Патент на изобретение №2373195
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) ПРОИЗВОДНЫЕ ИНДОЛАЛАНИНА
(57) Реферат:
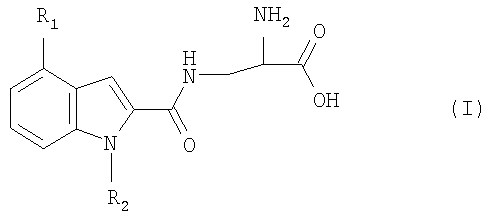
Изобретение относится к новым производным индолаланина формулы I:
где R1 означает фенил, нафтил, где фенил замещен одним или двумя атомами галогена, С1-6алкилами, C1-6алкокси или фенилС1-6алкилами; и R2 означает Н, С1-6алкил; в свободной форме, в форме гидрата или в форме фармацевтически приемлемой соли. Соединения I проявляют селективную агонистическую активность в отношении рецептора S1P4 по меньшей мере в 10 раз большую, чем селективность к одному или нескольким рецепторам S1P1, S1P2, S1P3 или S1P5, что позволяет использовать их в фармацевтической композиции. 4 н. и 3 з.п. ф-лы, 2 табл.
Настоящее изобретение относится к органическим соединениям, способу их получения и содержащим их фармацевтическим композициям. В одном варианте осуществления настоящее изобретение относится к соединению, которое является агонистом сфингозин-1-фостаного рецептора (S1P4), где соединение обладает селективностью к рецептору S1P4 по отношению к одному или нескольким рецепторам S1P1, S1P2, S1P3 или S1P5. Рецепторы S1P описаны, например, в WO 03/061567. Предпочтительно соединение проявляет селективность к рецептору S1P4 по отношению ко всем указанным выше рецепторам S1P. Соединение предпочтительно проявляет селективность по меньшей мере 10-кратную, более предпочтительно 20-кратную, наиболее предпочтительно 100-кратную к рецептору S1P4 по отношению к одному или нескольким указанным выше рецепторам S1P. Селективность может быть измерена определением соотношения ЕС50 соединения для рецептора S1P4 к EC50 соединения для рецепторов S1P1, S1P2, S1P3 или S1P5. Значения ЕС50 могут быть получены, например, в соответствии с анализом связывания с GTP В предпочтительном варианте осуществления настоящее изобретение относится к соединению формулы I:
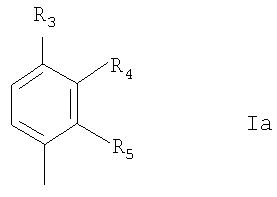
где R1 представляет собой фенил или нафтил, где фенил замещен одним или двумя атомами галогена, С1-6алкилами, C1-6алкокси или фенилС1-6алкилами; и R2 представляет собой водород или С1-6алкил; в свободной форме или в форме соли. Любая алкильная группа может быть линейной или разветвленной. C1-6алкил предпочтительно представляет собой С1-4алкил. С1-6алкокси предпочтительно представляет собой С1-4алкокси. Галоген может представлять собой F, Cl, Br или I. R1 предпочтительно представляет собой группу формулы Ia:
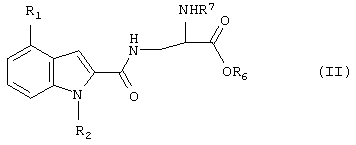
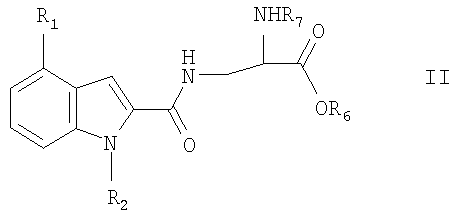
где один или два радикала из R3, R4 и R5 представляют собой водород; и один или два радикала R3, R4 и R5 представляют собой галоген, С1-6алкил, C1-6алкокси или фенилС1-6алкил; или R3 и R4, или R4 и R5, вместе с атомами углерода, к которым они присоединены, вместе образуют бензольное кольцо. В формуле I и Ia следующие значения предпочтительно представляют собой независимо, вместе или в любой комбинации или подкомбинации: а) один из R4 или R5 является отличным от водорода, более предпочтительно R5 является отличным от водорода и R4 представляет собой водород; б) R4 или R5 представляет собой бензил, этил, бутокси, пропил, изопропил или хлор, более предпочтительно R5 представляет собой одну из указанных выше групп и R4 представляет собой водород; в) R3 представляет собой водород или хлор, более предпочтительно водород; г) R2 представляет собой водород или метил. Соединения или формулы I также могут образовывать кислотные аддитивные соли с неорганическими или органическими кислотами, например хлористоводородной, бромистоводородной или малеиновой кислотой. Соединения формулы I также могут образовывать катионные соли, полученные из карбоксигруппы, в частности соли с щелочными или щелочноземельными металлами, например соли с натрием, калием, кальцием или магнием, и соли с аммонием, полученные из аммиака или органических аминов. Соединения формулы I содержат хиральный центр при атоме углерода с первичной аминогруппой. Соединение формулы I, следовательно, может существовать в двух энантиомерных формах. Следует понимать, что настоящее изобретение также включает рацемат формулы I и любую энантиомерную форму. Соединения формулы I также могут быть получены в форме их гидратов. Гидраты также составляют часть изобретения. Настоящее изобретение также относится к способу получения соединений формулы I, который включает снятие защитных групп с соединения формулы II
где R1 и R2 являются такими, как описано выше, R6 представляет собой С1-6алкил или бензил, R7 представляет собой защитную группу для аминогруппы, и и при необходимости превращение соединения формулы I в его соль. Способ может быть осуществлен с помощью обычных способов. Примерами подходящих защитных групп для аминогруппы являются, например, группы, описанные в книге “Protective Groups in Organic Synthesis”, T.W.Greene, J.Wiley & Sons NY, 2-ое Издание., глава 7, 1991, и в указанных там ссылках, например ацил, например трет-бутилоксикарбонил, бензилоксикарбонил, 9-флуоренилметоксикарбонил, трифторацетил, триметилсилилэтансульфонил и им подобные. Снятие защитных групп может осуществляться, например гидрогенолизом, когда R7 представляет собой бензилоксикарбонил и R6 представляет собой бензил. Реакцию затем можно осуществлять в органическом растворителе, таком как тетрагидрофуран, хлористый метилен или диоксан при комнатной температуре с помощью палладия на угле в качестве катализатора. Если R6 представляет собой С1-6алкил, способ обычно осуществляют в две стадии. На первой стадии с карбоксигруппы соединения формулы II могут быть удалены защитные группы мягким гидролизом, например с помощью водного раствора NaOH или основной ионообменной смолы в органическом растворителе, таком как тетрагидрофуран, диоксан, метанол или этанол, при комнатной температуре. На второй стадии с аминогруппы удаляют защитные группы, например с помощью йодтриметиллилана в метиленхлориде. Если R7 представляет собой бензилоксикарбонил, может использоваться гидрогенолиз. Двухстадийный способ обычно используют, когда один или два радикала R3-R5 представляют собой галоген. Необязательное образование соли может осуществляться обычным образом. Рацемическая смесь соединения формулы I может быть разделена известным способом, например с помощью оптически активной кислоты в качестве расщепляющего агента. Альтернативно, чистая энантиомерная форма может быть получена с помощью оптически активных исходных реагентов. Соединения формулы II, которые используются в качестве исходных реагентов, может быть получены, например реакцией соединения формулы III:
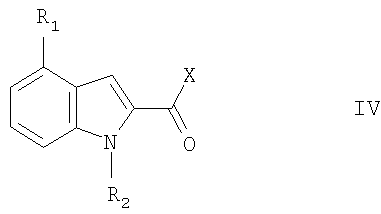
где R6 и R7 являются такими, как описано выше, с соединением формулы IV:
где R1 и R2 являются такими, как описано выше, и Х представляет собой уходящую группу. Реакция может осуществляться обычным способом. Например, реакция может проводиться в органическом растворителе, таком как метиленхлорид при температуре от -15° до 25°. Уходящая группа Х представляет собой, например галоген, особенно хлор или гидрокси. Обычно присутствует агент для связывания кислоты, например третичный амин, такой как пиридин. Несмотря на то, что получение исходных реагентов для указанных выше способов конкретно не описано, они могут быть получены аналогично методикам известных соединений или описанными здесь способами. В следующих примерах все температуры приведены в градусах Цельсия по стоградусной шкале и имеют значения без учета поправки. Значения [ Пример 1 3-(4-(2-Этилфенил)-2-карбоксамидоиндол)-D-аланин а) 4-(2-Этилфенил)индол-2-карбоновая кислота К смеси 0,28 г 4-броминдол-2-карбоновой кислоты и 0,069 г тетракистрифенилфосфинпалладия в 11 мл толуола и 2 мл 2М соды добавляли раствор 0,300 г 2-этилфенилборной кислоты в 3 мл этанола. Эту смесь кипятили с обратным холодильником в течение 16 ч, отфильтровывали и водную фазу подкисляли 2N HCl и экстрагировали этилацетатом. Концентрирование органической фазы приводило к получению продукта в виде коричневатого порошка, tпл 230-233°, достаточно чистого для следующей стадии. б) Метиловый эфир 3-(4-(2-этилфенил)-2-карбоксамидоиндол)-N(2)-Z-D-аланина К раствору 2,11 г 4-(2-этилфенил)индол-2-карбоновой кислоты в 32 мл пиридина добавляли 1,41 г карбонилдиимидазола. После прекращения выделения газа добавляли 2,29 г метил N(2)-Z-D-2,3-диаминопропионата и смесь перемешивали при комнатной температуре в течение 4 дней. После упаривания растворителя остаток экстрагировали смесью вода/этилацетат, органическую фазу высушивали, концентрировали и сырой продукт хроматографировали на силикагеле с помощью смеси изопропилацетат/толуол 4:1, получая желаемое соединение в виде желтого аморфного порошка. в) 3-(4-(2-Этилфенил)-2-карбоксамидоиндол)-D-аланин Смесь 2,03 г метилового эфира 3-(4-(2-этилфенил)-2-карбоксамидоиндол)-N(2)-Z-D-аланина, 4,1 мл 2N NaOH и 10 мл диоксана перемешивали при комнатной температуре в течение 2 ч. После обработки обычным способом полученную кислоту растворяли в 22 мл метиленхлорида и обрабатывали 1,03 мл йодтриметилсиланом при 0°С. После перемешивания в течение 50 мин смесь концентрировали досуха и остаток переносили в воду. Сырой продукт осаждался и его собирали фильтрацией, промывали водой и перекристаллизовывали из смеси вода/изопропанол при доведении рН до 6 добавлением 0,2N NaOH, приводя к получению указанного в заголовке соединения в виде белого порошка, tпл 258-265°. Соединения формулы I, где R1 и R2 являются такими, как показано в таблице 1, могут быть получены по одной из указанных выше методик, но используя соответствующие исходные реагенты.
Описанные здесь соединения, которые являются селективными агонистами рецептора S1P4 (далее указанные как соединения по изобретению), например соединения формулы I, в свободной форме или в форме фармацевтически приемлемой соли, проявляют ценные фармакологические свойства, например модулирующие свойства в отношении рециркуляции лимфоцитов, например как показано в тестах in vitro и in vivo, и, следовательно, подходят для терапии. В частности, соединения по изобретению являются полезными в качестве функциональных агонистов рецепторов S1P4 человека (EDG6), что показано в следующих испытаниях. Испытание для определения in vitro фармакологии соединений в отношении S1P4 а) Вектор экспрессии, кодирующий рецепторы S1P человека Вектор экспрессии для гена S1P4 человека (HSEDG4; GenBank, инвентарный номер AJ000479), соединенный с пептидной меткой с-myc на С-конце, получали как описано в статье Van Brooklyn и др., Blood, 2000, 95 (8), с.2624. Кодирующую ДНК для слитого белка S1P4-myc клонировали в векторе экспрессии pRc/CMV млекопитающего (Invitrogen), который имеет G418 резистентность для селекции стабильных клеток млекопитающего. Последовательности ДНК вставок S1P4 определяли в обеих цепочках. Никаких нуклеотидных различий, которые приводили бы к аминокислотным изменениям, по сравнению с данными GenBank для HSEDG4 обнаружено не было. S1P4-myc кДНК встраивали в вектор pRc/CMV с помощью HindIII/XbaI. Использовали следующие векторы. кДНК S1P1 (GenBank S1P5 человека (GI: 30171332 или GenBank б) Разработка стабильной клеточной линии СНО-К1, экспрессирующей S1P4 Для разработки стабильных клеточных линий, экспрессирующих с-myc-меченый рецептор S1P4, 5 мкг плазмиды pRc/CMV myc-S1P4 вырезали с помощью рестрикции эндонуклеазой PvuI, которая расщепляет плазмиду в одном месте. Выровненную плазмиду затем осаждали с помощью ацетата натрия с конечной концентрацией 0,3 М, рН 5,0 и 66% этанола. После центрифугирования и промывания остатки ДНК растворяли в 30 мкл воды. Для трансфекции СНО-K1 (АТСС номер: CCL-61) клетки помещали в среду MEM в) Сохранение клеточной культуры Парентеральные клетки СНО-K1 сохраняли в среде RPMI (Lifetechnologies) с 10% FBS и 10 мкг/мл гентамицина (Lifetechnologies). S1P4/myc трансфецированные СНО-К1 клетки, включая один отобранный в конце клон, хранили в альфа-среде MEM с 10% FBS, 10 мкг/мл гентамицина и 0,5 мг/мл G418 (Lifetechnologies). г) Определение уровней транскрипции S1P4 в стабильных клонах СНО Цельные РНК из клеток выделяли с помощью мининабора RNeasy (Qiagen). После разрушения клеток (выращенный в монослое) непосредственно в культуральных планшетах (на один 35 мм планшет использовали 350 мкл буфера, полученного в соответствии с протоколом Qiagen), лизат пипеткой наносили на вращающуюся колонку qiashredder и центрифугировали в течение 2 мин при 21’000 g. Добавляли 350 мкл 70% этанола в проточную смешанную ячейку, которой снабжена миниколонка RNeasy, и центрифугировали в течение 15 с при 10’000 g. Проточную ячейку разгружали, добавляли 700 мкл буфера RW1 (Qiagen) к RNeasy колонке и центрифугировали в течение 15 с для промывания колонки. Колонку затем промывали дважды добавлением 500 мкл буфера RPE (Qiagen) и центрифугировали в течение 15 с при 10000 g. РНК элюировали добавлением 30-50 мкл воды, не содержащей РНКазы, непосредственно в колонку с РНК-субстратом и центрифугировали в течение 1 мин при 10’000 g. РНК обратно транскрибировали с помощью набора Omniscript Qiagen (Qiagen). Около 2 мкг РНК смешивали с 2 мкл 10 х буфера, 2 мкл dNTP Mix, I мкл ингибитора РНКазы (10 единиц/мкл), 0,5 мкг случайного гексамера, 1 мкл обратной транскриптазы Omniscript и водой, не содержащей РНКазу, до конечного объема 20 мкл, и инкубировали при 37°С в течение 60 мин и дополнительно 30 мин при 42°С. Реакцию инактивировали в течение 5 мин при 93°С, охлаждали во льду и хранили при -20°С до использования. Для определения относительных уровней транскрипции S1P4 мРНК в выбранных клонах, использовали анализ PCR в реальном времени (полимеразная цепная реакция). Оптимальные концентрации праймера и пробы определяли как описано в User bulletin от РЕ (Perkin/Elmer) Biosystems с помощью S1P4 человека, содержащего плазмиду в качестве шаблона. Олигонуклеотиды S1P4 человека, а также их оптимальные концентрации для анализа PCR в реальном времени, являлись следующими (основываясь на HSEDG4; инвентарный номер GenBank AJ000479):
Первую молекулярную цепочку кДНК затем использовали для количественного анализа PCR на устройстве ABI7700 TaqMan (РЕ Biosystems). В качестве внутреннего контроля количества РНК, присутствующего в каждом образце, использовали 18S рибосомальную РНК реакцией многократной передачи S1P4 PCR с рибосомальной РНК PCR в той же пробирке, используя контрольные реагенты TaqMan рибосомальной РНК (РЕ Biosystems, P/N4308310, VIC-меченая проба). Хотя аналогичная эффективность двух независимых амплификации для S1P4 и рибосомальной РНК формально не определяли, полуколичественное определение уровней транскрипции S1P4 в различных клеточных клонах является достаточным, чтобы выбрать клеточные клоны для дальнейшей характеристики в функциональных анализах. д) Получение мембран Мембранные белки получали из СНО-K1 дикого типа и СНО клеточных клонов, экспрессирующих S1P4 человека. Для получения 10-30 мг мембранных белков клетки выращивали в одном большом культуральном планшете (500 см2) для клеточного клона до 80 и 90% слияния. Культуральную среду удаляли и клетки собирали на льду из планшета соскабливанием в 20 мл холодного 10 мМ HEPES (pH 7,5) с 0,1% бычьего сывороточного альбумина, не содержащего жирных кислота (БСА), и смесью ингибиторов протеазы (одна таблетка на 50 мл, Roche Diagnostics, Rotkreuz). Клетки центрифугировали при 750 g в течение 10 мин при 4°С и повторно суспендировали в 10 мл холодного мембранного буфера (20 мМ HEPES, pH 7,4; 100 мМ NaCl; 10 мМ MgCl2; 1 мМ EDTA; 0,1% БСА и смесь ингибиторов протеазы). Клеточную суспензию гомогенизировали во льду, используя гомогенизатор Polytron при 25000 об/мин с тремя интервалами по 20 секунд каждый. Гомогенизат центрифугировали при 26’900 g в течение 30 мин при 4°С и остаток мембранных белков повторно суспендировали при встряхивании в 2 мл холодного мембранного буфера. Концентрацию белка определяли с помощью анализа Bio Rad Protein и IgG человека в качестве стандарта. Объем суспензии мембранных белков доводили до конечной концентрации от 2 до 3 мг белка/мл. Раствор затем снова гомогенизировали (Polytron) во льду при 25000 об/мин в течение 20 с перед разделением на аликвоты в пробирки Эппендорфа объемом 0,8-1 мл. е) Измерение активности клеток, экспрессирующих hS1P4: е1) Анализ на связывание с GTP Выбор тестируемых клонов в анализе с GTP e2) Анализ мобилизации кальция Клетки СНО помещали в черные планшеты Costar (96 или 384-луночные, 50’000 клеток или 12,500 клеток, соответственно) в Определение in vitro специфичности и селективности агонистов S1P4. Соединения, указанные в данном описании, тестировали на специфичность, например на активность, которой они обладают, в родительских клеточных линиях до превращения с кДНК S1P4, и на селективность в отношении клеточных линий СНО, трансформированных S1P1, S1P2, S1P3 и S1P5. Выработку стабильных клеточных линий для других рецепторов S1P осуществляли как описано для S1P4, используя опубликованные последовательности кДНК человека, как описано выше. Предпочтительно отбирали соединения со стократной селективностью и специфичностью. Например, S1P4 специфичные и селективные соединения могут иметь значения EC50 (очевидные ЕС50), измеренные с S1P4, которые в 10 раз, предпочтительно в 100 раз ниже, чем значения, измеренные с клетками СНО дикого типа или с клетками, экспрессирующими любой из других четырех рецепторов S1P. Например, если значение EC50 200 нМ измерено для S1P4, значения ЕС50 в клетках СНО или в клетках СНО, экспрессирующих S1P 1, 2, 3, 5, предпочтительно составляют Значения ЕС50 (в мкМ) для соединений примера 2 в клетках СНО или в клетках СНО, экспрессирующих различные рецепторы S1P, показаны в таблице 2 далее.
Соединения по изобретению являются, следовательно, полезными для лечения и/или профилактики заболеваний или нарушений, опосредованных взаимодействиями лимфоцитов; например при трансплантации, таких как острое или хроническое отторжение алло- или ксенотрансплантатов клеток, тканей или органов, или задержка функции трансплантата, реакция хозяина против трансплантата, аутоиммунные заболевания, например ревматоидный артрит, системная красная волчанка, тиреоидит Хашимото, рассеянный склероз, бульбоспинальный паралич, диабеты типа I или II и связанные с ними заболевания, васкулит, пернициозная анемия, синдром Сьегрена, ювеит, псориаз, офтальмопатия Гравса, круговая алопеция и другие, аллергические заболевания, например аллергическая астма, атопический дерматит, аллергический ринит/коньюктивит, аллергический контактный дерматит, воспалительные заболевания, необязательно с сопутствующими аберрантными реакциями, например воспалительное кишечное заболевание, болезнь Крона или язвенный колит, инфекционно-аллергическая астма, воспалительная травма легкого, воспалительная травма печени, воспалительная гломерулярная травма, атеросклероз, остеоартрит, раздражающий контактный дерматит и другие экзематозные дерматиты, себорейный дерматит, кожные проявления иммунологически-опосредованных заболеваний, воспалительное заболевание глаз, кератоконьюктивит, миокардит или гепатит, травма при ишемии/реперфузии, например инфаркт миокарды, инсульт, кишечная ишемия, почечная недостаточность или геморрагический шок, травматический шок, другие, рак, например Т-клеточная лимфома или Т-клеточная лейкемия, инфекционные заболевания, например токсический шок (например, вызванный суперантигеном), септический шок, респираторный дистрессовый синдром у взрослых или вирусные инфекции, например СПИД, вирусный гепатит или хроническая бактериальная инфекция. Примеры трансплантатов клеток, тканей или целых органов включают, например, панкреатические островки, стволовые клетки, костный мозг, ткани роговой оболочки, нейронная ткань, сердце, легкое, комбинированное сердце-легкое, почка, печень, кишечник, поджелудочная железа, трахея или пищевод. Для указанных выше применений, необходимая дозировка будет конечно зависеть от способа введения, конкретного излечиваемого состояния и желаемого эффекта. Обычно, удовлетворительные результаты получают при систематическом введении суточных дозировок от около 0,03 до 2,5 мг/кг веса тела. Показанная суточная дозировка для более крупных млекопитающих, например людей, находится в области от около 0,5 мг до около 100 мг, обычно вводимая, например, отдельными дозами до четырех раз в день или в форме замедленного высвобождения. Подходящие единичные дозированные формы для перорального введения включают от около 1 до 50 мг активного ингредиента. Соединения по изобретению могут вводиться любым обычным способом, в частности энтерально, например перорально, например в форме таблеток или капсул, или парентерально, например в форме инъекционных растворов или суспензий, местно, например в форме лосьонов, гелей, мазей или кремов, или в форме назального или суппозиторного введения. Фармацевтические композиции, включающие соединение по изобретению в свободной форме или в форме фармацевтически приемлемой соли вместе с по крайней мере одним фармацевтически приемлемым носителем или разбавителем могут быть получены обычным способом смешения с фармацевтически приемлемым носителем или разбавителем. Соединения по изобретению могут вводиться в свободной форме или в форме фармацевтически приемлемой соли, например, как показано выше. Такие соли могут быть получены обычным способом и проявляют ту же активность, как и свободные соединения. В соответствии с вышеизложенным настоящее изобретение также относится к: 1,1) способу профилактики или лечения заболеваний или болезней, опосредованных лимфоцитами, например, таких, которые описаны выше, у субъекта, нуждающегося в таком лечении, который включает введение указанному субъекту эффективного количества соединения по изобретению, например, соединения формулы I или его фармацевтически приемлемой соли; 1,2) способу профилактики или лечения острого или хронического отторжения трансплантата или опосредованных Т-клетками воспалительных или аутоиммунных заболеваний, например указанных выше, у субъекта, нуждающегося в таком лечении, который включает введение указанному субъекту эффективного количества соединения по изобретению, например соединения формулы I, или его фармацевтически приемлемой соли; 2) соединению по изобретению, например соединению формулы I, в свободной форме или в форме фармацевтически приемлемой соли, для применения в качестве лекарственного средства, например для любого из способов, описанных выше в 1,1 или 1,2; 3) фармацевтической композиции, например для применения в любом из способов 1,1 или 1,2, включающей соединение по изобретению, например соединение формулы I, в свободной форме или в форме фармацевтически приемлемой соли, вместе с фармацевтически приемлемым разбавителем или носителем. Соединения по изобретению, например соединение формулы I, может вводиться в качестве единственного активного ингредиента или в комбинации, например в качестве адъюванта, с другими лекарственными средствами, например иммуносупрессорными или иммуномодулирующими агентами или другими противовоспалительными агентами, например для лечения или профилактики острого или хронического отторжения алло- или ксенотрансплантата или воспалительных или аутоиммунных заболеваний, или химиотерапевтическими агентами, например антипролиферативным агентом злокачественных клеток. Например, соединения по изобретению могут использоваться в комбинации с ингибитором кальциневрина, например циклоспорином А или FK 506; ингибитором mTOR, например рапамицином, 40-O-(2-гидроксиэтил)-рапамицином, CCI779 или АВТ578; аскомицином, обладающим иммуносупрессорными свойствами, например АВТ-281, ASM981 и т.д.; кортикостероидами; циклофосфамидом; азатиопреном; метотрексатом; лефлуномидом; мизорибином; микофенольной кислотой; микофенолятом мофетила; 15-дезоксиспергуалином или его иммуносупрессорными гомологами, аналогами или производными; иммуносупрессорными моноклональными антителами, например, моноклональными антителами для рецепторов лейкоцитов, например, МНС, CD2, CD3, CD4, CD7, CD8, CD25, CD28, CD40, CD45, CD58, CD80, CD86 или их лигандами; другими иммуномодуляторными соединениями, например рекомбинантной связывающей молекулой, имеющей по крайней мере часть внеклеточного домена CTLA4 или его мутанта, например по меньшей мере внеклеточную часть CTLA4 или его мутанта, связанного с не-CTLA4 белковой последовательностью, например CTLA4Ig (например, известная как АТСС 68629) или его мутанта, например LEA29Y; ингибиторами молекулы адгезии, например антагонистами LFA-1, антагонистами ICAM-1 или -3, антагонистами VCAM-4 или антагонистами VLA-4; или химиотерапевтическими агентами, например паклитакселем, гемцитабином, цисплатином, доксорубицином или 5-фторурацилом. В случаях, когда соединения по изобретению, например соединение формулы I, вводят в комбинации с другим иммуносупрессором/иммуномодулятором, противовоспалительным или химиотерапевтическим агентом, дозировки совместно вводимого иммунодепрессанта, иммуномодулятора, противовоспалительного или химиотерапевтического соединения будут конечно зависеть от типа совместно применяемого лекарственного средства, например, является ли он стероидом или ингибитором кальциневрина, от специфичности применяемого лекарственного средства, от состояния заболевания и так далее. В соответствии с вышеизложенным, настоящее изобретение относится к другому варианту осуществления: 4) способу, как описано выше, включающему совместное введение, например совместно или последовательно, терапевтически эффективного нетоксичного количества соединения по изобретению, например соединения формулы I, и по меньшей мере второго лекарственного вещества, например иммунодепрессанта, иммуномодулятора, противовоспалительного или химиотерапевтического лекарственного средства, например, описанного выше; 5) фармацевтической комбинации, например набору, включающему а) первый агент, который представляет собой соединение по изобретению, например соединение формулы I, как описано здесь, в свободной форме или в форме фармацевтически приемлемой соли, и б) по крайней мере один совместный агент, например иммунодепрессант, иммуномодулятор, противовоспалительное или химиотерапевтическое лекарственное средство. Набор может включать инструкции по его применению. Используемые здесь термины “совместное введение” или “объединенное введение” или им подобные подразумевают введение выбранных терапевтических агентов отдельному пациенту, и включают лечебные режимы, в которых агенты вводятся необязательно одним способом введения или в одно и то же время. Термин “фармацевтическая комбинация”, как здесь используется, обозначает продукт, который приводит к смешению или объединению более одного активного ингредиента и включает фиксированные и нефиксированные комбинации активных ингредиентов. Термин “фиксированная комбинация” обозначает, что активные ингредиенты, например соединение по изобретению и сопутствующий агент, оба вводятся пациенту одновременно в форме одного целого или в одной дозе. Термин “нефиксированная комбинация” обозначает, что активные ингредиенты, например соединение по изобретению и сопутствующий агент, оба вводятся пациенту в виде отдельных единиц совместно, одновременно или последовательно в неограниченных временных интервалах, где такое введение относится к терапевтически эффективным уровням двух соединений в организме пациента. Последнее также применимо к множественной терапии, например для введения 3 или более активных ингредиентов.
Формула изобретения
1. Соединение формулы I 2. Соединение по п.1, которое выбрано из 3. Соединение по п.2, которое представляет собой 3-(4-(2-этилфенил)-2-карбоксамидоиндол)-D-аланин, в свободной форме или в форме фармацевтически приемлемой соли. 4. Соединение по любому одному из пп.1-3 в свободной форме или в форме фармацевтически приемлемой соли проявляющее селективную агонистическую активность в отношении рецептора S1P4. 5. Фармацевтическая композиция, проявляющая селективную агонистическую активность в отношении рецептора S1P4, включающая соединение как определено в любом одном из пп.1-3 в свободной форме или в форме фармацевтически приемлемой соли вместе с фармацевтически приемлемым разбавителем или носителем. 6. Применение соединения по любому из пп.1-3 в свободной форме или в форме фармацевтически приемлемой соли, или фармацевтической композиции по п.5 для изготовления лекарственного средства для лечения или профилактики заболеваний или нарушений, опосредованных лимфоцитами, острого или хронического отторжения трансплантата, опосредованных Т-клетками воспалительных или аутоиммунных заболеваний, диабетов, аллергических заболеваний, миокардита, гепатита, травмы ишемии/реперфузии, почечной недостаточности, геморрагического шока, травматического шока, рака или инфекционных заболеваний, причем соединение или его фармацевтически приемлемую соль используют в количестве, эффективном для проявления селективной агонистической активности в отношении рецептора S1P4. 7. Способ получения соединения по п.1, который включает снятие защитных групп с соединения формулы II
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 21, p.2793-2798, 1997. – J.Biochemistry, JAPANESE Biochemical society, v.131,
21, p.2793-2798, 1997. – J.Biochemistry, JAPANESE Biochemical society, v.131, 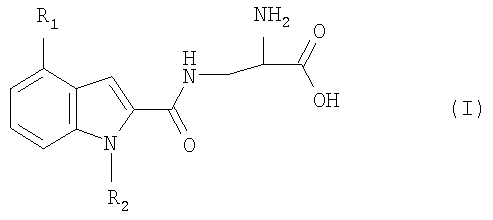
 35S или анализом мобилизации кальция, описанными далее. В другом предпочтительном варианте осуществления соединение также обладает значения ЕС50 для рецептора S1P4 1 мкМ или менее в анализе связывания с GTP
35S или анализом мобилизации кальция, описанными далее. В другом предпочтительном варианте осуществления соединение также обладает значения ЕС50 для рецептора S1P4 1 мкМ или менее в анализе связывания с GTP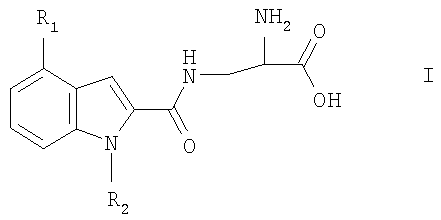
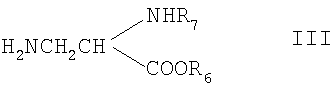
 ]D
]D 20 также приведены без учета поправки.
20 также приведены без учета поправки. , инвентарный номер М31210) и S1P3 (GenBank
, инвентарный номер М31210) и S1P3 (GenBank -лучи GTP
-лучи GTP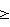 20 мкМ. Соединение примера 1 имеет значение ЕС50 для S1P4 в этом анализе 432 нМ.
20 мкМ. Соединение примера 1 имеет значение ЕС50 для S1P4 в этом анализе 432 нМ.