Патент на изобретение №2376013
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) ПЕРОРАЛЬНЫЙ ПРЕПАРАТ И СПОСОБ ЕГО ПОЛУЧЕНИЯ
(57) Реферат:
Настоящее изобретение относится к области лекарственных средств, в частности к пероральной композиции в форме сухого сиропа, содержащая: комплекс, полученный путем диспергирования или растворения лекарственного средства с неприятным вкусом (кларитромицин), и высокомолекулярного соединения, растворимого в желудочном соке (сополимер Е аминоалкилметакрилатов), в моностеарате глицерина, нагретом до и расплавленном при его температуре плавления или более высокой температуре, и гранулирования полученной смеси; и композицию покрытия, включающую сополимер этилакрилата, этилметакрилата хлорида триметиламмонийметилметакрилата и разрыхлитель, такой как натриевая соль карбоксиметилцеллюлозы, находящиеся в весовых пропорциях от 80:20 до 99:1, и полисорбат 80, нанесенную на указанный комплекс. Также изобретение относится к способу получения указанной композиции. Такие композиции способны равномерно диспергироваться в воде. 2 н. и 2 з.п. ф-лы, 2 табл.
Область техники Это изобретение относится к пероральным препаратам, а также к способу их получения, более точно, к пероральным препаратам, превосходно маскирующим неприятный вкус лекарственного средства и контролирующим кинетику высвобождения лекарственного средства, а также к способу их получения. Известные технологии На сегодняшний день известны многочисленные технологии нанесения покрытий, таких как пленочные покрытия, для покрытия лекарственных средств с целью маскировки неприятного вкуса и контроля кинетики высвобождения лекарственных средств. Например, описана композиция, содержащая слои лекарственного средства, такого как кетопрофен, на микрогранулах из инертного материала, и тонкие пленки из стеариновой кислоты и этилцеллюлозы или парафина и сополимера метакриловой кислоты на слоях лекарственного средства, которая имеет время замедленного высвобождения лекарственного средства от 4-х до 22-х и более часов (Патентный документ 1). Также был описан сухой сироп, отличающийся тем, что он содержит покрытые оболочкой частицы, полученные путем нанесения пленочных покрытий из любой из следующих комбинаций (а) стеариновой кислоты и поверхностно активного вещества, (б) стеариновой кислоты, поверхностно активного вещества и покрытия, растворимого в желудочном соке, или покрытия, растворимого в кишечнике, (в) гидрированного растительного масла и покрытия, растворимого в желудочном соке, или покрытия, растворимого в кишечнике, на сферические микрочастицы, сформированные из воскоподобного вещества, в котором диспергировано имеющее неприятный вкус лекарственное средство, легко растворимое в воде (Патентный документ 2). Также известны маскирующие вкус лекарственные препараты, содержащие покрытый оболочкой порошок или покрытые оболочкой гранулы. Покрытые оболочкой порошок или гранулы отличаются тем, что они содержат ядра в виде частиц, содержащие лекарственное средство с неприятным вкусом, и нанесенные на эти ядра пленочные покрытия, образованные веществом с низкой температурой плавления и низкомолекулярным водорастворимым веществом (Патентный документ 3). Описан также порошок, включающий содержащий лекарственное средство – препарат замедленного высвобождения и смешанный с ним препарат плацебо, содержащий подсластитель (Патентный документ 4). Между тем заявитель уже описывал технологии микрочастиц и тому подобного, сформированных из низкоплавких веществ или им подобных и лекарственного средства с неприятным вкусом и растворимого в желудочном соке высокомолекулярного соединения, с целью замаскировать лекарственное средство и контролировать профиль его высвобождения (Патентные документы 5-7). Сухие сиропы известны в качестве одной из лекарственных форм, пригодных для введения лекарственных средств детям. Сухой сироп применяется путем суспендирования или растворения его в воде. Если ребенок не хочет принимать сухой сироп, то он может быть добавлен к другому напитку, который нравится ребенку. Описанные выше микрочастицы или подобные им без каких-либо проблем растворяются в воде или других жидкостях и могут быть выпиты. Однако при приеме в форме, растворенной в напитках, содержащих кислоту, например, напитках, содержащих фруктовый сок, то лекарственное средство, находящееся в микрочастицах, может немедленно высвобождаться, что сделает напиток непригодным для употребления из-за неприятного вкуса. Патентный документ 1: JP-B-2518882 Патентный документ 2: JP-B-3247511 Патентный документ 3: JP-A-11-349473 Патентный документ 4: JP-A-2001-270821 Патентный документ 5: JP-B-3265680 Патентный документ 6: JP-B-2973751 Патентный документ 7: JP-A-2000-169364 Описание изобретения Целью настоящего изобретения является разработка технологии, которая сможет обеспечить маскировку лекарственного средства в указанных микрочастицах или им подобных и даже в вислых условиях сохранит профиль его высвобождения. Для достижения цели, описанной выше, авторы провели интенсивные исследования в данной области. В результате было обнаружено, что путем нанесения на комплекс, полученный путем диспергирования или растворения лекарственного средства, обладающего неприятным вкусом, вместе с высокомолекулярным единением, растворимым в желудочном соке, в веществе с низкой температурой плавления, нагретым до или расплавленным при его температуре плавления или более высокой температуре и гранулирования полученной смеси, покрытия из покровной композиции, содержащей в определенных весовых пропорциях нерастворимое высокомолекулярное соединение и разрыхлитель, лекарственное средство не высвобождается из полученного перорального препарата даже при растворении его в кислых условиях, в результате чего успешно маскируется неприятный вкус препарата, что и являлось задачей настоящего изобретения. Более точно, настоящее изобретение относится к пероральному препарату, содержащему: комплекс, полученный путем диспергирования или растворения лекарственного средства с неприятным вкусом и высокомолекулярного соединения, растворимого в желудочном соке, в веществе с низкой температурой плавления, нагретом до или расплавленном при его температуре плавления или более высокой температуре, и последующего гранулирования полученной смеси; и композицию покрытия, включающую высокомолекулярное нерастворимое соединение и разрыхлитель, в весовых пропорциях от 80:20 до 99:1, нанесенную на указанный комплекс. Настоящее изобретение также относится к способу получения перорального препарата, который включает: диспергирование или растворение лекарственного средства с неприятным вкусом и высокомолекулярного соединения, растворимого в желудочном соке, в веществе с низкой температурой плавления, нагретом до или расплавленном при его температуре плавления или более высокой температуре, гранулирование полученной смеси в комплекс, покрытие комплекса композицией покрытия, содержащей нерастворимое высокомолекулярное соединение и разрыхлитель, находящиеся в весовых соотношениях от 80:20 до 99:1, и затем утверждение при температуре от 35°С до 45°С для формирования пленочной оболочки на комплексе. Пероральный препарат по изобретению превосходно маскирует неприятный вкус лекарственного средства и сохраняет его профиль высвобождения даже в кислых условиях. Далее, композиция покрытия, используемая в способе по изобретению для получения перорального препарата, формирует пленку при низкой температуре, так что различные материалы с низкой температурой плавления могут быть использованы в качестве основы комплекса в пероральном препарате по изобретению. Подробное описание предпочтительных вариантов осуществления изобретения Пероральный препарат по изобретению состоит из комплекса, полученного путем диспергирования или растворения лекарственного средства с неприятным вкусом и высокомолекулярного соединения, растворимого в желудочном соке, в веществе с низкой температурой плавления, нагретом до и расплавленном при его температуре плавления или более высокой температуре, и последующего гранулирования полученной смеси (здесь и ниже просто называемого «комплекс»); и композиции покрытия, содержащей нерастворимое высокомолекулярное соединение и разрыхлитель, находящиеся в весовых соотношениях от 80:20 до 99:1 (здесь и ниже называемой «композиция покрытия»). В настоящем изобретении не накладывается никаких ограничений на лекарственное средство, обладающее неприятным вкусом, при условии, что имеет место неприятный вкус. Примерами являются макролидные антибиотики, такие как эритромицин, кларитромицин, китазамицин, джозомицин, мидекамицин, рокситромицин и азитромицин; В качестве высокомолекулярного соединения, растворимого в желудке, для использования в комплексе можно упомянуть полимеры, растворимые в желудочном соке. Примерами являются сополимер Е аминоалкилметакрилатов, поливинилацеталь диэтиламиноацетат (АЕА) и смеси этих веществ. Приведенные высокомолекулярные вещества, растворимые в желудке, являются доступными, например, под товарным наименованием «EUDRAGIT Е100» (продукт фирмы Rohm Pharma GmbH). Количество такого высокомолекулярного соединения, растворимого в желудке, в смеси для комплекса может варьировать от 1 до 60% по весу, желательно от 2 до 40% по весу. Веществом с низкой температурой плавления, используемым в качестве основы для комплекса, может быть такое вещество, которое используют в качестве вспомогательного вещества в лекарственных формах и имеющее температуру плавления в пределах от 40 до 120°С, предпочтительно от 45 до 100°С. Такими веществами с низкой температурой плавления могут быть углеводороды, такие как парафин, микрокристаллический воск и церезин; масла и жиры, такие как гидрированные растительные масла и масло какао; жирные кислоты, такие как миристиновая кислота, пальмитиновая кислота и стеариновая кислота; высшие спирты, такие как цетиловый и стеариновый спирты; поливодородные спирты, такие как макрогол 6000 (ПЭГ 6000) и макрогол 4000 (ПЭГ 4000); воски, такие как японский воск, воск пальмы карнауба и пчелиный воск; эфиры жирных кислот, такие как глицериновые эфиры жирных кислот, полипропилен гликолевые эфиры жирных кислот, эфиры сорбитана с высшими жирными кислотами (спены) и эфиры сахарозы и жирных кислот; и смеси этих соединений. Среди приведенных легкоплавких соединений наиболее предпочтительными являются глицериновые эфиры жирных кислот, стеариновый спирт, стеариновая кислота, гидрированные растительные масла, макрогол 6000 и макрогол 4000, воск пальмы карнауба, парафин, эфиры сахарозы и жирных кислот и смеси этих соединений. Термин «глицериновый эфир жирной кислоты», используемый здесь, означает химическое соединение, образованное глицерином и жирной кислотой, соединенными между собой одной или несколькими эфирными связями, предпочтительнее моноглицерид или триглицерид с одной или тремя молекулами жирной кислоты, связанными с одной молекулой глицерина. В качестве эфиробразующих жирных кислот могут быть упомянуты бегеновая кислота, стеариновая кислота, олеиновая кислота, пальмитиновая кислота, миристиновая кислота и лауриновая кислота. Количество такого низкоплавкого вещества в составе комплекса определенно не ограничивается, но предпочтительно может составлять от 2% до 98% по весу. Получать комплекс предпочтительнее, например, путем агломерации застывающего аэрозоля, что будет подробнее описано ниже. Использование агломерации застывающего аэрозоля для получения комплекса обеспечивает возможность как адекватно замаскировать неприятный вкус лекарственного средства, так и получить комплекс в мелкодисперсном состоянии и с превосходной биодоступностью. Более точно, получение путем агломерации застывающего аэрозоля может производиться по методике, описанной ниже. Сначала высокомолекулярное соединение, растворимое в желудочном соке, растворяют или диспергируют в веществе с низкой температурой плавления, нагретым до и расплавленным при его температуре плавления или более высокой температуре. Далее в полученном растворе или дисперсии диспергируют лекарственное средство с неприятным вкусом. Полученную таким образом дисперсию подвергают агломерации застывающего аэрозоля в заданных условиях, получая комплекс. Термин «агломерация застывающего аэрозоля», используемый здесь, описывает один из методов гранулирования, имеющих общее название «методы гранулирования при плавлении», и представляет собой образование капель жидкости с помощью распыления жидкости или суспензии и дальнейшее формирование из них сферических или гранулярных частиц при остывании. Особенностью этого метода является то, что в нем не используют органические растворители, и он отличается от сушки при распылении, типичного примера методов гранулирования при плавлении, так как присутствует охлаждение. Размер частиц комплекса, получаемых в результате агломерации застывающего аэрозоля, не является точно лимитированным, но преимущественно варьируется от 10 до 1000 мкм, наиболее предпочтительно, от 50 до 400 мкм. Полученный, как описано выше, комплекс затем покрывают композицией покрытия. Нерастворимое высокомолекулярное соединение, используемое в композиции покрытия, не растворяется в кислых условиях, например, в качестве рН-независимого полимера можно использовать этилцеллюлозу. В качестве нерастворимого высокомолекулярного соединения наиболее предпочтительным является аминоалкилметакрилат сополимер RS. Это сополимер этилакрилата, этилметакрилата и хлорида триметиламонийметилметакрилата, который является коммерчески доступным под товарным наименованием «EUDRAGIT RL30D» (продукт компании Rohm Pharma GmbH) или аналогичной ему. С другой стороны, разрыхлитель, используемый в композиции покрытия, является таким, который почти не изменяет вязкость жидкости, даже будучи растворенным или диспергированным в ней. Например, можно использовать крахмал, карбоксиметилцеллюлозу, гидроксипропилцеллюлозу слабозамещенную или аналогичные соединения. Среди предложенных разрыхлителей наиболее предпочтительна натриевая соль карбоксиметилцеллюлозы (CMC-Na). Она является коммерчески доступной под товарным наименованием «CELLOGEN PR-S» (производство компании Dai-ichi Kogyo Seiyaku Co., Ltd.) или аналогичной ей. Композиция покрытия для использования в настоящем изобретении может быть получена смешением вышеописанного нерастворимого высокомолекулярного соединения и разрыхлителя в весовых пропорциях от 80:20 до 99:1, предпочтительно, от 90:10 до 97:3. При температуре отвердевания от 35°С до 45°С, предпочтительно, от 38°С до 42°С, оболочка твердеет и формирует пленки. Предпочтительно также добавлять неионогенное поверхностно-активное вещество, например полисорбат 80, в качестве пластификатора к композиции покрытия. Это неионогенное поверхностно-активное вещество предпочтительно добавляют в количестве от 10 до 30 процентов от веса или около того в композицию покрытия. Далее к композиции покрытия может быть добавлен диоксид кремния или похожее вещество в качестве ингибитора агрегации. Количество его по весу может варьировать от 1 до 20%. Получение перорального препарата по изобретению с использованием комплекса и композиции покрытия может быть проведено, например, как описано ниже. Более точно, описываемый как пример комплекс, гранулированный, как описано, агломерацией застывающего аэрозоля, может быть покрыт от 0,1 до 2 г, предпочтительно от 0,5 до 1 г композиции покрытия на грамм комплекса, и отверждение затем может быть проведено при температуре от 35°С до 45°С, желательно от 38°С до 42°С, чтобы формировать пленку оболочки на комплексе. Вышеописанное нанесение покрытия может быть осуществлено, например, в токе воздуха при 40°С или около того в аппарате для нанесения покрытия, таком как аппарат Вюрстера (Wurster) для нанесения покрытия. С другой стороны, отверждение может быть осуществлено за время примерно от 5 до 50 часов при использовании аппарата для отверждения, такого как лотковая сушилка. Пероральный препарат, согласно настоящему изобретению, может быть получе, как описано выше. Однако после нанесения оболочки и до процесса отверждения желательно обработать поверхность оболочки одним или несколькими неорганическими соединениями. Этими неорганическими соединениями могут быть оксид магния, тальк или диоксид кремния. Процедура обработки неорганическим соединением (соединениями) препятствует слипанию после отверждения, что повышает продуктивность процесса. В этом случае желательно равномерно нанести неорганическое соединение (соединения) на оболочку. Неорганическое вещество (вещества) может быть взято в количестве примерно от 0,005 до 0,03 г на грамм комплекса. Пероральный препарат по изобретению, полученный, как было описано выше, может быть составлен в виде гранул, порошка, капсул, таблеток, сухого сиропа или им подобных форм с помощью известных per se технологических методов, тель-кель или после смешения с одной или более известными добавками, например, наполнителями, разрыхлителями, связующими лубрикантами, антиоксидантами, покровными агентами, красителями, корригентами, ПАВ, пластификаторами, вкусовыми добавками и т.п. по необходимости. Среди перечисленных форм сухой сироп является наиболее предпочтительным в настоящем изобретении. Сухой сироп обладает особенно предпочтительной диспергируемостью в воде. Примеры Здесь и далее настоящее изобретение будет описано на примерах и на тест-примерах, но настоящее изобретение никоим образом не должно быть ограничено этими вариантами. Пример 1 Получение сухого сиропа (1) Кларитромицин (180 г), моностеарат глицерина (360 г) и «EUDRAGIT E100» (продукт компании Rohm Pharma GmbH; 60 г) были расплавлены и диспергированы при 120°С, и затем был получен комплекс (размер частиц 100 мкм) методом агломерации застывающего аэрозоля в аппарате «Spray Dryer CL-12» (произведен компанией Ohkawara Kakohki Co., Ltd.). Полученный комплекс весом в 333 г был покрыт оболочкой следующего состава: «EUDRAGIT RL30D» (продукция компании Rohm Pharma GmbH; 600 г (твердая фаза составляет 200 г)), 18 г натриевой соли карбоксиметилцеллюлозы, 72 г полисорбата 80 и 700 г очищенной воды. Процесс был проведен методом Вюрстера (метод напыления снизу в псевдоожиженном слое) в аппарате «GLATT GPCG-1» производства компании Powrex Corporation. Пероральный препарат был получен после того, как к покрытому оболочкой комплексу добавили 10 г диоксида кремния и подвергли его процессу отвердевания в лотковой сушилке при температуре 40°С в течение 10 часов. С использованием 316,5 г перорального препарата, 40 г гукурузного крахмала, 133,5 г (D)-маннита и связующего раствора, полученного растворением 10 г гидроксипропилцеллюлозы в 190 г очищенной воды, осуществляли гранулирование и сушку в псевдоожиженном слое в аппарате «FLUIDIZED-BED GRANULATION DRYER FLO-1» (произведен Freund Corporation) с получением 10% сухого сиропа. Пример 2 Получение сухого сиропа (2) Кларитромицин (180 г), моностеарат глицерина (360 г) и «EUDRAGIT Е100» (продукция компании Rohm Pharma GmbH; 60 г) были расплавлены и диспергированы при 120°С, и затем был получен комплекс (размер частиц 120 мкм) методом агломерации застывающего аэрозоля в аппарате «Spray Dryer CL-12» (произведен компанией Ohkawara Kakohki Co., Ltd.). Полученный комплекс весом в 333 г был покрыт оболочкой следующего состава: «EUDRAGIT RL30D» (продукция компании Rohm Pharma GmbH; 600 г (твердая фаза составляет 200 г)), 18 г натриевой соли карбоксиметилцеллюлозы, 72 г полисорбата 80 и 700 г очищенной воды. Процесс был проведен методом Вюрстера (метод напыления снизу в псевдоожиженном слое) и аппарате «GLATT GPCG-1» производства компании Powrex Corporation. Пероральный препарат был получен после того, как к покрытому оболочкой комплексу добавили 10 г талька и подвергли его процессу отвердевания в лотковой сушилке при температуре 40°С в течение 10 часов. С использованием 316,5 г перорального препарата, 40 г кукурузного крахмала, 133,5 г (D)-маннита и связующего раствора, полученного растворением 10 г гидроксипропилцеллюлозы в 190 г очищенной воды, осуществляли гранулирование и сушку в псевдоожиженном слое в аппарате «FLUIDIZED-BED GRANULATION DRYER FLO-1» (произведен Freund Corporation) с получением 10% сухого сиропа. Пример 3 Получение сухого сиропа (3) Кларитромицин (180 г), моностеарат глицерина (360 г) и «EUDRAGIT E100» (продукция компании Rohm Pharma GmbH; 60 г) были расплавлены и диспергированы при 120°С, и затем был получен комплекс (размер частиц 100 мкм) методом агломерации застывающего аэрозоля в аппарате «Spray Dryer CL-12» (произведен компанией Ohkawara Kakohki Co., Ltd.). Полученный комплекс весом в 333 г был покрыт оболочкой следующего состава: «EUDRAGIT RL30D» (продукция компании Rohm Pharma GmbH; 600 г (твердая фаза составляет 200 г)), 9 г натриевой соли карбоксиметилцеллюлозы, 72 г полисорбата 80 и 700 г очищенной воды. Процесс был проведен методом Вюрстера (метод напыления снизу в псевдоожиженном слое) в аппарате «GLATT GPCG-1» производства компании Powrex Corporation. Пероральный препарат был получен после того, как к покрытому оболочкой комплексу добавили 10 г оксида магния и подвергли его процессу отвердевания в лотковой сушилке при температуре 40°С в течение 10 часов. С использованием 312 г перорального препарата, 40 г кукурузного крахмала, 138 г (D)-маннита и связующего раствора, полученного растворением 10 г гидроксипропилцеллюлозы в 190 г очищенной воды, осуществляли гранулирование и сушку в псевдоожиженном слое в аппарате «FLUIDIZED-BED GRANULATION DRYER FLO-1» (произведен Freund Corporation) с получением 10% сухого сиропа. Пример 4 Получение сухого сиропа (4) Кларитромицин (180 г), моностеарат глицерина (360 г) и «EUDRAGIT E100» (продукция компании Rohm Pharma GmbH; 60 г) были расплавлены и диспергированы при 120°С, и затем был получен Комплекс (размер частиц 100 мкм) методом агломерации застывающего аэрозоля в аппарате «Spray Dryer CL-12» (произведен компанией Ohkawara Kakohki Co., Ltd.). Полученный комплекс весом в 333 г был покрыт оболочкой следующего состава: «EUDRAGIT RL30D» (продукция компании Rohm Pharma GmbH; 480 г (твердая фаза составляла 160 г)), 14 г натриевой соли карбоксиметилцеллюлозы, 58 г полисорбата 80 и 560 г очищенной воды. Процесс был проведен методом Вюрстера (метод напыления снизу в псевдоожиженном слое) в аппарате «GLATT GPCG-1» производства компании Powrex Corporation. Пероральный препарат был получен после того, как к покрытому оболочкой комплексу добавили 10 г диоксида кремния и подвергли его процессу отвердевания в лотковой сушилке при температуре 40°С в течение 10 часов. С использованием 287,5 г перорального препарата, 40 г кукурузного крахмала, 162,5 г (D)-маннита и связующего раствора, полученного растворением 10 г гидроксипропилцеллюлозы в 190 г очищенной воды, осуществляли гранулирование и сушку в псевдоожиженном слое в аппарате «FLUIDIZED-BED GRANULATION DRYER FLO-1» (произведен Freund Corporation) с получением 10% сухого сиропа. Сравнительный пример 1 Получение сравнительного сухого сиропа Кларитромицин (180 г), моностеарат глицерина (360 г) и «EUDRAGIT Е100» (продукция компании Rohm Pharma GmbH; 60 г) были расплавлены и диспергированы при 120°С, и затем был получен комплекс (размер частиц 100 мкм) методом агломерации застывающего аэрозоля в аппарате «Spray Dryer CL-12» (произведен компанией Ohkawara Kakohki Co., Ltd.). Используя 166,5 г комплекса, 65 г кукурузного крахмала, 258,5 г (D)-маннита и связующего раствора, полученного растворением 10 г гидроксипропилцеллюлозы в 190 г очищенной воды, осуществляли гранулирование и сушку в псевдоожиженном слое в аппарате «FLUIDIZED-BED GRANULATION DRYER FLO-1» (произведен Freund Corporation) с получением 10% сухого сиропа. Тест-пример 1 Органолептический тест на горечь Ниже описано проведение органолептического теста на горечь в отношении каждого из примеров 1-4 и сравнительного примера 1. Сначала 1 г лекарственного препарата был помещен в 50 мл кислого водного раствора (рН 4) и перемешан с помощью шпателя. Временные интервалы рассчитывались с момента добавления препарата в раствор. В интервалах 10, 60, 120, 180 и 240 с пипеткой были отобраны образцы по 2 мл. Каждый образец был помещен в рот и удерживался в течение нескольких секунд для определения вкуса, затем образец выплевывался, и рот споласкивали водой для проверки вкуса следующего образца. Попутно, тестируемый раствор время от времени перемешивали лопаткой. Органолептическая оценка была проведена согласно нижеследующим стандартам, и ее результаты приведены в Таблице 1. Стандарты органолептической оценки
Тест-пример 2 Тест на диспергируемость В отношении препаратов примера 1 и сравнительного примера 1 был проведен тест на диспергируемость. Препарат в количестве 1 г был помещен в 50 мл воды при непрерывном перемешивании шпателем. Временные промежутки отсчитывались с момента добавления препарата. После 10, 20, 30 и 60 секунд визуально оценивалось дисперсное состояние. Оценка диспергируемости была приведена в соответствии с нижеследующими стандартами, и ее результаты приведены в Таблице 2. Стандарты оценки диспергируемости
Применение в промышленности Все приведенные в настоящем изобретении составы пероральных препаратов сохраняют способность маскировать вкус лекарственного средства и контролировать высвобождение лекарственного средства даже в кислых условиях. Таким образом, они могут быть широко использованы в качестве превосходных препаратов, на которые не влияют жидкости.
Формула изобретения
1. Пероральная композиция в форме сухого сиропа, содержащая комплекс, полученный путем диспергирования или растворения лекарственного средства с неприятным вкусом, такого как кларитромицин, и высокомолекулярного соединения, растворимого в желудочном соке, такого как сополимер Е аминоалкилметакрилатов, в моностеарате глицерина, нагретом до и расплавленном при его температуре плавления или более высокой температуре, и гранулирования полученной смеси; и композицию покрытия, включающую сополимер этилакрилата, этилметакрилата хлорида триметиламмонийметилметакрилата и разрыхлитель, такой как натриевая соль карбоксиметилцеллюлозы, находящиеся в весовых пропорциях от 80:20 до 99:1, и полисорбат 80, нанесенную на указанный комплекс. 2. Пероральная композиция по п.1, в котором размер частиц указанного комплекса составляет от 10 до 1000 мкм. 3. Способ получения пероральной композиции в форме сухого сиропа, который включает диспергирование или растворение лекарственного средства с неприятным вкусом, такого как кларитромицин, и высокомолекулярного соединения, растворимого в желудочном соке, такого как сополимер Е аминоалкилметакрилатов, в моностеарате глицерина, нагретом до и расплавленном при его температуре плавления или более высокой температуре, гранулирование полученной смеси в комплекс, нанесение на указанный комплекс композиции покрытия, содержащей сополимер этилакрилата, этилметакрилата хлорида триметиламмонийметилметакрилата и разрыхлитель, такой как натриевая соль карбоксиметилцеллюлозы, находящиеся в весовых пропорциях от 80:20 до 99:1, и полисорбат 80, и затем проведение отверждения при температуре от 35°С до 45°С для формирования покровной пленки на указанном комплексе. 4. Способ по п.3, дополнительно включающий перед вышеуказанным отверждением обработку поверхностей указанной композиции покрытия по крайней мере одним неорганическим соединением, выбранным из оксида магния, талька и диоксида кремния.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
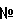 517
517 -лактамные антибиотики, такие как производные пенициллина и цефалоспорина; тетрациклиновые антибиотики, такие как тетрациклин; психотропные препараты, такие как хлорпромазин и карбонат лития; кардиотоники, такие как дигитоксин; антипиретики, такие как сульпирин; противоязвенные лекарственные вещества, такие как циметидин. Среди вышеперечисленных лекарственных средств с неприятным вкусом выделяются макролидные антибиотики, в особенности кларитромицин, как обдающий наиболее неприятным вкусом, так что пероральный препарат по изобретению высоко эффективен для кларитромицина. Количество подобного лекарственного средства с неприятным вкусом, которое необходимо добавить в комплекс, определяется в зависимости от желаемой дозировки лекарственного средства.
-лактамные антибиотики, такие как производные пенициллина и цефалоспорина; тетрациклиновые антибиотики, такие как тетрациклин; психотропные препараты, такие как хлорпромазин и карбонат лития; кардиотоники, такие как дигитоксин; антипиретики, такие как сульпирин; противоязвенные лекарственные вещества, такие как циметидин. Среди вышеперечисленных лекарственных средств с неприятным вкусом выделяются макролидные антибиотики, в особенности кларитромицин, как обдающий наиболее неприятным вкусом, так что пероральный препарат по изобретению высоко эффективен для кларитромицина. Количество подобного лекарственного средства с неприятным вкусом, которое необходимо добавить в комплекс, определяется в зависимости от желаемой дозировки лекарственного средства.