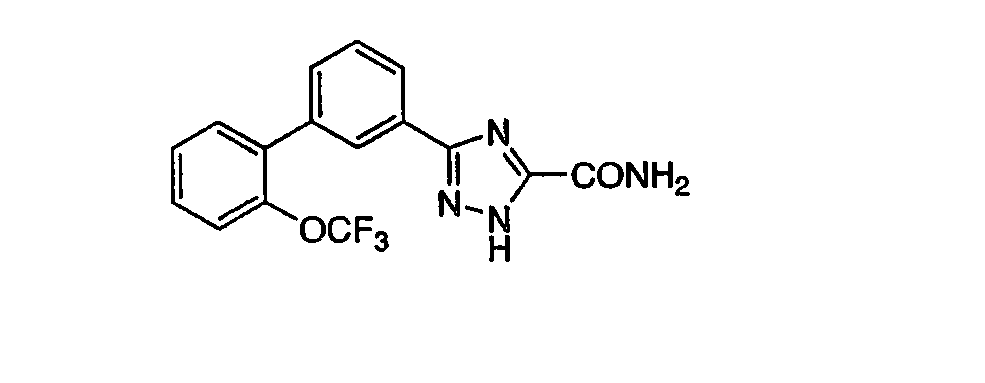
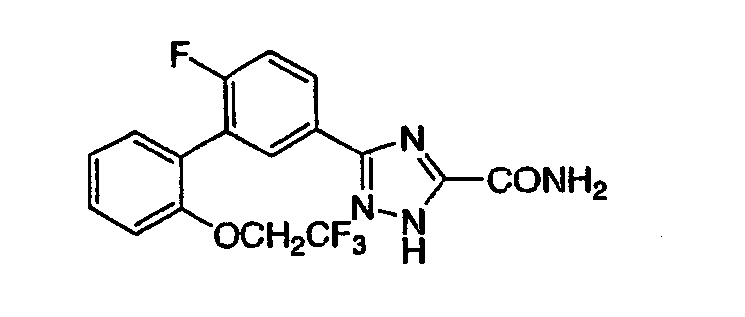
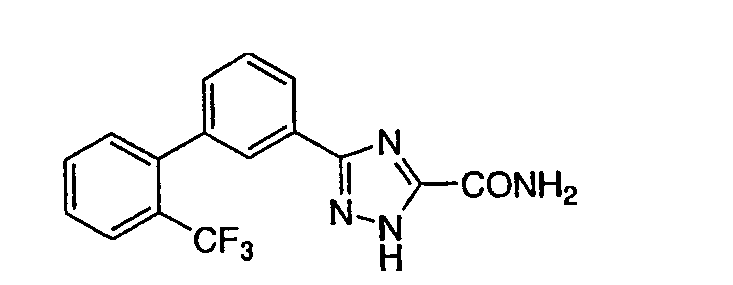
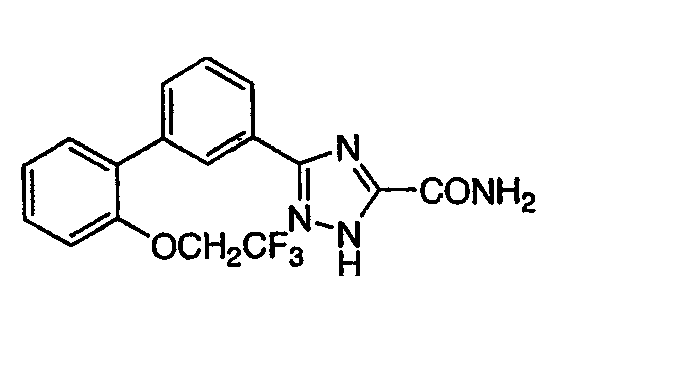
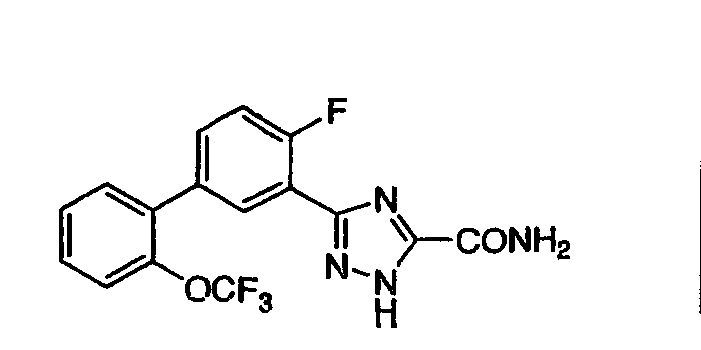
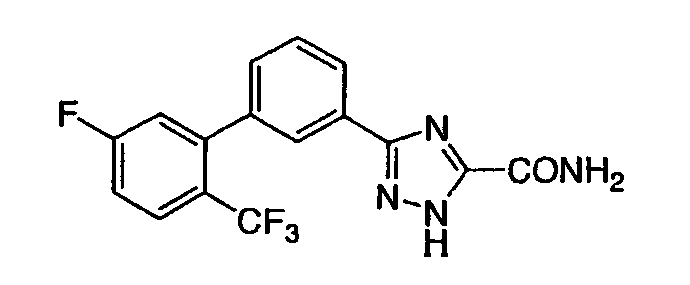
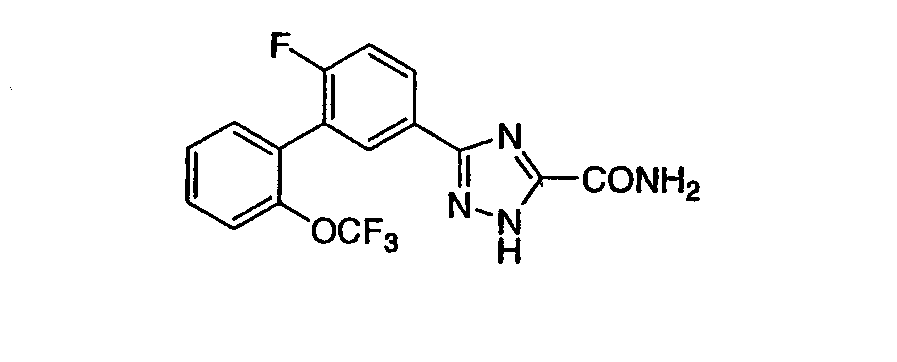
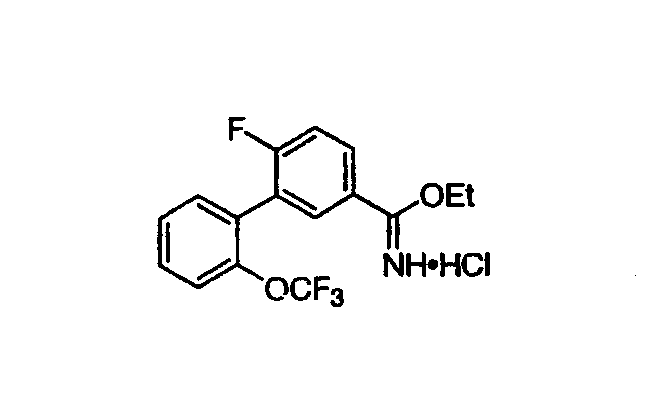
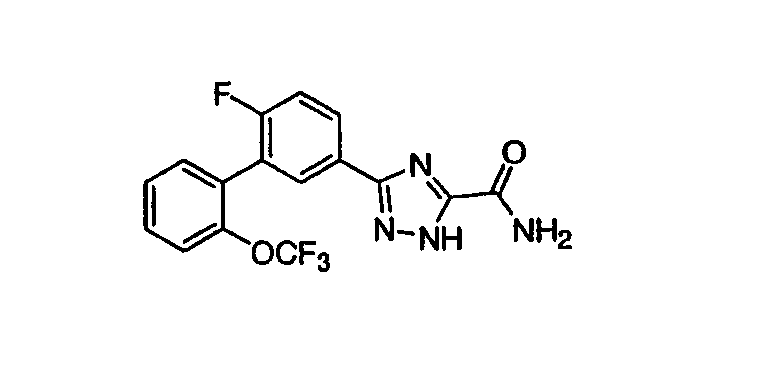
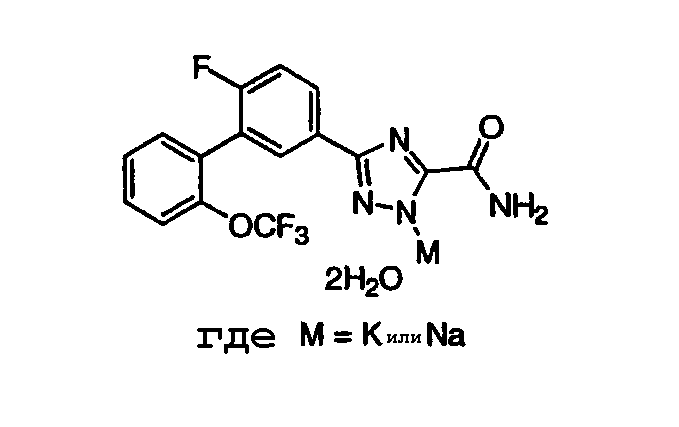
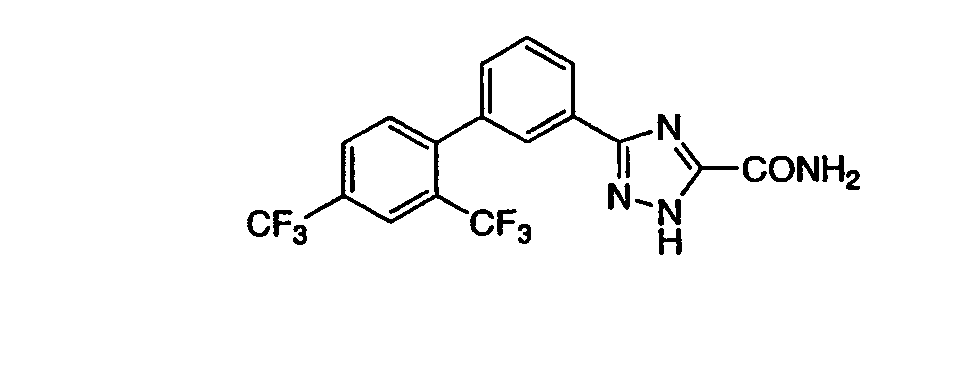
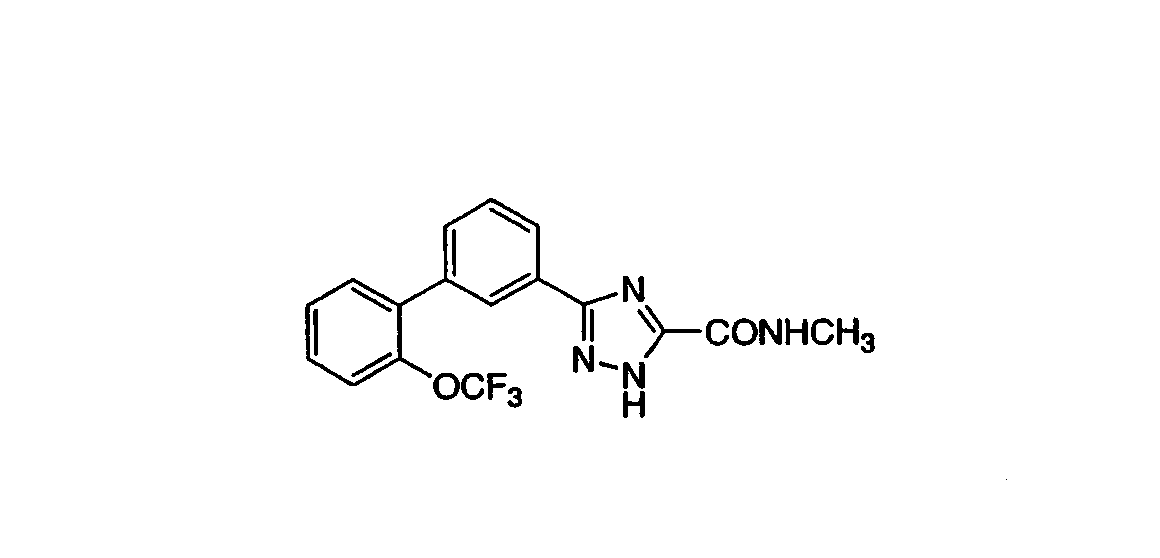
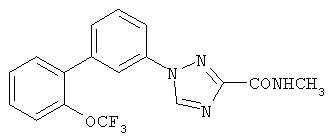
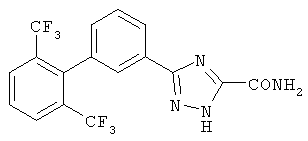
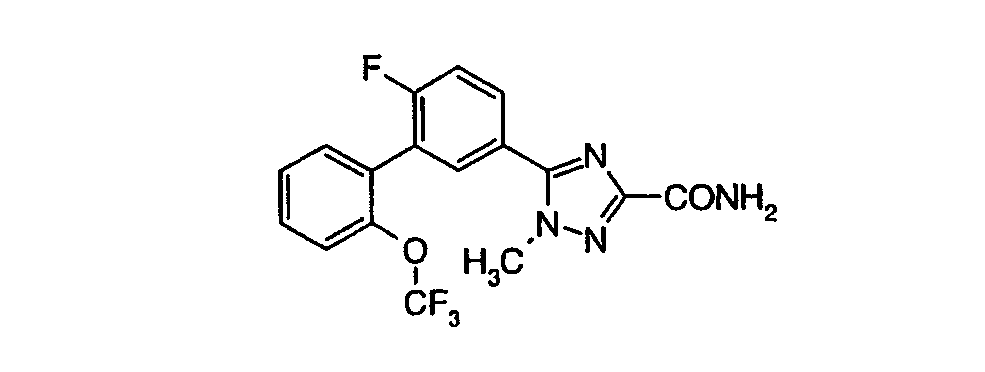
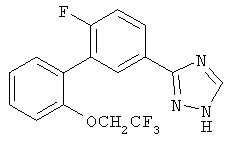
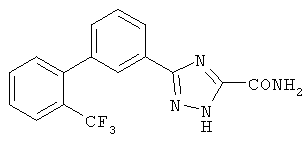
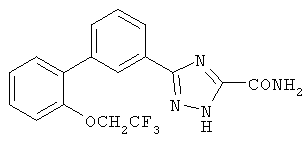
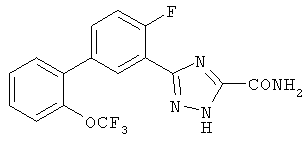
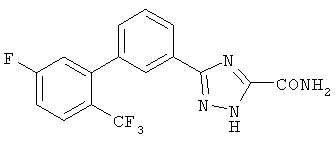
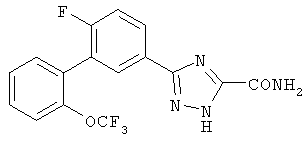
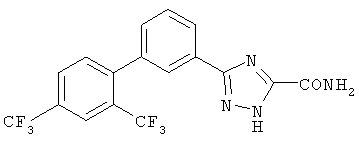
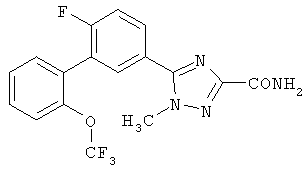
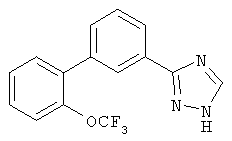
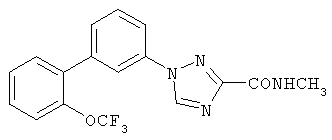
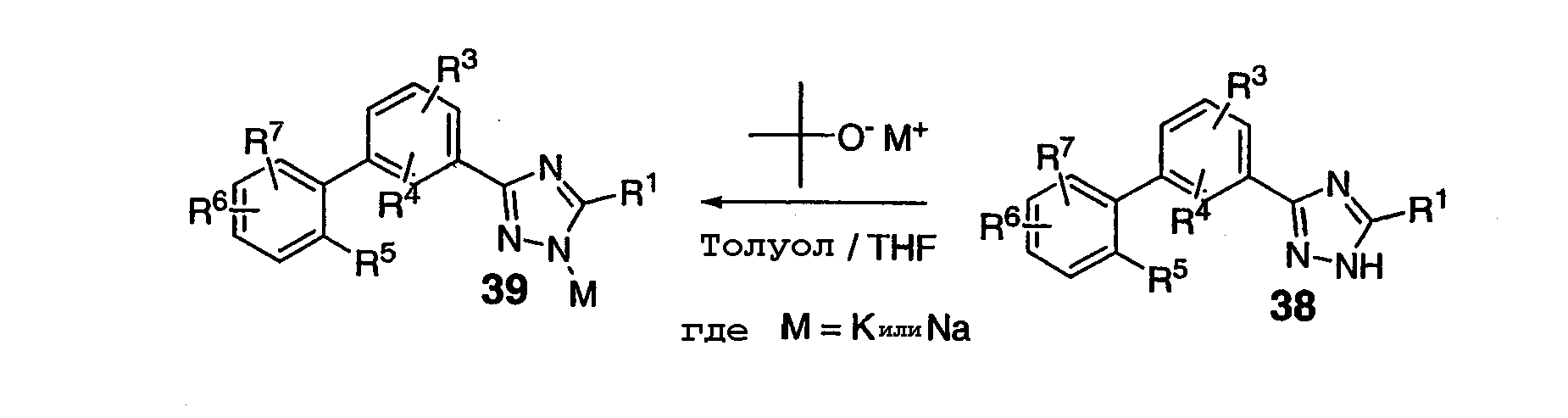Патент на изобретение №2356897
|
||||||||||||||||||||||||||||||||||||||||||||||
(54) БИАРИЛЗАМЕЩЕННЫЕ ТРИАЗОЛЫ КАК БЛОКАТОРЫ НАТРИЕВЫХ КАНАЛОВ
(57) Реферат:
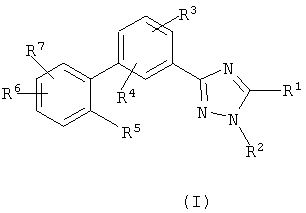
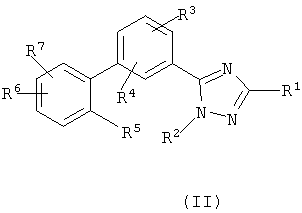
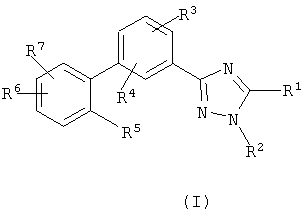
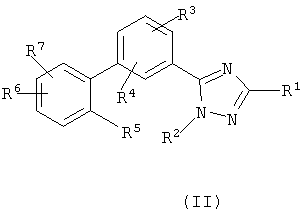
Описываются новые производные триазола общих формул (I) или (II)
или их фармацевтически приемлемые соли, где R1 представляет собой Н, C1-С6-алкил или группу CONRaRb, Ra означает водород, a Rb означает водород или C1-С6алкил; R2 представляет собой Н или С1-С6-алкил; R3 представляет собой Н или F, Cl, Br или I; R4 представляет собой Н; R5 представляет собой CF3 или O-(С1-С4)алкил, трижды замещенный F; R6 и R7 каждый независимо представляет собой Н, F или CF3, при условии, что один из R6 и R7 всегда означает Н; применение указанных соединений для приготовления лекарственного средства для лечения или профилактики боли или нарушения, вызванного болью; фармацевтическая композиция, содержащая новые соединения, и способ получения последних. 5 н. и 4 з.п. ф-лы.
Данное изобретение относится к биарилзамещенным производных триазола. В частности, данное изобретение относится к биарилзамещенным триазолам, которые являются блокаторами натриевых каналов и пригодны для лечения хронической и невропатической боли. Соединения данного изобретения также пригодны для лечения других состояний, включающих расстройства центральной нервной системы (ЦНС), такие как эпилепсия, маниакальная депрессия, биполярное расстройство, депрессивный синдром, состояние страха и диабетическая невропатия. Данное изобретение относится к биарилзамещенным триазолам, которые являются блокаторами натриевых каналов и пригодны для лечения хронической и невропатической боли. Соединения данного изобретения также пригодны для лечения других состояний, включающих расстройства центральной нервной системы (ЦНС), такие как эпилепсия, депрессивный синдром, состояние страха, маниакальная депрессия и биполярное расстройство. Данное изобретение также относится к фармацевтическим композициям, включающим соединение данного изобретения, либо индивидуально, либо в комбинации с одним или несколькими терапевтически активными соединениями, и фармацевтически приемлемый наполнитель. Данное изобретение включает также способы лечения острой боли, хронической боли, висцеральной боли, воспалительной боли, невропатической боли и расстройств ЦНС, включающих, но не ограниченных только ими, эпилепсию, депрессивный синдром, состояние страха, маниакальную депрессию и биполярное расстройство, с помощью введения соединений и фармацевтических композиций данного изобретения. Изобретение также относится к способу получения данных соединений. Подробное описание изобретения Данное изобретение включает соединения, представленные формулой (I) или (II)
или их фармацевтически приемлемые соли, где R1 представляет собой (a) Н; (b) C1-С6-алкил, С2-С4-алкенил, С2-С4-алкинил, С3-С6-циклоалкил или С1-С4-алкилен-[С3-С6-циклоалкил], любой из которых необязательно замещен одним или несколькими следующими заместителями: F, CF3, ОН, О-(С1-С4)алкил, O-CONRaRb, NRaRb, N (Ra)CONRaRb, COO-(С1-С4)алкил, COOH, CN, CONRaRb; (c) -С0-С4-алкилен-С1-С4-перфторалкил; (d) NRaRb, -N(CORa)Rb, -N(SO2Ra)Rb, -N(Ra)CON(Ra)2, -N(Ra)SO2Ra, -N(ORa)CONRaRb или -N(Ra)SO2N(Ra)2; (e) -CH(ORa)Ra, -C(ORb)CF3, -CH(NHRb)Ra, -C(=O)Ra, C(=O)CF3, -SOCH3, -SO2CH3, COORa, CN, CONRaRb, -COCONRaRb, -SO2NRaRb, -CH2O-SO2NRaRb, SO2N(Ra)ORa, -C(=NH)NH2, -CRa=N-ORa, CH=CHCONRaRb; (f) -CONRa(CH2)0-2C(Ra)(Rb)(CH2)0-2CONRaRb; (g) -C(Ra)=C(Rb)-COORa или -С(Ra)=C(Rb)-CONRaRb; Ra представляет собой (a) H; (b) С1-С4-алкил, необязательно замещенный одним или несколькими следующими заместителями: F, CF3, ОН, О-(С1-С4)алкил, S(О)0-2-(C1-С4)алкил, -OCONH2, -OCONH(С1-С4алкил), -OCON (С1-С4алкил) (С1-С4алкил), -OCONH(С1-С4алкиларил), -OCON(С1-С4алкил)(C1-С4алкиларил), NH2, NH(С1-С4алкил), N(С1-С4алкил)(С1-С4алкил), NH(С1-С4алкиларил), N(С1-С4алкил)(С1-С4алкиларил), NHCONH2, NHCONH(С1-С4алкил), NHCONH(С1-С4алкиларил), -NHCON (С1-С4алкил)(C1-С4алкил), NHCON(С1-С4алкил)(С1-С4алкиларил), N(С1-С4алкил)CON(С1-С4алкил)(С1-С4алкил), N(С1-С4алкил)CON(С1-С4алкил)(С1-С4алкиларил), COO-(С1-С4алкил), СООН, CN, CONH2, CONH(С1-С4алкил), CON(C1-С4алкил)(С1-С4алкил); (c) С0-С4-алкилен-(C1-C4)-перфторалкил; или (d) -С1-С4алкиленарил, где арил представляет собой фенил, пиридил, пиримидинил, фурил, тиенил, пирролил, триазолил, пиразолил, тиазолил, изоксазолил, оксазолил или оксадиазолил, любой арил из которых необязательно замещен 1-3 заместителями, выбранными из i) F, Cl, Br, I, ii) -CN, iii) -NO2, iv) -C(=O)(C1-С4алкил), v) -О(С1-С4алкил), vi) -N(С1-С4алкил)(С1-С4алкил), xiii) -C1-10алкил и xiv) -C1–10алкил, где один или несколько атомов углерода в алкиле могут быть замещены на -O-, -S(O)1-2-, -O-С(O), -С(O)-O-, -С(О)-, -СН(ОН)-, -С=С- или -C Rb представляет собой (a) Н или (b) C1-С6-алкил, необязательно замещенный одним или несколькими следующими заместителями: F, CF3, ОН, О-(С1-С4)-алкил, S(О)0-2-(C1-С4)алкил, -OCONH2, -OCONH(С1-С4алкил), NH2, NH(С1-С4алкил), N(C1-С4алкил)(С1-С4алкил), NHCONH2, NHCONH(С1-С4алкил), -NHCON(C1-С4алкил)(С1-С4алкил), COO-(С1-С4алкил), СООН, CN или CONH2; R2 представляет собой (a) Н; (b) -С1-С4-алкил, -С3-С6-циклоалкил или -С1-С4-алкилен-(С3-С6)циклоалкил, необязательно замещенный одним или несколькими следующими заместителями: F, CF3, ОН, О-(С1-С4)алкил, S(O)0-2-(C1-С4)алкил, O-CONRaRb, NRaRb, N(Ra)CONRaRb, COO-(C1-C4)алкил, СООН, CN, CONRaRb; (c) -С0-С4-алкилен-С1-С4-перфторалкил; (d) -C(=O)(Ra), -CONRaRb, -COO-(C1-C4)алкил, -SO2Ra, -SO2N(Ra)(Rb); R3 и R4 каждый независимо представляет собой (a) Н; (b) -C1-С6-алкил, -С2-С6-алкенил или -С2-С6-алкинил или -С3-С6циклоалкил, любой из которых необязательно замещен одним или несколькими следующими заместителями: F, CF3, -О-(С1-С4)алкил, CN, -N(Ra)(Rb), -N(Ra)СО-(С1-С4)алкил, COORb, CON(Ra)(Rb) или фенил; (c) -С0-С4-алкилен-С1-С4-перфторалкил или O-С0-С4-алкилен-С1-С4-перфторалкил; или (d) CN, NH2, NO2, F, Cl, Br, I, OH, OCON(Ra)(Rb), O(C1-C4-алкилен)CONRaRb, -OSO2N(Ra)(Rb), COORb, CON(Ra)(Rb) или арил, где арил необязательно замещен 1-3 заместителями, выбранными из i) F, Cl, Br, I, ii) -CN, iii) -NO2, iv) -С (=O)(Ra), v) -ORa, vi) -NRaRb, vii) -C0-4алкилен-CO-ORa, viii) -(С0-4алкилен)-NH-CO-ORa, ix) -(С0-4алкилен)СО-N(Ra)(Rb), x) -S(O)0-2Ra, xi) -SO2N(Ra)(Rb), xii) -NRaSO2Ra, xiii) -C1-10алкил и xiv) -C1-10алкил, где один или несколько атомов углерода в алкиле могут быть замещены на -NRa-, -O-, -S(O)1-2-, -O-С(O)-, -С(O)-O-, -C(O)-N(Ra)-, -N(Ra)-С(О) -, -N(Ra)-С(О)-N(Ra)-, -С(О)-, -СН(ОН)-, -С=С- или -С R5, R6 и R7 каждый независимо представляет собой (a) Н; (b) C1-С6-алкил, С2-С4-алкенил, или -С2-С4-алкинил, или -С3-С6-циклоалкил, любой из которых необязательно замещен одним или несколькими следующими заместителями: F, CF3, ОН, О-(С1-С4)алкил, OCON(Ra)(Rb), NRaRb, COORa, CN, CONRaRb, N(RaRb)CONRaRb; (c) -O-C1-С6-алкил или -O-С3-С6-циклоалкил, -S-C1-С6-алкил или -S-С3-С6-циклоалкил, любой из которых необязательно замещен одним или несколькими следующими заместителями: F, CF3, ОН, О-(C1-С4)алкил, NH2, NH(С1-С4алкил), N(С1-С4-алкил)2, СООН, CN, CONH2, CONH(С1-С4алкил), CON(С1-С4алкил)2, SO2NH2, SO2NH(С1-С4-алкил), тетразолил, триазолил, имидазолил, оксазолил, оксадиазолил, изоксазолил, тиазолил, фурил, тиенил, пиразолил, пирролил, пиридил, пиримидинил, пиразинил, фенил, пиперидинил, морфолинил, пирролидинил или пиперазинил; (d) -С0-С4-алкилен-С1-С4-перфторалкил или О-С0-С4-алкилен-С1-С4-перфторалкил; (e) CN, N(Ra)(Rb), NO2, F, Cl, Br, I, -ORa, -SRa, -OCON(Ra)(Rb), -OSO2N(Ra)(Rb), COORb, CON(Ra)(Rb), -N(Ra)CON(Ra)(Rb), -N(Ra)SO2N(Ra)(Rb), -C(ORb)Ra, -C(ORa)CF3, -C(NHRa)CF3, -C(=O)Ra, -C(=O)CF3, -SOCH3, -SO2CH3, -NHSO2(C1–6-алкил), NHSO2-арил, SO2N(Ra)(Rb), -CH2OSO2N(Ra)(Rb), SO2N(Rb)-ORa, -C(=NH)NH2, -CRa=NORa, CH=CH2 или арил, где арил необязательно замещен 1-3 заместителями, выбранными из i) F, Cl, Br, I, ii) -CN, iii) -NO2, iv) -C(=O)(Ra), v) -ORa, vi) -NRaRb, vii) -C0-4алкилен-CO-ORa, viii) -(С0-4алкилен)-NH-CO-ORa, ix) -(С0-4алкилен) -CO-N(Ra)(Rb), x) -S(O)0-2Ra, xi) -SO2N(Ra)(Rb), xii) -NRaSO2Ra, xiii) -С1-10алкил и xiv) -С1-10алкил, где один или несколько атомов углерода в алкиле могут быть замещены на -NRa-, -O-, -S(O)1-2-, -О-С(О)-, -С(O)-O-, -C(O)-N(Ra)-, -N(Ra)-С(О)-, -N(Ra)-C(O)-N(Ra)-, -С(O)-, -СН(ОН)-, -С=С- или -С Данное изобретение, кроме того, включает соединения формулы III
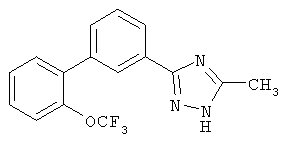
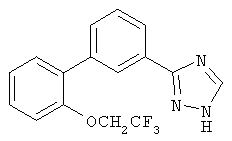
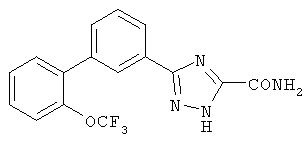
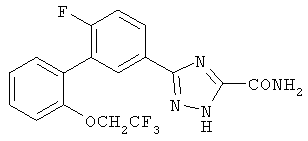
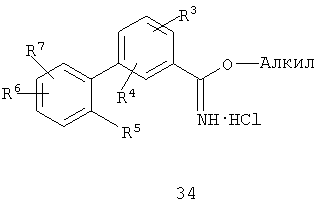
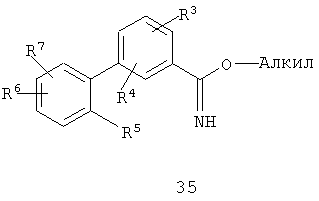
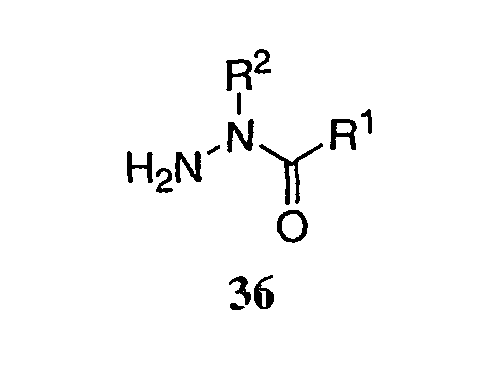
или их фармацевтические соли, где каждый R1-R7 принимает значения, определенные выше. Следующие соединения исключены из настоящего изобретения: 1-метил-3-(2-хлорфенил)-5-[3-(4-трифторметилфенил)фенил]-1,2,4-триазол, 1-метил-3-(2-хлор-6-фторфенил)-5-[3-(4-трифторметилфенил)-фенил]-1,2,4-триазол, 1-метил-3-(2,6-дифторфенил)-5-[3-(4-трифторметилфенил)-фенил]-1,2,4-триазол, 1-метил-3-(2,6-дифторфенил)-5-[3-(4-трифторметоксифенил)-фенил]-1,2,4-триазол, 1-(3-фенил)фенил-1,2,4-триазол. В первом аспекте данное изобретение относится к соединению, представленному химической формулой (I), или к его фармацевтически приемлемой соли, где R5 принимает значения, отличающиеся от Н, и все другие переменные принимают значения, определенные выше. В варианте осуществления указанного первого аспекта данное изобретение относится к соединению, представленному химической формулой (I), или к его фармацевтически приемлемой соли, где R5 представляет собой -ORa, и все другие переменные принимают значения, определенные выше. В другом варианте осуществления указанного первого аспекта данное изобретение относится к соединению, представленному химической формулой (I), или к его фармацевтически приемлемой соли, где R1 представляет собой необязательно замещенный C1-С6-алкил, необязательно замещенный С3-С6-циклоалкил, -C(=O)Ra или CONRaRb, и все другие переменные принимают значения, определенные выше. Во втором аспекте данное изобретение относится к соединению, представленному химической формулой (II), или к его фармацевтически приемлемой соли, где R5 принимает значения, отличающиеся от Н, и все другие переменные принимают значения, определенные выше. В варианте осуществления указанного второго аспекта данное изобретение относится к соединению, представленному химической формулой (II), или к его фармацевтически приемлемой соли, где R5 представляет собой -ORa, и все другие переменные принимают значения, определенные выше. В другом варианте осуществления указанного второго аспекта данное изобретение относится к соединению, представленному химической формулой (II), или к его фармацевтически приемлемой соли, где R1 представляет собой необязательно замещенный С1-С6-алкил, необязательно замещенный С3-С6-циклоалкил, -С(=O)Ra или CONRaRb, и все другие переменные принимают значения, определенные выше. В третьем аспекте данное изобретение относится к соединению, представленному химической формулой (III), или к его фармацевтически приемлемой соли, где R5 принимает значения, отличающиеся от Н, и все другие переменные принимают значения, определенные выше. В варианте осуществления указанного третьего аспекта данное изобретение относится к соединению, представленному химической формулой (III), или к его фармацевтически приемлемой соли, где R5 представляет собой -ORa, и все другие переменные принимают значения, определенные выше. В другом варианте осуществления указанного третьего аспекта данное изобретение относится к соединению, представленному химической формулой (III), или к его фармацевтически приемлемой соли, где R1 представляет собой необязательно замещенный С1-С6-алкил, необязательно замещенный С3-С6-циклоалкил, -С(=O)Ra или CONRaRb, и все другие переменные принимают значения, определенные выше. Данное изобретение также относится к способу получения соединения формулы (I) или его фармацевтически приемлемой соли, включающему взаимодействие соединения формулы 34 или 35:
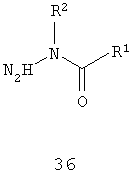
с соединением формулы 36:
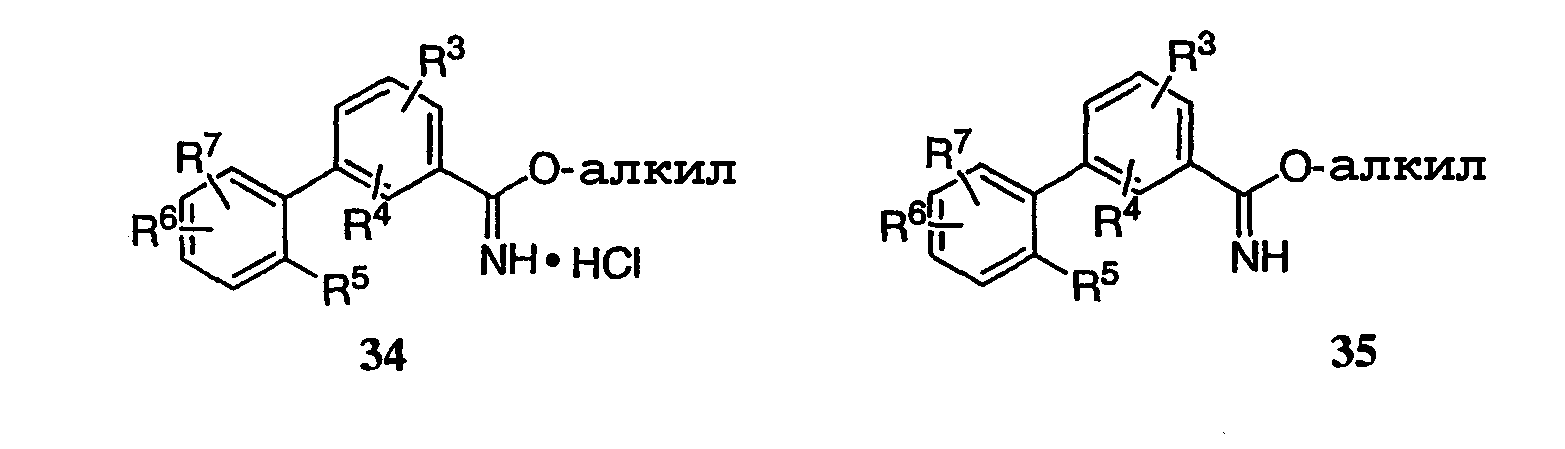
где каждый R1-R7 принимает значения, определенные выше, в присутствии основания для получения соединения формулы (I) или его фармацевтически приемлемой соли. В первом аспекте предлагаемого способа данное изобретение относится к способу получения соединения формулы (I) или его фармацевтически приемлемой соли, где R1 представляет собой Н, -C(=O)Ra, COORa, CONRaRb, C1-C6алкил или С3-С6циклоалкил, где указанный алкил или циклоалкил необязательно замещен одним или несколькими F, OH или NRaRb, R2 представляет собой Н, R5, R6 и R7 каждый независимо представляет собой Н, F, -ORa, C1-C6-алкил или -О-C1-C6-алкил, где указанный алкил необязательно замещен одним или несколькими F, CF3 или -О-(C1-C4)алкил, и все другие переменные принимают значения, определенные выше. Во втором аспекте предлагаемого способа данное изобретение относится к способу получения соединения формулы (I) или его фармацевтически приемлемой соли, где основание представляет собой ацетат металла, карбонат металла или третичный амин. В третьем аспекте предлагаемого способа данное изобретение относится к способу получения соединения формулы (I) или его фармацевтически приемлемой соли, где основание представляет собой ацетат калия. Данное изобретение также относится к способу получения соединения формулы (I) или его фармацевтически приемлемой соли, включающему взаимодействие соединения формулы 34 или 35:
с соединением формулы 36:
где каждый R1-R7 принимает значения, определенные выше, в присутствии основания и необязательно в спиртовом растворителе, и необязательно при нагревании, для получения соединения формулы (I) или его фармацевтически приемлемой соли. Используемый в данном описании термин Термин Термин Термин Термин Термин Термин Термин Термин Соединения, описанные в описании, могут содержать одну или несколько двойных связей и, следовательно, могут образовать цис/транс-изомеры так же, как и другие конформационные изомеры. Данное изобретение включает все такие возможные изомеры, так же как и смеси подобных изомеров, если не оговорено особо. Соединения, описанные в описании, могут содержать один или несколько асимметрических центров и, следовательно, могут образовать диастереоизомеры и оптические изомеры. Данное изобретение включает все возможные диастереоизомеры так же, как и их рацемические смеси, их по существу чистые разделенные энантиомеры, все возможные геометрические изомеры и их фармацевтически приемлемые соли. Вышеприведенные химические формулы показаны без указания стереохимии в некоторых положениях. Данное изобретение включает все стереоизомеры химических формул и их фармацевтически приемлемые соли. Кроме того, включены смеси стереоизомеров, так же как и выделенные специфические стереоизомеры. При проведении синтетических процедур, примененных для приготовления таких соединений, или использовании процедур рацемизации или эпимеризации, известных специалистам в данной области, продукты подобных процедур могут быть смесью стереоизомеров. Термин Если соединения данного изобретения являются основными, то их соответствующие соли удобно могут быть приготовлены из фармацевтически приемлемых нетоксичных кислот, включающих неорганические и органические кислоты. Такие кислоты включают, например, уксусную, бензолсульфоновую, бензойную, камфорсульфоновую, лимонную, этансульфоновую, фумаровую, глюконовую, глутаминовую, бромистоводородную, хлористоводородную, изэтионовую, молочную, малеиновую, яблочную, миндальную, метансульфоновую, слизевую, азотную, памовую, пантотеновую, фосфорную, янтарную, серную, винную, п-толуолсульфоновую кислоту и т.п. Фармацевтические композиции данного изобретения содержат соединение, представленное формулой I, II или III (или его фармацевтически приемлемую соль), в качестве активного ингредиента, фармацевтически приемлемый наполнитель и необязательно один или несколько дополнительных терапевтических средств или адъювантов. Такие дополнительные терапевтические средства могут включать, например, i) опиатные агонисты или антагонисты, ii) антагонисты кальциевых каналов, iii) агонисты 5НТ рецептора или антагонисты, iv) антагонисты натриевых каналов, v) агонисты NMDA рецептора или антагонисты, vi) селективные ингибиторы СОХ-2, vii) антагонисты NK1, viii) нестероидные противовоспалительные средства (НПВС, Данные соединения и композиции пригодны для лечения хронических, висцеральных, воспалительных и невропатических болевых синдромов. Они пригодны для лечения боли, полученной в результате травматического повреждения нерва, компрессии или ущемления нерва, постгерпетической невралгии, тригеминальной невралгии и диабетической невропатии. Данные соединения и композиции пригодны также для лечения хронической поясничной боли, фантомной боли, хронической тазовой боли, невромной боли, комплексного регионального болевого синдрома, хронической артритной боли и родственных невралгий и боли, связанной с раком, химиотерапией, ВИЧ и невропатией, вызванной лечением ВИЧ. Соединения данного изобретения могут быть также использованы как местные анестетики. Соединения данного изобретения пригодны для лечения синдрома раздраженной толстой кишки и родственных расстройств, а также болезни Крона. Данные соединения имеют клинические применения для лечения эпилепсии и частичных и генерализованных тонических припадков. Они также пригодны для нейрозащиты при ишемических состояниях, вызванных ударом или травмой нервов, и для лечения множественного склероза. Соединения данного изобретения пригодны для лечения депрессии, состояния страха, биполярного расстройства и тахиаритмий. Кроме того, подразумевается, что соединения данного изобретения могут быть введены в профилактически эффективных дозированных количествах для предотвращения вышеуказанных состояний и нарушений, а также для предотвращения других состояний и нарушений, связанных с активностью натриевых каналов. Пасты, мази, желеобразные формы, растворы или суспензии, содержащие данные соединения, могут быть применены для местного использования. Растворы и жидкости для полоскания ротовой полости включены в рамках локального применения для целей данного изобретения. Дозированные количества примерно от 0,01 мг/кг до примерно 140 мг/кг массы тела в сутки пригодны для лечения воспалительной и невропатической боли, или альтернативно примерно от 0,5 мг до примерно 7 г на пациента в сутки. Например, воспалительная боль может быть эффективно пролечена введением примерно от 0,01 мг до примерно 75 мг соединения на килограмм массы тела в сутки или альтернативно примерно от 0,5 мг до примерно 3,5 г на пациента в сутки. Невропатическая боль может быть эффективно пролечена введением примерно от 0,01 мг до примерно 125 мг соединения на килограмм массы тела в сутки или альтернативно примерно от 0,5 мг до примерно 5,5 г на пациента в сутки. Количество активного ингредиента, который может быть объединен с материалами-наполнителями для создания однократной лекарственной формы, будет меняться в зависимости от реципиента, подвергаемого лечению, и конкретного способа введения. Например, препарат, предназначенный для перорального введения людям, может просто содержать примерно от 0,5 мг до примерно 5 г активного ингредиента, смешанного с соответствующим и подходящим количеством материала-наполнителя, которое может изменяться примерно от 5 до примерно 95 процентов от общей композиции. Стандартные лекарственные формы большей частью будут содержать примерно от 1 мг до примерно 1000 мг активного ингредиента, обычно 25, 50, 100, 200, 300, 400, 500, 600, 800 или 1000 мг. Подразумевается, однако, что специфический уровень доз для любого конкретного пациента будет зависеть от множества факторов. Такие факторы, связанные с пациентом, включают возраст, массу тела, общее состояние здоровья, пол и диету пациента. Другие факторы включают время и путь введения, скорость выделения, комбинацию лекарственных средств и тяжесть конкретного заболевания, подвергающегося терапии. Практически соединения, представленные формулой I, II и III, или их фармацевтически приемлемые соли могут быть соединены как активные ингредиенты в однородной смеси с фармацевтическим наполнителем по традиционным фармацевтическим методикам смешивания. Наполнитель может быть в различных формах в зависимости от формы препарата, предназначенного для введения, например, перорального или парентерального (включая внутривенное). Таким образом, фармацевтические композиции данного изобретения могут быть представлены в виде дискретных единиц, подходящих для перорального введения, таких как капсулы, крахмальные облатки или таблетки, каждая из которых содержит предварительно определенное количество активного ингредиента. Кроме того, композиции могут быть представлены в виде порошка, гранул, раствора, суспензии в водной жидкости, неводной жидкости, эмульсии масло-в-воде или эмульсии вода-в-масле. В дополнение к вышеприведенному обычному набору лекарственных форм, соединения, представленные формулой I, II и III, или их фармацевтически приемлемые соли могут быть также введены средствами контролируемого высвобождения действующего вещества и/или устройствами для его высвобождения. Композиции могут быть приготовлены любым из способов фармации. По существу, такие способы включают стадию внесения активного ингредиента в ассоциацию с наполнителем, который содержит один или несколько необходимых ингредиентов. По существу, композиции готовят при постоянном и однородном перемешивании активного ингредиента с жидкими наполнителями или тонко размолотыми твердыми наполнителями или с теми и другими вместе. Продукту затем может быть придана удобная желаемая форма. Таким образом, фармацевтические композиции данного изобретения могут включать фармацевтически приемлемый наполнитель и соединение или фармацевтически приемлемую соль формулы I, II или III. Соединения формулы I, II и III или их фармацевтически приемлемые соли также могут быть включены в фармацевтические композиции в комбинации с одним или несколькими терапевтически активными соединениями. Примененный фармацевтический наполнитель может представлять собой, например, твердое вещество, жидкость или газ. Примеры твердых наполнителей включают лактозу, гипс, сахарозу, тальк, желатин, агар, пектин, аравийскую камедь, магния стеарат и стеариновую кислоту. Примеры жидких наполнителей представляют собой сахарный сироп, арахисовое масло, оливковое масло и воду. Примеры газообразных наполнителей включают диоксид углерода и азот. При приготовлении композиций для перорального введения лекарственной формы может быть применена любая удобная фармацевтическая среда. Например, вода, гликоли, масла, спирты, корригенты, консерванты, окрашивающие средства и т.п. могут быть применены для образования жидких препаратов для перорального введения, таких как суспензии, эликсиры и растворы; в то время как наполнители, такие как крахмалы, сахара, микрокристаллическая целлюлоза; разбавители, гранулирующие агенты, замасливатели, связующие вещества и дезинтегрирующие агенты могут быть применены для образования пероральных твердых препаратов, таких как порошки, капсулы и таблетки. Вследствие легкости их введения таблетки и капсулы являются предпочтительными стандартными дозировками для перорального введения, поэтому используют твердые фармацевтические наполнители. Необязательно, но таблетки могут быть с покрытием, выполненным по стандартным водным и неводным методикам. Таблетка, содержащая композицию данного изобретения, может быть приготовлена компрессией или формованием, необязательно с одним или несколькими дополнительными ингредиентами или адъювантами. Прессованные таблетки могут быть приготовлены в подходящей машине прессованием активного ингредиента в свободно-текучей форме, такой как порошок или гранулы, необязательно смешанного со связующим веществом, замасливателем, инертным разбавителем, поверхностно-активным или диспергирующим средством. Формованные таблетки могут быть приготовлены в подходящей машине формованием смеси порошкообразного соединения, увлажненного инертным жидким разбавителем. Каждая таблетка предпочтительно содержит примерно от 0,1 мг до примерно 500 мг активного ингредиента и каждая крахмальная облатка или капсула предпочтительно содержит примерно от 0,1 мг до примерно 500 мг активного ингредиента. Таким образом, таблетка, крахмальная облатка или капсула обычно содержит 0,1, 1, 5, 25, 50, 100, 200, 300, 400 или 500 мг активного ингредиента, принимаемого в виде одной или двух таблеток, крахмальных облаток или капсул один, два или три раза в сутки. Фармацевтические композиции данного изобретения, пригодные для парентерального введения, могут быть приготовлены в виде растворов или суспензий активного ингредиента в воде. Подходящий сурфактант может быть, например, гидроксипропилцеллюлозой. Могут быть также приготовлены дисперсии в глицерине, жидких полиэтиленгликолях и их смесях в маслах. Дополнительно может быть включен консервант для предотвращения вредного роста микроорганизмов. Фармацевтические композиции данного изобретения, пригодные для инъекций, включают стерильные водные растворы или дисперсии. Кроме того, композиции могут быть в форме стерильных порошков, приготовленных для немедленного приема таких стерильных инъецируемых растворов или дисперсий. Во всех случаях конечная инъецируемая форма должна быть стерильной и должна быть эффективно жидкой для легкого введения с помощью шприца. Фармацевтические композиции должны быть устойчивыми в условиях производства и хранения и, следовательно, должны быть законсервированы от заражающего действия микроорганизмов, таких как бактерии и грибки. Наполнитель может представлять собой растворитель или дисперсионную среду, содержащую, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль), растительные масла и подходящие смеси на их основе. Фармацевтические композиции данного изобретения могут иметь форму, подходящую для местного применения, такую как, например, аэрозоль, крем, мазь, лосьон и порошок для распыления. Кроме того, композиции могут иметь форму, пригодную для применения трансдермальными способами. Данные препараты могут быть приготовлены при использовании соединения формулы I, II или III или их фармацевтически приемлемых солей с помощью обычных методик. Например, крем или мазь готовят смешиванием гидрофильного материала и воды вместе с примерно от 5 мас.% до примерно 10 мас.% соединения для получения крема или мази желаемой консистенции. Фармацевтические композиции данного изобретения могут иметь форму, подходящую для ректального введения, где наполнитель представляет собой твердое вещество, например, когда смесь образует стандартную дозу в виде суппозиториев. Подходящие наполнители включают масло какао и другие материалы, обычно применяемые в данной области. Суппозитории могут быть приготовлены сначала смешиванием композиции с размягченным(и) или расплавленным(и) наполнителем(ями), затем охлаждением и формованием. В дополнение к вышеупомянутым ингредиентам-наполнителям фармацевтические препараты, описанные выше, могут включать, если целесообразно, один или несколько дополнительных ингредиентов-наполнителей, такие как разбавители, буферы, корригенты, связующие вещества, поверхностно-активные вещества, загустители, замасливатели и консерванты (включая антиоксиданты). Кроме того, могут быть включены другие адъюванты для придания препарату изотоничности с кровью предполагаемого реципиента. Композиции, содержащие соединение, описанное формулой I, II или III, или их фармацевтически приемлемые соли, также могут быть приготовлены в порошкообразной или жидкой концентрированной форме. Фармацевтические композиции данного изобретения предназначены для блокирования натриевых каналов. Соответственно, данное изобретение относится к лечению заболеваний у млекопитающих, интенсивность которых может снижаться через блокирование нейронных натриевых каналов, включающих, например, острую боль, хроническую боль, висцеральную боль, воспалительную боль и невропатическую боль, введением эффективного количества соединения данного изобретения. Термин Как используется в описании для целей способа по изобретению, термин Как указано в описании для целей способа по изобретению, термин Когда в способе по изобретению используется нагревание, то способ может быть осуществлен в температурном интервале примерно от 40°С до примерно 150°С, включаещем интервалы примерно от 50°С до примерно 140°С, примерно от 50°С до примерно 130°С, примерно от 60°С до примерно 120°С, примерно от 65°С до примерно 100°С или примерно от 65°С до примерно 85°С. Как используется в описании, термин Сокращения, используемые в описании, имеют следующие значения (сокращения, не приведенные здесь, имеют общепринятые значения, если не оговорено особо): Ас (ацетил), AIBN (2,2′-азобис(изобутиронитрил)), BINAP (1,1′-бис-2-нафтол), Bn (бензил), САМР (циклический аденозин-3′,5′-монофосфат), DAST ((диэтиламино)трифторид серы), DEAD (диэтилазодикарбоксилат), DBU (1,8-диазабицикло[5.4.0]ундец-7-ен), DIBAL (диизобутилалюминийгидрид), DMAP (4-(диметиламино)пиридин), DMF (N,N-диметилформамид), Dppf (1,1′-бис(дифенилфосфино)ферроцен), EDCI (1-(3-диметиаминопропил)-3-этилкарбодиимид гидрохлорид), Et3N (триэтиламин), GST (глутатионтрансфераза), HMDS (гексаметилдисилазид), LDA (литийдиизопропиламид), m-CPBA (метахлорпербензойная кислота), ММРР (монопероксифталевая кислота), Ms (метансульфонил; мезил или SO2Me), MsO (метансульфонат или мезилат), NBS (N-бромсукцинимид), NSAID (нестероидные противовоспалительные лекарственные средства), о-Tol (орто-толил), OXONE® (2KHSO5 Следующие анализы in vitro и in vivo были проведены для оценки биологической активности данных соединений. Оценка соединения (анализ in vitro) Идентификация ингибиторов натриевого канала основана на способности натриевых каналов вызывать деполяризацию клетки, когда ионы натрия проникают через каналы, модифицированные агонистом. В отсутствие ингибиторов воздействие на канал, модифицированный агонистом, ионами натрия будет вызывать деполяризацию клетки. Ингибиторы натриевых каналов будут препятствовать деполяризации клетки, вызываемой движением ионов натрия через натриевые каналы, модифицированные агонистом. Изменения в мембранном потенциале могут быть определены с помощью чувствительного к напряжению флуоресцентного резонансного переноса энергии (FRET) красильной парой с применением двух компонентов, где донором служит кумарин (СС2DMPE) и акцептором оксанол (DiSBAC2(3)). Оксанол представляет собой липофильный анион и распределяется вдоль мембраны в соответствии с мембранным потенциалом. В присутствии агониста натриевого канала, но в отсутствие натрия, внутренняя часть клетки является отрицательной по отношению к внешней части, оксанол аккумулируется на наружной части мембраны и возбуждение кумарина будет причиной осуществления FRET. Добавление натрия вызовет деполяризацию мембраны, приводящую к перераспределению оксанола на внутреннюю часть клетки и, как следствие, к уменьшению FRET. Таким образом, отношение изменения (донор/акцептор) увеличивается после мембранной деполяризации. В присутствии ингибитора натриевого канала деполяризация клетки не будет происходить и поэтому распределение оксанола и FRET будет оставаться неизменным. Клетки, стабильно трансфицированные натриевым каналом подтипа PN1 (HEK-PN1), выращивали в 96-луночных планшетах, покрытых полилизином, при плотности приблизительно 140000 клетки/лунка. Среду аспирировали, клетки промывали PBS буфером и инкубировали со 100 мкл, состоящими из 10 мкМ CC2-DMPE в 0,02% плурониевой кислоте. После инкубации при 25°С в течение 45 мин среду удаляли и клетки промывали 2 раза буфером. Клетки инкубировали со 100 мкл DiSBAC2(3) в TMA буфере, содержащем 20 мкМ вератридина, 20 нМ бреветоксина-3 и испытуемый образец. После инкубации при 25°С в течение 45 мин в темноте планшеты помещали в VIPR аппарат и регистрировали флуоресцентную эмиссию обоих компонентов, CC2-DMPE и DiSBAC2(3), в течение 10 с. В этот момент в лунки добавляли 100 мкл солевого (физиологического) буфера для определения степени зависимой от натрия деполяризации клеток и регистрировали флуоресцентную эмиссию обеих красок дополнительно в течение 20 с. Отношение CC2-DMPE/DiSBAC2(3) перед добавлением солевого буфера равно 1. В отсутствие ингибиторов данное отношение после добавления солевого буфера составляет >1,5. Когда натриевый канал полностью ингибирован или известным стандартом, или испытуемым соединением, то данное отношение остается равным 1. Поэтому возможно титровать активность ингибитора натриевого канала мониторингом изменений, зависимых от концентрации, во флуоресцентном отношении. Электрофизиологические анализы (анализы in vitro) Приготовление клеток: использовали собственную клеточную линию НЕК-293, стабильно экспрессирующую натриевые каналы подтипа PN1. Клетки культивировали в среде для роста МЕМ (Gibco) c 0,5 мг/мл G418, 50 единиц/мл Pen/Strep и 1 мл инактивированной нагреванием фетальной бычьей сыворотки при 37°С и в 10% СО2. Для электрофизиологической регистрации клетки помещали на 35 мм чашки, покрытые поли-D-лизином. Общая регистрация клеток: клетки НЕК-293, стабильно экспрессирующие подтип PN1 натриевых каналов, исследовали с помощью полного закрепления клеточного напряжения (Hamill, et al. Pfluegers Archives 391:85-100 (1981)) с применением усилителя ЕРС-9 и импульсного программного средства (Pulse software, HEKA Electronics, Lamprecht, Germany). Эксперименты проводили при комнатной температуре. Электроды были оплавлены до сопротивления 2-4 М Следующие протоколы использовали для оценки устойчивого сродства соединений в покое и инактивированном состоянии канала (Кr и Ki, соответственно): 1. 8 мс (миллисек) опытные импульсы применяли к деполяризующим напряжениям от -60 мВ до +50 мВ от исходного потенциала -90 мВ для построения зависимостей сила тока – напряжение (IV-кривые). Напряжение, близкое к пику IV-кривой (обычно -10 или 0 мВ), применяли в качестве опытного импульсного напряжения на всем протяжении остатка эксперимента. 2. Кривые равновесной инактивации (способности) строили измерением силы тока, активированного за счет 8 мс опытного импульса с последующими 10 сек импульсами к потенциалам в интервале от -120 мВ до -10 мВ. 3. Соединения применяли при исходном потенциале, при котором 20-50% каналов было инактивировано и блокада натриевых каналов контролировалась в течение 8 мс опытных импульсов с интервалом в 2 сек. 4. После того как устанавливалось равновесие для соединений, определяли зависимую от напряжения равновесную инактивацию в присутствии соединения по вышеприведенному протоколу 2). Соединения, которые блокируют состояние покоя канала, снижают силу тока, выявленную в течение опытных импульсов от всех исходных потенциалов, в то время как соединения, которые по существу блокируют инактивированное состояние, перемещают срединную точку кривой равновесной инактивации. Максимум силы тока при отрицательных исходных потенциалах (Imax) и разницу в срединных точках на кривых равновесной инактивации (
Если соединение не воздействовало на состояние покоя, Ki вычисляли по следующему уравнению:
Формалиновый тест на лапе крысы (анализ in vivo) Соединения оценивали по их способности ингибировать поведенческий ответ, вызванный 50 мкл инъекцией формалина (5%). Металлическую полоску фиксировали на левой задней лапе самцов крыс Sprague-Dawley (Charles River, 200-250 г) и каждую крысу содержали с полоской в течение 60 мин внутри пластмассового цилиндра (диаметр 15 см). Крысам давали дозу либо наполнителя, либо испытуемого соединения перед (локальным) или после (системного) введения формалина. Для локального введения соединения приготавливали в этаноле в качестве наполнителя, PEG400 и физиологического раствора (EPEGS) при их соотношении 1:4:5 и вводили путем инъекции подкожно в дорсальную поверхность левой задней лапы за 5 мин перед формалином. Для системного введения соединения приготавливали либо в наполнителе EPEGS, либо в наполнителе Tween 80 (10%)/стерильная вода (90%) и вводили путем инъекции внутривенно (через латеральную хвостовую вену через 15 мин после введения формалина) или перорально (за 60 мин перед введением формалина). Число подергиваний от боли считали непрерывно в течение 60 мин с помощью автоматического анализатора болевой рецепции (UCSD Anesthesiology Research, San Diego, CA). Статистическую значимость определяли путем сравнения общего числа подергиваний в ранней (0-10 мин) и поздней (11-60 мин) фазах с непарным t-тестом. Анализ in vivo c использованием модели CFA на крысе Одностороннее воспаление вызывали инъекцией 0,2 мл полного адъюванта Фрейнда (CFA: Mycobacterium tuberculosis, Sigma; суспендирован в эмульсии масло/физиологический раствор (1:1); 0,5 мг Mycobacterium/мл) в плантарную поверхность левой задней лапы. Данная доза CFA вызывала значительное распухание задней лапы, но животные проявляли нормальное поведение с чисткой тела и приростом в весе на протяжении всего эксперимента. Механическую гипералгезию оценивали через 3 дня после повреждения ткани с помощью теста Randall-Selitto. Повторно делали анализ вариантов (ANOVA), сопровождаемый Dunnet’s Post Hoc тестом. SNL: Механическая аллодиния (анализ in vivo) Осязательную аллодинию оценивали с помощью калиброванных филаментов Фрея, используя эффект подъем-падение перед и после двух недель вслед за нервным повреждением. Животных помещали в пластмассовые клетки с дном из проволочных отверстий и позволяли акклиматизироваться в течение 15 мин перед проведением каждого опыта. Для определения 50% порога ответной реакции применяли филаменты Фрея (интервал интенсивности от 0,4 до 28,8 г) к среднеплантарной поверхности в течение 8 с или до тех пор, пока не происходила реакция абстиненции. После положительного ответа оценивали значительно более слабый раздражитель. Если не возникал ответ на раздражитель, то брали намного более сильный раздражитель. После начального порогового перехода процедуру повторяли для представления четырех раздражителей на одно животное на проведение опыта. Механическую чувствительность оценивали 1 и 2 час после перорального введения испытуемого соединения. Соединения, описанные в данном изобретении, проявляют активность по блокированию натриевых каналов в концентрациях от примерно меньших, чем 0,05 мкМ, до примерно меньших, чем 50 мкМ, в опытах in vitro, приведенных выше. Преимущество состоит в том, что данные соединения проявляют активность по блокированию натриевых каналов в концентрациях, меньших, чем примерно 5 мкМ, в анализах in vitro. Большее преимущество состоит в том, что данные соединения проявляют активность по блокированию натриевых каналов в концентрациях, меньших, чем примерно 1 мкМ, в анализах in vitro. Еще большее преимущество состоит в том, что данные соединения проявляют активность по блокированию натриевых каналов в концентрациях, меньших, чем примерно 0,1 мкМ, в анализах in vitro. Самое большое преимущество состоит в том, что данные соединения проявляют активность по блокированию натриевых каналов в концентрациях, меньших, чем примерно 0,05 мкМ, в анализах in vitro. Данные соединения могут быть приготовлены в соответствии с общими схемами, представленным ниже, а также в соответствии с методиками, приведенными в примерах. Следующие схемы и примеры дополнительно раскрывают, но не ограничивают объем изобретения. Если не оговорено особо, то экспериментальные процедуры выполняли при следующих условиях. Все операции проводили при комнатной температуре или при температуре окружающей среды, т.е. в интервале температур 18-25°С. Выпаривание растворителя проводили с помощью роторного испарителя при пониженном давлении (600-4000 паскалей; 4,5-30 мм Hg) с температурой бани вплоть до 60°С. Ход реакции сопровождали тонкослойной хроматографией (TLC, ТСХ), и времена протекания реакции даны только для иллюстрации. Температуры плавления являются некорректированными, и ‘d’ означает разложение. Данные температуры плавления представляют собой температуры плавления, полученные для материалов, приготовленных, как описано. Полиморфизм в некоторых приготовлениях может быть результатом выделения материалов с различными температурами плавления. Структуру и чистоту всех целевых продуктов подтверждали, по меньшей мере, одним из следующих методов: ТСХ (TLC), масс-спектрометрией, спектрометрией ядерного магнитного резонанса (ЯМР (NMR)) или микроаналитическими данными. Если приведены выходы, то они представляют собой только иллюстрацию. Данные ЯМР, если они приведены, представлены в виде ( Способы синтеза Соединения данного изобретения могут быть приготовлены в соответствии с общими схемами, представленными ниже, а также в соответствии с методиками, приведенными в примерах. Заместители являются теми же самыми, что и в вышеприведенных формулах, за исключением случаев, где они определены иначе или в другом отношении очевидны для обычных специалистов-препараторов. Новые соединения данного изобретения могут быть легко синтезированы с помощью методик, известных специалистам в данной области, таких как, например, описанные в Advanced Organic Chemistry, March, 4th Ed., John Wiley and Sons, New York, NY, 1992; Advanced Organic Chemistry. Carey and Sundberg, Vol. A and B, 3rd Ed., Plenum Press, Inc., New York, NY, 1990; Protective groups in Organic Synthesis, Green and Wuts, 2nd Ed., John Wiley and Sons, New York, NY, 1991; Comprehensive Organic Transformations, Larock, VCH Publishers, Inc., New York, NY, 1988; Handbook of Heterocyclic Chemistry, Katritzky and Pozharskii, 2nd Ed., Pergamon, New York, NY, 2000, и в сносках, цитированных в них. Исходные материалы для данных соединений могут быть приготовлены при применении стандартных синтетических превращений химических предшественников, которые являются легко доступными от коммерческих источников, включающих Aldrich Chemical Co. (Milwaukee, WI); Sigma Chemical Co. (St. Louis, MO); Lancaster Synthesis (Windham, N.H.); Ryan Scientific (Columbia, S. C.); Maybridge (Cornwall, UK); Matrix Scientific (Columbia, S. C.); Arcos, (Pittsburgh, PA) and Trans World Chemicals (Rockville, MD). Методики, описанные в описании для синтеза соединений, могут включать одну или несколько стадий по манипуляциям с защитной группой или по очистке, такие как перекристаллизация, перегонка, колоночная хроматография, флэш-хроматография, тонкослойная хроматография (ТСХ, TLC) и высокоэффективная жидкостная хроматография (ВЭЖХ, HPLC). Продукты могут быть охарактеризованы с применением различных методов, хорошо известных в области химии, включающих протонный и углерод-13 ядерный магнитный резонанс (1Н и 13С ЯМР), инфракрасную и ультрафиолетовую спектроскопию (ИК и УФ), рентгеновскую кристаллографию, элементный анализ и ВЭЖХ и масс-спектрометрию (ЖХ-МС). Способы снятия защитной группы, очистки, идентификации структуры и определения количества хорошо известны каждому специалисту в области химического синтеза. Подразумевается, что функциональные группы, присутствующие в соединениях, представленных на схемах ниже, могут быть далее превращены, когда целесообразно, с помощью стандартных методик превращения функциональных групп, доступных для специалистов в данной области, для получения целевых соединений, описанных в данном изобретении. Другие изменения или модификации, которые будут очевидными для специалистов в данной области, охватываются объемом и прописями данного изобретения. Данное изобретение не следует ограничивать, кроме тех случаев, которые излагаются в следующей формуле изобретения. Схема 1:
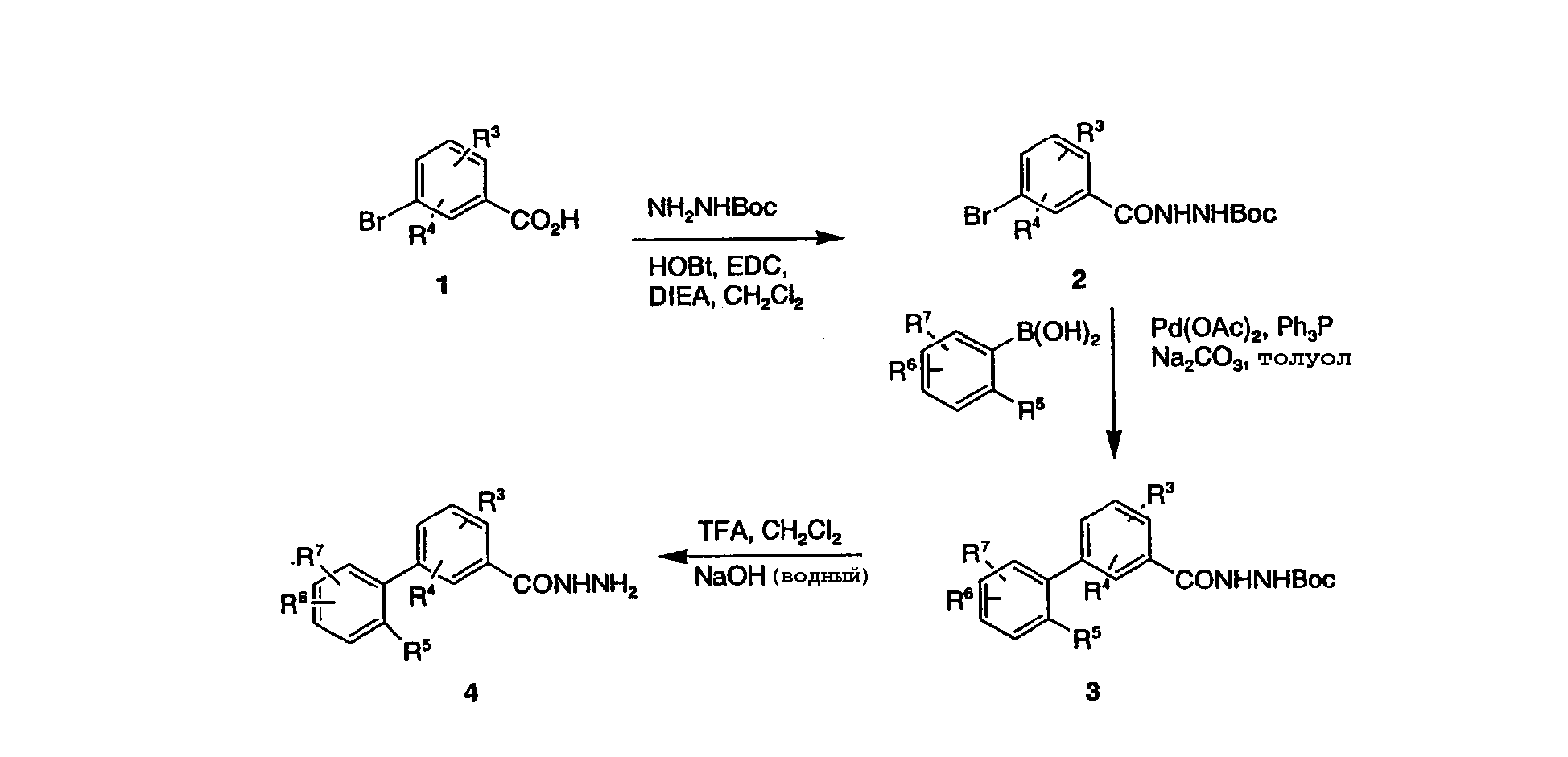
В одном из протоколов 3-бромбензойную кислоту 1 вводят во взаимодействие с трет-бутилкарбазатом активированием с помощью HOBt (гидроксибензотриазол) в присутствии подходящего карбодиимида, такого как EDC [1-(3-диметиламинопропил)-3-этилкарбодиимид], и диизопропилэтиламина (DIEA)) в дихлорметане или ТГФ для получения защищенного гидразида 2. Существует ряд других подходящих способов активирования карбоновых кислот для взаимодействия (см. March J. Схема 2
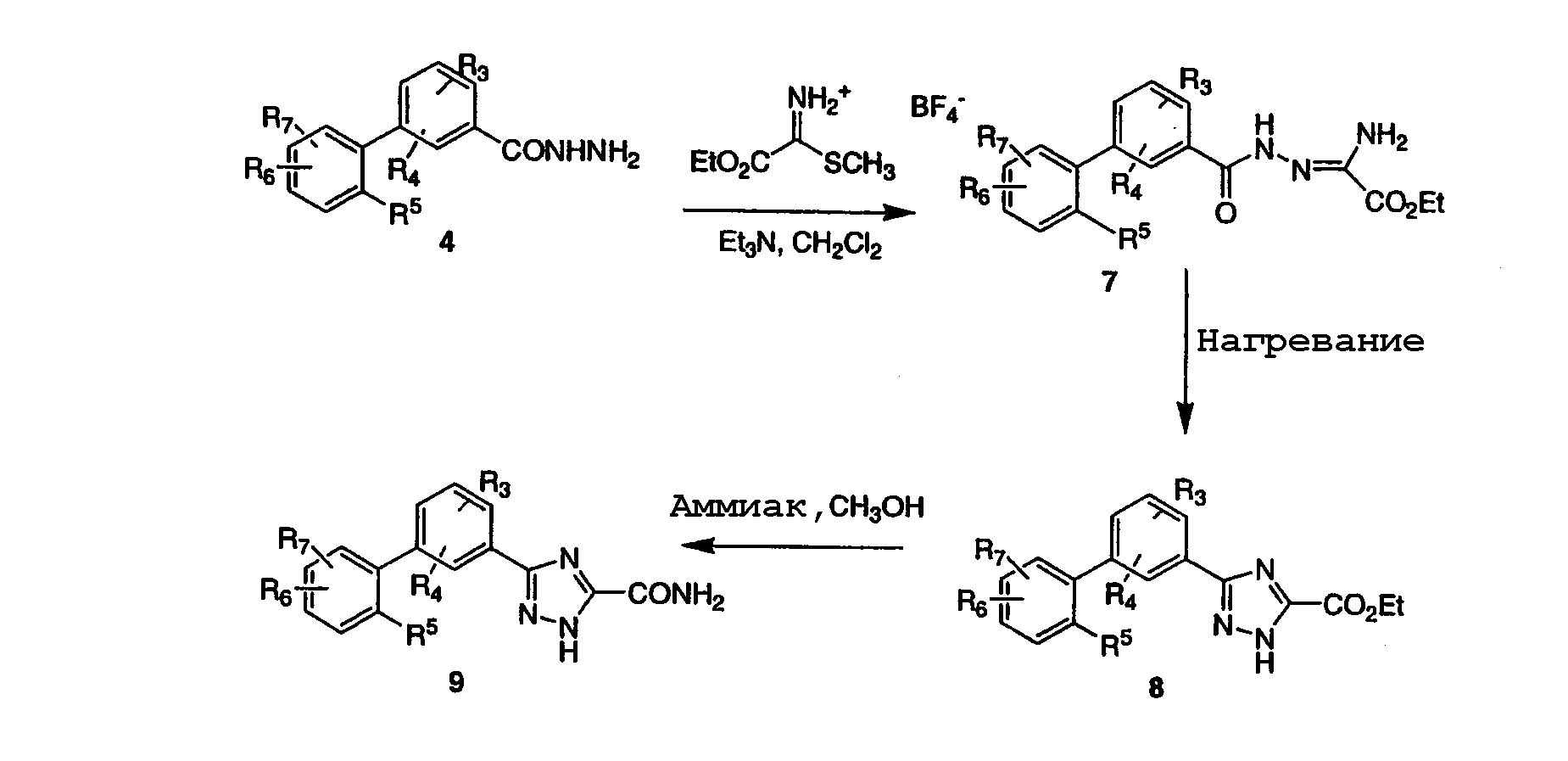
На схеме 2 представлен способ приготовления производных 5-бифенил-3-замещенного-1,2,4-триазола, где замещение может представлять собой остатки эфиров, кислот, амидов и т.д. (Catarzi, et al. J. Med. Chem., 38, 2196-2201, 1995). Взаимодействие гидразида 4 с тетрафторборатом карбэтокси-S-метилтиоформимидия и триэтиламином в дихлорметане дает оксамидразонат 7, который циклизуется в триазоловый эфир 8. Реагент карбэтокси-S-метилтиоформимидий тетрафторбората получали взаимодействием этил-2-тиооксамата с триметилоксоний тетрафторборатом (см. Catarzi, et al., выше) в дихлорметане. Эфир 8 может быть превращен в амид нагреванием его с соответствующим амином, в данном случае с аммиаком, в растворителе, таком как метанол. Эфир 8 может быть подвергнут гидролизу до соответствующей кислоты в стандартных условиях, и образовавшаяся кислота может быть превращена в амид в различных условиях, таких, как описано на схеме 1. Кроме того, эфир 8 может быть восстановлен до первичного спирта, например, борогидридом натрия (NaBH4) для получения соединений формулы (I), где R1 представляет собой гидроксиметил. Альтернативно, эфир 8 может быть превращен во вторичный спирт взаимодействием со смесью борогидрида лития и реактива Гриньяра в апротонном растворителе, таком как ТГФ. Такой первичный или вторичный спирт, полученный из эфира 8, может быть затем превращен в производное с помощью ряда способов, включающих окисление до кетона окисляющим реагентом, таким как хромсодержащий реагент. Такой спирт может быть также превращен во фторпроизводное, например, взаимодействием диэтиламинотрифторида серы (DAST) в дихлорметане при пониженных температурах. Схема 3
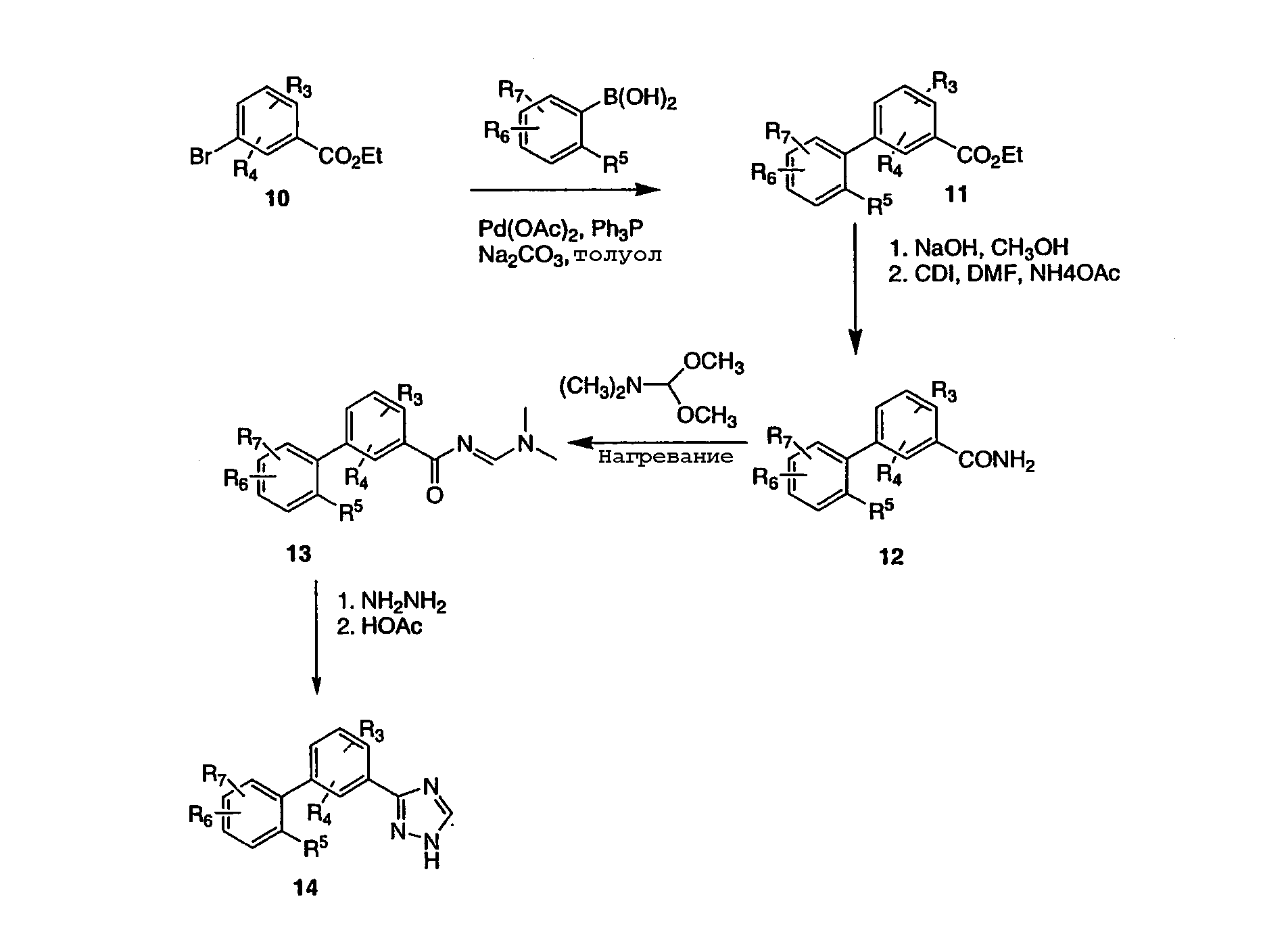
На схеме 3 описан способ приготовления незамещенной 3-триазоловой циклической системы (Lin, et al., J. Org. Chem., 44(23), 4160-4165, 1979). Этил-3-бромбензоат 10 подвергают взаимодействию с арилбороновой кислотой, как описано на схеме 1, для получения бифенильного эфира 11. Эфир 11 обеспечивает предварительное образование бифенильного промежуточного продукта, который затем может быть превращен в соединение 4 или родственные производные, как описано на более ранних схемах 1-2. По схеме 3 эфир 11 превращают в амид 12 в стандартных условиях. В частности, эфир 11 гидролизуют в соответствующую кислоту, которую затем активируют карбонилдиимидазолом (CDI) в ДМФА (DMF) c последующим добавлением аммиака в виде ацетата аммония, получая амид 12. Амид 12 нагревают в диметилацетале диметилформамида, получая промежуточный продукт 13, который при нагревании с гидразином в уксусной кислоте дает триазол 14. Схема 4
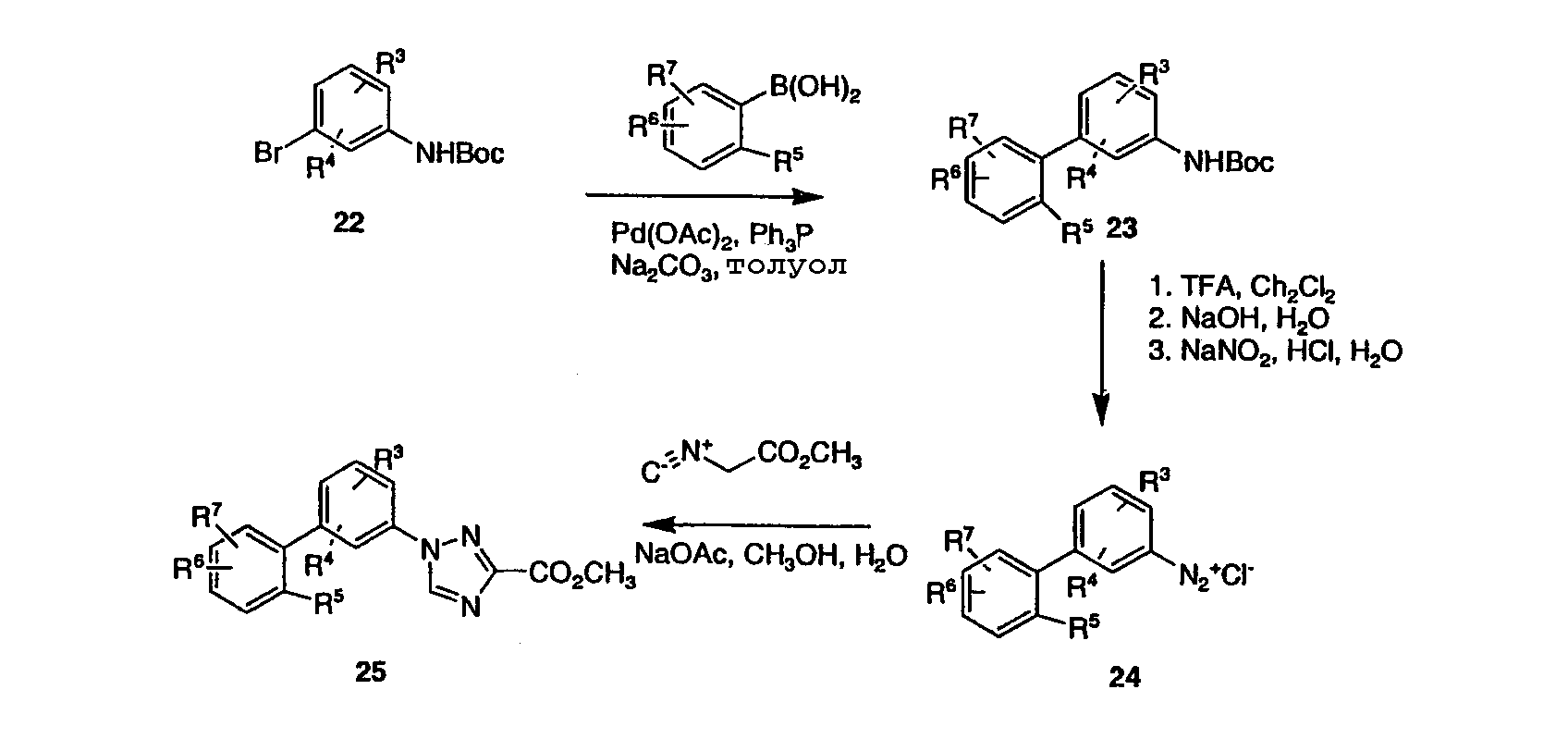
В протоколе для приготовления производных 1-бифенил-3-замещенного-1,2,4-триазола, исходный броманилин 22, где аминогруппа защищена с помощью Вос-группы, и арилбороновую кислоту превращают в ряд несимметричных бифенильных промежуточных продуктов 23, как представлено на схеме 1. Вос защитную группу соединения 23 удаляют, как описано ранее, и превращают в его диазониевую соль 24 стандартным взаимодействием с нитритом натрия и HCl в воде. Добавление соединения 24 к смеси метилизоцианоацетата и ацетата натрия в метаноле и воде приводит к триазоловому эфиру 25. Ключевое промежуточное соединение 25 затем может быть превращено в ряд полезных производных с помощью способов, представленных на схемах 1-3. Схема 5
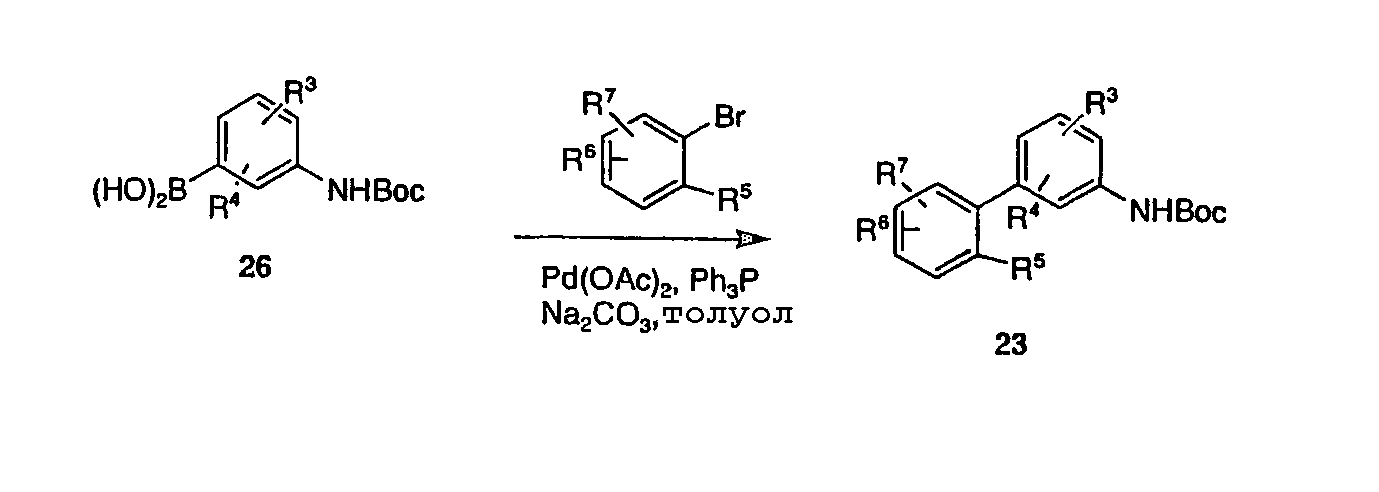
В варианте протоколов, описанных выше на схемах 1, 3 и 4, анилин 26, защищенный Вос-группой и содержащий группу бороновой кислоты или боронатного эфира, и арилбромид, йодид или трифлат превращали в ряд несимметричных бифенильных промежуточных продуктов 23, как представлено на схеме 1. Схема 6
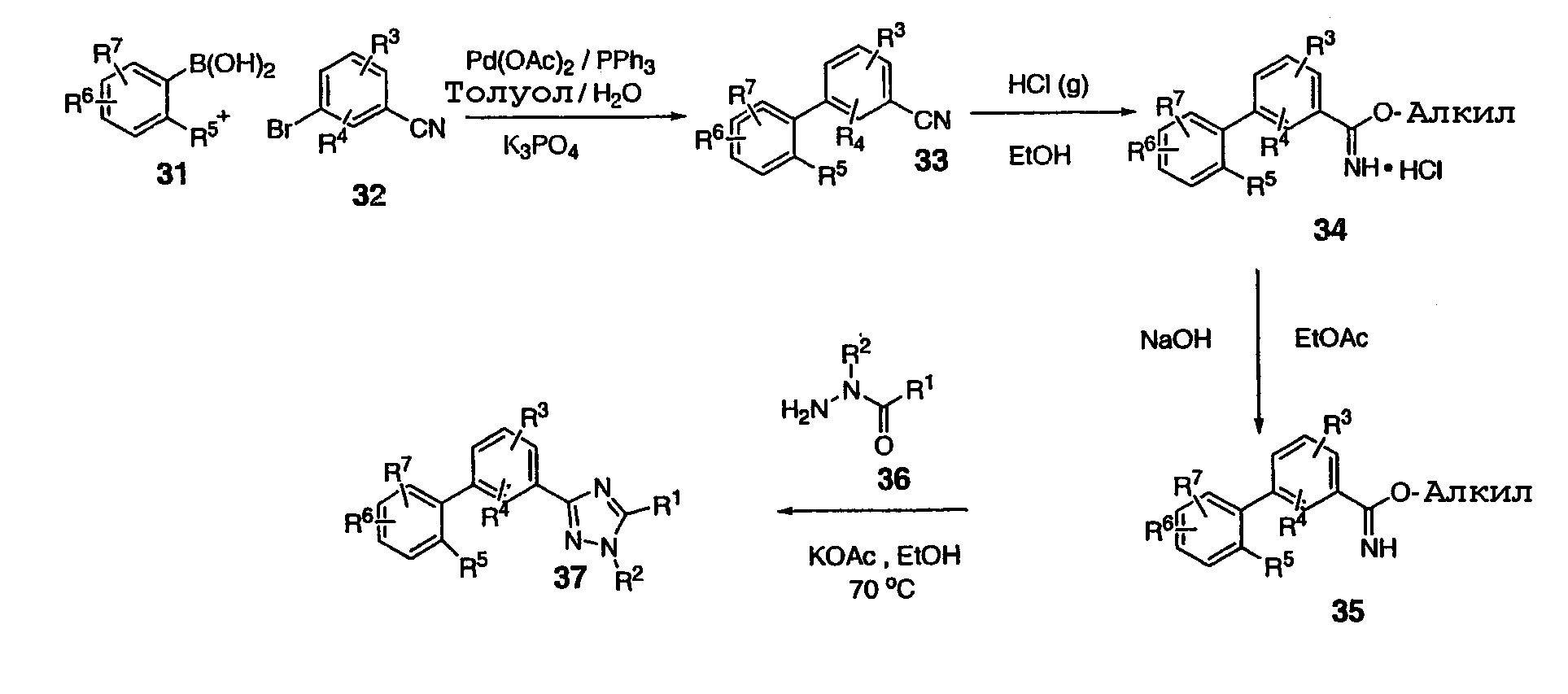
В соответствии со схемой 6, 31 и 32 могут быть превращены в бифенильный промежуточный продукт 33, например, по реакции Suzuki-Miyaura c применением каталитической системы ацетат палладия/трифенилфосфин. Бифенильный промежуточный продукт 33 может быть превращен в HCl соль имидата 34 в стандартных условиях Пиннера, например, с помощью концентрированного раствора хлористоводородной кислоты в этаноле. Имидат 34 может быть превращен в триазол 37 подщелачиванием с применением бифазной смеси, такой как EtOAc и NaOH, через получение этилимидата 35 с последующей обработкой гидразидом оксаминовой кислоты 36, спиртовым растворителем, таким как этанол, и любым соответствующим основанием, одним из многих, включая ацетат металла, таких как ацетат калия, третичный амин или карбонат металла, при нагревании. Схема 7
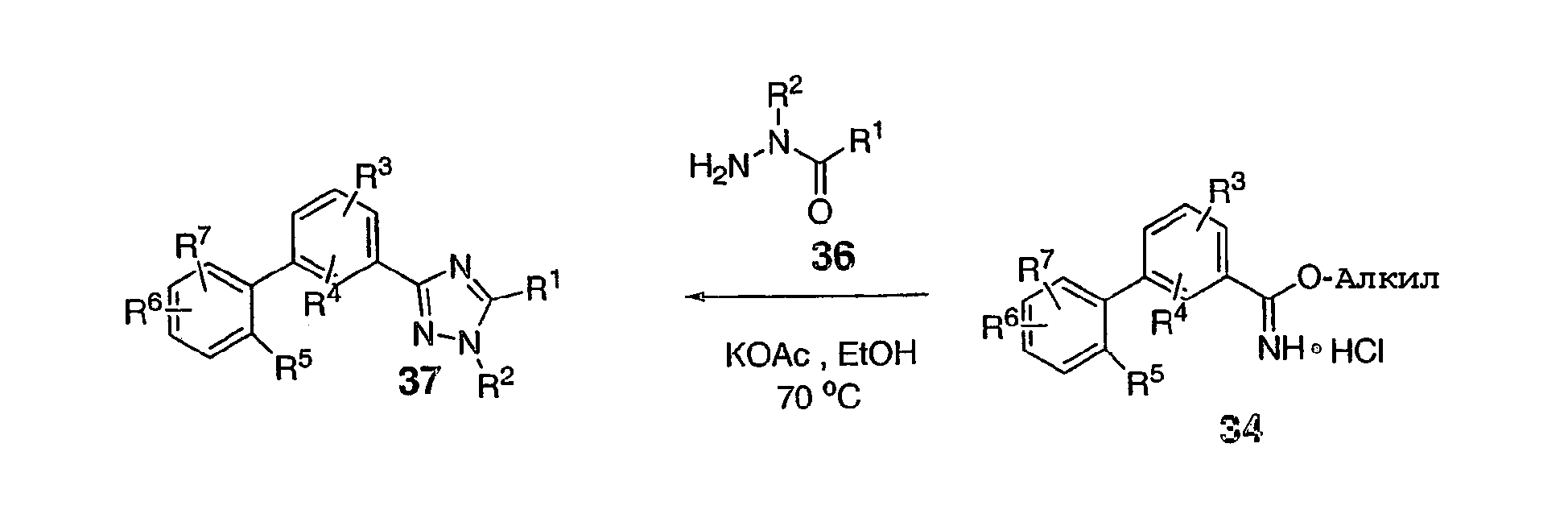
В варианте к схеме 6 промежуточный продукт 34 превращают в триазол 37 прямым добавлением гидразида оксаминовой кислоты 36, спиртового растворителя, такого как этанол, и любого соответствующего основания, одного из многих, включая ацетат металла, таких как ацетат калия, третичный амин или карбонат металла, при нагревании. Схема 8
В соответствии со схемой 8 соединения формул (I) или (II), где R2 представляет собой Н, 38, могут быть превращены депротонизацией с помощью основания алкоксида металла, такого как трет-бутоксид калия или трет-бутоксид натрия, в соответствующей смеси растворитель/сорастворитель в соль 39. ПРИМЕР 1
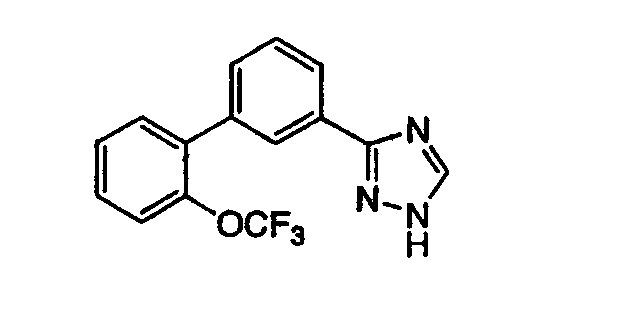
3-[3-(2-Трифторметоксифенил)фенил]-1,2,4-триазол Стадия А: 2-Трифторметоксифенилбороновая кислота К перемешиваемому раствору 2 г (9,5 ммоль) 1-бром-2-трифторметоксибензола в 28 мл тетрагидрофурана (THF, ТГФ) при -78°С осторожно добавляли 5,9 мл 1,7 М раствора трет-бутиллития в гексане (9,5 ммоль). Реакционную смесь перемешивали 45 мин при -78°С. К реакционной смеси при -78°С добавляли 2,58 мл (11,1 ммоль) триизопропилбората и данную смесь медленно нагревали при комнатной температуре (RT) в течение 16 час. Реакционную смесь разбавляли водой и обрабатывали 2N NaOH до щелочной реакции. Реакционную смесь промывали EtOAc. Водную фракцию подкисляли раствором 2N HCl и перемешивали 1 час при RT. Реакционную смесь экстрагировали EtOAc и органические фракции промывали водой, насыщенным раствором NaCl (рассолом), сушили над Na2SO4 и фильтровали. Фильтрат концентрировали, получая указанное в заголовке соединение в виде белого твердого вещества. 1H ЯМР (CDCl3) ( Стадия В: Этил-3-(2-трифторметоксифенил)бензоат К раствору 0,94 г (4,58 ммоль) этил-3-бромбензоата в 14,5 мл толуола при RT добавляли 0,25 г (0,218 ммоль) тетракис(трифенилфосфин)палладия(0), 0,94 г (4,58 ммоль) 2-трифторметоксифенилбороновой кислоты, 2,22 мл (4,45 ммоль) водного 2М раствора карбоната натрия и 7 мл этанола. Реакционную смесь нагревали с обратным холодильником в течение 18 час. Реакционную смесь охлаждали и разбавляли этилацетатом и водой. Органическую фракцию отделяли и промывали насыщенным раствором NaCl (рассолом), сушили над MgSO4, фильтровали и фильтрат концентрировали до получения масла, которое очищали хроматографией (двуокись кремния, 1%, 5%, 30% последовательно этилацетат:гексаны), получая указанное в заголовке соединение. 1H ЯМР (CD3OD) ( Стадия С: 3-(2-Трифторметоксифенил)бензойная кислота Раствор 0,3 г (4,19 ммоль) этил-3-(2-трифторметоксифенил)бензоата и 8,3 мл (8,3 ммоль) 1N раствора NaOH в 12,5 мл метанола перемешивали 18 час при RT. Реакционную смесь концентрировали и доводили до рН 2 с помощью 1N раствора HCl. Смесь экстрагировали этилацетатом (EtOAc) и органическую фракцию сушили над MgSO4, фильтровали и фильтрат концентрировали, получая указанное в заголовке соединение в виде белого твердого вещества, которое использовали без дополнительной очистки. Стадия D: 3-(2-Трифторметоксифенил)бензамид К раствору 0,94 г (3,36 ммоль) 3-(2-трифторметоксифенил)бензойной кислоты в 17 мл ДМФА (DMF) добавляли 0,55 г (3,36 ммоль) карбонилдиимидазола (CDI) и реакционную смесь перемешивали при RT 4 час. К реакционной смеси добавляли 2,6 г (33,6 ммоль) ацетата аммония и смесь перемешивали в течение ночи при RT. Реакционную смесь распределяли между этилацетом и водой и органическую фракцию промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и фильтрат концентрировали. Остаток очищали хроматографией (двуокись кремния, 30%, 50% последовательно EtOAc:гексаны), получая указанное в заголовке соединение. Масс-спектр (ESI) m/e (M+1): 282,2. Стадия Е: 3-[3-(2-Трифторметоксифенил)фенил]-1,2,4-триазол Раствор 0,137 г (0,48 ммоль) 3-(2-трифторметоксифенил)бензамида в 1 мл диметилацеталя N,N-диметилформамида нагревали 2 час при 120°С, одновременно реакционную смесь концентрировали в вакууме. К данному продукту в 2,3 мл уксусной кислоты добавляли 0,028 г (0,55 ммоль) гидразингидрата и реакционную смесь нагревали 2 час при 90°С. Затем реакционную смесь концентрировали и распределяли между EtOAc и насыщенным раствором NaHCO3. Органическую фракцию промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и фильтрат концентрировали. Остаток очищали хроматографией (двуокись кремния, 30:1, 9:1, 3:1 последовательно СН2Cl2:ацетон), получая указанное в заголовке соединение. 1H ЯМР (CD3OD) ( ПРИМЕР 2
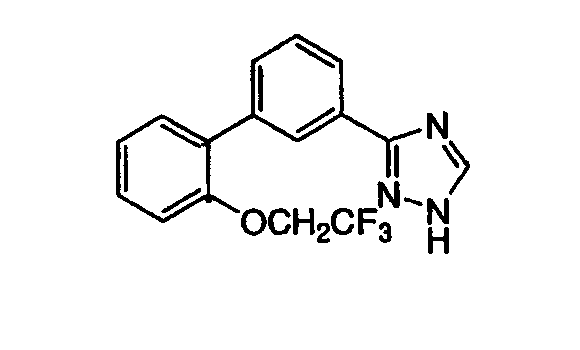
3-[3-(2-(2,2,2-Трифторэтоксифенил)фенил]-1,2,4-триазол Стадия А: 2-(2,2,2-Трифторэтокси)фенилбромид Раствор 0,35 г (2 ммоль) 2-бромфенола, 0,63 г (3 ммоль) 2,2,2-трифторэтилйодида, 0,55 г (4 ммоль) карбоната калия в 2 мл ДМФА подвергали взаимодействию при 150°С в микроволновой системе (Personal Chemistry, Smithcreator) 30 мин. После охлаждения до RT реакционную смесь разбавляли водой и экстрагировали этилацетатом. Органическую фракцию сушили над MgSO4, фильтровали и фильтрат концентрировали. Остаток очищали хроматографией (5%, 10% последовательно EtOAc:гексаны), получая указанное в заголовке соединение. Стадия В: Этил 3-(2-(2,2,2-трифторэтоксифенил)бензоат К раствору 2,5 г (9,8 ммоль) 2-трифторэтоксифенилбромида в 33 мл толуола при RT добавляли 0,57 г (0,49 ммоль) тетракис(трифенилфосфин)палладия(0), 0,2 г (10,3 ммоль) 3-этоксикарбонилфенилбороновой кислоты, 5,9 мл (11,8 ммоль) 2М водного раствора карбоната натрия и 17 мл этанола. Реакционную смесь нагревали с обратным холодильником в течение 18 час. Реакционную смесь охлаждали и разбавляли этилацетатом и водой. Органическую фракцию отделяли и промывали насыщенным раствором NaCl (рассолом), сушили над MgSO4, фильтровали и фильтрат концентрировали до получения масла, которое очищали хроматографией (двуокись кремния, 1%, 5%, 30% последовательно этилацетат:гексаны), получая указанное в заголовке соединение. Масс-спектр (ESI) m/e (M+1): 325,1. Стадия С: 3-[3-(2-(2,2,2-Трифторэтоксифенил)фенил]-1,2,4-триазол Указанное в заголовке соединение может быть приготовлено с помощью процедур, аналогичных процедурам, описанным в примере 1, стадии С-Е. ПРИМЕР 3
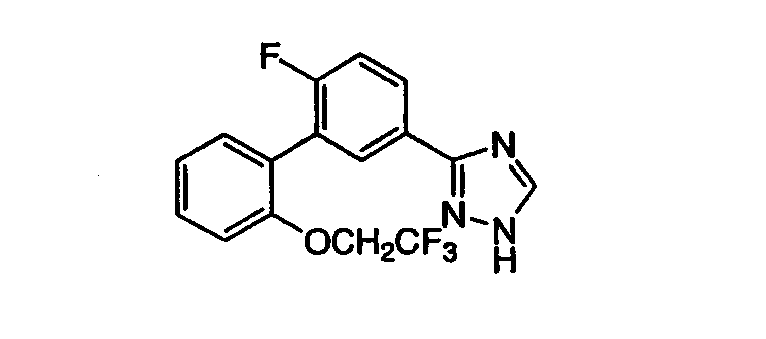
3-[3-((2-(2,2,2-Трифторэтокси)фенил)-(4-фтор)фенил]-1,2,4-триазол Масс-спектр (ESI) m/e (M+1): 338,0. Стадия А: Метил 3-((2-гидрокси)фенил)-4-фторбензоат К раствору 2 г (8,45 ммоль) метил-3-бром-4-фторбензоата в 28 мл толуола при RT добавляли 0,49 г (0,42 ммоль) тетракис(трифенилфосфин)палладия(0), 1,95 г (8,9 ммоль) 2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенола, 5,1 мл (10,15 ммоль) 2М водного раствора карбоната натрия и 14 мл н-пропанола. Реакционную смесь нагревали с обратным холодильником в течение 18 час. Реакционную смесь охлаждали и разбавляли этилацетатом и водой. Органическую фракцию отделяли и промывали насыщенным раствором NaCl (рассолом), сушили над MgSO4, фильтровали и фильтрат концентрировали до масла, которое очищали хроматографией (двуокись кремния, 90:1, 30:1 последовательно СН2Cl2:ацетон), получая указанное в заголовке соединение. Масс-спектр (ESI) m/e (M+1): 247,0. Стадия В: Метил 3-((2-(2,2,2-трифторэтокси)фенил)-4-фторбензоат Смесь 1,7 г (7,1 ммоль) метил 3-((2-гидрокси)фенил)-4-фторбензоата, 2,46 г (10,6 ммоль) 2,2,2-трифторэтил трифторметансульфоната и 3,45 г (10,6 ммоль) карбоната цезия в 35 мл ДМФА перемешивали 18 час при 60°С. Охлажденную реакционную сиесь распределяли между EtOAc и водой. Водный слой экстрагировали EtOAc и объединенные органические фракции промывали водой, насыщенным раствором NaCl, сушили над MgSO4 и фильтровали. Фильтрат концентрировали и очищали хроматографией (двуокись кремния, 5%, 30% последовательно EtOAc:гексаны), получая указанное в заголовке соединение. Масс-спектр (ESI) m/e (M+1): 329,0. Стадия С: 3-[3-((2-(2,2,2-Трифторэтокси)фенил)-(4-фтор)фенил]-1,2,4-триазол Указанное в заголовке соединение может быть приготовлено с помощью процедур, аналогичных процедурам, описанным в примере 1, стадии С-Е. ПРИМЕР 4
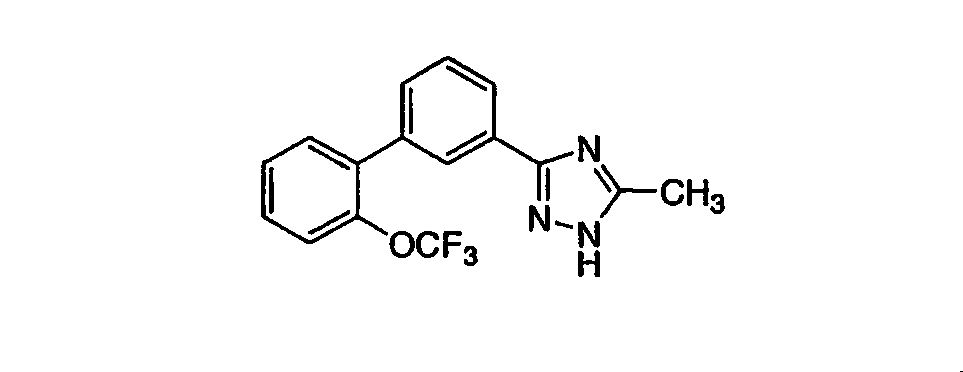
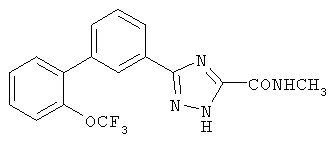
5-Метил-3-[3-((2-трифторметокси)фенил)фенил]-1,2,4-триазол Стадия А: 3-Бромфенилкарбонил-(N-трет-бутоксикарбонил)гидразид Раствор 1 г (4,97 ммоль) 3-бромбензойной кислоты, 0,59 г (4,52 ммоль) трет-бутилкарбазата, 0,95 г (4,97 ммоль) EDC [1-(3-диметиламинопропил)3-этилкарбодиимид), 0,67 г (4,97 ммоль) гидроксибензотриазола (HOBt) и 3,15 мл (18,1 ммоль) диизопропилэтиламина в 23 мл CH2Cl2 перемешивали 18 час при RT. Реакционную смесь разбавляли CH2Cl2 и промывали 1N раствором HCl, насыщенным раствором NaHCO3 и насыщенным раствором NaCl. Раствор сушили над MgSO4, фильтровали и фильтрат концентрировали. Остаток очищали хроматографией (двуокись кремния, 30:1, 9:1, 3:1 последовательно СН2Cl2:ацетон), получая указанное в заголовке соединение. Масс-спектр (ESI) m/e (М): 314,0 (M+2): 316,0. Стадия В: 3-((2-Трифторметокси)фенил)фенилгидразид Раствор 0,22 г (1,07 ммоль) 2-трифторметоксифенилбороновой кислоты и 0,32 г (1,02 ммоль) 3-бромфенилкарбонил-N-трет-бутоксикарбонилгидразида в 5 мл толуола и 2,5 мл н-пропанола перемешивали 30 мин. К реакционной смеси добавляли 0,0007 г (0,003 ммоль) ацетата палладия, 0,0024 г (0,009 ммоль) трифенилфосфина и 0,61 мл (1,2 ммоль) 2М водного раствора карбоната натрия и смесь нагревали с обратным холодильником в течение 18 час. Реакционную смесь охлаждали и разбавляли EtOAc и водой. Органическую фракцию сушили над Mg2SO4, фильтровали и фильтрат концентрировали. Остаток очищали хроматографией (двуокись кремния, 30:1, 9:1 последовательно, СН2Cl2:ацетон), получая защищенный гидразид, который затем растворяли в смеси 2,1 мл TFA и 2,1 мл CH2Cl2. Реакционную смесь перемешивали 2 час, после чего ее концентрировали, растворяли в CH2Cl2 и промывали 1N раствором NaOH. Органическую фракцию сушили над Mg2SO4, фильтровали и фильтрат концентрировали, получая указанное в заголовке соединение в виде белого твердого вещества. Масс-спектр (ESI) m/e (M+1): 297,1. Стадия С: 5-Метил-3-[3-((2-трифторметокси)фенил)фенил]-1,2,4-триазол К раствору 0,093 г (0,98 ммоль) гидрохлорида ацетамидина в 1,1 мл этанола добавляли 0,22 мл (0,98 ммоль) 25% раствора метилата натрия в метаноле и реакционную смесь перемешивали 30 мин, после чего ее фильтровали. К фильтрату добавляли 0,19 г (0,66 ммоль) 3-((2-трифторметокси)фенил)фенилгидразида и реакционную смесь перемешивали в течение ночи. Реакционную смесь концентрировали и очищали хроматографией (двуокись кремния, 3%, 10%, 30% последовательно, метанол:CH2Cl2), получая белое твердое вещество. Белое твердое вещество нагревали (осторожно) при его температуре плавления 30 мин. Реакционную смесь охлаждали до RT, растворяли в CH2Cl2 и концентрировали. Остаток очищали хроматографией (двуокись кремния, 3%, 10%, последовательно, метанол:СН2Cl2), получая указанное в заголовке соединение в виде белого твердого вещества. 1H ЯМР (CD3OD) ( ПРИМЕР 5
3-[3-((2-Трифторметокси)фенил)фенил]-1,2,4-триазол-5-карбоксамид Стадия А: Этил N1-3-(2-трифторметокси)бензоил-N2-оксамидразонат К раствору 0,45 г (1,54 ммоль) 3-(2-трифторметоксифенил)фенилгидразида (пример 4, стадия В) в 20 мл СН2Cl2 добавляли 0,54 г (2,3 ммоль) тетрафторбората карбэтокси-S-метилтиоформимидия и 0,43 мл (3,08 ммоль) триэтиламина и реакционную смесь нагревали с обратным холодильником при перемешивании 4 час. Реакционную смесь охлаждали до RT, промывали водой, сушили над Na2SO4, фильтровали и фильтрат концентрировали до затвердевания. Добавляли 2 мл CH2Cl2 и образовавшийся твердый продукт выделяли фильтрованием. Масс-спектр (ESI) m/e (M+1): 396,1. Стадия В: Этил 3-[3-((2-трифторметокси)фенил)фенил]-1,2,4-триазол-5-карбоксилат Твердый этил N1-3-(2-трифторметокси)бензоил-N2-оксамидразонат со стадии А (0,25 г, 0,616 ммоль) нагревали на масляной бане при температуре выше его температуры плавления 20 мин. После охлаждения до RT остаток растворяли в CH2Cl2 и концентрировали, получая желтое твердое вещество. Его очищали хроматографией (двуокись кремния, 10%, 30%, 50% последовательно, EtOAc:гексаны), получая белое твердое вещество. Масс-спектр (ESI) m/e (M+1): 378,1. Стадия С: 3-[3-((2-трифторметокси)фенил)фенил]-1,2,4-триазол-5-карбоксамид Раствор 0,13 г (0,34 ммоль) этил 3-[3-((2-трифторметокси)фенил)фенил]-1,2,4-триазол-5-карбоксилата (со стадии В) в 2 мл метанола в пробирке насыщали аммиаком. Пробирку герметизировали и реакционную смесь нагревали при 60°С в течение ночи. Реакционную смесь затем концентрировали и остаток очищали хроматографией (двуокись кремния, 3%, 10%, 20% последовательно, метанол:СН2Cl2), получая указанное в заголовке соединение. 1H ЯМР (CD3OD) ( ПРИМЕР 6
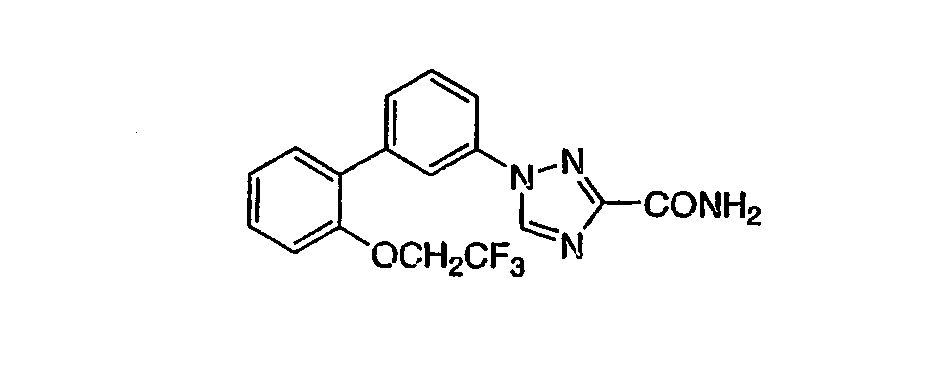
3-[3-((2-(2,2,2-Трифторэтокси)фенил)-4-фторфенил]-1,2,4-триазол-5-карбоксамид Стадия А: 3-((2-(2,2,2-Трифторэтокси)фенил)-4-фторбензойная кислота К раствору 0,75 г (2,29 ммоль) метил 3-((2-(2,2,2-трифторэтокси)фенил)-4-фторбензоата (пример 3, стадия В) в 11,5 мл смеси ТГФ:вода 3:1 добавляли 0,164 г (6,86 ммоль) LiOH и реакционную смесь перемешивали 18 час при RT. Реакционную смесь концентрировали и доводили до рН 2 с помощью 1N раствора HCl. Смесь экстрагировали EtOAc и объединенные органические фракции промывали насыщенным раствором NaCl, сушили над MgSO4, фильтровали и фильтрат концентрировали, получая указанное в заголовке соединение, которое использовали без дополнительной очистки. Стадия В: 3-[3-((2-(2,2,2-Трифторэтокси)фенил)-4-фторфенил]-1,2,4-триазол-5-карбоксамид Указанное в заголовке соединение получали из 3-((2-трифторэтокси)фенил)-4-фторбензойной кислоты в соответствии с методиками, описанными в примере 4, стадии A и B, и примере 4. 1H ЯМР (CD3OD) ( Следующие примеры 7-10 выполняли в соответствии с методиками, описанными в примерах 5 и 6.
ПРИМЕР 11
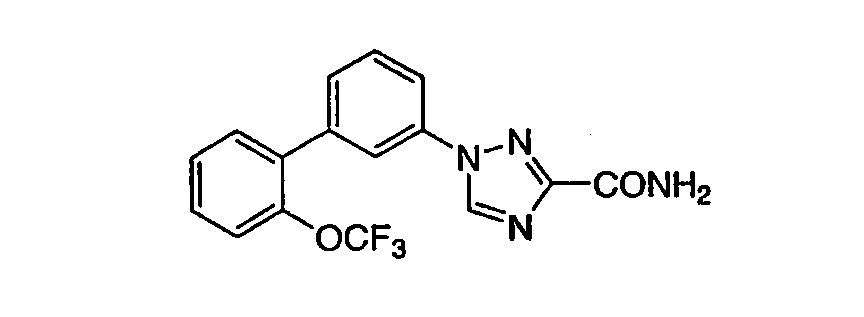
3-[3-((2-Трифторметокси)фенил)-(4-фтор)фенил]-1,2,4-триазол-5-карбоксамид Масс-спектр (ESI) m/e (M+1): 367,0. Стадия А: Приготовление 4-фтор-3-(2-трифторметоксифенил)бензонитрила 3-Бром-4-фторбензонитрил (1,0 эквивалент), 2-(трифторметокси)бензолбороновую кислоту (1,25 эквивалента), ацетат палладия (0,005 эквивалента) и трифенилфосфин (0,01 эквивалента) последовательно загружали в колбу с несколькими отверстиями. После продувания колбы азотом добавляли толуол (5 мл/г 3-бром-4-фторбензонитрила) и образовавшуюся взвесь перемешивали, барботируя азот вглубь реакционной смеси примерно 20 минут. В отдельной колбе готовили водный раствор фосфата калия растворением твердого фосфата калия (2,0 эквивалент) в воде (2 мл/г фосфата калия). Полученный раствор дезоксигенировали барботированием азота вглубь реакционной смеси при перемешивании примерно 30 минут. Водный раствор фосфата калия добавляли к толуольной взвеси и реакционную смесь нагревали при 60-65°С с паровым обогревом. Ход реакции контролировали ВЭЖХ и температуру реакционной смеси поддерживали в интервале 63-69°С. Когда 3-бром-4-фторбензонитрил был израсходован, нагревание прекращали и реакционную смесь охлаждали до RT на ледяной бане. Водный слой сифонировали из сосуда и добавляли в реакционный сосуд Ecosorb C-941 (0,5 г/г 3-бром-4-фторбензонитрила, коммерчески доступный от Graver Technologies, Глазго, Делавэр). Образовавшуюся черную взвесь перемешивали при RT 15 часов. Уголь удаляли фильтрованием взвеси через рыхлый слой солкафлок на фильтровальном тигле. Осадок на фильтре промывали толуолом (4 мл/г 3-бром-4-фторбензонитрила). Объединенные фильтраты дозированно концентрировали (40-50°С) до получения биарилнитрильного продукта в виде густого светло-оранжевого масла (выход 94%). Стадия В: Приготовление:
Абсолютный этанол (1,8 мл/г биарилнитрила) помещали в круглодонную колбу. Перемешиваемый этанол охлаждали на бане лед/ацетон и барботировали вглубь раствора газообразный хлористый водород, при этом внутреннюю температуру поддерживали менее 20°С. Добавление HCl контролировали титрованием протона с помощью Metrohm 808 Titrando (коммерчески доступный от Metrohm Ltd.) для определения концентрации HCl в растворе этанола. Добавление прекращали после того, как концентрация HCl достигала 38% (7,5 эквивалентов). Биарилнитрил со стадии А (1,0 зквивалент) добавляли в виде неразбавленного масла к охлажденному этанольному раствору HCl и реакционному раствору позволяли нагреваться до RT при перемешивании в течение ночи (>15 час). Реакцию считали завершенной, когда анализ ВЭЖХ не показывал присутствия исходного биарилнитрила. Полученную реакционную смесь затем разбавляли толуолом (8 мл/г биарилнитрила). Для того чтобы вызвать кристаллизацию, загрузку растворителей переключали на толуол совместной перегонкой с этанолом (<35°С внутренняя температура), которая азеотропно удаляла этанол. Данную процедуру выполняли до тех пор, пока содержание этанола в маточной жидкости было меньше, чем 1 мол.%, по отношению к толуолу. Данную конечную точку определяли протонным ЯМР маточной жидкости в CDCl3. Твердые вещества отделяли фильтрованием, промывали толуолом (2,3 мл/г биарилнитрила). Белую кристаллическую HCl соль этилимидатного продукта сушили в токе азота. Стадия C: Приготовление
HCl соль имидата со стадии В (1,0 эквивалент) и EtOAc (4 мл/г HCl соль имидата) добавляли в колбу с последующим перемешиванием для образования гетерогенной взвеси. Данную взвесь охлаждали до 10°С с последующим медленным добавлением 5,0N раствора NaOH (3,3 эквивалента). Скорость добавления раствора NaOH периодически регулировали для установления температуры реакционной смеси ниже 15°С. После завершения добавления NaOH бифазной реакционной смеси позволяли нагреваться до температуры окружающей среды, при которой все твердые вещества растворялись (приблизительно 30 мин). Перемешивание прекращали и оставляли два слоя для полного разделения. Нижний водный слой удаляли и отбрасывали. В колбу, содержащую органический слой, добавляли воду (2 мл/г HCl соли имидата) и образовавшуюся бифазную смесь перемешивали 5 минут. Слои оставляли для разделения и нижний водный слой удаляли и отбрасывали. Затем следовало добавление в колбу 10% водного раствора NaCl (2 мл/г HCl соли имидата). Образовавшиеся два слоя перемешивали 5 минут и нижний водный слой удаляли и отбрасывали. Верхний органический слой переносили в круглодонную колбу, которую присоединяли к периодическому концентратору. Затем проводили замену растворителя с EtOAc на EtOH удалением EtOAc совместной перегонкой с EtOH. После завершения замены растворителя (<3% EtOAc по данным 1Н ЯМР, концентрация имидата приблизительно 185 мг/мл) этанольный раствор, содержащий свободный основной имидат, переносили в новую колбу. Гидразид оксаминовой кислоты (1,1 эквивалента) и ацетат калия (5,0 эквивалентов) добавляли к этанольному раствору, содержащему вышеуказанный имидат. Образовавшуюся гетерогенную смесь затем нагревали в интервале 60-70°С на паровой бане с сильным перемешиванием. Реакцию контролировали ВЭЖХ анализом по превращению имидата в целевой триазоловый продукт. После завершения реакции реакционную смесь охлаждали до температуры окружающей среды, затем следовало медленное добавление воды (4 мл/г HCl соли имидата) для того, чтобы вызвать кристаллизацию целевого продукта из реакционной смеси. Целевой продукт затем отделяли фильтрацией под вакуумом в виде не совсем белого твердого вещества (выход 85%). Стадия D: Приготовление
Триазоловый продукт стадии С (1,0 эквивалент) добавляли в реакционный сосуд с последующим добавлением толуола (5,3 мл/г триазола) и ТГФ (1,7 мл/г триазола). Образовавшуюся гетерогенную смесь перемешивали при температуре окружающей среды. К данной взвеси добавляли раствор трет-бутилата калия (1,1 эквивалента). Скорость добавления основания периодически регулировали для установления температуры реакционной смеси ниже 35°С. По мере добавления реакционная смесь становилась менее гетерогенной. После завершения добавления основания полученный гомогенный светло-желтый раствор перемешивали при температуре окружающей среды 30 мин. В данный момент к загрузке медленно добавляли (приблизительно 10 мин) воду (2,5 эквивалента) с незначительным экзотермическим эффектом. Примерно через 15 мин реакционная смесь становилась сильно мутной и температура содержимого стала медленно подниматься. Приблизительно через 20 мин после добавления воды начали образовываться твердые вещества, что указывало на начало кристаллизации. Примерно через 30 мин температура загрузки достигала своего максимума (24,9°С). Гетерогенное содержимое перемешивали дополнительно 30 минут. Содержимое фильтровали в фильтровальный резервуар, используя маточную жидкость для получения любых остаточных твердых веществ в реакционном сосуде. Влажный осадок промывали прохладным толуолом (4 мл/г триазола). Загрузку переносили в вакуумный термостат и сушили при 45°С и 100 мм рт. ст. в течение 24 часов до получения дигидрата калиевой соли продукта в виде белого твердого вещества (выход 95%). ПРИМЕР 12
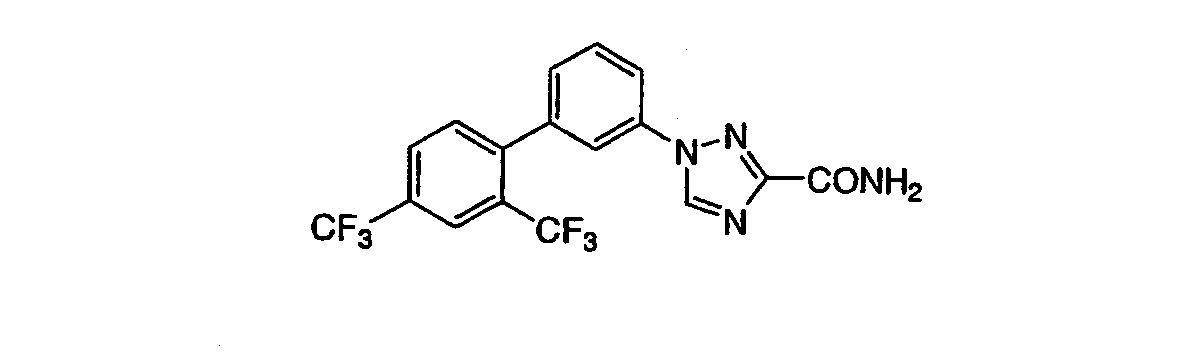
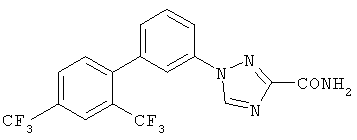
3-[3-((2,4-Бис(трифторметил)фенил)фенил]-1,2,4-триазол-5-карбоксамид Масс-спектр (ESI) m/e (M+1): 401,0. ПРИМЕР 13
N-метил-3-[3-((2-трифторметокси)фенил)фенил]-1,2,4-триазол-5-карбоксамид К раствору 0,083 г (0,24 ммоль) 3-[3-((2-трифторметокси)фенил)фенил]-1,2,4-триазол-5-карбоновой кислоты (полученной из этил 3-[3-((2-трифторметокси)фенил)фенил]-1,2,4-триазол-5-карбоксилата примера 5, следуя методикам примера 6) в 1,2 мл ДМФА при RT добавляли 0,038 г (0,24 ммоль) карбонилдиимидазола (CDI) и реакционную смесь перемешивали 3 час. К реакционной смеси добавляли 1,2 мл (2,4 ммоль) 2М раствора метиламина в ТГФ и реакционную смесь перемешивали в течение ночи при RT. Реакционную смесь концентрировали и распределяли между EtOAc и водой. Водную фракцию экстрагировали EtOAc и объединенные органические фракции промывали водой и насыщенным солевым раствором, сушили над MgSO4, фильтовали и фильтрат концентрировали. Остаток очищали хроматографией (двуокись кремния, 30%, 50% последовательно, EtOAc:гексаны), получая указанное в заголовке соединение в виде пенистого белого твердого вещества. Масс-спектр (ESI) m/e (M+1): 363,0. ПРИМЕР 14
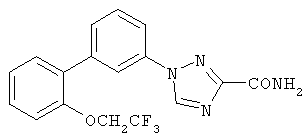
1-[3-((2-(2,2,2-Трифторэтокси)фенил)фенил]-1,2,4-триазол-3-карбоксамид Стадия А: 3-((2-(2,2,2-Трифторэтокси)фенил)анилин К раствору 1,0 г (3,93 ммоль) 2-(2,2,2-трифторэтокси)фенилбромида (пример 2, стадия А) в 39 мл толуола добавляли 0,136 г (0,118 ммоль) тетракис(трифенилфосфин)палладия(0), 0,56 г (4,31 ммоль) 3-аминофенилбороновой кислоты, 47 мл (94,1 ммоль) 2М раствора карбоната натрия и 8 мл этанола и реакционную смесь нагревали 22 час при 90°С. Реакционную смесь охлаждали до RT и распределяли между водой и EtOAc. Водную фракцию экстрагировали EtOAc и объединенные органические фракции промывали водой и насыщенным солевым раствором, сушили над Na2SO4, фильтровали и фильтрат концентрировали. Остаток очищали хроматографией (двуокись кремния, 4:1 гексаны:EtOAc), получая указанное в заголовке соединение. Масс-спектр (ESI) m/e (M+1): 268,1. Стадия В: Метил 1-[3-((2-(2,2,2-трифторэтокси)фенил)фенил]-1,2,4-триазол-3-карбоксилат К раствору 0,923 г (3,45 ммоль) 3-((2-трифторэтокси)фенил)анилина в 6 мл 1N раствора HCl при 0°С добавляли 0,238 г (3,45 ммоль) нитрита натрия и 1 мл воды и реакционную смесь перемешивали 20 мин, получая раствор диазониевой соли. К раствору 0,27 г (2,76 ммоль) метилизоцианоацетата в 15 мл метанола и 2 мл воды при 0°С добавляли 1,8 г (22,08 ммоль) ацетата натрия. К данной реакционной смеси добавляли по каплям раствор диазониевой соли и реакционную смесь перемешивали 1 час при 0°С. Реакционную смесь затем разбавляли метанолом и концентрировали. Остаток разбавляли EtOAc и 0,5N раствором HCl. Водный слой экстрагировали EtOAc и объединенные органические фракции промывали 5% раствором NaHCO3, насыщенным солевым раствором, сушили над Na2SO4, фильтровали и фильтрат концентрировали. Остаток очищали хроматографией (двуокись кремния, 1:1 EtOAc:гексаны), получая указанное в заголовке соединение. Масс-спектр (ESI) m/e M+1: 378,1. Стадия С: 1-[3-((2-(2,2,2-Трифторэтокси)фенил)фенил]-1,2,4-триазол-3-карбоновая кислота Раствор 0,29 г (0,769 ммоль) метил-1-[3-((2-трифторэтокси)фенил)фенил]-1,2,4-триазол-3-карбоксилата и 2,2 мл (2,2 ммоль) 1М раствора NaOH в воде перемешивали 18 час при RT. Реакционную смесь концентрировали. Остаток разбавляли водой и рН доводили до 2-4 1N раствором HCl. Смесь экстрагировали EtOAc и объединенные органические фракции промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и фильтрат концентрировали, получая указанное в заголовке соединение. Масс-спектр (ESI) m/e M+1: 363,9. Стадия D: 1-[3-((2-(2,2,2-Трифторэтокси)фенил)фенил]-1,2,4-триазол-3-карбоксамид К раствору 0,225 г (0,619 ммоль) 1-[3-((2-трифторэтокси)фенил)фенил]-1,2,4-триазол-3-карбоновой килоты в 3,1 мл ДМФА добавляли 0,1 г (0,19 ммоль) CDI и реакционную смесь перемешивали 4 час при RT. К реакционной смеси добавляли 0,477 г (6,19 ммоль) ацетата аммония и реакционную смесь перемешивали 19 час. Реакционную смесь разбавляли водой и EtOAc и водный слой экстрагировали EtOAc. Объединенные органические фракции промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и фильтрат концентрировали. Остаток очищали хроматографией (двуокись кремния, 1:1 EtOAc:гексаны, 1% метанол:СН2Cl2, 10% метанол:CH2Cl2), получая указанное в заголовке соединение. Масс-спектр (ESI) m/e M+1: 363,1. ПРИМЕР 15
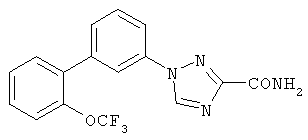
1-[3-((2-Трифторметокси)фенил)фенил]-1,2,4-триазол-3-карбоксамид Стадия А: 1-N-Трет-бутоксикарбониламино-3-бромбензол Раствор 10 г (58,13 ммоль) 3-броманилина и 15,2 г (69,75 ммоль) Вос2О в 300 мл толуола нагревали в течение ночи при 70°С. Реакционную смесь концентрировали и разбавляли EtOAc и 0,5N раствором HCl. Органическую фракцию промывали 0,5N раствором HCl и насыщенным солевым раствором. Фракцию сушили над Na2SO4, фильтровали и фильтрат концентрировали. Остаток очищали хроматографией (гексаны, 9:1 гексаны:EtOAc последовательно), получая указанное в заголовке соединение. Стадия В: 1-N-Трет-бутоксикарбонил-3-((2-трифторметокси)фенил)анилин 1-N-Трет-бутоксикарбониламино-3-бромбензол подвергали взаимодействию с 2-трифторметоксифенилбороновой кислотой в соответствии с методиками примера 14, стадия А. Стадия С: 3-((2-Трифторметокси)фенил)анилин Раствор 0,977 г (2,77 ммоль) 1-N-трет-бутоксикарбонил-3-((2-трифторметокси)фенил)анилина в 7 мл TFA и 7 мл CH2Cl2 перемешивали 1 час при RT. Реакционную смесь концентрировали и остаток разбавляли 1N раствором NaOH и EtOAc. Органическую фракцию промывали 1N раствором NaOH и насыщенным солевым раствором, сушили над Na2SO4, фильтровали и фильтрат концентрировали, получая указанное в заголовке соединение. Масс-спектр (ESI) m/e M+1 254,1. Стадия D: 1-[3-((2-Трифторметокси)фенил)фенил]-1,2,4-триазол-3-карбоксамид Указанное в заголовке соединение получали из 3-((2-трифторметокси)фенил)анилина, следуя методикам примера 14. Масс-спектр (ESI) m/e M+1 349,1. Следующие примеры 16-17 получали, используя методики примеров 14 или 15. ПРИМЕР 16
1-[3-((2,4-Бис-трифторметокси)фенил)фенил]-1,2,4-триазол-3-карбоксамид Масс-спектр (ESI) m/e М+1 401,1. ПРИМЕР 17
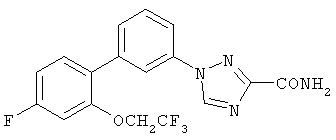
1-[3-((2-(2,2,2-Трифторэтокси-4-фтор)фенил)фенил]-1,2,4-триазол-3-карбоксамид Масс-спектр (ESI) m/e М+1 381,2. Следующий ПРИМЕР 18 получали, используя методики примера 13, исходя из 3-[3-((2-трифторметокси)фенил)фенил]-1,3,4-триазол-5-карбоновой кислоты.
Пример 19
3-[3-((2,6-Бис-трифторметил)фенил)фенил]-1,2,4-триазол-5-карбоксамид Указанное в заголовке соединение получали, используя методики примеров 5 и 6. Пример 20
Масс-спектр (ESI) m/e M+1 381.
Формула изобретения
1. Соединение, представленное формулой (I) или (II) 2. Соединение по п.1, представленное химической формулой (I), или его фармацевтически приемлемая соль, где R1-R7 принимают значения, определенные выше. 3. Соединение по п.1, представленное химической формулой (II), или его фармацевтически приемлемая соль, где R1-R7 принимают значения, определенные выше. 4. Соединение по п.1, выбранное из
или их фармацевтически приемлемые соли. 5. Соединение, представленное формулой (III) 6. Соединение по п.5, выбранное из
или их фармацевтически приемлемые соли. 7. Фармацевтическая композиция, проявляющая активность по блокированию натриевых каналов, содержащая терапевтически эффективное количество соединения по п.1 или его фармацевтически приемлемую соль и фармацевтически приемлемый наполнитель. 8. Применение соединения по п.1 для приготовления лекарственного средства для лечения или профилактики боли или нарушения, вызванного болью. 9. Способ получения соединения формулы (I):
PD4A – Изменение наименования обладателя патента СССР или патента Российской Федерации на изобретение
(73) Новое наименование патентообладателя:
Адрес для переписки:
Извещение опубликовано: 20.04.2010 БИ: 11/2010
|
||||||||||||||||||||||||||||||||||||||||||||||
 C-;
C-;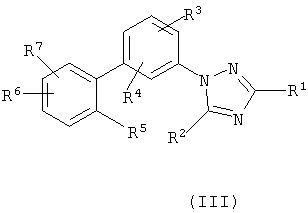
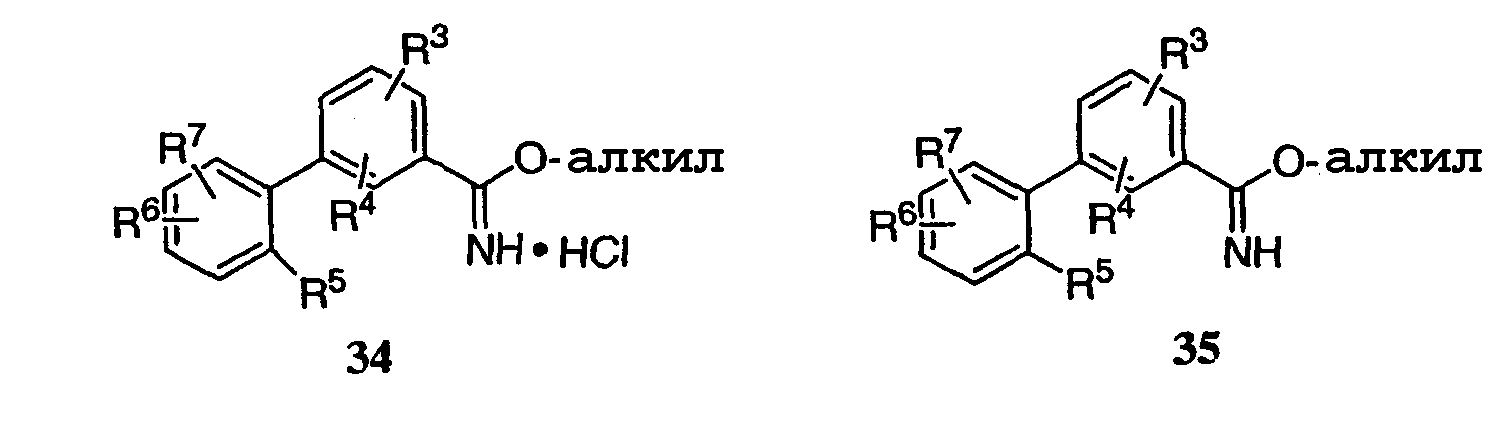
 алкил
алкил так же, как и другие группы, содержащие префикс
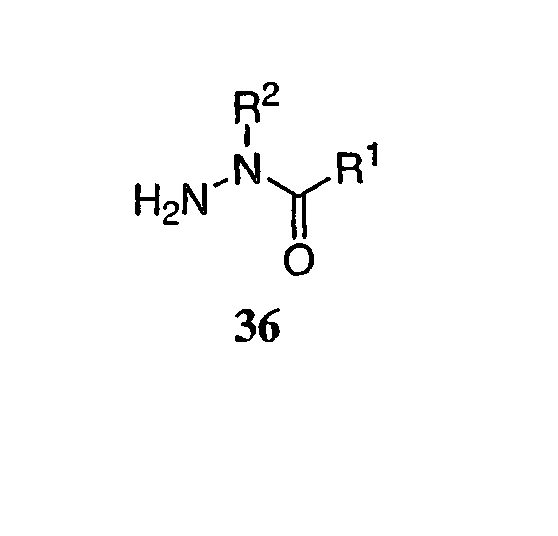
так же, как и другие группы, содержащие префикс  KHSO4
KHSO4 . Ошибки напряжения были минимизированы серией компенсационного сопротивления, и ложный сигнал электрической емкости был исключен с помощью ЕРС-9 компонента в электрической цепи. Данные получали при 50 кГц и фильтровали при 7-10 кГц. Раствор для промывания состоял из 40 мМ NaCl, 120 мМ NMDG Cl, 1 мМ KCl, 2,7 мМ CaCl2, 0,5 мМ MgCl2, 10 мМ NMDG HEPES, pH 7,4, и внутренний (пипеточный) раствор содержал 110 мМ Cs-метансульфоната, 5 мМ NaCl, 20 мМ CsCl, 10 мМ CsF, 10 мМ ВАРТА (тетра Cs соль), 10 мМ Cs HEPES, pH 7,4.
. Ошибки напряжения были минимизированы серией компенсационного сопротивления, и ложный сигнал электрической емкости был исключен с помощью ЕРС-9 компонента в электрической цепи. Данные получали при 50 кГц и фильтровали при 7-10 кГц. Раствор для промывания состоял из 40 мМ NaCl, 120 мМ NMDG Cl, 1 мМ KCl, 2,7 мМ CaCl2, 0,5 мМ MgCl2, 10 мМ NMDG HEPES, pH 7,4, и внутренний (пипеточный) раствор содержал 110 мМ Cs-метансульфоната, 5 мМ NaCl, 20 мМ CsCl, 10 мМ CsF, 10 мМ ВАРТА (тетра Cs соль), 10 мМ Cs HEPES, pH 7,4. 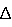 V) в контроле и в присутствии соединения использовали для расчета Kr и Ki по следующим уравнениям:
V) в контроле и в присутствии соединения использовали для расчета Kr и Ki по следующим уравнениям:
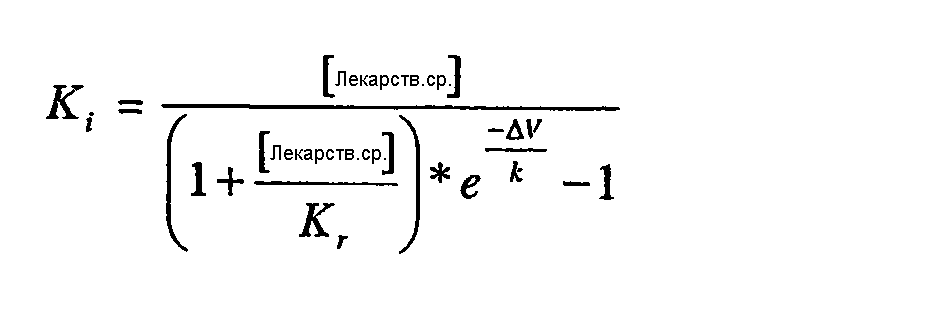
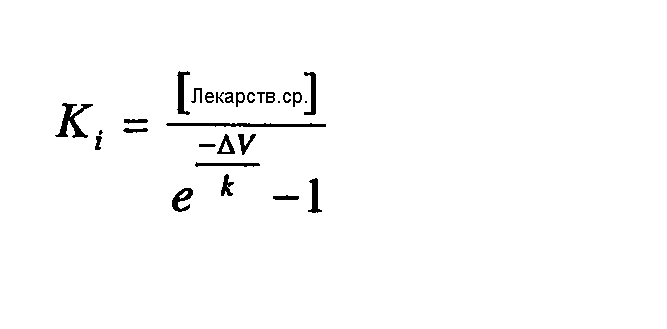
 ) величин для большинства диагностируемых протонов с размерностью в частях на миллион (ppm) относительно тетраметилсилана (TMS), как внутреннего стандарта, определенных при 300 МГц, 400 МГц или 500 МГц с применением указанного растворителя. Обычные сокращения, примененные для конфигурации сигналов, представляют собой: s. синглет; d. дуплет; t. триплет; m. мультиплет; br. уширение и т.д. Дополнительно,
) величин для большинства диагностируемых протонов с размерностью в частях на миллион (ppm) относительно тетраметилсилана (TMS), как внутреннего стандарта, определенных при 300 МГц, 400 МГц или 500 МГц с применением указанного растворителя. Обычные сокращения, примененные для конфигурации сигналов, представляют собой: s. синглет; d. дуплет; t. триплет; m. мультиплет; br. уширение и т.д. Дополнительно,