Патент на изобретение №2357968
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНОЙ ИМИДАЗОЛА
(57) Реферат:
Изобретение относится к практически чистым полиморфным формам А и В N-[({2-[4-(2-этил-4,6-диметил-1Н-имидазо[4,5-с]пиридин-1-ил)фенил]этил}-амино)карбонил]-4-метилбензолсульфонамида, к фармацевтическим композициям, включающим данные полиморфные формы, а также к их применению и применению содержащих их фармацевтических композиций. Также изобретение относится к способу терапии боли, воспаления, остеоартрита или ревматоидного артрита, который включает введение вышеописанных полиморфных форм А и В. Технический результат: получены новые кристаллические формы N-[({2-[4-(2-этил-4,6-диметил-1Н-имидазо[4,5-с]пиридин-1-ил)фенил]этил}-амино)карбонил]-4-метилбензолсульфонамида, которые являются наиболее стабильными из идентифицированных форм. 10 н. и 4 з.п. ф-лы, 2 табл., 6 ил.
Настоящее изобретение относится к новым кристаллическим формам N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)-фенил]этил}амино)карбонил]-4-метилбензолсульфонамида (альтернативное название 2-этил-4,6-диметил-1-(4-{2-[({[(4-метилфенил)сульфонил]амино}карбонил)амино]этил}фенил)-1H-имидазо[4,5-c]пиридин).
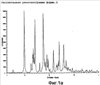
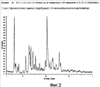
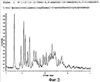
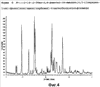
Более конкретно изобретение относится к полиморфным формам, известным как A и B, способам получения таких полиморфов, применению содержащих их композиций. N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида раскрыт в WO-A-02/32900 в качестве антагониста EP4 рецептора, который применяют для терапии или облегчения боли и воспаления и других, связанных с воспалением заболеваний, таких как артрит, в частности, остеоартрит. Использование N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфон- амида для лечения ревматоидного артрита также раскрыто в WO-A-02/32422. Кроме того, использование N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)- карбонил]-4-метилбензолсульфонамида при лечении заболеваний, связанных с ИЛ-6, таких как алкогольный цирроз, амилоидоз, атеросклероз, заболевание сердца, склероз и реакции при трансплантации органов, раскрыто в WO-A-03/086371. Ранее известные способы получения N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)- карбонил]-4-метилбензолсульфонамида, описанные в WO-A-02/32900, приводили к получению нестабильного сольвата, который может быть изоморфной формой кристаллической формы C. Целью этого изобретения является разработка фармацевтически приемлемой, практически чистой, кристаллической формы N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}-амино)карбонил]-4-метилбензолсульфонамида, которая может быть легко, экономически эффективно и воспроизводимо получена для применения в фармацевтической композиции с постоянными характеристиками, такими как стабильность и биодоступность. Неожиданно было обнаружено, что эта цель может быть достигнута с помощью настоящего изобретения, которое предлагает практически чистые, кристаллические полиморфные формы N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида, известные как полиморфная форма A и полиморфная форма B и изобретенный способ для получения каждого из них. Было обнаружено, что полиморфная форма A является самой стабильной из идентифицированных форм. Она является безводной, кристаллической, негигроскопической, с высокой температурой плавления и обладает приемлемыми свойствами для разработки твердой лекарственной формы. Было также обнаружено, что полиморфная форма B является стабильной и подходящей для использования в фармацевтическом составе. Соответственно, настоящее изобретение предлагает практически чистую, кристаллическую полиморфную форму A N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}-амино)карбонил]-4-метилбензолсульфонамида, которая характеризуется порошковой рентгенограммой (PXRD), полученной при использовании Cu K Полиморфная форма A N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида дополнительно характеризуется дифференциальной сканирующей калориметрией (DSC), при которой она проявляет эндотермический эффект при 160 Полиморфная форма А N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида еще дополнительно характеризуется инфракрасным (ИК) спектром (в KBr), который включает полосы поглощения при 2985, 2920, 2871, 1706, 1641, 1596, 1515, 1456, 1369, 1340, 1294, 1249, 1224, 1164, 1124, 1091, 1016, 902, 815, 659, 574 и 549 см-1. Настоящее изобретение дополнительно предлагает практически чистую, кристаллическую полиморфную форму В N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)- карбонил]-4-метилбензолсульфонамида, которая характеризуется порошковой рентгенограммой, полученной с использованием Cu K Полиморфная форма В N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метил-бензолсульфонамида дополнительно характеризуется дифференциальной сканирующей калориметрией (DSC), при которой она проявляет эндотермический эффект при 178 Полиморфная форма B N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида еще дополнительно характеризуется инфракрасным (ИК) спектром (в KBr), который включает специфические полосы поглощения при 3443, 3296 и 1704 см-1. Дополнительно описаны практически чистые, кристаллические формы C, D и G N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида. Следует иметь в виду, что эти кристаллические формы не являются только синтетическими промежуточными продуктами, которые могут быть далее превращены в полиморфные формы A и B N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил] -этил}амино)карбонил]-4-метилбензолсульфонамида, но они также обладают аналогичными терапевтическими свойствами. Однако кристаллические формы C, D и G N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида являются не такими удобными для использования при получении фармацевтических составов, как полиморфные формы A и B, в основном из-за того, что кристаллические формы являются менее стабильными. Кристаллические формы C, D и G N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида характеризуются рентгенограммой и дифференциальной сканирующей калориметрией (DSC), приведенными в таблице 1.
Используемое здесь выражение “практически чистый” означает, по меньшей мере, чистоту 95% по массе. Более предпочтительно, чтобы выражение “практически чистый” означало, по меньшей мере, чистоту 98% по массе и наиболее предпочтительно, по меньшей мере, чистоту 99% по массе. В качестве дополнительного аспекта изобретения предлагается полиморфная форма A или B N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)- карбонил]-4-метилбензолсульфонамида для использования в качестве лекарственного препарата. В качестве еще одного аспекта изобретения предлагается использование полиморфной формы A или B N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида в производстве лекарственного препарата для лечения заболевания, для которого применяют антагонист EP4 рецептора, в частности, для лекарственной, профилактической или паллиативной терапии боли, воспаления, остеоартрита или ревматоидного артрита. В качестве другого аспекта предлагается способ лечения любого заболевания, для которого применяют антагонист EP4 рецептора, в частности, для лекарственной, профилактической или паллиативной терапии боли, воспаления, остеоартрита или ревматоидного артрита, включающий введение терапевтически эффективного количества полиморфной формы A или B N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида млекопитающему, включая человека, нуждающемуся в таком лечении. Полиморфные формы A и B N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида настоящего изобретения используют для общей терапии боли. Физиологическая боль является важным защитным механизмом, предназначенным для предупреждения об опасности потенциально опасного воздействия из внешней среды. Система воздействует через специфический набор первичных сенсорных нейронов и активируется в результате вредного воздействия через периферийные трансдуцирующие механизмы (смотрите обзор Millan, 1999, Prog. Neurobiol., 57, 1-164). Эти сенсорные волокна известны в качестве ноцирецепторов, и они являются обычно нейритами небольшого диаметра с низкими скоростями проводимости. Ноцирецепторы кодируют интенсивность, продолжительность и качество вредного воздействия, и посредством их топографически организованного проецирования на спинной мозг, расположение воздействия. Ноцирецепторы основываются на ноцицептивных нервных волокнах, которые могут быть двух главных типов, A-дельта волокна (миелинизированные) и C волокна (немиелинизированные). Активность, генерируемая поступлением ноцирецептора, после сложной обработки в спинном мозге, переносится или непосредственно, или через передающее ядро ствола мозга к вентробазальному таламусу и затем на кортикальный слой, где генерируется ощущение боли. В целом боль может классифицироваться как острая или хроническая. Острая боль начинается внезапно и продолжается недолго (обычно двенадцать недель или меньше). Она обычно ассоциируется со специфической причиной, такой как конкретное повреждение, и является часто резкой и сильной. Она является видом боли, который может происходить после конкретных повреждений в результате хирургической операции, стоматологических вмешательств, растяжения или вывиха. Острая боль обычно не приводит к какой-либо устойчивой психологической реакции. В отличие от этого хроническая боль является продолжительной болью, которая обычно длится в течение более чем трех месяцев и приводит к существенным психологическим и эмоциональным проблемам. Типичными примерами хронической боли являются невропатическая боль (например, болевая диабетическая невропатия, постгерпетическая невралгия), синдром карпального канала, боль в пояснице, головная боль, боль при раке, боль при артрите и хроническая послеоперационная боль. Когда в результате болезни или травмы происходит значительное повреждение ткани организма, изменяются характеристики активации ноцирецептора и наблюдается сенсибилизация на периферии, локально вокруг повреждения и центрально, где ноцирецепторы оканчиваются. Эти эффекты ведут к повышенному болевому ощущению. При острой боли эти механизмы могут быть полезными для ускорения защитных реакций, которые могут способствовать процессам заживления. Естественно было бы ожидать, что чувствительность должна возвращаться к нормальному уровню, как только повреждение зажило. Однако при многих состояниях хронической боли гиперчувствительность значительно превышает по времени процесс заживления и, чаще всего, вследствие повреждения нервной системы. Это повреждение обычно ведет к нарушениям в волокнах чувствительного нерва, связанным с недостаточной приспособленностью и аберрантной активностью (Woolf & Salter, 2000, Science, 288, 1765-1768). Клиническая боль присутствует тогда, когда среди симптомов пациента характерной чертой являются дискомфорт и аномальная чувствительность. Так как пациенты обычно сильно отличаются друг от друга, то у них могут проявляться различные симптомы боли. Такие симптомы включают: 1) спонтанную боль, которая может быть тупой, жгучей или пронизывающей; 2) ненормально увеличенные болевые реакции на опасное воздействие (гипералгезия) и 3) боль, вызываемую обычно безопасным воздействием (аллодиния) (Meyer et al., 1994, Textbook of Pain, 13-44). Несмотря на то, что пациенты, страдающие от различных форм острой и хронической боли, могут иметь одинаковые симптомы, лежащие в основе механизмы могут быть различными и требовать, соответственно, различных схем лечения. Поэтому боль может также быть подразделена на ряд различных подтипов, согласно отличающейся патофизиологии, включая ноцицептивную боль, боль при воспалении и невропатическую боль. Ноцицептивная боль вызывается повреждением ткани или сильным воздействием со способностью вызывать повреждение. Афферентные боли активируются трансдукцией воздействия с помощью ноцирецепторов на месте поражения и активируют нейроны в спинном мозге на уровне их окончания. Это затем передается на спинномозговые пути в мозг, где боль воспринимается (Meyer et al., 1994, Textbook of Pain, 13-44). Активация ноцирецепторов активирует два типа афферентных нервных волокон. Миелинизированные A-дельта волокна являются быстропроводящими и ответственны за ощущения резкой и пронзительной боли, в то время как немиелинизированные C волокна имеют более низкую скорость проводимости и передают тупую или ноющую боль. Ноцицептивная боль, от умеренной до сильной и острой, является характерным признаком боли при травме центральной нервной системы, растяжениях/вывихах, ожогах, инфаркте миокарда и остром панкреатите, послеоперационной боли (боль, возникающая после любого типа хирургического вмешательства), посттравматической боли, почечной колики, боли при раке и боли в пояснице. Боль при раке может быть хронической болью, такой как боль, связанная с новообразованием (например, боль в костях, головная боль, лицевая боль или висцеральная боль), или болью, связанной с терапией рака (например, синдром после химиотерапии, синдром хронической послеоперационной боли или синдром после лучевой терапии). Боль при раке может также возникать в ответ на химиотерапию, иммунотерапию, гормональную терапию или радиотерапию. Боль в пояснице может быть связана с грыжей или разрывом межпозвоночных дисков или нарушений поясничных фасетных суставов, крестцово-повздошных сочленений, параспинальных мышц или задних продольных связок. Боль в пояснице может устраняться самопроизвольно, но у некоторых пациентов, когда она продолжается в течение 12 недель, она становится хроническим состоянием, которое может частично ухудшать здоровье. Невропатическую боль в настоящее время определяют как боль, инициируемую или вызванную первичным поражением или нарушением функции нервной системы. Поражение нерва может быть вызвано травмой и болезнью, и, таким образом, термин “невропатическая боль” включает в себя много заболеваний с разнообразными этиологиями. Они включают, но этим не ограничивая, периферическую невропатию, диабетическую невропатию, постгерпетическую невралгию, невралгию тройничного нерва, боль в пояснице, невропатию при раке, невропатию при ВИЧ, фантомные боли в ампутированных конечностях, синдром карпального канала, боль после инсульта центрального происхождения и боль, связанную с хроническим алкоголизмом, гипотиреозом, уремией, множественным склерозом, повреждением спинного мозга, болезнью Паркинсона, эпилепсией и авитаминозом. Невропатическая боль является патологической, так как она не выполняет защитной функции. Она часто продолжается значительно дольше того момента, как отпала первоначальная причина, часто многие годы, значительно понижая качество жизни пациента (Woolf and Mannion, 1999, Lancet, 353, 1959-1964). Симптомы невропатической боли трудно выявлять, так как они являются часто разнородными даже между пациентами с одной и той же болезнью (Woolf & Decosterd, 1999, Pain Supp., 6, S141-S147; Woolf and Mannion, 1999, Lancet, 353, 1959-1964). Они включают спонтанную боль, которая может быть постоянной, и приступообразную или атипичную приобретенную боль, такую как гипералгезия (повышенная чувствительность к вредному воздействию) или аллодиния (чувствительность к нормальному безвредному воздействию). Воспалительный процесс является сложным рядом биохимических и клеточных явлений, активируемых в ответ на повреждение ткани или присутствие чужеродных веществ, которые приводят к распуханию и боли (Levine and Taiwo, 1994, Textbook of Pain, 45-56). Боль при артрите является самой распространенной болью при воспалении. Ревматоидный артрит является одним из самых распространенных хронических воспалительных состояний в развитых странах, и ревматоидный артрит является частой причиной потери трудоспособности. Точная этиология ревматоидного артрита неизвестна, но в настоящее время предполагают, что могут быть важными как генетические, так и микробиологические факторы (Grennan & Jayson, 1994, Textbook of Pain, 397-407). По оценкам, почти 16 миллионов американцев страдают симптоматическим остеоартритом (OA) или дегенеративным поражением суставов, большинство из которых старше 60 лет, и ожидается, что это количество увеличится до 40 миллионов по мере старения населения, делая эту проблему здравоохранения громадного масштаба (Houge & Mersfelder, 2002, Ann Pharmacother., 36, 679-686; McCarthy et al., 1994, Textbook of Pain, 387-395). Большинство пациентов с остеоартритом нуждается в медицинской помощи из-за связанной с ним боли. Артрит имеет значительное воздействие на психосоциологическую и физическую функцию, и известно, что он является основной причиной нетрудоспособности в более позднем возрасте. Анкилозирующий спондилоартрит является также ревматическим заболеванием, которое вызывает артрит позвоночника и крестцово-повздошных сочленений. Он проявляется от перемежающихся приступов боли в пояснице, которые протекают в течение всей жизни, до тяжелого хронического заболевания, которое поражает позвоночник, периферические суставы и другие органы организма. Другим типом боли при воспалении является висцеральная боль, которая включает боль, связанную с воспалительной болезнью кишечника (IBD). Висцеральная боль является болью, связанной с внутренними органами, которые включают органы брюшной полости. Эти органы включают половые органы, селезёнку и часть пищеварительной системы. Боль, связанная с внутренними органами, может быть подразделена на висцеральную боль, связанную с пищеварительной системой, и висцеральную боль, не связанную с пищеварительной системой. Обычно встречающиеся желудочно-кишечные (ЖК) заболевания, которые вызывают боль, включают функциональное заболевание кишечника (ФЗК) и воспалительную болезнь кишечника (ВБК). Эти ЖК заболевания включают широкий диапазон болезненных состояний, которые в настоящее время контролируют только в определенной степени, включая, что касается ФЗК, гастроэзофагеальный рефлюкс, диспепсию, синдром раздражённой толстой кишки (IBS) и функциональный синдром боли в животе (FAPS), и, что касается ВБК, болезнь Крона, илеит и неспецифический язвенный колит, все, которые являются источником систематической висцеральной боли. Другие типы висцеральной боли включают боль, связанную с дисменореей, воспалением мочевого пузыря и панкреатитом, и боль в тазовой области. Следует отметить, что некоторые виды боли имеют множество этиологий и, таким образом, могут быть отнесены к более чем одной области, например, боль в пояснице и боль при раке имеют как ноцицептивные, так и невропатические компоненты. Другие типы боли включают: – боль, обусловленную костно-мышечными заболеваниями, включая миалгию, фибромиалгию, спондилит, сероотрицитальные (неревматоидные) артропатии, фиброзит, дистофинопатию, гликогенолиз, полимиозит и пиомиозит; – сердечную и сосудистую боль, включая боль, вызванную ангиной, инфарктом миокарда, стенозом митрального клапана, перикардитом, феноменом Рейно, склередомой взрослых и ишемией скелетной мышцы; – головную боль, такую как мигрень (включая мигрень с состоянием, предшествующим эпилептическому припадку, и мигрень без состояния, предшествующего эпилептическому припадку), гистаминовую головную боль, сдавливающую головную боль, смешанную головную боль и головную боль, связанную с сосудистыми заболеваниями; и – челюстную боль, включая зубную боль, ушную боль, синдром жжения во рту и боль височнонижнечелюстного сустава. Полиморфные формы N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида настоящего изобретения могут также быть использованы для лечения заболевания или состояния, выбранного из группы, состоящей из боли, лихорадки или воспаления, связанных с ревматической атакой, гриппом или другими вирусными инфекциями, простудой, болью в нижней части спины и шее, скелетной болью, послеродовой болью, дисменореей, головной болью, мигренью, зубной болью, растяжениями и вывихами, миозитом, невралгией, фибромиалгией, синовитом, артритом, включая ревматоидный артрит, дегенеративными поражениями суставов (остеоартритом), подагрой и анкилозирующим спондилоартритом, бурситом, ожогами, включая радиационное и разъедающее химическое повреждения, солнечной эритемой, болью после хирургического и стоматологического вмешательства или перелома кости, иммунными и аутоиммунными заболеваниями, такими как системная красная волчанка; СПИДом (синдромом приобретённого иммунодефицита), раками желудочно-кишечного тракта, такими как рак толстой кишки; клеточными малигнизациями или метастазированием; диабетической ретинопатией, опухолевым ангиогенезом; индуцированным простаноидами сокращением гладкой мышцы, связанным с дисменореей, преждевременными родами, аллергическим ринитом, атопическим дерматитом, астмой или заболеваниями, имеющими отношение к эозинофилу, гипериммуноглобулинемией, болезнью Кастлемена, миеломой; болезнью Альцгеймера, нарушениями сна, эндокринными нарушениями; глаукомой; разрежением кости; остеопорозом; активацией остеогенеза; болезнью Педжета; цитопротекцией при язве желудка и двенадцатиперстной кишки, гастритом, регионарным энтеритом, неспецифическим язвенным колитом, дивертикулитом или другими желудочно-кишечными нарушениями; желудочно-кишечным кровотечением и пациентами, подвергающимися химиотерапии; расстройствами коагуляции, выбранными из гипопротромбинемии, гемофилии и других проблем кровотечения; заболеванием почек; тромбозом; оклюзивным сосудистым заболеванием; подготовкой к операции; и антикоагуляцией. Полиморфные формы N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида настоящего изобретения также используют при лечении заболеваний, связанных с ИЛ-6, выбираемых из группы, состоящей из алкогольного цирроза, амилоидоза, атеросклероза, болезни сердца, такой как стенокардия, инфаркт миокарда, миокардиопатии и миокардита, склероза, такого как множественный склероз, и реакций при трансплантации органов. Способы синтезов для получения N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)- карбонил]-4-метилбензолсульфонамида описаны в WO-A-02/32900 и в приводимых ниже примерах. Полиморфная форма A N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида может быть получена путем кристаллизации из раствора N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида в этилацетате. В качестве варианта, полиморфная форма A N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)-карбонил]-4-метилбензолсульфонамида может быть получена путем кристаллизации из раствора N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида в ацетоне. В качестве варианта, полиморфная форма A N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)-карбонил]-4-метилбензолсульфонамида может быть получена путем кристаллизации из раствора полиморфной формы B в смеси ацетонитрила и этилацетата. В качестве варианта, полиморфная форма A N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)-карбонил]-4-метилбензолсульфонамида может быть получена путем кристаллизации из раствора полиморфной формы B в смеси дихлорметана и ацетона с использованием затравочного кристалла полиморфной формы A. В качестве варианта, полиморфная форма A N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)-карбонил]-4-метилбензолсульфонамида может быть получена путем кристаллизации из раствора полиморфной формы B в этаноле. Полиморфная форма B N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида может быть получена путем кристаллизации из раствора N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида в дихлорметане и ацетоне. В качестве варианта, полиморфная форма B N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)-карбонил]-4-метилбензолсульфонамида может быть получена путем кристаллизации из раствора N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида в дихлорметане путем частичной замены дихлорметана ацетоном. Полиморфные формы N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида настоящего изобретения могут быть введены отдельно или в комбинации с одним или более другими лекарственными средствами (или в виде любой их комбинации). Обычно их вводят в виде лекарственной формы совместно с одним или более фармацевтически приемлемыми наполнителями. Термин “наполнитель” используют здесь для обозначения любого ингредиента, отличного от соединения (соединений) изобретения. Выбор наполнителя будет зависеть в большой степени от таких факторов, как конкретный способ введения, влияние наполнителя на растворимость и стабильность и природа лекарственной формы. Таким образом, в качестве дополнительного аспекта настоящего изобретения предлагается фармацевтическая композиция, включающая полиморфную форму A или B N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино) карбонил]-4-метилбензолсульфонамида и один или более подходящих наполнителей. Композицию используют для терапии боли, воспаления, остеоартрита или ревматоидного артрита. Следует иметь в виду, что применяемый здесь термин “терапия” включает лечебную, паллиативную и профилактическую терапию. Для введения композиции животному, не принадлежащему к человеческому роду, используемый здесь термин “фармацевтический” может быть заменен термином “ветеринарный”. Фармацевтические композиции, используемые для доставки полиморфных форм изобретения, и способы их получения очевидны для специалистов в этой области. Такие композиции и способы их получения могут быть найдены, например, в монографии Remington’s Pharmaceutical Sciences, 19th Edition (Mack Publishing Company, 1995). ПЕРОРАЛЬНОЕ ВВЕДЕНИЕ Полиморфные формы изобретения могут быть введены перорально. Пероральное введение может включать проглатывание с тем, чтобы соединение попадало в желудочно-кишечный тракт, и/или буккальное, лингвальное или сублингвальное введение, с помощью которых соединение попадает в кровоток непосредственно изо рта. Лекарственные формы, используемые для перорального введения, включают твердые, полутвердые и жидкие системы, такие как таблетки; мягкие или твердые капсулы, содержащие множество твердых частиц или наночастиц, жидкости или порошки; пастилки (включая наполненные жидкостью); жевательные резинки; гели; быстро диспергирующиеся лекарственные формы; пленки; овули; спреи; и буккальные/приклеиваемые во рту пластыри. Жидкие лекарственные формы включают суспензии, растворы, сиропы и эликсиры. Такие формы могут быть использованы в виде наполнителей в мягких или твердых капсулах (изготовленных, например, из желатина или гидроксипропилметилцеллюлозы), и они обычно включают носитель, например, воду, этанол, полиэтиленгликоль, пропиленгликоль, метилцеллюлозу или подходящее масло, и один или более эмульгаторов и/или суспендирующих веществ. Жидкие формы могут также быть получены путем растворения твердого вещества, например, из пакетика. Полиморфные формы изобретения могут также быть использованы в быстро растворимых, быстро распадающихся лекарственных формах, таких как описанные в заключении эксперта в Therapeutic Patents, 11 (6), 981-986, by Liang and Chen (2001). Для лекарственных форм в виде таблеток, в зависимости от дозы, лекарственное средство может составлять от 1 до 80 мас.% от лекарственной формы, более характерно – от 5 до 60 мас.% от массы лекарственной формы. В дополнение к лекарственному средству, таблетки обычно содержат вещество, вызывающее распад. Примеры веществ, вызывающих распад, включают натрия крахмалгликолят, карбоксиметилцеллюлозу натрия, карбоксиметилцеллюлозу кальция, натрия кроскармелозу, кросповидон, поливинилпирролидон, метилцеллюлозу, микрокристаллическую целлюлозу, замещенную низшим алкилом гидроксипропилцеллюлозу, крахмал, пептизированный крахмал и альгинат натрия. Обычно вещество, вызывающее распад, составляет от 1 до 25 мас.%, предпочтительно от 5 до 20 мас.% от массы лекарственной формы. Связующие обычно используют для придания когезионных качеств форме в виде таблетки. Подходящие связующие включают микрокристаллическую целлюлозу, желатин, сахара, полиэтиленгликоль, природные и синтетические камеди, поливинилпирролидон, пептизированный крахмал, гидроксипропилцеллюлозу и гидроксипропилметилцеллюлозу. Таблетки могут также содержать разбавители, такие как лактоза (моногидрат, высушенный распылением моногидрат, безводная и другая подобная), маннит, ксилит, декстроза, сахароза, сорбит, микрокристаллическая целлюлоза, крахмал и дигидрат двухосновного фосфата кальция. Таблетки могут также необязательно включать поверхностно-активные вещества, такие как натрийлаурилсульфат и полисорбат 80, и вещества, способствующие скольжению, такие как диоксид кремния или тальк. В случае их присутствия поверхностно-активные вещества могут составлять от 0,2 до 5 мас.% от массы таблетки, и вещества, способствующие скольжению, могут составлять от 0,2 до 1 мас.% от массы таблетки. Таблетки также обычно содержат скользящие вещества, такие как стеарат магния, стеарат кальция, стеарат цинка, натрия стеарилфумарат и смеси стеарата магния с натрийлаурилсульфатом. Скользящие вещества обычно составляют от 0,25 до 10 мас.%, предпочтительно от 0,5 до 3 мас.% от массы таблетки. Другие возможные ингредиенты включают антиоксиданты, красители, ароматизаторы, консерванты и вещества, исправляющие вкус лекарственного средства. Типичные таблетки содержат до 80% лекарственного средства, от 10 до 90 мас.% связующего, от 0 до 85 мас.% разбавителя, от 2 до 10 мас.% вещества, вызывающего распад, и от 0,25 до 10 мас.% скользящего вещества. Смеси для изготовления таблеток могут быть подвергнуты прессованию непосредственно или с помощью валика с получением таблеток. Смеси для изготовления таблеток или части смесей могут, в качестве варианта, быть подвергнуты мокрому гранулированию, сухому гранулированию или гранулированию из расплава, застыванию из расплава или экструзии перед таблетированием. Конечная форма может включать один или более слоев и может быть с покрытием или без покрытия; она может даже быть инкапсулированной. Получение таблеток обсуждается в монографии Pharmaceutical Dosage Forms: Tablets, Vol. 1, by H. Lieberman and Л. Lachman (Marcel Dekker, New York, 1980). Расходуемые пероральные пленки для использования человеком и в ветеринарии являются обычно лекарственными формами в виде гибкой водорастворимой или набухаемой в воде тонкой пленки, которая может быстро растворяться или прилипать во рту, и они обычно включают полиморфную форму согласно изобретению, пленкообразующий полимер, связующее, растворитель, увлажнитель, пластификатор, стабилизатор или эмульгатор, модификатор вязкости и растворитель. Некоторые компоненты лекарственной формы могут выполнять более чем одну функцию. Полиморфные формы изобретения могут быть растворимыми или нерастворимыми в воде. Водорастворимое соединение обычно составляет от 1 до 80 мас.%, более характерно – от 20 до 50 мас.% от массы растворов. Менее растворимые соединения могут составлять большую часть композиции, обычно до 88 мас.% от массы растворов. В качестве варианта, полиморфные формы изобретения могут быть в форме гранул из множества частиц. Пленкообразующий полимер может быть выбран из природных полисахаридов, белков или синтетических гидроколлоидов и обычно присутствует в интервале от 0,01 до 99 мас.%, более характерно – в интервале от 30 до 80 мас.%. Другие возможные ингредиенты включают антиоксиданты, красители, ароматизаторы и усилители аромата, консерванты, стимуляторы слюноотделения, охлаждающие средства, сорастворители (включая масла), смягчающие вещества, наполнители, пеногасители, поверхностно-активные вещества и вещества, исправляющие вкус лекарственного средства. Пленки согласно изобретению обычно получают испарительной сушкой тонких водных пленок, нанесенных на отслаивающийся поддерживающий носитель или бумагу. Это может быть осуществлено в сушильной печи или камере, обычно с помощью совмещенной машины для нанесения покрытия с сушкой, или путем лиофильной сушки, или путем вакуумирования. Твердые лекарственные формы для перорального введения могут быть получены в виде формы с немедленным высвобождением лекарственного средства и/или с модифицированным высвобождением. Формы с модифицированным высвобождением лекарственного средства включают замедленное высвобождение, длительное высвобождение, импульсное высвобождение, регулируемое высвобождение, целенаправленное высвобождение и программированное высвобождение. Подходящие лекарственные формы с модифицированным высвобождением для целей изобретения описаны в Патенте США 6106864. Детали других подходящих технологий высвобождения, таких как высокоэнергетические дисперсии и осмотические и с нанесенным покрытием частицы можно найти в Pharmaceutical Technology On-line, 25(2), 1-14, by Verma et al (2001). Использование жевательной резинки для достижения контролируемого высвобождения описано в патентной заявке WO 00/35298. ПАРЕНТЕРАЛЬНОЕ ВВЕДЕНИЕ Полиморфные формы изобретения могут также быть введены непосредственно в кровоток, в мышцу или во внутренний орган. Подходящие методы для парентерального введения включают внутривенное, внутриартериальное, интраперитонеальное, интратекальное, интравентрикулярное, интрауретральное, надчревное, интракраниальное, внутримышечное, внутрисуставное и подкожное введение. Подходящие устройства для парентерального введения включают игольчатые (включая микроигольчатые) шприцы, безигольчатые шприцы и техническое оснащение для инфузии. Парентеральные лекарственные формы обычно являются водными растворами, которые могут содержать наполнители, такие как соли, углеводы и буферы (предпочтительно для pH от 3 до 9), но, для ряда применений, их может быть более удобно приготавливать в виде стерильного неводного раствора или в виде высушенной формы, используемой в сочетании с подходящим носителем, таким как стерильная апирогенная вода. Приготовление парентеральных лекарственных форм в стерильных условиях, например, путем лиофилизации, может быть легко осуществлено с использованием стандартных фармацевтических способов, хорошо известных специалистам в этой области. Формы для парентерального введения могут быть получены в виде формы с немедленным высвобождением лекарственного средства и/или с модифицированным высвобождением. Формы с модифицированным высвобождением лекарственного средства включают замедленное высвобождение, длительное высвобождение, импульсное высвобождение, регулируемое высвобождение, целенаправленное высвобождение и программированное высвобождение. Таким образом, полиморфные формы изобретения могут быть получены в виде суспензии или твердого вещества, полутвердого вещества или тиксотропной жидкости для введения в качестве имплантированного депо, обеспечивающего модифицированное высвобождение активного соединения. Примеры таких лекарственных форм включают покрытые лекарственным средством стенты и полутвердые вещества и суспензии, включающие заполненные лекарственным средством микросферы из сополимера молочной и гликолевой кислот (PGLA). МЕСТНОЕ ВВЕДЕНИЕ Полиморфные формы изобретения могут также быть введены местно, (интра)дермально или трансдермально на коже или слизистой оболочке. Типичные формы для этой цели включают гели, гидрогели, лосьоны, растворы, кремы, мази, присыпки, повязки, пены, пленки, пластыри на кожу, пластинки, имплантанты, тампоны, волокно, бинты и микроэмульсии. Могут также быть использованы липосомы. Типичные носители включают спирт, воду, минеральное масло, вазелиновое масло, медицинский вазелин, глицерин, полиэтиленгликоль и пропиленгликоль. Могут быть введены вещества, способствующие проникновению, – смотрите, например, J Pharm Sci, 88 (10), 955-958, by Finnin and Morgan (October 1999). Другие способы местного введения включают доставку с помощью электропорации, ионтофореза, фонофореза, сонофореза и микроигольчатой и безигольчатой (например, Powderject Лекарственные формы для местного введения могут быть получены в виде формы с немедленным высвобождением лекарственного средства и/или с модифицированным высвобождением. Формы с модифицированным высвобождением лекарственного средства включают замедленное высвобождение, длительное высвобождение, импульсное высвобождение, регулируемое высвобождение, целенаправленное высвобождение и программированное высвобождение. Полиморфные формы изобретения могут также быть введены интраназально или путем ингаляции, обычно в форме сухого порошка (или отдельно, в виде смеси, например, сухой смеси с лактозой, или в виде компонента смеси в форме частицы, например, смешанной с фосфолипидами, такими как фосфатидилхолин) из ингалятора сухого порошка, в форме струи аэрозоля из контейнера под давлением, насоса, спрея, пульверизатора (предпочтительно пульверизатора, использующего электрогидродинамику для получения мелкодисперсного тумана), или распылителя с использованием или без использования соответствующего пропеллента, такого как 1,1,1,2-тетрафторэтана или 1,1,1,2,3,3,3-гептафторпропана, или в виде каплей для носа. Для интраназального применения порошок может включать биоадгезивное вещество, например, хитозан или циклодекстрин. Контейнер под давлением, насос, спрей, пульверизатор или распылитель содержит раствор или суспензию полиморфной формы согласно изобретению, включающий, например, этанол, водный этанол или подходящее альтернативное вещество для диспергирования, солюбилизации или продолжительного высвобождения активного вещества, пропеллент (пропелленты) в виде растворителя и необязательное поверхностно-активное вещество, такое как сорбитантриолеат, олеиновая кислота или олигомолочная кислота. Перед использованием лекарственной формы в виде сухого порошка или суспензии готовую лекарственную форму измельчают до размера, подходящего для доставки ингаляцией (обычно меньше, чем 5 микрон). Это может быть достигнуто с помощью соответствующего способа измельчения, такого как размол на спиральной струйной мельнице, струйной мельнице в псевдоожиженном слое, обработка в сверхкритической среде с образованием наночастиц, гомогенизация при высоком давлении или распылительная сушка. Могут быть приготовлены капсулы (изготовленные, например, из желатина или гидроксипропилметилцеллюлозы), блистеры и картриджи для использования в ингаляторе или инсуффляторе, содержащие порошковую смесь соединения изобретения, подходящую порошковую основу, такую как лактоза или крахмал, и модификатор свойств, такой как Л-лейцин, маннит или стеарат магния. Лактоза может быть безводной или в форме моногидрата, предпочтительно в форме моногидрата. Другие подходящие наполнители включают декстран, глюкозу, мальтозу, сорбит, ксилит, фруктозу, сахарозу и трегалозу. Соответствующая форма в виде раствора для применения в пульверизаторе с использованием электрогидродинамики для получения мелкодисперсного тумана может содержать от 1 мкг до 20 мг соединения изобретения на одну загрузку, и объем загрузки может изменяться от 1 мкл до 100 мкл. Типичная форма может включать полиморфную форму согласно изобретению, пропиленгликоль, стерильную воду, этанол и хлорид натрия. Альтернативные растворители, которые могут быть использованы вместо пропиленгликоля, включают глицерин и полиэтиленгликоль. Подходящие ароматизаторы, такие как ментол или левоментол, или подсластители, такие как сахарин или сахарин натрия, могут быть добавлены в эти лекарственные формы изобретения, предназначенные для ингаляционного/интраназального введения. Лекарственные формы для ингаляционного/интраназального введения могут быть получены в виде формы с немедленным высвобождением лекарственного средства и/или с модифицированным высвобождением с использованием, например, сополимера молочной и гликолевой кислот. Формы с модифицированным высвобождением лекарственного средства включают замедленное высвобождение, длительное высвобождение, импульсное высвобождение, регулируемое высвобождение, целенаправленное высвобождение и программированное высвобождение. В случае ингаляторов сухого порошка и аэрозольных баллончиков введение разовой дозы осуществляется с помощью клапана, который отпускает дозированное количество. Приборы в соответствии с изобретением обычно приспособлены для введения отмеренной дозы или “вдувания”, содержащего от 1 мкг до 20 мг соединения формулы 1. Суммарная дневная доза обычно составляет от 1 мкг до 100 мг, которая может быть введена однократно или, чаще всего, в виде разделенных доз в течение дня. РЕКТАЛЬНОЕ/ ИНТРАВАГИНАЛЬНОЕ ВВЕДЕНИЕ Полиморфные формы изобретения могут быть введены ректально или вагинально, например, в форме суппозитория, вагинального суппозитория или спринцовки. Масло какао является традиционной основой суппозитория, но по обстоятельствам могут быть использованы различные варианты. Лекарственные формы для ректального/вагинального введения могут быть получены в виде формы с немедленным высвобождением лекарственного средства и/или с модифицированным высвобождением. Формы с модифицированным высвобождением лекарственного средства включают замедленное высвобождение, длительное высвобождение, импульсное высвобождение, регулируемое высвобождение, целенаправленное высвобождение и программированное высвобождение. ГЛАЗНОЕ/УШНОЕ ВВЕДЕНИЕ Полиморфные формы изобретения могут быть также введены непосредственно в глаз или ухо, обычно в форме капель микроизмельченной суспензии или раствора в изотоническом, скорректированном по рН, стерильном физиологическом растворе. Другие формы, подходящие для глазного и ушного введения, включают мази, гели, биоразлагаемые (например, рассасывающиеся гелевые тампоны, коллаген) и небиоразлагаемые (например, силиконовые) имплантанты, пластинки, линзы и корпускулярные и везикулярные системы, такие как ниосомы или липосомы. Полимер, такой как сшитая полиакриловая кислота, поливиниловый спирт, гиалуроновая кислота, целлюлозный полимер, например, гидроксипропилметилцеллюлоза, гидроксиэтилцеллюлоза или метилцеллюлоза, или гетерополисахаридный полимер, например, gelan gum, могут быть введены вместе с консервантом, таким как бензалконий хлорид. Такие лекарственные формы могут также быть введены с помощью ионтофореза. Лекарственные формы для глазного/ушного введения могут быть получены в виде формы с немедленным высвобождением лекарственного средства и/или с модифицированным высвобождением. Формы с модифицированным высвобождением лекарственного средства включают замедленное высвобождение, длительное высвобождение, импульсное высвобождение, регулируемое высвобождение, целенаправленное высвобождение и программированное высвобождение. ДРУГИЕ МЕТОДЫ Полиморфные формы изобретения могут быть объединены с растворимыми макромолекулярными структурами, такими как циклодекстрин и подходящими его производными или полимерами, содержащими полиэтиленгликоль, для улучшения их растворимости, скорости растворения, улучшения вкуса, биологической усвояемости и/или стабильности при использовании в любой из вышеупомянутых форм введения. Обнаружено, что, например, комплексы “лекарственное средство-декстрин” обычно подходят для большинства лекарственных форм и способов введения. Могут быть использованы как комплексы включения, так и комплексы, не являющиеся комплексами включения. В качестве варианта непосредственного комплексообразования с лекарственным средством, циклодекстрин может быть использован в качестве вспомогательной добавки, то есть в качестве носителя, разбавителя или солюбилизатора. Наиболее часто используемыми для этих целей являются альфа-, бета- и гамма-циклодекстрины, примеры которых могут быть найдены в Международных патентных заявках WO 91/11172, WO 94/02518 и WO 98/55148. НАБОР При необходимости введения комбинации активных соединений, например, для целей лечения конкретного заболевания или состояния, в объем настоящего изобретения могут входить две или более фармацевтические композиции, по меньшей мере, одна из которых содержит полиморфную форму согласно изобретению, могут удобно быть объединены в виде набора, подходящего для совместного введения композиций. Таким образом, набор изобретения включает две или более отдельных фармацевтических композиций, по меньшей мере, одна из которых содержит полиморфную форму согласно изобретению, и средства для раздельного сохранения указанных композиций, такого как контейнер, флакон с несколькими отделениями или упаковка из фольги с несколькими отделениями. Примером такого набора является известная блистерная упаковка, используемая для расфасовки таблеток, капсул и другие подобные наборы. Набор изобретения особенно подходит для введения различных лекарственных форм, например, пероральных и парентеральных, для введения отдельных композиций при различных временных интервалах между введением лекарственного средства, или для определения титра отдельных композиций относительно друг друга. Для облегчения соответствия набор обычно содержит инструкции по применению, и он может быть обеспечен так называемой памяткой. ДОЗА Для введения людям суммарная дневная доза полиморфных форм изобретения обычно составляет от 0,1 до 3000 мг в зависимости, разумеется, от способа введения. Например, пероральное введение может потребовать суммарной дневной дозы от 1 до 3000 мг, в то время как доза внутривенно может составлять от 0,1 до 300 мг. Суммарная дневная доза может быть введена однократно или в виде разделенных доз и может, по усмотрению врача, выходить за пределы приведенного здесь обычного интервала. Предпочтительно, чтобы суммарная дневная доза составляла от 0,1 до 500 мг. Эти дозы определены для среднего обследуемого человека с массой от 60 до 70 кг. Врач может легко определить дозы для субъектов, чья масса выходит за пределы этого интервала, таких как дети младшего возраста и пожилые люди. Следует иметь в виду, что применяемый здесь термин “терапия” включают лечебную, паллиативную и профилактическую терапию. Полиморфные формы настоящего изобретения могут также, необязательно, объединены с другим фармакологически активным соединением или с двумя или более другими фармакологически активными соединениями, в частности, при лечении боли. Например, полиморфные формы настоящего изобретения, определенные выше, могут быть введены одновременно, последовательно или раздельно в комбинации с одним или более средствами, выбранными из: – опиоидного аналгетика, например, морфина, героина, гидроморфона, оксиморфона, леворфанола, леваллорфана, метадона, меперидина, фентанила, кокаина, кодеина, дигидрокодеина, оксикодона, гирокодона, пропоксифена, налмефена, налорфина, налоксона, налтрексона, бупренорфина, буторфанола, налбуфина или пентазоцина; – нестероидного противовоспалительного лекарственного средства (NSAID), например, аспирина, диклофенака, дифлузинала, этодолака, фенбуфена, фенопрофена, флуфенизала, флурбипрофена, ибупрофена, индометацина, кетопрофена, кеторолака, меклофенамовой кислоты, мефенамовой кислоты, мелоксикама, набуметона, напроксена, нимесулида, нитрофлюрбипрофена, олсалазина, оксапрозина, фенилбутазона, пироксикама, сульфасалазина, сулиндака, толметина или зомепирака; – барбитуратного седативного средства, например, амобарбитала, апробарбитала, бутабарбитала, бутабитала, мефобарбитала, метарбитала, метогекситала, пентобарбитала, фенобарбитала, секобарбитала, талбутала, теамилала или тиопентала; – бензодиазепина, имеющего седативное действие, например, хлордиазепоксида, клоразепата, диазепама, флуразепама, лоразепама, оксазепама, темазепама или триазолама; – H1 антагониста, имеющего седативное действие, например, дифенгидрамина, пириламина, прометазина, хлорфенирамина или хлорциклизина; – седативного средства, такого как глютетимида, мепробамата, метаквалона или дихлоралфеназона; – релаксанта скелетной мышцы, например, баклофена, карисопродола, хлорзоксазона, циклобензаприна, метокарбамола или орфренадина; – антагониста NMDA рецептора, например, декстрометорфана ((+)-3-гидрокси-N-метилморфинана) или его метаболита декстрорфана ((+)-3-гидрокси-N-метилморфинана), кетамина, мемантина, пирролохинолин хинина, цис-4-(фосфонометил)-2- пиперидинкарбоновой кислоты, будипина, EN-3231 (MorphiDex®, комбинированной формы морфина и декстрометорфана), топирамата, нерамексана или перзинфотела, включая NR2B антагонист, например, ифенпродил, траксопродил или (-)-(R)-6-{2-[4-(3-фторфенил)-4-гидрокси-1-пиперидинил]-1-гидроксиэтил-3,4-дигидро-2(1H)-хиноли-нон; – альфа-адренергического средства, например, доксазозина, тамсулозина, клонидина, гуанфацина, дексметатомидина, модафинила или 4-амино-6,7-диметокси-2-(5-метансульфонамидо-1,2,3,4-тетрагидроизохинол-2-ил)-5-(2-пиридил) хиназолина; – трициклического антидепрессанта, например, дезипрамина, имипрамина, амитриптилина или нортриптилина; – противосудорожного средства, например, карбамазепина, ламотригина, топиратмата или вальпроата; – антагониста тахикинина (NK), в частности, антагониста NK-3, NK-2 или NK-1, например, ( – мускаринового антагониста, например, оксибутинина, толтеродина, пропиверина, тропсиум хлорида, дарифенацина, солифенацина, темиверина и ипратропия; – селективного ингибитора COX-2, например целекоксиба, рофекоксиба, парекоксиба, валдекоксиба, деракоксиба, эторикоксиба или лумиракоксиба; – анальгетика, полученного из каменноугольной смолы, в частности, парацетамола; – нейролептика, такого как дроперидол, хлорпромазин, галоперидол, перфеназин, тиоридазин, мезоридазин, трифторперазин, фторфеназин, клозапин, оланзапин, рисперидон, зипразидон, кветиапин, сертиндол, арипипразол, сонепипразол, блонансерин, илоперидон, пероспирон, раклоприд, зотепин, бифепрунокс, азенапин, лурасидон, амисульприд, балаперидон, палиндор, эпливансерин, осанетант, римонабант, меклинертант, Мираксион® или саризотан; – агониста ванилоидного рецептора (например, резинфератоксина) или антагониста (например, капсазепина); – бета-адренергического средства, такого как пропранолол; – анестезирующего средства местного действия, такого как мексилетин; – кортикостероида, такого как дексаметазон; – агониста или антагониста 5-HT рецептора, в частности, 5-HT1B/1D агониста, такого как элетриптан, суматриптан, наратриптан, золмитриптан или ризатриптан; – агониста 5-HT2A рецептора, такого как R(+)-alpha-(2,3-диметоксифенил)-1-[2-(4-фторфенилэтил)]-4-пиперидинметанол (MDL-100907); – холинергического (никотинового) анальгетика, такого как изпрониклин (TC-1734), (E)-N-метил-4-(3-пиридинил)-3-бутен-1-амин (RJR-2403), (R)-5-(2-азетидинилметокси)-2-хлорпиридин (ABT-594) или никотин; – Трамадола ®; – ингибитора PDEV, такого как 5-[2-этокси-5-(4-метил-1-пиперазинилсульфонил)фенил]-1-метил-3-н-пропил-1,6-дигидро-7H-пиразоло[4,3-d]пиримидин-7-он (силденафил), (6R,12aR)-2,3,6,7,12,12a-гексагидро-2-метил-6-(3,4-метилендиоксифенил)-пиразино[2′,1′:6,1]-пиридо[3,4-b]индол-1,4-дион (IC-351 или тадалафил), 2-[2-этокси-5-(4-этилпиперазин-1-ил-1-сульфонил)-фенил]-5-метил-7-пропил-3H-имидазо[5,1-f][1,2,4]триазин-4-он (варденафил), 5-(5-ацетил-2-бутокси-3-пиридинил)-3-этил-2-(1-этил-3-азетидинил)-2,6-дигидро-7H-пиразоло[4,3-d]пиримидин-7-он, 5-(5-ацетил-2-пропокси-3-пиридинил)-3-этил-2-(1-изопропил-3-азетидинил)-2,6-дигидро-7H-пиразоло[4,3-d]пиримидин-7-он, 5-[2-этокси-5-(4-этилпиперазин-1-илсульфонил)пиридин-3-ил]-3-этил-2-[2-метоксиэтил]-2,6-дигидро-7H-пиразоло[4,3-d]пиримидин-7-он, 4-[(3-хлор-4-метоксибензил)амино]-2-[(2S)-2-(гидроксиметил)-пирролидин-1-ил]-N-(пиримидин-2-илметил)пиримидин-5-карбоксамид, 3-(1-метил-7-оксо-3-пропил-6,7-дигидро-1H-пиразоло[4,3-d]- пиримидин-5-ил)-N-[2-(1-метилпирролидин-2-ил)этил]-4-пропокси-бензолсульфонамид; – альфа-2-дельта лиганда, такого как габапентин, прегабалин, 3-метилгабапентин, (1 – каннабиноида; – антагониста метаботропных глутаматных рецепторов подтипа 1 (mGluR1); – ингибитора повторного поглощения серотонина, такого как сертралин, метаболит сертралина диметилсертралин, флуоксетин, норфлуоксетин (метаболит дезметилфлуоксетин), флувоксамин, пароксетин, циталопрам, метаболит циталопрама дезметилциталопрам, эсциталопрам, d,l- фенфлурамин, фемокситин, ифоксетин, цианодотиепин, литоксетин, дапоксетин, нефазодон, церикламин и тразодон; – ингибитора повторного поглощения норадреналина (норэпинефрина), такого как мапротилин, лофепрамин, миртазепин, оксапротилин, фезоламин, томоксетин, миансерин, бупроприон, метаболит бупроприона гидроксибупроприон, номифензин и вилоксазин (Вивалан®), особенно из селективного ингибитора повторного поглощения норадреналина, такого как ребоксетин, в частности, (S,S)-ребоксетин; – двойного ингибитора повторного поглощения серотонина-норадреналина, такого как венлафаксин, метаболит венлафаксина O-дезметилвенлафаксин, кломипрамин, метаболит кломипрамина дезметилкломипрамин, дулоксетин, милнаципран и имипрамин; – ингибитора, индуцируемого синтазой окиси азота (iNOS), такого как S-[2-[(1-иминоэтил)амино]этил]-Л-гомоцистеин, S-[2-[(1-иминоэтил)амино]этил]-4,4-диоксо-Л-цистеин, S-[2-[(1-иминоэтил)-амино]этил]-2-метил-Л-цистеин, (2S,5Z)-2-амино-2-метил-7-[(1-иминоэтил)амино]-5-гептановая кислота, 2-[[(1R,3S)-3-амино-4-гидрокси-1-(5-тиазолил)бутил]тио]-5-хлор-3-пиридин-карбонитрил; 2-[[(1R,3S)-3-амино-4-гидрокси-1-(5-тиазолил)-бутил]тио]-4-хлорбензонитрил, (2S,4R)-2-амино-4-[[2-хлор-5-(трифторметил)-фенил]тио]-5-тиазолбутанол, 2-[[(1R,3S)-3-амино-4-гидрокси-1-(5-тиазолил)бутил]тио]-6-(трифторметил)-3-пиридин-карбонитрил, 2-[[(1R,3S)-3-амино-4-гидрокси-1-(5-тиазолил)-бутил]тио]-5-хлорбензонитрил, N-[4-[2-(3-хлорбензиламино)этил]-фенил]тиофен-2-карбоксамидин или гуанидиноэтилдисульфид; – ингибитора ацетилхолинэстеразы, такого как донепезил; – антагониста лейкотриена B4, такого как 1-(3-бифенил-4-илметил-4-гидроксихроман-7-ил)циклопентанкарбоновая кислота (CP-105696), 5-[2-(2-карбоксиэтил)-3-[6-(4-метоксифенил)-5E- гексенил]оксифенокси]валериановая кислота (ONO-4057) или DPC-11870; – ингибитора 5-липоксигеназы, такого как зилеутон, 6-[(3-фтор-5-[4-метокси-3,4,5,6-тетрагидро-2H-пиран-4-ил])фенокси-метил]-1-метил-2-хинолон (ZD-2138) или 2,3,5-триметил-6-(3-пиридилметил),1,4-бензохинон (CV-6504); – блокатора натриевых каналов, такого как лидокаин; – антагониста 5-HT3, такого как ондансетрон, и из их фармацевтически приемлемых солей и сольватов. Настоящее изобретение распространяется на комбинацию, включающую полиморфную форму A или B N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]-пиридин-1-ил)фенил]этил}амино)карбо-нил]-4-метилбензолсульфонамида и одно или более перечисленных выше терапевтических средств для одновременного, раздельного или последовательного использования в лечебной, профилактической или паллиативной терапии боли, воспаления, остеоартрита или ревматоидного артрита. Таким образом, изобретение предлагает: I. Практически чистые, кристаллические полиморфные формы A и B N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)-фенил]этил}амино)карбонил]-4-метилбензолсульфонамида. II. Способы получения практически чистых, кристаллических полиморфных форм A и B N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)-фенил]этил}амино)карбонил]-4-метилбензолсульфонамида. III. Фармацевтическую композицию, включающую практически чистые, кристаллические полиморфные формы A и B N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)-фенил]этил}амино)-карбонил]-4-метилбензолсульфонамида и один или более фармацевтически приемлемые наполнители. IV. Практически чистые, кристаллические полиморфные формы A и B N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)-фенил]этил}амино)карбонил]-4-метилбензолсульфонамида для применения в качестве лекарственного препарата. V. Применение практически чистых, кристаллических полиморфных форм A и B N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)-фенил]этил}амино)карбонил]-4-метилбензолсульфонамида в производстве лекарственного препарата для терапии боли, воспаления, остеоартрита или ревматоидного артрита. VI. Способ терапии боли, воспаления, остеоартрита или ревматоидного артрита, который включает введение эффективного количества практически чистых, кристаллических полиморфных форм A и B N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)-фенил]этил}амино)карбонил]-4-метилбензолсульфонамида, или фармацевтически приемлемой их композиции животному, включая человека, в случае необходимости такой терапии. ПРИМЕРЫ Следующий пример приводится только в качестве иллюстрации. ПРИМЕР 1 2-Этил-5,7-диметил-3-(4-{2-[({[(4-метилфенил)сульфонил]-амино}карбонил)амино]этил}фенил)-3H-имидазо[4,5-b]пиридин СТАДИЯ 1. 4,6-Диметил-3-нитро-2(1H)-пиридинон Смесь этилнитроацетата (80,0 г, 601 ммоль) в гидроксиде аммония (25% NH3 в воде, 400 мл) перемешивали при комнатной температуре в течение 3 дней и затем раствор концентрировали сушкой на воздухе. Остаток растворяли в воде (450 мл). К раствору добавляли 2,4-пентандион (73,1 г, 730 ммоль), пиридин (16,2 мл, 200 ммоль) и уксусную кислоту (11,4 мл, 200 ммоль) и смесь перемешивали в течение еще 7 дней. Получающиеся осадки собирали фильтрацией и сушили под пониженным давлением с получением 35,0 г (35%) названного соединения в виде твердых веществ желтого цвета: 1H-ЯМР (DMSO-d6) СТАДИЯ 2. 2-Хлор-4,6-диметил-3-нитропиридин Смесь 4,6-диметил-3-нитро-2(1H)-пиридинона (стадия 1, 10,0 г, 29,7 ммоль) в оксихлориде фосфора (35 мл, 187,3 ммоль) перемешивали при 95°C в течение 3 часов, затем охлаждали до 45°C. Избыточное количество оксихлорида фосфора удаляли отгонкой при пониженном давлении при 45°C. Остаток охлаждали до комнатной температуры и разбавляли дихлорметаном (75 мл). Получаемый раствор охлаждали до 0°C и в раствор добавляли по каплям 2N хлористоводородную кислоту (50 мл). Органический слой отделяли и промывали 2N соляной кислотой (4 x 25 мл), 2N водным NaOH (2 x 50 мл) и рассолом (50 мл). Органическую фазу сушили (MgSO4) и концентрировали при пониженном давлении с получением 10,0 г (90%) названного соединения в виде твердого вещества белого цветаа: 1H-ЯМР (CDCl3) СТАДИЯ 3. 2-{4-[(4,6-Диметил-3-нитро-2-пиридинил)амино]- фенил}этанол Смесь 2-хлор-4,6-диметил-3-нитропиридина (стадия 2, 1,3 г, 7,0 ммоль) и 4-аминофенилэтилового спирта (1,4 г, 10,2 ммоль) помещали в закупоренную трубку и нагревали при 150°C в течение 3 часов. Реакционную смесь охлаждали и очищали колонной флэш хроматографией на силикагеле с элюированием смесью гексан/этилацетат (2:1) с получением 1,6 г (80%) названного соединения в виде твердого вещества оранжевого цвета: 1H-ЯМР (CDCl3) СТАДИЯ 4. 2-{4-[(3-Амино-4,6-диметил-2-пиридинил)амино]-фенил}этанол К перемешиваемому раствору 2-{4-[(4,6-диметил-3-нитро-2-пиридинил)амино]фенил}этанола (стадия 3, 1,6 г, 5,6 ммоль) в этилацетате (15 мл) добавляли 10% Pd-C (160 мг). Смесь перемешивали при комнатной температуре в течение 6 часов в атмосфере водорода. Палладиевый катализатор удаляли фильтрованием и промывали этанолом (100 мл). Фильтрат концентрировали при пониженном давлении с получением 1,3 г (92%) названного соединения в виде вещества бледно-желтого цвета: 1H-ЯМР (CDCl3) СТАДИЯ 5. 2-[4-(2-Этил-5,7-диметил-3H-имидазо[4,5-b]-пиридин-3-ил)фенил]этил пропионат К перемешиваемой суспензии 2-{4-[(3-амино-4,6-диметил-2-пиридинил)амино]фенил}этанола (стадия 4, 1,3 г, 5,1 ммоль) в толуоле (30 мл) добавляли по каплям хлористый пропионил (990 мг, 10,7 ммоль) при 0°C и реакционную смесь нагревали при температуре кипения с обратным холодильником в течение 2 часов. После охлаждения смесь выливали в воду (50 мл) и экстрагировали этилацетатом (100 мл). Органический слой промывали 2N водным NaOH (50 мл) и рассолом (50 мл), затем сушили (MgSO4). После удаления растворителя получали 1,8 г (количественно) названного соединения в виде твердого вещества коричневого цвета: 1H-ЯМР (CDCl3) СТАДИЯ 6. 2-[4-(2-Этил-5,7-диметил-3H-имидазо[4,5-b]пиридин -3-ил)фенил]этанол К раствору 2-[4-(2-этил-5,7-диметил-3H-имидазо[4,5-b]пиридин-3-ил)фенил]этил пропионата (стадия 5, 1,75 г, 5,1 ммоль) в смеси метанол/ТГФ (об./об., 1:1, 28 мл) добавляли 4N водный LiOH (4,6 мл, 18,4 ммоль) и полученную смесь перемешивали при комнатной температуре. После перемешивания в течение 3 часов смесь концентрировали. Остаток растворяли в воде (30 мл) и экстрагировали этилацетатом (100 мл). Органический слой промывали рассолом (50 мл), сушили (MgSO4) и концентрировали. Очистка колонной флэш хроматографией на силикагеле с элюированием смесью гексан/этилацетат (градиентное элюирование от 2:1 до 0:1) давала 1,3 г (86%) названного соединения в виде твердого вещества бледно-коричневого цвета: 1H-ЯМР (CDCl3) СТАДИЯ 7. 3-[4-(2-Хлорэтил)фенил]-2-этил-5,7-диметил-3H-имидазо[4,5-b]пиридин К раствору 2-[4-(2-этил-5,7-диметил-3H-имидазо[4,5-b]-пиридин-3-ил)фенил]этанола (стадия 6, 2,2 г, 7,4 ммоль) в толуоле (40 мл) добавляли тионилхлорид (2,0 мл, 23,6 ммоль) и образующуюся смесь перемешивали при 80°C в течение 3 часов. Летучие компоненты удаляли при пониженном давлении и остаток очищали колонной флэш хроматографией на силикагеле с элюированием смесью гексан/этилацетат (градиентное элюирование от 2:1 до 1:1) с получением 2,1 г (90%) названного соединения в виде твердого вещества белого цвета: 1H-ЯМР (CDCl3) СТАДИЯ 8. 2-[4-(2-Этил-5,7-диметил-3H-имидазо[4,5-b]пиридин -3-ил)фенил]этил азид К перемешиваемому раствору 3-[4-(2-хлорэтил)фенил]-2-этил-5,7-диметил-3H-имидазо[4,5-b]пиридина (стадия 7, 2,8 г, 9,0 ммоль) и KI (1,5 г, 9,0 ммоль) в ДМФ (50 мл) добавляли азид натрия (1,2 г, 18,0 ммоль) и затем полученную смесь перемешивали в течение ночи при 100°C. Реакционную смесь выливали в воду (100 мл) и экстрагировали этилацетатом (100 мл). Органический слой промывали водой (50 мл) и рассолом (50 мл), затем сушили (Na2SO4). После удаления растворителя неочищенный продукт очищали колонной флэш хроматографией на силикагеле с элюированием смесью гексан/этилацетат (1:1) с получением 2,35 г (85%) названного соединения в виде твердого вещества белого цвета: 1H-ЯМР (CDCl3) СТАДИЯ 9. 2-[4-(2-Этил-5,7-диметил-3H-имидазо[4,5-b]пиридин -3-ил)фенил]этиламин К раствору 2-[4-(2-этил-5,7-диметил-3H-имидазо[4,5-b]-пиридин-3-ил)фенил]этил азида (стадия 8, 2,35 г, 7,3 ммоль) в метаноле (50 мл) добавляли 10% Pd-C (200 мг). Образующуюся смесь перемешивали в течение 4 часов в атмосфере водорода. Смесь фильтровали через слой целлита и фильтрат концентрировали. Остаток очищали колонной флэш хроматографией на силикагеле с элюированием смесью дихлорметан/метанол/триэтиламин (100:5:1) с получением 2,01 г (94%) названного соединения в виде твердого вещества белого цвета: 1H-ЯМР (CDCl3) СТАДИЯ 10. 2-Этил-5,7-диметил-3-(4-{2-[({[(4-метилфенил) сульфонил]амино}карбонил)амино]этил}фенил)-3H-имидазо[4,5-b]- пиридин
К раствору 2-[4-(2-этил-5,7-диметил-3H-имидазо[4,5-b]-пиридин-3-ил)фенил]этиламина (стадия 9, 1,2 г, 4,0 ммоль) в дихлорметане (15 мл) добавляли п-толуолсульфонилизоцианат (805 мг, 4,0 ммоль). Получающуюся смесь перемешивали при комнатной температуре в течение 3 часов. После удаления растворителя остаток очищали колонной флэш хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (20:1) с получением 1,10 г (56%) названного соединения в виде вещества белого цвета: 1H-ЯМР (CDCl3) Следующий пример иллюстрирует ранее известный способ получения N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин -1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида, описанный в патентной заявке WO-A-02/32900. ПРИМЕР 2 2-Этил-4,6-диметил-1-(4-{2-[({[(4-метилфенил)сульфонил]-амино}карбонил)амино]этил}фенил)-1H-имидазо[4,5-c]пиридин СТАДИЯ 1. 2-{4-[(2,6-Диметил-3-нитро-4-пиридинил)амино]- фенил}этанол Названное соединение получали согласно методике, описанной для стадии 3 в примере 1, из 4-хлор-2,6-диметил-3-нитропиридина (Tanaka, A.; et al. J.Med.Chem., 1999, 41, 4408.) и 4-аминофенилэтилового спирта. 1H-ЯМР (CDCl3) СТАДИЯ 2. 2-{4-[(3-Амино-2,6-диметил-4-пиридинил)амино]- фенил} этанол Названное соединение получали согласно методике, описанной для стадии 4 примера 1, из 2-{4-[(2,6-диметил-3-нитро-4-пиридинил)амино]фенил}этанола (стадия 1). 1H-ЯМР (CDCl3) СТАДИЯ 3. 2-[4-(2-Этил-4,6-диметил-1H-имидазо[4,5-c]пиридин -1-ил)фенил]этил пропионат Смесь 2-{4-[(3-амино-2,6-диметил-4-пиридинил) амино]фенил}- этанола (стадия 2, 2,4 г, 9,3 ммоль) пропионового ангидрида (13 мл, 101 ммоль) и пропионовой кислоты (13 мл, 174 ммоль) перемешивали при 120°C в течение 16 часов. После охлаждения смесь разбавляли 2N водным NaOH (150 мл) и экстрагировали дихлорметаном (3 x 150 мл). Объединенные органические экстракты промывали рассолом (50 мл), сушили (MgSO4) и концентрировали. Очистка колонной флэш хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (градиентное элюирование от 20:1 до 10:1) давала 2,3 г (69 %) названного соединения в виде масла коричневого цвета: 1H-ЯМР (CDCl3) СТАДИЯ 4. 2-[4-(2-Этил-4,6-диметил-1H-имидазо[4,5-c]пиридин -1-ил)фенил]этанол Названное соединение получали согласно методике, описанной для стадии 6 примера 1, из 2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил пропионата (стадия 3). 1H-ЯМР (CDCl3) СТАДИЯ 5. 1-[4-(2-Хлорэтил)фенил]-2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин Названное соединение получали согласно методике, описанной для стадии 7 примера 1, из 2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этанола (стадия 4). ТСХ Rf = 0,1 (этилацетат). СТАДИЯ 6. 1-[4-(2-Азидоэтил)фенил]-2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин Названное соединение получали согласно методике, описанной для стадии 8 примера 1, из 1-[4-(2-хлорэтил)фенил]-2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридина (стадия 5). 1H-ЯМР (CDCl3) СТАДИЯ 7. 2-[4-(2-Этил-4,6-диметил-1H-имидазо[4,5-c]пиридин -1-ил)фенил]этиламин Названное соединение получали согласно методике, описанной для стадии 9 примера 1, из 1-[4-(2-азидоэтил)фенил]-2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридина (стадия 6). 1H-ЯМР (CDCl3) СТАДИЯ 8. 2-Этил-4,6-диметил-1-(4-{2-[({[(4-метилфенил)- сульфонил]амино}карбонил)амино]этил}фенил)-1H-имидазо[4,5-c]-пиридин Названное соединение получали согласно методике, описанной для стадии 10 примера 1, из 2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этиламина (стадия 7). Температура плавления 143°C; MS (ESI) m/z 492,12 (M + H)+; 1H-ЯМР (CDCl3) ПРИМЕР 3 Полиморфная форма A N-[({2-[4-(2-Этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метил-бензолсульфонамида (альтернативное название 2-Этил-4,6-диметил-1-(4-{2-[({[(4-метилфенил)сульфонил]амино}карбонил)амино]этил}-фенил)-1H-имидазо [4,5-c]пиридин) СТАДИЯ 1: Неочищенный аморфный продукт 4-горловую круглодонную колбу объемом 20 л, снабженную механической мешалкой, термометром и двумя 300 мл капельными воронками, помещали в водяную баню (температура водяной бани 18°C). В колбу к раствору 508 г 2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этанамина и 480 мл триэтиламина в 3,5 л CH2Cl2 добавляли 270,4 мл п-тозилизоцианата по каплям медленно из одной из капельных воронок в течение 1,0 часа при поддержании внутренней температуры ниже 28°C. Образующийся раствор перемешивали при комнатной температуре в течение 1 часа, затем добавляли по каплям 6,0 л 1,0 M водного раствора лимонной кислоты в течение 30 минут при поддержании внутренней температуры ниже 22°C. Получающуюся смесь интенсивно перемешивали при комнатной температуре в течение 30 минут, затем добавляли по каплям 1,70 л 2,0 N водного раствора NaOH. После того, как добавление было закончено, значение pH раствора доводили до 5-5,5. Затем разделяли слои, водный слой повторно экстрагировали 1,0 л CH2Cl2 и органический слой объединяли. Органический слой промывали смесью 3,0 л 0,3 M водного раствора лимонной кислоты и 1,2 л 2,0 N водного раствора NaOH. После разделения слоев водный слой повторно экстрагировали 1,0 Л CH2Cl2 и органический слой объединяли. К полученному объединенному органическому слою добавляли 300 г Na2SO4 и 15,0 г активированного угля и смесь осторожно перемешивали при комнатной температуре в течение 12 часов. После фильтрования смеси через слой целита (1 кг) фильтрат объединяли с 70 г фильтрата, полученного на опытной установке до проведения этого эксперимента. Объединенные фильтраты концентрировали с получением 1,03 кг неочищенного продукта в виде бледно-желтого аморфного твердого вещества. СТАДИЯ 2: Превращение в полиморфную форму A и ее очистка Круглодонную 4-горлую колбу объемом 10 л, снабженную механической мешалкой, термометром и обратным холодильником, погружали в водяную баню. В колбе 5,15 л горячего (40°C) ацетона добавляли к неочищенному N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метил бензолсульфонамиду (стадия 1, 1,03 кг). Смесь перемешивали при 50°C в течение 7 часов в атмосфере азота, затем медленно охлаждали до комнатной температуры в течение 16 часов. Добавляли 515 мл ацетона и смесь перемешивали при комнатной температуре в атмосфере азота в течение 7 часов. Отфильтровывали кристаллы на бумажном фильтре, промывали 515 мл ацетона и сушили в токе газообразного азота в течение 15 часов с получением кристаллов указанного соединения (859 г, 1,75 моль), которые дополнительно очищали по следующей методике. 3-горловый реактор из нержавеющей стали объемом 10 л, снабженный механической мешалкой, термометром и обратным холодильником, погружали в водяную баню. В колбе смесь (суспензию) 859 г названного выше соединения в 1,72 л ацетона перемешивали при 50°C в течение 8 часов, затем охлаждали до комнатной температуры в течение 14 часов. Реакционную смесь перемешивали при 50°C в течение 8 часов, затем охлаждали до комнатной температуры в течение 14 часов. Отбирали аликвоту и собирали кристаллы путем отсасывания для получения образца для ВЭЖХ анализа с целью определения чистоты кристалла. Смесь перемешивали при комнатной температуре в атмосфере азота в течение 8 часов. Кристаллы отфильтровывали на бумажном фильтре, промывали 172 мл ацетона, сушили в токе газообразного азота в течение 15 часов и сушили при пониженном давлении при 40°C в течение 20 часов (837 г, 1,70 моль). Продукт дополнительно очищали по следующей методике. Круглодонную 4-горлую колбу емкостью 12 л, снабженную механической мешалкой, термометром и обратным холодильником, погружали в водяную баню. В колбу к упомянутым выше кристаллам (836 г) добавляли 3,34 л ацетона. Смесь перемешивали при 50°C в течение 4 часов в атмосфере азота, затем медленно охлаждали до комнатной температуры в течение 15 часов. Отбирали аликвоту и собирали кристаллы путем отсасывания для получения образца для ВЭЖХ анализа с целью определения чистоты кристалла. Реакционную смесь перемешивали при 50°C в течение 9 часов, затем охлаждали до комнатной температуры в течение 15 часов. Отбирали аликвоту и собирали кристаллы путем отсасывания для получения образца для ВЭЖХ анализа с целью определения чистоты кристалла. Реакционную смесь перемешивали при 50°C в течение 8 часов и затем охлаждали до комнатной температуры в течение 64 часов. Отбирали аликвоту и собирали кристаллы путем отсасывания для получения образца для ВЭЖХ анализа с целью определения чистоты кристалла. Реакционную смесь перемешивали при 50°C в течение 9 часов и затем охлаждали до комнатной температуры в течение 14 часов. Отбирали аликвоту и собирали кристаллы путем отсасывания для получения образца для ВЭЖХ анализа с целью определения чистоты кристалла. Смесь перемешивали при комнатной температуре в атмосфере азота в течение 6 часов. Кристаллы отфильтровывали на бумажном фильтре, промывали 1,67 л ацетона, сушили в токе газообразного азота в течение 22 часов и сушили при пониженном давлении при 40°C в течение 17 часов с получением полиморфной формы A названного соединения (771 г, 1,57 моль). В качестве варианта, полиморфная форма A может быть получена согласно следующей методике. ПРИМЕР 4 Полиморфная форма A N-[({2-[4-(2-Этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида (альтернативное название 2-Этил-4,6-диметил-1-(4-{2-[({[(4-метилфенил)сульфонил]амино}карбонил)амино]этил}-фенил)-1H-имидазо [4,5-c]пиридин) В чистую и сухую 3-горловую круглодонную колбу емкостью 12 л загружали 2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этанамин (422 г, 1,43 молей) и CH2Cl2 (3,37 л). Добавляли в реакционную среду тозилизоцианат (217 мл, 1,43 моль), растворенный в CH2Cl2 (851 мл) при поддержании температуры ниже 21°C, и перемешивали в течение, по меньшей мере, 90 минут. После окончания реакции, которое определялось с помощью ВЭЖХ, добавляли активированный уголь (DARCO KB-B 4,2 г). Полученную смесь фильтровали через 0,5-микронный фильтр в не содержащую пылинки 3-горловую круглодонную колбу емкостью 5 л и фильтр промывали CH2Cl2 (421 мл). Реакционную смесь концентрировали при атмосферном давлении до минимального объема, при котором было возможно перемешивание, и замещали удаленный растворитель не содержащим пылинки ацетоном до тех пор, пока внутренняя температура не достигала 58 – 62°C и конечный объем не достигал ~1,3 л. Реакционную смесь охлаждали, по меньшей мере, до 30°C и добавляли затравку полиморфной формы A N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]-этил}амино)карбонил]-4-метилбензолсульфонамида. Реакцию проводили при температуре между 20 и 25°C в течение, по меньшей мере, 10 часов. После охлаждения реакционной смеси до температуры между 0 и 5°C и кристаллизации в течение, по меньшей мере, 2 часов реакционную смесь фильтровали на не содержащем пылинок фильтре. Твердое вещество промывали два раза не содержащим пылинок ацетоном (1,68 л), охлажденным до температуры между 0 и 5°C. Влажный осадок на фильтре возвращали в не содержащую пылинок 3-горлую круглодонную колбу емкостью 12 л и добавляли не содержащий пылинок этилацетат (4,4 л). Суспензию нагревали до, по меньшей мере, 75°C и выдерживали в течение, по меньшей мере, 2 часов. Реакционную смесь охлаждали до, по меньшей мере, 30°C и твердое вещество отфильтровывали на не содержащем пылинок фильтре. Твердое вещество промывали не содержащим пылинок этилацетатом (1,6 л). Влажный осадок на фильтре возвращали в ту же самую не содержащую пылинок 3-горловую круглодонную колбу емкостью 12 л и добавляли не содержащий пылинок этилацетат (4,4 л). Суспензию нагревали до, по меньшей мере, 75°C и выдерживали в течение, по меньшей мере, 2 часов. Реакционную смесь охлаждали до, по меньшей мере, 30°C и твердое вещество отфильтровывали на не содержащем пылинок фильтре. Твердое вещество промывали не содержащим пылинок этилацетатом (1,6 л). Продукт сушили при температуре между 45 и 50°C в течение, по меньшей мере, 24 часов, получая в результате 289 г названного продукта, полиморфную форму A (выход 41,4%, чистота по ВЭЖХ 98,68%). Размер частиц, достигаемый с помощью данной методики, не требует измельчения. С помощью простого ручного просеивания удаляются любые комочки. Продукт (289 г) подвергали просеиванию вручную через несодержащее пылинки ручное сито #25 с размером отверстий 0,0278 дюйма. Получали 277 г материала. В качестве варианта, полиморфная форма A может быть получена путем превращения полиморфной формы B согласно следующим методикам. ПРИМЕР 5 Полиморфная форма A N-[({2-[4-(2-Этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метил-бензолсульфонамида Добавляли ацетонитрил (79,50 л, 62,01 кг, 1510 моль) к полиморфной форме B N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метил-бензолсульфонамида (5,300 кг, 10,78 моль) при поддержании температуры между 20 и 30 ПРИМЕР 6 Полиморфная форма A N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида Примерно 30 объемов этанола добавляли к одному грамму полиморфной формы B N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида. Материал растворяли с получением раствора при комнатной температуре. Приблизительно половину растворителя отгоняли на роторном испарителе до момента, когда начинало образовываться масло на стенках колбы. В раствор вводили затравку кристаллов полиморфной формы A и перемешивали при комнатной температуре в течение ночи. На следующий день раствор охлаждали на бане с ледяной водой и фильтровали под вакуумом. Выход составлял около 63%. ПРИМЕР 7 Полиморфная форма B N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида В продутый инертным газом футерованный стеклом реактор емкостью 100 л загружали 2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этиламин (4,24 кг) и затем CH2Cl2 (34 л) с получением слегка мутного раствора. Для удаления следов соответствующей хлористоводородной соли раствор фильтровали через встроенный фильтр и реактор/фильтр промывали CH2Cl2 (4 л). Переносили в капельную воронку толуолсульфонилизоцианат (2,78 кг) и линии промывали CH2Cl2 (2 л). Изоцианатный раствор добавляли в течение 30 минут к раствору исходного материала при поддержании температуры в интервале 17-22 В качестве варианта, полиморфные формы B и A могут быть получены согласно следующим методикам. ПРИМЕР 8 Полиморфная форма B и ее превращение в полиморфную форму A N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)- фенил]этил}амино)карбонил]-4-метилбензолсульфонамида СТАДИЯ 1: Бензил-4-(2,6-диметил-3-нитропиридин-4-иламино) фенетилкарбамат гидрохлорид В чистую и сухую, продутую азотом круглодонную колбу емкостью 1 л загружали тетра-N-бутиламмонийбромид (1,24 г, 3,85 ммоль) и 2-метилтетрагидрофуран (350 мл). Затем добавляли в виде расплава 4-(2-аминоэтил)бензенамин (35,0 г, 257 ммоль). Затем добавляли раствор гидроксида натрия (51,4 г, 1280 ммоль) в воде (350 мл) при поддержании температуры ниже 30°C. Реакционную смесь затем охлаждали до температуры между -5°C и 5°C. После выдержки в течение, по меньшей мере, 30 минут при температуре между -5°C и 5°C добавляли бензилхлорформиат (43,8 г, 257 ммоль) при поддержании температуры между -5°C и 5°C. Реакционную смесь нагревали до температуры между 0°C и 10°C и выдерживали в течение, по меньшей мере, 30 минут до момента окончания реакции, которое определялось с помощью ВЭЖХ. После подогрева реакционной смеси до температуры между 20 и 30°C реакционной смеси давали возможность расслоиться в течение, по меньшей мере, 60 минут и затем фазы разделяли. К органическому слою добавляли лимонную кислоту (54,0 г, 257 ммоль), растворенную в воде (350 мл). После перемешивания в течение, по меньшей мере, 60 минут при температуре между 20 и 30°C фазам давали возможность расслоиться в течение, по меньшей мере, 2 часов. Фазы разделяли и органический раствор бензил-4-аминофенетилкарбамата оставляли для последующего использования. Далее в пустую колбу добавляли 4-хлор-2,6-диметил-3-нитропиридин (40,0 г, 214 ммоль), затем добавляли органический слой, содержащий бензил-4-аминофенетилкарбамат и 2-пропанол (350 мл). Реакционную смесь нагревали до температуры между 70 и 80°C в течение, по меньшей мере, 2 часов до момента окончания реакции, которое определялось с помощью ВЭЖХ. Замещали 2-пропанол при атмосферном давлении 2-метилтетрагидрофураном до конечного объема около 350 мл. Реакционную смесь затем охлаждали до температуры между 15 и 25°C и отбирали образец для KF и газовой хроматографии в свободном пространстве над продуктом. Реакционную смесь выдерживали при температуре между 15 и 25°C в течение, по меньшей мере, 6 часов и реакционную смесь фильтровали и промывали 2-метилтетрагидрофураном (105 мл). После продувки осадка азотом в течение, по меньшей мере, 2 часов и сушки под вакуумом с продуванием азотом при температуре от 40 и до 50°C в течение, по меньшей мере, 24 часов, выделяли 76,7 г (168 ммоль) гидрохлорида бензил-4-(2,6-диметил-3-нитропиридин-4-иламино)фенетилкарбамата с выходом 78,4%. СТАДИЯ 2: Гидрохлорид бензил-4-(3-амино-2,6-диметил-пиридин-4-иламино)фенетилкарбамата В чистый, продутый азотом реактор гидрирования емкостью 1 л загружали 5% Pt/C (625 мг) гидрохлорид бензил-4-(2,6-диметил-3-нитропиридин-4-иламино)фенетилкарбамата (25,0 г, 54,7 ммоль) и метанол (550 мл). Реакционную смесь перемешивали в течение, по меньшей мере, 15 минут и затем продували последовательно азотом и водородом. Повышали давление реакционной смеси с помощью водорода до величины между 18 и 25 psi при температуре между 22 и 28°C до тех пор, пока анализ с помощью ВЭЖХ не показывал, что исходный материал полностью израсходован. Реакционную смесь продували азотом и катализатор отфильтровывали на смоченном водой, покрытом целлитом фильтре, осадок промывали метанолом (125 мл). Полученный раствор гидрохлорида бензил-4-(3-амино-2,6-диметилпиридин-4-иламино)фенетилкарбамата переносили в чистую и сухую, продутую азотом круглодонную колбу емкостью 1 л. Метанол замещали при атмосферном давлении 2-пропанолом до конечного объема приблизительно 125 мл. Реакционную смесь затем охлаждали до температуры между 15 и 25°C в течение, по меньшей мере, 60 минут и отбирали пробу для KF. Реакционную смесь затем фильтровали и промывали 2-пропанолом (75 мл). После продувания осадка азотом в течение, по меньшей мере, 15 минут и сушки в вакууме при температуре от 40 и до 50°C в токе азота в течение, по меньшей мере, 12 часов выделяли 21,0 г (49,2 ммоль) гидрохлорида бензил-4-(3-амино-2,6-диметилпиридин-4-иламино)фенетилкарбамата с выходом 89,9%. СТАДИЯ 3: Гидрохлорид 2-(4-(2-Этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил)этанамина В чистую и сухую, продутую азотом круглодонную колбу емкостью 1 л загружали гидрохлорид бензил-4-(3-амино-2,6-диметилпиридин-4-иламино)фенетилкарбамата (30,0 г, 70,3 ммоль), 2-метилтетрагидрофуран (150 мл), триэтиламин (14,9 г, 148 ммоль) и пропионовый ангидрид (11,9 г, 91,3 ммоль) при температуре между 20 и 25°C. Реакционную смесь затем нагревали до температуры между 70 и 80°C в течение, по меньшей мере, 6 часов. После охлаждения до температуры между 20 и 25°C отбирали образец реакционной смеси на анализ с помощью ВЭЖХ на полноту протекания реакции. Реакцию затем останавливали гидроксидом натрия (8,4 г, 211 ммоль), растворенным в воде (150 мл), поддерживая температуру ниже 30°C. После выдерживания реакционной смеси при температуре между 20 и 25°C в течение, по меньшей мере, 30 минут реакционной смеси давали возможность расслоиться в течение, по меньшей мере, 60 минут. Фазы разделяли и использовали 2-метилтетрагидрофуран для удаления при атмосферном давлении следов триэтиламина до тех пор, пока конечный объем не достигал приблизительно 180 мл. Полученный раствор бензил-2-(4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил)этилкарбамата переносили в чистый, продутый азотом реактор гидрирования емкостью 1 л, содержащий 10% Pd/C (1,5 г) и метанол (120 мл). Реакционную смесь перемешивали в течение, по меньшей мере, 15 минут и затем продували последовательно азотом и водородом. Реакционную смесь нагревали до температуры между 45 и 55°C и затем повышали давление с помощью водорода до величины между 45 и 55 psi. После перемешивания при этих температуре и давлении в течение 18 часов реакционную смесь охлаждали и отбирали пробу для определения окончания реакции. Проводили анализ реакционной смеси с помощью ВЭЖХ на завершение реакции и смесь фильтровали через смоченный водой покрытый целлитом фильтр. Фильтр затем промывали метанолом (195 мл) и полученный раствор 2-(4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил)этанамина переносили в чистую и сухую, продутую азотом круглодонную колбу емкостью 1 л, затем добавляли концентрированную соляную кислоту (6,3 мл, 73,8 ммоль). Метанол удаляли затем при атмосферном давлении с помощью 2-метилтетрагидрофурана до тех пор, пока не достигались низкие концентрации воды. Затем корректировали содержание растворителей до требуемого соотношения 4:1 2-метилтетрагидрофуран:метанол (суммарно 10 объемов). Реакционную смесь затем нагревали до температуры между 60 и 70°C в течение, по меньшей мере, 2 часов. После охлаждения до температуры между 15 и 25°C в течение, по меньшей мере, 60 минут реакционную смесь выдерживали при температуре между 15 и 25°C в течение, по меньшей мере, 60 минут. Реакционную смесь затем фильтровали и осадок промывали 2-метилтетрагидрофураном (90 мл). После продувания осадка азотом в течение, по меньшей мере, 2 часов твердое вещество возвращали в тот же самый реактор и добавляли 2-метилтетрагидрофуран (240 мл) и метанол (60 мл). Реакционную смесь нагревали до температуры между 60 и 70°C и выдерживали при этой температуре в течение, по меньшей мере, 2 часов. После охлаждения до температуры между 15 и 25°C в течение как минимум 60 минут реакционную смесь выдерживали при температуре в течение, по меньшей мере, 60 минут. Реакционную смесь затем фильтровали и осадок промывали 2-метилтетрагидрофураном (90 мл). После сушки в вакууме при температуре между 40 и 50°C в течение, как минимум, 12 часов в небольшом токе азота выделяли 21,0 г (63,5 ммоль) гидрохлорида 2-(4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил)-этанамина с выходом 90,3%. СТАДИЯ 4: Полиморфная форма B N-[({2-[4-(2-Этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил] -4-метилбензолсульфонамида В чистую и сухую, продутую азотом круглодонную колбу емкостью 500 мл загружали гидрохлорид 2-(4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил)этанамина (40,0 г, 121 ммоль), дихлорметан (400 мл) и гидроксид натрия (9,7 г, 242 ммоль), разбавленный водой (400 мл) при поддержании температуры ниже 30°C. Реакционную смесь выдерживали при этой температуре в течение как минимум 30 минут и затем давали возможность реакционной смеси расслоиться в течение, по меньшей мере, 60 минут. Фазы разделяли, водный слой смешивали с дихлорметаном (400 мл) в течение, по меньшей мере, 30 минут при температуре ниже 30°C и затем давали возможность реакционной смеси расслоиться в течение, по меньшей мере, 60 минут. Фазы разделяли и использовали дихлорметан для удаления при атмосферном давлении избытка воды из объединенных органических слоев до конечного объема приблизительно 800 мл. Реакционную смесь фильтровали и переносили в круглодонную колбу емкостью 2 л. К фильтрату добавляли толуолсульфонилизоцианат (23,8 г, 121 ммоль) в течение как минимум 30 минут при поддержании температуры между 10 и 25°C. После выдерживания реакционной смеси при температуре между 10 и 25°C в течение, как минимум, 30 минут определяли окончание реакции с помощью ВЭЖХ. Использовали ацетон для вытеснения при атмосферном давлении дихлорметана до низких концентраций, и конечный объем реакционной смеси составлял приблизительно 240 мл. Реакционную смесь охлаждали до температуры между 20 и 30°C в течение, как минимум, 2 часов и отбирали пробу реакционной смеси. Анализ показал присутствие полиморфа B, и реакционную смесь затем охлаждали до температуры между -5 и -25°C в течение, как минимум, 2 часов. Реакционную смесь фильтровали и промывали ацетоном (80 мл, 2 раза), охлажденным до температуры между -5 и -25°C. Осадок выдерживали на фильтре в потоке азота в течение, по меньшей мере, 12 часов. В течение этого времени осадок становился сухим, и получали 49,6 г (101 ммоль) полиморфной формы B N-[({2-[4-(2-Этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида с выходом 83,4%. СТАДИЯ 5: Полиморфная форма A N-[({2-[4-(2-Этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил] -4-метилбензолсульфонамида В чистую и сухую, продутую азотом, круглодонную колбу емкостью 1 л загружали полиморфную форму B N-[({2-[4-(2-Этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)-карбонил]-4-метилбензолсульфонамида (30,0 г, 61,0 ммоль) и ацетонитрил (600 мл). Реакционную смесь затем нагревали до несильного кипения с обратным холодильником для достижения гомогенности. После кипячения реакционной смеси с обратным холодильником в течение, по меньшей мере, 60 минут реакционную смесь концентрировали приблизительно до объема 450 мл. Реакционную смесь затем охлаждали до температуры между 20 и 30°C в течение менее чем 90 минут. Реакционную смесь выдерживали при температуре 30°C в течение ночи, и ее анализ показал превращение в форму A. Ацетонитрил вытесняли этилацетатом под вакуумом до остаточного конечного объема около 300 мл. Реакционную смесь нагревали до несильного кипения с обратным холодильником и давали возможность осаждаться твердому веществу при этой температуре в течение, по меньшей мере, 6 часов. Реакционную смесь охлаждали до температуры между 18 и 22°C и перемешивали при этой температуре в течение, по меньшей мере, 2 часов. Реакционную смесь фильтровали и промывали этилацетатом (90 мл). После сушки под вакуумом при температуре между 40 и 50°C в течение, как минимум, 12 часов в легком токе азота, получали 27,0 г (54,9 ммоль) полиморфной формы A N-[({2-[4-(2-Этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)-фенил]этил}амино)карбонил]-4-метилбензолсульфонамида с выходом 90,0%. ПРИМЕР 9 Непосредственное выделение полиморфной формы A N-[({2-[4-(2-Этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}-амино)карбонил]-4-метилбензолсульфонамида В чистую и сухую, продутую азотом круглодонную колбу емкостью 250 мл загружали 2-(4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил)этанамин (11,0 г, 37,2 ммоль) и дихлорметан (110 мл). Добавляли толуолсульфонилизоцианат (7,3 г, 37,2 ммоль) в течение как минимум 30 минут при поддержании температуры между 10°C и 25°C. После выдерживания реакционной смеси при температуре между 10°C и 25°C в течение как минимум 30 минут определяли окончание реакции с помощью ВЭЖХ. Добавляли активированный уголь (DARCO, 110 мг) и перемешивали в течение, по меньшей мере, 15 минут. Реакционную смесь фильтровали и переносили в круглодонную колбу емкостью 150 мл. Использовали ацетон для замещения при атмосферном давлении дихлорметана до низких концентраций, и конечный объем реакционной смеси составлял приблизительно 30 мл. Реакционную смесь охлаждали до температуры между 20 и 30°C и вносили затравку полиморфной формы A N-[({2-[4-(2-Этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)-фенил]этил}амино)карбонил]-4-метилбензолсульфонамида (219 мг). Давали возможность произойти осаждению в течение, по меньшей мере, 12 часов при температуре между 20 и 30°C. Реакционную смесь затем охлаждали до температуры от -5 до 5°C в течение как минимум 2 часов. Реакционную смесь фильтровали и промывали ацетоном (40 мл), охлажденным до температуры между -5 и 5°C. Осадок на фильтре выдерживали в токе азота в течение, по меньшей мере, 12 часов. После этого получали 1,8 г (3,66 ммоль) полиморфной формы A N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида с выходом 9,8%. ПРИМЕР 10 Кристаллическая форма C N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)-фенил]этил}амино)карбонил]-4-метилбензолсульфонамида Кристаллическую форму C получали кристаллизацией из раствора N-[({2-[4-(2-Этил-4,6-диметил-1H-имидазо[4,5-c]пиридин -1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида в смеси хлороформа и этилацетата. ПРИМЕР 11 Кристаллическая форма D N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида Кристаллическую форму D получали кристаллизацией из водной суспензии N-[({2-[4-(2-Этил-4,6-диметил-1H-имидазо[4,5-c]- пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида. ПРИМЕР 12 Кристаллическая форма G N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида Кристаллическая форма G обнаруживалась при повышенных температурах при анализе кристаллической формы D методом порошковой рентгенографии. Кристаллическую форму G не обнаруживали при комнатной температуре. ДАННЫЕ АНАЛИЗА Рентгеновская порошковая дифрактометрия Сравнительные анализы методом порошковой рентгенографии полиморфных форм A и B и контрольного образца из патентной заявки WO 02/32900, пример 42, стадия 8 (см. пример 2) (см. фиг.6) проводили с использованием порошкового рентгеновского дифрактометра Rigaku RINT-TTR с Cu-K Рентгеноструктурный анализ порошков полиморфных форм A и B и кристаллических форм C, D и G, приведенный на фигурах 1-5, проводили согласно следующей методике. Рентгеноструктурный анализ порошков Образцы порошков регистрировали с использованием дифрактометра Bruker D5000 с медным излучением, установленными щелями (1,0, 1,0, 0,6 мм) и твердотельным детектором Kevex или Sol-X. Данные регистрировали от 3,0 до 40,0 градусов два тета с шагом 0,04 градуса и временным шагом 1,0 сек. Рентгеновский анализ монокристалла Данные регистрировали при комнатной температуре с использованием дифрактометра Bruker D5000 с медным излучением и графитовыми монохроматорами. Структуры рассчитывали прямыми методами. Использование компьютерной библиотеки SHELXTL, предоставляемой фирмой Bruker AXS, Inc, облегчало выполнение всех необходимых кристаллографических вычислений и изображение молекулы. Компьютерная библиотека SHELXTL разрабатывается и модернизируется уже в течение длительного периода времени. Самой последней версией является следующая: SHELXTLTM Reference Manual, Version 5.1, Bruker AXS, Madison, Wisconsin, USA (1997.) Расчет порошковой рентгенограммы из данных для монокристалла Для сравнения результатов для монокристалла и порошкового образца может быть получена рассчитанная порошковая рентгенограмма из данных для монокристалла. Эти вычисления могут быть осуществлены с помощью компьютерных программ XFOG и XPOW, предоставляемых в качестве компонентов компьютерной библиотеке SHELXTL. Сравнение вычисленной порошковой рентгенограммы с экспериментальной порошковой рентгенограммой позволяет подтвердить соответствие порошковой рентгенограммы приписываемой структуре монокристалла. Эта методика была применена для полиморфной формы A и кристаллической формы D N-[({2-[4-(2-Этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)-фенил]этил}амино)карбонил]-4-метилбензолсульфонамида. Совместимость этих двух рентгенограмм указывало на соответствие между порошковой рентгенограммой и соответствующей структурой монокристалла. На фигуре 1a приведена порошковая рентгенограмма полиморфной формы A N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида. На фигуре 1b приведена рассчитанная порошковая рентгенограмма полиморфной формы A N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида. На фигуре 2 приведена порошковая рентгенограмма полиморфной формы В N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида. На фигуре 3 приведена порошковая рентгенограмма кристаллической формы С N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида. На фигуре 4a приведена порошковая рентгенограмма кристаллической формы D N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида. На фигуре 4b приведена рассчитанная порошковая рентгенограмма кристаллической формы D N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида. На фигуре 5 приведена порошковая рентгенограмма кристаллической формы G N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида. На фигуре 6 приведено сравнение порошковых рентгенограмм полиморфных форм A и B и контрольного продукта, приготовленного способом получения N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида, описанным в патентной заявке WO 02/32900, пример 42, стадия 8 (см. пример 2). На этой сравнительной диаграмме видно, что полиморфные формы A и B не соответствуют контрольному продукту, описанному в патентной заявке WO 02/32900, и являются поэтому индивидуальными новыми полиморфными формами. Перечень пиков для упомянутых выше фигур приводится в таблице 2. Интенсивность пика зависит от морфологии и размера частиц образца и может изменяться, причем пики с низкой интенсивностью (интенсивность меньше 10%) в некоторых случаях не приводятся.
Дифференциальная сканирующая калориметрия Проводили дифференциальную сканирующую калориметрию (ДСК) с использованием прибора для термического анализа, дифференциального сканирующего калориметра 2920. Образец помещали в алюминиевый тигель для ДСК и точно регистрировали массу. Использовали или открытый тигель, или тигель, закрытый крышкой. Каждый образец приводили в равновесие при 25 Фурье ИК-спектроскопия Инфракрасные спектры полиморфной формы B регистрировали на ИК-спектрофотометре с преобразованием Фурье Magna-IR 860® (фирмы Thermo Nicolet), снабженном средним/дальним ИК-источником Ever-Glo, светоделителем с расширенной областью из бромида калия (KBr) и детектором из дейтерированного триглицинсульфата (DTGS). При исследовании образцов использовали устройство для диффузного отражения (CollectorTM, Thermo Spectra-Tech). Каждый спектр представляет собой 256 добавленных вместе сканов, регистрируемых при спектральной чувствительности 4 см-1. Приготовление образца состояло из помещения образца в кювету диаметром 3 или 13 мм. Набор исходных данных получали с помощью юстировочного зеркала на месте. Log 1/R (R = отражение) спектра получали путем взятия отношения этих двух наборов данных относительно друг друга. Градуировку по длинам волн осуществляли по полистиролу. Полученные значения являются округленными, и поэтому их следует рассматривать как приблизительные. Инфракрасные спектры полиморфной формы A регистрировали на инфракрасном спектрофотометре с Фурье преобразованием Shimadzu FTIR-8200PC, оборудованным источником луча из покрытой чернью нагретой проволоки, светоделителем из бромида калия (KBr) с нанесенным на него слоем германия и высокочувствительным пироэлектрическим детектором (DLATGS). Каждый спектр представляет собой 40 добавленных вместе сканов, регистрируемых при спектральной чувствительности 4 см-1. Приготовление образца состояло из помещения диска из KBr, полученного из 1 мг образца и 150 мг KBr. Набор исходных данных получали с помощью не содержащего образцы диска из KBr. Log 1/R (R = отражение) спектра получали путем взятия отношения этих двух наборов данных относительно друг друга. Градуировку по длинам волн осуществляли по полистиролу. Полученные значения являются округленными, и поэтому их следует рассматривать как приблизительные.
Формула изобретения
1. Практически чистая, кристаллическая, полиморфная форма A N-[({2-[4-(2-Этил-4,6-диметил-1Н-имидазо[4,5-с]пиридин-1-ил)-фенил]этил}амино)карбонил]-4-метилбензолсульфонамида, которая характеризуется порошковой рентгенограммой, полученной при Cu K 2. Полиморфная форма N-[({2-[4-(2-Этил-4,6-диметил-1Н-имидазо[4,5-с]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метил-бензолсульфонамида по п.1, которая дополнительно характеризуется дифференциальной сканирующей калориметрией (ДСК) с эндотермическим эффектом при температуре около 160°С. 3. Фармацевтическая композиция, обладающая антагонистической активностью в отношении ЕР4 рецептора, включающая полиморфную форму А N-[({2-[4-(2-Этил-4,6-диметил-1Н-имидазо[4,5-с]пиридин-1-ил)-фенил]этил}амино)карбонил]-4-метилбензолсульфонамида по пп.1 и 2, вместе с одним или более фармацевтически приемлемыми наполнителями. 4. Полиморфная форма А N-[({2-[4-(2-Этил-4,6-диметил-1Н-имидазо[4,5-с]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метил-бензолсульфонамида по пп.1 и 2 для применения в качестве лекарственного препарата. 5. Применение полиморфной формы А N-[({2-[4-(2-Этил-4,6-диметил-1Н-имидазо[4,5-с]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида по пп.1 и 2 для получения лекарственного препарата для лечебной, паллиативной или профилактической терапии боли, воспаления, остеоартрита или ревматоидного артрита. 6. Применение фармацевтической композиции по п.3, для получения лекарственного препарата для лечебной, паллиативной или профилактической терапии боли, воспаления, остеоартрита или ревматоидного артрита. 7. Способ терапии боли, воспаления, остеоартрита или ревматоидного артрита, который включает введение эффективного количества полиморфной формы А N-[({2-[4-(2-Этил-4,6-диметил-1Н-имидазо[4,5-с]пиридин-1-ил)фенил]этил)амино)карбонил]-4-метил-бензолсульфонамида по пп.1 и 2 или фармацевтической композиции по п.3 животному, включая человека, в случае необходимости такой терапии. 8. Практически чистая, кристаллическая, полиморфная форма В N-[({2-[4-(2-Этил-4,6-диметил-1Н-имидазо[4,5-с]пиридин-1-ил)-фенил]этил}амино)карбонил]-4-метилбензолсульфонамида, которая характеризуется порошковой рентгенограммой, полученной при Cu K 9. Полиморфная форма В N-[({2-[4-(2-Этил-4,6-диметил-1Н-имидазо[4,5-с]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метил-бензолсульфонамида по п.8, которая дополнительно характеризуется дифференциальной сканирующей калориметрией (ДСК) с эндотермическим эффектом при температуре около 178°С. 10. Фармацевтическая композиция, обладающая антагонистической активностью в отношении ЕР4 рецептора, включающая полиморфную форму В N-[({2-[4-(2-Этил-4,6-диметил-1Н-имидазо[4,5-с]пиридин-1-ил)-фенил]этил}амино)карбонил]-4-метилбензолсульфонамида по пп.8 и 9 вместе с одним или более фармацевтически приемлемыми наполнителями. 11. Полиморфная форма В N[({2-[4-(2-Этил-4,6-диметил-1Н-имидазо[4,5-с]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метил-бензолсульфонамида по пп.8 и 9 для применения в качестве лекарственного препарата. 12. Применение полиморфной формы В N-[({2-[4-(2-Этил-4,6-диметил-1Н-имидазо[4,5-с]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамида по пп.8 и 9 для получения лекарственного препарата для лечебной, паллиативной или профилактической терапии боли, воспаления, остеоартрита или ревматоидного артрита. 13. Применение фармацевтической композиции по п.10 для получения лекарственного препарата для лечебной, паллиативной или профилактической терапии боли, воспаления, остеоартрита или ревматоидного артрита. 14. Способ терапии боли, воспаления, остеоартрита или ревматоидного артрита, который включает введение эффективного количества полиморфной формы В N-[({2-[4-(2-Этил-4,6-диметил-1Н-имидазо[4,5-с]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метил-бензолсульфонамида по пп.8 и 9 или фармацевтической композиции по п.10 животному, включая человека, в случае необходимости такой терапии.
РИСУНКИ
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
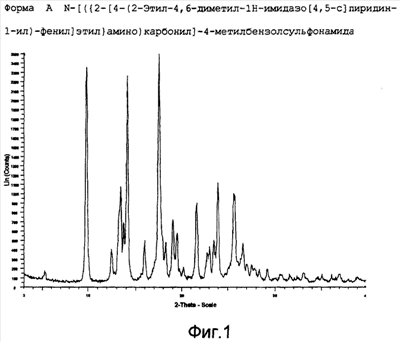
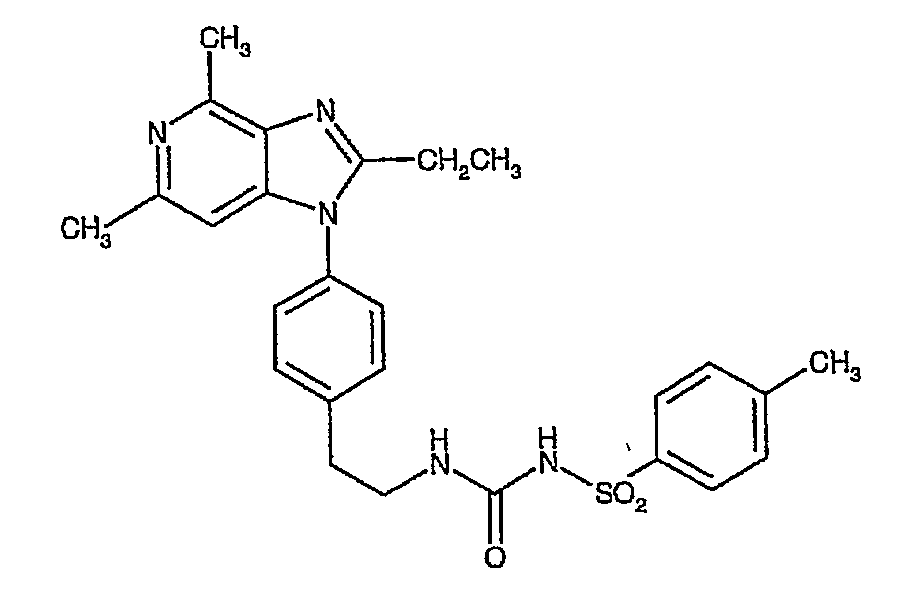
 излучения, которая включает основные пики при 2-тета 9,8, 13,2, 13,4, 13,7, 14,1, 17,5, 19,0, 21,6, 24,0 и 25,7 +/- 0,2.
излучения, которая включает основные пики при 2-тета 9,8, 13,2, 13,4, 13,7, 14,1, 17,5, 19,0, 21,6, 24,0 и 25,7 +/- 0,2. C.
C. , Bioject
, Bioject 12,44 (1H, br.s), 6,06 (1H, s), 2,19 (3H, s), 2,13 (3H, s).
12,44 (1H, br.s), 6,06 (1H, s), 2,19 (3H, s), 2,13 (3H, s).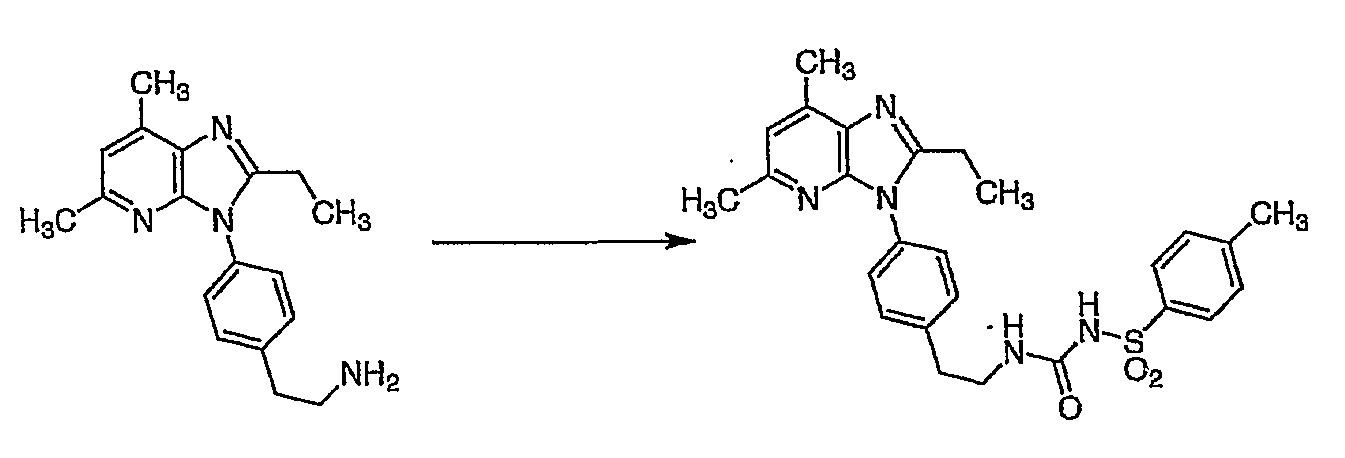
 . Для проверки точности установки прибора анализировали кремниевый эталон. Данные регистрировали и анализировали с использованием рентгеновской системы Rigaku. Приготавливали образцы для анализа путем помещения их в алюминиевый держатель образца, который горизонтально вращается со скоростью 60 об/мин в процессе регистрации данных.
. Для проверки точности установки прибора анализировали кремниевый эталон. Данные регистрировали и анализировали с использованием рентгеновской системы Rigaku. Приготавливали образцы для анализа путем помещения их в алюминиевый держатель образца, который горизонтально вращается со скоростью 60 об/мин в процессе регистрации данных.