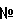Патент на изобретение №2373208
|
||||||||||||||||||||||||||||||||||||||||
(54) ПРОИЗВОДНЫЕ ИМИДАЗО[1,2-C]ПИРИМИДИНИЛУКСУСНОЙ КИСЛОТЫ
(57) Реферат:
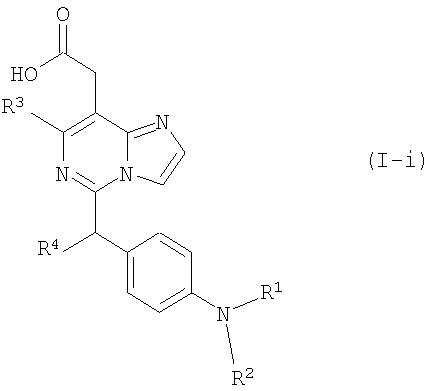
Изобретение относится к новым производным имидазо[1,2-с]пиримидинилуксусной кислоты формулы (I) или к его солям:
где R1 представляет собой
в которой n представляет собой целое число от 0 до 6; Y представляет собой арил, где указанный арил является необязательно замещенным в замещаемом положении одним или несколькими заместителями, выбранными из группы, состоящей из галогена или C1-6алкила, необязательно, замещенного моно-, ди- или тригалогеном; R2 представляет собой водород; R3 представляет собой водород или галоген; и R4 представляет собой водород. Изобретение также относится к производным имидазо[1,2-с]пиримидинилуксусной кислоты формулы (I-i) или к его солям, к лекарственному средству, к применению соединения по п.1, а также к лекарственному средству в форме стандартной единичной дозировки. Технический результат – получение новых биологически активных соединений, обладающих активностью по отношению к CRTH2. 5 н. и 18 з.п. ф-лы.
Настоящее изобретение относится к производному пиримидинилуксусной кислоты, которое является пригодным в качестве активного ингредиента фармацевтических препаратов. Производное пиримидинилуксусной кислоты по настоящему изобретению имеет антагонистическую активность по отношению к CRTH2 (G-белок-связанный рецептор хемоаттрактанта, экспрессируемый в клетках Th2) и может использоваться для профилактики и лечения заболеваний, связанных с активностью CRTH2, в частности, для лечения аллергических заболеваний, таких как астма, аллергический ринит, атопический дерматит и аллергический конъюнктивит; заболеваний, связанных с эозинофилами, таких как синдром Чарджа-Штрауса и синусит; заболеваний, связанных с базофилами, таких как базофильная лейкемия, хроническая уртикария и базофильный лейкоцитоз у людей и других млекопитающих; и воспалительных заболеваний, характеризуемых Т-лимфоцитами и профузными инфильтратами лейкоцитов, таких как псориаз, экзема, воспалительное заболевание кишечника, язвенный колит, болезнь Крона, COPD (хроническая обструктивное пульмонарное расстройство) и артрит. CRTH2 представляет собой G-белок-связанный рецептор хемоаттрактанта, экспрессируемый в клетках Th2 (Nagata et al. J. Immunol., 162, 1278-1286, 1999), эозинофилах и базофилах (Hirai et al., J. Exp. Med., 193, 255-261, 2001). Th2-поляризация наблюдается при аллергических заболеваниях, таких как астма, аллергический ринит, атопический дерматит и аллергический конъюнктивит (Romagnani S. Immunology Today, 18, 263-266, 1997; Hammad H. et al., Blood, 98, 1135-1141, 2001). Клетки Th2 регулируют аллергические заболевания посредством продуцирования цитокинов Th2, таких как IL-4, EL-5 и DL-13 (Oriss et al., J. Immunol., 162, 1999-2007, 1999; Viola et al., Blood, 91, 2223-2230, 1998; Webb et al., J. Immunol, 165, 108-113, 2000; Dumont F.J., Exp. Opin. Ther. Pat., 12, 341-367, 2002). Эти цитокины Th2 непосредственно или опосредованно индуцируют миграцию, активирование, праймирование и пролонгированное выживание эффекторных клеток, таких как эозинофилы и базофилы, при аллергических заболеваниях (Sanz et al., J. Immunol., 160, 5637-5645, 1998; Pope et al, J. Allergy Clin. Immunol, 108, 594-601, 2001; Teran L.M, Clin. Exp. Allergy, 29, 287-290, 1999). PGD2, лиганд для CRTH2, продуцируется из тучных клеток и других важных эффекторных клеток при аллергических заболеваниях (Nagata et al, FEBS Lett. 459, 195-199, 1999; Hirai et al, J. Exp. Med, 193, 255-261, 2001). PGD2 индуцирует миграцию и активирование клеток Th2 эозинофилов и базофилов в клетках человека посредством CRTH2 (Hirai et al, J. Exp. Med, 193, 255-261, 2001; Gervais et al., J. Allergy Clin. Immunol., 108, 982-988, 2001; Sugimoto et al., J. Pharmacol. Exp. Ther, 305, (1), 347-52, 2003). По этой причине антагонисты, которые ингибируют связывание CRTH2 и PGD2, должны быть пригодными для лечения аллергических заболеваний, таких как астма, аллергический ринит, атопический дерматит и аллергический конъюнктивит. В дополнение к этому несколько линий экспериментальных доказательств демонстрируют вклад эозинофилов в синусит (Hamilos et al., Am. J. Respir. Cell и Mol. Biol., 15, 443-450, 1996; Fan et al., J. Allergy Clin. Immunol., 106, 551-558, 2000), и синдром Чарджа-Штрауса (Coffin et al., J. Allergy Clin. Immunol., 101, 116-123, 1998; Kurosawa et al., Allergy, 55, 785-787, 2000). В тканях этих пациентов может наблюдаться совместная локализация тучных клеток с эозинофилами (Khan et al., J. Allergy Clin. Immunol., 106, 1096-1101, 2000). Это говорит о том, что продуцирование PGD2 из тучных клеток индуцирует рекрутирование эозинофилов. По этой причине антагонисты CRTH2 также являются пригодными для лечения других заболеваний, связанных с эозинофилами, таких как синдром Чарджа-Штрауса и синусит. Антагонисты CRTH2 также могут быть пригодными для лечения некоторых заболеваний, связанных с базофилами, таких как базофильная лейкемия, хроническая уртикария и базофильный лейкоцитоз, из-за высокой экспрессии CRTH2 на базофилах. A. F. Kluge описывает синтез аналога имидазо[1,2-c]пиримидина, представленного общей формулой
где Rа1 представляет собой
(Journal of Heterocyclic Chemistry (1978), 15(1), 119-21). Однако производные имидазо[1,2-c]пиримидинилуксусной кислоты, обладающие антагонистической активностью по отношению к CRTH2 не были описаны ранее. Разработка соединения, которое имеет эффективную антагонистическую активность по отношению к CRTH2 и может использоваться для профилактики и лечения заболеваний, связанных с активностью CRTH2, является желательной. Настоящее изобретение раскрывает производные имидазо[1,2-c]пиримидинилуксусной кислоты формулы (I), их таутомерные и стереоизомерные формы и их соли:
где R1 представляет собой
в которой n представляет собой целое число от 0 до 6; Q1 представляет собой -NH-, -N(C1-6алкил) – или -O-; Y представляет собой водород, С3-8циклоалкил, необязательно замещенный C1-6алкилом, С3-8циклоалкил, конденсированный с бензолом, арил или гетероарил, где указанный арил и гетероарил являются необязательно замещенными в замещаемом положении одним или несколькими заместителями, выбранными из группы, состоящей из циано, галогена, нитро, гуанидино, пирролила, сульфамоила, C1-6алкиламиносульфонила, ди (C1-6алкил) аминосульфонила, фенилокси, фенила, амино, C1-6алкиламино, ди(C1-6алкил) амино, C1-6алкоксикарбонила, C1-6алканоила, C1-6алканоиламино, карбамоила, C1-6алкилкарбамоила, ди(C1-6алкил) карбамоила, C1-6алкилсульфонила, C1-6алкила, необязательно замещенного моно-, ди- или тригалогеном, C1-6алкокси, необязательно замещенной моно-, ди- или тригалогеном и C1-6алкилтио, необязательно замещенной моно-, ди- или тригалогеном, или арил, конденсированный с 1,3-диоксоланом; R2 представляет собой водород или C1-6алкил; R3 представляет собой водород, галоген, C1-6алкил, необязательно замещенный моно-, ди- или тригалогеном, C1-6алкокси, необязательно замещенную моно-, ди- или тригалогеном,
в которой R3a и R3b независимо представляют собой C3-8циклоалкил или C1-6алкил, необязательно замещенный карбокси, C3-8циклоалкилом, карбамоилом, C1-6алкилкарбамоилом, арил-замещенный C1-6алкилкарбамоил, C1-6алкилкарбамоил, ди(C1-6алкил)карбамоил, C3-8циклоалкилкарбамоил, C3-8гетероциклокарбонил, C1-6алкиламино, ди(C1-6алкил)амино или C1-6алкокси,
в которой q представляет собой целое число от 1 до 3; R3c представляет собой водород, гидрокси, карбокси, или C1-6алкил, необязательно замещенный гидрокси, карбокси, или (фенил-замещенный C1-6алкил)карбамоил; Xa представляет собой -O-, -S- или -N(R3d)- в котором R3d представляет собой водород или C1-6алкил; и R4 представляет собой водород или C1-6алкил. В одном из вариантов осуществления, соединения формулы (I) представляют собой соединения, где: R1 представляет собой
в которой n представляет собой целое число от 0 до 2; Q1 представляет собой -NH-, -N(C1-6алкил)- или -O-; Y представляет собой С3-8циклоалкил, необязательно замещенный C1-6алкилом, С3-8циклоалкил, конденсированный с бензолом, арил, выбранный из группы, состоящей из фенила и нафтила или гетероарил, выбранный из группы, состоящей из индолила, хинолила, бензофуранила, фуранила и пиридила, где указанный арил и гетероарил являются необязательно замещенными в замещаемом положении одним или несколькими заместителями, выбранными из группы, состоящей из циано, галогена, нитро, пирролила, сульфамоила, С1-6алкиламиносульфонила, ди(C1-6алкил) аминосульфонила, фенилокси, фенила, C1-6алкиламино, ди(C1-6алкил) амино, C1-6алкоксикарбонила, C1-6алканоиламино, карбамоила, C1-6алкилкарбамоила, ди(C1-6алкил)карбамоила, C1-6алкилсульфонила, C1-6алкила, необязательно замещенного моно-, ди- или тригалогеном, C1-6алкокси, необязательно замещенной моно-, ди- или тригалогеном, и C1-6алкилтио, необязательно замещенной моно-, ди- или тригалогеном; и R2 представляет собой водород. В другом варианте осуществления, соединения формулы (I) представляют собой соединения, где: R3 представляет собой водород, галоген, C1-6алкил, необязательно замещенный моно-, ди- или тригалогеном, C1-6алкокси, необязательно замещенную моно-, ди- или тригалогеном,
в которой R3a и R3b независимо представляют собой C1-6алкил, необязательно замещенный карбокси, C3-8циклоалкилом, карбамоилом, C1-6алкилкарбамоилом, ди(C1-6алкил)карбамоилом, C3-8циклоалкилкарбамоилом, C3-8гетероциклокарбонилом, C1-6алкиламино, ди(C1-6алкил)амино или C1-6алкокси,
в которой R3c представляет собой водород, гидрокси, карбокси или C1-6алкил, необязательно замещенный гидрокси, карбокси или (фенил-замещенный C1-6алкил)карбамоил; Xa представляет собой -O-, -S- или -N(R3d)-, в котором R3d представляет собой C1-6алкил. В другом варианте осуществления, соединения формулы (I-i) представляют собой
где R1 представляет собой
в которой n представляет собой целое число от 0 до 2; Q1 представляет собой -NH-, -N(C1-6 алкил)- или -O-; Y представляет собой фенил, нафтил, индолил, хинолил, бензофуранил, фуранил или пиридил, где указанный фенил, нафтил, индолил, хинолил, бензофуранил, фуранил и пиридил являются необязательно замещенными в замещаемом положении одним или двумя заместителями, выбранными из группы, состоящей из циано, галогена, нитро, фенилокси, фенила, C1-6алкила, необязательно замещенного моно-, ди- или тригалогеном, C1-6алкокси, необязательно замещенной моно-, ди- или тригалогеном, и C1-6алкилтио, необязательно замещенной моно-, ди- или тригалогеном; R2 представляет собой водород или C1-6алкил; R3 представляет собой водород, галоген, C1-6алкил, необязательно замещенный моно-, ди- или тригалогеном, C1-6алкокси,
в которой R3a и R3b независимо представляют собой C3-8циклоалкил или C1-6алкил, необязательно замещенный C3-8циклоалкилом, карбамоилом, C1-6алкилкарбамоилом, фенил-замещенным C1-6алкилкарбамоилом, C1-6алкилкарбамоилом, ди(C1-6алкил)карбамоилом, C3-8циклоалкилкарбамоилом, C3-8гетероциклокарбонилом, C1-6алкиламино, ди(C1-6алкил)амино или C1-6алкокси,
R3c представляет собой водород, гидрокси, карбокси или C1-6алкил, необязательно замещенный гидрокси, карбокси, или (фенил-замещенный C1-6алкил)карбамоил; и R4 представляет собой водород или метил. Предпочтительными являются соединения формулы (I), в которых R2 представляет собой водород. Предпочтительными являются соединения формулы (I), в которых R3 представляет собой водород и галоген, предпочтительно, хлор. Предпочтительными являются соединения формулы (I), в которых R4 представляет собой водород. Предпочтительными являются соединения формулы (I), в которых R1 представляет собой одну из групп
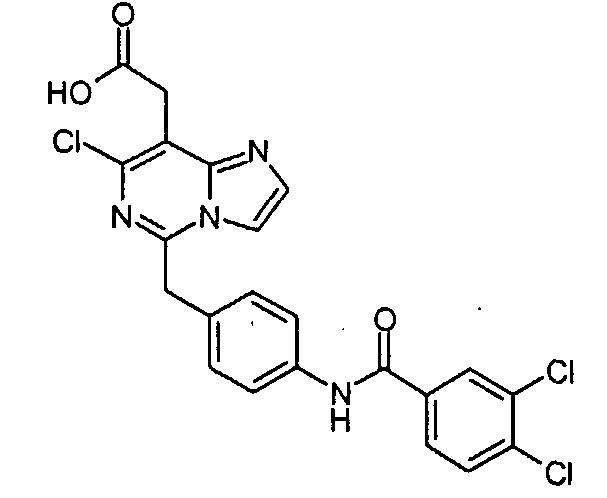
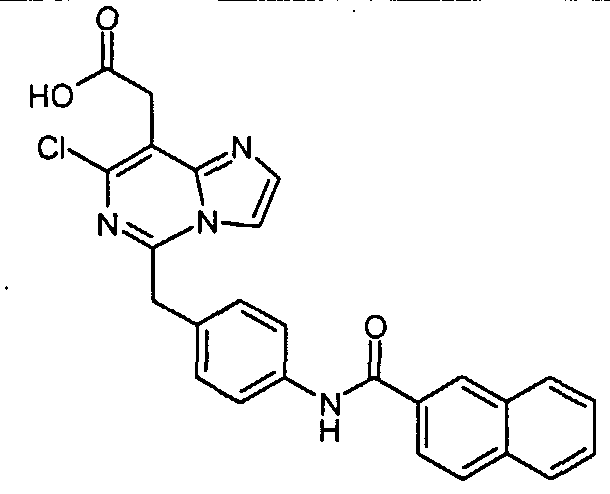
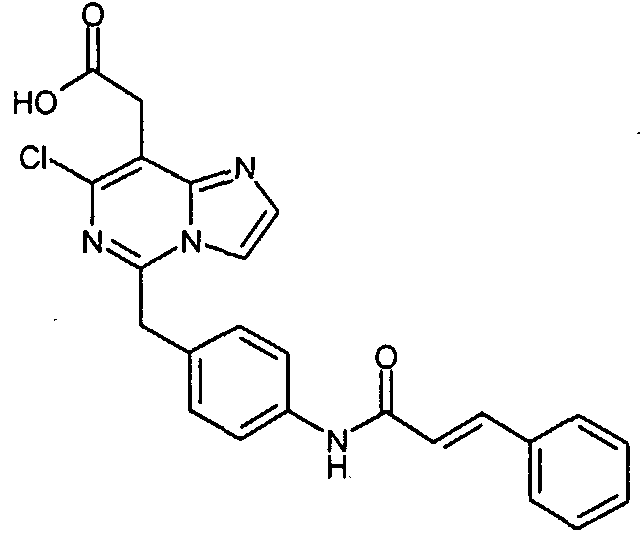
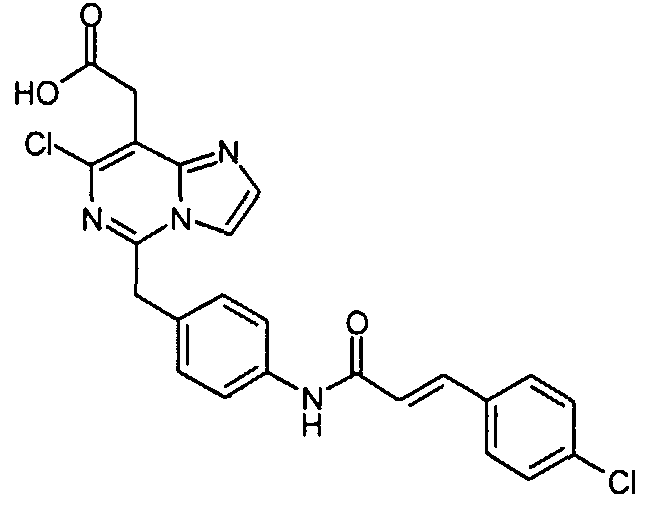
Предпочтительные соединения по настоящему изобретению представляют собой следующие соединения: [7-хлор-5-(4-{[4-(трифторметил)бензоил]амино}бензил)имидазо[1,2-c]пиримидин-8-ил]уксусную кислоту; (7-хлор-5-{4-[(3,4-дихлорбензоил)амино]бензил}имидазо[1,2-c]пиримидин-8-ил)уксусную кислоту; {7-хлор-5-[4-(2-нафтоиламино)бензил]имидазо[1,2-c]пиримидин-8-ил}уксусную кислоту; [7-хлор-5-(4-{[(2E)-3-фенилпроп-2-еноил]амино}бензил)имидазо[1,2-c]пиримидин-8-ил]уксусную кислоту; [7-хлор-5-(4-{[(2E)-3-(4-хлорфенил)проп-2-еноил]амино}бензил)имидазо[1,2-c]пиримидин-8-ил]уксусную кислоту; (5-{4-[(3,4-дихлорбензоил)амино]бензил}имидазо[1,2-c]пиримидин-8-ил)уксусную кислоту; [5-(4-{[4-(трифторметил)бензоил]амино}бензил)имидазо[1,2-c]пиримидин-8-ил]уксусную кислоту и их таутомерные и стереоизомерные формы, и их соли. Производные имидазо[1,2-c]пиримидинилуксусной кислоты формулы (I) демонстрируют превосходную антагонистическую активность по отношению к CRTH2. По этой причине, они являются пригодными, в частности, для профилактики и лечения заболеваний, связанных с активностью CRTH2. Более конкретно, производные имидазо[1,2-c]пиримидинилуксусной кислоты формулы (I) и (I-i) являются эффективными для лечения или профилактики аллергических заболеваний, таких как астма, аллергический ринит, атопический дерматит и аллергический конъюнктивит. Соединения формулы (I) и (I-i) также являются пригодными для лечения или профилактики заболеваний, таких как синдром Чарджа-Штрауса, синусит, базофильная лейкемия, хроническая уртикария и базофильный лейкоцитоз, поскольку такие заболевания являются также связанными с активностью по отношению к CRTH2. Кроме того, настоящее изобретение предусматривает медикамент, который содержит одно из соединений, описанных выше, и, необязательно, фармацевтически приемлемые наполнители. Алкил, сам по себе, и “алк” и “алкил” в алкокси, алканоиле, алкиламино, алкиламинокарбониле, алкиламиносульфониле, алкилсульфониламино, алкоксикарбониле, алкоксикарбониламино и алканоиламино представляет собой алкильный радикал с прямой или разветвленной цепью, имеющий, как правило, 1-6, предпочтительно, 1-4, а наиболее предпочтительно, 1-3 атома углерода, он иллюстративно и предпочтительно представляет собой метил, этил, н-пропил, изопропил, трет-бутил, н-пентил и н-гексил. Алкокси иллюстративно и предпочтительно представляет собой метокси, этокси, н-пропокси, изопропокси, трет-бутокси, н-пентокси и н-гексокси. Алканоил иллюстративно и предпочтительно представляет собой ацетил и пропаноил. Алкиламино представляет собой алкиламино радикал, имеющий один или два (независимо выбранных) алкильных заместителя, иллюстративно и предпочтительно представляет собой метиламино, этиламино, н-пропиламино, изопропиламино, трет-бутиламино, н-пентиламино, н-гексиламино, N,N-диметиламино, N,N-диэтиламино, N-этил-N-метиламино, N-метил-N-н-пропиламино, N-изопропил-N-н-пропиламино, N-трет-бутил-N-метиламино, N-этил-N-н-пентиламино и N-н-гексил-N-метиламино. Алкиламинокарбонил или алкилкарбамоил представляет собой алкиламинокарбонил радикал, имеющий один или два (независимо выбранных) алкильных заместителей, он иллюстративно и предпочтительно представляет собой метиламинокарбонил, этиламинокарбонил, н-пропиламинокарбонил, изопропиламинокарбонил, трет-бутиламинокарбонил, н-пентиламинокарбонил, н-гексиламинокарбонил, N,N-диметиламинокарбонил, N,N-диэтиламинокарбонил, N-этил-N-метиламинокарбонил, N-метил-N-н-пропиламинокарбонил, N-изопропил-N-н-пропиламинокарбонил, N-трет-бутил-N-метиламинокарбонил, N-этил-N-н-пентиламинокарбонил и N-н-гексил-N-метиламинокарбонил. Алкиламиносульфонил представляет собой алкиламиносульфонильный радикал, имеющий один или два (независимо выбранных) алкильных заместителя, он иллюстративно и предпочтительно представляет собой метиламиносульфонил, этиламиносульфонил, н-пропиламиносульфонил, изопропиламиносульфонил, трет-бутиламиносульфонил, н-пентиламиносульфонил, н-гексиламиносульфонил, N,N-диметиламиносульфонил, N,N-диэтиламиносульфонил, N-этил-N-метиламиносульфонил, N-метил-N-н-пропиламиносульфонил, N-изопропил-N-н-пропиламиносульфонил, N-трет-бутил-N-метиламиносульфонил, N-этил-N-н-пентиламиносульфонил и N-н-гексил-N-метиламиносульфонил. Алкилсульфониламино иллюстративно и предпочтительно представляет собой метилсульфониламино, этилсульфониламино, н-пропилсульфониламино, изопропилсульфониламино, трет-бутилсульфониламино, н-пентилсульфониламино и н-гексилсульфониламино. Алкоксикарбонил иллюстративно и предпочтительно представляет собой метоксикарбонил, этоксикарбонил, н-пропоксикарбонил, изопропоксикарбонил, трет-бутоксикарбонил, н-пентоксикарбонил и н-гексоксикарбонил. Алкоксикарбониламино иллюстративно и предпочтительно представляет собой метоксикарбониламино, этоксикарбониламино, н-пропоксикарбониламино, изопропоксикарбониламино, трет-бутоксикарбониламино, н-пентоксикарбониламино и н-гексоксикарбониламино. Алканоиламино иллюстративно и предпочтительно представляет собой ацетиламино и этилкарбониламино. Циклоалкил сам по себе и в циклоалкиламино, и в циклоалкилкарбониле представляет собой циклоалкильную группу, имеющую, как правило, 3-8, а предпочтительно, 5-7 атомов углерода, он иллюстративно и предпочтительно представляет собой циклопропил, циклобутил, циклопентил, циклогексил и циклогептил. Циклоалкиламино представляет собой циклоалкиламино радикал, имеющий один или два (независимо выбранных) циклоалкильных заместителей, он иллюстративно и предпочтительно представляет собой циклопропиламино, циклобутиламино, циклопентиламино, циклогексиламино и циклогептиламино. Циклоалкилкарбонил иллюстративно и предпочтительно представляет собой циклопропилкарбонил, циклобутилкарбонил, циклопентилкарбонил, циклогексилкарбонил и циклогептилкарбонил. Арил сам по себе и в ариламино, и в арилкарбониле представляет собой моно- – трициклический ароматический карбоциклический радикал, имеющий, как правило, 6-14 атомов углерода, он иллюстративно и предпочтительно представляет собой фенил, нафтил и фенантренил. Ариламино представляет собой ариламино радикал, имеющий один или два (независимо выбранных) арильных заместителя, он иллюстративно и предпочтительно представляет собой фениламино, дифениламино и нафтиламино. Арилкарбонил иллюстративно и предпочтительно представляет собой фенилкарбонил и нафтилкарбонил. Гетероарил сам по себе и в гетероариламино, и гетероарилкарбониле представляет собой ароматический моно- или бициклический радикал, имеющий, как правило, 5-10, а предпочтительно, 5 или 6 атомов в кольце и до 5, а предпочтительно, до 4 гетероатомов, выбранных из группы, состоящей из S, O и N, он иллюстративно и предпочтительно представляет собой тиенил, фурил, пирролил, тиазолил, оксазолил, имидазолил, пиридил, пиримидил, пиридазинил, индолил, индазолил, бензофуранил, бензотиофенил, хинолинил, изохинолинил. Гетероариламино представляет собой гетероариламино радикал, имеющий один или два (независимо выбранных) гетероарильных заместителя, он иллюстративно и предпочтительно представляет собой тиениламино, фуриламино, пирролиламино, тиазолиламино, оксазолиламино, имидазолиламино, пиридиламино, пиримидиламино, пиридазиниламино, индолиламино, индазолиламино, бензофураниламино, бензотиофениламино, хинолиниламино, изохинолиниламино. Гетероарилкарбонил иллюстративно и предпочтительно представляет собой тиенилкарбонил, фурилкарбонил, пирролилкарбонил, тиазолилкарбонил, оксазолилкарбонил, имидазолилкарбонил, пиридилкарбонил, пиримидилкарбонил, пиридазинилкарбонил, индолилкарбонил, индазолилкарбонил, бензофуранилкарбонил, бензотиофенилкарбонил, хинолинилкарбонил, изохинолинилкарбонил. Гетероциклил сам по себе и в гетероциклилкарбониле представляет собой моно- или полициклический, предпочтительно моно- или бициклический, неароматический гетероциклический радикал, имеющий, как правило, 4-10, а предпочтительно, 5-8 атомов в кольце и до 3, а предпочтительно до 2 гетероатомов и/или гетерогрупп, выбранных из группы, состоящей из N, O, S, SO и SO2. Гетероциклильные радикалы могут быть насыщенными или частично ненасыщенными. Предпочтение отдается 5-8-членным моноциклическим насыщенным гетероциклильным радикалам, имеющим до двух гетероатомов, выбранных из группы, состоящей из O, N и S, таким как, иллюстративно и предпочтительно, тетрагидрофуран-2-ил, пирролидин-2-ил, пирролидин-3-ил, пирролинил, пиперидинил, мофолинил, пергидроазепинил. Гетероциклилкарбонил иллюстративно и предпочтительно представляет собой тетрагидрофуран-2-карбонил, пирролидин-2-карбонил, пирролидин-3-карбонил, пирролинкарбонил, пиперидинкарбонил, морфолинкарбонил, пергидроазепинкарбонил. Соединения формулы (I) по настоящему изобретению могут, но, не ограничиваясь этим, быть получены посредством объединения различных известных способов. В некоторых вариантах осуществления, один или несколько заместителей, таких как амино группа, карбоксильная группа и гидроксильная группа, соединений, используемых в качестве исходных материалов или промежуточных соединений, преимущественно защищаются защитной группой, известной специалистам в данной области. Примеры защитных групп описываются в “Protective Groups in Organic Synthesis (3rd Edition)” by Greene and Wuts, John Wiley and Sons, New York 1999. Соединения формулы (I) по настоящему изобретению могут, но, не ограничиваясь этим, быть получены посредством способов [A], [B] [C], [D], [E], [F], [G], [H] или [I], ниже. [Способ A]
Соединения формулы (I-a) (где R3 и R4 являются такими, как определено выше, и R1a представляет собой
в которых n и Y являются такими, как определено выше) могут, например, быть получены посредством следующих процедур, в две стадии. На Стадии A-1, соединения формулы (IV) (где R1a, R3 и R4 являются такими, как определено выше, и Z1 представляет собой C1-6алкил, бензил, 4-метоксибензил или 3,4-диметоксибензил) могут быть получены посредством взаимодействия соединений формулы (II) (где R3, R4 и Z1 являются такими, как определено выше) с соединениями формулы (III) (где R1a является таким, как определено выше, и L1 представляет собой уходящую группу, включающую в себя, например, атом галогена, такой как атом хлора, брома и йода, азол, такой как имидазол и триазол, и гидрокси). Реакция может осуществляться в растворителе, включая, например, галогенированные углеводороды, такие как дихлорметан, хлороформ и 1,2-дихлорэтан; простые эфиры, такие как простой диэтиловый эфир, простой изопропиловый эфир, диоксан и тетрагидрофуран (ТГФ), и 1,2-диметоксиэтан; ароматические углеводороды, такие как бензол, толуол и ксилол; нитрилы, такие как ацетонитрил; амиды, такие как N, N-диметилформамид (ДМФ), N,N-диметилацетамид (DMAC) и N-метилпирролидон (NMP); мочевины, такие как 1,3-диметил-2-имидазолидинон (DMI); сульфоксиды, такие как диметилсульфоксид (ДМСО); и другие. Необязательно, два или более из растворителей, выбранных из тех, которые перечислены выше, могут смешиваться и использоваться. Температура реакции может необязательно устанавливаться, в зависимости от соединений, которые должны взаимодействовать. Температура реакции обычно составляет, но, не ограничиваясь этим, примерно 0°C-180°C, предпочтительно, примерно 20°C-100°C. Реакция может осуществляться, обычно, в течение от 30 минут до 24 часов, а предпочтительно, от 1 до 12 часов. Реакция может преимущественно осуществляться в присутствии основания, включая, например, карбонат натрия, карбонат калия, пиридин, триэтиламин и N,N-диизопропилэтиламин, диметиланилин, диэтиланилин и другие. В случае, когда L1 в соединениях формулы (III) (где R1a представляет собой
в которой n и Y являются такими, как определено выше) представляет собой гидрокси, соединения формулы (IV) (где R3, R4 и Z1 являются такими, как определено выше, и R1a представляет собой
в которой n и Y являются такими, как определено выше) могут быть получены посредством взаимодействия соединений формулы (II) (где R3, R4 и Z1 являются такими, как определено выше) с соединениями формулы (III), с использованием связывающего агента включая, например, карбодимиды, такие как N,N-дициклогексилкарбодимид и 1-(3-диметиламинопропил)-3-этилкарбодимид, бензотриазол-1-ил-окси-трис-пирролидинофосфоний гексафторфосфат (PyBOP), дифенилфосфорилазид. N-гидроксисукцинимид, 1-гидроксибензотиазол моногидрат (HOBt), и тому подобное, могут использоваться в качестве ускорителя реакции. Реакция может осуществляться в растворителе, включая, например, галогенированные углеводороды, такие как дихлорметан, хлороформ и 1,2-дихлорэтан; простые эфиры, такие как простой диэтиловый эфир, простой изопропиловый эфир, диоксан и тетрагидрофуран (ТГФ), и 1,2-диметоксиэтан; ароматические углеводороды, такие как бензол, толуол и ксилол; нитрилы, такие как ацетонитрил; амиды, такие как N, N-диметилформамид (ДМФ), N,N-диметилацетамид (DMAC) и N-метилпирролидон (NMP); мочевины, такие как 1,3-диметил-2-имидазолидинон (DMI); сульфоксиды, такие как диметилсульфоксид (ДМСО); и другие. Необязательно, два или более из растворителей, выбранных из тех, которые перечислены выше, могут смешиваться и использоваться. Температура реакции может необязательно устанавливаться, в зависимости от соединений, которые должны взаимодействовать. Температура реакции обычно составляет, но, не ограничиваясь этим, примерно 0°C-180°C, предпочтительно, примерно 20°C-100°C. Реакция может осуществляться, обычно, в течение от 30 минут до 24 часов, а предпочтительно, от 1 до 12 часов. На Стадии A-2, соединения формулы (I-a) (где R1a, R3 и R4 являются такими, как определено выше) могут быть получены посредством удаления защитной группы Z1 соединений формулы (IV) (где R1a, R3, R4 и Z1 являются такими, как определено выше). Удаление защитной группы Z1 может осуществляться посредством использования основания, включая, например, гидроксид натрия, гидроксид лития и гидроксид калия, или кислоты, включая, например, HCl, HBr, трифторуксусную кислоту и BBr3. Снятие защиты может также осуществляться посредством гидрирования с использованием катализатора, включая, например, палладий на угле и гидроксид палладия, когда Z1 представляет собой бензил, 4-метоксибензил или 3,4-диметоксибензил. Также, снятие защиты может осуществляться посредством использования реагента, такого как церий аммоний нитрат (CAN) или 2,3-дихлор-5,6-дициано-1,4-бензохинон (DDQ), когда Z1 представляет собой 4-метоксибензил или 3,4-диметоксибензил. Реакция может осуществляться в растворителе, включая, например, галогенированные углеводороды, такие как дихлорметан, хлороформ и 1,2-дихлорэтан; простые эфиры, такие как простой диэтиловый эфир, простой изопропиловый эфир, диоксан и тетрагидрофуран (ТГФ), и 1,2-диметоксиэтан; ароматические углеводороды, такие как бензол, толуол и ксилол; диметилформамид (ДМФ), диметилацетамид (DMAC), 1,3-диметил-3,4,5,6-тетрагидро-2(1H)-пиримидинон (DMPU), 1,3-диметил-2-имидазолидинон (DMI), N-метилпирролидинон (NMP), сульфоксиды, такие как диметилсульфоксид (ДМСО), спирты, такие как метанол, этанол, 1-пропанол, изопропанол и трет-бутанол, воду и другие. Необязательно, два или более из растворителей, выбранных из тех, которые перечислены выше, могут смешиваться и использоваться. Температура реакции может необязательно устанавливаться, в зависимости от соединений, которые должны взаимодействовать. Температура реакции обычно составляет, но, не ограничиваясь этим, примерно 0°C-180°C, предпочтительно, примерно 20°C-100°C. Реакция может осуществляться, обычно, в течение от 30 минут до 24 часов, а предпочтительно, от 1 до 12 часов. Соединения формулы (III) являются коммерчески доступными или могут синтезироваться посредством обычных способов. [Способ B]
Соединения формулы (I-b) (где R3 и R4 являются такими, как определено выше, и Z2 представляет собой
в которых n и Y являются такими, как определено выше) могут, например, быть получены посредством следующих процедур, в две стадии. На Стадии B-1, соединения формулы (VI) (где R3, R4, Z1 и Z2 являются такими, как определено выше) могут быть получены посредством взаимодействия соединений формулы (II) (где R3, R4 и Z1 являются такими, как определено выше) с соединениями формулы (V) (где Z2 является таким как определено выше) с использованием восстанавливающего агента, такого как натрий триацетоксиборгидрид. Реакция может преимущественно осуществляться в присутствии кислоты, такой как уксусная кислота или хлористоводородная кислота, или дегидратирующего агента, такого как молекулярные сита. Реакция может осуществляться в растворителе, включая, например, галогенированные углеводороды, такие как 1,2-дихлорэтан, простые эфиры, такие как простой диэтиловый эфир, простой изопропиловый эфир, диоксан и тетрагидрофуран (ТГФ) и 1,2-диметоксиэтан; ароматические углеводороды, такие как бензол, толуол и ксилол, и другие. Необязательно, два или более растворителей, выбранных из тех, которые перечислены выше, могут смешиваться и использоваться. Температура реакции может необязательно устанавливаться, в зависимости от соединений, которые должны взаимодействовать. Температура реакции обычно составляет, но, не ограничиваясь этим, примерно 0°C-180°C, предпочтительно, примерно 20°C-100°C. Реакция может осуществляться, обычно, в течение от 30 минут до 24 часов, а предпочтительно, от 1 до 12 часов. На Стадии B-2, соединения формулы (I-b) (где R3, R4 и Z2 являются такими, как определено выше) могут быть получены посредством удаления защитной группы Z1 соединений формулы (VI) (где R3, R4, Z1 и Z2 являются такими, как определено выше) способом, подобным Стадии A-2, для получения соединений формулы (I-a). Соединения формулы (V) являются коммерчески доступными или могут синтезироваться посредством обычных способов. [Способ C]
Соединения формулы (I-c) (где n, R3, R4 и Y являются такими, как определено выше) могут, например, быть получены посредством следующих процедур, в две стадии. На Стадии C-1, Соединения формулы (VIII) (где n, R3, R4, Y и Z1 являются такими, как определено выше) могут быть получены посредством взаимодействия соединений формулы (II) (где R3, R4 и Z1 являются такими, как определено выше) с соединениями формулы (VII) (где n и Y являются такими, как определено выше). Реакция может осуществляться в растворителе, включая, например, галогенированные углеводороды, такие как дихлорметан, хлороформ и 1,2-дихлорэтан; простые эфиры, такие как простой диэтиловый эфир, простой изопропиловый эфир, диоксан и тетрагидрофуран (ТГФ), и 1,2-диметоксиэтан; ароматические углеводороды, такие как бензол, толуол и ксилол; нитрилы, такие как ацетонитрил; амиды, такие как N,N-диметилформамид (ДМФ), N,N-диметилацетамид (DMAC) и N-метилпирролидон (NMP); мочевины, такие как 1,3-диметил-2-имидазолидинон (DMI); сульфоксиды, такие как диметилсульфоксид (ДМСО); и другие. Необязательно, два или более растворителей, выбранных из тех, которые перечислены выше, могут смешиваться и использоваться. Температура реакции может необязательно устанавливаться, в зависимости от соединений, которые должны взаимодействовать. Температура реакции обычно составляет, но, не ограничиваясь этим, примерно 0°C-180°C, предпочтительно, примерно 20°C-100°C. Реакция может осуществляться, обычно, в течение от 30 минут до 24 часов, а предпочтительно, от 1 до 12 часов. На Стадии C-2, соединения формулы (I-c) (где n, R3, R4 и Y являются такими, как определено выше) могут быть получены посредством удаления защитной группы Z1 соединений формулы (VIII) (где n, R3, R4, Y и Z1 являются такими, как определено выше) способом, подобным Стадии A-2, для получения соединений формулы (I-a). Соединения формулы (VII) являются коммерчески доступными или могут синтезироваться посредством обычных способов. [Способ D]
Соединения формулы (I-d) (где n, R3, R4 и Y являются такими, как определено выше) могут, например, быть получены посредством следующих процедур, в две стадии. На Стадии D-1, Соединения формулы (X) (где n, R3, R4, Y и Z1 являются такими, как определено выше) могут быть получены посредством взаимодействия соединений формулы (II) (где R3, R4 и Z1 являются такими, как определено выше) с соединениями формулы (IX) (где n и Y являются такими, как определено выше и L2 представляет собой уходящую группу, включая, например, атом галогена, такой как хлор и бром). Реакция может осуществляться в растворителе, включая, например, галогенированные углеводороды, такие как дихлорметан, хлороформ и 1,2-дихлорэтан; простые эфиры, такие как простой диэтиловый эфир, простой изопропиловый эфир, диоксан и тетрагидрофуран (ТГФ), и 1,2-диметоксиэтан; ароматические углеводороды, такие как бензол, толуол и ксилол; нитрилы, такие как ацетонитрил; амиды, такие как N,N-диметилформамид (ДМФ), N,N-диметилацетамид (DMAC) и N-метилпирролидон (NMP); мочевины, такие как 1,3-диметил-2-имидазолидинон (DMI); сульфоксиды, такие как диметилсульфоксид (ДМСО); и другие. Необязательно, два или более растворителей, выбранных из тех, которые перечислены выше, могут смешиваться и использоваться. Температура реакции может необязательно устанавливаться, в зависимости от соединений, которые должны взаимодействовать. Температура реакции обычно составляет, но, не ограничиваясь этим, примерно 0°C-180°C, предпочтительно, примерно 20°C-100°C. Реакция может осуществляться, обычно, в течение от 30 минут до 24 часов, а предпочтительно, от 1 до 12 часов. На Стадии C-2, соединения формулы (I-d) (где n, R3, R4 и Y являются такими, как определено выше) могут быть получены посредством удаления защитной группы Z1 соединений формулы (VIII) (где n, R3, R4, Y и Z1 являются такими, как определено выше) способом, подобным Стадии A-2, для получения соединений формулы (I-a). Соединения формулы (IX) являются коммерчески доступными или могут синтезироваться посредством обычных способов. [Способ E]
Соединения формулы (I-e) (где n, R3, R4 и Y являются такими, как определено выше и Q1 представляет собой -NH-, -N(C1-6 алкил)-, или -O-) могут, например, быть получены посредством следующих процедур, в две стадии. На Стадии E-1, Соединения формулы (XII) (где n, Q1, R3, R4, Y и Z1 являются такими, как определено выше) могут быть получены посредством взаимодействия соединений формулы (II) (где R3, R4 и Z1 являются такими, как определено выше), соединения формулы (XI) (где n, Q1 и Y являются такими, как определено выше) и агента, включая, например, производное арилформиата, такое как фенил хлорформиат; производное галогенкарбонила, такое как фосген, дифосген и трифосген; производное карбонилдиазола, такое как 1,1-карбонилдиимидазол (CDI) и 1,1′-карбонилди(1,2,4-триазол) (CDT), и тому подобное. Реакция может осуществляться в растворителе, включая, например, галогенированные углеводороды, такие как дихлорметан, хлороформ и 1,2-дихлорэтан; простые эфиры, такие как простой диэтиловый эфир, простой изопропиловый эфир, диоксан и тетрагидрофуран (ТГФ) и 1,2-диметоксиэтан; ароматические углеводороды, такие как бензол, толуол и ксилол; нитрилы, такие как ацетонитрил; амиды, такие как N,N-диметилформамид (ДМФ), N,N-диметилацетамид (DMAC) и N-метилпирролидон (NMP); мочевины, такие как 1,3-диметил-2-имидазолидинон (DMI); и другие. Необязательно, два или более растворителей, выбранных из тех, которые перечислены выше, могут смешиваться и использоваться. Температура реакции может необязательно устанавливаться, в зависимости от соединений, которые должны взаимодействовать. Температура реакции обычно составляет, но, не ограничиваясь этим, примерно 0°C-180°C, предпочтительно, примерно 20°C-100°C. Реакция может осуществляться, обычно, в течение от 30 минут до 24 часов, а предпочтительно, от 1 до 12 часов. Реакция может преимущественно осуществляться в присутствии основания, включая, например, органические амины, такие как пиридин, триэтиламин и N,N-диизопропилэтиламин, диметиланилин, диэтиланилин, 4-диметиламинопиридин и другие. На Стадии E-2, соединения формулы (I-e) (где n, Q1, R3, R4 и Y являются такими, как определено выше) могут быть получены посредством удаления защитной группы Z1 соединений формулы (XII) (где n, Q1, R3, R4, Y и Z1 являются такими, как определено выше) способом, подобным Стадии A-2, для получения соединений формулы (I-a). Соединения формулы (XI) являются коммерчески доступными или могут синтезироваться посредством обычных способов. [Способ F]
Соединения формулы (I-f) (где R1 и R4 являются такими, как определено выше), могут быть получены посредством следующих процедур. На Стадии F-1, соединения формулы (XIII) (где R1, R4 и Z1 являются такими, как определено выше) могут быть получены посредством взаимодействия соединений формулы (II-a) (где R4 и Z1 являются такими, как определено выше) способом, подобным тому, который описан на Стадии A-1, B-1, C-1, D-1 и E-1. На Стадии F-2, соединения формулы (XIV) (где R1, R4 и Z1 являются такими, как определено выше) могут быть получены посредством восстановления соединений формулы (XIII) (где R1, R4 и Z1 являются такими, как определено выше) посредством гидрирования, с использованием катализатора, включая, например, палладий на угле и платину на угле, в присутствии основания, такого как ацетат калия. Реакция может осуществляться в растворителе, включая, например, простые эфиры, такие как простой диэтиловый эфир, простой изопропиловый эфир, диоксан, тетрагидрофуран (ТГФ) и 1,2-диметоксиэтан, ароматические углеводороды, такие как бензол, толуол и ксилол, спирты, такие как метанол, этанол, 1-пропанол, изопропанол и трет-бутанол, воду и другие. Температура реакции может необязательно устанавливаться, в зависимости от соединений, которые должны взаимодействовать. Температура реакции обычно составляет, но, не ограничиваясь этим, примерно 0°C-180°C, предпочтительно, примерно 20°C-100°C. Реакция может осуществляться, обычно, в течение от 30 минут до 24 часов, а предпочтительно, от 1 до 12 часов. На Стадии F-3, соединения формулы (I-f) (где R1 и R4 являются такими, как определено выше) могут быть получены посредством удаления защитной группы Z1 соединений формулы (XIV) (где R1, R4 и Z1 являются такими, как определено выше) способом, подобным Стадии A-2, для получения соединений формулы (I-a). [Способ G]
Соединения формулы (I-g) (где R1 и R4 являются такими, как определено выше, и R3′ имеет такое же значение как R3, как определено выше, исключая водород и галоген), могут быть получены посредством следующих процедур. На Стадии G-1, соединения формулы (XVI) (где R1, R3′, R4 и Z1 являются такими, как определено выше) могут быть получены посредством взаимодействия соединений формулы (XIII) (где R1, R4 и Z1 являются такими, как определено выше) с соединениями формулы (XV) (где R3′ является таким как определено выше). Реакция может осуществляться без растворителя или в растворителе, включая, например, простые эфиры, такие как простой диэтиловый эфир, простой изопропиловый эфир, диоксан и тетрагидрофуран (ТГФ) и 1,2-диметоксиэтан; ароматические углеводороды, такие как бензол, толуол и ксилол; нитрилы, такие как ацетонитрил; амиды, такие как N, N-диметилформамид (ДМФ), N,N-диметилацетамид (DMAC) и N-метилпирролидон (NMP); мочевины, такие как 1,3-диметил-2-имидазолидинон (DMI); сульфоксиды, такие как диметилсульфоксид (ДМСО); и другие. Необязательно, два или более растворителей, выбранных из тех, которые перечислены выше, могут смешиваться и использоваться. Температура реакции может необязательно устанавливаться, в зависимости от соединений, которые должны взаимодействовать. Температура реакции обычно составляет, но, не ограничиваясь этим, примерно 0°C-180°C, предпочтительно, примерно 20°C-100°C. Реакция может осуществляться, обычно, в течение от 30 минут до 24 часов, а предпочтительно, от 1 до 12 часов. Реакция может преимущественно осуществляться в присутствии основания, включая, например, органические амины, такие как пиридин, триэтиламин, N,N-диизопропилэтиламин, диметиланилин, диэтиланилин и другие. На Стадии G-2, соединения формулы (I-g) (где R1, R3′ и R4 являются такими, как определено выше) могут быть получены посредством удаления защитной группы Z1 соединений формулы (XVI) (где R1, R3′, R4 и Z1 являются такими, как определено выше) способом, подобным Стадии A-2, для получения соединений формулы (I-a). [Способ H]
Соединения формулы (I-h) (где R3 и R4 являются такими, как определено выше, R1a представляет собой
в которых n и Y являются такими, как определено выше, и Z3 представляет собой водород или C1-5 алкил) могут также быть получены посредством следующих процедур. На Стадии H-1, соединения формулы (XVIII) (где R3, R4, Z1 и Z3 являются такими, как определено выше) могут быть получены посредством взаимодействия соединений формулы (II) (где R3, R4 и Z1 являются такими, как определено выше) с соединениями формулы (XVII) (где Z3 является таким как определено выше) способом, подобным тому, который описан на Стадии B-1, для получения соединений формулы (VI). На Стадии H-2, соединения формулы (XIX) (где R1a, R3, R4, Z1 и Z3 являются такими, как определено выше) могут быть получены посредством взаимодействия соединений формулы (XVIII) (где R3, R4, Z1 и Z3 являются такими, как определено выше) с соединениями формулы (III) (где R1a и L1 являются такими, как определено выше) способом, подобным тому, который описан на Стадии A-1, для получения соединений формулы (IV). На Стадии H-3, соединения формулы (I-h) (где R1a, R3, R4 и Z3 являются такими, как определено выше) могут быть получены посредством удаления защитной группы Z1 соединений формулы (XIX) (где R1a, R3, R4, Z1 и Z3 являются такими, как определено выше) способом, подобным Стадии A-2, для получения соединений формулы (I-a). Соединения формулы (XVII) являются коммерчески доступными или могут синтезироваться посредством обычных способов. [Способ I]
Соединения формулы (I) (где R1, R2, R3 и R4 являются такими, как определено выше) могут быть получены посредством следующих процедур. На Стадии 1-1, соединения формулы (XXII) (где R1, R2, R3, R4 и Z1 являются такими, как определено выше) могут быть получены посредством взаимодействия соединений формулы (XX) (где R1, R2, R3, R4 и Z1 являются такими, как определено выше) с соединениями формулы (XXI). Реакция может осуществляться в растворителе, включая, например, галогенированные углеводороды, такие как дихлорметан, хлороформ и 1,2-дихлорэтан; простые эфиры, такие как простой диэтиловый эфир, простой изопропиловый эфир, диоксан и тетрагидрофуран (ТГФ) и 1,2-диметоксиэтан; ароматические углеводороды, такие как бензол, толуол и ксилол; нитрилы, такие как ацетонитрил; амиды, такие как N, N-диметилформамид (ДМФ), N,N-диметилацетамид (DMAC) и N-метилпирролидон (NMP); мочевины, такие как 1,3-диметил-2-имидазолидинон (DMI); сульфоксиды, такие как диметилсульфоксид (ДМСО); и другие. Необязательно, два или более растворителей, выбранных из тех, которые перечислены выше, могут смешиваться и использоваться. Реакция может преимущественно осуществляться в присутствии основания, включая, например, органические амины, такие как триэтиламин и N,N-диизопропилэтиламин и другие. Температура реакции может необязательно устанавливаться, в зависимости от соединений, которые должны взаимодействовать. Температура реакции обычно составляет, но, не ограничиваясь этим, примерно 0°C-180°C, предпочтительно, примерно 20°C-100°C. Реакция может осуществляться, обычно, в течение от 30 минут до 24 часов, а предпочтительно, от 1 до 12 часов. На Стадии I-2, соединения формулы (XXIII) (где R1, R2, R3, R4 и Z1 являются такими, как определено выше) могут быть получены посредством взаимодействия соединений формулы (XXII) (где R1, R2, R3, R4 и Z1 являются такими, как определено выше). Реакция может осуществляться в присутствии агента, включая, например, кислоту, такую как хлористоводородная кислота и трифторуксусная кислота, трифторуксусный ангидрид, или POCl3, и тому подобное. Температура реакции может необязательно устанавливаться, в зависимости от соединений, которые должны взаимодействовать. Температура реакции обычно составляет, но, не ограничиваясь этим, примерно 0°C-180°C, предпочтительно, примерно 20°C-100°C. Реакция может осуществляться, обычно, в течение от 30 минут до 24 часов, а предпочтительно, от 1 до 12 часов. На Стадии 1-3, соединения формулы (I) (где R1, R2, R3 и R4 являются такими, как определено выше) могут быть получены посредством удаления защитной группы Z1 соединений формулы (XXIII) (где R1, R2, R3, R4 и Z1 являются такими, как определено выше) способом, подобным Стадии A-2, для получения соединений формулы (I-a). Соединения формулы (XX) и (XXI) являются коммерчески доступными или могут синтезироваться посредством обычных способов. Получение исходных соединений
Соединения формулы (II-c) (где R3′, R4 и Z1 являются такими, как определено выше) могут, например, быть получены посредством следующих процедур, в две стадии. На Стадии i-1, соединения формулы (XXIV) (где R4 и Z1 являются такими, как определено выше) взаимодействуют с соединениями формулы (XV) (где R3′ является таким, как определено выше), с получением соединений формулы (XXV) (где R3′, R4 и Z1 являются такими, как определено выше). Реакция может осуществляться без растворителя или в растворителе, включая, например, простые эфиры, такие как простой диэтиловый эфир, простой изопропиловый эфир, диоксан и тетрагидрофуран (ТГФ) и 1,2-диметоксиэтан; ароматические углеводороды, такие как бензол, толуол и ксилол; нитрилы, такие как ацетонитрил; амиды, такие как N, N-диметилформамид (ДМФ), N,N-диметилацетамид (DMAC) и N-метилпирролидон (NMP); мочевины, такие как 1,3-диметил-2-имидазолидинон (DMI); сульфоксиды, такие как диметилсульфоксид (ДМСО); и другие. Необязательно, два или более растворителей, выбранных из тех, которые перечислены выше, могут смешиваться и использоваться. Температура реакции может необязательно устанавливаться, в зависимости от соединений, которые должны взаимодействовать. Температура реакции обычно составляет, но, не ограничиваясь этим, примерно 0°C-180°C, предпочтительно, примерно 20°C-100°C. Реакция может осуществляться, обычно, в течение от 30 минут до 24 часов, а предпочтительно, от 1 до 12 часов. Реакция может преимущественно осуществляться в присутствии основания, включая, например, органические амины, такие как пиридин, триэтиламин, N,N-диизопропилэтиламин, диметиланилин, диэтиланилин и другие. На Стадии i-2, соединения формулы (II-c) (где R3′, R4 и Z1 являются такими, как определено выше) могут быть получены посредством восстановления нитро-группы соединений формулы (XXV) (где R3′, R4 и Z1 являются такими, как определено выше) с использованием агента, включая, например, металлы, такие как цинк и железо, в присутствии кислоты включая, например, хлористоводородную кислоту и уксусную кислоту, и хлорида олова, или посредством гидрирования, с использованием катализатора, включая, например, палладий на угле и платину на угле. Реакция может осуществляться в растворителе, включая, например, простые эфиры, такие как простой диэтиловый эфир, простой изопропиловый эфир, диоксан, тетрагидрофуран (ТГФ) и 1,2-диметоксиэтан, ароматические углеводороды, такие как бензол, толуол и ксилол, спирты, такие как метанол, этанол, 1-пропанол, изопропанол и трет-бутанол, воду и другие. Температура реакции может необязательно устанавливаться, в зависимости от соединений, которые должны взаимодействовать. Температура реакции обычно составляет, но, не ограничиваясь этим, примерно 0°C-180°C, предпочтительно, примерно 20°C-100°C. Реакция может осуществляться, обычно, в течение от 30 минут до 24 часов, а предпочтительно, от 1 до 12 часов. Соединения формулы (II-a) (где R4 и Z1 являются такими, как определено выше) могут быть получены посредством восстановления нитро-группы соединений формулы (XXIV) (где R4 и Z1 являются такими, как определено выше) способом, подобным тому, который описан на Стадии i-2, для получения соединений формулы (II-c), как показано на Стадии i-3. Соединения формулы (II-b) (где R4 и Z1 являются такими, как определено выше) могут быть получены посредством восстановления группы хлора и нитро-группы соединений формулы (XXIV) (где R4 и Z1 являются такими, как определено выше) посредством гидрирования, с использованием катализатора, включая, например, палладий на угле и платину на угле, в присутствии основания, такого как ацетат калия, как показано в Стадии i-4. Реакция может осуществляться в растворителе, включая, например, простые эфиры, такие как простой диэтиловый эфир, простой изопропиловый эфир, диоксан, тетрагидрофуран (ТГФ) и 1,2-диметоксиэтан, ароматические углеводороды, такие как бензол, толуол и ксилол, спирты, такие как метанол, этанол, 1-пропанол, изопропанол и трет-бутанол, воду и другие. Температура реакции может необязательно устанавливаться, в зависимости от соединений, которые должны взаимодействовать. Температура реакции обычно составляет, но, не ограничиваясь этим, примерно 0°C-180°C, предпочтительно, примерно 20°C-100°C. Реакция может осуществляться, обычно, в течение от 30 минут до 24 часов, а предпочтительно, от 1 до 12 часов. Получение соединений формулы (XXIV)
Соединения формулы (XXIV) (где R4 и Z1 являются такими, как определено выше) могут, например, быть получены посредством следующих процедур. На Стадии ii-1, соединения формулы (XXVII) (где R4 и Z1 являются такими, как определено выше) могут быть получены посредством взаимодействия соединений формулы (XXVI) (где R4 и Z1 являются такими, как определено выше) с соединениями формулы (XXI) способом, подобным тому, который описан на Стадии I-1, для получения соединений формулы (XXII). На Стадии ii-2, соединения формулы (XXIV) (где R4 и Z1 являются такими, как определено выше) могут быть получены посредством взаимодействия соединений формулы (XXVII) (где R4 и Z1 являются такими, как определено выше) способом, подобным тому, который описан на Стадии I-2, для получения соединений формулы (XXIII). Получение соединений формулы (XXVI)
Соединения формулы (XXVI) (где R4 и Z1 являются такими, как определено выше) могут, например, быть получены посредством следующих процедур. На Стадии iii-1, соединения формулы (XXVDI) (где R4 и Z1 являются такими, как определено выше) могут быть получены посредством взаимодействия соединений формулы (XXIX) (где R4 является таким как определено выше) с соединениями формулы (XXX) (где Z1 является таким как определено выше, и Z4 представляет собой C1-6алкил). Реакция может преимущественно осуществляться в присутствии основания, такого как метоксид натрия. Реакция может осуществляться в растворителе, включая, например, спирты, такие как метанол, этанол, 1-пропанол, изопропанол и трет-бутанол. Температура реакции может необязательно устанавливаться, в зависимости от соединений, которые должны взаимодействовать. Температура реакции обычно составляет, но, не ограничиваясь этим, примерно 0°C-180°C, а предпочтительно, примерно 20°C-100°C. Реакция может осуществляться, обычно, в течение от 30 минут до 24 часов, а предпочтительно, от 1 час до 12 часов. На Стадии iii-2, соединения формулы (XXVI) (где R4 и Z1 являются такими, как определено выше) могут быть получены, например, посредством взаимодействия соединений формулы (XXVIII) (где R4 и Z1 являются такими, как определено выше) с соответствующим галогенирующим реагентом, включая, например, POCl3, PCl5, и тому подобное. Реакция может осуществляться без растворителя или в растворителе, включая, например, галогенированные углеводороды, такие как дихлорметан, хлороформ и 1,2-дихлорэтан, ароматические углеводороды, такие как бензол, толуол и ксилол, и другие. Необязательно, два или более растворителей, выбранных из тех, которые перечислены выше, могут смешиваться и использоваться. Реакция может преимущественно осуществляться в присутствии основания, включая, например, пиридин, триэтиламин и N,N-диизопропилэтиламин, N,N-диметиланилин, диэтиланилин и другие. Температура реакции обычно составляет, но, не ограничиваясь этим, примерно 40°C-200°C, а предпочтительно, примерно 20°C-180°C. Реакция может осуществляться, обычно, в течение от 30 минут до 48 часов, а предпочтительно, от 2 часов до 12 часов. Соединения формулы (XXIX) являются коммерчески доступными или могут синтезироваться посредством обычных способов. Типичные соли соединения, показанного посредством формулы (I), включают в себя соли, полученные посредством взаимодействия соединений по настоящему изобретению с минеральной или органической кислотой, или с органическим или с неорганическим основанием. Такие соли известны как соли добавления кислот и соли добавления оснований соответственно. Кислоты для образования солей добавления кислот включают в себя неорганические кислоты, такие как, без ограничения, серная кислота, фосфорная кислота, хлористоводородная кислота, бромистоводородная кислота, йодистоводородная кислота, и тому подобное, и органические кислоты, такие как, без ограничения, п-толуолсульфоновая кислота, метансульфоновая кислота, щавелевая, п-бромфенилсульфоновая кислота, угольная кислота, янтарная кислота, лимонная кислота, бензойная кислота, уксусная кислота и тому подобное. Соли добавления оснований включают в себя соли, которые получают из неорганических оснований, таких как, без ограничения, гидроксид аммония, гидроксид щелочного металла, гидроксиды щелочноземельных металлов, карбонаты, бикарбонаты, и тому подобное, и органических оснований, таких как, без ограничения, этаноламин, триэтиламин, трис(гидроксиметил)аминометан, и тому подобное. Примеры неорганических оснований включают в себя, гидроксид натрия, гидроксид калия, карбонат калия, карбонат натрия, бикарбонат натрия, бикарбонат калия, гидроксид кальция, карбонат кальция и тому подобное. Соединение по настоящему изобретению или его соли, в зависимости от его заместителей, могут модифицироваться, с образованием сложных эфиров низших алкилов или других известных сложных эфиров; и/или гидратов или других сольватов. Такие сложные эфиры, гидраты и сольваты включаются в рамки настоящего изобретения. Соединение по настоящему изобретению может вводиться в пероральных формах, таких как, без ограничения, нормальные таблетки и таблетки с ентеральным покрытием, капсулы, пилюли, порошки, гранулы, эликсиры, микстуры, растворы, суспензии, сиропы, твердые и жидкие аэрозоли и эмульсии. Они могут также вводиться в парентеральных формах, таких как, без ограничения, внутривенная, внутрибрюшинная, подкожная, внутримышечная и тому подобное, формы, хорошо известные специалистам в области фармацевтики. Соединения по настоящему изобретению могут вводиться в интраназальной форме посредством местного использования соответствующих интраназальных носителей, или посредством чрескожных способов, с использованием систем чрескожной доставки, хорошо известных специалистам в данной области. Режим дозировки при использовании соединений по настоящему изобретению выбирается одним из специалистов в данной области, с учетом разнообразных факторов, включая, без ограничения, возраст, массу, пол и медицинское состояние пациента, тяжесть состояния, которое должно лечиться, способ введения, уровень метаболической и экскреторной функции пациента, используемую форму дозировки, конкретное используемое соединение и его соли. Соединения по настоящему изобретению предпочтительно приготавливаются перед введением вместе с одним или несколькими фармацевтически приемлемыми наполнителями. Наполнители представляют собой инертные вещества, такие как, без ограничения, носители, разбавители, ароматизирующие агенты, подсластители, смазывающие вещества, солюбилизаторы, суспендирующие агенты, связующие вещества, разрыхляющие агенты и инкапсулирующий материал для таблеток. Еще одни вариант осуществления настоящего изобретения представляет собой фармацевтический препарат, содержащий соединение по настоящему изобретению и один или несколько фармацевтически приемлемых наполнителей, которые являются совместимыми с остальными ингредиентами препарата и не вредными для пациента, который его принимает. Фармацевтические препараты по настоящему изобретению получают посредством объедения терапевтически эффективного количества соединений по настоящему изобретению вместе с одним или несколькими фармацевтически приемлемыми наполнителями для них. При приготовлении композиций по настоящему изобретению активный ингредиент может смешиваться с разбавителем или заключаться в носитель, который может находиться в форме капсулы, саше, бумаги или другого контейнера. Носитель может служить в качестве разбавителя, который может представлять собой твердый, полутвердый, или жидкий материал, который действует в качестве растворителя, или может находиться в форме таблеток, пилюль, порошков, лепешек, эликсиров, суспензий, эмульсий, растворов, сиропов, аэрозолей, мазей, содержащих, например, до 10 мас.% активного соединения, мягких и твердых желатиновых капсул, суппозиториев, стерильных растворов для инъекций и стерильных упакованных порошков. Для перорального введения активный ингредиент может объединяться с пероральным и нетоксичным фармацевтически приемлемым носителем, таким как, без ограничения, лактоза, крахмал, сахароза, глюкоза, карбонат натрия, маннитол, сорбитол, карбонат кальция, фосфат кальция, сульфат кальция, метилцеллюлоза и тому подобное; необязательно, вместе, с разрыхляющими агентами, такими как, без ограничения, кукурузный крахмал, метилцеллюлоза, агар, бентонит, ксантановая смола, альгиновая кислота и тому подобное; и необязательно, со связывающими агентами, например, без ограничения, желатином, природными сахарами, бета-лактозой, кукурузными подсластителями, природными и синтетическими смолами, смолой акации, смолой трагаканта, альгинатом натрия, карбоксиметилцеллюлозой, полиэтиленгликолем, восками, и тому подобное; и, необязательно, со смазывающими агентами, например, без ограничения, стеаратом магния, стеаратом натрия, стеариновой кислотой, олеатом натрия, бензоатом натрия, ацетатом натрия, хлоридом натрия, тальком и тому подобное. В форме порошка носитель может представлять собой мелкодисперсный твердый материал, который находится в смеси с мелкодисперсным активным ингредиентом. Активный ингредиент может смешиваться с носителем, имеющим связывающие свойства, в соответствующих пропорциях, и компактироваться в форме и размерах, желаемых для производства таблеток. Порошки и таблетки предпочтительно содержат примерно от 1 примерно до 99 процентов массовых активного ингредиента, который представляет собой новую композицию по настоящему изобретению. Пригодные для использования твердые носители представляют собой магний карбоксиметилцеллюлозу, легкоплавкие воски и масло какао. Стерильные жидкие препараты включают в себя суспензии, эмульсии, сиропы и эликсиры. Активный ингредиент может растворяться или суспендироваться в фармацевтически приемлемом носителе, таком как стерильная вода, стерильный органический растворитель, или в смеси как стерильной воды, так и стерильного органического растворителя. Активный ингредиент может также растворяться в соответствующем органическом растворителе, например, в водном растворе пропиленгликоля. Другие композиции могут приготавливаться посредством диспергирования мелкодисперсного активного ингредиента в водном растворе крахмала или натрий карбоксиметилцеллюлозы или в соответствующем масле. Препарат может находиться в форме стандартной единичной дозировки, которая представляет собой физически отдельную единицу, содержащую единичную дозу, пригодную для введения людям или другим млекопитающим. Форма стандартной единичной дозировки может представлять собой капсулу или таблетку, или ряд капсул или таблеток. “Стандартная единичная дозировка” представляет собой заданное количество активного соединения по настоящему изобретению, рассчитанное для оказания желаемого терапевтического воздействия, в сочетании с одним или несколькими наполнителями. Количество активного ингредиента в стандартной единичной дозировке может изменяться или устанавливаться примерно от 0,1 примерно до 1000 миллиграмм или более в соответствии с конкретным соответствующим лечением. Типичные пероральные дозировки соединения по настоящему изобретению, когда оно используется для указанных воздействий, будет находиться в пределах примерно от 1 мг/кг/день примерно до 10 мг/кг/день. Соединения по настоящему изобретению могут вводиться в виде одной ежедневной дозы, или же общая ежедневная доза может вводиться в разделенных дозах, два, три или более раз в день. Когда доставка осуществляется посредством чрескожных форм, разумеется, введение является непрерывным. ПРИМЕРЫ Настоящее изобретение будет описываться как форма примеров, но они не должны, ни в коем случае, рассматриваться в качестве пределов и границ настоящего изобретения. В примерах, ниже, все количественные данные, если не указано иного, относятся к процентам массовым. Масс спектры получают с использованием технологий ионизации электрораспылением (ES) (Micromass Platform LC). Температуры плавления не корректируются. Данные жидкостной хроматографии – масс-спектрометрии (LC-MS) регистрируют на Micromass Platform LC с колонкой Shimadzu Phenomenex ODS (диаметр 4,6 мм × 30 мм) с элюированием смесью ацетонитрил-вода (от 9:1 до 1:9) при скорости потока 1 мл/мин. ТСХ осуществляют на пластинке с предварительно нанесенным силикагелем (Merck silica gel 60 F-254). Силикагель (WAKO-gel C-200 (75-150 мкм)) используют для всех разделений с помощью колоночной хроматографии. Все химикалии имеют химическое качество и покупаются от Sigma-Aldrich, Wako pure chemical industries, Ltd., Great Britain, Tokyo kasei kogyo Co., Ltd., Nacalai tesque, Inc., Watanabe Сhemical Ind. Ltd., Maybridge plc, Lancaster Synthesis Ltd., Merck KgaA, Germany, Kanto Chemical Co., Ltd. Спектры 1H NMR регистрируют с использованием либо спектрометра Bruker DRX-300 (300 МГц для 1H), либо Brucker 500 UltraShield Все исходные материалы являются коммерчески доступными или могут быть получены с использованием способов, цитируемых в литературе. Воздействия настоящих соединений исследуют с помощью следующих далее анализов и фармакологических исследований. ПРИМЕР 1 [Получение линии клеток L1.2, трансфицированных CRTH2 человека] кДНК CRTH2 человека амплифицируют из кДНК эозинофилов человека с помощью ген-специфичных праймеров, содержащих сайты рестрикции для клонировании в вектор pEAK (Edge Bio Systems). кДНК CRTH2 человека клонируют в вектор экспрессии pEAK млекопитающего. Эту плазмиду экспрессии (40 мкг) трансфицируют в клетки L1.2, при плотности клеток 1×107 клеток/500 мкл, посредством использования устройства для порообразования (Gene Pulser II, BioRad), при 250 В/1000 мкФ. Через день после трансфекции, пуромицин (1 мкг/мл, Sigma) добавляют в чашки с культурами клеток. Через два недели после трансфекции, выращенные клетки отбирают для дальнейшего роста. [Измерение мобилизации Ca2+ линии клеток L1.2, трансфицированных CRTH2 человека] (Анализ 1) Загрузочный буфер Ca2+ приготавливают посредством смешивания 5 мкл Fluo-3AM (2 мМ в ДМСО, конечная концентрация 1 мкM, (Molecular Probes) и 10 мкл Pluronic F-127 (Molecular Probes) и разбавления полученной смеси в 10 мл буфера Ca2+ для анализа (20 мМ HEPES pH 7,6, 0,1% BSA, 1 мМ пробенецида, раствор Хэнкса). Клетки, трансфицированные CRTH2, которые получают в Примере 1, промывают PBS, повторно суспендируют в загрузочном буфере Ca2+, при 1×107 клеток/мл, и инкубируют в течение 60 мин при комнатной температуре. После инкубирования клетки промывают и повторно суспендируют в буфере Ca2+ для анализа, затем распределяют в 96-луночные планшеты с прозрачным дном (#3631, Costar), при 2×105 клеток/лунка. Клетки инкубируют с различными концентрациями исследуемого соединения в течение 5 минут, при комнатной температуре. Флуоресценцию, испускаемую на 480 нм, измеряют на FDSS6000, устройстве для измерений Ca2+ (Hamamatsu Photonics, Hamamatsu, Japan). Трансфектант демонстрирует индуцируемую PGD2 мобилизацию Ca2+ концентрационно-зависимым образом. [Анализ связывания рецептора CRTH2 человека] (Анализ 2) Трансфектанты CRTH2 промывают один раз PBS и повторно суспендируют в буфере для связывания (50 мМ Трис-HCl, pH 7,4, 40 мМ MgCl2, 0,1% BSA, 0,1% NaN3). Затем 100 мкл суспензии клеток (2×105 клеток), [3H]-меченный PGD2 и различные концентрации исследуемого соединения смешивают в 96-луночном полипропиленовом планшете с U-образным дном и инкубируют в течение 60 мин при комнатной температуре, чтобы сделать возможным осуществление связывания. После инкубирования суспензию клеток переносят в фильтровальный планшет (#MAFB, Millipore) и промывают 3 раза в буфере для связывания. В фильтровальный планшет добавляют сцинтиллирующее вещество, и радиоактивность, остающуюся на фильтре, измеряют посредством TopCount (Packard), счетчика сцинтилляций. Неспецифическое связывание определяют посредством инкубирования суспензии клеток и [3H]-меченного PGD2 в присутствии 1 мкM немеченого PGD2. Устойчивые к пуромицину трансфектанты L1.2 связываются с [3H]-меченным PGD2 с высоким сродством (KD=6,3 нM). [Анализ миграции эозинофилов человека] (Анализ 3) Полиморфоядерные клетки человека выделяют из гепаринизированной венозной крови здоровых доноров посредством наслаивания крови на Моно-Poly Resolving Medium (ICN Biomedicals, Co.Ltd) и центрифугирования ее при 400×g в течение 30 мин, при комнатной температуре. После центрифугирования эозинофилы очищают от нижнего слоя полиморфоядерных клеток посредством CD16-отрицательной селекции с использованием магнитных шариков, конъюгированных с анти-CD16 (Miltenyi Biotech GmbH). Эозинофилы человека промывают PBS и повторно суспендируют в буфере для хемотаксиса (20 мМ HEPES pH 7,6, 0,1% BSA, раствор Хэнкса), при 6×106 клеток/мл. Затем пятьдесят мкл суспензии клеток (3×105 клеток/лунку) распределяют в верхнюю камеру и 30 мкл раствора лиганда (PGD2, конечная концентрация 1 нM) добавляют в нижнюю камеру 96-луночной камеры для хемотаксиса (диаметр = 5 мкм, #106-5, Neuro Probe). Клетки предварительно инкубируют при различных концентрациях исследуемого соединения, при 37°C, в течение 10 минут. Затем позволяют осуществляться хемотаксису в увлажненном инкубаторе при 37°C, 5% CO2, в течение 2 часов. Количество клеток, мигрирующих в нижнюю камеру, считают с помощью FACScan (Becton-Dickinson). [Анализ миграции Т-клеток CD4+ человека] (Анализ 4) Моноядерные клетки человека выделяют из гепаринизированной венозной крови здоровых доноров посредством наслаивания крови на Mono-Poly Resolving Medium (ICN Biomedicals, Co.Ltd) и центрифугирования ее при 400×g в течение 30 мин, при комнатной температуре. После центрифугирования Т-лимфоциты CD4+ очищают от моноядерных клеток посредством использования набора для выделения T-клеток CD4+ (Miltenyi Biotec GmbH). Т-лимфоциты CD4+ человека промывают PBS и повторно суспендируют в буфере для хемотаксиса (20 мМ HEPES pH 7,6, 0,1% BSA, раствор Хэнкса), при 6×106 клеток/мл. Затем пятьдесят мкл суспензии клеток (3×105 клеток/лунка) распределяют в верхнюю камеру, а 30 мкл раствора лиганда (PGD2, конечная концентрация 10 нM) добавляют в нижнюю камеру 96-луночной камеры для хемотаксиса (диаметр = 3 мм, #106-3, Neuro Probe). Клетки предварительно инкубируют при различных концентрациях исследуемого соединения при 37°C в течение 10 минут. Затем позволяют осуществляться хемотаксису в увлажненном инкубаторе при 37°C, 5% CO2, в течение 4 часов. Количество клеток, мигрирующих в нижнюю камеру, считают с помощью FACScan (Becton-Dickinson). Результаты анализа в анализе 1 показаны в примерах и таблицах, в разделе Примеры, ниже. Данные соответствуют соединениям, как они получены посредством твердофазного синтеза, и, таким образом, уровням чистоты примерно от 40 до 90%. По практическим причинам соединения группируются в четыре класса активности следующим образом: IC50=A(< или =)10 нM Соединения по настоящему изобретению также демонстрируют превосходную селективность и сильную активность в анализах 2, 3 и 4, описанных выше. Z, используемое для температуры плавления, в следующем разделе, означает разложение. Все неорганические кислоты и основания представляют собой водные растворы, если не утверждается иного. Концентрации элюента выражаются как % об./об. Получение соединений Метил [4,6-дихлор-2-(4-нитробензил)пиримидин-5-ил]ацетат
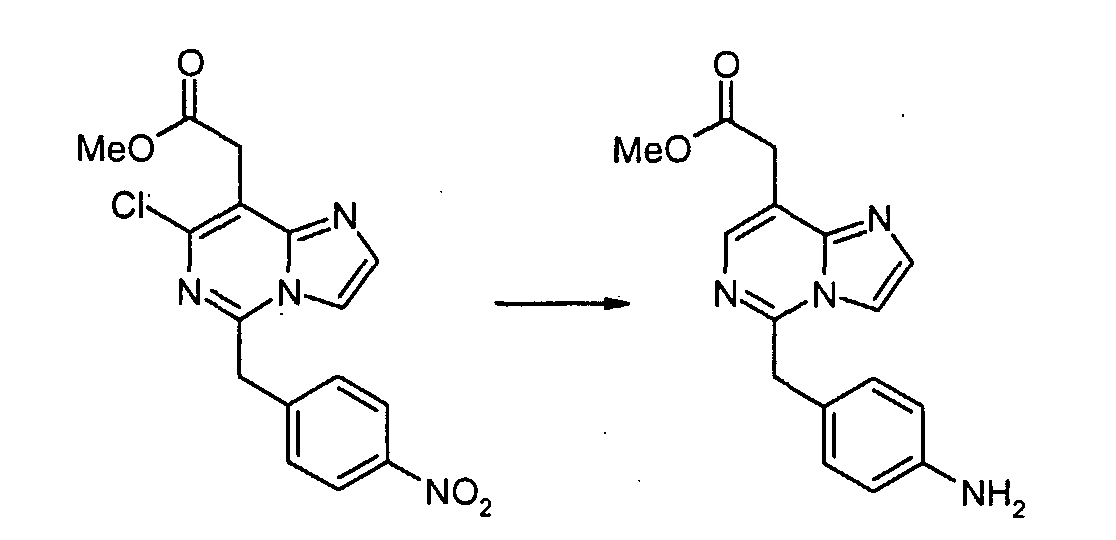
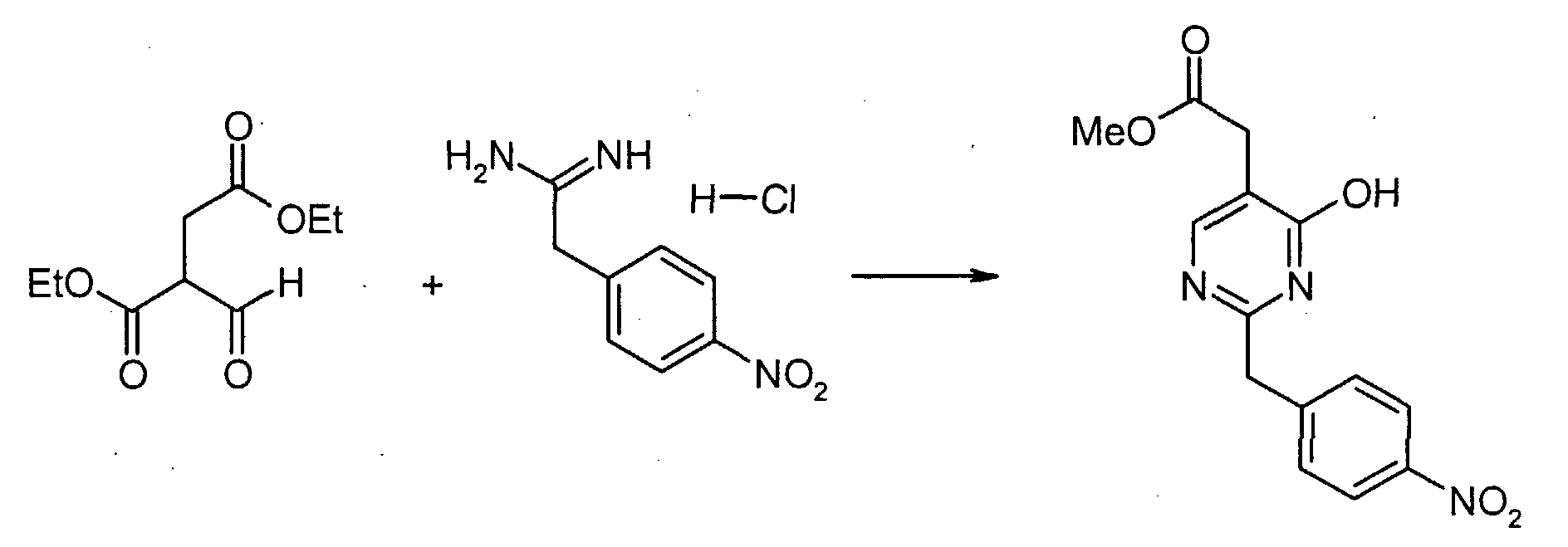
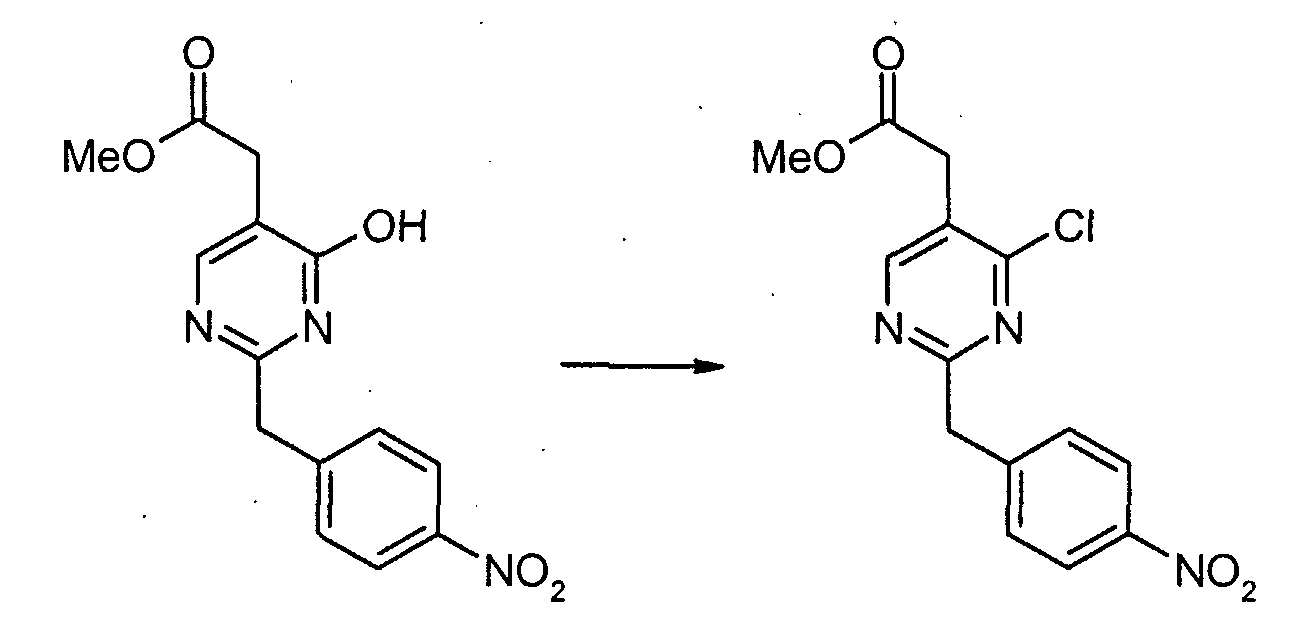
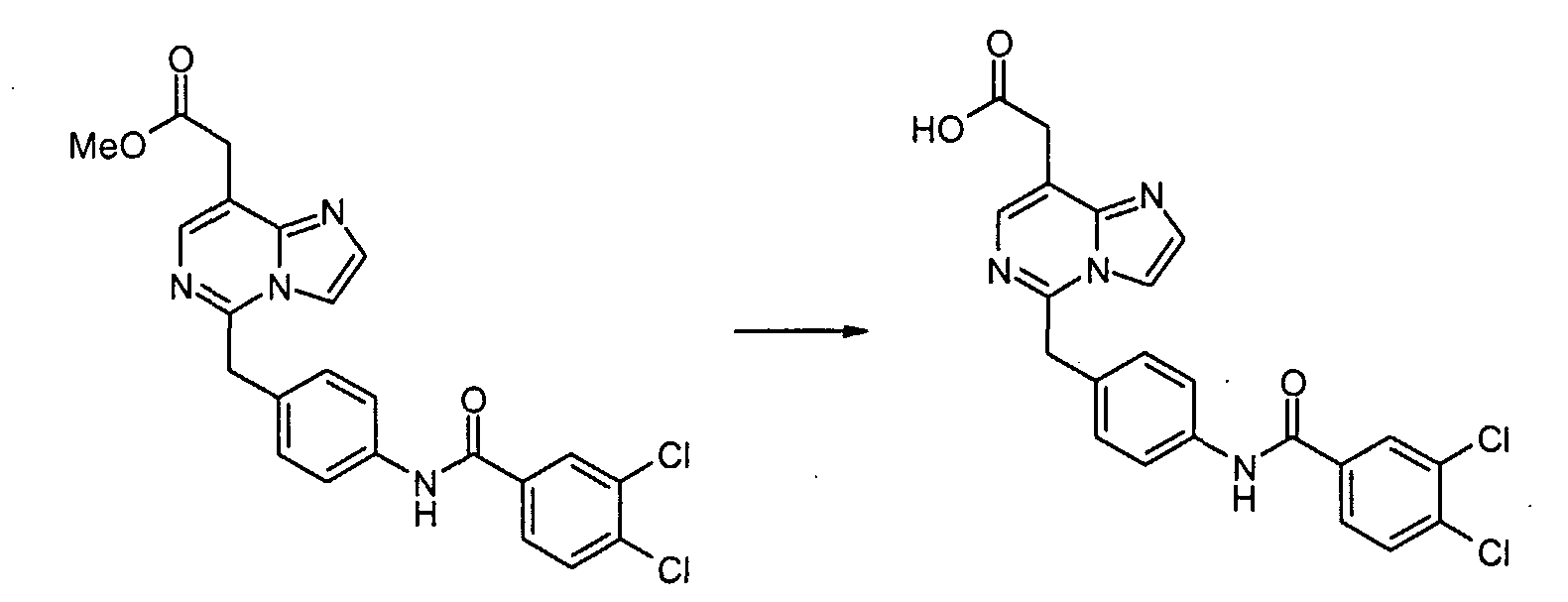
4-Нитрофенилацетонитрил (81,07 г, 500 ммоль) суспендируют в EtOH (300 мл) и добавляют диоксан (300 мл). После растворения всех твердых продуктов, сухой газообразный HCl барботируют через реакционную смесь в течение 1 часа, затем перемешивают при комнатной температуре в течение 15 часов. Затем добавляют Et2O, и выделившиеся твердые продукты собирают посредством отсоса и промывают Et2O. Это промежуточное соединение растворяют в EtOH, насыщенным NH3, и раствор, полученный таким образом, перемешивают при комнатной температуре в течение 14 часов. Избыток растворителя удаляют в вакууме, с получением 2-(4-нитрофенил)этанимидамид гидрохлорида (73,65 г, выход 68%) в виде белого порошка. К смеси триэтил 1,1,2-этантрикарбоксилата (3,51 мл, 15,30 ммоль) и 2-(4-нитрофенил)этанимидамид гидрохлорида (46,95 г, 217,72 ммоль) в безводном MeOH (300 мл) при комнатной температуре добавляют NaOMe (3,8,82 г, 718,49 ммоль), и полученную суспензию нагревают с обратным холодильником в течение 16 часов. После охлаждения до комнатной температуры реакционную смесь охлаждают до 0°C, подкисляют с помощью 6 н. HCl, и выделившиеся твердые продукты собирают посредством отсоса и промывают холодной водой. Затем сушка в высоком вакууме при 45°C в течение 6 часов дает метил [4,6-дигидрокси-2-(4-нитробензил)пиримидин-5-ил]ацетат (56,48 г, 81% выход) в виде тускло-белого порошка. К суспензии метил[4,6-дигидрокси-2-(4-нитробензил)пиримидин-5-ил]ацетата (4,12 г, 12,89 ммоль) в POCl3 (24 мл) при комнатной температуре и в атмосфере Ar добавляют N,N-диметиланилин (8,17 мл, 64,44 ммоль), и полученную темную суспензию нагревают с обратным холодильником в течение 16 часов. После охлаждения до комнатной температуры избыток POCl3 выпаривают, и оставшийся темный остаток растворяют в EtOAc. Затем этот органический слой промывают последовательно насыщенным NaHCO3 водой, и насыщенным раствором соли, сушат над безводным MgSO4, фильтруют и концентрируют в вакууме. Сырой продукт, полученный таким образом, растворяют в CH2Cl2 и пропускают через тонкий слой силикагеля, с получением чистого метил[4,6-дихлор-2-(4-нитробензил)пиримидин-5-ил]ацетата (2,98 г, выход 65%) в виде беловатого порошка. Метил[7-хлор-5-(4-нитробензил)имидазо[1,2-c]пиримидин-8-ил]ацетат
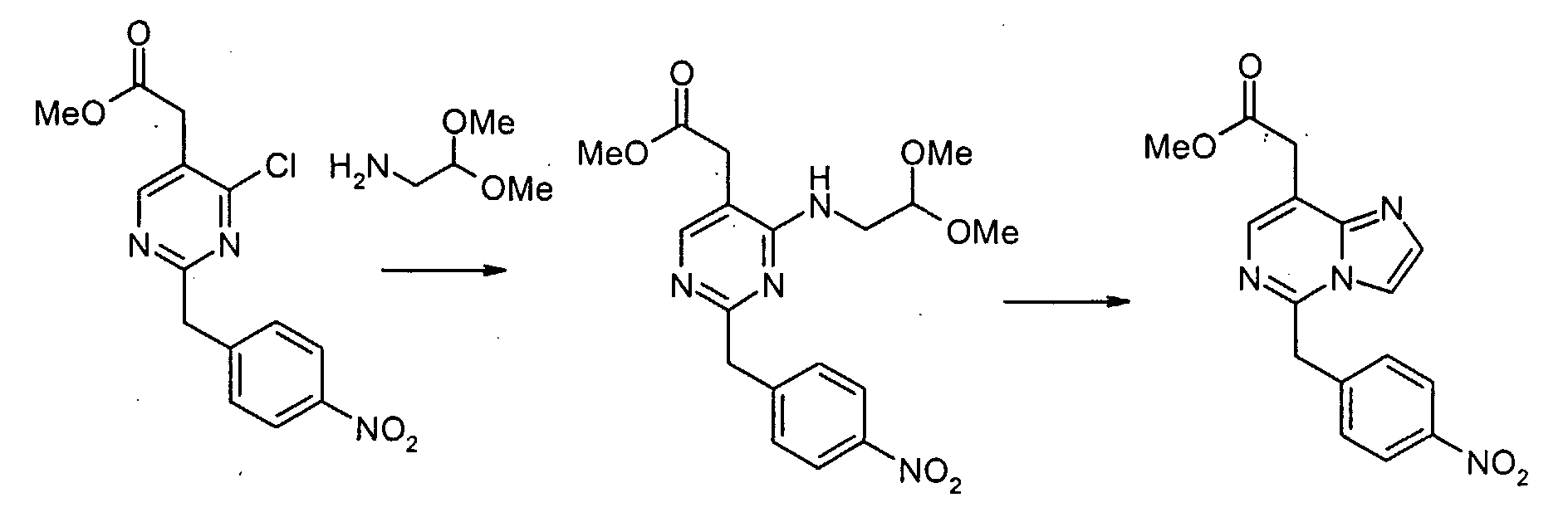
Смесь метил[4,6-дихлор-2-(4-нитробензил)пиримидин-5-ил]ацетата (2,0 г, 5,6 ммоль), аминоацетальдегид диметилацеталя (0,68 г, 6,5 ммоль) и диизопропилэтиламина (1,5 мл, 8,4 ммоль) в 1,4-диоксане (50 мл) перемешивают при 80°C в течение 2 часов. Смесь концентрируют при пониженном давлении. Остаток экстрагируют этилацетатом. Экстракты промывают водным раствором NaHCO3 и насыщенным раствором соли, сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Сырой продукт очищают с помощью препаративной MPLC (жидкостной хроматографии средней эффективности) (силикагель, гексан: этилацетат, 2/1), с получением метил[4-хлор-6-[(2,2-диметоксиэтил)амино]-2-(4-нитробензил)пиримидин-5-ил]ацетата (1,89 г, 79%) в виде серого твердого продукта. Метил[4-хлор-6-[(2,2-диметоксиэтил)амино]-2-(4-нитробензил)пиримидин-5-ил]ацетат (100 мг, 0,19 ммоль) обрабатывают 50% трифторуксусной кислотой/дихлорметаном (5 мл) при комнатной температуре в течение ночи. Смесь выливают в воду и экстрагируют дихлорметаном. Экстракты промывают водным раствором NaHCO3 и насыщенным раствором соли, сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении. Сырой продукт растворяют в дихлорметане (5 мл). К раствору добавляют трифторуксусный ангидрид (0,053 мл, 0,38 ммоль). Смесь перемешивают при комнатной температуре в течение 3 часов. Смесь выливают в воду и экстрагируют дихлорметаном. Экстракты промывают водным раствором NaHCO3 и насыщенным раствором соли, сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении. Сырой продукт очищают с помощью препаративной ТСХ (силикагель, хлороформ: этанол, 19/1), с получением метил[7-хлор-5-(4-нитробензил)имидазо[1,2-c]пиримидин-8-ил]ацетата (22 мг, 25 %) в виде бесцветной пленки. Метил[7-хлор-5-(4-аминобензил)имидазо[1,2-c]пиримидин-8-ил]ацетат
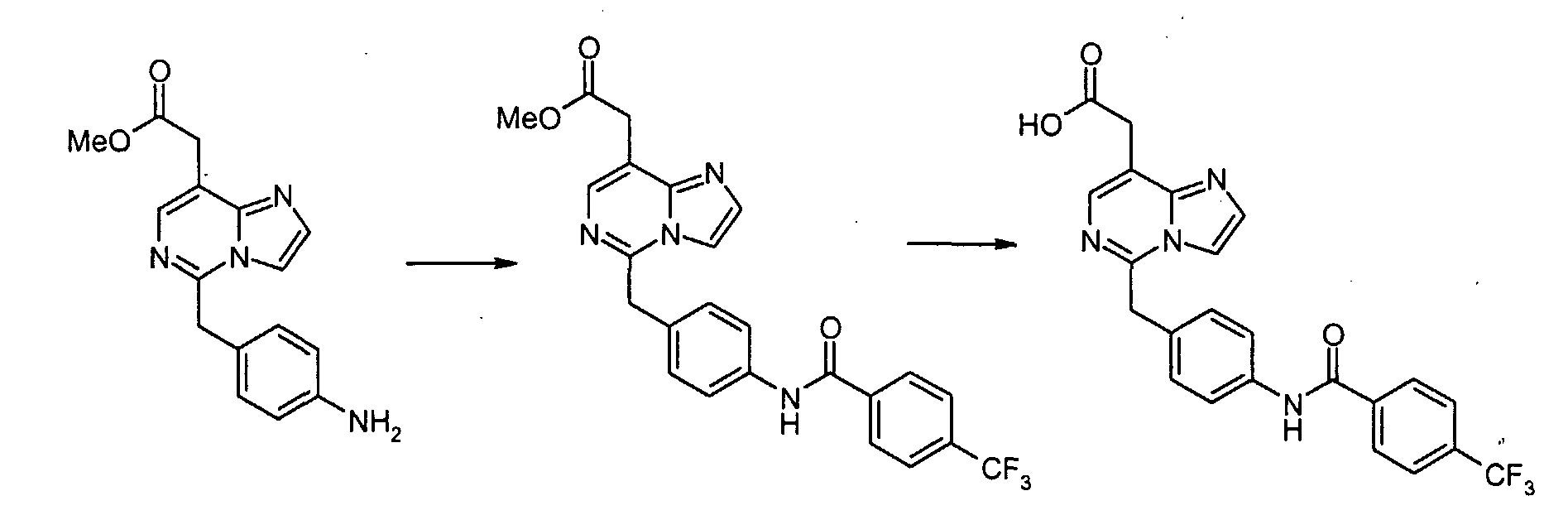
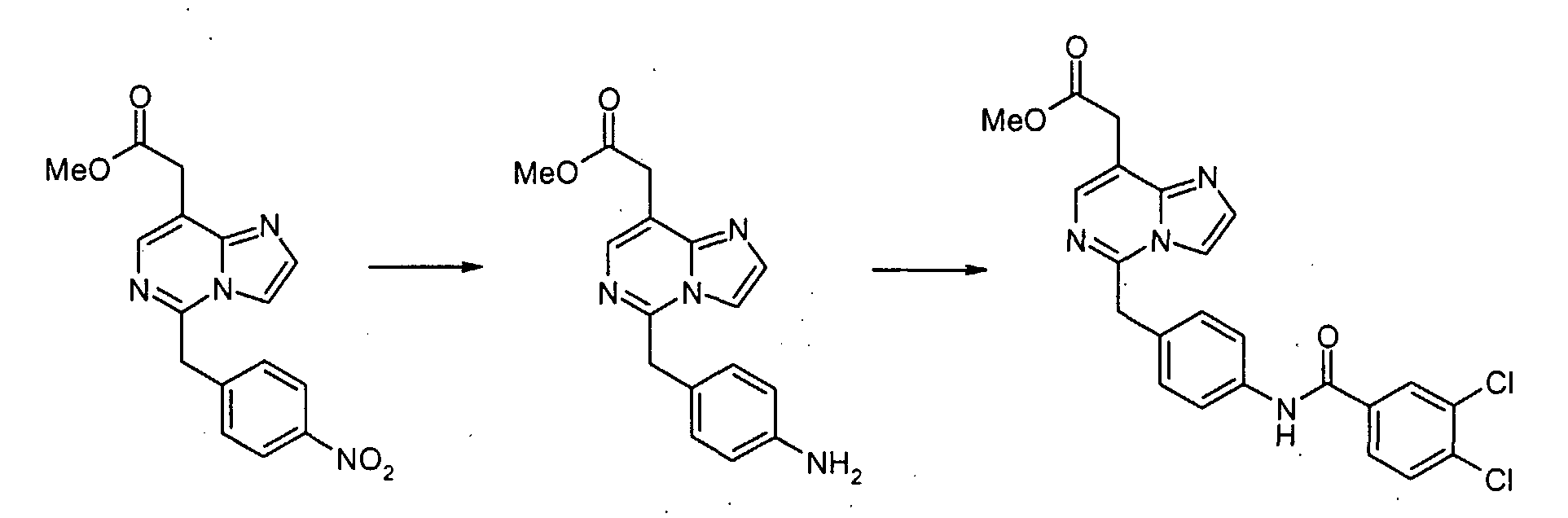
Смесь метил[7-хлор-5-(4-нитробензил)имидазо[1,2-c]пиримидин-8-ил]ацетата (0,383 г, 1,06 ммоль) и SnCl2·2H2O (1,44 г, 6,37 ммоль) суспендируют в EtOH (5 мл) при температуре дефлегмации в течение 1 часа. После охлаждения до комнатной температуры реакционную смесь выливают в насыщенный водный раствор NaHCO3 и EtOAc, и полученную белую суспензию фильтруют через слой Целита. Водный слой отделяют и экстрагируют EtOAc. Объединенные органические экстракты промывают насыщенным раствором соли, сушат над безводным MgSO4, фильтруют и концентрируют в вакууме, с получением метил[7-хлор-5-(4-аминобензил)имидазо[1,2-c]пиримидин-8-ил]ацетата (0,322 г, 92%). Пример 1-1 [7-Хлор-5-(4-{[4-(трифторметил)бензоил]амино}бензил)имидазо[1,2-c]пиримидин-8-ил]уксусная кислота
К раствору метил[7-хлор-5-(4-аминобензил)имидазо[1,2-c]пиримидин-8-ил]ацетата (0,081 г, 0,243 ммоль), 3,4-дихлорбензойной кислоты (0,070 г, 0,365 ммоль) и WSCI (0,070 г, 0,365 ммоль) в CH2Cl2 (5 мл) добавляют N,N-диизопропилэтиламин (0,127 мл, 0,730 ммоль). После перемешивания при комнатной температуре в течение 20 часов, реакционную смесь конденсируют при пониженном давлении, с получением сырого продукта в виде густого масла, которое очищают с помощью хроматографии на силикагеле, элюируя 30% EtOAc в CHCl3, с получением метил[7-хлор-5-(4-{[4-(трифторметил)бензоил]амино}бензил)имидазо[1,2-c]пиримидин-8-ил]ацетата в виде белого твердого продукта (0,100 г, 82%). К раствору метил[7-хлор-5-(4-{[4-(трифторметил)бензоил]амино}бензил)имидазо[1,2-c]-пиримидин-8-ил]ацетата (0,100 г, 0,199 ммоль) в диоксане (1,00 мл) добавляют 6 н. водный раствор HCl (0,5 мл) и нагревают до 100°C в течение 22 часов. После охлаждения до комнатной температуры, летучие продукты удаляют при пониженном давлении, с получением осадка, который суспендируют в воде, собирают посредством отсоса, промывают водой и простым диэтиловым эфиром и сушат в высоком вакууме, с получением [7-хлор-5-(4-{[4-(трифторметил)бензоил]амино}бензил)имидазо[1,2-c]пиримидин-8-ил]уксусной кислоты в виде белого порошка (0,020 г, 20%). 1H-ЯMР (500 MГц, ДMСO-d6): Температура плавления: 201 Z°C Молекулярная масса: 488,85 Масс-спектрометрия: 489 Уровень активности in vitro: B Способом, подобным тому, который описан в Примере 1-1, синтезируют соединения в Примерах 1-2 – 1-5, как показано в таблице 1.
Метил [5-(4-аминобензил)имидазо[1,2-c]пиримидин-8-ил]ацетат
Смесь метил[7-хлор-5-(4-нитробензил)имидазо[1,2-c]пиримидин-8-ил]ацетата (150 мг, 0,42 ммоль) и Pd/C (влажный, 45 мг) в метаноле (10 мл) – ТГФ (5 мл) перемешивают в атмосфере водорода (0,4 MПa) в течение 2 дней. После удаления Pd/C посредством фильтрования через слой Целита, фильтрат концентрируют в вакууме. Сырой продукт очищают с помощью препаративной ТСХ (силикагель, хлороформ:метанол, 95/5, 0,1% триэтиламина), с получением метил[5-(4-аминобензил)имидазо[1,2-c]пиримидин-8-ил]ацетата (16 мг, 13%) в виде чуть желтоватого твердого продукта. Пример 2-1 [5-(4-{[4-(трифторметил)бензоил]амино}бензил)имидазо[1,2-c]пиримидин-8-ил]уксусная кислота
Смесь метил [5-(4-аминобензил)имидазо[1,2-c]пиримидин-8-ил]ацетата (17 мг, 0,06 ммоль), п-трифторметилбензоилхлорида (0,01 мл, 0,07 ммоль) и триэтиламина (0,016 мл, 0,11 ммоль) в дихлорметане (1 мл) перемешивают при комнатной температуре в течение 1 часа. Реакцию гасят водой и экстрагируют хлороформом. Экстракты промывают водой и насыщенным раствором соли, сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Сырой продукт очищают с помощью препаративной ТСХ (силикагель, хлороформ:метанол, 95/5), с получением метил [5-(4-{[4-(трифторметил)бензоил]амино}бензил)имидазо[1,2-c]пиримидин-8-ил]ацетата (21,5 мг, 80%) в виде чуть желтоватого твердого продукта. К раствору метил [5-(4-{[4-(трифторметил)бензоил]амино}бензил)имидазо[1,2-c]-пиримидин-8-ил]ацетата (20 мг, 0,04 ммоль) в метаноле (1 мл) добавляют 1M водный раствор NaOH (0,2 мл) при комнатной температуре, и смесь перемешивают в течение 1 часа. После удаления метанола при пониженном давлении к остатку добавляют воду. Раствор промывают простым диэтиловым эфиром и нейтрализуют с помощью водного раствора хлористоводородной кислоты. Полученные преципитаты собирают посредством фильтрования и сушат при пониженном давлении, с получением[5-(4-{[4-(трифторметил)бензоил]амино}бензил)имидазо[1,2-c]пиримидин-8-ил]уксусной кислоты (14,2 мг, 73%) в виде чуть коричневатого твердого продукта. 1H-ЯMР (500 MГц, ДMСО-d6): Температура плавления: 205-207°C Молекулярная масса: 454,41 Масс-спектрометрия: 453 (M-H)–, 455 (M+H)+ Уровень активности in vitro: B Диэтил 2-формилсукцинат
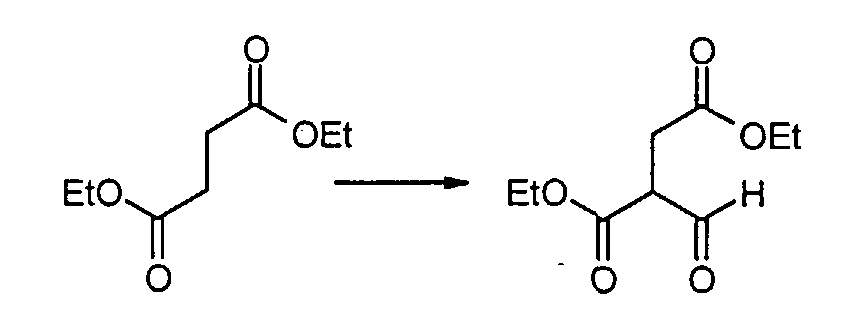
К смеси натрия (1,66 г, 72,13 ммоль) в простом диэтиловом эфире (35 мл) добавляют диэтилсукцинат (10 мл, 60,11 ммоль) и этилформиат (8,25 мл, 102,18 ммоль), при комнатной температуре, и реакционную смесь нагревают с обратным холодильником в течение 5 часов. После охлаждения до комнатной температуры воду добавляют к смеси до тех пор, пока соль натрия не растворится полностью, и водный слой отделяют. Водный слой нейтрализуют с помощью 6M хлористоводородной кислоты (10,8 мл) и экстрагируют простым диэтиловым эфиром. Экстракты промывают насыщенным раствором NaHCO3, сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Сырой продукт очищают с помощью дистилляции (115-120°C, 10 мм рт.ст.), с получением диэтил 2-формилсукцината (6,55 г, 54%) в виде бесцветного масла. Метил [4-гидрокси-2-(4-нитробензил)пиримидин-5-ил]ацетат
К смеси 2-(4-нитрофенил)этанимидамид гидрохлорида (6,06 г, 28,12 ммоль) и диэтил 2-формилсукцината (6,54 г, 32,34 ммоль) в метаноле (100 мл) добавляют метоксид натрия (28% в метаноле, 11,2 мл, 56,25 ммоль), при комнатной температуре, и реакционную смесь перемешивают при 90°C в течение ночи. После охлаждения до комнатной температуры реакцию гасят уксусной кислотой (3,38 мл, 59,06 ммоль) и добавляют к ней воду (100 мл). Полученные преципитаты собирают посредством фильтрования и промывают водой и ацетоном/простым диизопропиловым эфиром (3/2), с получением метил [4-гидрокси-2-(4-нитробензил)пиримидин-5-ил]ацетата (5,41 г, 64%) в виде коричневого твердого продукта. Метил [4-хлор-2-(4-нитробензил)пиримидин-5-ил]ацетат
Смесь метил[4-гидрокси-2-(4-нитробензил)пиримидин-5-ил]ацетата (4,0 г, 13,19 ммоль), оксихлорида фосфора (6,15 мл, 65,95 ммоль) и N,N-диметиланилина (2,51 мл, 19,78 ммоль) перемешивают при 150°C в течение 3 часов. После удаления избытка оксихлорида фосфора остаток растворяют с помощью этилацетата. Органический слой промывают водой и насыщенным раствором соли, сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Сырой продукт очищают с помощью колоночной хроматографии на силикагеле (гексан: этилацетат, 7/3), с получением метил[4-хлор-2-(4-нитробензил)пиримидин-5-ил]ацетата (2,70 г, 64%) в виде оранжевого твердого продукта. Метил [5-(4-нитробензил)имидазо[1,2-c)пиримидин-8-ил]ацетат
К раствору метил[4-хлор-2-(4-нитробензил)пиримидин-5-ил]ацетата (2,70 г, 8,39 ммоль) и аминоацетоальдегид диметилацеталя (1,83 мл, 16,78 ммоль) в диоксане (40 мл) добавляют N,N-диизопропилэтиламин (1,46 мл, 8,39 ммоль). Смесь перемешивают при 85°C в течение 1 дня. После охлаждения до комнатной температуры реакцию гасят водой и экстрагируют этилацетатом. Экстракты промывают водой и насыщенным раствором соли, сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Сырой продукт очищают с помощью колоночной хроматографии на силикагеле, (этилацетат), с получением метил[4-[(2,2-диметоксиэтил)амино]-2-(4-нитробензил)-пиримидин-5-ил]ацетата (2,41 г, 74%) в виде коричневого масла. Раствор метил[4-[(2,2-диметоксиэтил)амино]-2-(4-нитробензил)пиримидин-5-ил]ацетата (1,99 г, 2,56 ммоль) и HCl (1,0 M в воде, 3,84 мл, 3,85 ммоль) в диоксане (10 мл) перемешивают при 85°C в течение 1 часа. После удаления растворителя в вакууме к остатку добавляют воду. Водный раствор нейтрализуют с помощью 1M NaOH и экстрагируют этилацетатом. Экстракты промывают насыщенным раствором соли, сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. К полученному сырому продукту добавляют оксихлорид фосфора (1,4 мл) и смесь перемешивают при 85°C в течение 3 часов. После удаления избытка оксихлорида фосфора остаток растворяют с помощью этилацетата. Органический слой промывают водой, насыщенным раствором NaHCO3 и насыщенным раствором соли, сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Сырой продукт очищают с помощью колоночной хроматографии на силикагеле, (хлороформ:метанол, 98/2), с получением метил[5-(4-нитробензил)имидазо[1,2-c]пиримидин-8-ил]ацетата (28 мг, 3%) в виде коричневого масла. Метил (5-{4-[(3,4-дихлорбензоил)амино]бензил}имидазо[1,2-c]пиримидин-8-ил)ацетат
Смесь метил[5-(4-нитробензил)имидазо[1,2-c]пиримидин-8-ил]ацетата (28 мг, 0,09 ммоль) и Pd/C (влажный, 3 мг) в метаноле (1 мл) перемешивают в атмосфере водорода в течение 1 часа. После удаления Pd/C посредством фильтрования через слой Целита, фильтрат концентрируют в вакууме. Полученный сырой метил[5-(4-aминoбензил)имидазо[1,2-c]пиримидин-8-ил]ацетат (26 мг, 0,09 ммоль) растворяют с помощью дихлорметана (1 мл). К этому раствору добавляют 3,4-дихлорбензойную кислоту (20,1 мг, 0,11 ммоль), 1-гидроксибензотриазол (14,2 мг, 0,11 ммоль), триэтиламин (0,037 мл, 0,26 ммоль) и 1-этил-3-(3-диметиламинопропил)карбодимид гидрохлорид (21,9 мг, 0,11 ммоль), при комнатной температуре. Смесь перемешивают при комнатной температуре в течение ночи и разбавляют этилацетатом. Раствор промывают водой и насыщенным раствором соли, сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Сырой продукт очищают с помощью препаративной ТСХ (силикагель, хлороформ:метанол, 19:1), с получением метил(5-{4-[(3,4-дихлорбензоил)амино]бензил}имидазо[1,2-c]пиримидин-8-ил)ацетата (29 мг, 70%) в виде слегка желтого масла. Пример 2-2 (5-{4-[(3,4-Дихлорбензоил)амино]бензил}имидазо[1,2-c]пиримидин-8-ил)уксусная кислота
К раствору метил(5-{4-[(3,4-дихлорбензоил)амино]бензил}имидазо[1,2-c]пиримидин-8-ил)ацетата (25 мг, 0,05 ммоль) в метаноле (1 мл) добавляют 1M водный раствор NaOH (0,2 мл), при комнатной температуре и смесь перемешивают в течение 1 часа. После удаления метанола при пониженном давлении к остатку добавляют воду. Раствор промывают простым диэтиловым эфиром и нейтрализуют с помощью водного раствора хлористоводородной кислоты. Полученные преципитаты собирают посредством фильтрования и сушат при пониженном давлении, с получением (5-{4-[(3,4-дихлорбензоил)амино]бензил}имидазо[1,2-c]пиримидин-8-ил)уксусной кислоты (17,2 мг, 71%) в виде чуть желтоватого твердого продукта. 1H-ЯMР (500 MГц, ДMСO-d6): Температура плавления: 185-188°C Молекулярная масса: 455,30 Масс-спектрометрия: 455 (M+H)+ Уровень активности in vitro: C.
Формула изобретения
1. Производные имидазо[1,2-с]пиримидинилуксусной кислоты формулы (I) или его соль 2. Производное имидазо[1,2-с]пиримидинилуксусной кислоты формулы (I) или его соль по п.1, 3. Производное имидазо[1,2-c]пиримидинилуксусной кислоты формулы (I-i) или его соль 4. Производное имидазо[1,2-с]пиримидинилуксусной кислоты формулы (I) или его соль по п.1, где указанное производное имидазо [1,2-c]-пиримидинилуксусной кислоты формулы (I) выбирают из группы, состоящей из: 5. Лекарственное средство, которое обладает активностью по отношению к CRTH2, содержащее производное имидазо[1,2-с]пиримидинилуксусной кислоты или его физиологически приемлемую соль по п.1, в качестве активного ингредиента. 6. Лекарственное средство по п.5, дополнительно содержащее один или несколько фармацевтически приемлемых наполнителей. 7. Лекарственное средство по п.5, где указанное производное имидазо[1,2-с]пиримидинилуксусной кислоты формулы (I) или его физиологически приемлемая соль представляет собой антагонист CRTH2. 8. Лекарственное средство по п.5 для лечения и/или профилактики расстройства или заболевания, связанного с активностью CRTH2. 9. Лекарственное средство по п.8, где указанное расстройство или заболевание выбирают из группы, состоящей из астмы, аллергического ринита, атопического дерматита и аллергического конъюнктивита. 10. Лекарственное средство по п.8, где указанное расстройство или заболевание выбирают из группы, состоящей из синдрома Чарджа-Штрауса, синусита, базофильной лейкемии, хронической уртикарии и базофильного лейкоцитоза. 11. Применение соединения по п.1 для производства лекарственного средства для лечения и/или профилактики расстройства или заболевания, связанного с активностью CRTH2. 12. Лекарственное средство по п.6, где наполнитель выбирают из носителей, разбавителей, ароматизирующих агентов, подсластителей, смазывающих веществ, солюбилизаторов, суспендирующих агентов, связующих веществ, разрыхляющих агентов и инкапсулирующего материала для таблеток. 13. Лекарственное средство по п.12, где наполнителем является носитель, выбранный из лактозы, крахмала, сахарозы, глюкозы, карбоната натрия, маннита, сорбита, карбоната кальция, фосфата кальция, сульфата кальция и метилцеллюлозы. 14. Лекарственное средство по п.13, где лекарственное средство, кроме того, включает разрыхляющий агент, выбранный из кукурузного крахмала, метилцеллюлозы, агара, бентонита, ксантановой смолы и альгиновой кислоты. 15. Лекарственное средство по п.14, где носитель является в форме, выбранной из таблетки, пилюли, порошка, пастилки, эликсира, суспензии, эмульсии, раствора, сиропа, аэрозоли, мази, мягкой или твердой желатиновой капсулы, суппозитории, стерильного инъецируемого раствора и стерильного упакованного порошка. 16. Лекарственное средство по п.12, где наполнителем является связующее вещество, выбранное из желатина, природного сахара, бета-лактозы, подсластителя на основе кукурузы, природной или синтетической смолы, аравийской, камеди, трагаканта, альгината натрия, карбоксиметилцеллюлозы, полиэтиленгликоля и восков. 17. Лекарственное средство по п.12, где наполнителем является смазывающее вещество, выбранное из стеарата магния, стеарата натрия, стеариновой кислоты, олеата натрия, бензоата натрия, ацетата натрия, хлорида натрия и талька. 18. Лекарственное средство по п.5, где количество активного ингредиента составляет от около 1 до около 99 вес.% в расчете на общий вес фармацевтической композиции. 19. Лекарственное средство в форме стандартной единичной дозировки, содержащее производное имидазо[1,2-с]пиримидинилуксусной кислоты или его физиологически приемлемую соль определенного по п.1, в качестве активного агента, где количество активного агента составляет от 0,1 до около 1000 ммг. 20. Применение по п.11, где указанное расстройство или заболевание представляет собой астму, аллергический ринит, атонический дерматит или аллергический конъюнктивит. 21. Применение по п.11, где указанное расстройство или заболевание представляет собой синдром Чарджа-Штрауса, синусит, базофильную лейкемию, хроническую уртикарию, базофильный лейкоцитоз. 22. Производное имидазо[1,2-с]пиримидинилуксусной кислоты по пп.1-4, представляющее собой соединение формулы (I). 23. Производное имидазо[1,2-с]пиримидинилуксусной кислоты по пп.1-4, в форме соли.
|
||||||||||||||||||||||||||||||||||||||||
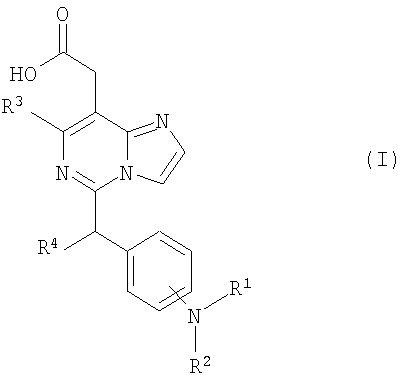
 ,
, 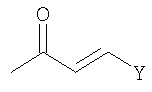 ,
,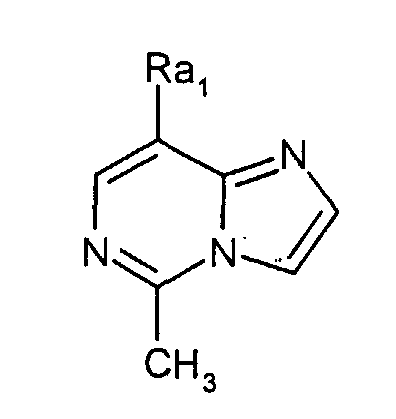
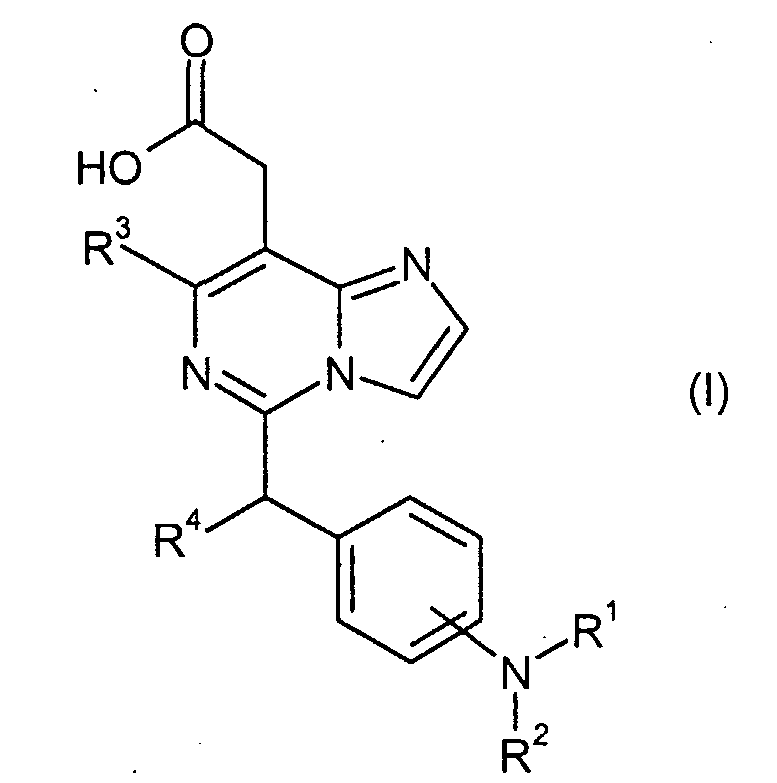
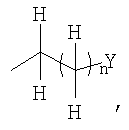
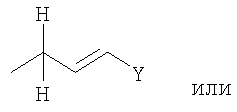
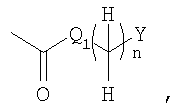
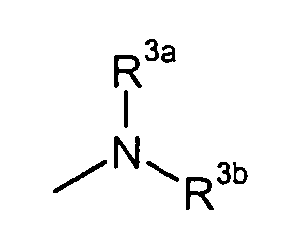
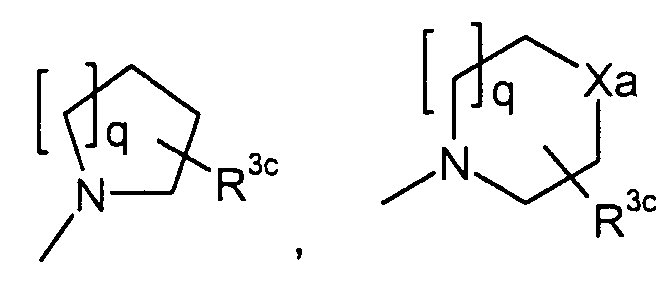
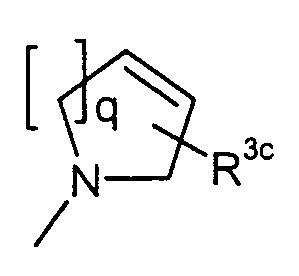
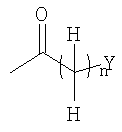 ,
, 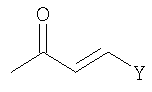 или
или 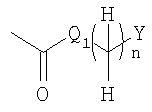
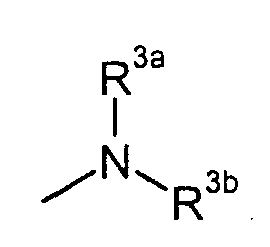
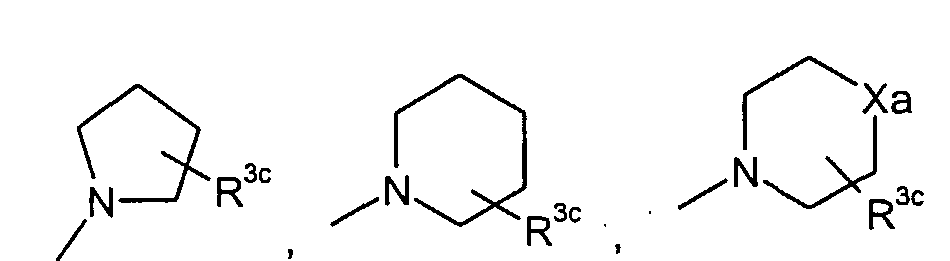
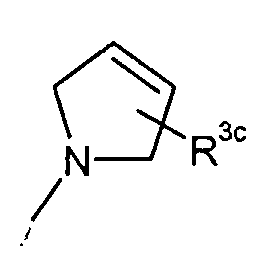
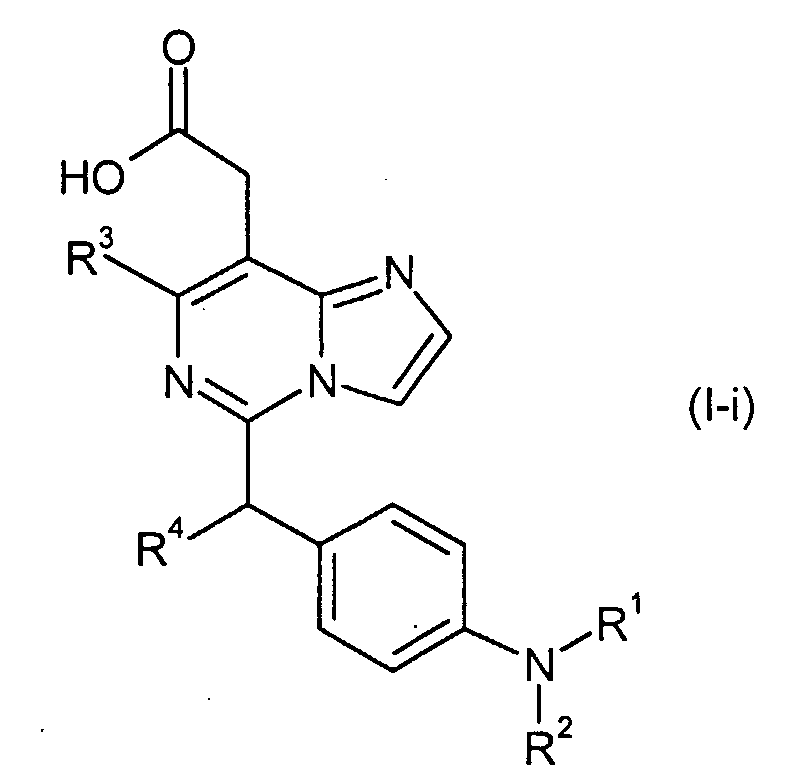
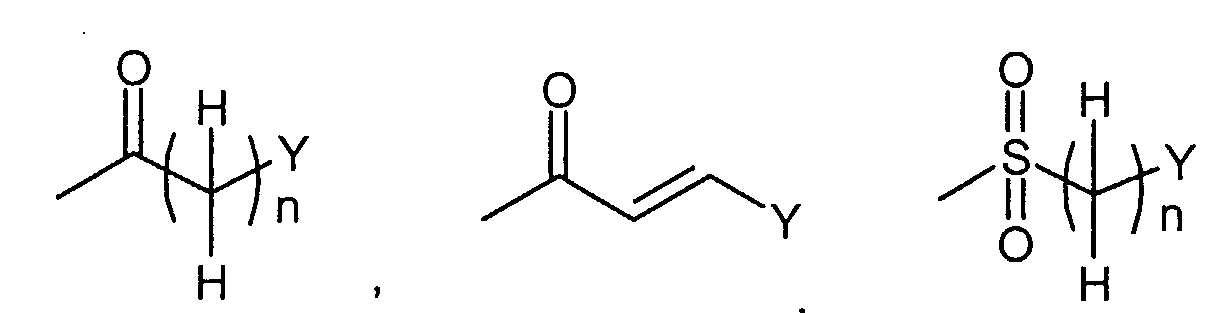
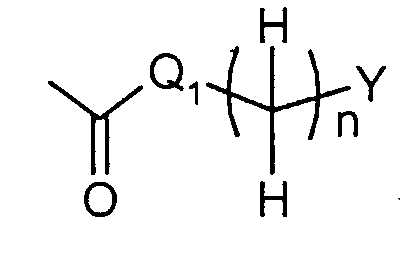
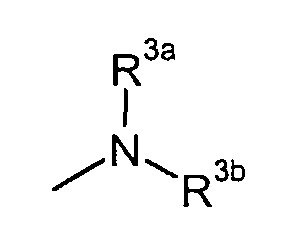
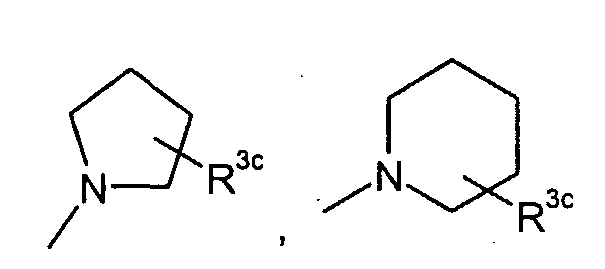
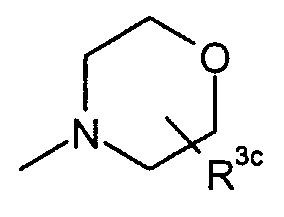
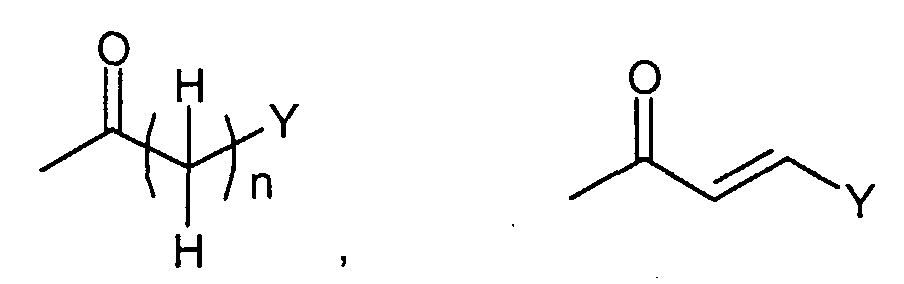
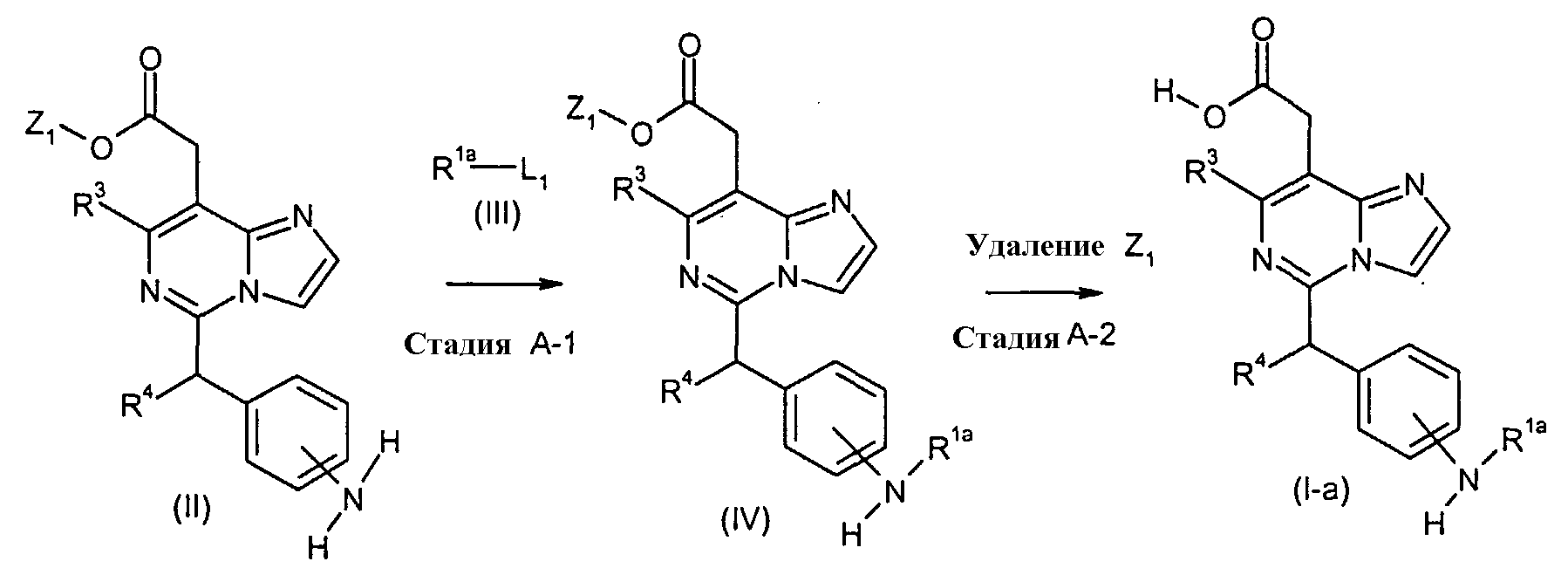
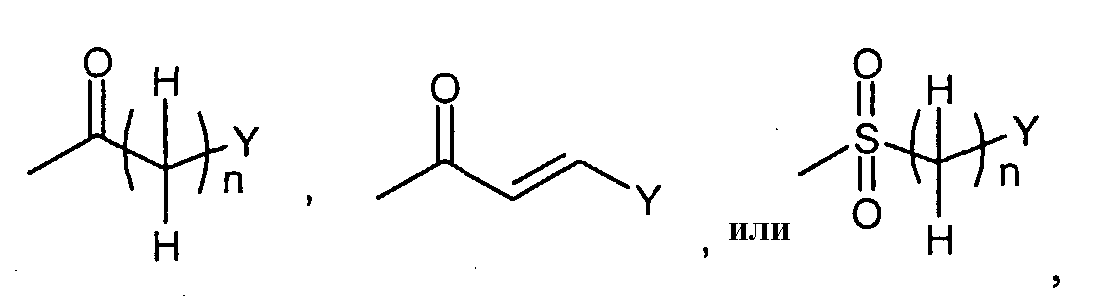
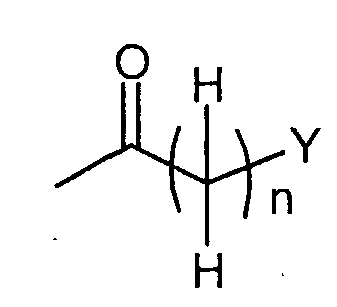 или
или 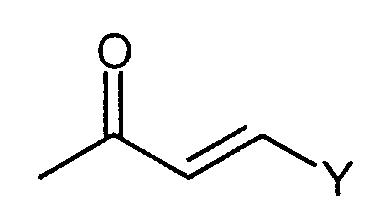
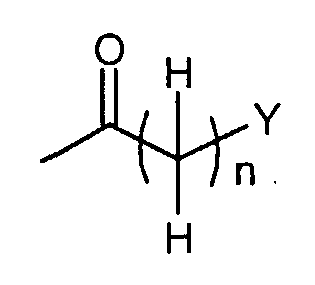 или
или 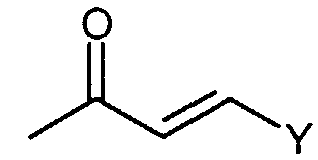
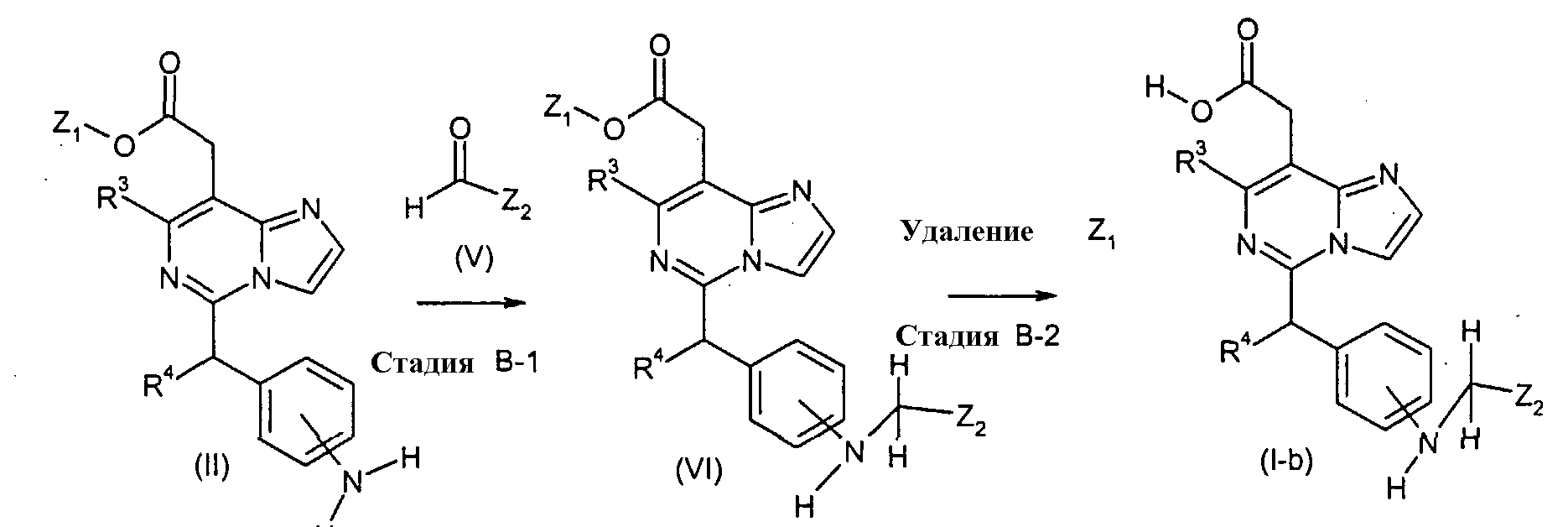
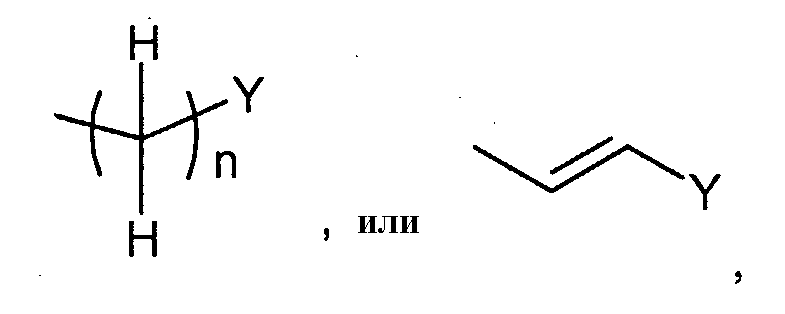
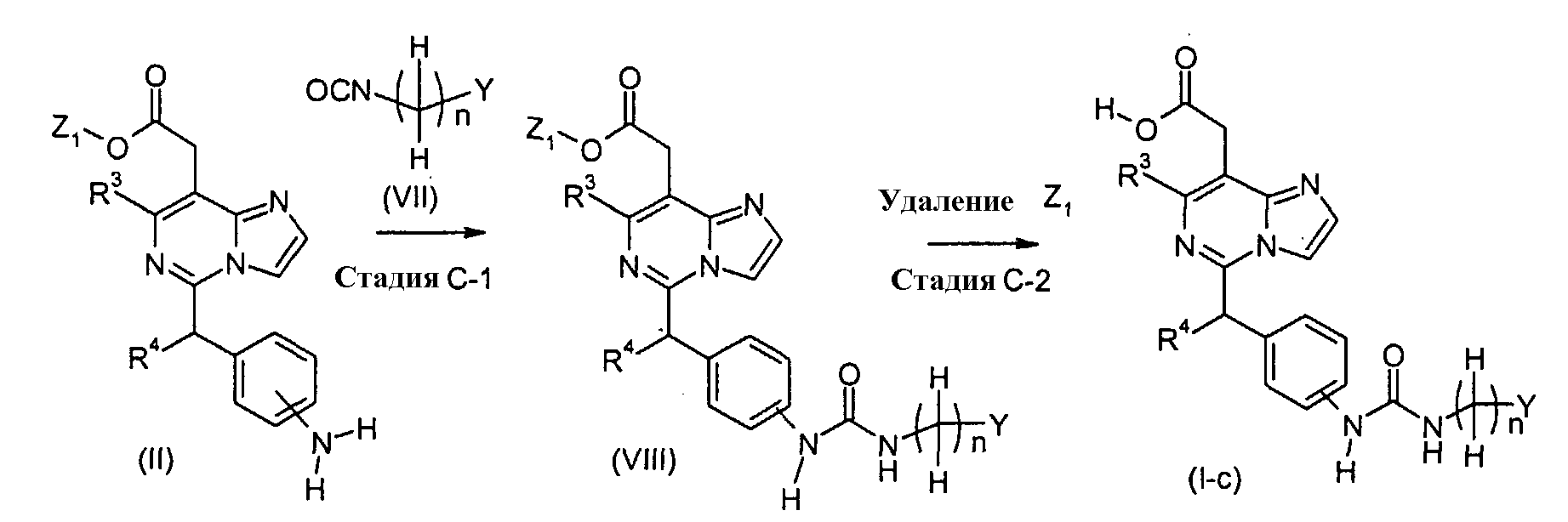
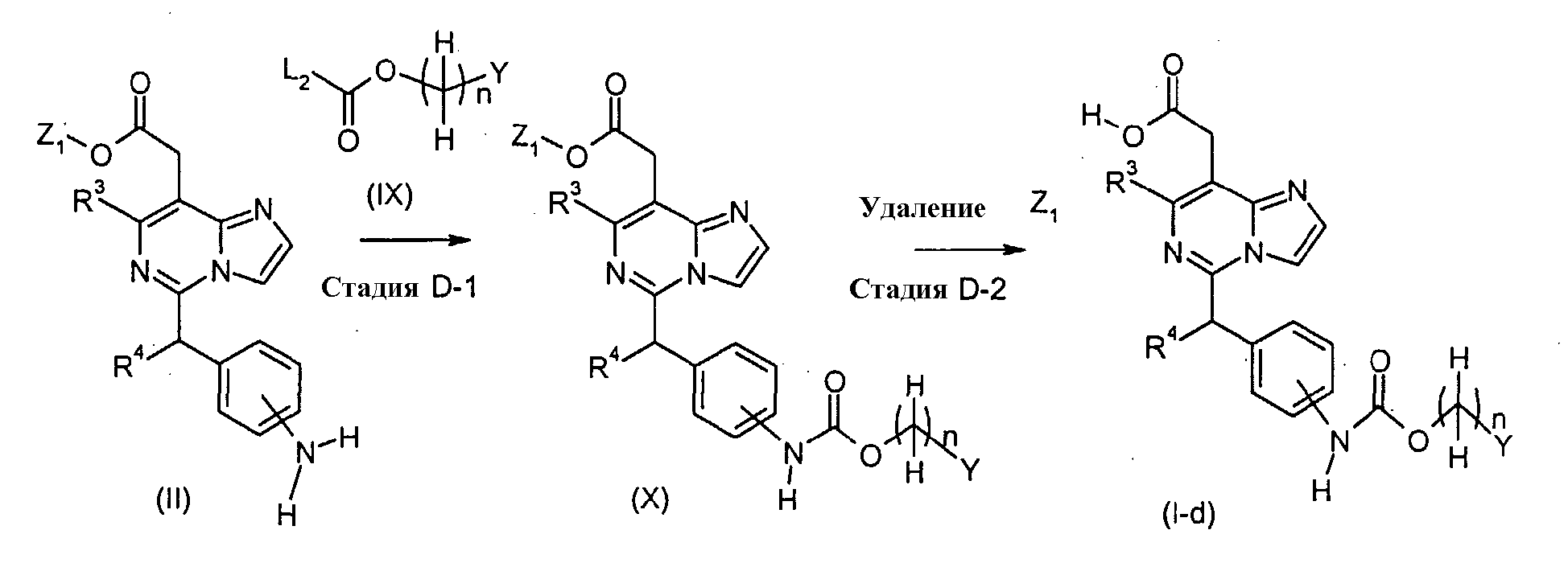
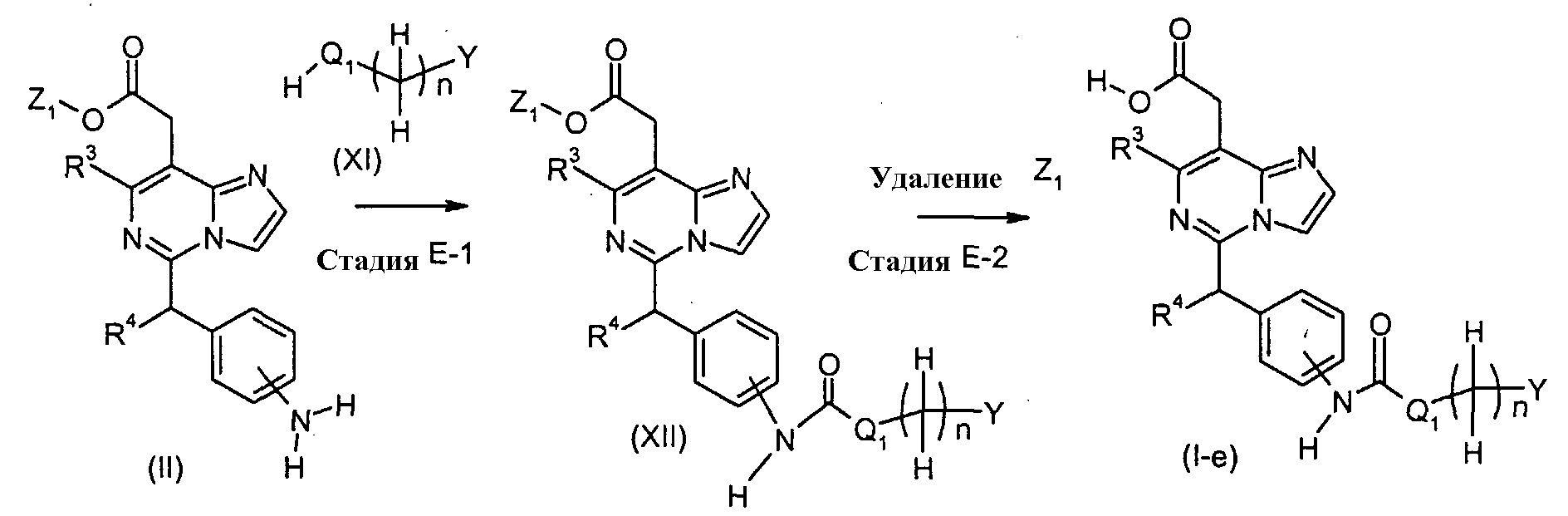
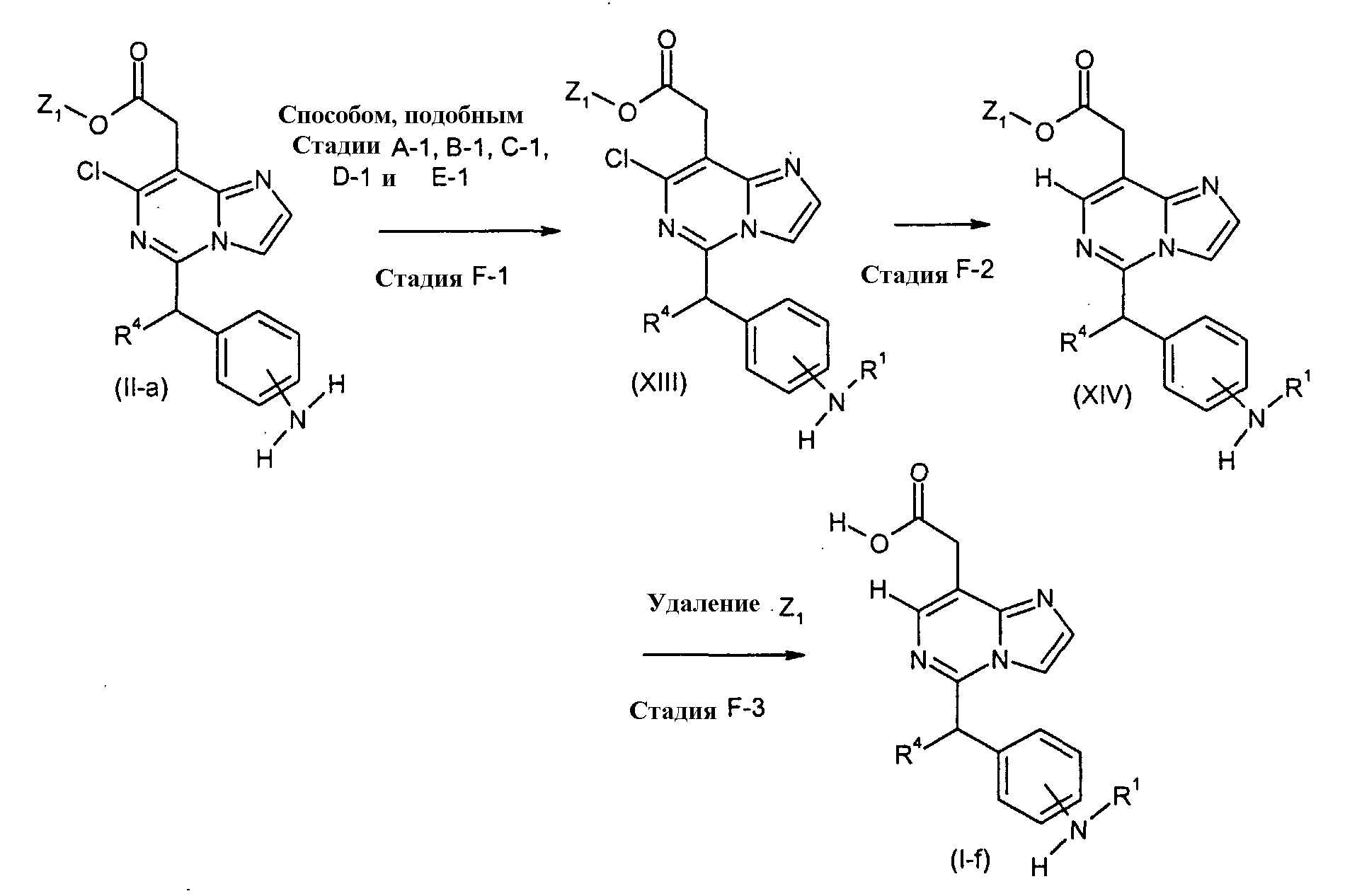
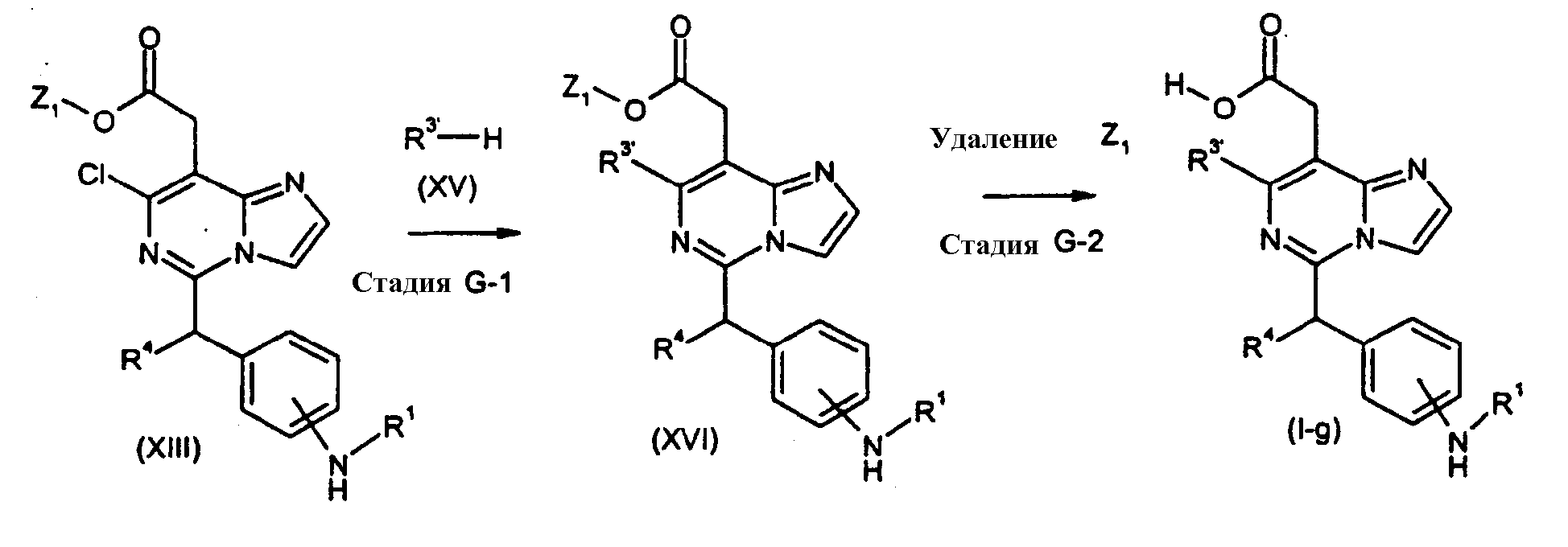
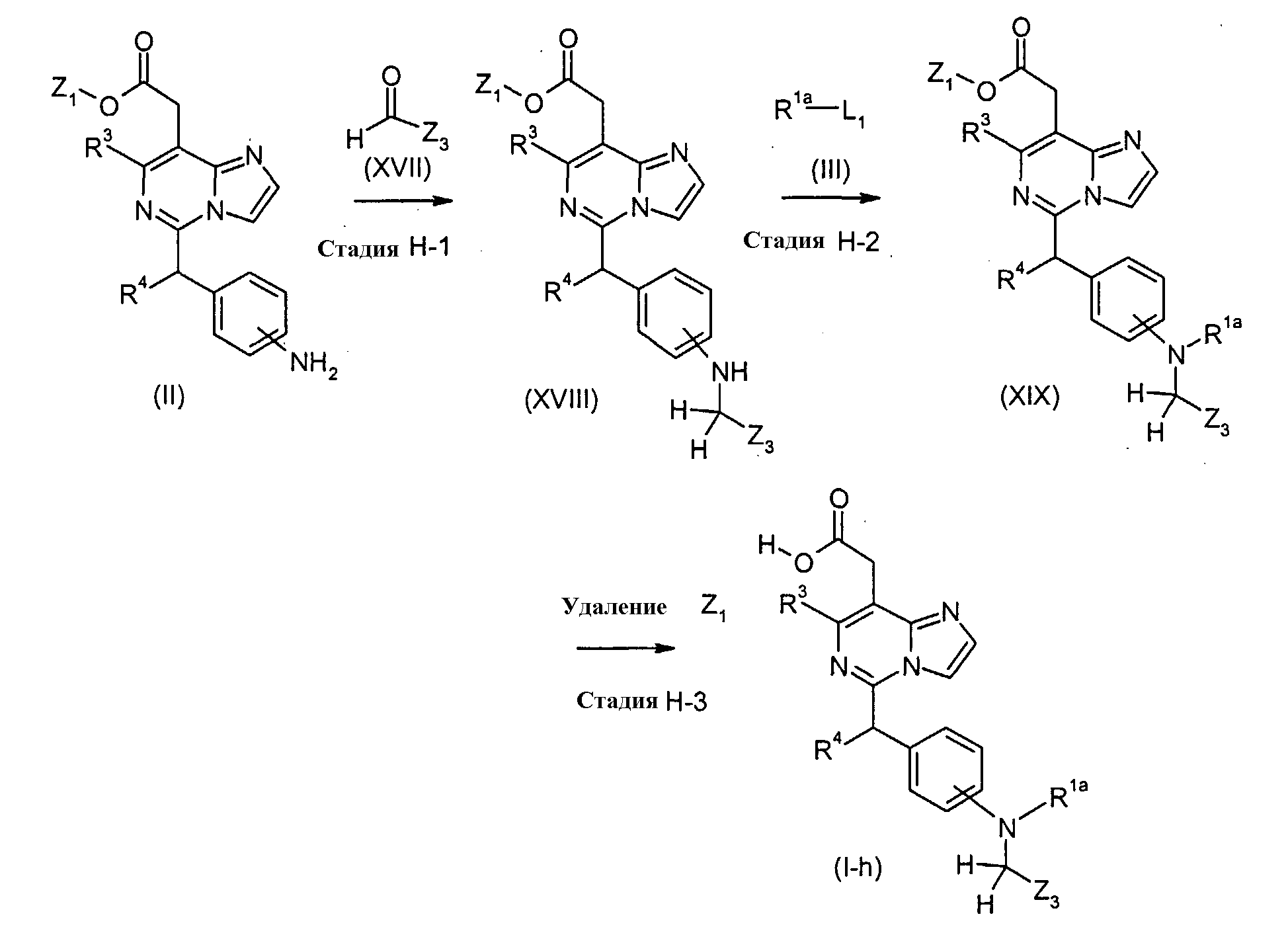
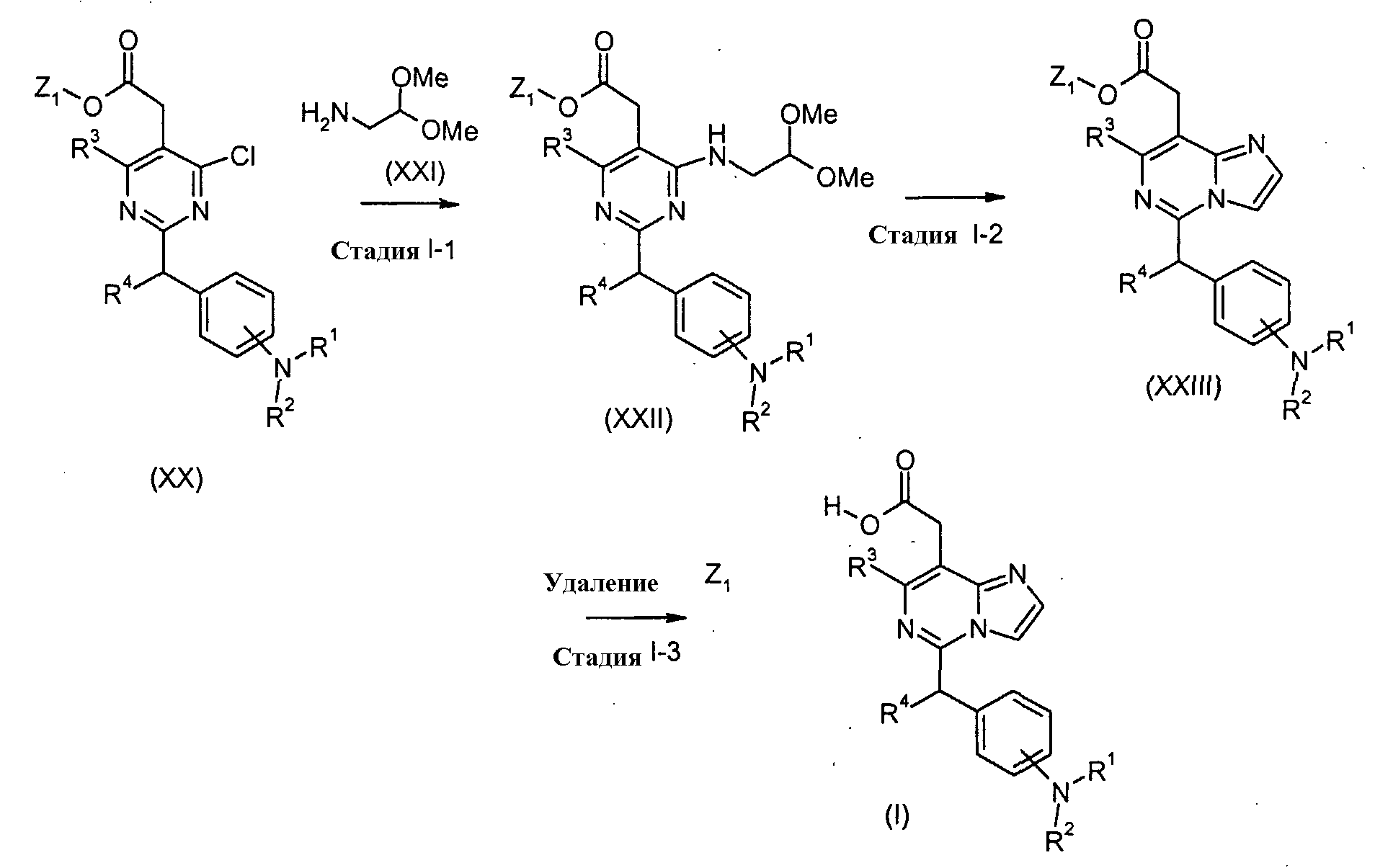
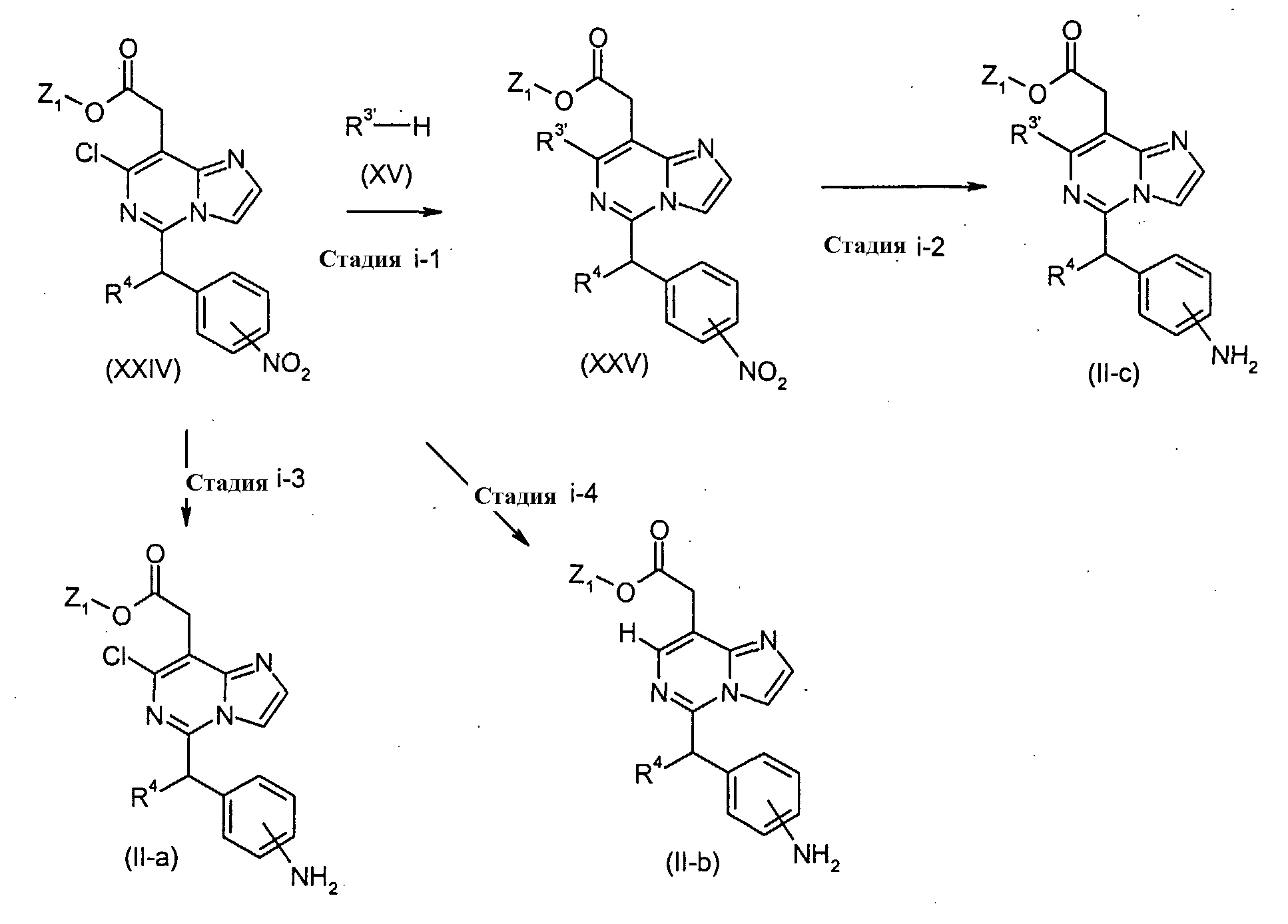
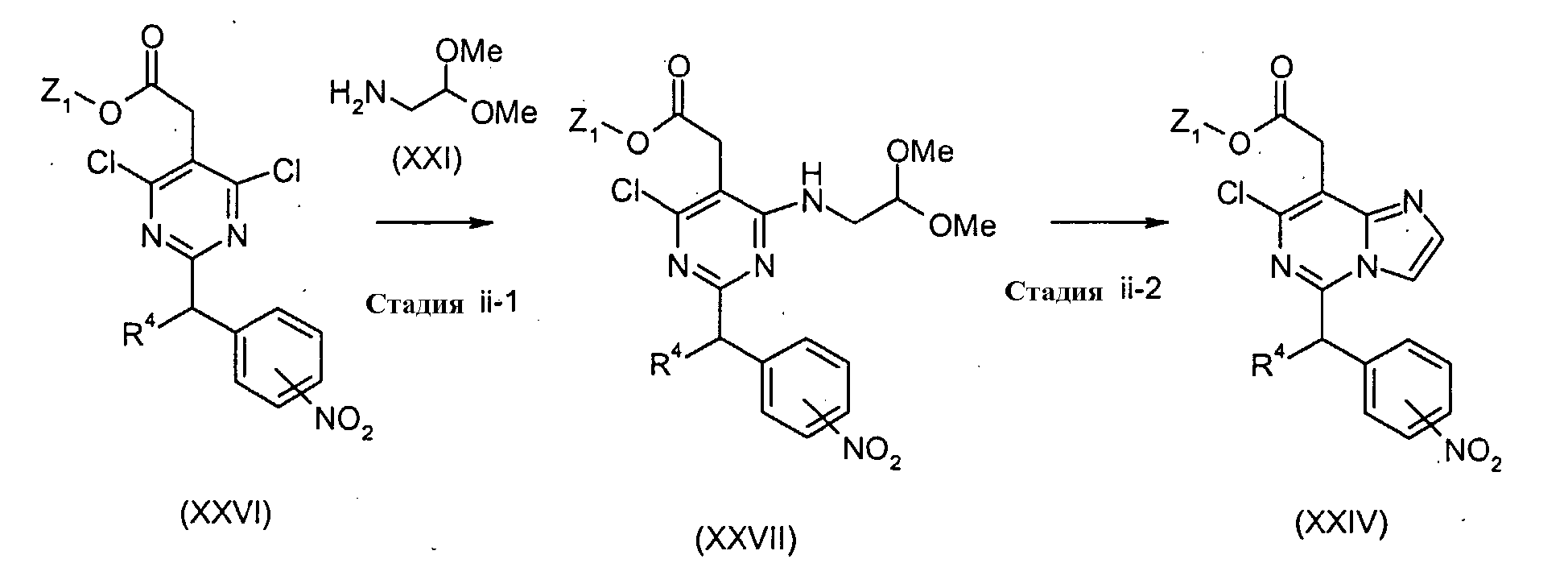
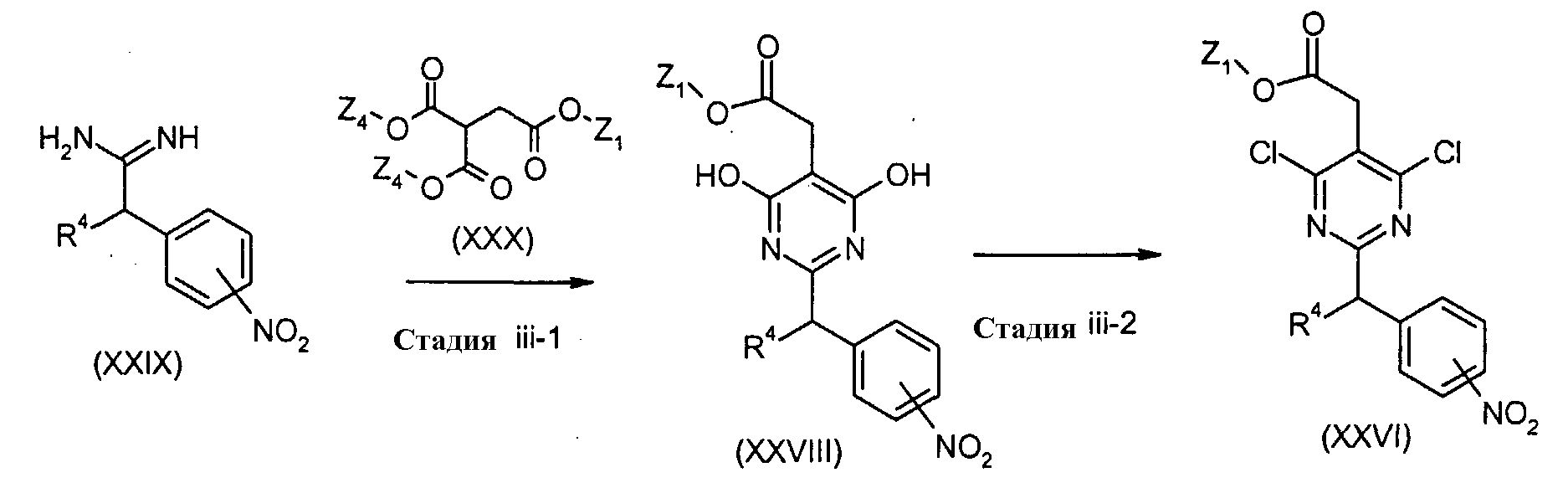
 (500 МГц для 1H). Химические сдвиги регистрируются в миллионных долях (м.д.), с тетраметилсиланом (TMS), в качестве внутреннего стандарта при ноле м.д. Константы связывания приводятся (J) в герцах, и сокращения с, д, т, кв, м, и шир. относятся к синглету, дублету, триплету, квартету, мультиплету и уширенному пику соответственно. Определения массы осуществляют посредством MAT95 (Finnigan MAT).
(500 МГц для 1H). Химические сдвиги регистрируются в миллионных долях (м.д.), с тетраметилсиланом (TMS), в качестве внутреннего стандарта при ноле м.д. Константы связывания приводятся (J) в герцах, и сокращения с, д, т, кв, м, и шир. относятся к синглету, дублету, триплету, квартету, мультиплету и уширенному пику соответственно. Определения массы осуществляют посредством MAT95 (Finnigan MAT).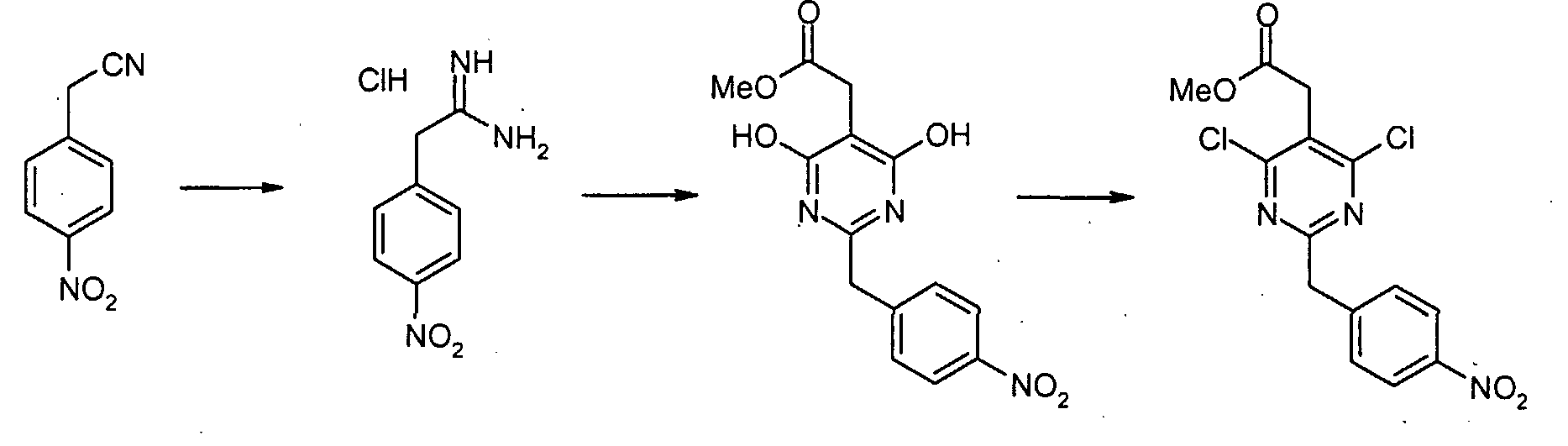
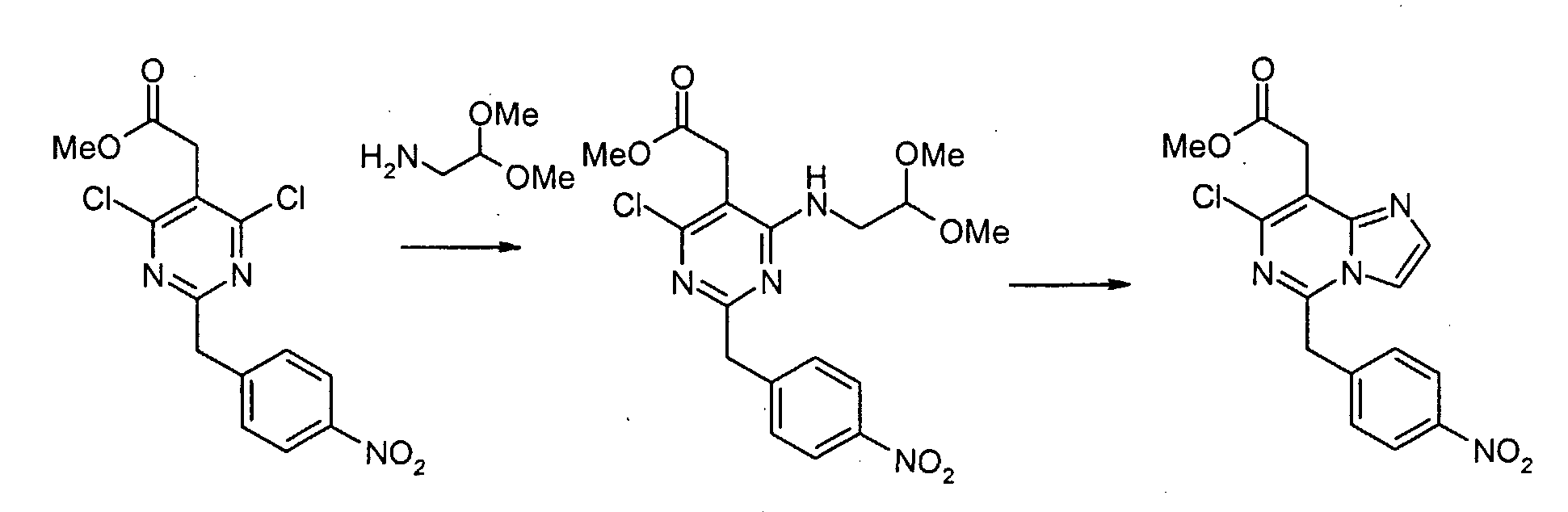
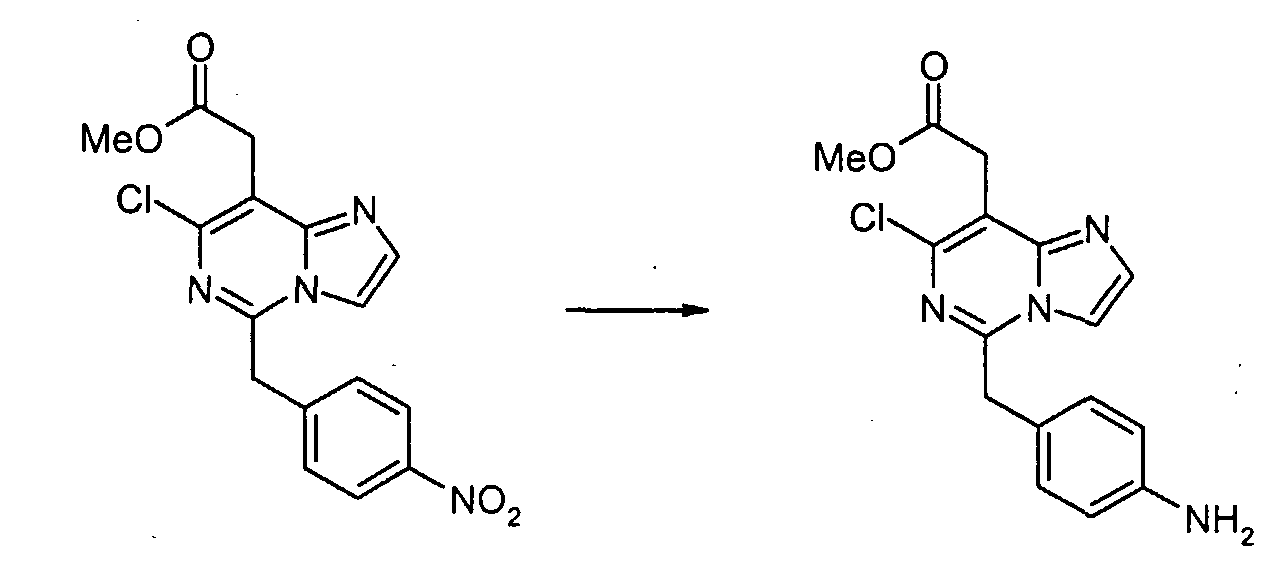
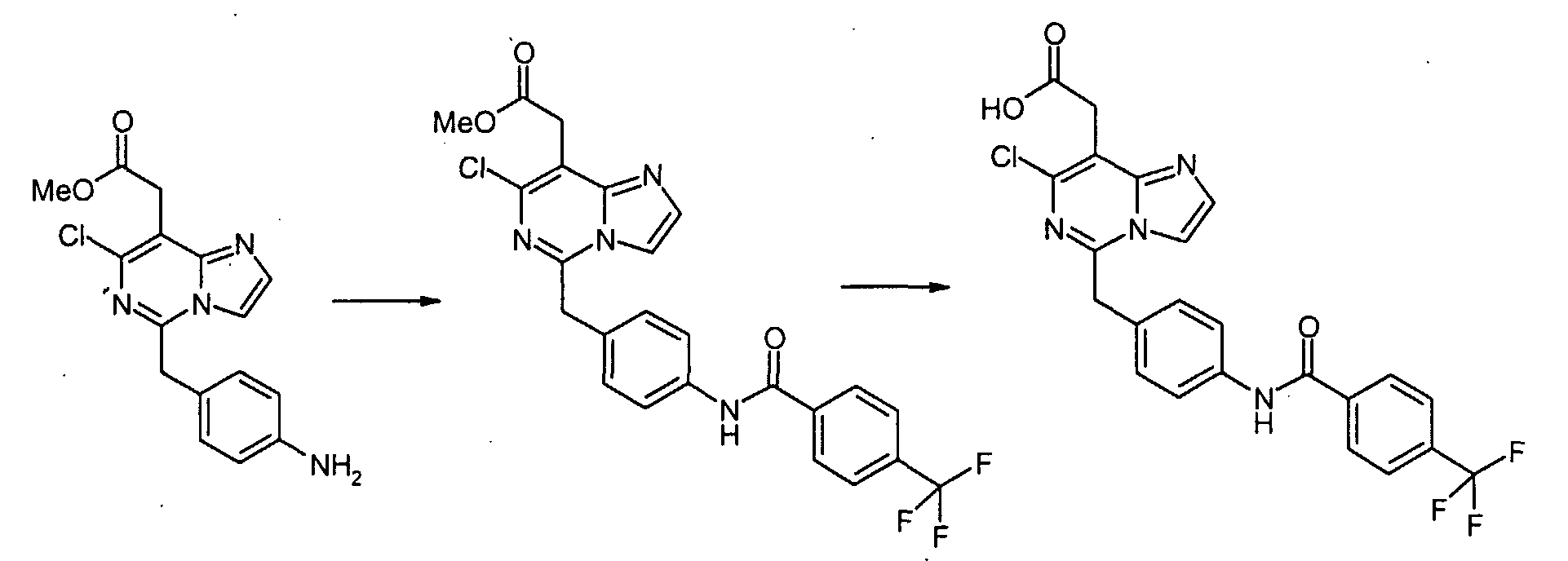
 3,96 (с, 2H), 4,52 (с, 2H), 7,39 (д, J=8 Гц, 2H), 7,73 (д, J=1,5 Гц, 1H), 7,74 (д, J=8 Гц, 2H), 7,91 (д, J=8 Гц, 2H), 8,13 (д, J=8 Гц, 2H), 8,19 (д, J=1,5 Гц, 1H), 10,48 (с, 1H), 12,71 (шир.с, 1H)
3,96 (с, 2H), 4,52 (с, 2H), 7,39 (д, J=8 Гц, 2H), 7,73 (д, J=1,5 Гц, 1H), 7,74 (д, J=8 Гц, 2H), 7,91 (д, J=8 Гц, 2H), 8,13 (д, J=8 Гц, 2H), 8,19 (д, J=1,5 Гц, 1H), 10,48 (с, 1H), 12,71 (шир.с, 1H)