Патент на изобретение №2357970
|
||||||||||||||||||||||||||
(54) СПОСОБ ПОЛУЧЕНИЯ КЛОПИДОГРЕЛА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ИСПОЛЬЗУЕМЫЕ В СПОСОБЕ
(57) Реферат:
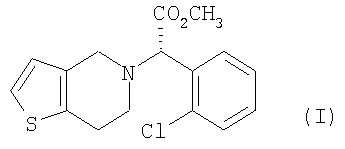
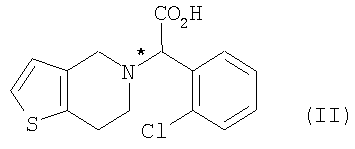
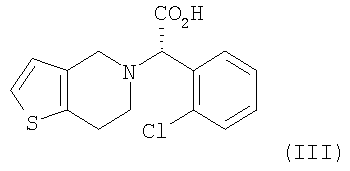
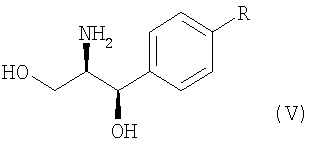
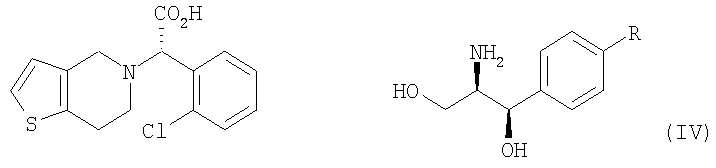
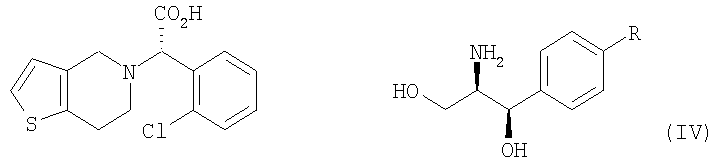
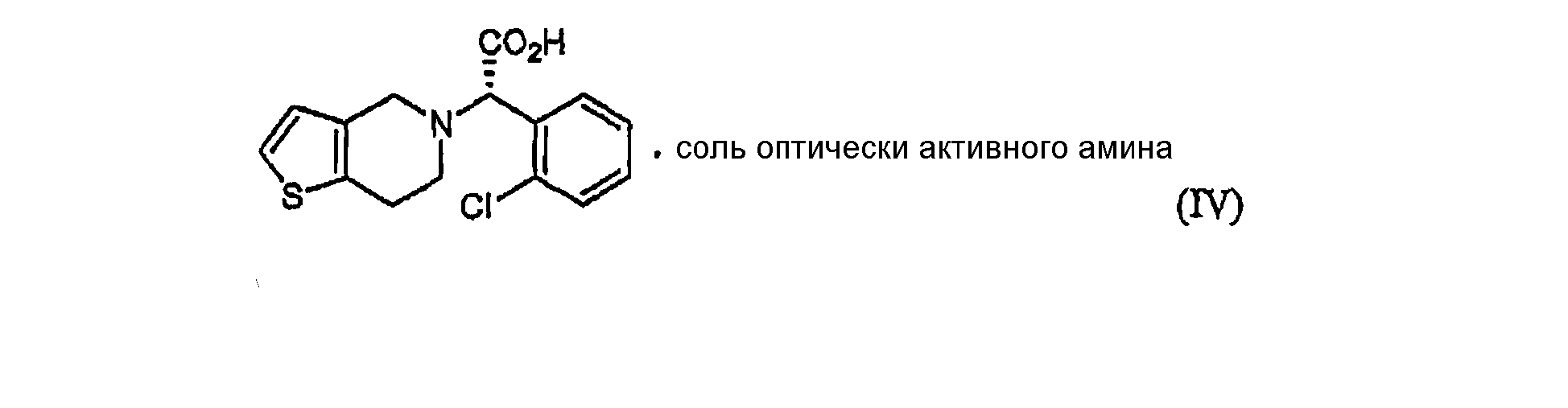
Настоящее изобретение относится к способу получения клопидогрела путем оптического разделения его рацемической формы с использованием оптически активного амина формулы V с получением оптически активной формы соединения формулы III или его кислотно-аддитивной соли и последующего метилирования соединения III или его соли. Описан также промежуточный продукт формулы IV, образующийся при взаимодействии рауемической формы клопидогрела с амином V.
Настоящий способ позволяет получить целевой продукт с высоким выходом. 2 н. и 20 з.п. ф-лы.
Область техники, к которой относится изобретение Настоящее изобретение относится к способу получения оптически чистого клопидогрела с высоким выходом и новому промежуточному соединению, используемому в этом способе. Уровень техники Клопидогрел, метил-(S)-(+)-
Различные способы получения клопидогрела описаны в европейских патентах Например, в европейском патенте Схема реакции 1 Европейский патент
Европейский патент
Европейский патент
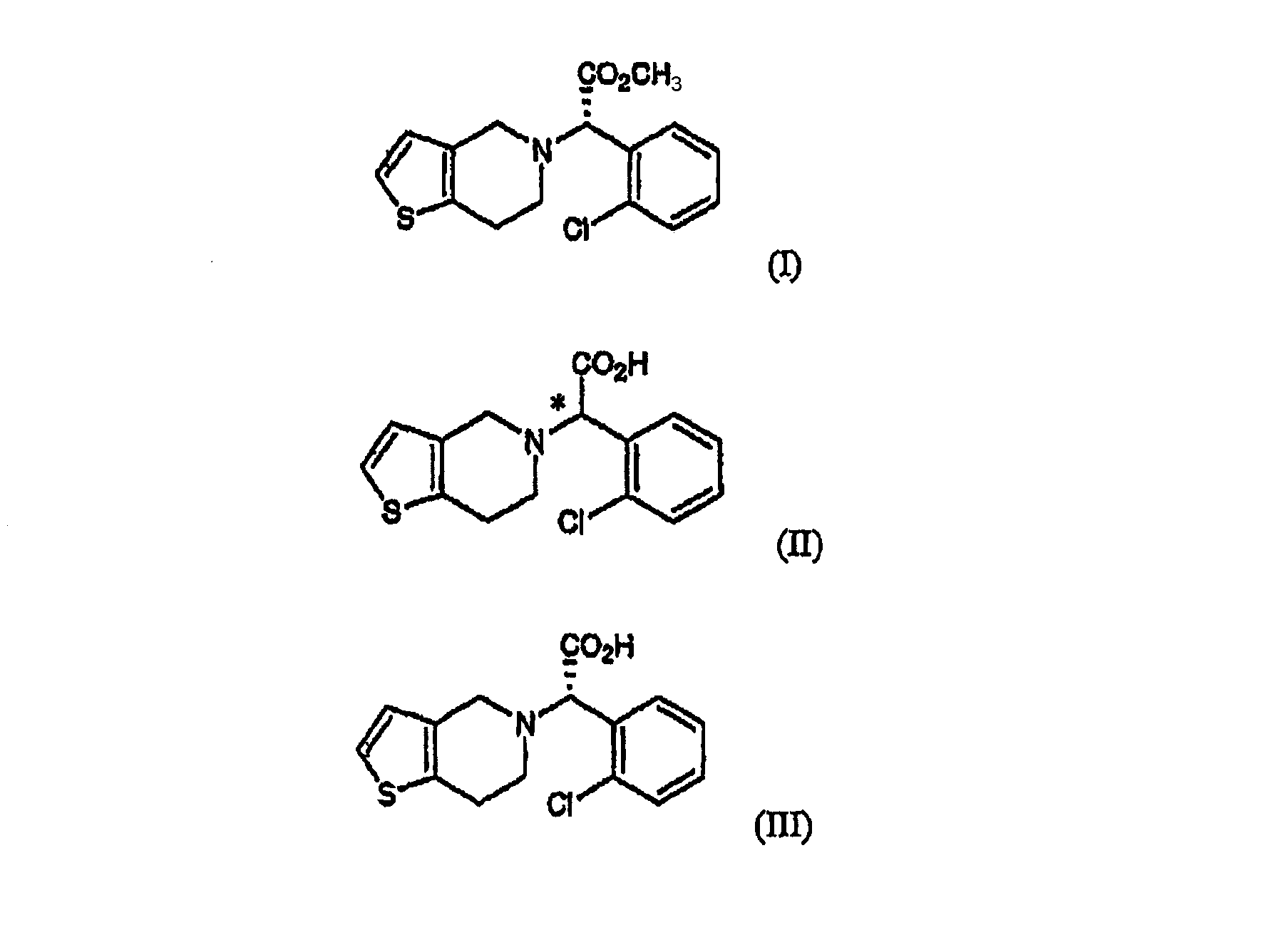
Однако эти способы имеют недостаток, заключающийся в том, что соль клопидогрела или его промежуточное соединение, образованные при селективной кристаллизации из рацемата клопидогрела или его промежуточного соединения и (1R)-(-)-10-камфорсульфокислоты, имеет недостаточную оптическую чистоту и требует дополнительной очистки, причем очистка приводит к снижению выхода. Кроме того, трудно извлечь (1R)-(-)-10-камфорсульфокислоту из реакционного раствора с целью ее повторного использования, вследствие ее высокой растворимости в воде. Сущность изобретения Соответственно, главной задачей настоящего изобретения является разработка простого, обеспечивающего высокий выход способа получения оптически чистого клопидогрела. Другой задачей настоящего изобретения является разработка нового промежуточного соединения, используемого при получении клопидогрела. В соответствии с одним аспектом настоящего изобретения предлагается способ получения клопидогрела формулы (I), включающий стадии: (a) оптического разделения рацематной формы соединения формулы (II) с использованием оптически активного амина с образованием оптически активной формы соединения формулы (III) или его кислотно-аддитивной соли; и (b) метилирования соединения формулы (III) или его кислотно-аддитивной соли, полученных на стадии (a):
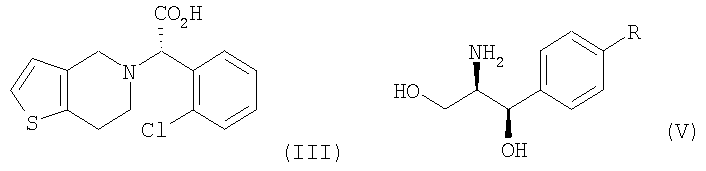
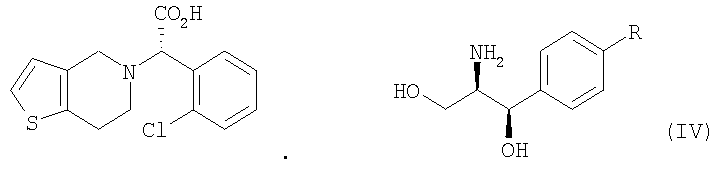
В соответствии с другим аспектом настоящего изобретения предлагается соль формулы (IV), используемая в качестве промежуточного соединения при получении клопидогрела:
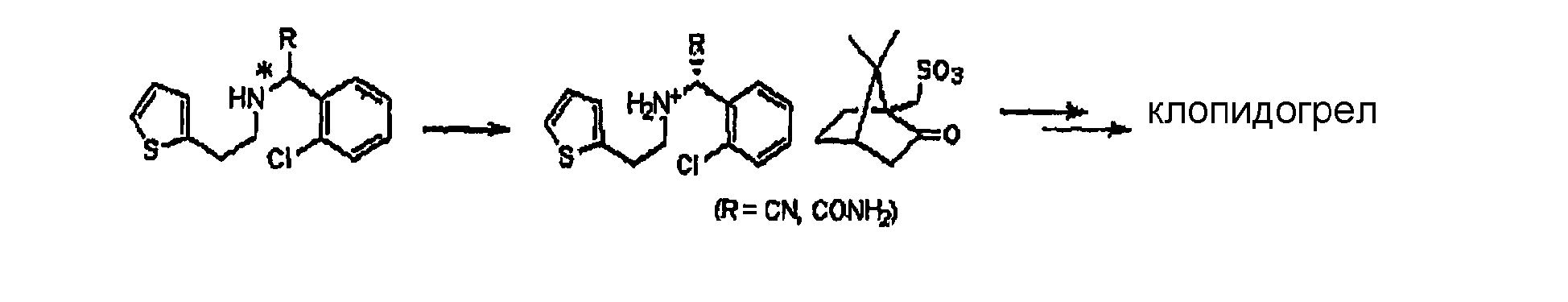
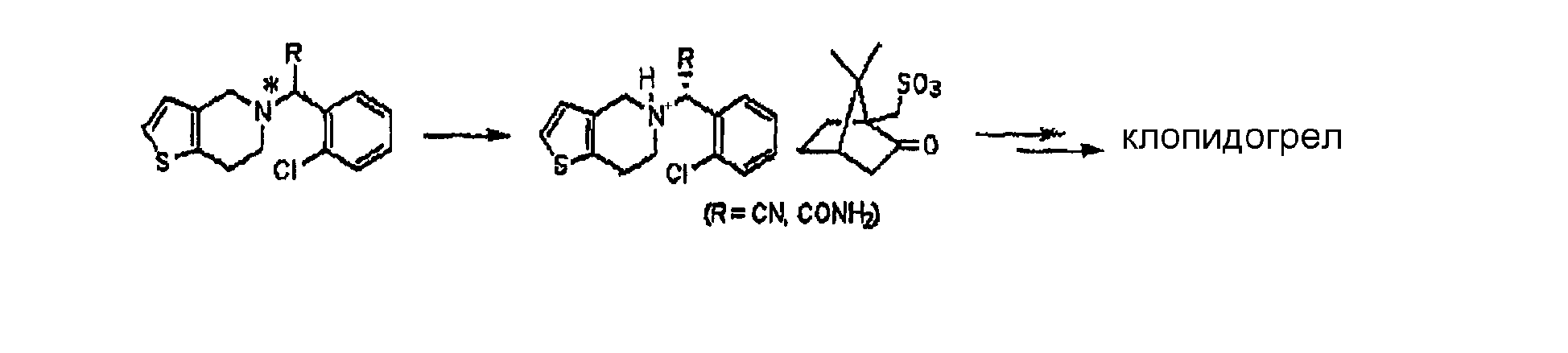
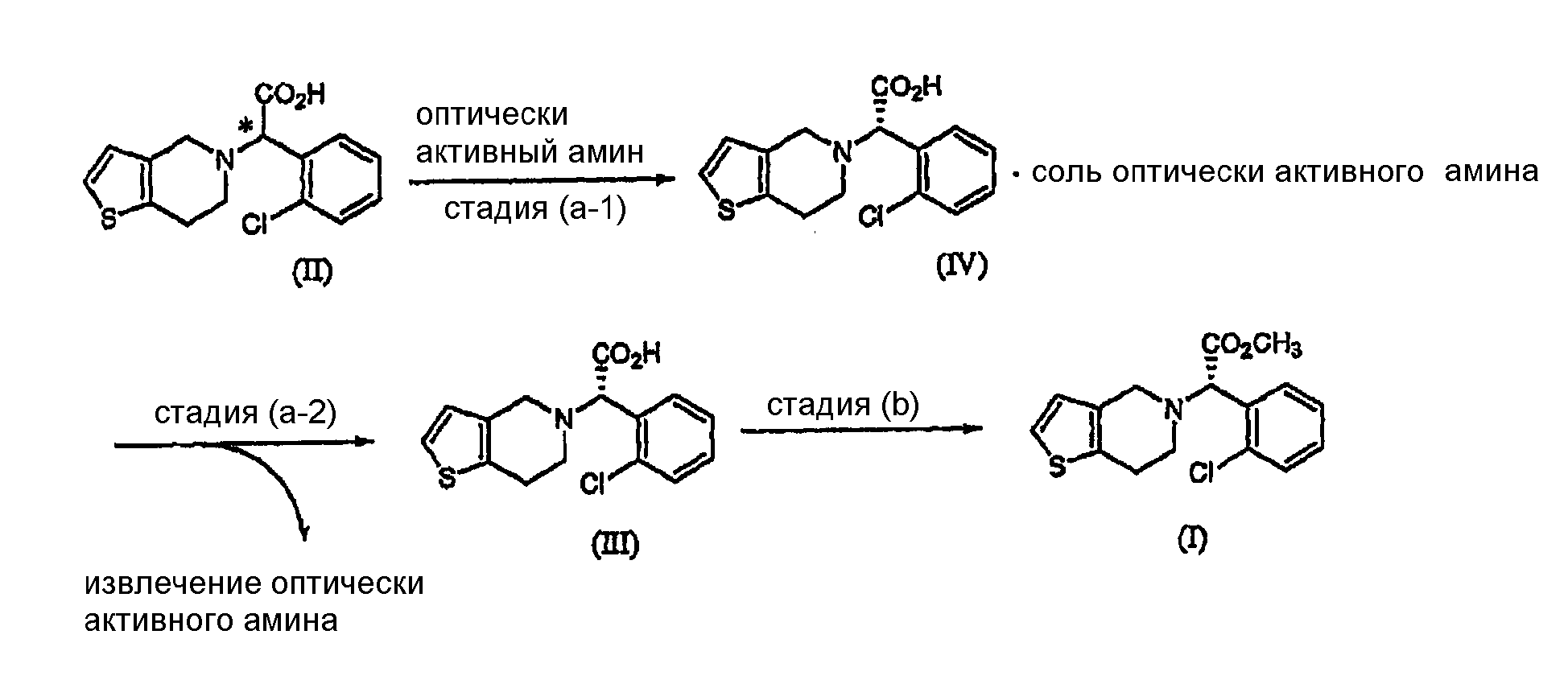
Подробное описание изобретения Способ настоящего изобретения включает процесс оптического разделения (стадия (a), состоящая из подстадий (a-1) и (a-2)), и процесс этерификации (стадия (b)), как показано на схеме реакции 2. Схема реакции 2
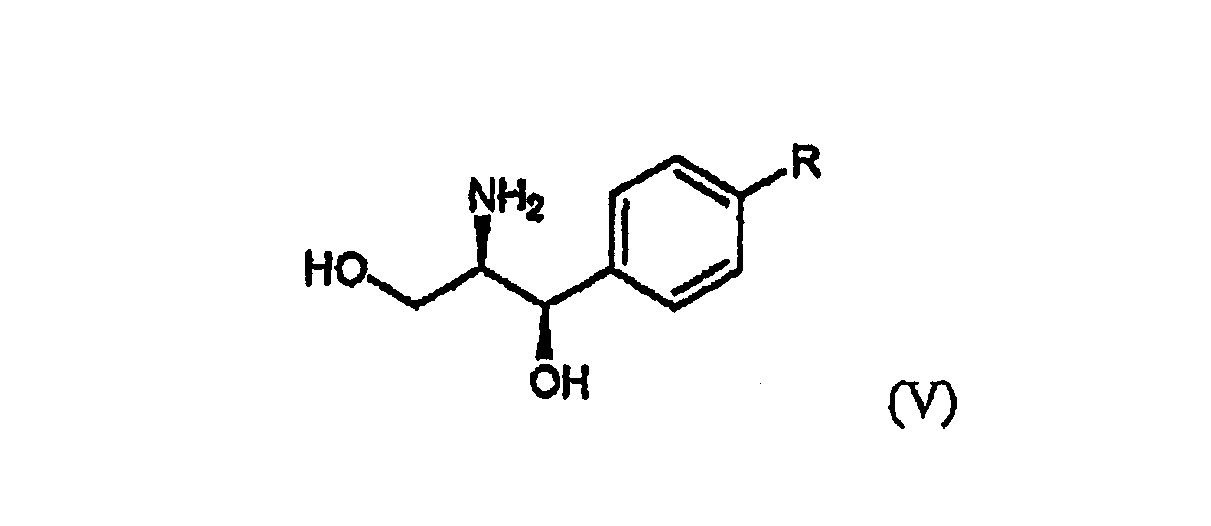
Стадия (a) Процесс оптического разделения (стадия (a)) в соответствии с настоящим изобретением включает взаимодействие рацемической формы соединения формулы (II) с оптически активным амином с кристаллизацией соли формулы (IV) (стадия (a-1)), и удаление фрагмента оптически активного амина из соли с образованием оптически активной формы соединения формулы (III) (стадия (a-2)). Стадия (a-1) Кристаллизация соли формулы (IV) на стадии (a-1) может быть осуществлена путем добавления соединения формулы (II) и оптически активного амина в подходящий растворитель, перемешивания полученного раствора при температуре в интервале от комнатной температуры до температуры кипения растворителя, и охлаждения смеси до температуры порядка от 0°C до комнатной температуры. Оптически активный амин, используемый в настоящем изобретении, может быть одним из любых агентов оптического разделения, используемых для оптического разделения рацемической карбоновой кислоты (см. [E.L. Eliel, S.L. Eliel и L.N. Mander, Stereochemistry of Organic Chemistry, 1994, John Wiley & Son, New York, pp.329-337]). Такой амин выбирают из группы, состоящей из эфедрина, 2-амино-1,2-дифенилэтанола,
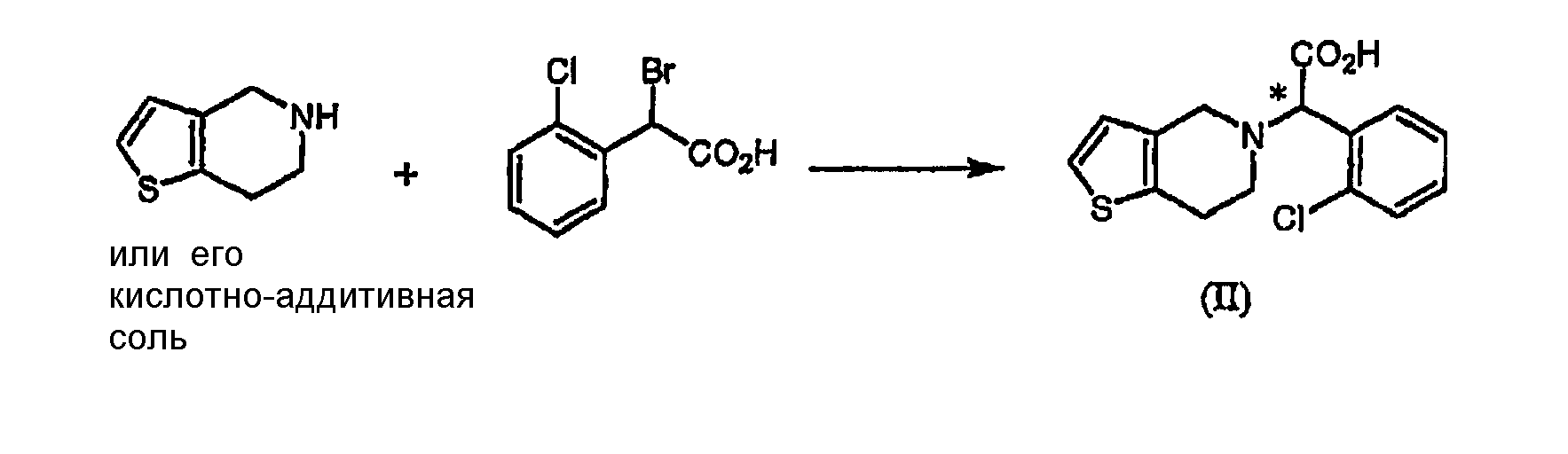
где R является водородом, галогеном, нитро, метилом или метокси. Оптически активный амин может быть использован в количестве, изменяющемся в интервале от 0,4 до 1,1 моль-эквивалентов по отношению к количеству соединения формулы (II). Подходящим для использования в качестве растворителя в настоящем изобретении является органический растворитель, такой как метанол, этанол, изопропанол, ацетон, этилацетат, ацетонитрил, тетрагидрофуран, 1,4-диоксан и N,N-диметилформамид, или смесь одного из органических растворителей и воды. Полученная таким образом соль формулы (IV) имеет удовлетворительную оптическую чистоту, которая не требует дополнительной очистки, но, если необходимо, она может быть дополнительно перекристаллизована из любого из перечисленных выше растворителей для повышения ее чистоты. Стадия (a-2) На стадии (a-2) соединение формулы (III) может быть получено путем подщелачивания водного раствора соли формулы (IV) с помощью основания с последующей фильтрацией или экстракцией для отделения оптически активного амина от соли формулы (IV) для дальнейшего повторного использования и подкислением фильтрата, который подвергают экстракции органическим растворителем. Основанием, используемым в настоящем изобретении, может быть гидроксид натрия или гидроксид калия, и, предпочтительно, чтобы его количество находилось в интервале от 1 до 3 моль-эквивалентов по отношению к количеству соединения формулы (IV). Выделение оптически активного амина, высвобождающегося из соли, достигается с помощью фильтрации; а когда амин является жидкостью, он может быть выделен экстракцией с использованием традиционного органического растворителя. Выделенный таким образом оптически активный амин может быть повторно использован в процессе кристаллизации стадии (a-1) в результате простой методики очистки, включающей перекристаллизацию или стадию дистилляции. После удаления фрагмента оптически активного амина фильтрат подкисляют для того, чтобы его pH составляло от 2 до 5, и затем подвергают экстракции органическим растворителем, таким как этилацетат, хлороформ и дихлорметан. Полученный экстракт концентрируют с получением соединения формулы (III) в форме пены. Предпочтительно, чтобы полученное таким образом соединение формулы (III) превращали в форму кристаллов чистой кислотно-аддитивной соли, например, кристаллов гидрохлорида. Рацемическая форма соединения формулы (II) может быть получена традиционным способом (см. европейский патент Схема реакции 3
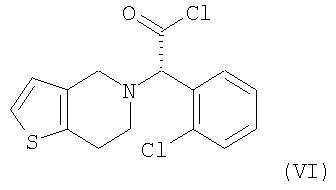
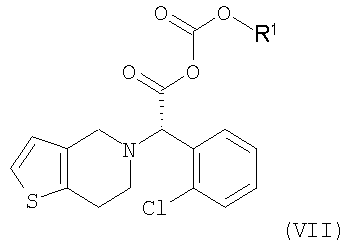
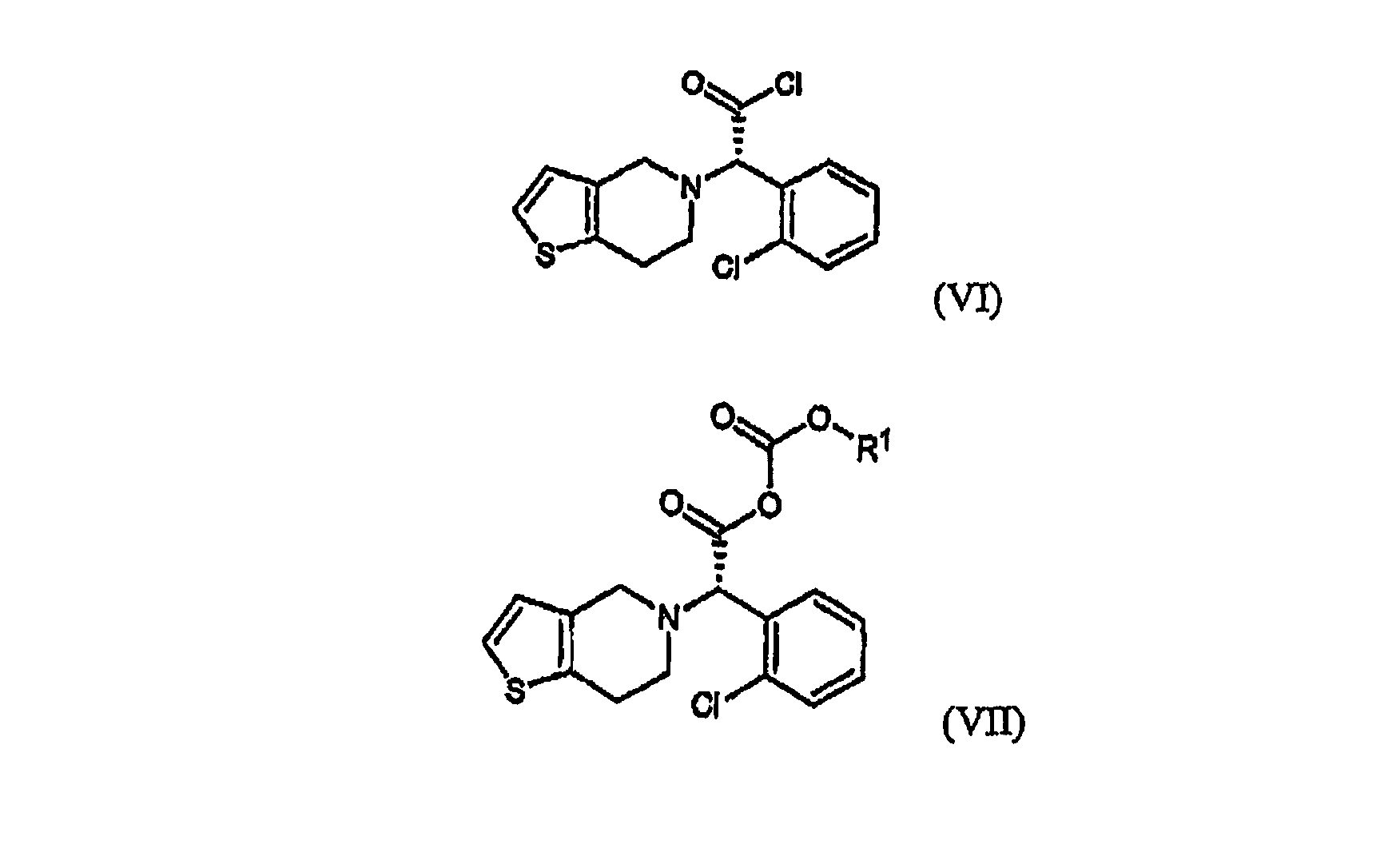
В частности, соединение формулы (II) может быть получено взаимодействием Типичные примеры растворителя, который может быть использован в настоящем изобретении, включают воду, метанол, этанол, н-пропанол, изопропанол, ацетон, тетрагидрофуран, 1,4-диоксан, ацетонитрил и их смесь; и типичные примеры основания, используемого в настоящем изобретении, включают гидроксид натрия, гидроксид калия, карбонат натрия, карбонат калия и их смесь. По отношению к количеству 4,5,6,7-дигидро-4H-тиено[3,2-c]пиридина или его кислотно-аддитивной соли количество используемого основания находится в интервале от 1 до 10 моль-эквивалентов, предпочтительно от 2 до 4 моль-эквивалентов, и количество Стадия (b) Стадия метилирования (b) может быть осуществлена путем (i) взаимодействия соединения формулы (III) или его кислотно-аддитивной соли с хлорирующим агентом в инертном растворителе с образованием хлорированного соединения формулы (VI) или его кислотно-аддитивной соли, которую обрабатывают метанолом; или (ii) взаимодействия соединения формулы (III) или его кислотно-аддитивной соли с алкилхлорформиатом в инертном растворителе в присутствии основания с получением смешанного ангидрида формулы (VII), который обрабатывают метанолом; или (iii) взаимодействия соединения формулы (III) или его кислотно-аддитивной соли с метанолом в присутствии кислотного катализатора:
где R1 является C1-4алкилом. На стадии (b) реакция соединения формулы (III) или его кислотно-аддитивной соли с хлорирующим агентом может быть проведена в инертном растворителе, таком как дихлорметан, хлороформ, бензол и толуол при температуре в интервале от -30 до 40°C, предпочтительно от -15°C до комнатной температуры, с получением хлорированного соединения формулы (VI) или его кислотно-аддитивной соли. Типичные примеры хлорирующего агента, используемого в настоящем изобретении, включают тионилхлорид, пентахлорид фосфора, хлористый фосфорил, фосген, дифосген, трифосген, оксалилхлорид и их смесь, где оксалилхлорид является предпочтительным. Предпочтительно, чтобы количество используемого хлорирующего агента составляло от 1 до 5 моль-эквивалентов по отношению к количеству соединения формулы (III) или его кислотно-аддитивной соли. Затем может быть осуществлена реакция хлорированного соединения формулы (VI) или его кислотно-аддитивной соли с метанолом при температуре в интервале от -15°C до комнатной температуры. Предпочтительно, чтобы количество используемого метанола находилось в интервале от 1 до 10 моль-эквивалентов по отношению к количеству соединения формулы (III) или его кислотно-аддитивной соли. Взаимодействие соединения формулы (III) или его кислотно-аддитивной соли с алкилхлорформиатом может быть осуществлено в инертном растворителе, таком как дихлорметан, хлороформ, тетрагидрофуран, ацетонитрил, бензол и толуол, в присутствии основания при температуре в интервале от -30 до 40°C, предпочтительно от -15°C до комнатной температуры, с получением смешанного ангидрида формулы (VII). Подходящим для использования на этой стадии является алкилхлорформиат, такой как метилхлорформиат, этилхлорформиат, н-пропилхлорформиат, изопропилхлорформиат и изобутилхлорформиат. Типичные примеры используемого основания на указанной стадии включают триэтиламин, диизопропилэтиламин, трибутиламин, пиридин, пиколин, лутидин, диметиламинопиридин и их смесь. Предпочтительно, чтобы количества алкилхлорформиата и основания находились в интервале от 1 до 3 моль-эквивалентов и от 1 до 4 моль-эквивалентов, соответственно, по отношению к количеству соединения формулы (III) или его кислотно-аддитивной соли. Взаимодействие смешанного ангидрида формулы (VII) с метанолом может быть осуществлено путем добавления метанола к смеси, полученной на предыдущей стадии, и перемешивания полученной смеси при температуре в интервале от -15 Взаимодействие соединения формулы (III) или его кислотно-аддитивной соли с метанолом может быть проведено в метаноле в присутствии кислотного катализатора, такого как безводная хлористоводородная кислота, серная кислота, метансульфоновая кислота и парра-толуолсульфоновая кислота. Предпочтительно, чтобы количество используемого кислотного катализатора находилось в интервале от 1 до 4 моль-эквивалентов по отношению к количеству соединения формулы (III) или его кислотно-аддитивной соли. Предпочтительно, чтобы эту реакцию осуществляли при температуре кипения метанола, используемого в качестве растворителя. Полученный таким образом оптически активный метиловый эфир клопидогрела формулы (I) практически не содержит левовращающий изомер, и он может быть превращен в форму его кислотно-аддитивной соли традиционным способом (см. европейский патент Клопидогрел или его кислотно-аддитивная соль, полученные в соответствии со способом изобретения, имеют высокую оптическую чистоту, по меньшей мере, 98% ee, что удовлетворяет минимальной фармацевтической чистоте, требуемой Фармакопеей США. Кроме того, соль формулы (IV), используемая в качестве промежуточного соединения при получении клопидогрела согласно настоящему изобретению, является новым соединением, содержащим оптически активный амин, такой как эфедрин, 2-амино-1,2-дифенилэтанол, Следующие примеры приводятся только в целях иллюстрации, и предполагается, что они не ограничивают объем изобретения. Пример 1: Получение (±)- 50,0 г гидрохлорида 4,5,6,7-тетрагидротиено[3,2-c]пиридина и 74,6 г Т.пл.: 114-116°C. Содержание воды: 5,6% (по методу Карла Фишера). 1H-ЯМР (CDCl3, ppm): 13C-ЯМР (CDCl3, ppm): ИК (KBr, см-1): 3394, 3079, 1668, 1638. MS (ESI, m/z): 308,2 (M+H.) Пример 2: Получение соли (S)-(+)- 167,0 г моногидрата (±)- Т.пл.: 199-201°C. Оптическое вращение: [ Оптическая чистота: 99,0% ee (ВЭЖХ, на соединение формулы (III)). 1H-ЯМР (ДМСО-d6, ppm): 13C-ЯМР (ДМСО-d6, ppm): ИК (KBr, см-1) 3419, 1920, 1606, 1556, 1518, 1350 40,0 г полученного таким образом соединения перекристаллизовывали из 280 мл 90% изопропанола с получением 35,0 г указанного в заголовке соединения более высокой чистоты. Т.пл.: 207-208°C. Оптическое вращение: [ Оптическая чистота: 99,9% ee (ВЭЖХ, на соединение формулы (III)). Пример 3: Получение соли (S)-(+)- 130,0 г моногидрата (±)- Т.пл.: 201-202°C. Оптическое вращение: [ Оптическая чистота: 99,4% ee (ВЭЖХ, на соединение формулы (III)). Пример 4: Получение соли (S)-(+)- 10,0 г моногидрата (±)- Т.пл.: 181-183°C. Оптическая чистота: 99,0% ee (ВЭЖХ, на соединение формулы (III)). 1H-ЯМР (ДМСО-d6, ppm): 13C-ЯМР (ДМСО-d6, ppm): ИК (KBr, см-1): 3455, 3069, 1561 Пример 5: Получение (S)-(+)- 113,0 г соли, полученной в примере 2 или 3, добавляли к 250 мл воды и охлаждали до температуры ниже 5°C. Добавляли 17,4 г растворенного в 125 мл дистиллированной воды гидроксида натрия и перемешивали при этой температуре в течение 30 мин. Полученные осажденные кристаллы отфильтровывали и промывали 125 мл дистиллированной воды. Отделяли и извлекали из нее 43,0 г (степень извлечения: 93%) (1R,2R)-(-)-2-амино-1-(4-нитрофенил)-1,3-пропандиола (формула (V): R является нитро). С другой стороны, к фильтрату добавляли 113 г хлорида натрия и 340 мл хлороформа, доводили pH полученной смеси до 3,5 с помощью 6N HCl, и органический слой отделяли. Водный слой затем подвергали экстракции 340 мл хлороформа. Хлороформные слои объединяли, концентрировали при пониженном давлении с получением 68 г указанного в заголовке соединения в виде пенообразного материала. Оптическая чистота: 99,0% ee (ВЭЖХ). Пример 6: Получение гидрохлорида (S)-(+)- 61 г (S)-(+)- Т.пл.: 200-201°C. Оптическое вращение: [ Оптическая чистота: 99,5% ee (ВЭЖХ). 1H-ЯМР (ДМСО-d6, ppm): 13C-ЯМР (ДМСО-d6, ppm): ИК (KBr, см-1): 3435, 3114, 1728 Пример 7: Получение метил-(S)-(+)- 8,9 г (S)-(+)- Оптическая чистота: 98,5% ee (ВЭЖХ). 1H-ЯМР (ДМСО-d6, ppm): Пример 8: Получение метил-(S)-(+)- 8,9 г (S)-(+)- Оптическая чистота: 98,5% ee (ВЭЖХ). Пример 9: Получение метил-(S)-(+)- 8,9 г (S)-(+)- Оптическая чистота: 98,1% ee (ВЭЖХ). Пример 10: Получение метил-(S)-(+)- 41,8 г гидрохлорида (S)-(+)- Оптическая чистота: 98,5% ee (ВЭЖХ). Пример 11: Получение метил-(S)-(+)- 7,9 г (S)-(+)- Оптическая чистота: 98,0% ee (ВЭЖХ). Ссылочный пример 1: Получение кислого сульфата клопидогрела (кислый сульфат формулы (I)) Согласно способу, раскрытому в европейском патенте Оптическая чистота: 98,5% ee (ВЭЖХ). Ссылочный пример 2: Получение 1,5-нафталиндисульфонатаклопидогрела (1,5-нафталиндисульфонат формулы (I)) Согласно способу, раскрытому в международной заявке PCT/KR2004/002665, 35,0 г клопидогрела, полученного в примере 9, растворяли в 200 мл ацетона. К раствору клопидогрела добавляли в течение 30 мин 20,2 г тетрагидрата 1,5-нафталиндисульфоновой кислоты, растворенной в смеси 140 мл ацетона и 10 мл воды, и полученную смесь перемешивали при комнатной температуре в течение 12 час и затем при температуре 0-5°C в течение 4 час. Осажденные кристаллы отфильтровывали, промывали 100 мл холодного ацетона и сушили при 50°C с получением 46,7 г кристаллов указанного в заголовке соединения в виде белого порошка (выход: 90%). Т.пл.: 223-225°C. Содержание воды: 1,91% (по методу Карла Фишера). Оптическая чистота: 99,8% ee (ВЭЖХ). Как показано выше, способ настоящего изобретения позволяет получить оптически чистый клопидогрел с высоким выходом. Несмотря на то, что изобретение описано со ссылкой на конкретные варианты осуществления, следует понимать, что специалистами в этой области могут быть сделаны различные модификации и изменения изобретения, которые также подпадают под объем изобретения, определяемый прилагаемой формулой изобретения.
Формула изобретения
1. Способ получения клопидогрела формулы (I), включающий стадии: 2. Способ по п.1, в котором оптическое разделение на стадии (а) включает взаимодействие рацемической формы соединения формулы (II) с оптически активным амином формулы (V) с кристаллизацией соли формулы (IV) и извлечение из нее оптически активного амина формулы (V) с получением оптически активной формы соединения формулы (III): 3. Способ по п.2, в котором оптически активный амин выбирают из группы, состоящей из (1R,2R)-(-)-2-амино-1-фенил-1,3-пропандиола и (1R,2R)-(-)-2-амино-1-(4-нитрофенил)-1,3-пропандиола. 4. Способ по п.2, в котором кристаллизацию соли формулы (IV) осуществляют путем добавления соединения формулы (II) и оптически активного амина формулы (V) в растворитель, перемешивания полученного раствора при температуре в интервале от комнатной температуры до температуры кипения растворителя и охлаждения полученной смеси до температуры в интервале от 0°С до комнатной температуры. 5. Способ по п.4, в котором оптически активный амин используют в количестве от 0,4 до 1,1 моль-эквивалентов по отношению к количеству соединения формулы (II). 6. Способ по п.4, в котором растворителем является органический растворитель, выбранный из группы, состоящей из метанола, этанола, изопропанола, ацетона, этилацетата, ацетонитрила, тетрагидрофурана, 1,4-диоксана, N,N-диметилформамида и их смеси, или смеси одного из органических растворителей и воды. 7. Способ по п.2, в котором извлечение оптически активного амина осуществляют путем подщелачивания водного раствора соли формулы (IV) с помощью основания. 8. Способ по п.7, в котором основание используют в количестве от 1 до 3 моль-эквивалентов по отношению к количеству соли формулы (IV). 9. Способ по п.7, в котором основанием является гидроксид натрия или гидроксид калия. 10. Способ по п.1, в котором рацемическую форму соединения формулы (II) получают путем проведения реакции замещения 11. Способ по п.10, в котором реакцию замещения проводят в присутствии основания. 12. Способ по п.11, в котором основание выбирают из группы, состоящей из гидроксида натрия, гидроксида калия, карбоната натрия, карбоната калия и их смеси. 13. Способ по п.11, в котором основание используют в количестве от 2 до 4 моль-эквивалентов по отношению к количеству 4,5,6,7-дигидро-4Н-тиено[3,2-с] пиридина или его кислотно-аддитивной соли. 14. Способ по п.1, в котором осуществляют стадию метилирования (b) путем (i) взаимодействия соединения формулы (III) или его кислотно-аддитивной соли с хлорирующим агентом в инертном растворителе с получением хлорированного соединения формулы (VI) или его кислотно-аддитивной соли, которое обрабатывают метанолом; или (ii) взаимодействия соединения формулы (III) или его кислотно-аддитивной соли с алкилхлорформиатом в инертном растворителе в присутствии основания с получением смешанного ангидрида формулы (VII), который обрабатывают метанолом; или (iii) взаимодействия соединения формулы (III) или его кислотно-аддитивной соли с метанолом в присутствии кислотного катализатора: 15. Способ по п.14, в котором хлорирующий агент выбирают из группы, состоящей из тионилхлорида, пентахлорида фосфора, хлористого фосфорила, фосгена, дифосгена, трифосгена, оксалилхлорида и их смеси. 16. Способ по п.14, в котором хлорирующим агентом является оксалилхлорид. 17. Способ по п.14, в котором хлорирующий агент используют в количестве от 1 до 5 моль-эквивалентов по отношению к количеству соединения формулы (III) или его кислотно-аддитивной соли. 18. Способ по п.14, в котором алкилхлорформиат выбирают из группы, состоящей из метилхлорформиата, этилхлорформиата, н-пропилхлорформиата, изопропилхлорформиата, изобутилхлорформиата и их смеси. 19. Способ по п.14, в котором основание выбирают из группы, состоящей из триэтиламина, диизопропилэтиламина, трибутиламина, пиридина, пиколина, лутидина, диметиламинопиридина и их смеси. 20. Способ по п.14, в котором кислотный катализатор выбирают из группы, состоящей из безводной хлористоводородной кислоты, безводной серной кислоты, метансульфоновой кислоты, пара-толуолсульфоновой кислоты и их смеси. 21. Соль формулы (IV), предназначенная для использования в качестве промежуточного соединения в способе по п.1 или 2: 22. Соль по п.21, в которой R является водородом или нитро.
|
||||||||||||||||||||||||||
 -(2-хлорфенил)-6,7-дигидротиено[3,2-a]пиридин-5(4H)ацетат формулы (I), является ингибитором агрегации тромбоцитов и эффективен при лечении заболеваний периферических артерий, таких как удар, тромбоз и эмболия, а также заболеваний коронарных артерий, таких как инфаркт миокарда и стенокардия:
-(2-хлорфенил)-6,7-дигидротиено[3,2-a]пиридин-5(4H)ацетат формулы (I), является ингибитором агрегации тромбоцитов и эффективен при лечении заболеваний периферических артерий, таких как удар, тромбоз и эмболия, а также заболеваний коронарных артерий, таких как инфаркт миокарда и стенокардия: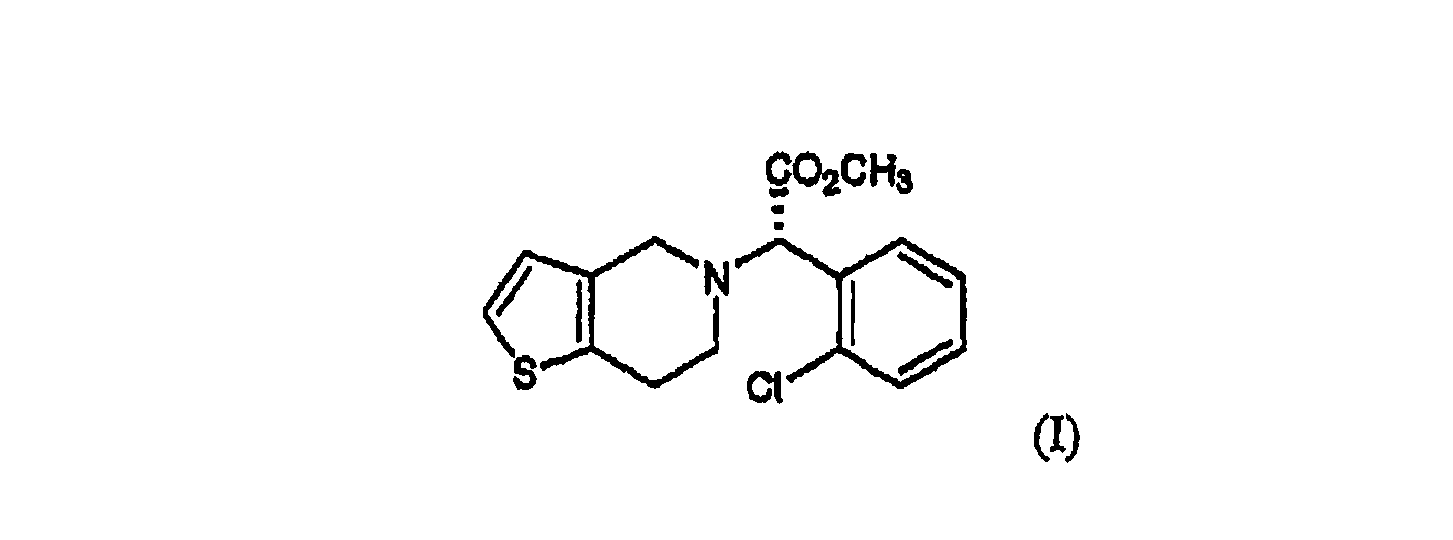
 0281459, 0466569, 0971915, 0099802, 1021449, 1404681 и 1353928 и в международной заявке WO
0281459, 0466569, 0971915, 0099802, 1021449, 1404681 и 1353928 и в международной заявке WO 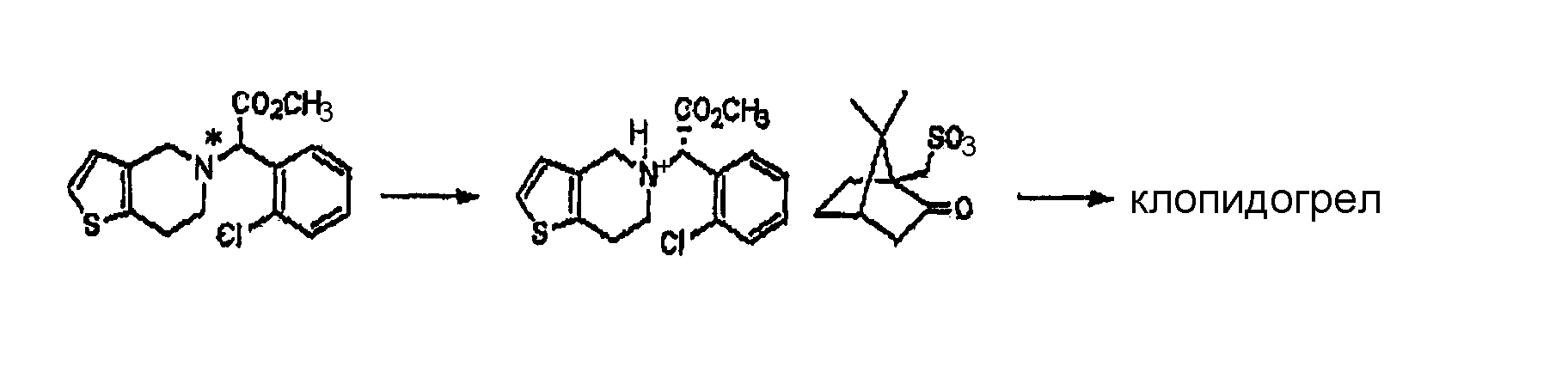
 C до комнатной температуры. Предпочтительно, чтобы количество используемого метанола находилось в интервале от 1 до 10 моль-эквивалентов по отношению к количеству соединения формулы (III) или его кислотно-аддитивной соли.
C до комнатной температуры. Предпочтительно, чтобы количество используемого метанола находилось в интервале от 1 до 10 моль-эквивалентов по отношению к количеству соединения формулы (III) или его кислотно-аддитивной соли. 3,00 (ушир.с, 2H), 3,16-3,56 (м, 2H), 4,05-4,28 (м, 2H), 5,14 (д, 1H, J=4,4 Гц), 6,04 (ушир.с, H), 6,55-6,68 (м, 1H), 7,04-7,31 (м, 1H), 7,31-7,41 (м, 1H), 7,85-7,98 (м, 1H)
3,00 (ушир.с, 2H), 3,16-3,56 (м, 2H), 4,05-4,28 (м, 2H), 5,14 (д, 1H, J=4,4 Гц), 6,04 (ушир.с, H), 6,55-6,68 (м, 1H), 7,04-7,31 (м, 1H), 7,31-7,41 (м, 1H), 7,85-7,98 (м, 1H) 20+9,53° (c=1%, ДМФ).
20+9,53° (c=1%, ДМФ).