Патент на изобретение №2163910
|
||||||||||||||||||||||||||
(54) АНТАГОНИСТЫ РИЛИЗИНГ-ФАКТОРА ЛЮТЕИНИЗИРУЮЩЕГО ГОРМОНА (ЛГ-РФ) (ВАРИАНТЫ), СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ПРИГОТОВЛЕНИЯ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
(57) Реферат: Изобретение относится к новым антагонистам ЛГ-РФ, прежде всего к пептидомиметикам и пептидам с модифицированной боковой цепью, формулы V, где D-Xxx, R1 – R8 охарактеризованы в описании, и их солям с фармацевтически приемлемыми кислотами, а также в нем описывается их терапевтическое применение в качестве аналога рилизинг-фактора лютеинизирующего гормона (ЛГ-РФ), обладающего высокой антагонистической активностью и не обнаруживающего нежелательных побочных действий, в особенности не стимулирующего отеков. Описывается также способ получения соединения формулы V путем твердофазного метода или частично твердофазного метода и получение терапевтического состава, обладающего высоким сродством к рецептору рилизинг-фактора лютеинизирующего гормона, включающего в качестве активного вещества соединения по пп. 1 – 4. Также описывается способ приготовления лекарственных препаратов, содержащих соединения по пп.1 – 9, характеризующийся тем, что данные соединения смешивают с вспомогательными веществами. 6 с. и 11 з.п.ф-лы, 7 табл., 6 ил. Ac-D-Nal(2)1-D-(pCl)Phe2-D-Phe(3)3-Ser4-Tyr5-D-Xxx6-heu7-Arg8-Pro9-D-Ala10-NH2 (V). Изобретение относится к новым антагонистам ЛГ-РФ, прежде всего к пептидомиметикам и пептидам с модифицированной боковой цепью и их солям с фармацевтически допустимыми кислотами, а также к способам синтеза антагонистов ЛГ-РФ и их солей. Заявленные согласно изобретению пептиды являются аналогами рилизинг-фактора лютеинизирующего гормона (ЛГ-РФ), который имеет следующую структуру: p-Glu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2, [ЛГ-РФ, гонадорелин]. Исследования потенциальных селективных антагонистов декапептидов ЛГ-РФ проводятся уже более 20 лет [M.Karten und J.E.Rivier, Endocrine Reviews 7, 44-66 (1986)]. Повышенный интерес к таким антагонистам связан с потребностью в этих препаратах в ряде областей медицины: эндокринологии, гинекологии, контрацепции и онкологии. Было синтезировано большое количество соединений, представляющих собой потенциальные антагонисты ЛГ-РФ. Самый большой интерес среди синтезированных к настоящему времени соединений вызывают соединения с модифицированной структурой ЛГ-РФ. Первая серия потенциальных антагонистов была получена путем введения эфиров ароматических аминокислот в положения 1, 2, 3 и 6 или 2, 3 и 6. Общепринятым способом описания таких модифицированных аналогов является следующий: прежде всего указывают аминокислотный остаток, которым заменяют имеющийся остаток в ЛГ-РФ, причем цифрами в надстрочном индексе указывают номер позиции аминокислотного остатка, в которой произошла замена. Далее, при описании аналогов, в структуре которых были произведены замены, будет использоваться сокращение “LH-RH” (ЛГ-РФ). Известными антагонистами являются: [Ac-D-Phe(4-Cl)1,2, D-Trp3,6] LH-RH (D.H.Coy et al., In: Gross, E. and Meienhofer, J. (Eds) Peptides: Proceedings of the 6th American Peptid Symposium, S. 775-779, Pierce Chem.Co., Rockville III. (1979); [Ac-Pro1, D-Phe(4-Cl)2, D-Nal(2)3,6] LH-RH (US-Patent Nr.4.419.347) и [Ac-Pro1, D-Phe(4-Cl)2, D-Trp3,6] LH-RH (J.L.Pineda, et al., J.Clin. Endocrinol.Metab. 56, 420, 1983). Позднее с целью повышения растворимости антагонистов в воде было осуществлено введение остатков основных аминокислот, например, D-Arg в положение 6. Например, [Ac-D-Phe(4-Cl)1,2, D-Trp3, D-Arg6, D-Ala10] LH-RH (ORG-30276) (D. H.Coy, et al., Endocrinology 100, 1445, 1982) и [Ac-D-Nal(2)1, D-Phe(4-F)2, D-Trp3, D-Arg6] LH-RH (ORF 18260) (J.E. Rivier et al., in: Vickery B.H. Nestor, Jr.J.J., Hafez, E.S.E (Eds). LHRH and its Analogs, S.11-22 MTP Press, Lancaster, UK 1984). Такие аналоги характеризуются не только ожидаемой повышенной растворимостью в воде, но и повышенной антагонистической активностью. Однако, эти в высшей степени гидрофильные аналоги, содержащие D-Arg и другие основные боковые цепи в положении 6, вызывают временные отеки лица и конечностей при подкожном введении крысам в дозе 1,25 или 1,5 мг/кг (F.Schmidt, et al., Contraception 29, 283, 1984: J.E. Morgan, et al., Int.Archs.Allergy Appl. lmmun. 80, 70 (1986). Другие потенциальные антагонисты ЛГ-РФ описаны в заявках WO 92/19651, WO 94/19370, WO 92/17025, WO 94/14841, WO 94/13313, US-A 5,300,492, US-A 5,140,009 и EP 0 413 209 А1. Из-за стимуляции отека, обнаруженной при испытании некоторых из указанных антагонистов на крысах, возникли сомнения в безопасности применения этих соединений по отношению к человеку и в связи с этим задерживалось их внедрение в клиническую практику в качестве лекарственных средств.Таким образом существует необходимость в получении антагонистических пептидов, которые не обладают побочным действием. Согласно изобретению эту проблему решают с использованием соединений общей формулы I 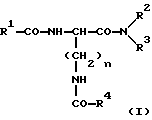 где n = 4, R1– алкилоксигруппа, арильная группа, аралкильная группа, гетероаралкильная группа, аралкилокси группа или гетероаралкилокси группа, в каждом конкретном случае незамещенная или замещенная, R2 и R3– независимо друг от друга в зависимости от необходимости означают атом водорода, алкильную группу, аралкильную группу или гетероаралкильную группу, в зависимости от необходимости незамещенную или замещенную, причем замещение, в свою очередь, может включать арильную группу или гетероарильную группу, или -NR2R3 представляет собой аминокислотный остаток, причем R4 – группа формулы (II) 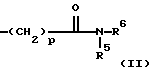 где p – целое число от 1 до 4, R5 – водород или алкильная группа и R6 – незамещенная или замещенная арильная группа или гетероарильная группа, причем заместитель, в свою очередь, может включать арильную группу или гетероарильную группу, или R4 представляет собой кольцо общей формулы (III): 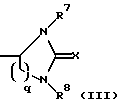 в которой q – число 1 или 2, R7 и R8 являются атомами водорода, а Х представляет собой атом серы, причем ароматические или гетероароматические остатки могут существовать в полностью гидрированной форме, причем хиральные атомы углерода имеют L-конфигурацию; а также соли указанных соединений с фармацевтически приемлемыми кислотами. Предпочтительными комбинациями остатков от R1 до R4 являются следующие: a) R1 – бензилокси, R2 – водород и R3 – водород, б) R1 – бензилокси, R2 – водород и R4 – группа формулы II, в которой p – 2 или 3, R5 – водород и R6 – 4-амидинофенильная группа, а также в) R2 – водород, R3 – водород и R4 – группа формулы II, в которой p – 2, R5 – водород и R6 – 4-амидинофенильная группа. Алкильными группами предпочтительно являются метил-, этил-, н-пропил-, изопропил-, н-бутил-, изобутил-, трет-бутил-, 2-этилгексил-, додецил- и гексадецил группы. Арильными группами предпочтительно являются фенил-, нафтил-, фенантранил- и флуоренил группы. Гетероарильными группами предпочтительно являются 2-, 3- и 4-пиридил-, 2- и 4-пиримидил-, имидазолил-, имидазопиридил-, 5- и 6-индолил-, 5- и 6-индазолил-, триазолил-, тетразолил-, бензимидазолил-, хинолил-, 2,6- дихлорпиридо-3-ил- и фурил- группы. Гидрированными гетероарильными группами предпочтительно являются пиперидин-, пиперазинил-, морфолино- и пирролидинил-группы. Аралкильными группами и гетероаралкильными группами являются группы, которые связаны в соответствующем положении через алкиленовую группу, предпочтительно метилен-, этилен-, н-пропилен- или н-бутилен- группы. Предпочтительными заместителями наряду с названными арил- и гетероарил-группами являются атомы галогена, такие как: фтор, хлор, бром и иод, а также метил-, этил-, изопропил-, трет-бутил-, циано-, нитро-, остатки карбоновой кислоты, амида карбоновой кислоты, метилового эфира карбоновой кислоты, этилового эфира карбоновой кислоты, этилового эфира кротоновой кислоты, трифторметил-, бензоил-, метокси-, бензилокси-, пиридилокси-, амино-, диметиламино-, изопропиламино-, амидино- и хинолилметокси- группы. В дальнейшем, согласно изобретению, используют соединения общей формулы (V) [Ac-D-Nal(2)1-D(pCl)Phe2-D-Pal(3)3-Ser4– Tyr5-D-Xxx6-Leu7-Arg8-Pro9-D- Ala10-NH2, где D-Xxx – остаток аминокислоты общей формулы (VI) 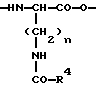 а также n, p, q, R4, R5, R6, R7, R8 и X – как указано выше и их соли с фармацевтически приемлемыми кислотами с вышеназванными свойствами. Заявленные согласно изобретению соединения характеризуются высокой антагонистической активностью и не обладают нежелательным побочным действием, прежде всего не вызывают отек. Если они используются не в виде солей с плохорастворимыми в воде фармацевтически приемлемыми кислотами, то эти соединения, кроме того, обладают повышенной растворимостью в воде. Эти соединения характеризуются высоким сродством к рецептору ЛГ-РФ человека, то есть являются высокоактивными при ингибировании высвобождения гонадотропинов из гипофиза млекопитающих, включая человека: при испытании на крысах обнаруживают пролонгированное ингибирование высвобождения тестостерона, а также вызывают минимальное высвобождение гистамина in vitro. Соединениями общей формулы (1) предпочтительно являются:  – N-Z-[ – N-Z-[ -N’-4-(4-амидинофенил)-амино-1,4-диоксобутил]-лизинамид, – N-Z-[-N’-(имидазолидин-2-он-4-ил)-формил]- лизинамид.
Пептидами формулы V предпочтительно являются такие соединения, в которых Xxx – [– N’-4-(4-амидинофенил)-амино-1,4- диоксобутил]-лизил-группа или [-N’-(имидазолидин-2-он-4- ил)-формил]-лизил-группа. Предпочтительно соли с фармацевтически приемлемыми кислотами плохо растворимы в воде. Особенно предпочтительны соли 4,4′-метилен-бис(3-гидрокси-2-нафтойевой кислоты), известной также как эмбоновая или памоевая кислота.
Номенклатура, применяемая для обозначения пептида, соответствует правилам биохимической номенклатуры, опубликованным Административным советом ИЮПАК(IUPAC-IUB) (European J. Biochem., 1984, 138, 9-37), согласно которой стандартным способом представления пептида является указание N- концевого аминокислотного остатка слева, а C-концевого – справа. Заявленные согласно изобретению пептиды и пептиды-миметики, являющиеся антагонистами ЛГ-РФ, содержат как природные аминокислоты, так и синтетические аминокислоты, причем к первым относятся Ala, Val, Leu, Ile, Ser, Thr, Lys, Arg, Asp, Asn, Glu, Gln, Cys, Met, Phe, Tyr, Pro, Trp и His. Аббревиатура отдельных аминокислотных остатков основывается на тривиальных названиях аминокислот: Ala – аланин, Arg – аргинин, Gly – глицин, Leu – лейцин, Lys – лизин, Pal(3) – 3-(3-пиридил)аланин, Nal(2) – 3-(2-нафтил)аланин, Phe – фенилаланин, (pCl)Phe – 4-хлорфенилаланин, Pro – пролин, Ser – серин, Thr – треонин, Trp – триптофан и Tyr – тирозин. Все вышеприведенные аминокислоты относятся к L-ряду, если не указано иное. Например, аббревиатура D-Nal(2) является сокращением от 3-(2-нафтил)-D-аланина и Ser – сокращением L-серина. Ниже другие используемые сокращения: Boc – трет-бутилоксикарбонил; Вор – бензотриазол-1-ил-окси-трис-(диметиламино)-фосфонийгексафторфосфат; DCC – дициклогексилкарбодиимид; DCM – дихлорметан; Ddz – диметоксифенил-диметилметиленоксикарбонил (диметоксидиметил-Z), DIC – диизопропилкарбодиимид; DIPEA- N,N-диизопропилэтиламин, DMF – диметилформамид; Fmoc – флуоренилметилоксикарбонил; HF – жидкая безводная фтористоводородная кислота; HOBt – 1-гидроксибензотриазол; HPLC – жидкостная хроматография высокого давления (ЖХВД); РуВор – бензотриазол-1-ил-окси-трис-пирролидинофосфоний гексафторфосфат; TFA – трифторуксусная кислота; Z – бензилоксикарбонил.
Согласно изобретению соединения общей формулы (I) получают следующим образом: прежде всего обеспечивают защиту двух из трех функциональных групп ( – амино-, – -аминогруппа и – карбоксильная группа), а свободная незащищенная группа участвует в реакции конденсации. При известных условиях, если это возможно и приводит к наилучшим результатам, на первой стадии вводят промежуточные защитные группы, которые удаляют после второй стадии, чтобы получить требуемые функциональные группы. Походящие защитные группы и методы их введения известны в специальной литературе. Например, защитные группы описаны с следующих руководствах “Principles of Peptide Synthesis”, Springer Verlag 1984), Lehrbuch “Solid Phase Peptide Synthesis” J.M.Steward and J.D. Young, Pierce Chem.Company, Rockford, III, 1984, и G.Barany and R.B.Merrifild “Peptides”, Ch.1, S.1-285, 1979, Academic Press Inc.
Синтез соединений формулы (V) можно проводить либо при использовании классического метода конденсации фрагментов, либо с помощью твердофазного метода Меррифилда с постепенным наращиванием пептидной цепи и применением в качестве исходного соединения D-лизина, ацилированного по боковой группе карбоновой кислотой общей формулы VII, а также путем обеспечения реакции декапептидной единицы с соответствующими карбоновыми кислотами с образованием амидной связи в боковой цепи D-лизина6. В связи с этим, согласно изобретению, имеется два альтернативных способа для получения соединения общей формулы (V).
Первый альтернативный способ для получения соединения общей формулы (V) включает следующие стадии: -N’-4-(4-амидинофенил)-амино-1,4-диоксобутил]-лизинамид, – N-Z-[-N’-(имидазолидин-2-он-4-ил)-формил]- лизинамид.
Пептидами формулы V предпочтительно являются такие соединения, в которых Xxx – [– N’-4-(4-амидинофенил)-амино-1,4- диоксобутил]-лизил-группа или [-N’-(имидазолидин-2-он-4- ил)-формил]-лизил-группа. Предпочтительно соли с фармацевтически приемлемыми кислотами плохо растворимы в воде. Особенно предпочтительны соли 4,4′-метилен-бис(3-гидрокси-2-нафтойевой кислоты), известной также как эмбоновая или памоевая кислота.
Номенклатура, применяемая для обозначения пептида, соответствует правилам биохимической номенклатуры, опубликованным Административным советом ИЮПАК(IUPAC-IUB) (European J. Biochem., 1984, 138, 9-37), согласно которой стандартным способом представления пептида является указание N- концевого аминокислотного остатка слева, а C-концевого – справа. Заявленные согласно изобретению пептиды и пептиды-миметики, являющиеся антагонистами ЛГ-РФ, содержат как природные аминокислоты, так и синтетические аминокислоты, причем к первым относятся Ala, Val, Leu, Ile, Ser, Thr, Lys, Arg, Asp, Asn, Glu, Gln, Cys, Met, Phe, Tyr, Pro, Trp и His. Аббревиатура отдельных аминокислотных остатков основывается на тривиальных названиях аминокислот: Ala – аланин, Arg – аргинин, Gly – глицин, Leu – лейцин, Lys – лизин, Pal(3) – 3-(3-пиридил)аланин, Nal(2) – 3-(2-нафтил)аланин, Phe – фенилаланин, (pCl)Phe – 4-хлорфенилаланин, Pro – пролин, Ser – серин, Thr – треонин, Trp – триптофан и Tyr – тирозин. Все вышеприведенные аминокислоты относятся к L-ряду, если не указано иное. Например, аббревиатура D-Nal(2) является сокращением от 3-(2-нафтил)-D-аланина и Ser – сокращением L-серина. Ниже другие используемые сокращения: Boc – трет-бутилоксикарбонил; Вор – бензотриазол-1-ил-окси-трис-(диметиламино)-фосфонийгексафторфосфат; DCC – дициклогексилкарбодиимид; DCM – дихлорметан; Ddz – диметоксифенил-диметилметиленоксикарбонил (диметоксидиметил-Z), DIC – диизопропилкарбодиимид; DIPEA- N,N-диизопропилэтиламин, DMF – диметилформамид; Fmoc – флуоренилметилоксикарбонил; HF – жидкая безводная фтористоводородная кислота; HOBt – 1-гидроксибензотриазол; HPLC – жидкостная хроматография высокого давления (ЖХВД); РуВор – бензотриазол-1-ил-окси-трис-пирролидинофосфоний гексафторфосфат; TFA – трифторуксусная кислота; Z – бензилоксикарбонил.
Согласно изобретению соединения общей формулы (I) получают следующим образом: прежде всего обеспечивают защиту двух из трех функциональных групп ( – амино-, – -аминогруппа и – карбоксильная группа), а свободная незащищенная группа участвует в реакции конденсации. При известных условиях, если это возможно и приводит к наилучшим результатам, на первой стадии вводят промежуточные защитные группы, которые удаляют после второй стадии, чтобы получить требуемые функциональные группы. Походящие защитные группы и методы их введения известны в специальной литературе. Например, защитные группы описаны с следующих руководствах “Principles of Peptide Synthesis”, Springer Verlag 1984), Lehrbuch “Solid Phase Peptide Synthesis” J.M.Steward and J.D. Young, Pierce Chem.Company, Rockford, III, 1984, и G.Barany and R.B.Merrifild “Peptides”, Ch.1, S.1-285, 1979, Academic Press Inc.
Синтез соединений формулы (V) можно проводить либо при использовании классического метода конденсации фрагментов, либо с помощью твердофазного метода Меррифилда с постепенным наращиванием пептидной цепи и применением в качестве исходного соединения D-лизина, ацилированного по боковой группе карбоновой кислотой общей формулы VII, а также путем обеспечения реакции декапептидной единицы с соответствующими карбоновыми кислотами с образованием амидной связи в боковой цепи D-лизина6. В связи с этим, согласно изобретению, имеется два альтернативных способа для получения соединения общей формулы (V).
Первый альтернативный способ для получения соединения общей формулы (V) включает следующие стадии:а) связывание D-аланина, содержащего блокированную аминогруппу, с подходящей подложкой для твердофазного синтеза, б) удаление защитной группы с аминогруппы аланина, в) обеспечение реакции конденсации аланина, связанного с подложкой, с пролином, содержащим защищенный атом азота, г) удаление защитной группы с атома азота пролина, д) повторение стадий с) и г) с использованием аминокислот с первой по восьмую согласно общей формуле (V) в порядке от восьмой до первой, е) удаление соединения, полученного на стадии д), с подложки, ж) обеспечение реакции конденсации с карбоновой кислотой общей формулы (VII); R4COOH, – где R4 имеет значения, как указано выше, з) при необходимости обеспечение реакции конденсации с фармацевтически приемлемой кислотой, предпочтительно эмбоновой кислотой, с образованием соли. Второй альтернативный способ синтеза соединения общей формулы (V) включает следующие стадии: а) связывание D-аланина, содержащего блокированную аминогруппу, с подходящей подложкой для твердофазного синтеза, б) удаление защитной группы с аминогруппы аланина, в) обеспечение реакции конденсации аланина, связанного с подложкой, с пролином, содержащим защищенный атом азота, г) удаление защитной группы с атома азота пролина, д) повторение стадий в) и г) с использованием аминокислот с шестой до восьмой согласно общей формуле (V) в порядке от восьмой до шестой, е) удаление защитной группы с – аминогруппы D-лизина или D-орнитина в положении 6 и обеспечение конденсации с карбоновой кислотой формулы (VII) 4-COOH, где R4 имеет значения, как указано выше,ж) удаление защитной группы с – аминогруппы D-лизина или D-орнитина,з) повторение стадий в) и г) с использованием аминокислот с первой до пятой согласно общей формуле (IV) в порядке от пятой до первой, и) удаление соединения, полученного на стадии з), с подложки и очистка (например, ЖХВД), к) при необходимости обеспечение реакции конденсации с фармацевтически приемлемой кислотой, предпочтительно эмбоновой кислотой, с образованием соли. Третий альтернативный способ синтеза соединения общей формулы (V) включает следующие стадии: а) связывание D-аланина, содержащего блокированную аминогруппу, с подходящей подложкой для твердофазного синтеза, б) удаление защитной группы с аминогруппы аланина, в) обеспечение реакции конденсации аланина, связанного с подложкой, с пролином, содержащим защищенный атом азота, г) удаление защитной группы с атома азота пролина, д) повторение стадий в) и г) с использованием аминокислот с шестой до восьмой согласно общей формуле (V) в порядке от восьмой до шестой, е) удаление защитной группы с – аминогруппы D-лизина или D-орнитина в положении 6 и обеспечение конденсации с карбоновой кислотой формулы (Vll) 4-COOR, где R4 имеет значения, как указано выше,ж) удаление защитной группы с – аминогруппы D-лизина или D-орнитина,з) повторение стадий в) и г) с использованием аминокислот с первой до пятой согласно общей формуле (IV) в порядке от пятой до первой, и) удаление соединения, полученного на стадии з), с подложки и очистка (например, ЖХВД), к) при необходимости обеспечение реакции конденсации с фармацевтически приемлемой кислотой, предпочтительно эмбоновой кислотой, с образованием соли. Предпочтительными карбоновыми кислотами формулы (VII) являются: имидазолидин-2-он-4-карбоновая кислота и N-(4-амидинофенил)-амино-4-оксомасляная кислота. Соединения формулы (V) синтезируют в соответствии с известными методиками, например, при использовании твердофазного метода, частично твердофазного метода или классического синтеза в растворе (см. M.Bodanszky, “Principles of Peptide Synthesis” Springer Verlag 1984). Например, методы твердофазного синтеза описаны в руководствах Lehrbuch “Solid Phase Peptide Synthesis” J. M. Steward and J.D.Young, Pierce Chem.Company, Rockford, III, 1984, и G.Barany and R.B.Merrifild “Peptides”, Ch.1, S.1-285, 1979, Academic Press Inc. Методы классического синтеза в растворе подробно описаны в руководстве “Methoden der Organischen Chemie (Houben-Weyl), Synthese von Peptiden” E.Wunsch (Herausgeber) 1974, Georg Thieme Verlag, Stuttgart, BRD. Ступенчатый синтез осуществляют, например, следующим образом: прежде всего C-концевую аминокислоту с защищенной – аминогруппой ковалентно присоединяют к общеизвестной нерастворимой подложке, удаляют защитную группу с – аминогруппы указанной аминокислоты и к полученной таким образом свободной аминогруппе присоединяют следующую аминокислоту с заблокированной аминогруппой, затем таким же образом, стадия за стадией присоединяют остальные аминокислоты в нужном порядке и после присоединения всех аминокислот готовый пептид удаляют с подложки. Ступенчатая конденсация осуществляется по стандартной методике с использованием подходящих аминокислот с аминогруппами, заблокированными в соответствии с общепринятым способом. Возможно также использование и автоматического синтезатора пептидов, например, типа Labortec SP 650 (Bachem, Швейцария) с применением коммерческих препаратов защищенных аминокислот.
Связывание аминокислот друг с другом осуществляют в соответствии с общепринятыми методиками, в частности могут быть использованы следующие методы:– метод симметричных ангидридов в присутствии дициклогексилкарбодиимида или диизопропилкарбодиимида (DCC, DIC); – общий карбодиимидный метод; – карбодиимид-гидроксибензотриазольный метод (см. The Peptides, Volume 2, Ed. E.Gross and J.Meienhofer). Для присоединения аргинина предпочтительно используют карбодиимидный метод. Для остальных аминокислот в основном применяют методы симметричных или смешанных ангидридов. Для соединения фрагментов предпочтительно используют азидный метод, не сопровождающийся рацемизацией, или DCC-1-гидроксибензотриазольный, или DCC-3-гидрокси-4-оксо-3,4-дигидро-1,2,3-бензотриазиновый методы. Для соединения фрагментов можно также использовать и метод активированных эфиров. При ступенчатой конденсации особенно предпочтительно применять активированные эфиры N-защищенных аминокислот, например, N-гидроксисукцинимидный эфир или 2,4,5- трихлорфениловый эфир. Для катализа аминолиза эффективно использовать N-гидроксисоединения, кислотность которых приблизительно равна кислотности уксусной кислоты, например, 1 -гидроксибензотриазол. В качестве промежуточных защитных групп применяют группы, удаляемые гидрогенолизом, например, бензилоксикарбонильную группу (Z-радикал) или группы, отщепляемые в слабокислой среде. В качестве защитных групп – аминогрупп применяют, например, трет- бутилоксикарбонильные группы, карбобензокси-группы или карбобензотио- группы (при необходимости в каждом случае содержащие п-бром- или п-нитробензил-группу), трифторацетил-, фталил-, о-нитрофеноксиацетил-, тритил-, п-толуолсульфонил-, бензил-группы, замещенные в бензоловом ядре бензильные группы (п-бром или п-нитробензил), – фенилэтил-группы (см. руководства Jesse P. Greenstein und Milton Winitz, Chemistry of Amino Acids, New York, 1961, John Wiley and Sons, Inc., Volume 2, P.883 и следующие, a также The Peptides, Volume 2, Ed. E.Gross and J.Meienhofer, Academic Press, New York). Эти же защитные группы используют для блокировки функциональных групп боковых цепей (ОН-, NH2-групп) соответствующих аминокислот.
Для защиты гидроксигрупп аминокислот (серина, треонина) предпочтительно используют бензильные или сходные с ними группы. Для защиты других боковых групп (например,  – аминогруппы, гуанидиновой группы аргинина) предпочтительно используют ортогональный метод.
Реакцию конденсации аминокислот проводят в общепринятом для этих целей инертном растворителе или в суспензии (например, в дихлорметане), причем при известных условиях для увеличения растворимости можно добавлять диметилформамид.
Для введения R4-CO-группы путем конденсации аминогруппы лизина с карбоновой кислотой общей формулы (VII) могут быть использованы вышеуказанные методы конденсации аминокислот. Однако особенно предпочтительно проводить конденсацию в присутствии карбодиимида, например, 1-этил-3-(3-диметиламинопропил)- карбодиимида и 1-гидроксибензотриазола.
В качестве синтетических подложек используют нерастворимые полимеры, например, набухающую в органическом растворителе полистирольную смолу в виде гранул (например, сополимер полистирола и дивинилбензола (1%)). Синтез защищенного амида декапептида проводят на метилбензгидриламидной смоле (МВНА-смола, то есть полистирол, содержащий метилбензагидриламидные группы), причем после отщепления от подложки в присутствии HF получают пептид с необходимой С-концевой амидной группой в соответствии со схемой A (см. в конце описания).
Для синтеза пептида аминокислоты с N--Boc-защитными группами добавляют в трехкратном молярном избытке в присутствии диизопропилкарбодиимида (DIC) и 1-гидроксибензотриазола (HOBt) в CH2Cl2/DMF, конденсацию проводят в течение 90 мин и Boc-группу удаляют путем обработки 50% трифторуксусной кислотой (TFA) в CH2Cl2 в течение 30 мин. Для контроля полноты конверсии используют хлораниловый тест по Кристенсену и нингидриновый тест по Кайзеру. Остатки свободных аминогрупп блокируют ацетилированием с использованием ацетилимидазола (пятикратный избыток) в CH2Cl2.
На схеме приведена последовательность стадий синтеза пептида. На стадии отщепления пептида от смолы конечный продукт твердофазного синтеза сушат в вакууме над P2O5 и обрабатывают его после высушивания 500-кратным избытком смеси HF/анизол в соотношении 10:1/(об./об.) в течение 60 мин при 0oC.
После удаления HF и анизола в вакууме и обработки остатка безводным этиловым эфиром амид пептида выпадает в виде белого твердого продукта, отделение пептида от выпавшего совместно с ним полимерного носителя проводят промывкой осадка 50%-ным раствором уксусной кислоты в воде. С помощью осторожного упаривания уксуснокислых растворов в вакууме можно получить соответствующий пептид в виде высоковязкого масла, которое после добавления к нему абсолютного эфира на холоду превращается в белый твердый продукт.
Дальнейшую очистку проводят с помощью препаративной ЖХВД.
Получение кислых солей пептидов осуществляют стандартным способом с помощью их обработки кислотами. Свободные пептиды можно получить обработкой их кислых солей основаниями. Эмбонаты пептидов можно получить обработкой солей трифторуксусной кислоты (TFA-соли) свободной эмбоновой (памоевой) кислотой или соответствующей динатриевой солью эмбоновой кислоты. Для этого водный раствор TFA-соли пептида смешивают с раствором динатриевой соли эмбоновой кислоты в диполярном апротонном растворителе, предпочтительно диметилацетамиде, и выделяют образующийся светложелтый осадок.
Для лучшего понимания сущности изобретения приведены следующие примеры, не ограничивающие область изобретения.
Пример 1 – аминогруппы, гуанидиновой группы аргинина) предпочтительно используют ортогональный метод.
Реакцию конденсации аминокислот проводят в общепринятом для этих целей инертном растворителе или в суспензии (например, в дихлорметане), причем при известных условиях для увеличения растворимости можно добавлять диметилформамид.
Для введения R4-CO-группы путем конденсации аминогруппы лизина с карбоновой кислотой общей формулы (VII) могут быть использованы вышеуказанные методы конденсации аминокислот. Однако особенно предпочтительно проводить конденсацию в присутствии карбодиимида, например, 1-этил-3-(3-диметиламинопропил)- карбодиимида и 1-гидроксибензотриазола.
В качестве синтетических подложек используют нерастворимые полимеры, например, набухающую в органическом растворителе полистирольную смолу в виде гранул (например, сополимер полистирола и дивинилбензола (1%)). Синтез защищенного амида декапептида проводят на метилбензгидриламидной смоле (МВНА-смола, то есть полистирол, содержащий метилбензагидриламидные группы), причем после отщепления от подложки в присутствии HF получают пептид с необходимой С-концевой амидной группой в соответствии со схемой A (см. в конце описания).
Для синтеза пептида аминокислоты с N--Boc-защитными группами добавляют в трехкратном молярном избытке в присутствии диизопропилкарбодиимида (DIC) и 1-гидроксибензотриазола (HOBt) в CH2Cl2/DMF, конденсацию проводят в течение 90 мин и Boc-группу удаляют путем обработки 50% трифторуксусной кислотой (TFA) в CH2Cl2 в течение 30 мин. Для контроля полноты конверсии используют хлораниловый тест по Кристенсену и нингидриновый тест по Кайзеру. Остатки свободных аминогрупп блокируют ацетилированием с использованием ацетилимидазола (пятикратный избыток) в CH2Cl2.
На схеме приведена последовательность стадий синтеза пептида. На стадии отщепления пептида от смолы конечный продукт твердофазного синтеза сушат в вакууме над P2O5 и обрабатывают его после высушивания 500-кратным избытком смеси HF/анизол в соотношении 10:1/(об./об.) в течение 60 мин при 0oC.
После удаления HF и анизола в вакууме и обработки остатка безводным этиловым эфиром амид пептида выпадает в виде белого твердого продукта, отделение пептида от выпавшего совместно с ним полимерного носителя проводят промывкой осадка 50%-ным раствором уксусной кислоты в воде. С помощью осторожного упаривания уксуснокислых растворов в вакууме можно получить соответствующий пептид в виде высоковязкого масла, которое после добавления к нему абсолютного эфира на холоду превращается в белый твердый продукт.
Дальнейшую очистку проводят с помощью препаративной ЖХВД.
Получение кислых солей пептидов осуществляют стандартным способом с помощью их обработки кислотами. Свободные пептиды можно получить обработкой их кислых солей основаниями. Эмбонаты пептидов можно получить обработкой солей трифторуксусной кислоты (TFA-соли) свободной эмбоновой (памоевой) кислотой или соответствующей динатриевой солью эмбоновой кислоты. Для этого водный раствор TFA-соли пептида смешивают с раствором динатриевой соли эмбоновой кислоты в диполярном апротонном растворителе, предпочтительно диметилацетамиде, и выделяют образующийся светложелтый осадок.
Для лучшего понимания сущности изобретения приведены следующие примеры, не ограничивающие область изобретения.
Пример 1Получение Ac-D-Nal(2)-D(pCl)Phe-D-Pal(3)-Ser-Туr-D-[ – N’- (имидазолидин-2-он-4-ил)формил]-Lys-Leu-Arg-Pro-D-Ala-NH2Синтез проводят в соответствии с приведенной выше схемой с использованием 5 г смолы mBHA (нагрузка полимера растущими пептидными цепями составляет 1,08 мМоля пептида на 1 г полимера). Лизин присоединяют к полимеру в виде Fmoc-D-Lys(Boc)-OH, который после удаления Boc-защитной группы в боковой цепи лизина ацилируют трехкратным избытком имидазолидин-2-он-4-карбоновой кислоты. Затем удаляют Fmoc-защитную группу в присутствии 20% пиперидин/DMF и наращивают пептидную цепь по направлению к N-концу согласно схеме. Синтезированный пептид отщепляют от полимера и получают 5,2 г сырого продукта. Пептид подвергают очистке препаративной HPLC и после лиофильного высушивания получают 2,1 г пептида, который по данным ЖХВД представляет собой гомогенный продукт общей формулы C74H97N18O15Cl с молекулярной массой, скорректированной по данным FAB-масс-спектроскопии – 1514 (М+H+) (рассчитанная – 1512,7) и с соответствующим 1H-ЯМР-спектром. 1H-ЯМР (500 МГц, DMSO- d6,  в ppm): 8,56, m, аром.H; 8,08, m, 1H, аром. H; 7,81, m, 1H, аром.H: 7,73 m, 2H, аром.H; 7,66, m, 1H, аром.H; 7,60, s, 1H, аром.H; 7,44, m, 2H, аром.H; 7,30, d, 1H, аром.H; 7,25, и 7,18, 2 d, 2 x 2H, аром.H p-Cl-Phe; 6,97 и 6,60, 2 d, 2 x 2H, аром.H Tyr; 9,2-6,3, некоторые сигналы, амид-NH; 4,8-4,0, многочисленные m, C – H и алифатич.H; 2,1-1,1, многочисленные m, остаточный алифатич.H; 1,70, s, 3H, ацетил: 1,22, d, 3H, С в ppm): 8,56, m, аром.H; 8,08, m, 1H, аром. H; 7,81, m, 1H, аром.H: 7,73 m, 2H, аром.H; 7,66, m, 1H, аром.H; 7,60, s, 1H, аром.H; 7,44, m, 2H, аром.H; 7,30, d, 1H, аром.H; 7,25, и 7,18, 2 d, 2 x 2H, аром.H p-Cl-Phe; 6,97 и 6,60, 2 d, 2 x 2H, аром.H Tyr; 9,2-6,3, некоторые сигналы, амид-NH; 4,8-4,0, многочисленные m, C – H и алифатич.H; 2,1-1,1, многочисленные m, остаточный алифатич.H; 1,70, s, 3H, ацетил: 1,22, d, 3H, С  – H Ala: 0,85, dd, С – H Leu.
Пример 2 – H Ala: 0,85, dd, С – H Leu.
Пример 2Получение Ac-D-Nal(2)-D(pCl) Phe-D-Pal(3)-Ser-Tyr-D-[ -N’-4-(4- амидинофенил)-амино-1,4-диоксобутил]-Lys-Leu-Arg-Pro-D-Alа-N20,7 мМоля (1,03 г) декапептида Ac-D-Nal-D(pCl)Phe-D-Pal-Ser-Tyr-D-Lys-Leu-Arg-Pro-D-Ala-NH2 конденсируют с 0,1 мМоля (0,27 г) (4-амидинофенил)амино- 4-оксомасляной кислоты в присутствии 1,0 мМоля (0,16 г) 1-этил-3- (3-диметиламинопропил)-карбодиимида и 1,0 мМоля (0,16 г) 1- гидроксибензотриазола в свежеперегнанном DMF. После инкубировании реакционной смеси в течение 24 ч растворитель удаляют в вакууме, полученный остаток растворяют в воде и высушивают лиофильно. Полученный сырой продукт (1,63 г) подвергают очистке с помощью обратнофазовой ЖХВД и получают 0,61 г пептида, который по данным ЖХВД представляет собой гомогенный продукт суммарной формулы C81H104N19O15Cl с молекулярной массой, скорректированной по данным FAB-масс-спектроскопии – 1618,7 (М+H+) (рассчитанная – 1617,7) и с соответствующим 1H-ЯМР-спектром. 1H-ЯМР (500 МГц, DMCO-d6, в ррm):10,4, s, 1H у. 9,15, s, 2H, u 8,8, s, 1H, NH группы 4-амидиноанилина; 8,60, m, 2H, аром. H; 8,20, m, 1H, аром. H; 7,80, m, 1H, аром.H; 7,73, m, аром. H; 7,61, s, 1H, аром.H; 7,44, m, 2H, аром.H; 7,30, d, 1H, аром.H; 7,25 u 7,20, 2d, 4H, аром. H (pCl)Phe; 7,0 u 6,6, 2d,4H, аром.H Tyr: 8,3-7,2, некоторые сигналы, амид-NH; 4,73-4,2, некоторые мультиплеты, C -H; 4,13, m, 1H, C -H; Ala; 3,78-2,4, некоторые мультиплеты, C -H и алифатич.Н; 1,72, s, 3H, ацетил; 1,22, d, 3Н, C Ala: 0,85, dd, 6Н, C Leu.
Пример 3К 0,5 г (0,3 мМоля) пептидного антагониста ЛГ-РФ, полученного согласно примеру 1, растворенного в 50 мл воды, добавляют раствор 0,130 г (0,3 мМоля) динатриевой соли эмбоновой кислоты в 2 мл воды, при этом получают эмбонат пептида, который быстро выпадает из раствора в виде желтого осадка. Получают 0,281 г тонкокристаллического порошка желто- зеленого цвета. Содержание эмбоновой кислоты составляет 33%. Пример 4 К 0,3 г (0,17 мМоля) пептидного антагониста ЛГ-РФ, полученного согласно примеру 2, растворенного в 5 мл диметилацетамида, добавляют раствор 0,195 г (0,45 мМоля) динатриевой соли эмбоновой кислоты в 2 мл воды, при этом получают эмбонат пептида, который выпадает из раствора в виде желтого осадка после добавления 50 мл воды. Получают 0,330 г тонкокристаллического порошка желтого цвета. Содержание эмбоновой кислоты составляет 20%. Соединения общей формулы I могут быть получены согласно следующим схемам 1, 3, 4 и 5, причем три функциональные группы R1, R3 и R4 систематически варьируют. На схеме 1 приведен синтез соединения в соответствии с примером 1. Общие принципы синтеза соединений общей формулы I согласно схеме 1. Карбоновую кислоту R4-COOH, содержащую замещенный остаток R4 и лежащую в основе формулы I и схемы синтеза 1, и которая в случае основного остатка R4 может существовать также в виде соли, например, в виде хлоргидрата, гидросульфата или ацетата, растворяют или суспендируют в безводной среде при перемешивании в неполярном или диполярном апротонном органическом растворителе, например, в тетрагидрофуране, диоксане, метил-трет-бутиловом эфире, толуоле, диметилформамиде, диметилацетамиде, N-метилпирролидоне, диметилсульфоксиде или хлористом метилене и соединяют при перемешивании с основанием, которое применяют для удаления кислоты, например, с диизопропиламином, триэтиламином, N-метилморфолином, диметиламинопиридином или пиридином. Затем добавляют смесь Z-(L) -лизинамида гидрохлорида в разбавителе, причем в качестве разбавителя применяют растворитель, использованный для растворения карбоновой кислоты R4-COOH, содержащей заместитель R4. Затем pH реакционной смеси поддерживают с помощью основания, которое добавляют для связывания кислоты, в диапазоне 6,5-9,0, предпочтительно 7,0-8,5, особенно 7,0-7,5. Наконец, к реакционной смеси добавляют при перемешивании раствор конденсирующего агента, например, бензотриазол-1-ил-окси-трис(диметиламино)- фосфонийгексафторфосфат (ВОР) или бензотриазол-1-ил-окси-трис- пирролидинофосфонийгексафторфосфат (РуВОР) или дициклогексил карбодиимид (DCC) и через непродолжительный период времени снова доводят pH реакционной смеси в вышеуказанном диапазоне. Суспензию перемешивают, например, в течение 1-15 ч при 0-80oC, предпочтительно при 10-50oC, особенно при 20-30oC, затем отфильтровывают, твердый продукт промывают и фильтрат высушивают в вакууме. Полученный остаток кристаллизуют путем растирания в органическом растворителе, например, толуоле, тетрагидрофуране, метилэтилкетоне или изопропиловом спирте, или очищают перекристаллизацией, перегонкой или колоночной или флэш-хроматографией на силикагеле или окиси алюминия. В качестве подвижной фазы используют, например, смесь, включающую хлористый метилен, метанол, аммиак (25%) в соотношении 85: 15:1 (б/б), или смесь, включающую хлористый метилен, метанол, аммиак (25%) в соотношении 80:25:5 (б/б). Синтез трифторацетата Соединение, очищенное как указано выше, растворяют в протонном или апротонном растворителе, например, в спиртах, таких как метанол, этанол, изопропанол, или в циклических эфирах, например, тетрагидрофуране или диоксане, и pH среды доводят до 10-11 раствором 2 н. едкого натрия. Выпавший твердый продукт отфильтровывают, промывают, сушат в вакууме и раствор в этаноле смешивают со стехиометрическим количеством или 2-4-кратным избытком трифторуксусной кислоты при температуре от 10 до 80oC, предпочтительно от 20 до 40oC. Через 24 ч инкубирования реакционной смеси при 0-4oC трифторацетат выпадает в осадок, который отфильтровывают и высушивают в вакууме. Соединения, приведенные в таблице 1, синтезируют, как описывается в примере 5, согласно вышеуказанному общему методу, который является основой схемы синтеза 1. Пример 5 Получение – N-[бензилоксикарбонил]- N-[5-[(4-амидинофенил)амино]-5-оксопентаноил]-L-лизинамид трифторацетат5 г (17,5 мМоля) гидрохлорида 5-[[4-(аминоиминометил)фенил]-амино]- 5- оксопентановой кислоты суспендируют при перемешивании в 200 мл безводного диметилформамида и добавляют 3,85 мл (35,0 мМоля) N- метилморфолина, а затем к суспензии добавляют смесь 5,53 г (17,5 мМоля) гидрохлорида Z-(L)-лизинамида в 100 мл диметилформамида и pH среды доводят до 7,0-7,5 N-метилморфолином (NMM). На конечном этапе добавляют раствор 9,73 г (21,9 мМоля) бензотриазол-1-ил- окси-трис-(диметиламино)фосфоний-гексафторфосфата (ВОР) и после инкубирования в течение 10-15 мин снова устанавливают pH среды до 7,0-7,5. Суспензию желтого цвета перемешивают при постоянном контроле значения pH, которое должно составлять 7,0-7,5, в течение 3-4 ч при комнатной температуре, отфильтровывают бесцветный осадок. Осадок промывают дважды диметилформамидом и фильтрат желтого цвета упаривают досуха. Маслянистый остаток обрабатывают метилэтилкетоном (5 x 40 мл) и после каждой из пяти обработок растворителем фазу метилэтилкетона отделяют и отбрасывают. Выделившийся в кристаллической форме сырой продукт промывают 30 мл метилэтилкетона и сушат в вакууме при комнатной температуре. Полученный твердый продукт растворяют приблизительно в 50 мл этанола и pH раствора доводят до 10-11 2 н. раствором едкого натрия. Выпавшее основание отфильтровывают, промывают водой и этанолом, сушат в вакууме при 35oC. Выход: 5,5 г (62% от теоретического) Трифторацетат: Суспензию 5,5 г основания в этаноле смешивают с пятикратным молярным избытком трифторуксусной кислоты при 60oC. Раствор хранят в течение ночи при 4oC, полученный трифторацетат отфильтровывают и сушат в вакууме при 35oC. Выход: 5,9 г (87,7% от теории) Температура плавления: 185oC Данные элементного анализа: Рассчитано: C 53,84 H 5,65 N 13,45 Найдено: C 54,11 H 5,74 N 13,33 Структура соединений с общей формулой I, синтезированных по вышеприведенному методу, приведена в таблице 1, причем для всех примеров n равно 4. 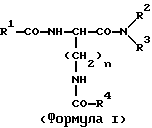 Температура плавления соединений, полученных согласно вышеприведенным примерам, приведена в таблице 2. Предшественники для синтеза соединений общей формулы I, полученных согласно схеме синтеза 1, структура которых приведена в таблице 1. В качестве исходного вещества для синтеза соединений в соответствии с примерами 5-34 используют коммерческий препарат Z-(L)-лизинамида. Замещенные производные “арил”- или “гетероариламино-оксо-предельных кислот”, полученные согласно схеме 1 и используемые для дальнейшего синтеза в качестве промежуточных производных, можно синтезировать и аналогично схеме 2 по известному в литературе методу (P.R.Bovy, J.Organ.Chem. 58, 7948 (1993)). В качестве ароматических или гетероароматических аминов (A-NH2) исходных соединений для синтеза по схеме 2, используют коммерческие препараты. Аминоимидазо[1,2а] пиридин, являющийся исходным соединением для синтеза согласно примеру 28, можно синтезировать аналогично известным в литературе методам (R.Westwood, J.Med.Chem 31, 1098 (1988)). “Арил” или соответственно “гетероариламино-оксо-предельные кислоты”, используемые в качестве исходных соединений, можно также синтезировать с помощью реакции аминолиза монометилового эфира предельной дикарбоновой кислоты, например, монометилового эфира субериновой кислоты и монометилового эфира азелаиновой кислоты с использованием ароматического или гетероароматического амина в кипящем этаноле или бутаноле, или по известной методике в ароматическом растворителе, например, толуоле или ксилоле при температуре кипения растворителя или в автоклаве при температуре кипения растворителя под давлением до 50 бар. Затем реакционную смесь концентрируют в вакууме и остаток кристаллизуют из метанола или этанола или подвергают очистке методом колоночной хроматографии. В качестве подвижной фазы используют, например, смесь, включающую хлористый метилен, метанол, аммиак (25%), в соотношении 85:15:1 (б/б) или хлористый метилен, метанол, аммиак (25%) в соотношении 80: 25:5 (б/б). Альтернативным методом получения соединений общей формулы I, где R1 – бензилоксикарбонильная группа, a R2 и R3– атом водорода, является следующий: 1. Амидируют – карбоксильную группу.
2. В – аминогруппу вводят Z-защитную группу.
3. В аминогруппу вводят Boc-защитную группу, что обеспечивает селективность при более позднем удалении защитных групп с NH2-групп.
4. Удаляют Z-группу с -аминогруппы.
5. В – аминогруппу вводят необходимый R4-CO-заместитель.
6. С – аминогруппы удаляют Boc-защитную группу.
7. В – аминогруппу вводят Z- защитную группу.
Другие соединения общей формулы I получают согласно схеме 3, на которой представлен синтез соединения, описанного в примере 35.
Общие принципы синтеза соединений общей формулы I согласно схеме 3.
1-я стадияВ качестве диполярного апротонного или неполярного органического растворителя используют, например, тетрагидрофуран, диметилсульфоксид, диметилформамид, ацетонитрил, этиловый эфир уксусной кислоты, диметилацетамид, N- метилпирролидон, диоксан, толуол, простой эфир, хлористый метилен или хлороформ при температуре от -30oC до 30oC, предпочтительно от -20oC до 20oC, особенно от -15oC до 5oC в случае использования Z-Lys(BOC)-OH, а также основание, например, триэтиламин, диизопропиламин, N-метилморфолин, N-этилпиперидин и алифатический или ароматический хлорангидгид карбоновой кислоты, например, ацетилхлорид, изобутироилхлорид, изовалероилхлорид, пивалоилхлорид, бензоилхлорид, или 4-метоксибензоилхлорид. Через некоторое время, например, от 30 мин до 3 ч добавляют предварительно охлажденный до -10oC раствор или суспензию амина в диполярном апротонном растворителе или неполярном органическом растворителе, например, тетрагидрофуране, диметилсульфоксиде, диметилформамиде, ацетонитриле, этиловом эфире уксусной кислоты, диметилацетамиде, N-метилпирролидоне, диоксане, толуоле, простом эфире, хлористом метилене или хлороформе при интенсивном перемешивании. Суспензию перемешивают при температуре от -30oC до 30oC, предпочтительно от -20oC до 20oC, в особенности от -15oC до 5oC в течение периода времени от 1 ч до 2 ч. После завершения реакции основание в виде хлорангидрида отфильтровывают под вакуумом и отгоняют растворитель. Маслянистый остаток обрабатывают апротонным или неполярным органическим растворителем, например, простым эфиром, диизопропиловым эфиром, метил-трет-бутиловым эфиром, петролейным эфиром, толуолом, ксилолом, пентаном, гексаном. Раствор перемешивают, например, в течение от 30 мин до 3 ч до выпадения белого порошкообразного осадка. Осадок отфильтровывают и сушат. 2-я стадия Z-Lys(BOC)-амид, полученный на 1-й стадии, как описано выше, растворяют в трифторуксусной кислоте при температуре от -20oC до 30oC, предпочтительно от -10oC до 20oC, в особенности от -5oC до 5oC, и перемешивают в течение от 15 мин до 1 ч. Избыточную трифторуксусную кислоту отгоняют и маслянистый остаток смешивают с диполярным апротонным или неполярным органическим растворителем, например, диметилформамидом, хлористым метиленом, тетрагидрофураном, ацетонитрилом, N-метилпирролидином, этиловым эфиром уксусной кислоты. Затем добавляют требуемую кислоту, основание, например, диизопропилэтиламин, N-метилморфолин и подходящий конденсирующий реагент, например, ВОР, PyBOP, DCC в диполярном апротонном или неполярном органическом растворителе, например, диметилформамиде, хлористом метилене, тетрагидрофуране, ацетонитриле, N-метилпирролидине, этиловом эфире уксусной кислоты. Реакцию проводят при температуре от -10oC до 100oC, предпочтительно от 0oС до 80oС, в особенности в интервале между 10oC и 35oC. После инкубирования реакционной смеси при указанных температурных условиях в течение периода времени от 1 до 5 ч и при комнатной температуре в течение 24 ч отгоняют растворитель. Остаток обрабатывают водой или органическим растворителем, например, изопропанолом, хлористым метиленом или простым эфиром. Сырой продукт подвергают хроматографической очистке на колонке с силикагелем. Согласно приведенному общему описанию 1-й и 2-й стадий, которые являются основой схемы 3 синтеза, получают соединения, приведенные в таблице 3, причем для всех соединений без исключения n = 4. 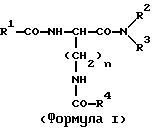 Пример 35: Получение N-[ -N-[бензилоксикарбонил] –– N-[4-[(4-амидинофенил)-aмино] -4-оксо- бутаноил]]-L-лизин-N-(3-пиридилметил))-амида1-я стадия Получение N-[ – N-[бензилоксикарбонил]-– N-[трет-бутилоксикарбонил]]- L-лизин-N-(3-пиридилметил))-амидаК 60 мл тетрагидрофурана добавляют при -15oC 4 г (10 мМолей) Z-Lys(BOC)-OH (коммерческий препарат), 1 г (10 мМолей) триэтиламина и 1,26 г (10 мМолей) пивалоилхлорида. Через 30 мин к смеси добавляют при интенсивном перемешивании предварительно охлажденный до -10oC раствор 1,08 г (10 мМолей) 3-(аминометил) пиридина в 20 мл тетрагидрофурана. Суспензию перемешивают при -15oC в течение от 1 до 2 ч. Гидрохлорид триэтиламина отфильтровывают при низкой температуре и тетрагидрофуран отгоняют. Маслянистый остаток смешивают с 100 мл диэтилового эфира. Раствор перемешивают до выпадения белого порошкообразного осадка, осадок отфильтровывают и сушат. Выход: 4 г (85% от теории). 2-я стадия Получение N-[ – N-[бензилоксикарбонил] –-N-[4-[(4-амидинофенил)-амино] -4-оксо- бутаноил]]-L-лизин-N-(3-пиридилметил))-амида2 г (4,25 мМоля) N-[ – N-бензилоксикарбонил] –– N-[трет- бутилоксикарбонил] ] -L-лизин-N-(3-пиридилметил))-амида растворяют при 0oC в 20 мл трифторуксусной кислоты (TFA) и перемешивают в течение 20 мин. Избыток TFA отгоняют и маслянистый остаток обрабатывают 10 мл DMF. После этого добавляют 4,6 мл (42,5 мМоля) N-метилморфолина, 1,15 г (4,25 мМоля) 4-[[4- (аминоиминометил)фенил] амино] -4-оксомасляной кислоты гидрохлорида, 2,35 г (5,3 мМоля) ВОР и 20 мл DMF. Полученную смесь перемешивают при комнатной температуре в течение 24 ч. DMF отгоняют, остаток обрабатывают дважды 40 мл воды, отфильтровывают и сушат. Сырой продукт подвергают хроматографической очистке на колонке с силикагелем с использованием элюента 89b (70% HCCl3, 40% MeOH, 10% CH3COO–Na+ в растворе NH4OH (1 Моль/л, 25%). Выход: 340 мг (14% от теоретического).
Синтез соединений согласно примерам 36-55 проводили по аналогии с примером 35.
Результаты даны в табл. 4.
Соединения общей формулы I получают в соответствии с приведенными ниже схемами 4 и 5.
Ацилирование карбоновыми кислотами или эфирами хлорангидридов муравьиной кислоты в соответствии со схемами 4 и 5:H-Lys(Boc)-NH2 конденсируют с карбоновой кислотой в диполярном апротонном растворителе (DMF, DMSO) в присутствии основания (DIPEA, NММ) и конденсирующего агента (DCC, DIC, EDCI) и получают производное амида. После удаления растворителя остаток смешивают с водой и выделяют вязкий маслянистый сырой продукт, который очищают кристаллизацией из спирта (например, метанола, этанола или изопропанола) или эфира (например, этилового эфира уксусной кислоты) или кетона (например, метилэтилкетона). Взаимодействие H-Lys(Boc)-NH2 c xлорангидридами карбоновых кислот проводят в водно-щелочном растворе (условия Шоттен-Баумана), выход целевых продуктов составляет 90-95%. Сырой продукт отфильтровывают и подвергают очистке путем перекристаллизации из спирта (метанол/этанол/изопропанол) или из этилацетата или метилэтилкетона. 2. Удаление Вос-защитной группы с помощью TFA. Количественное удаление Вос-защитной группы проводят при комнатной температуре в смеси дихлорметан : ТFA (2: 1) в течение 60 мин. Полученный R1-Lys-NH2 используют немедленно без дальнейшей очистки. 3. Ацилирование гидрохлоридом 4-((4-аминоиминометил)фенил)амино)- 4-оксомасляной кислоты. Реакцию с использованием соответствующей карбоновой кислоты (R4) проводят в диполярном апротонном растворителе (DMF, DMSO) при комнатной температуре в присутствии основания (NMM, DIPEA) и конденсирующего агента (такого как EDCI, Вор или РуВор). После удаления растворителя остаток обрабатывают водой и получают сырой продукт в виде осадка. Полученный продукт подвергают очистке методом препаративной ЖХВД на колонке RP18 с использованием элюента следующего состава: вода, ацетонитрил и TFA. Конечный продукт представляет собой TFA-соль. Согласно указанным общим принципам синтеза, представленным в схемах 4 и 5, получают соединения, синтез которых описан в примере 56, а структура – в таблице 5. Пример 56 К 120 мл сухого дегазированного N,N-диметилформамида (DMF) добавляют при комнатной температуре 32 ммоль гидрохлорида Z-лизинамида и 32 ммоль гидрохлорида 4-((4-(аминоиминометил)фенил)-амино)-4-оксо- масляной кислоты. Добавленные реагенты быстро растворяются при перемешивании; после добавления 104 ммоль диизопропилэтиламина и 40 ммоль ВОР реакционную смесь перемешивают при комнатной температуре в течение 16 ч. Растворитель и избыточный DIPEA упаривают на роторном испарителе при остаточном давлении, составляющем приблизительно 10 мбар, и нагревании на водяной бане при 50-55oC. Маслянистый остаток обрабатывают 250 мл воды, гомогенизируют в ультразвуковой ванне и охлаждают. Полученный сырой продукт отфильтровывают на нутч-фильтре и промывают водой. После высушивания под вакуумом над хлористым кальцием получают приблизительно 16 г бежевого порошкообразного продукта, который представляет собой хлоргидрат, чистота которого составляет приблизительно 90% (ЖХВД). Для получения соответствующего трифторацетата продукт суспендируют в 100 мл воды и к суспензии добавляют 32 ммоль (2,45 мл) TFA (99%). Избыток кислоты удаляют на роторном испарителе в течение короткого периода времени, затем водную суспензию лиофилизируют. После перекристаллизации сырого продукта из спирта (этанол/метанол) полученный продукт можно еще раз лиофилизировать для увеличения растворимости. Выход: 5,26 г. Т.пл.: 210-213oC. Данные 1Н-ЯМР (500 МГц, DMSO-d6, в ppm):10,47, с. , 6, 1H, NH анилид, 9,14 и 8,82с., NH амидин, 7,82, м., 1H, Lys- -NH и 7,79 и 7,46, 2с., ароматич. H, 7,27 и 6,93 2с., 2H, CONH2, 7,20, д., 1H, уретан. NH, 5,0, s, 2H, бензил., H, 3,89, м., 1H, С – H, 3,0 и 2,58 и 2,40, 3 м., все 6H, алифат.H, 1,60 – 1,20, 4 м., все 6H, остаточный алифатич.H.
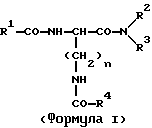 Соли соединений общей формулы I Полученные согласно изобретению соединения могут существовать в виде солей различных кислот, скажем, в виде солей минеральных кислот, таких как, например, соляной, серной, фосфорной кислоты, солей органических кислот, таких как, например, уксусной, трифторуксусной, молочной, малоновой, малеиновой, фумаровой, глюконовой, глюкуроновой, лимонной, эмбоновой, метансульфоновой, гидроксиэтансульфоновой, пировиноградной и янтарной. Соединения общей формулы I, так же, как и их соли, проявляют биологическую активность. Соединения общей формулы I могут применяться как в свободной форме, так и в форме солей, образованных приемлемыми кислотами. Соединения могут быть введены пероральным, парентеральным, внутривенным, подкожным или ингаляционным путем. Изобретение также относится к способу приготовления лекарственных препаратов, содержащих антагонисты ЛГ-РФ, соответствующие изобретению. Для приготовления лекарственных препаратов соединения по изобретению смешивают с вспомогательными веществами. В качестве вспомогательных веществ могут быть использованы обычные наполнители и другие дополнительные компоненты, традиционно применяемые в уровне техники. Изобретение также относится к фармацевтическим препаратам, содержащим по крайней мере одно соединение общей формулы I или его соль, образованную физиологически приемлемой неорганической или органической кислотой, или его смесь с фармацевтически приемлемым носителем и/или разбавителем/вспомогательным материалом. Пример 83. Сродство цетрореликса, соединений, полученных согласно примерам 1, 2, 5, 35 и 56, к рецептору ЛГ-РФ на клетках человека (Цетрореликс: Ac-D-Nal(2)-D-pCl-Phe-D-Pal(3)-Ser-Tyr-D-Cit-Leu-Arg- Pro-D-Ala-NH2). Способ определения сродства к рецептору (константы диссоциации Kd): Определение сродства пептидов к рецептору проводят методом конкурентного связывания (Beckers et al. Eur.J.Biochem. 231, 535-543, 1995). В качестве радиоактивно меченого лиганда используют [125J] цетрореликс (удельная активность 5-10 x 105 dpm/пкМоль; раствор содержит 20% (б/б) ацетонитрила, 0,2% (в/о) альбумина, 0,1% (в/о) TFA, примерно 80% (б/б) воды)). Способность к связыванию с рецептором у иодированных пептидов составляет 60 и 85%. В качестве немеченых тестируемых соединений используют цетрореликс (соединения, полученные согласно примерам 1, 2 и 56). Соединения используют в диапазоне концентраций 0,01 нМ-1000 нМ (цетрореликс, соединения, полученные согласно примерам 1 и 2) и 0,01 мкМ – 10 мкМ (соединения, полученные согласно примерам 5, 35, 56). В эксперименте используют клетки человека (клон L 3.5/78), сверхэкспрессирующие рецептор ЛГ-РФ. Клетки отмывают от не достигших плотности монослоя слоя клеток буфером PBS/EDTA (PBS без Ca2+/Mg2+/1mM EDTA), определяют число клеток и ресуспендируют в соответствующем объеме инкубационной среды (среда Игла в модификации Дальбекко: 4,5 г/л глюкозы, 10 мМ ХЕПЕС, pH 7,5, 0,5% (ес/б) БСА, 1 г/л бацитрацин, 0,1 г/л SBTI, 0,1% (ес/б) NaN3). В специальную реакционную пробирку на 400 мкл (Fa.Rerner Тур Beckman) помещают 200 мкл силикон/парафиновой смеси (84/16% (по объему)) и с помощью пипетки добавляют 50 мкл клеточной суспензии (2,5 x 105 клеток). Затем к клеточной суспензии на силикон-парафиновом слое прибавляют 50 мкл раствора [125J]-цетрореликса и тестируемого вещества в соответствующей концентрации. Далее смесь инкубируют при медленном вращении в термостате при 37oC в течение 60 мин. Затем центрифугируют (Biofuge Heraeus 15R, ротор НТА 13,8) в течение 2 мин при 9000 об/мин при комнатной температуре. Клетки проходят через силикон/парафиновый слой и таким образом отделяются от раствора с лигандами. После центрифугирования реакционную пробирку быстро замораживают жидким азотом и нижнюю часть пробирки с осадком отрезают щипцами. Нижнюю часть пробирки с осадком (связанный лиганд [125J] -цетрореликс), а также верхний слой (несвязанный лиганд [125J]-цетрореликс) помещают в кювету для счетчика импульсов. При определении максимального связывания (Во) конкурентных соединений не добавляют. Для определения неспецифического связывания добавляют 1 мкМ немеченого цетрореликса в качестве конкурирующего соединения. Неспецифическое связывание составляет не более 10% от полного связывания (Во). Подсчет импульсов осуществляют при использовании гамма-счетчика, результаты обрабатывают в соответствии с программой EBDA/Ligand V3.0 (McPherson, J.Pharmacol. Methods 14, 213-228, 1985). По диаграмме доза/эффект оценивают IC50 (концентрацию, вызывающую 50%-ное ингибирование связывания с рецептором). На основании этих данных с помощью программы EBDA/Ligand рассчитывают константу диссоциации Kd [нМ]. Результаты: Из графика конкурентного связывания (см.фиг. 1) очевидно, что все тестированные соединения конкурируют с радиоактивно меченым лигандом – [125J] цетрореликсом за связывание с рецептором ЛГ-РФ. Показаны также величины связывания (% от полного связывания Во) в зависимости от концентрации конкурирующего вещества. Для соединений, приведенных на фиг. 1, рассчитаны константы диссоциации Kd [нМ], отражающие степень сродства к рецептору: цетрореликс (SB-75): 0,214 нМ, соединение, полученное согласно примеру 1: 0,305 нМ, соединение, полученное согласно примеру 2: 0,104 нМ, соединение, полученное согласно примеру 5: 108 нМ, соединение, полученное согласно примеру 35: 300 нМ и соединение, полученное согласно примеру 56: 1082 нМ. Средние значения сродства с рецептором приведены в таблице 7. Пример 84 Функциональное тестирование антагонистического действия соединений, полученных согласно примерам 2 и 56 в отношении рецептора ЛГ-РФ клеток человека. Способ определения IP3 (D-миоинозит-1,3,5-трифосфата): Достигшую состояния субмонослоя культуру клеток человека (клон L 3.5/78), экспрессирующих рецептор ЛГ-РФ, промывают однократно PBS, клетки суспендируют в PBS/EDTA и собирают клеточную суспензию. Затем клетки суспендируют в соответствующем объеме инкубационной среды (среда Игла в модификации Дальбекко: 4,5 г/л глюкоза, 10 мМ ХЕПЕС, pH 7,5, 0,5% (ес/б) БСА, 5 мМ LiCl, 1 г/л бацитрацина, 0,1 г/л SBTI), отбирают аликвотную часть в пробирку объемом 1,5 мл и инкубируют при 37oC в течение 30 мин. В каждом измерении используют 4 x 106 клеток в 500 мкл. После предварительной инкубации в клеточную суспензию вносят ЛГ-РФ (0.5 мМ исходный раствор в буфере: 10 мМ трис, pH 7.5, 1 мМ дитиотреитол, 0,1% (ес/б) БСА/Bachem Art # H4005) до конечной концентрации 10 нМ. Одновременно к клеточной суспензии добавляют антагонисты в соответствующей концентрации (соединение, полученное согласно примеру 2: 0.0316; 0.1; 0,316 и т.д. до 100 нМ). В качестве контроля используют клеточную суспензию, инкубированную в отсутствие ЛГ-РФ. После инкубации образцов в течение 15 мин при 37oC образующийся IP3 выделяют экстракцией трихлоруксусной кислотой (TFA). К клеточной суспензии добавляют 500 мкл охлажденной до 0oC 15%-ной (ес/б) ТХУ. Осадок отделяют центрифугированием (Biofuge 15R Heraeus) при 2000 g и 4oC в течение 15 мин. Надосадочную жидкость объемом 950 мкл трижды экстрагируют 10 объемами охлажденного водного диэтилового эфира в пробирке объемом 15 мл. После завершающей экстракции pH раствора доводят до 7,5 с помощью разбавленного NaHCO3. Определение концентрации IP3 проводят с помощью чувствительного теста на конкурентное связывание с IP3-связывающим белком в присутствии меченого [3H] -IP3 и немеченого IP3. При измерении используют набор для тестирования TRK 1000 производства Amersham. Используют стандартную методику, прилагаемую к набору. После выполнения необходимых операций добавляют 2 мл сцинтиллятора для водных проб (Rotiszint Ecoplus), тщательно смешивают с ресуспендированным осадком, содержащим связанный [3H]-IP3, и проводят измерения при использовании бета-сцинтилляционного счетчика. Количество внутриклеточного IP3 вычисляют по калибровочной кривой и строят график дозовой зависимости. IC50 рассчитывают по конечной точке на графике дозовой зависимости. Результаты: На фиг. 1 представлены соответствующие графики дозовой зависимости для пептидных антагонистов, указанных в примере 2 (фиг.2), а также для пептидомиметиков, указанных в примере 56 (фиг.3). Определено стимулирующее действие 10 нМ ЛГ-РФ и ингибирующее действие на образование IP3 в зависимости от концентрации вещества. Соединения, упомянутые в примерах 2 и 56, не проявляют свойств агонистов, т.е. действие этих веществ не стимулирует синтеза IP3. В не приведенных здесь контрольных экспериментах было показано, что ЛГ-РФ не стимулирует образование IP3 у нетрасфицированных клеток. Концентрации IP3, определенные при действии высоких концентраций ЛГ-РФ, контролируют с величинами и соответствующими таковым для нестимулированных клеток. Таком образом в примерах 2 и 56 речь идет о функциональных антагонистах ЛГ-РФ. Кроме того, тестируемые вещества различаются по активности. В условиях эксперимента IC50 для соединений, полученных согласно примеру 2, составляет примерно 0,4 нМ; для соединений, полученных согласно примеру 56, IC50 составляет примерно 4 мкМ. Эта величина активности хорошо коррелирует с данными о сродстве к рецептору (Kd=0,109 нМ для соединения, полученного согласно примеру 2, и Kd = 1,08 мкМ для соединения, полученного согласно примеру 56), установленными в опытах in vitro по конкурентному связыванию в присутствии [125J]-цетрореликса. Пример 85 Гормон-супрессорное действие соединений, полученных согласно примерам 1, 2 и 56, определенное в опытах на здоровых самцах крысы. Для определения супрессии тестостерона в крови здоровых самцов крыс тестируемые соединения вводят подкожно в правый бок. Доза в случае соединений, полученных согласно примерам 1 и 2, составляет 1,5 мг/кг, а в случае соединения, полученного согласно примеру 56, – 10 мг/кг. Для определения концентрации тестостерона у подопытных животных отбирают пробы крови из подъязычной вены объемом 300 мкл спустя 0, 2, 4, 8 (только для соединения, полученного согласно примеру 56), 24, 48, 72 и 96 часов и затем каждые 3 дня до окончания супрессии. Низкий уровень тестостерона (не более 1 нг/мл) после введения соединения, полученного согласно примеру 1, сохраняется до 264 часов (одно животное), до 336 часов (2 животных), до 384 часов (одно животное) (фиг. 4). Для соединения, полученного согласно примеру 2, уровень тестостерона снижается в период времени до 408 часов (два животных), до 648 часов (4 животных) (фиг.5). Для соединения, полученного согласно примеру 56 (введение 10 мг/кг подкожно), уровень тестостерона снижается у всех пяти животных уже после двух часов, и это действие сохраняется в течение 8 часов. В следующей временной точке (24 ч) уровень тестотерона снова возрастает (фиг.6). Таблица 7 Биологические данные Сродство к рецептору ЛГ-РФ клеток человека (выраженное через константы диссоциации Kd [нМ] ; данные анализа рассчитаны по программе EBDA/Ligand. Приведены средние значения, полученные в различных экспериментах (количество экспериментов указано в скобках). В табл. 7 приведены также результаты опытов in vivo по супрессии тестостерона, определению выброса гистамина in vitro и растворимости в воде различных соединений по сравнению с цетрореликсом SB-75. Формула изобретения
Ac-D-Nal(2)1-D-(pCl)Phe2-D-Pal(3)3-Ser4-Tyr5-D-Xxx6-Leu7-Arg8-Pro9-D-Ala10-NH2 причем D-Xxx представляет собой аминокислотную группу общей формулы VI 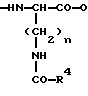 где n = 4; R4 – группа формулы II 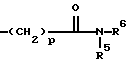 где p = 1 – 4, целое число, R5 – атом водорода или алкильная группа, R6 – незамещенная или замещенная арильная или гетероарильная группа, или R4 – кольцевая структура общей формулы III 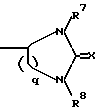 где q = 1 или 2, R7 и R8 – атом водорода, X – атом серы, и их соли с фармацевтически приемлемыми кислотами. 2. Соединение по п.1, отличающееся тем, что Xxx – [ -N-4(4-амидинофенил)-амино-1,4-диоксобутил]-лизил-группа.
3. Соединение по п.1, отличающееся тем, что Xxx – [-N-(имидазолидин-2-он-4-ил)-формил]-лизил-группа.
4. Соединение по пп.1 – 3, отличающееся тем, что представляет собой соли эмбоновой кислоты.
5. Соединение общей формулы I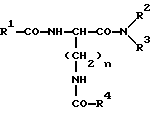 где n = 4; R1 – арил-, алкилокси, аралкил-, гетероаралкил-, аралкилокси- или гетероаралкилоксигруппа, незамещенная или замещенная; причем R2 и R3 независимо друг от друга могут быть представлены атомом водорода, алкильной группой, аралкильной группой или гетероаралкильной группой, незамещенными или замещенными, причем заместитель может быть представлен арильной или гетероарильной группой, или -NR2R3 является аминокислотной группой, а R4 – группа формулы II -(CH2)p-CO-NR5R6, где p = 1 – 4; R5 – атом водорода или алкильная группа; R6 – незамещенная или замещенная арильная или гетероарильная группа, причем заместители могут быть представлены арильной или гетероарильной группой; или R4 – кольцевая структура общей формулы III 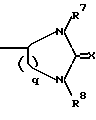 где q = 1, 2; R7 и R8 – атом водорода; X – атом серы, причем ароматические остатки могут быть полностью гидрированы и могут быть представлены хиральными углеродными атомами с конфигурацией L, а также их соли с фармацевтически приемлемыми кислотами. 6. Соединение по п.5, отличающееся тем, что представляет собой -N-[бензилоксикарбонил] –-N-[5-[(4-амидинофенил)амино]-5-оксо-пентаноил]-L-лизинамид-трифторацетат.
7. Соединение по п.5, отличающееся тем, что представляет собой -N-[бензилоксикарбонил] –-N-[5-[(4-амидинофенил)амино] -4-оксо-бутаноил]-L-лизинамид-трифторацетат.
8. Соединение по п.5, отличающееся тем, что представляет собой -N-[бензилоксикарбонил] –-N-[4-[(4-амидинофенил)амино] -4-оксо-бутаноил]-L-лизин-N-(3-пиридилметил)амид-трифторацетат.
9. Соединение по пп.5 – 8, отличающееся тем, что представляет собой соли эмбоновой кислоты.
10. Соединение по пп.1 – 9, отличающееся тем, что оно предназначено для получения лекарственных препаратов для лечения гормонзависимых опухолей, в особенности карциномы простаты или рака молочной железы, а также незлокачественных опухолей, лечение которых требует супрессии ЛГ-РФ.
11. Фармацевтическая композиция, обладающая высоким сродством к рецептору рилизинг-фактора лютеинизирующего гормона, содержащая в качестве активного вещества соединения по пп.1 – 4.
12. Способ получения соединения по п.1, формулы V, включающий следующие стадии: (а) связывание D-аланина, содержащего блокированную аминокислоту, с подходящей подложкой для твердофазного синтеза, (б) удаление защитной группы с аминогруппы аланина, (в) обеспечение реакции конденсации связанного с подложкой аланина с пролином, содержащим защитную группу на атоме азота, (г) удаление защитной группы с атома азота пролина, (д) повторение стадий (в) и (г) с аминокислотами 1 – 8 согласно формуле (V) в порядке от восьмой до первой, (е) удаление соединения, полученного на стадии (д), с подложки, (ж) обеспечение реакции конденсации с карбоновой кислотой общей формулы VIIR4-COOH, (VII) где R4 имеет значения по п.1, (з) при необходимости обеспечения реакции конденсации с фармацевтически приемлемой кислотой, предпочтительно эмбоновой кислотой, с образованием соли. 13. Способ получения соединения по п.1 формулы V, включающий следующие стадии: (а) связывание D-аланина, содержащего блокированную аминогруппу, с подходящей подложкой для твердофазного синтеза, (б) удаление защитной группы с аминогруппы аланина, (в) обеспечение реакции конденсации связанного с подложкой аланина с пролином, содержащим защитную группу на атоме азота, (г) удаление защитной группы с атома азота пролина, (д) повторение стадий (в) и (г) с аминокислотами 6 – 8 согласно общей формуле (V) в порядке от восьмой до шестой, (е) удаление защитной группы с -аминогруппы D-лизина или D-орнитина в положении 6 и обеспечение конденсации с карбоновой кислотой общей формулы VIIR4-COOH, (VII) где R4 имеет значения в соответствии с п.1, (ж) удаление защитной группы с -аминогруппы D-лизина и D-орнитина, (з) повторение стадий (в) и (г) с аминокислотами 1 – 5 согласно общей формуле (IV) в порядке от пятой до первой, (и) удаление соединения, полученного на стадии (з), с подложки и очистка, в особенности методом ЖХВД, (к) при необходимости обеспечения реакции конденсации с фармацевтически приемлемой кислотой, предпочтительно эмбоновой кислотой, с образованием соли.
14. Способ по пп.12 и 13, отличающийся тем, что в качестве карбоновой кислоты общей формулы VII используется N-(4-амидино-фенил)-амино-4-оксо-масляная кислота.
15. Способ по пп.12 и 13, отличающийся тем, что в качестве карбоновой кислоты общей формулы VII используется имидазолидин-2-он-4-карбоновая кислота.
16. Способ по пп.12 – 15, отличающийся тем, что в качестве фармацевтически приемлемой кислоты используется эмбоновая кислота.
17. Способ приготовления лекарственных препаратов, содержащих соединения по пп.1 – 9, характеризующийся тем, что данные соединения смешивают с вспомогательными веществами.
РИСУНКИ
PC4A – Регистрация договора об уступке патента Российской Федерации на изобретение
Номер и год публикации бюллетеня: 13-2002
(73) Патентообладатель:
Дата и номер государственной регистрации перехода исключительного права: 25.03.2002 № 14190
Извещение опубликовано: 10.05.2002
PD4A – Изменение наименования обладателя патента Российской Федерации на изобретение
Номер и год публикации бюллетеня: 20-2004
(73) Новое наименование патентообладателя:
Извещение опубликовано: 20.07.2004
MM4A Досрочное прекращение действия патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 15.11.2005
Извещение опубликовано: 20.10.2006 БИ: 29/2006
|
||||||||||||||||||||||||||