. Реакционную смесь промывают насыщенным раствором соли и водную фазу снова экстрагируют этилацетатом. Объединенные органические фазы сушат над сульфатом натрия и концентрируют досуха. Продукт очищают на силикагеле с применением в качестве элюента смеси 1:1 гексана и этилацетата. Выходы составляют приблизительно 63-80%. Хотя стадия реакции, показанная на схеме 3, конкретно иллюстрирует восстановление четырех синтонов (11a-11d) в четыре транс-спирта 12a-12d, эту стереоселективную синтетическую стадию можно применять для синтеза различных транс-спиртов для синтеза аналогов син-аминокислот, пригодных для получения изображений опухолей. Схема 4 ниже иллюстрирует этот аспект изобретения.
На схеме 5 иллюстрируются стадии синтеза син-FACBC.
а) бензилбромид, Hg2Cl2, 150°С; b) nBuLi, CH3S(O)CH2SCH3, THF then 35% HClO4/Et2O; с) NH4CO3)2, NH4Cl, KCN, 1:1 EtOH:Н2O,60°C d) 3N NaOH, 180°C then Boc2O, 9:1 CH3OH:Et3N; e) (CH3)3SiCHN2, 1:1 CH3OH:THF; f) 10% Pd/C, H2, CH3O; g) оксалилхлорид, DMSO; h) L-селектрид, ZnСl2); i)(СF3SО2)2O пиридин, СН2Сl2; j) K18F, K222, K2CO3, 90°C; k) 4N HCl, 120°C. На схеме 6 иллюстрируется синтез аналога аминокислоты, [18F]-1-амино-4-фторциклогексан-1-карбоновой кислоты (FACHC), которую можно синтезировать с применением описанного здесь стереоселективного синтетического метода.
а) МН4(СО3)2, NH4Cl, KCN. 1:1 EtOH:H2O,60°C b) 3N NaOH, 180°C then Boc2O, 9:1 СН3ОН:Еt3N; с) (СН3)3SiCHN2, 1:1 CH3OH:THF; d) 10% Pd/C. H2, СН3О; е)оксалил-хлорид, DMSO; 1)L-селектрид. ZnCl2); g) (СF3SО2)2О, пиридин, СН2Сl2; h) K18F, К222, К2СО3, 90°C, i) 4N НСl, 120°С. На схеме 7 показан синтез син/анти-1-амино-3-бензилоксициклобутан-1-карбоновой кислоты 20, которая является ключевым синтоном, применяемым в описанном здесь стереоселективном методе.
На схеме 8 показаны синтезы метилового эфира 1-[N-(трет-бутоксикарбонил)амино]-4-циклогексанон-1-карбоновой кислоты (24), метилового эфира 1-амино-4-циклогексанон-1-карбоновой кислоты (25), которые являются ключевыми циклогексаноновыми промежуточными соединениями, применяемыми в описанном здесь стереоселективном синтетическом методе.
На схеме 9 показан синтез метиловых эфиром син/анти-1-[N-замещенный амин]-4-гидроксициклогексан-1-карбоновых кислот 27a-d, полученных в описанном здесь стереоспецифическом синтетическом способе.
ПРИМЕРЫ Нижеследующие описания предоставляют примерные синтезы предпочтительных вариантов осуществления настоящего изобретения. Однако среднему специалисту в данной области должно быть понятно, что на практике изобретения можно применять исходные вещества, реагенты, растворители, температуру, твердые субстраты, синтетические методы, методы очистки, аналитические методы и другие условия реакции, отличающиеся от условий, конкретно указанных в качестве примеров, без применения излишнего экспериментирования. Предполагается, что все известные в данной области функциональные эквиваленты любых таких веществ и методов включены в данное изобретение. Термины и выражения, которые использовали, применяют в качестве терминов описания, а не ограничения, без намерения при применении таких терминов и выражений исключить любые эквиваленты показанных и описанных признаков или их частей, но признается, что в пределах объема изобретения, описанного в формуле изобретения, возможны различные модификации. Таким образом, должно быть понятно, что хотя настоящее изобретение конкретно описано предпочтительными вариантами осуществления и необязательными признаками, специалисты в данной области могут прибегнуть к модификации и варианту описанных здесь сущностей изобретения и считается, что такие модификации и варианты находятся в пределах объема данного изобретения, определяемого прилагаемой формулой изобретения. Пример 1: синтез син- и анти-[18F]-1-амино-3-фторциклобутан-1-карбоновой кислоты (FACBC) (схемы 1, 2 и 5) Нижеследующие методы применяли в описанных здесь методиках. [18F]-Фторид получали из циклотрона Seimens с применением 18O(p,n)18F-реакции с протоном 11 МэВ на 95% обогащенной [18О] воде. Все растворители и химические соединения были аналитического сорта и их применяли без дополнительной очистки. Точки плавления соединений определяли в капиллярных трубках с применением аппаратуры Buchi SP. Анализ тонкослойной хроматографией (ТСХ) проводили с применением 250-мм плотных слоев силикагеля G PF-254, нанесенных на алюминий (получен от Analtech, Inc. Newark, DE). Колоночную хроматографию проводили с применением силикагеля 60-200 меш (Sigma-Aldrich, St. Louis, МО). Инфракрасные спектры (ИК) регистрировали на спектрофотометре Beckman 18A с пластинками NaCl. Спектры протонного ядерного магнитного резонанса (1Н ЯМР) получали при 300 МГц на приборе Nicolet высокого разрешения. Синтез 1-бром-2-бензилокси-3-бромпропана 1 В колбе, снабженной холодильником, смесь, состоящую из бензилбромида (83 мл, 0,70 моль), эпибромгидрина (60 мл, 0,70 моль) и хлорида ртути (I) (120 мг, 0,25 ммоль) нагревают с перемешиванием при 150°С на протяжении ночи. Продукт выделяют посредством вакуумной дистилляции при помощи холодильника Vigreux 30 см (110-115°С, 0,5 мм Нg), получая при этом 1 (152 г, 70%) в виде бесцветной жидкости: 1Н ЯМР (СDСl3) Синтез 3-бензилоксициклобутанона 2 Получение циклобутанона 2 основано на методике, описанной Ogura et al. (1984) Bull, Chem. Soc. Jpn. 57; 1637-42. Порцию 2,4 экв. н-бутиллития (1,6 М в гексане, 243 мл) по каплям добавляют к раствору, содержащему 2,4 экв. метилметилсульфинилметилсульфида (41 мл, 0,39 ммоль) в 400 мл тетрагидрофурана при -10°С. Реакционную смесь затем перемешивают при -10°С в течение 2 часов и затем охлаждают до -70°С. Желтую реакционную смесь выдерживают при -70°С и по каплям добавляют 1 эквивалент дибромсоединения 1 (50 г, 0,16 ммоль) в 85 мл тетрагидрофурана. Реакционной смеси дают возможность нагреться до комнатной температуры на протяжении ночи. Реакционную смесь добавляют к насыщенному раствору соли и экстрагируют дважды этилацетатом. Объединенные органические слои подвергают обычной обработке, получая при этом ~60 мл темной красно-коричневой жидкости. Эту смесь промежуточных соединений, S-оксидов син- и анти-дитиокеталей, очищают в виде трех порций посредством колоночной хроматографии на силикагеле (90 г диоксида кремния). Менее полярные примеси элюируют сначала смесью 3:7 этилацетат-гексан с последующим элюированием продукта чистым этилацетатом. Этим способом получают всего 23,8 грамм промежуточного соединения. Во втором синтезе получают 24,6 грамм 2 с применением идентичных условий. Промежуточные соединения, S-оксиды син- и анти-дитиокеталей (48,4 г, 0,18 моль), растворяют в 1200 мл диэтилового простого эфира и раствор обрабатывают 68 мл 35% перхлорной кислотой. После перемешивания на протяжении ночи реакционную смесь нейтрализуют бикарбонатом натрия с последующей обычной обработкой. Очистка посредством колоночной хроматографии на силикагеле (смесь 15:85 этилацетат:гексан) дает кетон 2 (23,6 г, выход 41% от 1) в виде оранжево-желтой жидкости: 1H ЯМР Синтез цис/транс-3-(3-бензилоксициклобутан)гидантоина 3 К раствору 10 экв. карбоната аммония (125 г, 1,3 моль) и 4 экв. хлорида аммония (27,8 г, 0,52 моль) в 900 мл воды добавляют 1 экв. циклобутанона 2 (23,6 г, 0,13 моль) в 900 мл этанола. После перемешивания при комнатной температуре в течение 30 минут добавляют 4,5 экв. порцию цианида калия (38 г, 0,58 моль) и реакционную смесь нагревают при 60°С на протяжении ночи. Растворитель удаляют при пониженном давлении и сырое желтое твердое вещество тщательно промывают приблизительно 1 литром воды для удаления солей. Получают белый кристаллический продукт (16,4 г, 51%) в виде смеси 5:1 син:анти-изомеров. Основной изомер выделяют посредством колоночной хроматографии на силикагеле (смесь 2:98 метанол:дихлорметан). С применением этой методики очистка 1,0 г смеси на 95 г силикагеля дает 500-600 мг чистого 3 за одно пропускание, син-5-(3-бензилоксициклобутан)гидантоин (3): 1Н ЯМР CDCl3) Синтез син/анти-1-(N-(трет-бутоксикарбонил)амино)-3-бензилоксициклобутан-1-карбоновой кислоты 5 Суспензию соединения 3 (1,35 г, 5,5 ммоль) в 30 мл 3 н. гидроксида натрия нагревают при 180°С на протяжении ночи в герметизированном сосуде из нержавеющей стали. После охлаждения реакционную смесь нейтрализуют до рН 6-7 концентрированной хлористоводородной кислотой. После выпаривания воды при пониженном давлении образовавшееся твердое вещество экстрагируют 4×30 мл горячего этанола. Объединенные этанольные экстракты концентрируют и остаток растворяют в 50 мл смеси 9:1 метанол:триэтиламин. К раствору добавляют порцию 1,3 экв. ди-трет-бутилдикарбоната (1,56 г) и раствор перемешивают при комнатной температуре на протяжении ночи. Растворитель удаляют при пониженном давлении и сырой продукт перемешивают в смеси охлажденного льдом 80 мл этилацетата и охлажденной льдом 80 мл 0,2 н. хлористоводородной кислоты в течение пяти минут. Органический слой сохраняют и водную фазу экстрагируют 2×80 мл охлажденного льдом этилацетата. Объединенные органические слои промывают 3×60 мл воды с последующей обычной обработкой. N-Вос-кислоту 5 (1,27 г, 72%) получают в виде белого твердого вещества, подходящего для применения в следующей стадии без дополнительной очистки. 1H ЯМР (CDCl3) Синтез метилового эфира син/анти-1-(N-(трет-бутоксикарбонил)амино)-3-бензилоксициклобутан-1-карбоновой кислоты 6 1,5 экв. часть 2,0 М триметилсилилдиазометана в гексане (1,4 мл) добавляют по каплям к раствору N-Вос-кислоты 5 (600 мг, 1,87 ммоль) в 10 мл смеси 1:1 метанол-тетрагидрофуран. Во время экзотермического добавления имеет место значительное выделение газа. После 20 минут перемешивания реакционную смесь концентрируют при пониженном давлении и сырой продукт очищают посредством колоночной хроматографии на силикагеле (смесь 2:8 этилацетат:гексан). N-Вос-метиловый эфир 6 (0,45 г, 72%) получают в виде белого кристаллического твердого вещества. 1Н ЯМР (CDCl3) 61,42 (9Н, с), 2,24-2,36 (2Н, ушир. м), 2,88-2,96 (2Н, м), 3,75 (3Н, с), 4,16-4,23 (1Н, м), 4,44 (2Н, с), 5,13 (1Н, с), 7,27-7,36 (5Н, м). Синтез метилового эфира син/анти-1-(N-(трет-бутоксикарбонил)амино)-3-гидроксициклобутан-1-карбоновой кислоты 7 К раствору 6 (450 мг, 1,34 ммоль) в 10 мл СН3ОН в атмосфере аргона добавляют 200 мг 10% Pd/C. Реакционную смесь перемешивают на протяжении ночи при комнатной температуре в атмосфере водорода. Суспензию затем фильтруют через целитR и концентрируют при пониженном давлении. Очистка посредством колоночной хроматографии на силикагеле (смесь 6:4 этилацетат:гексан) дает спирт 7 (200 мг, 61%) в виде белого кристаллического твердого вещества: 134-135°С (128-130°С по данным Shoup and Goodman, J. Labelled Compd Radiopharm, 1999; 42: 215-225. 1H ЯМР (CDCl3) Синтез метилового эфира 1-[N-(трет-бутоксикарбонил)амино]циклобутан-3-он-1-карбоновой кислоты 11 с К 1,1 экв. части оксалилхлорида (1,05 мл 2 М раствора в дихлорметане) в 4 мл дихлорметана при температуре от -50 до -60°С в атмосфере аргона добавляют по каплям 2,2 экв. диметилсульфоксида (290 мкл) в 1 мл дихлорметана. Данный раствор перемешивают в течение 3 минут с последующим добавлением по каплям изомерно чистого 7 (458 мг, 1,9 ммоль), растворенного в 2 мл дихлорметана и 0,8 мл диметилсульфоксида. Реакционную смесь перемешивают при температуре от -50 до -60°С в течение 20 минут и затем добавляют 5 экв. триэтиламина (1,3 мл). Реакционную смесь перемешивают в течение 5 минут, охлаждающую баню убирают и раствор перемешивают в течение дополнительных 15 минут. Сырой продукт очищают посредством колоночной хроматографии на силикагеле (смесь 1:4 этилацетат-гексан), получая при этом 11с (456 мг, выход 100%) в виде белого твердого вещества: 118-119°С (смесь этилацетат/гексан): 1Н ЯМР (CDCl3) Синтез метилового эфира анти-1-(N-(трет-бутоксикарбонил)амино)-3-гидроксициклобутан-1-карбоновой кислоты 12с К раствору кетона (11с, 16,4 мг, 0,067 ммоль) в 1 мл ТГФ (безв.) добавляют ZnCl2 (18 мг, 0,134 ммоль, в ТГФ) при к.т. в атмосфере Аr. Раствор перемешивают при к.т. в течение 30 мин с последующим добавлением L-селектрида (19 мг, 0,10 ммоль, в ТГФ) при -78°С. Смесь перемешивают при -78°С в течение 2 час, затем при к.т. на протяжении ночи. Добавляют NH4Cl (1 н. водный, 3 эквивалента) и смесь перемешивают при к.т. в течение 30 мин. Реакционную смесь промывают насыщенным раствором соли и водную фазу реэкстрагируют этилацетатом. Объединенные органические фазы сушат над сульфатом натрия и концентрируют досуха. Продукт очищают на силикагеле с применением смеси 1:1 гексана и этилацетата в качестве элюента. Продуктом (12с, 16 мг, 100%) является белое твердое вещество: 1Н ЯМР (CDCl3) Синтез метилового эфира анти-1-(N-(трет-бутоксикарбонил)амино)-3-трифторметилсульфоноксициклобутан-1-карбоновой кислоты 13 Раствор 9 (10 мг, 0,04 ммоль) растворяют в 2 мл дихлорметана в атмосфере аргона. При охлаждении баней со льдом добавляют 100 мкл часть пиридина с последующим добавлением 4,5 экв. части трифторметансульфонового ангидрида (30 мкл). После перемешивания в течение 15 минут растворитель удаляют при пониженном давлении и при комнатной температуре. Сырой продукт очищают посредством колоночной хроматографии на силикагеле (смесь 3:7 этилацетат:гексан), получая при этом предшественник для мечения 13. Синтез син-[18F]-1-амино-3-фторциклобутан-1-карбоновой кислоты (FACBC) 15 [18F}-Фторид получают с применением 18O(р,n)18F-реакции с протонами 11 МэВ на 95% обогащенной [18О] воде. После выпаривания воды и сушки фторида выпариванием ацетонитрила трифлат защищенной кислоты 13 (20 мг) вводят в раствор ацетонитрила (1 мл). Без добавления носителя (NCA) реакцию фторирования проводят при 85°С в течение 5 мин в герметизированном сосуде в присутствии карбоната калия и криптофикса (товарный знак Aldrich Chemical Co., Milwaukee, WI). Непрореагировавший 18F удаляют разбавлением реакционной смеси метиленхлоридом с последующим пропусканием через силикагель Seppak, что дает 18F-меченый продукт 14. Снятие защиты у 14 достигают с применением 1 мл 6 н. HCl при 115°С в течение 15 мин и затем водный раствор, содержащий син-[18F]FACBC 15, пропускают через ионзадерживающую смолу (AG 11A8, 50-100 меш). Синтез анти-[18F]FACBC 10 [18F]-Фторид получают с применением 18O(р,n]18F-реакции с протонами 11 МэВ на 95% обогащенной [18О] воде. После выпаривания воды и сушки фторида выпариванием ацетонитрила трифлат защищенной аминокислоты, метиловый эфир син-1-(N-(трет-бутоксикарбонил)амино)-3-трифторметансульфоноксициклобутан-1-карбоновой кислоты (20 мг) вводят в раствор ацетонитрила (1 мл). Без добавления носителя (NCA) реакцию фторирования проводят при 85°С в течение 5 мин в герметизированном сосуде в присутствии карбоната калия и криптофикса (товарный знак Aldrich Chemical Co., Milwaukee, WI). Непрореагировавший 18F удаляют разбавлением реакционной смеси метиленхлоридом с последующим пропусканием через силикагель Seppak, что дает 18F-меченый продукт, метиловый эфир син-1-(N-(тpeт-бyтoкcикapбoнил)aминo)-3-[18F]фтopциклoбyтaн-1-карбоновой кислоты с выходом Е.О.В. 42%. Снятие защиты у метилового эфира син-1-(N-(трет-бyтoкcикapбoнил)aминo)-3-[18F]фтopциклoбyтaн-1-карбоновой кислоты проводят с применением 1 мл 4 н HCl при 115°С в течение 15 мин и затем водный раствор, содержащий 18FACBC 13, пропускают через ионзадерживающую смолу (AG 11A8, 50-100 меш). Синтез завершают за 60 мин по Е.О.В. с общим радиохимическим выходом 12% (17,5% Е.О.В.). Для подробностей см. выше McConathy et al. (2003). Пример 2: синтез эфиров син- и анти-1-амино-4-гидроксициклогексан-1-карбоновой кислоты (схемы 7-9) 4-Этиленацетальциклогексанол (16) К раствору моноэтиленацеталя 1,4-циклогександиона (3,41 г, 21,8 ммоль) в 50 мл метанола, охлажденному до 0°С, добавляют порциями борогидрид натрия (0,826 г, 21,8 ммоль). Реакционную смесь перемешивают в течение дополнительных 1,5 час перед установлением рН 7 добавлением 1 н. HCl. Смесь распределяют между этилацетатом и насыщенным раствором соли. Водный слой концентрируют до точки, в которой начинается образование осадка, и этот слой экстрагируют дважды этилацетатом. Объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют. Этот сырой спирт (3,28 г, 95,2%) применяют без дополнительной очистки. 1Н ЯМР (CDCl3) 1-Этиленацеталь-4-бензилоксициклогексан (17) К суспензии гидрида натрия (410 мг, 17,1 ммоль) в 15 мл ТГФ при O°C добавляют 4-этиленацетальциклогексанол (1) (1,36 г, 8,61 ммоль) в 5 мл ТГФ. Реакционную смесь перемешивают при 0°С в течение 1,5 час и добавляют бензилбромид (1,75 г, 10,2 ммоль). Реакционную смесь перемешивают при к.т. на протяжении ночи. Реакцию гасят хлоридом аммония (насыщ.). Продукт экстрагируют этилацетатом и органическую фазу промывают насыщенным раствором соли, сушат над сульфатом натрия, фильтруют и концентрируют. Сырой продукт очищают хроматографией на силикагеле (20% этилацетат в гексане), получая при этом 2,17 г (100%) бензилового простого эфира. 1H ЯМР (CDCl3) 4-Бензилоксициклогексанон (18) К раствору 1-этиленацеталь-4-бензилоксициклогексана (17) (3,13 г, 12,6 ммоль) в 50 мл ТГФ при к.т. добавляют водную хлористоводородную кислоту (1 н., 30 мл). Реакционную смесь перемешивают на протяжении ночи и нейтрализуют бикарбонатом натрия (насыщ.). Продукт экстрагируют этилацетатом и органическую фазу промывают насыщенным раствором соли, сушат над сульфатом натрия, фильтруют и концентрируют. Очистка хроматографией на силикагеле (20% этилацетат в гексане) дает 2,45 г (95,2%) указанного в заголовке кетона. 1Н ЯМР (CDCl3) син/анти-6-(4-Бензилоксициклогексан)гидантоины (19) К раствору 4-бензилоксициклогексанона (18) (2,45 г, 12 ммоль) в 100 мл этанола добавляют раствор карбоната аммония (4,6 г, 48 ммоль) и хлорид аммония (1,28 г, 24 ммоль) в 100 мл воды. Смесь перемешивают при к.т. в течение 15 мин и затем добавляют цианид калия (940 мг, 14,4 ммоль). Реакционную смесь перемешивают при к.т. на протяжении ночи. Растворитель удаляют при пониженном давлении. Образовавшееся твердое вещество промывают многократно водой и собирают фильтрованием. Эту сырую син/анти-смесь гидантоинов (3,02 г, 91,8%) применяют без дополнительной очистки. 1Н ЯМР (CD2OD) син/анти-1-Амино-4-бензилоксициклогексан-1 -карбоновые кислоты (20) син/анти-Гидантоины (19) (2,72 г, 9,93 ммоль) суспендируют в 30 мл 3 н. NaOH и герметизируют в стальном цилиндре, который нагревают при 120°С в течение 1 дня. После охлаждении до к.т. рН реакционной смеси устанавливают до 7 добавлением концентрированного раствора хлористоводородной кислоты. Сырой продукт син/анти-аминокислот получают концентрированием досуха при пониженном давлении. Продукт применяют без дополнительной очистки. син/анти-1-[N]-(трет-Бутоксикарбонил)амино1-4-бензилоксициклогексан-1-карбоновые кислоты (21) К суспензии син/анти-1-амино-4-бензилоксициклогексан-1-карбоновых кислот (20) из указанного выше получения в 50 мл смеси 9:1 МеОН/триэтиламин добавляют ди-трет-бутилдикарбонат (3,25 г, 14,9 ммоль). Реакционную смесь перемешивают при к.т. в течение 24 час. Растворитель удаляют при пониженном давлении. Образовавшийся остаток растворяют в 50 мл охлажденной льдом смеси 1:1 вода/этилацетат. рН раствора устанавливают до 2-3 добавлением 3 н. HCl. Органический слой сохраняют, тогда как водный слой насыщают хлоридом натрия и экстрагируют этилацетатом (3×25 мл). Объединенные органические слои сушат над сульфатом магния и растворитель удаляют при пониженном давлении. Этот продукт (3,46 г, 100%) применяют без дополнительной очистки. 1H ЯМР (CD3OD) Метиловые эфиры син/анти-1-[N]-(трет-бутоксикарбонил)амино]-4-бензилоксициклогексан-1-карбоновых кислот (22) син/анти-1-[N-(трет-Бутоксикарбонил)амино]-4-бензилоксициклогексан-1-карбоновые кислоты (21) (1,14 г, 3,26 ммоль) растворяют в 40 мл бензола и 10 мл метанола и при к.т. добавляют триметилсилилдиазометан (558 мг, 4,88 ммоль, 2,5 мл 2 М раствора в гексане). Реакционную смесь перемешивают при к.т. в течение 30 мин, затем растворитель удаляют при пониженном давлении. Очистка флэш-хроматографией с 20% этилацетатом в гексане дает 1,03 г (87,2%) чистого продукта в виде масла. 1Н ЯМР (СD3ОD) Метиловые эфиры син/анти-1-[М-(трет-бутоксикарбонил)амино1-4-гидроксициклогексан-1-карбоновых кислот (23) Суспензию бензиловых простых эфиров (22) (947 мг, 2,6 ммоль) и 10% палладия на угле (142 мг) в 50 мл этанола перемешивают в атмосфере водорода на протяжении ночи. Реакционную смесь фильтруют через целитR и фильтрат концентрируют при пониженном давлении. Очистка посредством колоночной хроматографии на силикагеле (50% этилацетат в гексане) дает (23) (701 мг, 98,4%), отношение анти/син составляет 34:66. 1H ЯМР (CD3OD) Метиловый эфир 1-[N-трет-бутоксикарбонил)амино1-4-циклогексанон-1-карбоновой кислоты (24) Перрутенат тетрапропиламмония (26 мг, 0,075 ммоль) добавляют в виде одной порции к перемешиваемой смеси спиртов (23) (410 мг, 1,5 ммоль), N-оксида N-метилморфолина (264 мг, 2,25 ммоль) и 750 мг молекулярных сит 4 А в 15 мл 10% ацетонитрила в дихлорметане в атмосфере аргона. Реакционную смесь перемешивают при к.т. в течение 1 час, затем растворитель удаляют при пониженном давлении. Образовавшийся остаток переносят в дихлорметан и очищают колоночной хроматографией на силикагеле (30% этилацетат в гексане). Получают 372 мг (91,6%) кетона (24) в виде белого твердого вещества. 1Н ЯМР (CD3OD) Метиловый эфир 1-амино-4-циклогексанон-1-карбоновой кислоты (25) К раствору кетона (24) (325 мг, 1,2 ммоль) в 5 мл дихлорметана добавляют трифторуксусную кислоту (1,37 г, 12 ммоль). Реакционную смесь перемешивают при к.т. на протяжении ночи. Растворитель и реагент удаляют при пониженном давлении. Образовавшееся белое твердое вещество применяют без дополнительной очистки. Метиловый эфир 1-[N-(фталоил)амино1-4-циклогексанон-1-карбоновой кислоты (26b) К суспензии амина (25) (80 мг, 0,47 ммоль) и триэтиламина (476 мг, 4,7 ммоль) в 10 мл толуола добавляют фталевый ангидрид (77 мг, 0,52 ммоль). Смесь кипятят с обратным холодильником при 120°С в течение 5 час. Реакционную смесь промывают насыщенным раствором соли и водный слой экстрагируют этилацетатом. Объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют. Сырой продукт очищают флэш-хроматографией с применением смеси 1:4 этилацетата и гексана, получая при этом кетон (26b) (37,6 мг, 26,6%, 2 стадии) в виде белого твердого вещества. 1H ЯМР (CD3OD) Метиловый эфир 1-[N-(трифторацетил)амино1-4-циклогексанон-1-карбоновой кислоты (26с) К суспензии амина (25) (14 мг, 0,082 ммоль) и триэтиламина (166 мг, 1,64 ммоль) в 1 мл дихлорметана, охлажденной до -10°С, добавляют трифторуксусный ангидрид (86 мг, 0,41 ммоль). Смесь нагревают до к.т. и перемешивают на протяжении ночи. Добавляют несколько капель 1 н. хлорида аммония и смесь перемешивают в течение 30 мин. Реакционную смесь промывают насыщенным раствором соли и водный слой экстрагируют этилацетатом. Объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют. Сырой продукт очищают флэш-хроматографией с применением смеси 1:2 этилацетата и гексана, получая при этом кетон (26с) (17,5 мг, 79,9%) в виде прозрачного масла. 1H ЯМР (СD3ОD) Метиловый эфир 1-[N-(бензоил)амино1-4-циклогексанон-1-карбоновой кислоты (26d) К суспензии амина (25) (50 мг, 0,29 ммоль) и пиридина (934 мг, 11,8 ммоль) в 3 мл дихлорметана, охлажденной до 0°С, добавляют бензоилхлорид (62 мг, 0,44 ммоль). Смесь нагревают до к.т. и перемешивают на протяжении ночи. Реакционную смесь промывают насыщенным раствором соли и водный слой экстрагируют этилацетатом. Объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют. Сырой продукт очищают флэш-хроматографией с применением смеси 1:2 этилацетата и гексана, получая при этом кетон (26d) (22 мг, 27,6%) в виде белого твердого вещества. 1H ЯМР (CD2OD) Метиловые эфиры син/анти-1-[N-(трет-бутоксикарбонил)амино1-4-гидроксициклогексан-1 -карбоновых кислот (27а) К раствору кетона (26а) (18 мг, 0,066 ммоль) в 1 мл ТГФ добавляют хлорид цинка (18 мг, 0,13 ммоль, 264 мкл 0,5 М раствора в ТГФ) при к.т. и смесь перемешивают в течение 30 мин. Реакционную смесь охлаждают до -78°С и добавляют L-селектрид (19 мг, 0,10 ммоль, 100 мкл 1 М раствора в ТГФ). Смесь перемешивают при -78°С в течение 2 час и при комнатной температуре на протяжении ночи. Добавляют несколько капель 1 н. хлорида аммония и смесь перемешивают в течение 30 мин. Реакционную смесь промывают насыщенным раствором соли и водный слой экстрагируют этилацетатом. Объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют. Сырой продукт очищают флэш-хроматографией с применением смеси 1:1 этилацетата и гексана, получая при этом спирты (27а) (12,7 мг, 70,5%) в виде прозрачного масла, отношение анти-изомера к син-изомеру 67:33. 1H ЯМР (CD3OD) Метиловые эфиры син/анти-1-[N-(трет-бутоксикарбонил)амино]-4-гидроксициклогексан-1-карбоновых кислот (27а) (получение в отсутствие хлорида цинка) К раствору кетона (24) (21,7 мг, 0,08 ммоль) в 1 мл ТГФ, охлажденному до -78°С, добавляют L-селектрид (22,8 мг, 0,12 ммоль, 120 мкл 1 М раствора в ТГФ). Смесь перемешивают при -78°С в течение 2 час и при комнатной температуре на протяжении ночи. Добавляют несколько капель 1 н. хлорида аммония и смесь перемешивают в течение 30 мин. Реакционную смесь промывают насыщенным раствором соли и водный слой экстрагируют этилацетатом. Объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют. Сырой продукт очищают флэш-хроматографией с применением смеси 1:1 этилацетата и гексана, получая при этом спирты (27а) (3 мг, 13,7%) в виде прозрачного масла, отношение анти-изомера к син-изомеру 11:89. 1H ЯМР (СD3СD) Метиловые эфиры син/анти-1-[N-(фталоил)амино1-4-гидроксициклогексан-1-карбоновых кислот (27b) К раствору кетона (26b) (20 мг, 0,066 ммоль) в 1 мл ТГФ добавляют хлорид цинка (18 мг, 0,13 ммоль, 260 мкл 0,5 М раствора в ТГФ) при к.т. и смесь перемешивают в течение 30 мин. Реакционную смесь охлаждают до -78°С и добавляют L-селектрид (19 мг, 0,10 ммоль, 100 мкл 1 М раствора в ТГФ). Смесь перемешивают при -78°С в течение 2 час и при комнатной температуре на протяжении ночи. Добавляют несколько капель 1 н. хлорида аммония и смесь перемешивают в течение 30 мин. Реакционную смесь промывают насыщенным раствором соли и водный слой экстрагируют этилацетатом. Объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют. Сырой продукт очищают флэш-хроматографией с применением смеси 1:1 этилацетата и гексана, получая при этом спирты (27b) (13,2 мг, 66%) в виде прозрачного масла, отношение анти-изомера к син-изомеру 52:48. 1Н ЯМР (CD3OD) Метиловые эфиры син/анти-1-[N-(фталоил)амино1-4-гидроксициклогексан-1-карбоновых кислот (27b) (получение в отсутствие хлорида цинка) К раствору кетона (26b) (18 мг, 0,059 ммоль) в 1 мл ТГФ, охлажденного до -78°С, добавляют L-селектрид (17 мг, 0,09 ммоль, 90 мкл 1 М раствора в ТГФ). Смесь перемешивают при -78°С в течение 2 час и при комнатной температуре на протяжении ночи. Добавляют несколько капель 1 н. хлорида аммония и смесь перемешивают в течение 30 мин. Реакционную смесь промывают насыщенным раствором соли и водный слой экстрагируют этилацетатом. Объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют. Сырой продукт очищают флэш-хроматографией с применением смеси 1:1 этилацетата и гексана, получая при этом спирты (27b) (13,2 мг, 66%) в виде прозрачного масла, отношение анти-изомера к син-изомеру 52:48. Метиловые эфиры син/анти-1-[N-(трифторацетил)амино1-4-гидроксициклогексан-1-карбоновых кислот (27с) К раствору кетона (26с) (17 мг, 0,064 ммоль) в 1 мл ТГФ добавляют хлорид цинка (17 мг, 0,13 ммоль, 256 мкл 0,5 М раствора в ТГФ) при к.т. и смесь перемешивают в течение 30 мин. Реакционную смесь охлаждают до -78°С и добавляют L-селектрид (18 мг, 0,096 ммоль, 96 мкл 1 М раствора в ТГФ). Смесь перемешивают при -78°С в течение 2 час и при комнатной температуре на протяжении ночи. Добавляют несколько капель 1 н. хлорида аммония и смесь перемешивают в течение 30 мин. Реакционную смесь промывают насыщенным раствором соли и водный слой экстрагируют этилацетатом. Объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют. Сырой продукт очищают флэш-хроматографией с применением смеси 1:1 этилацетата и гексана, получая при этом спирты (27с) (13,5 мг, 78,4%) в виде прозрачного масла, отношение анти-изомера к син-изомеру 66:34. 1H ЯМР (CD3OD) Метиловые эфиры син/анти-1-[N-(бензоил)амино1-4-гидроксициклогексан-1 -карбоновых кислот (27d) К раствору кетона (26d) (22 мг, 0,08 ммоль) в 1 мл ТГФ добавляют хлорид цинка (22 мг, 0,16 ммоль, 320 мкл 0,5 М раствора в ТГФ) при к.т. и смесь перемешивают в течение 30 мин. Реакционную смесь охлаждают до -78°С и добавляют L-селектрид (23 мг, 0,12 ммоль, 120 мкл 1 М раствора в ТГФ). Смесь перемешивают при -78°С в течение 2 час и при комнатной температуре на протяжении ночи. Добавляют несколько капель 1 н. хлорида аммония и смесь перемешивают в течение 30 мин. Реакционную смесь промывают насыщенным раствором соли и водный слой экстрагируют этилацетатом. Объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют. (Продукт не мог быть детектирован). Пример 3: анализы поглощения аминокислот in vitro и in vivo Опухолевые клетки сначала выращивали в виде монослоев в Т-колбах, содержащих модифицированную по способу Дульбекко среду Игла (DMEM) в увлажненных условиях инкубатора (37°С, 5% СO2/95% воздуха). Среды для выращивания дополняли 10% фетальной бычьей сывороткой и антибиотиками (10000 единиц/мл пенициллина и 10 мг/мл стрептомицина). Среды для выращивания заменяли три раза в неделю и клетки пассивировали, чтобы клетки могли достичь состояния конфлюэнтности за недельное время. Когда монослои стали конфлюэнтными, клетки приготовляли для экспериментирования следующим образом. Среды для выращивания удаляли из Т-колбы и клетки монослоев подвергали действию смеси 1 Х трипсин:ЕDТА в течение ~1 минуты для ослабления прикрепления белка клеток к колбе. По колбе затем похлопывали, чтобы вызвать высвобождение клеток. Добавляли дополненные среды для ингибирования протеолитического действия трипсина и клетки аспирировали через иглу 18 Ga до тех пор, пока они не были монодиспергированными. Образец клеток подсчитывали под микроскопом с применением гемоцитометра и фракции живых/погибших клеток оценивали при помощи окрашивания трипаном синим (>98% жизнеспособность). Оставшуюся часть клеток помещали в центрифужные пробирки, центрифугировали при 75×g в течение 5 минут и супернатант удаляли. Клетки затем снова суспендировали в системе аминокислота/бессывороточные соли DMEM. В данном исследовании приблизительно 4,55×105 клеток подвергали действию либо [18F]10 (анти-FACBC), либо [18F]15 (син-FACBC, 5 мкКи) в 3 мл среды без аминокислот ± ингибиторы транспортера (10 мМ) в течение 30 минут в условиях инкубатора в стеклянных сосудах 12×75 мм. Анализ при каждом условии проводили в двух повторностях. После инкубации клетки дважды центрифугировали (75×g в течение 5 минут) и промывали охлажденной льдом системой аминокислота/бессывороточные соли DMEM для устранения остаточной активности в супернатанте. Сосуды помещали в счетчик Packard Cobra II Auto-Gamma, исходное число импульсов корректировали на разложение (распад) метки и определяли активность на число клеток. Данные этих исследований (выраженные как процентное поглощение относительно контроля) наносили на график с применением Excel со статическими сравнениями между группами, анализированными с применением однофакторного анализа ANOVA (пакет программного обеспечения GraphPad Prism). Для проверки гипотезы, что [18F]10 и [18F]15 входят в клетки преимущественно посредством системы переноса аминокислоты L-типа, проводили анализы поглощения аминокислот с применением культивированных клеток глиосаркомы 9L и различных раковых клеточных линий человека в присутствии и в отсутствие двух хорошо описанных ингибиторов переноса аминокислот. N-MeAlB является селективным конкурирующим ингибитором системы переноса аминокислоты А-типа, тогда как 2-аминобицикло[2.2.1]гептан-2-карбоновую кислоту (ВСН) обычно применяют в качестве ингибитора для натрий-независимой системы переноса L-типа, хотя это соединение также конкурентно ингибирует поглощение аминокислот посредством натрий-зависимых систем переноса В0,+ и В0. Системы переноса аминокислот А- и L-типа участвовали в поглощении in vivo меченых радиоактивными изотопами аминокислот, применяемых для получения изображения опухоли. В отсутствие ингибиторов как [18F]10, так и [18F]15 проявили подобные уровни поглощения в клетках глиосаркомы 9L и различных раковых клеточных линиях человека с внутриклеточными накоплениями 0,43% и 0,50% начальной дозы на миллион клеток после 30 минут инкубации соответственно. Для облегчения сравнения действия ингибиторов данные выражали как процентное поглощение относительно контрольного условия (без ингибитора), как показано в таблице. В случае [18F]10 и [18F]15 ВСН блокировала >50% активности поглощения относительно контролей. Снижение поглощения [18F]10 и [18F]15 из-за ВСН по сравнению с контролями было статистически значимым (р<0,05, р<0,01 соответственно по однофакторному анализу ANOVA). Эти исследования ингибирования показывают, что [18F]10 и [18F]15 являются субстратами для системы переноса аминокислот L-типа в исследованных раковых клетках на основании ингибирования поглощения обоих соединений в присутствии ВСН.
Индуцирование опухоли и подготовка животного Все эксперименты на животных проводили в гуманных условиях и одобрены Institutional Animal Use and Care Committee (IUCAC) at Emory University (Комитет по использованию и уходу за животными Университета Эмори). Клетки глиосаркомы 9L крыс имплантировали в головной мозг самцов крыс Fischer. Вкратце, анестезированным мышам, помещенным в стереотаксическую головную камеру, инъецировали суспензию 4×104 клеток глиосаркомы 9L крыс (1×107 на мл) в место на 3 мм правее от средней линии и на 1 мм впереди от брегмы на глубину 5 мм наружной пластинки черепной кости. Инъекцию проводили в течение 2 минут и иголку удаляли на протяжении 1 минуты для минимизации противотока опухолевых клеток. Трепанационное отверстие и разрез скальпа закрывали и животных возвращали в их первоначальную колонию после выздоровления от процедуры. У крыс с опухолью развивались интракраниальные опухоли, которые вызывали потерю массы, апатию и принятие сгорбленного положения тела, животных применяли для изучения через 17-19 дней после имплантации. Из 30 животных, которым имплантировали опухолевые клетки, у 25 развивались опухоли, видимые невооруженным глазом при рассечении, их применяли в исследовании. На фиг.1-3 показаны результаты этих исследований. Исследования биораспределения радиоактивности у грызунов Распределение радиоактивности в ткани определяли у 16 нормальных самцов крыс Фишера 344 (200-250 г) после внутривенной инъекции ~85 мкКи [18F]10 или [18F]15 в 0,3 мл стерильной воды. Перед экспериментом животным давали корм и воду ad libitum. Инъекции в вену хвоста вводили бодрствующим животным с применением устройства для закрепления грызунов RTV-190 (Braintree Scientific), чтобы избежать смертность, сопровождающую анестезию в присутствии интракраниальной массы. Группы из четырех крыс погибали через 5 минут, 30 минут, 60 минут и 120 минут после инъекции дозы. Животных вскрывали и выбранные ткани взвешивали и подсчитывали вместе со стандартами доз в счетчике Packard Cobra II Auto-Gamma. Исходное число импульсов корректировали на распад метки и число импульсов нормализовали как процент общей инъецированной дозы на грамм ткани (% ID/г). Для сравнения поглощения активности в опухолевой ткани соответствующую область головного мозга, контралатеральную к опухоли, вырезали и применяли для сравнения, в каждый момент времени анализ проводили с применением однофакторного анализа ANOVA (пакет программного обеспечения GraphPad Prism). На фиг.1-3 ниже показаны результаты этих исследований. Как видно на фиг.1-3 у крыс, которым имплантировали интракраниально клетки глиосаркомы 9L, сохранение радиоактивности в опухолевой ткани было высоким через 60 минут после внутривенной инъекции [18F]10 и [18F]15, тогда как поглощение радиоактивности в ткани головного мозга, контралатеральной к опухоли, сохранялось низким (<0,3% дозы/г). Отношения поглощения опухолью к поглощению нормальным головным мозгом для [18F]10 было 6,5:1 через 60 и 120 минут, тогда как для [18F]15 отношение было 5,3:1 в тот же момент времени. Эти результаты показывают, что подобно анти-[18F]FАСВС, [18F]10, син-[18F]FACBC, [18F]15 является превосходным кандидатом для получения изображений опухолей головного мозга. Соединения, полученные способом изобретения, могут быть сольватированными, особенно гидратированными. Гидратирование может происходить во время получения соединений или композиций, включающих в себя соединения, или гидратация может происходить на протяжении времени вследствие гигроскопической природы соединений. Кроме того, соединения настоящего изобретения могут существовать в несольватированных формах, а также в формах, сольватированных фармацевтически приемлемыми растворителями, такими как вода, этанол и тому подобное. В общем, сольватированные формы считаются эквивалентными несольватированным формам для целей настоящего изобретения. Когда соединения изобретения нужно применять в качестве агентов для получения изображения, их нужно метить подходящими радиоактивными изотопами галогена, такими как 123I, 131I, 18F, 76Br и 77Br. Радиогалогенированные соединения данного изобретения можно легко предоставить в наборах с материалами, необходимыми для получения изображения опухоли. Например, набор может содержать конечный продукт, меченый подходящим изотопом (например, 18F), готовый для применения для получения изображения, или промежуточное соединение и метку (например, K[18F]F) с реагентами (например, растворителем, агентом для снятия защиты), так чтобы конечный продукт можно было получить на месте или во время применения. В первой стадии способа получения изображения опухоли меченое соединение изобретения вводят в ткань или в организм пациента в детектируемом количестве. Соединение обычно является частью фармацевтической композиции и его вводят в ткань или пациенту методами, хорошо известными специалисту в данной области. Например, соединение можно вводить либо пероральным, ректальным, парентеральным (внутривенно, внутримышечно или подкожно), внутриполостным, внутривагинальным, внутрибрюшинным, внутрипузырным, местным (порошки, мази или капли) путем или в виде спрея для трансбуккального или назального введения. В способе получения изображения изобретения меченое соединение вводят пациенту в детектируемом количестве и после времени, прошедшего после введения и достаточного для того, чтобы соединение стало ассоциированным с опухолевыми тканями или клетками, меченое соединение детектируют неинвазивно внутри пациента. В другом варианте осуществления изобретения меченое соединение вводят пациенту, причем время после введения должно быть достаточным, чтобы соединение стало ассоциированным с опухолевыми тканями, и затем берут образец ткани пациента и меченое соединение в ткани детектируют вне пациента. В альтернативном случае у пациента берут образец ткани и меченое соединение изобретения вводят в образец ткани. После времени, достаточного для того, чтобы соединение стало связанным с тканями образца, соединение детектируют. Термин «ткань» означает часть тела пациента. Примеры тканей включают в себя головной мозг, сердце, печень, кровеносные сосуды и артерии. Детектируемым количеством является количество меченого соединения, необходимого для детектирования выбранным методом детектирования. Количество меченого соединения, которое вводят пациенту, чтобы обеспечить его детектирование, может легко определить специалист в данной области. Например, увеличивающиеся количества меченого соединения можно давать пациенту до тех пор, пока соединение не будет детектировано выбранным методом детектирования. Метку вводят в соединения для обеспечения детектирования соединений. Введение меченого соединения пациенту можно проводить путем общего или местного введения. Например, меченое соединение можно вводить пациенту так, чтобы он доставлялся по всему телу. В альтернативном случае меченое соединение можно вводить в определенный, представляющий интерес орган или ткань. Специалисты в данной области знакомы с определением продолжительности времени для соединения, достаточного для того, чтобы оно стало ассоциированным с опухолью. Продолжительность необходимого времени можно легко определить введением детектируемого количества меченого соединения пациенту и затем детектированием меченого соединения в различные моменты времени после введения. Специалисты в данной области знакомы с различными путями для детектирования меченых соединений. Для детектирования радиомеченых соединений можно применять, например, магнитно-резонансную томографию (МРI), позитронно-эмиссионную томографию (ПЭТ) или однофотонную эмиссионную компьютерную томографию (ОЭКТ). ПЭТ и ОЭКТ являются предпочтительными, когда соединения изобретения применяют в качестве агентов для получения изображения опухолей. Метка, которую вводят в соединение, будет зависеть от требуемого метода детектирования. Например, если ПЭТ выбран в качестве метода детектирования, соединение должно иметь испускающий позитроны атом, такой как 11С или 18F. Радиоактивный диагностический агент должен иметь достаточную радиоактивность и концентрацию радиоактивности, которая может гарантировать надежный диагноз. Например, в случае, когда радиоактивным металлом является технеций-99m, его обычно включают в количестве 0,1-50 мКи приблизительно в 0,5-5,0 мл во время введения. Количество соединения формулы может быть таким, которое достаточно для образования стабильного хелатного соединения с радиоактивным металлом. Соединение изобретения в качестве радиоактивного диагностического агента является достаточно стабильным и поэтому его можно вводить непосредственно как таковое или сохранять до его применения. Когда нужно, радиоактивный диагностический агент может содержать любую добавку, такую как регулирующие рН агенты (например, кислоты, основания, буферы), стабилизаторы (например, аскорбиновую кислоту) или изотонизирующие агенты (например, хлорид натрия). Получение изображения опухоли можно также проводить количественно с применением соединений здесь так, чтобы терапевтический агент для данной опухоли можно было оценить на его эффективность. Предпочтительные соединения для получения изображения включают в себя радиоизотоп, такой как 123I, 124I, 125I, 131I, 18F, 76Вr, 77Вr или 11С. Синтетические схемы, описанные здесь, представляют собой примерные синтезы предпочтительных вариантов осуществления настоящего изобретения. Однако среднему специалисту в данной области будет понятно, что на практике изобретения можно применять исходные вещества, реагенты, растворители, температуру, твердые субстраты, синтетические методы, методы очистки, аналитические методы и прочие условия реакции, другие, чем условия, конкретно приведенные в примерах, без применения излишнего экспериментирования. Предполагается, что все известные в данной области функциональные эквиваленты любого из таких веществ и методов включены в данное изобретение. Термины и выражения, которые использовали, применяют в качестве терминов описания, а не ограничения и без намерения при применении таких терминов и выражений исключить любые эквиваленты показанных или описанных признаков или их частей, но признается, что в пределах объема изобретения, описанного в формуле изобретения, возможны различные модификации. Таким образом, должно быть понятно, что хотя настоящее изобретение конкретно описано предпочтительными вариантами осуществления и необязательными признаками, специалисты в данной области могут прибегнуть к модификации и варианту описанных здесь сущностей изобретения и считается, что такие модификации и варианты находятся в пределах объема данного изобретения, определяемого прилагаемой формулой изобретения. Когда здесь описана группа заместителей, понятно, что все отдельные члены этой группы и всех подгрупп, включающие любые изомеры и энантиомеры членов группы, описаны по отдельности. Предполагается, что когда здесь применяют группу Маркуша или другую группу, все отдельные члены группы и все возможные комбинации и субкомбинации группы включены в описание по отдельности. Предполагается, что, когда здесь описывают соединение так, что конкретный изомер или энантиомер соединения не указывается, например, в формуле или в химическом названии, данное описание включает в себя каждый изомер и энантиомер соединения, описанного индивидуально или в любой комбинации. Кроме того, предполагается, если не оговорено особо, что все изотопные варианты соединений, описанных здесь, включены в настоящее описание. Например, должно быть понятно, что любой один или несколько атомов водорода в описанной молекуле могут быть заменены дейтерием или тритием. Изотопные варианты молекулы обычно являются пригодными в качестве стандартов в анализах молекулы и в химическом и биологическом исследовании, относящемся к молекуле или ее применению. Предполагается, что определенные названия соединений являются примерными, так как известно, что средний специалист в данной области может назвать те же соединения иначе. Многие из молекул, описанных здесь, содержат одну или несколько ионизуемых групп [групп, у которых можно удалить протон (например, -СООН) или добавить протон (например, амины) или которые можно кватернизовать (например, амины)]. Предполагается, что все возможные ионные формы таких молекул и их солей включены по отдельности в данное описание. Что касается солей описанных соединений, средний специалист в данной области может выбрать среди большого разнообразия доступных противоионов противоионы, которые являются подходящими для получения солей настоящего изобретения для данного применения. Каждый препарат или комбинацию компонентов, описанную или приведенную в качестве примеров здесь, можно применять на практике изобретения, если не оговорено особо. Предполагается, что всякий раз, когда в описании указывается диапазон, например диапазон температуры, диапазон времени, диапазон чистоты или диапазон состава или концентрации, все промежуточные диапазоны и поддиапазоны, а также все индивидуальные величины, включенные в данные диапазоны, включены в описание. Все патенты и публикации, указанные в описании изобретения, являются показателем уровней квалификации специалистов в данной области, к которой относится изобретение. Ссылки, цитированные здесь, включены здесь в качестве ссылки во всей их полноте для характеристики состояния данной области к дате их подачи и предполагается, что эту информацию можно применять здесь, если необходимо, для исключения определенных вариантов осуществления, которые находятся в известном уровне техники. Например, когда соединение заявлено, должно быть понятно, что соединения, известные и доступные в известном уровне техники до изобретения заявителей, включая соединения, для которых включающее их описание представлено в ссылках, цитированных здесь, не предназначены для включения здесь в пункты формулы изобретения, относящиеся к химическим соединениям. Применяемый здесь термин «включающий в себя» является синонимом термину «включающий», «содержащий» или «характеризующийся», включен в любом его значении и не исключает дополнительных неуказанных элементов или стадий способа. В данном контексте термин «состоящий из» исключает любой элемент, любую стадию или любой ингредиент, не указанные в пунктах, относящихся к элементу. В данном контексте термин «состоящий по существу из» не исключает вещества или стадии, которые фактически не влияют на базовые и новые характеристики этого пункта. В каждом случае здесь любой из терминов «содержащий», «состоящий по существу из» и «состоящий из» можно заменить любым из двух других терминов. Изобретение, иллюстративно описанное здесь, подходящим образом можно осуществить на практике в отсутствие любого элемента или элементов, ограничения или ограничений, которые конкретно здесь не описаны. Все ссылки, цитированные здесь, таким образом включены в качестве ссылки до степени, которая является не совместимой с данным описанием. В частности, патенты США 5808146, 5817776 и WO 03/093412 цитируют здесь и включают в качестве ссылки здесь для предоставления примеров аналогов аминокислот, которые можно получить с применением изобретения и подробных синтетических способов. Некоторые ссылки, представленные здесь, включены в качестве ссылки для предоставления подробностей, относящихся к источникам исходных соединений, дополнительных исходных соединений, дополнительных реагентов, дополнительных способов синтеза, дополнительных методов анализа и дополнительных применений изобретения.
Формула изобретения
1. Способ синтеза по существу чистой син-аминокислоты формулы II 2. Способ по п.1, где Y и Z в аминокислоте представляют собой СН2. 3. Способ по п.1, где аминокислотой является син-[18F]-1-амино-3-фторциклобутан-1-карбоновая кислота (син-3-[18F]FACBC). 4. По существу чистое соединение формулы I 5. Соединение по п.4, где R1, R2 и R3 представляют собой водород и Y и Z представляют собой СН2. 6. Соединение по п.4, где R1, R2 и R3 представляют собой водород и Y и Z представляют собой С2Н4. 7. По существу чистая син-аминокислота формулы II, указанная в п.1. 8. Аминокислота по п.7, представляющая собой син-3-[18F]FАСВС. 9. Фармацевтическая композиция для получения изображения опухоли, включающая в себя син-аминокислоту по п.7 в эффективном количестве и фармацевтически приемлемый носитель. 10. Композиция по п.9, где аминокислотой является син-3-[18F]FACBC. 11. Способ получения изображения опухоли позитронно-эмиссионной томографией или однофотонной эмиссионной компьютерной томографией, включающий в себя а) введение субъекту с подозрением на появление опухоли образующего изображение опухоли количества меченого соединения по п.7; b) предоставление достаточного времени для того, чтобы меченое соединение стало ассоциированным с опухолью, и с) измерение распределения меченого соединения в организме субъекта ПЭТ и ОЭКТ. 12. Способ по п.11, где меченым соединением является син-3-[18F]FACBC.
РИСУНКИ
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 735
735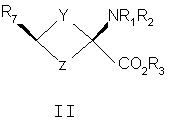 и
и 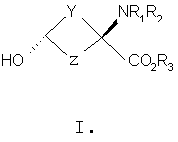
 -аминоизомасляной кислоты (AIB), таких как 2-амино-3-фтор-2-метилпропановая кислота (FAMP) и 3-фтор-2-метил-2-(метиламино)пропановая кислота (N-MeFAMP), подходящие для мечения при помощи 18F и применения при получении изображения ПЭТ. AIB является неметаболизируемой
-аминоизомасляной кислоты (AIB), таких как 2-амино-3-фтор-2-метилпропановая кислота (FAMP) и 3-фтор-2-метил-2-(метиламино)пропановая кислота (N-MeFAMP), подходящие для мечения при помощи 18F и применения при получении изображения ПЭТ. AIB является неметаболизируемой 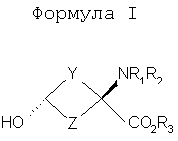
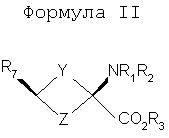
 -излучатели). Диапазон пригодных изотопов больше, чем для РЭТ, но ОЭКТ обеспечивает более низкое трехмерное разрешение. Тем не менее, ОЭКТ широко применяют для получения клинически значимой информации о скоростях связывания, локализации и выведения аналогов. Подходящим изотопом для получения изображения ОЭКТ является [123I],
-излучатели). Диапазон пригодных изотопов больше, чем для РЭТ, но ОЭКТ обеспечивает более низкое трехмерное разрешение. Тем не менее, ОЭКТ широко применяют для получения клинически значимой информации о скоростях связывания, локализации и выведения аналогов. Подходящим изотопом для получения изображения ОЭКТ является [123I], 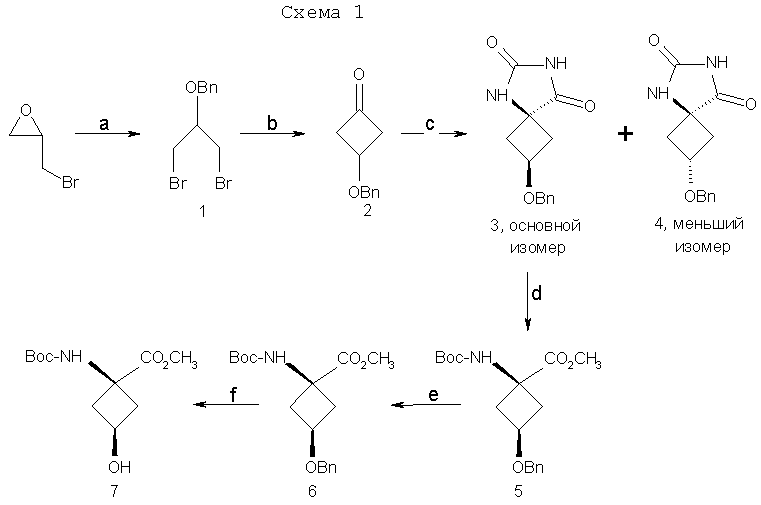
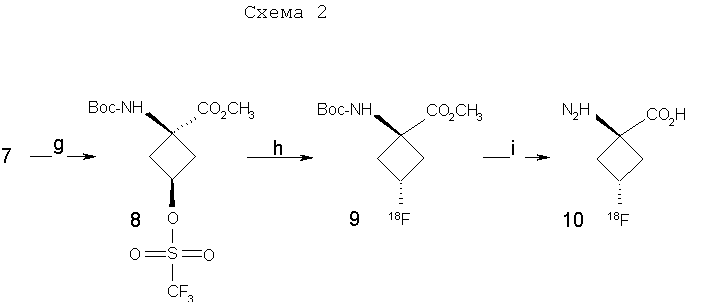
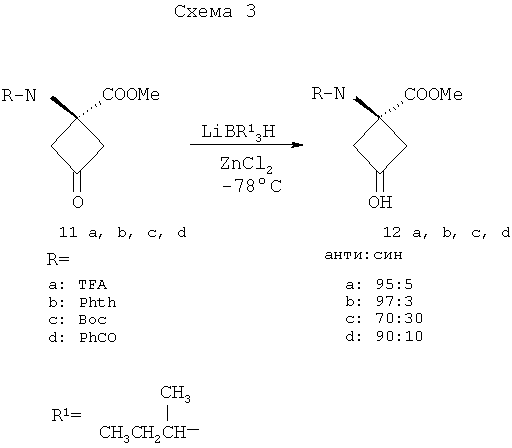
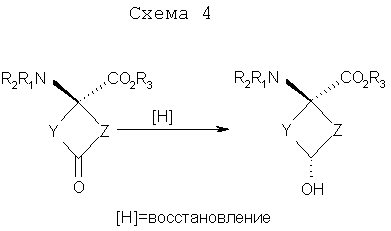
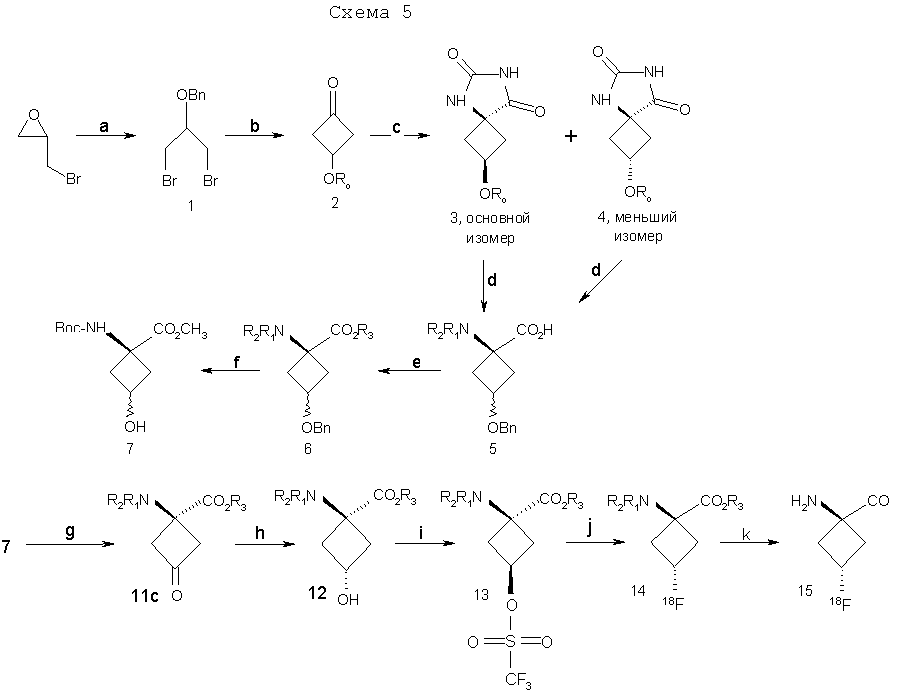
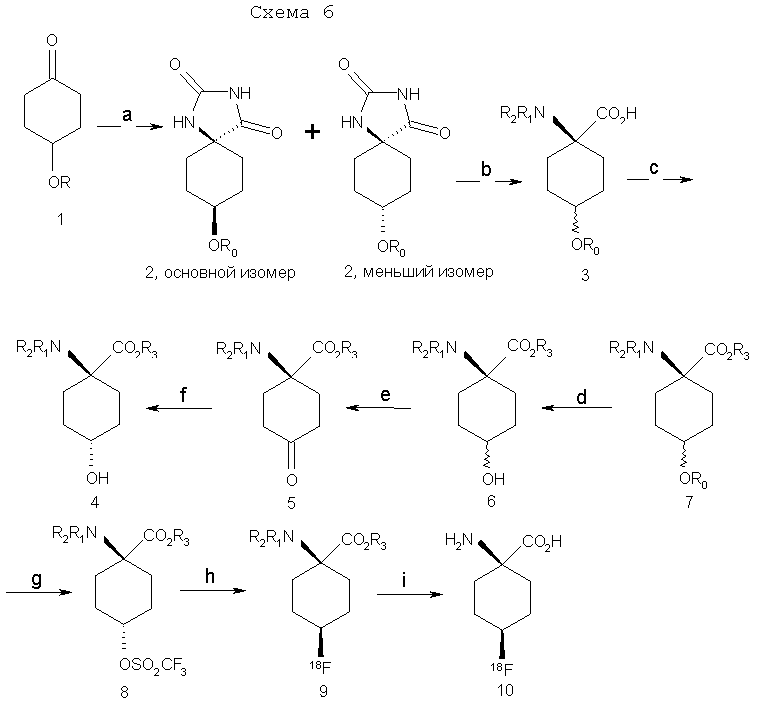
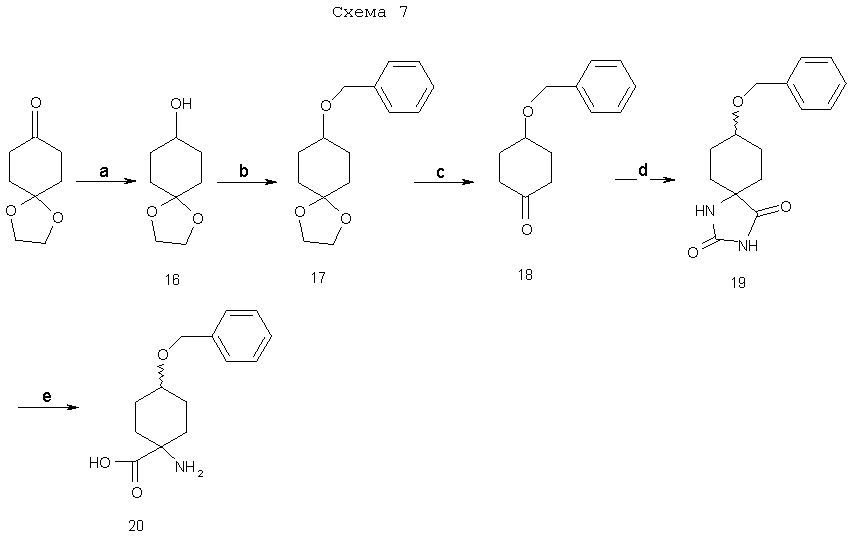
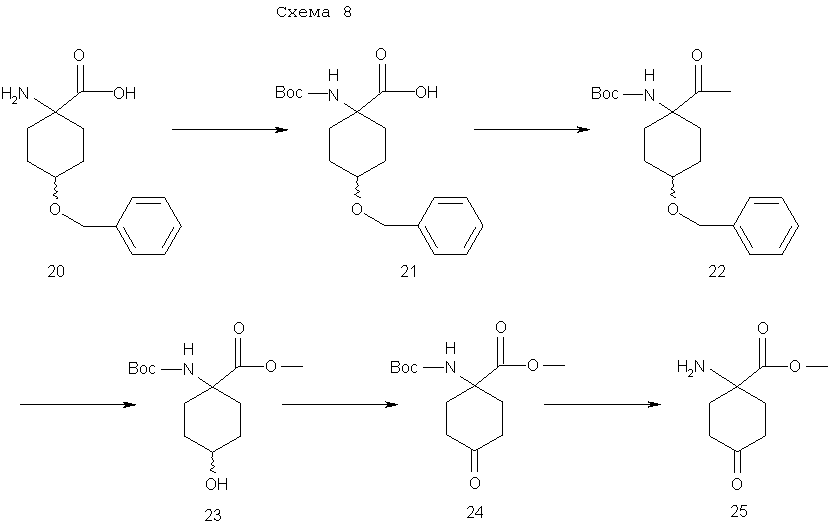
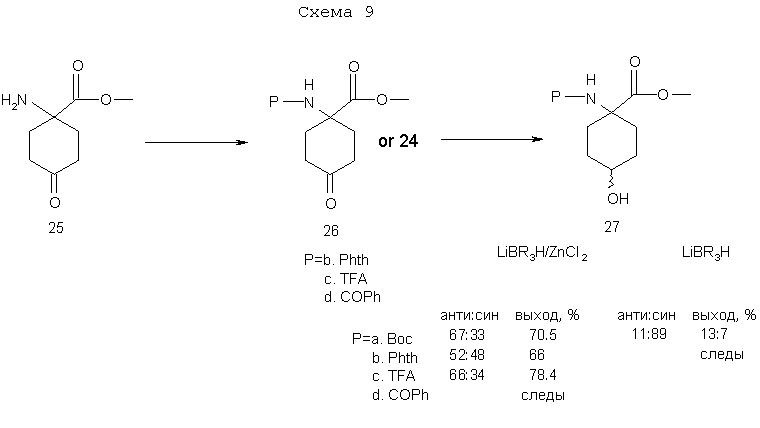
 3,45 (4Н, д, J=5,2), 3,66-3,71 (1Н, м), 4,55 (2Н, с), 7,19-7,27 (5Н, м).
3,45 (4Н, д, J=5,2), 3,66-3,71 (1Н, м), 4,55 (2Н, с), 7,19-7,27 (5Н, м). 3,11-3,29 (4Н, м), 4,35-4,42 (1Н, м), 4,53 (2Н, с), 7,30-7,40 (5Н, м).
3,11-3,29 (4Н, м), 4,35-4,42 (1Н, м), 4,53 (2Н, с), 7,30-7,40 (5Н, м).
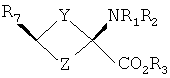 ,
,