Патент на изобретение №2357964
|
||||||||||||||||||||||||||||||||||||||
(54) БИЦИКЛО-3.1.1-ГЕПТАН-ЗАМЕЩЕННЫЕ БЕНЗИМИДАЗОЛОН- И ХИНАЗОЛИНОН-ПРОИЗВОДНЫЕ АГОНИСТЫ ORL1 РЕЦЕПТОРОВ ЧЕЛОВЕКА
(57) Реферат:
Настоящее изобретение относится к группе гидронопол-замещенных бензимидазолон- и хиназолинон-производных, которые являются агонистами ORL1 рецепторов (ноцирецепторов) человека. Изобретение относится также к получению указанных соединений, к фармацевтическим композициям, содержащим фармакологически активное количество, по меньшей мере, одного из указанных новых бензимидазолон- и хиназолинон-производных в качестве активного ингредиента, а также к применению указанных фармацевтических композиций для лечения расстройств, в которые вовлечены ORL1 рецепторы. Изобретение относится к соединениям общей формулы (1), где символы принимают значения, указанные в описании. 4 н. и 3 з.п. ф-лы, 2 табл.
Настоящее изобретение относится к группе гидронопол-замещенных бензимидазолон- и хиназолинон-производных, которые являются агонистами ORL1 рецепторов (ноцирецепторов) человека. Изобретение относится также к способу получения данных соединений, к фармацевтических композициям, содержащим фармакологически активное количество, по меньшей мере, одного из указанных новых бензимидазолон- и хиназолинон-производных в качестве активного ингредиента, а также к способу применения указанных фармацевтических композиций для лечения расстройств, в которые вовлечены ORL1 рецепторы. Рецептор, подобный опиоидному рецептору 1 (Opioid Receptor-like 1 (ORL1)), идентифицирован из библиотеки комплементарных ДНК человека. Было установлено, что гомология данного «орфан-рецептора» близка к гомологии µ-, Среди патентных публикаций предшествующего уровня наиболее близкой к настоящему изобретению является WO 01/39775. Однако оказалось, что производные бензимидазолона, описанные в указанной публикации, не удовлетворяют критериям, которые, как общеизвестно, важны для применения в качестве терапевтических средств, связанных с воздействием на ORL1. Они характеризуются следующим: (1) умеренная активность (сродство в отношении ORL1 рецепторов в интервале от 166 до 1252 нМ); (2) отсутствие селективности в отношении µ-опиатных рецепторов (сродство в интервале от 19 до 457 нМ); (3) отсутствие доказательства биологической доступности при пероральном введении, (4) отсутствие доказательства биологической доступности для ЦНС. В настоящее время было неожиданно установлено, что в ряду гидронопил-замещенных производных бензимидазолона и хиназолинона группа соединений показывает очень высокое сродство в отношении ORL1 рецепторов человека. Кроме того, указанные соединения показали прекрасную селективность в отношении ORL1 рецепторов по сравнению с µ-опиатными рецепторами, они легко доступны при пероральном введении и проникают через гематоэнцефалический барьер. Настоящее изобретение относится к соединениям общей формулы (1)
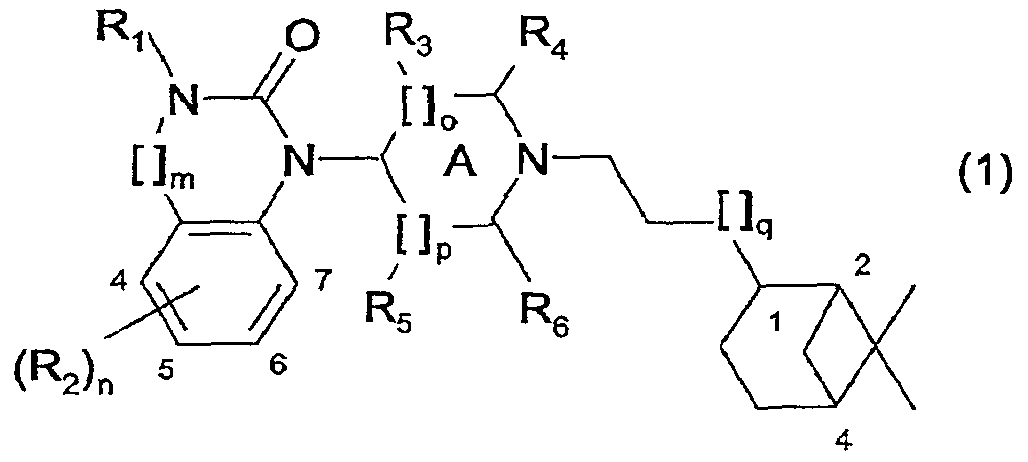
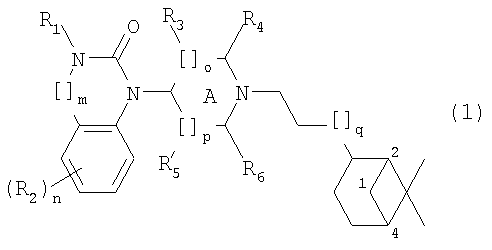
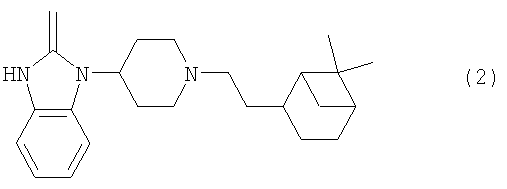
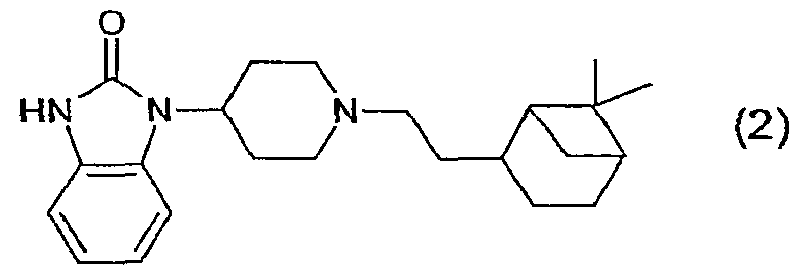
где R1 представляет собой Н, алкил(1-6С), алкил(1-3С)циклоалкил(3-6С), карбалкокси(2-7С) или ацил(2-7С), []m означает -(СН2)m-, где m равно 0 или 1, R2 представляет собой галоген, CF3, алкил(1-6С), алкил(1-3С)циклоалкил(3-6С), фенил, амино, аминоалкил(1-3С), алкил(1-3С)амино, диалкил(1-3С)амино, циано, цианоалкил(1-3C), гидрокси, гидроксиалкил(1-3С), (1-3С)алкокси, OCF3, ацил(2-7С), трифторацетил, аминокарбоксил, (1-3С)алкилсульфонил или трифторметилсульфонил, и n представляет собой целое число от 0 до 4, при условии, что когда n равно 2, 3 или 4, R2 заместители могут быть одинаковыми или разными, А представляет собой насыщенный или частично ненасыщенный цикл, []o и []p представляют собой -(СН2)o– и -(СН2)р соответственно, при условии, что возможно также значение -СН-, когда А представляет собой частично ненасыщенный цикл, и о и р независимо представляют собой 0, 1 или 2, R3, R4, R5 и R6 независимо представляют собой водород, алкил(1-3С), алкил(1-3С)циклоалкил(3-6С), СН2ОН, или (R3 и R5), или (R3 и R6), или (R4 и R5), или (R4 и R6) вместе могут образовывать алкиленовый мостик, включающий от 1 до 3 атомов углерода, при условии, что когда o равно 2, R3 представляет собой водород, и, когда р равно 2, R5 представляет собой водород, []q означает -(СН2)q-, где q представляет собой целое число от 0 до 2, и их фармацевтически приемлемым солям и пролекарствам. К изобретению относятся все соединения формулы (1), рацематы, смеси диастереомеров и отдельные стереоизомеры. Таким образом, соединения, в которых заместители на потенциально асимметричных атомах углерода имеют R- или S-конфигурацию, также относятся к настоящему изобретению. Кроме того, к изобретению относятся пролекарства, т.е. соединения, которые при введении людям любым известным способом в ходе обмена веществ превращаются в соединения формулы (1). В частности, изобретение относится к соединениям с первичными или вторичными амино- или гидроксильными группами. Такие соединения могут подвергаться взаимодействию с органическими кислотами с получением соединений формулы (1), в которых присутствует дополнительная группа, легко удаляемая после введения, например, но без ограничения, амидина, енамина, основания Манниха, гидроксиметилен-производного, О-(ацилоксиметиленкарбамат)-производного, карбамата, сложного эфира, амида или енаминона. Пролекарство представляет собой неактивное соединение, которое при абсорбции подвергается превращению в активную форму (Medicinal Chemistry: Principles and Practice, 1994, ISBN 0-85186-494-5, Ed.: F.D. King, p. 216). Настоящее изобретение, в частности, относится к соединениям формулы (1), где: А представляет собой насыщенный цикл, R1 представляет собой водород, алкил(1-3С) или ацил(2-4С), R3, R4, R5 и R8 независимо представляют собой водород или алкил(1-3С), или (R3 и R5), или (R3 и R6), или (R4 и R5), или (R4 и R6) вместе могут образовывать алкиленовый мостик, включающий от 1 до 3 атомов углерода, при условии, что когда о равно 2, R3 представляет собой водород, когда р равно 2, R5 представляет собой водород, и R2, m, n, o, p и q принимают значения, указанные выше. Точнее, настоящее изобретение относится к соединениям формулы (1), где: А представляет собой насыщенный цикл, m=0, n=0 или 1, о=1, р=1, q=0, R1=H или ацетил, R2 представляет собой галоген, CF3, алкил(1-3С), амино, гидрокси, циано, ОСН3 или OCF3, R3, R4, R5 и R6 независимо представляют собой водород или алкил(1-2С), или (R4 и R5) вместе могут образовывать алкиленовый мостик, включающий от 1 до 2 атомов углерода. Более предпочтительным является соединение формулы (2) и все его стереоизомеры.
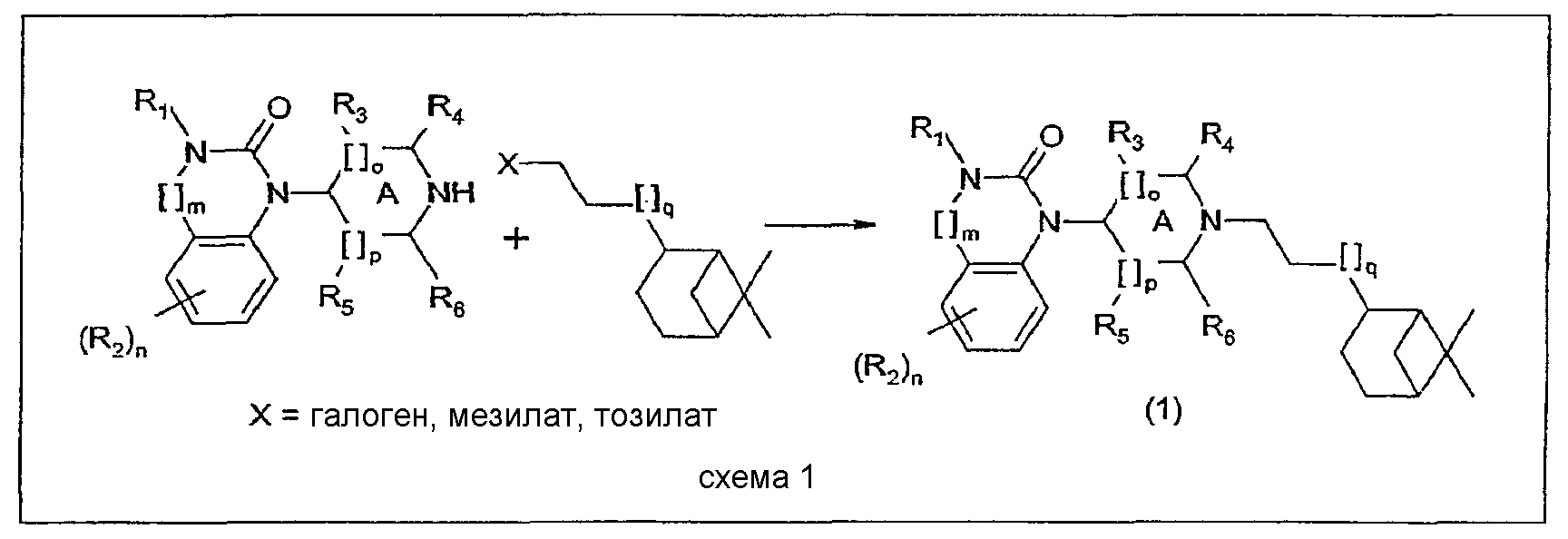
Соединения настоящего изобретения и их соли могут быть получены согласно общему способу, представленному ниже.
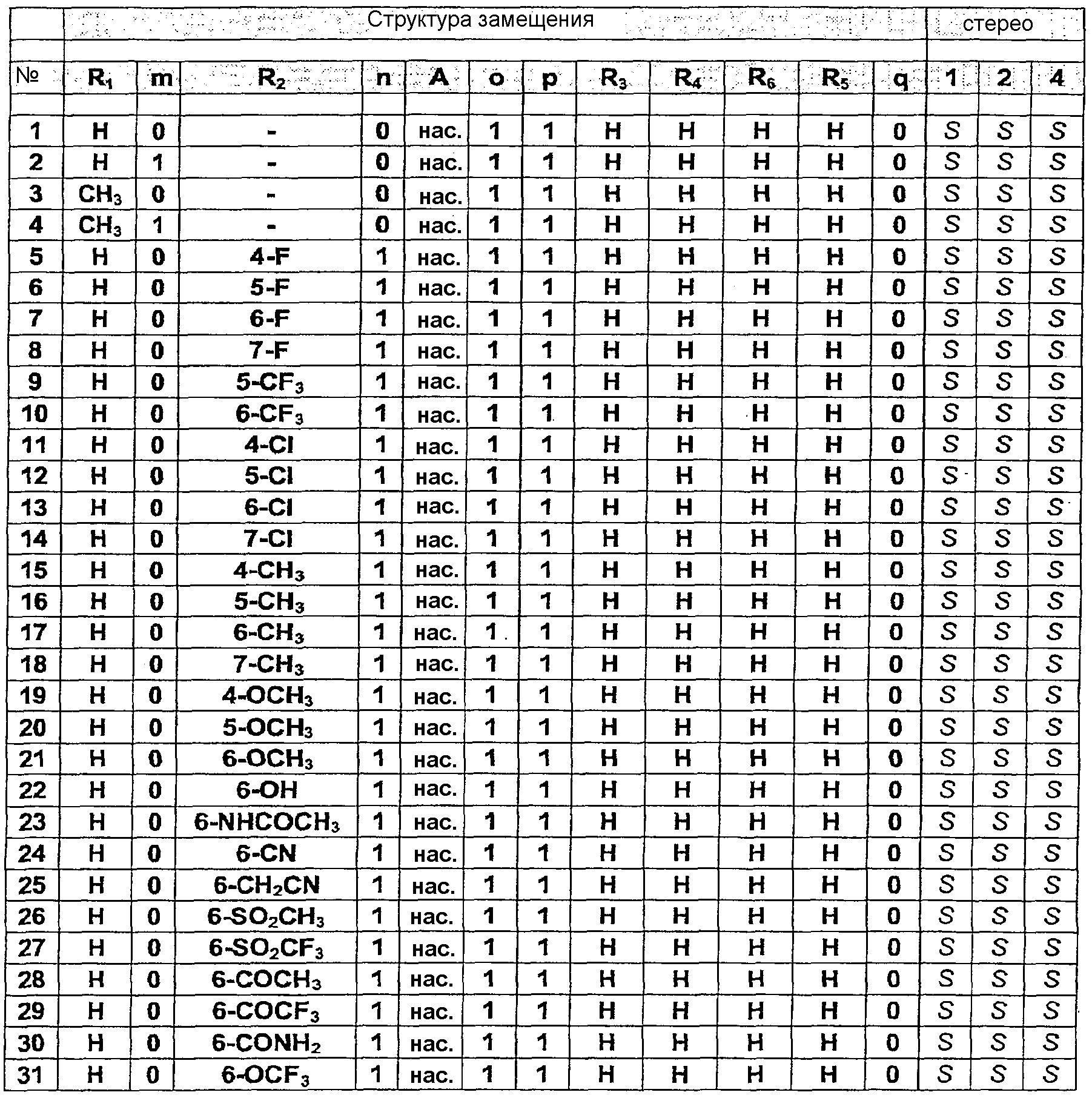
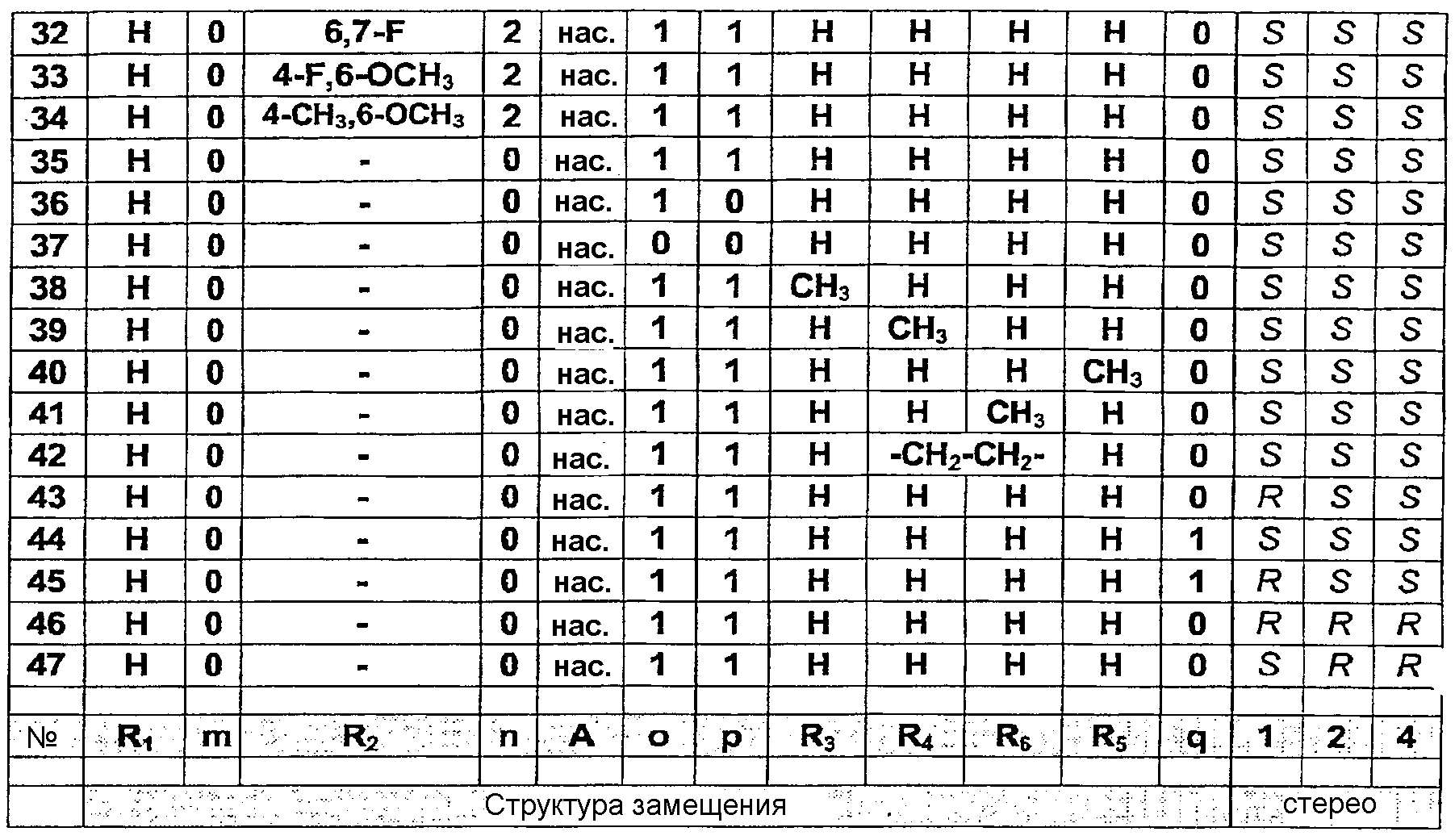
Исходные соединения для данного общего способа получены следующим образом. Бензимидазолоны (m=0) могут быть синтезированы в соответствии со способами, описанными в публикациях J.Med. Chem., 30, 814-819, 1987 и WO 99/36421 (Pfizer). Хиназолоны (m=1) могут быть синтезированы в соответствии с методикой публикации Chem. Pharm. Bull., 33, 1116-1128, 1985. Гидронопол-производные, содержащие Х в качестве удаляемой группы (галоген, мезилат, тозилат), могут быть синтезированы из соответствующих спиртов с помощью стандартных методик. Соответствующие спирты могут быть синтезированы следующим образом: – для цис-аналога с q=0 – из (-)- – для транс-аналога с q=0 – из транс-миртанола, как описано в публикации Bull. Soc. Chim. Fr., 196, 1958, или в соответствии со слегка измененной методикой (бромирование спирта, замещение циано-группой, превращение в сложный этиловый эфир и восстановление до целевого спирта); – для цис- и транс-аналогов с q=1 или 2 – посредством одного или двух циклов методики гомологизации аналогов, как описано для синтеза транс-аналога с q=0 из транс-миртанола. Фармацевтически приемлемые соли могут быть получены с использованием стандартных методик, хорошо известных в данной области техники, например смешением соединения настоящего изобретения с подходящей кислотой, например неорганической кислотой, такой как соляная кислота, или с органической кислотой. Соединения настоящего изобретения общей формулы (1), а также их соли обладают ORL1 антагонистической активностью. Они могут применяться для лечения расстройств, в которые вовлечены ORL1 рецепторы или которые могут лечиться посредством воздействия на данные рецепторы. Например, при острой и хронической боли, расстройствах центральной нервной системы, особенно, но без ограничения, для облегчения симптомов расстройств, связанных с состоянием беспокойства и стресса, депрессии, различных форм эпилепсии, удара, расстройств, характеризующихся ухудшением познавательной способности и памяти, таких как болезнь Альцгеймера, болезнь Крейтцфельда-Якоба, болезнь Хантингтона, болезнь Паркинсона, нейрореабилитация (посттравматические повреждения головного мозга); острое повреждение головного или спинного мозга; расстройства, связанные с некоторыми веществами, включая расстройства, вызванные их неправильным применением (такие как зависимость и злоупотребление), и расстройства, вызванные веществами (такие как абстинентный синдром); расстройства питания, такие как анорексический невроз и булимический невроз, ожирение; при желудочно-кишечных расстройствах, в частности синдроме раздраженной толстой кишки, воспалении толстой кишки (болезнь Крона и неспецифический язвенный колит), воспалении мочевых путей, почечные расстройства, характеризующиеся нарушением равновесия удержания/выделения воды или выделения соли; при сердечно-сосудистых расстройствах, таких как инфаркт миокарда, аритмия, гипертензия, тромбоз, анемия, атеросклероз, стенокардия; при кожных заболеваниях, таких как крапивница, красная волчанка и зуд; при офтальмологических расстройствах, таких как глаукома; при респираторных расстройствах, включая кашель, хроническое обструктивное заболевание легких, бронхит и муковисцидоз; при заболеваниях иммунной системы и вирусных инфекциях. Антагонистические свойства соединений данного изобретения в отношении ORL1 рецептора в условиях in vitro и in vivo были изучены с использованием способов, представленных ниже. Сродство в отношении ORL1 рецепторов человека Сродство соединений в отношении ORL1 рецепторов человека определяют с использованием метода количественной оценки связывания рецептора in vitro, описанного в публикации Ardati et al., Mol. Pharmacol., 51, 816, 1997. Кратко, мембранные препараты получают из клеток яичника китайского хомячка (Chinese Hamster Ovary (CHO)), в которые был стабильно экспрессирован ORL1 рецептор человека. Мембраны инкубируют с [3Н]-ноцицептином в отсутствие или в присутствии опытных соединений, взятых в различных концентрациях, с разбавлением в подходящем буфере. Неспецифическое связывание определяют как связывание, сохранившееся в присутствии 10-6М ноцицептина. Разделение связанной и свободной радиоактивностей выполняют фильтрованием через фильтры из стекловолокна Packard GF/B и несколькими промывками буфером, охлажденным льдом, с использованием сборщика клеток Packard. Связанную радиоактивность количественно определяют с помощью сцинтилляционного счетчика (Topcount, Packard) с использованием жидкого сцинтилляционного коктейля (Microscint 0, Packard). Результаты количественного определения радиоактивности представляют в виде графика зависимости от концентрации замещающих опытных веществ, и кривые замещения вычисляют с использованием логистической регрессии с четырьмя параметрами, получая значения IC50, т.е. концентрации замещающего соединения, при которой замещается 50% радиолиганда. Значения сродства рК1 вычисляют корректировкой значений IC50 с учетом концентрации радиолиганда и его сродства в отношении ORL1 рецептора человека в соответствии с уравнением Ченга-Прусоффа: рК1 = -log(IC50/(1+S/Kd)), в котором IC50 имеет значение, описанное выше, S представляет собой концентрацию [3H]-ноцицептина, используемую в испытании и выраженную в моль/л (обычно 0,2 нМ), и Кd представляет собой константу равновесной диссоциации [3H]-ноцицептина для ORL1 рецепторов человека (0,4 нМ). Соединения настоящего изобретения демонстрируют высокое сродство в отношении ORL1 рецепторов в биологическом испытании, описанном выше. Это свойство делает их применимыми для лечения расстройств, в которые вовлечены ORL1 рецепторы или которые могут лечиться воздействием на эти рецепторы. Сродство в отношении µ-опиатных рецепторов Сродство соединений в отношении µ-опиатных рецепторов определяют с использованием биологического испытания связывания рецептора in vitro, описанного в публикации Childers et al., Eur. J. Pharm.,55, 11, 1979. Кратко, мембранные препараты получают из СНО клеток, в которые был стабильно экспрессирован µ-опиатный рецептор, и инкубируют с [3Н]-налоксоном в отсутствие или в присутствии опытных соединений при разбавлении до концентраций в интервале от 10 мкМ до 0,1 нМ подходящим буфером. Неспецифическое связывание определяют как связывание, сохранившееся в присутствии 10-7 М леваллорфан-тартрата. Разделение связанной и свободной радиоактивностей осуществляют, как описано выше, и сродство соединений вычисляют аналогичным образом, используя концентрацию (S) 1 нМ [3H]-налоксона и значение Kd, равное 1,3 нМ. Большинство соединений настоящего изобретения проявляют низкое сродство в отношении µ-опиатных рецепторов в биологическом испытании связывания, описанном выше: обычно фактор 100 ниже их сродства в отношении ORL1 рецепторов. Следовательно, маловероятно, что они вызывают нежелательные побочные эффекты, которые, как известно, имеют место с опиатами, подобными морфину. Агонизм в отношении ORL1 рецептора in vitro Активация G-белок-связанного ORL1 рецептора ингибирует активность аденилатциклазы и снижает внутриклеточную концентрацию второго информационного АМФ. Количественное определение активности соединений в отношении ORL1 рецепторов проводят в соответствии с методикой, описанной в публикации Jenck et al., Proc. Natl. Acad. Sci. USA, 97, 4938-4943, 2000. Полученные результаты показывают, что соединения являются сильными агонистами со значениями рЕС50, соответствующими их значениям рКi. Агонизм в отношении ORL1 рецептора in vivo Соединения настоящего изобретения при испытании в соответствии с методикой кондиционированной ультразвуковой дистресс-вокализации, описанной в публикации Molewijk et al., Psychopharmacology, 117, 32-40, 1995, при интраперитонеальном и/или пероральном введении проявляют высокую активность. Это показывает не только то, что соединения обладают хорошей биодоступностью после перорального введения, но также и то, что они преодолевают гематоэнцефалический барьер. Пептид ноцицептин также активен в данном биологическом опыте, но для демонстрации его эффекта необходимо вводить его прямо в мозг (посредством интрацеребро-вентикулярной инъекции). КОНКРЕТНЫЕ ПРИМЕРЫ СИНТЕЗА Пример синтеза 1 (см. таблицу, представленную ниже) Стадия 1. Раствор (-)-цис-гидронопола (20 г, 0,12 моль) и триэтиламина (41,6 мл, 0,30 моль) в дихлорметане (150 мл) охлаждают до 0°C и при охлаждении на ледяной бане по каплям добавляют раствор мезилхлорида (11,2 мл, 0,15 моль) в дихлорметане (50 мл). Смесь перемешивают при комнатной температуре в течение 16 часов, после чего добавляют раствор HCl (1N, 100 мл). Водный слой промывают дважды 70 мл дихлорметана, объединенные органические слои сушат над сульфатом магния и концентрируют в вакууме. Мезилат получают в виде масла желтовато-оранжевого цвета (27,7 г, 11 моль, выход 92%). Стадия 2. Раствор 4-(1-бензимидазолон)пиперидина (ACROS, 6,51 г, 30 ммоль), мезилата, полученного на предыдущей стадии (8,9 г, 36 ммоль), карбоната калия (16,8 г, 120 ммоль) и йодида натрия (5,4 г, 36 ммоль) в метилэтилкетоне (800 мл) при перемешивании нагревают в атмосфере N2 до 80°C и выдерживают при данной температуре в течение 16 часов. Реакционную смесь концентрируют в вакууме, затем добавляют дихлорметан (500 мл) и водный NaHCO3 (5%, 300 мл). Водный слой промывают дихлорметаном (2 раза, 80 мл) и объединенные органические слои сушат над MgSO4 и концентрируют в вакууме. Сырой продукт очищают колоночной хроматографией (силикагель), используя смесь дихлорметан:метанол:аммиак (94,5:5:0,5) в качестве элюента. Раствор чистого продукта, полученный после концентрирования в вакууме (7,5 г, 20 ммоль, выход 68%), растворяют в растворе HCl в абсолютированном этаноле (60 мл). Концентрирование в вакууме полученного раствора при 30°C приводит к получению HCl соли соединения примера 1 (8,25 г, 20 ммоль, количественный выход) в виде твердого белого аморфного вещества с M+ 368 m/z и температурой плавления 167-174°C. Пример синтеза 43 Стадия 1. Трифенилфосфин (116 г, 0,44 моль) растворяют в ацетонитриле (1 литр) и охлаждают на ледяной бане в атмосфере N2. К полученному раствору по каплям добавляют бром (22,5 мл, 0,44 моль). Температуру экзотермической реакции поддерживают ниже 10°C. После завершения добавления ледяную баню удаляют и медленно добавляют (-)-транс-миртанол (68,8 г, 0,44 моль, Aldrich), растворенный в 250 мл ацетонитрила. После завершения добавления раствор светло-желтого цвета кипятят с обратным холодильником в течение 3 часов при использовании насадки Дина-Старка с удалением из водоотделителя 200 мл растворителя. Полученную реакционную смесь концентрируют в вакууме. Сырой продукт очищают колоночной хроматографией (силикагель) с использованием смеси дихлорметан-диэтиловый эфир (1:1 об./об.) в качестве элюента. Чистый продукт получают в виде масла светло-желтого цвета (87,8 г, 41 ммоль, выход 93%). Стадия 2. Полученный миртанилбромид (87,8 г, 0,41 моль) растворяют в 1 литре диметилформамида. В полученный раствор добавляют цианид натрия (40 г, 0,81 моль) и полученную смесь кипятят с обратным холодильником при перемешивании в течение 5 часов. После охлаждения смесь разбавляют водой (3 литра) и экстрагируют метил-трет-бутиловым эфиром (3 раза, 1,5 литра). Органический слой промывают насыщенным раствором соли (300 мл), сушат над Na2SO4 и концентрируют в вакууме. Сырой продукт очищают колоночной хроматографией (силикагель) с использованием смеси дихлорметан-гептан (1:1 об./об.) в качестве элюента. Чистый продукт получают в виде бесцветной жидкости (52,4 г, 0,32 моль, выход 78%). Стадия 3. Серную кислоту (190 мл) по каплям добавляют к 500 мл этанола, охлажденного на ледяной бане. К раствору добавляют раствор полученного выше миртанилцианида (52,4 г, 0,32 моль) в этаноле (100 мл) и полученную смесь кипятят с обратным холодильником при перемешивании в течение 16 часов. После охлаждения и добавления 1,5 литра воды смесь экстрагируют три раза 1,5 литрами метил-трет-бутилового эфира. Органический слой промывают насыщенным водным раствором NaHCO3 (1 литр), сушат над Na2SO4 и концентрируют в вакууме. Сырой сложный эфир (54,2 г, 0,26 моль, выход 81%) получают в виде почти бесцветной жидкости. Стадия 4. Сложный эфир, полученный на предыдущей стадии (54,2 г, 0,26 моль), добавляют к суспензии алюмогидрида лития (20 г, 0,52 моль) в тетрагидрофуране (1 литр). После завершения добавления смесь кипятят с обратным холодильником в течение 1 часа. Реакционную смесь охлаждают на ледяной бане, затем осторожно добавляют 1 литр водной HCl (1N). После завершения добавления смесь разбавляют 1 литром воды и три раза экстрагируют метил-трет-бутиловым эфиром (1,5 л). Органический слой промывают насыщенным раствором соли (250 мл), сушат над Na2SO4 и концентрируют в вакууме. Сырую смесь очищают перегонкой Kugelrohr (т.кип. 85°C при 3,10-2 мбар), получая 35 г (0,17 моль, 65%) (-)-транс-гидронопола в виде бесцветного масла. Стадия 5. Раствор полученного (-)-транс-гидронопола (5 г, 28 ммоль) и триэтиламина (9,4 мл, 68 ммоль) в дихлорметане (50 мл) охлаждают до 0°C. К полученному раствору при охлаждении на ледяной бане по каплям добавляют раствор мезилхлорида (2,7 мл, 35 ммоль) в дихлорметане (13 мл). Реакционную смесь перемешивают при комнатной температуре в течение 16 часов, после чего добавляют раствор HCl (1N, 50 мл). Водный слой промывают дихлорметаном (2 раза, 30 мл), объединенные органические слои сушат над сульфатом магния и концентрируют в вакууме. Полученный мезилат очищают колоночной хроматографией (силикагель) с использованием смеси дихлорметан-метанол (90:10) в качестве элюента. Чистый продукт получают в виде бесцветной жидкости (5,6 г, 23 ммоль, выход 81%). Стадия 6. Раствор 4-(1-бензимидазолон)пиперидина (ACROS, 1,8 г, 8,3 ммоль), мезилата, полученного на предыдущей стадии синтеза (2,5 г, 10 ммоль), карбоната калия (4,7 г, 34 ммоль) и йодида натрия (1,5 г, 10 ммоль) в метилэтилкетоне (300 мл) нагревают в атмосфере N2 до 80°C и выдерживают при данной температуре в течение 16 часов. Реакционную смесь концентрируют в вакууме, затем добавляют диизопропиловый эфир (300 мл). Образующийся осадок светло-желтого цвета отфильтровывают, промывают петролейным эфиром (100 мл) и сушат в вакууме. Осадок снова растворяют в кипящем эфире (300 мл) и фильтруют для удаления исходного вещества. Осадок, полученный после охлаждения, собирают фильтрованием и сушат в вакууме, получая 1,85 г (5 ммоль, выход 61%) чистого продукта в виде твердого белого аморфного вещества с M+ 368 m/z и температурным интервалом плавления 220-239°C. Пример синтеза 17 Стадия 1. Раствор 3-фтор-4-нитротолуола (Aldrich, 4,65 г, 30 ммоль), 4-амино-1-бензилпиперидина (Aldrich, 6,1 мл, 30 ммоль) и K2CO3 (6,63 г, 48 ммоль) в диметилформамиде (50 мл) перемешивают при 65°C в атмосфере N2 в течение 18 часов. После охлаждения до комнатной температуры смесь выливают в смесь воды (200 мл) и дихлорметана (350 мл). Водный слой экстрагируют дихлорметаном (два раза, 70 мл), объединенные органические слои промывают водой (два раза, 50 мл), сушат над MgSO4 и концентрируют в вакууме. Полученный сырой продукт очищают колоночной хроматографией (силикагель) с использованием смеси дихлорметан-метанол (97:3) в качестве элюента. После концентрирования в вакууме чистый продукт получают в виде маслянистого вещества желтого цвета (9,1 г, 28 ммоль, выход 93%). Стадия 2. Порцию никеля Ренея (Aldrich R 2800 [7440-02-0], приблизительно 600 мг) промывают 96% этанолом (дважды, 10 мл) и затем в атмосфере N2 добавляют к раствору продукта, полученному на предыдущей стадии синтеза (9,1 г, 28 ммоль), в 96% этаноле (200 мл). Раствор гидрируют при комнатной температуре и давлении 1 атмосфера в течение 18 часов. Смесь фильтруют через Hyflo, промывают 96% этанолом (3 раза, 100 мл), фильтрат концентрируют в вакууме и дважды подвергают совместному выпариванию с этилацетатом с получением восстановленного продукта в виде маслянистого вещества пурпурного цвета (8,3 г, 28 ммоль, выход 100%). Стадия 3. К раствору продукта, полученного на предыдущей стадии синтеза (8,3 г, 28 ммоль), в ацетонитриле (100 мл), перемешивая в атмосфере азота при комнатной температуре, добавляют порцию 1,1′-карбонилдиимидазола (ACROS, 6,5 г, 40 ммоль). Осадок, который начинает образовываться через 5 минут и образуется в течение 3 часов, собирают фильтрованием, промывают ацетонитрилом (20 мл) и диизопропиловым эфиром (100 мл) и сушат в вакууме. Сырой продукт (5,5 г) очищают колоночной хроматографией (силикагель) с использованием смеси дихлорметан-метанол (95:5) в качестве элюента. После концентрирования в вакууме чистый продукт получают в виде твердого белого вещества (5,0 г, 15 ммоль, выход 55%). Стадия 4. К раствору 5,0 г (15 ммоль) продукта, полученного на предыдущей стадии синтеза, в 300 мл метанола при перемешивании в атмосфере N2 добавляют 1N спиртовой раствор HCl (полученный из 1,22 г (15 ммоль) ацетилхлорида в 50 мл абсолютированного этанола). После добавления 10% Pd/C (приблизительно 500 мг) смесь гидрируют при комнатной температуре и давлении 1 атмосфера в течение 2,5 часов. Смесь фильтруют через Hyflo, промывают метанолом (2 раза, 100 мл) и фильтрат концентрируют в вакууме, получая 4,15 г (15 ммоль, выход 100%) продукта в виде твердого белого вещества. Стадия 5. Раствор продукта, полученного на предыдущей стадии синтеза (4,15 г, 15 ммоль), мезилата, полученного на стадии 1 примера 1 (8,9 г, 16 ммоль), карбоната калия (10,4 г, 75 ммоль) и йодида натрия (2,4 г, 16 ммоль) в метилэтилкетоне (250 мл) при перемешивании нагревают в атмосфере N2 до 80°C и выдерживают при данной температуре в течение 16 часов. После концентрирования реакционной смеси в вакууме добавляют дихлорметан (500 мл) и водный NaHCO3 (5%, 300 мл). Водный слой промывают дихлорметаном (2 раза, 80 мл), объединенные органические слои сушат над MgSO4 и концентрируют в вакууме. Сырой продукт очищают колоночной хроматографией (силикагель) с использованием смеси дихлорметан- метанол-аммиак (92:7,5:0,5) в качестве элюента. Концентрирование в вакууме приводит к получению чистого продукта (5,0 г, 13 ммоль) в виде маслянистого вещества. После добавления к полученному продукту 100 мл диизопропилового эфира и перемешивания смеси при комнатной температуре в течение 30 минут продукт выпадает в осадок в виде твердого белого вещества. Твердый продукт собирают фильтрованием и сушат в вакууме, получая 3,3 г (8,6 ммоль, выход 57%) с M+ 382 m/z и температурой плавления 214-217°C. С помощью описанных выше и аналогичных способов синтезируют 45 специфических примеров соединений. Они предназначены для более подробного дополнительного пояснения изобретения и поэтому, как подразумевается, никоим образом не ограничивают область настоящего изобретения. Структуры этих соединений общей формулы (1) представлены в таблице, приведенной ниже.
ПРИМЕР ПРЕПАРАТА СОЕДИНЕНИЯ, ИСПОЛЬЗУЕМОГО В ИССЛЕДОВАНИИ НА ЖИВОТНЫХ Препарат соединения примера 1 Для перорального (р.о.) введения: к нужному количеству (0,5-5 мг) твердого соединения примера 1 в стеклянной пробирке добавляют несколько стеклянных шариков и твердое вещество измельчают вихревым перемешиванием в течение 2 минут. После добавления 1 мл раствора 1% метилцеллюлозы в воде и 2% (об./об.) Poloxamer 188 (Lutrol F68) соединение суспендируют вихревым смешением в течение 10 минут. Значение рН доводят до 7 добавлением нескольких капель водного раствора NaOH (0,1N). Частицы, сохранившиеся в суспензии, дополнительно суспендируют с использованием ультразвуковой бани. Для интраперитонеального (i.p.) введения: к нужному количеству (0,5-15 мг) твердого соединения примера 1 в стеклянной пробирке добавляют несколько стеклянных шариков и твердое вещество измельчают вихревым перемешиванием в течение 2 минут. После добавления 1 мл раствора, содержащего 1% метилцеллюлозы и 5% маннита в воде, соединение суспендируют вихревым смешением в течение 10 минут. И наконец, значение рН доводят до 7. ФАРМАКОЛОГИЧЕСКИЕ ДАННЫЕ
Формула изобретения
1. Соединение общей формулы (1) 2. Соединения по п.1 общей формулы (1), где А представляет собой насыщенный цикл, 3. Соединения по п.1 общей формулы (1), 4. Соединение по п.1 формулы (2) и его стереоизомеры: 5. Фармацевтические композиции, обладающие ORL1 антагонистической активностью, содержащие фармакологически активное количество, по меньшей мере, одного из соединений по любому из пп.1-4. 6. Соединение по любому из пп.1-4 или его соль для получения фармацевтической композиции, обладающей ORL1 антагонистической активностью для лечения расстройств, в которые вовлечены ORL1 рецепторы или которые могут лечиться посредством воздействия на эти рецепторы. 7. Применение соединения по любому из пп.1-4, для лечения состояния острой или хронической боли, состояния стресса, расстройств, характеризующихся ухудшением памяти, расстройства, связанные с некоторыми веществами, расстройства питания, желудочно-кишечные расстройства, почечные расстройства, сердечно-сосудистые расстройства и заболевания иммунной системы.
|
||||||||||||||||||||||||||||||||||||||
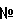 517
517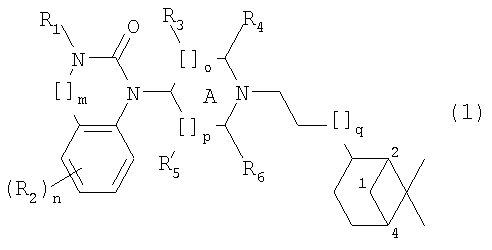
 – и
– и  -опиоидных рецепторов (Mollereau et al., FEBS Lett., 341, 33-38, 1994; Bunzow et al., FEBS Lett., 347, 284-288, 1994). Несмотря на близкое последовательное и структурное сходство с опиоидными рецепторами, лиганды классических опиоидных рецепторов не взаимодействуют с ORL1 рецепторами. В 1995 году из экстрактов головного мозга был получен чистый нейропептид, образованный 17 аминокислотами, и затем было показано, что он является природным лигандом G-белок-связанного ORL1 рецептора (Reinscheid et al., Science, 270, 792-794, 1995; Meunier et al., Nature, 377, 532-535, 1995). Этот пептид был назван орфанином FQ или ноцицептином, и он не связывается с тремя традиционными опиоидными рецепторами. Полученные результаты послужили импульсом для серьезных исследований функциональной роли и изыскания новых лигандов ORL1 рецептора. Результатом этих исследований стали несколько сотен публикаций, включая несколько обзоров (см., например, Grond et al., Anaesthesist, 51, 996-1005, 2002) и несколько дюжин заявок на патенты, в которых описываются как пептидные, так и непептидные лиганды, отличающиеся по силе и селективности (в отношении ORL1 по сравнению с µ-опиатными). Поскольку µ-опиатные рецепторы широко распространены по всему организму, отсутствие селективности могло бы приводить к ряду нежелательных опиат-подобных побочных эффектов, например седативному эффекту, угнетению дыхания, толерантности и зависимости (Drug News Perspect, 14, 335, 2001). Шесть из заявок на патенты, касающихся ORL1, относятся к производным бензимидазолона: WO 98/54168, WO 99/36421, WO 00/006545, WO 00/08013, WO 01/39775 и заявка на патент США 20020128288.
-опиоидных рецепторов (Mollereau et al., FEBS Lett., 341, 33-38, 1994; Bunzow et al., FEBS Lett., 347, 284-288, 1994). Несмотря на близкое последовательное и структурное сходство с опиоидными рецепторами, лиганды классических опиоидных рецепторов не взаимодействуют с ORL1 рецепторами. В 1995 году из экстрактов головного мозга был получен чистый нейропептид, образованный 17 аминокислотами, и затем было показано, что он является природным лигандом G-белок-связанного ORL1 рецептора (Reinscheid et al., Science, 270, 792-794, 1995; Meunier et al., Nature, 377, 532-535, 1995). Этот пептид был назван орфанином FQ или ноцицептином, и он не связывается с тремя традиционными опиоидными рецепторами. Полученные результаты послужили импульсом для серьезных исследований функциональной роли и изыскания новых лигандов ORL1 рецептора. Результатом этих исследований стали несколько сотен публикаций, включая несколько обзоров (см., например, Grond et al., Anaesthesist, 51, 996-1005, 2002) и несколько дюжин заявок на патенты, в которых описываются как пептидные, так и непептидные лиганды, отличающиеся по силе и селективности (в отношении ORL1 по сравнению с µ-опиатными). Поскольку µ-опиатные рецепторы широко распространены по всему организму, отсутствие селективности могло бы приводить к ряду нежелательных опиат-подобных побочных эффектов, например седативному эффекту, угнетению дыхания, толерантности и зависимости (Drug News Perspect, 14, 335, 2001). Шесть из заявок на патенты, касающихся ORL1, относятся к производным бензимидазолона: WO 98/54168, WO 99/36421, WO 00/006545, WO 00/08013, WO 01/39775 и заявка на патент США 20020128288.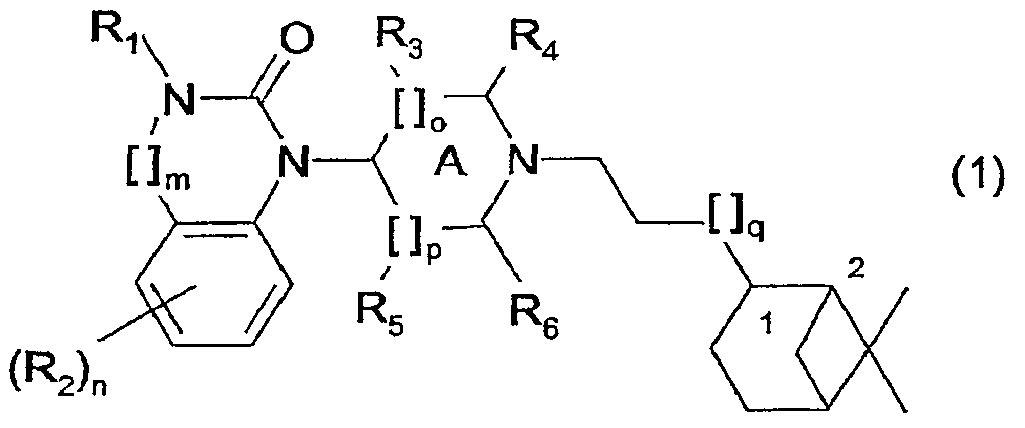
 -пинена, как описано в публикации J. Amer. Chem. Soc. 68, 638, 1946 и в патентах США
-пинена, как описано в публикации J. Amer. Chem. Soc. 68, 638, 1946 и в патентах США