Патент на изобретение №2352390
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) МАССЫ ОКСИДОВ МЕТАЛЛОВ
(57) Реферат:
Настоящее изобретение относится к массе оксидов металлов, предназначенной как катализатор для гетерогенно-катализируемого частичного окисления и/или аммокисления, по меньшей мере, одного насыщенного и/или ненасыщенного углеводорода, к способу ее получения, способу окисления и способу аммокисления, по меньшей мере, одного насыщенного или ненасыщенного углеводорода. Описана масса оксидов металлов, предназначенная как катализатор для гетерогенно-катализируемого частичного окисления и/или аммокисления, по меньшей мере, одного насыщенного и/или ненасыщенного углеводорода, общей стехиометрии I
где М1 = означает Те; М2 = означает Nb; M3 = означает, по меньшей мере, один из элементов группы, включающей Pb, Ni, Co, Bi и Pd; а=0,05 до 0,6, b=0,01 до 0,5, с=0,01 до 0,5, d=0,0005 до 0,5 и n равно числу, которое определяется валентностью и количеством отличных от кислорода элементов в (I), рентгеновская дифрактограмма которых имеет дифракционные рефлексы h, i и k, пики которых лежат под углами дифракции (20 – дифракционный рефлекс h в пределах рентгеновской дифрактограммы является самым интенсивным и имеет полуширину пика максимально 0,5°, – интенсивность Рi дифракционного рефлекса i и интенсивность Рk дифракционного рефлекса k выполняют отношение 0,65 R=Pi/(Pi+Pk) соотношением интенсивностей, и – полуширина пика дифракционного рефлекса i и дифракционного рефлекса k каждая составляет
Настоящее изобретение относится к массам оксидов металлов общей стехиометрии I
где М1 = означает Те М2 = означает Nb, М3 = означает, по меньшей мере, один из элементов группы, включающей Pb, Ni, Co, Bi, Pd, a=0,05 до 0,6, b=0,01 до 0,5, с=0,01 до 0,5, d=0,0005 до 0,5 и n = равно числу, которое определяется валентностью и количеством отличных от кислорода элементов в (I), рентгеновская дифрактограмма которых имеет дифракционные рефлексы h, i и k, пики которых лежат под углами дифракции (2 – дифракционный рефлекс h в пределах рентгеновской дифрактограммы является самым интенсивным и имеет полуширину пика максимально 0,5°, – интенсивность Рi дифракционного рефлекса i и интенсивность Рk дифракционного рефлекса k выполняют соотношение 0,65 R=Pi/(Pi+Pk) соотношением интенсивностей, и – полуширина пика дифракционного рефлекса i и дифракционного рефлекса k каждая составляет по меньшей мере, одна масса оксидов металлов (I) представляет собой такую, рентгеновская дифрактограмма которой не имеет дифракционного рефлекса с положением пика 2 Далее настоящее изобретение относится к получению масс оксидов металлов (I) и к их применению для гетерогенно-катализируемого частичного окисления и/или аммокисления насыщенных и/или ненасыщенных углеводородов. Массы оксидов металлов общей стехиометрии (I) и со стехиометрическим коэффициентом d=0, получаемые таким путем, что хорошо перемешанную (однородную) сухую смесь их элементарных компонентов кальцинируют в содержащей кислород атмосфере, известны, например, из ЕР-А318295. Они пригодны, например, для гетерогенно-катализируемого аммокисления (окисления в присутствии аммиака) пропана или изобутана для получения акрилнитрила, соответственно метакрилнитрила и отличаются тем, что они имеют высокое содержание аморфной структуры. Из ЕР-А 512846 известно, что характеристика масс оксидов металлов из ЕР-А 318295 в отношении их применения в качестве катализаторов для частичного аммокисления насыщенных углеводородов может быть улучшена добавкой промоторных элементов М3. Из документов ЕР-А 529853, ЕР-А 603836, ЕР-А 608838, ЕР-А767164, ЕР-А 895809 и ЕР-А 962253 известны массы оксидов металлов общей стехиометрии (I) и со стехиометрическим коэффициентом d=0, которые получают таким образом, что хорошо перемешанную (однородную) сухую смесь их элементарных компонентов кальцинируют в основном в свободной от кислорода атмосфере. Они, по сравнению с массами оксидов металлов из ЕР-А 318295 и ЕР-А 512846, еще лучше пригодны в качестве катализаторов для гетерогенно-катализируемого частичного аммокисления и/или окисления насыщенных углеводородов. Последнее действительно особенно тогда, когда тщательно перемешанная сухая смесь получена в качестве форконтакта (специального предварительного катализатора) посредством распылительной сушки. В приведенных документах это обосновывается тем, что эти массы оксидов металлов имеют специфическую кристаллическую структуру, которая отличается тем, что ее рентгеновская дифрактограмма в положении пиков 2 Из документов DE-A 19835247, ЕР-А 1090684 и WO 0206199 известно, что вышеприведенная кристаллическая структура образует кристаллическую фазу, в которой могут иметься массы оксидов металлов. Эта кристаллическая фаза в вышеприведенных документах обозначается как k-фаза. Вторую специфичную кристаллическую структуру, которую могут иметь массы оксидов металлов, обозначают, как правило, i-фазой. Ее рентгеновская дифрактограмма согласно вышеприведенным документами отличается, среди прочего, тем, что она в положении пиков 2 Например, согласно документам ЕР-А 529853, ЕР-А 608838, также ЕР-А 603836 k-фаза отвечает за каталитическую активность приведенных в ней масс оксидов металлов. При вышеописанных методах обычно не получают ни чистой k-фазы, ни чистой i-фазы, а получают смешанные кристаллические структуры, которые представляют собой сросшуюся смесь из k-фазы и i-фазы. В документах ЕР-А 1192987, ЕР-А 1192986, ЕР-А 1192983 и ЕР-А 1192982 описывается получение таких масс оксидов металлов со смешанной кристаллической структурой и показывается то, что их характеристика в отношении их применения в качестве катализаторов для частичного аммокисления и/или окисления насыщенных углеводородов может быть улучшена добавкой промоторных элементов М3, причем k-фазе приписывается решающая роль. В отличие от этого в JP-A 11-169716 решающее значение для каталитической активности таких масс оксидов металлов со смешанной кристаллической структурой при частичном аммокислении насыщенных углеводородов приписывается как k-фазе, так и i-фазе. В соответствие с этим k-фаза ответственна за удовлетворительную селективность образования нитрила и i-фаза за достаточную конверсию насыщенных углеводородов. В публикации Из документов JP-A 7-232071 и WO 0206199 в противоположность этому известно, что также имеющие исключительно структуру i-фазы массы оксидов металлов пригодны в качестве катализаторов для гетерогенно-катализируемого частичного аммокисления и/или окисления насыщенных углеводородов. Уже проводились исследования, которыми было показано, что имеющие исключительно структуру k-фазы массы оксидов металлов являются каталитически инактивными, и которые поддерживают утверждения в JP-A 11-169716, согласно которым i-фаза ответственна за активность и k-фаза только за оптимизацию селективности. Из международных заявок WO 00/29106, WO 00/29105, WO 00/38833 и WO 00/69802 известны содержащие промоторы массы оксидов металлов, имеющие в основном аморфную структуру, которая в рентгеновской дифрактограмме изображена в форме очень широких дифракционных рефлексов и которые также рекомендуются в качестве катализаторов для частичного окисления. Из DE-A 10118814, соответственно РСТ/ЕР/02/04073 известно, что чисто i-фазные массы оксидов металлов представляют собой также пригодные катализаторы для частичного окисления ненасыщенных углеводородов. Из JP-А 8-57319 известно, что содержащие молибден Мо и/или ванадий V массы оксидов металлов могут быть активированы обработкой кислотой. Недостатком приведенных известных решений является, однако, то, что они, с одной стороны, оставляют открытым вопрос встроены ли промоторы как в i-фазу, так и в k-фазу и влияют ли они на каталитическую активность обеих фаз, и, с другой стороны, их массы оксидов металлов не в полном объеме удовлетворяют требованиям в отношении селективности образования целевого продукта при применении в качестве катализатора для гетерогенно-катализируемого частичного окисления и/или аммокисления насыщенных и/или ненасыщенных углеводородов. Задачей настоящего изобретения поэтому является предоставление ответа на открытый вопрос и разработка усовершенствованных масс оксидов металлов. В соответствие с этим были разработаны приведенные в начале описания массы оксидов металлов (I) (в настоящей заявке все относящиеся к рентгеновской дифрактограмме данные касаются полученной при применении Cu-К Предпочтительно согласно изобретению, когда 0,67 Наряду с дифракционными рефлексами h, i и k рентгеновская дифрактограмма масс оксидов металлов (I) согласно изобретению, как правило, содержит еще другие дифракционные рефлексы, пики которых лежат под углом дифракции (2 9,0±0,4° (I), 6,7±0,4° (о) и 7,9±0,4° (р). Далее предпочтительным является такое выполнение, при котором рентгеновская дифрактограмма содержит дополнительно дифракционный рефлекс, пик которого находится под углом дифракции (2 Часто рентгеновская дифрактограмма масс оксидов металлов (I) содержит также еще дифракционные рефлексы 29,2±0,4° (m) и 35,4±0,4° (n) (положения пиков кривых). Если дифракционному рефлексу h придается интенсивность 100, то согласно изобретению имеет преимущество, если дифракционные рефлексы i, l, m, n, о, р, q на одной шкале интенсивности имеют следующие значения: i: 5 до 95, часто от 5 до 80, частично от 10 до 60; l: 1 до 30; m: 1 до 40; n: 1 до 40; о: 1 до 30; р: 1 до 30 и q: 5 до 60. Если рентгеновская дифрактограмма масс оксидов металлов (I) согласно изобретению имеет вышеприведенный дополнительный дифракционный рефлекс, то его полуширина пика, как правило, составляет Удельная поверхность масс оксидов металлов (I) согласно изобретению составляет многократно от 1 до 40 м2/г, часто от 11, соответственно от 12 до 40 м2/г и часто от 15, соответственно от 20 до 40, соответственно до 30 м2/г (определено по методу БЭТ, азот). Согласно изобретению стехиометрический коэффициент а масс оксидов металлов (I) составляет независимо от предпочтительных интервалов для других стехиометрических коэффициентов масс оксидов металлов (I) от 0,05 до 0,6, особенно предпочтительно, от 0,1 до 0,6 или 0,5. Независимо от предпочтительных интервалов для других стехиометрических коэффициентов масс оксидов металлов (I), стехиометрический коэффициент b составляет предпочтительно от 0,01 до 1, и особенно предпочтительно от 0,01, соответственно от 0,1 до 0,5 или 0,4. Стехиометрический коэффициент с масс оксидов металлов (I), независимо от предпочтительных интервалов для других стехиометрических коэффициентов масс оксидов металлов (I), составляет от 0,01 до 1 и особенно предпочтительно от 0,01, соответственно от 0,1 до 0,5 или 0,4. Очень предпочтительным интервалом для стехиометрического коэффициента с, который независимо от предпочтительных интервалов для других стехиометрических коэффициентов масс оксидов металлов (I), может комбинироваться со всеми другими предпочтительными интервалами в настоящей заявке, является интервал от 0,05 до 0,2. Согласно изобретению стехиометрический коэффициент d масс оксидов металлов (I) согласно изобретению, независимо от предпочтительных интервалов для других стехиометрических коэффициентов масс оксидов металлов (I), составляет от 0,00005, соответственно от 0,0005 до 0,5, особенно предпочтительно от 0,001 до 0,5, часто от 0,002 до 0,3 и зачастую от 0,005, соответственно от 0,01 до 0,1. Особенно предпочтительны массы оксидов металлов (I) согласно изобретению, стехиометрические коэффициенты которых а, b, с и d имеют одновременно следующие значения: а = от 0,05 до 0,6; b = от 0,01 до 1 (соответственно от 0,01 до 0,5); с = от 0,01 до 1 (соответственно от 0,01 до 0,5); и d = от 0,0005 до 0,5 (соответственно от 0,001 до 0,3). В особенной степени предпочтительны массы оксидов металлов (I) согласно изобретению, стехиометрические коэффициенты которых а, b, с и d имеют одновременно следующие значения: а = от 0,1 до 0,6; b = от 0,1 до 0,5; с = от 0,1 до 0,5; и d = от 0,001 до 0,5, соответственно от 0,002 до 0,3, соответственно от 0,005 до 0,1. М1 означает предпочтительно Те. Вышеприведенное действительно прежде всего тогда, когда М2, по меньшей мере, на 50 мол.% своего общего количества является Nd и особенно предпочтительно тогда, когда М2, по меньшей мере, на 75 мол.% своего общего количества соответственно на 100 мол.% своего общего количества является Nb. Это действительно, независимо от значения М2, прежде всего также и тогда, когда М3 является, по меньшей мере, одним элементом из группы, включающей Ni, Co, Bi, Pd, Ag, Au, Pb и Ga или, по меньшей мере, одним элементом из группы, включающей Ni, Со, Pd и Bi. Все вышеизложенное действительно, прежде всего, также и тогда, когда М2, по меньшей мере, на 50 мол.% своего общего количества или, по меньшей мере, на 75 мол.%, или на 100 мол.% является ниобием и М3 является элементом из группы, включающей Ni, Со, Bi, Pd, Ag, Au, Pb и Ga. Все вышеизложенное действительно, прежде всего, также и тогда, когда М2, по меньшей мере, на 50 мол.% своего общего количества или, по меньшей мере, на 75 мол.%, или на 100 мол.% является Nb и М3 является элементом из группы, включающей Ni, Со, Pd и Bi. В особенной степени предпочтительно все вышеизложенное относительно стехиометрических коэффициентов действительно тогда, когда М1=Те, М2=Nb и М3 = по меньшей мере, элемент из группы, включающей Ni, Co и Pd. Другими пригодными согласно изобретению стехиометриями являются такие, которые раскрыты в вышеприведенных документах уровня техники для масс оксидов металлов стехиометрии (I). Принцип нацеленного способа для получения масс оксидов металлов (I) согласно изобретению раскрыт, например, в WO 0206199 и в цитируемых в этом документе публикациях. Согласно этому уровню техники сначала известным образом получают массу оксидов металлов, которая имеет стехиометрию (I), однако является, как правило, хорошо перемешанной сросшейся смешанной системой кристаллов из i-фазы и других фаз (например, k-фазы). Из этой смеси доля i-фазы может выделяться за счет того, что другие фазы, например, k-фазу, вымывают пригодными жидкостями. В качестве таких жидкостей пригодны, например, водные растворы органических кислот (например, щавелевой кислоты, муравьиной кислоты, уксусной кислоты, лимонной кислоты и винной кислоты), неорганических кислот (например, азотной кислоты), спирты и водные растворы перекиси водорода. Далее JP-A 7-232071 описывает способ получения i-фазных масс оксидов металлов. Смешанные системы кристаллов из i-фазы и k-фазы, как правило, получают описанными в уровне техники способами (см., например, DE-A 19835247, ЕР-А 529853, ЕР-А 603836, ЕР-А 608838, ЕР-А 895809, DE-A 19835247, ЕР-А 962253, ЕР-А 1080784, ЕР-А 1090684, ЕР-А 1123738, ЕР-А 1192987, ЕР-А 1192986, ЕР-А 1192982, ЕР-А 1192983 и ЕР-А 1192988). Согласно этим способам из подходящего источника элементарных компонентов масс оксидов металлов получают по возможности хорошо перемешанную, предпочтительно тонкодисперсную (порошковую) сухую смесь и ее термически обрабатывают при температуре от 350 до 700°С, соответственно от 400 до 650°С или от 400 до 600°С. Термическая обработка может в принципе осуществляться как под окисляющей, восстанавливающей, так и под инертной атмосферой. В качестве окисляющей атмосферы пригоден воздух, обогащенный молекулярным кислородом воздух или обедненный кислородом воздух. Предпочтительно термическую обработку осуществляют под инертной атмосферой, т.е. под молекулярным азотом и/или благородным газом. Обычно термическая обработка проводится при нормальном давлении (1 атм). Само собой разумеется термическая обработка может осуществляться также и в вакууме или при избыточном давлении. Если термическая обработка осуществляется под газообразной атмосферой, она может быть как неподвижной, так и текущей. Предпочтительно она является текущей. В общем термическая обработка может занимать до 24 часов или более. Термическая обработка предпочтительно осуществляется сначала под окисляющей (содержащей кислород) атмосферой (например, под воздухом) при температуре от 150 до 400°С, соответственно от 250 до 350°С (т.е. предварительная стадия разложения). Затем термическую обработку продолжают под инертным газом при температуре от 350 до 700°С, соответственно от 400 до 650°С или от 450 до 600°С. Само собой разумеется, термическую обработку можно проводить таким образом, что массу форконтакта (специального предварительного катализатора) перед ее термической обработкой сначала (в случае необходимости, после измельчения до порошкообразного состояния) таблетируют (в случае необходимости, при добавке от 0,5 до 2 вес.% тонкодисперсного графита), потом термически обрабатывают и затем снова измельчают. Хорошее перемешивание исходных соединений может осуществляться в сухой или во влажной форме. Если это производится в сухой форме, исходные соединения целесообразным образом применяются как тонкодисперсный порошок и после смешения и, в случае необходимости, уплотнения подвергаются кальцинированию (термической обработке). Предпочтительно хорошее смешивание производится во влажной форме. Обычно исходные соединения перемешиваются друг с другом в форме водного раствора (в случае необходимости, при участии комплексообразователей, см., например, документ DE-A 10145958) и/или суспензии. После этого водную массу сушат и после сушки кальцинируют. Целесообразным образом при водной массе речь идет о водном растворе или водной суспензии. Предпочтительно процесс сушки осуществляется непосредственно после получения водной смеси (в частности, в случае водного раствора; см., например, JP-A 7-315842) распылительной сушкой (выходные температуры составляют, как правило, от 100 до 150°С; распылительная сушка может осуществляться в параллельном токе или в противотоке), которая обуславливает особенно хорошо перемешанную смесь, прежде всего тогда, когда при подлежащей распылительной сушке массе речь идет о водном растворе или суспензии. Однако она может сушиться также и упариванием в вакууме, сублимационной сушкой или обычным выпариванием. В качестве источника элементарных компонентов в рамках проведения вышеописанного способа получения i-/k-фазных масс оксидов металлов пригодны все те, которые при нагреве (в случае необходимости на воздухе) могут образовывать оксиды и/или гидроксиды. Само собой разумеется, в качестве таких исходных соединений могут также применяться еще оксиды и/или гидроксиды элементарных компонентов или исключительно только они. Это означает то, что пригодны все названные в документах уровня техники исходные соединения. Пригодными согласно изобретению источниками для элемента Мо являются, например, оксиды молибдена, такие как триоксид молибдена, молибдаты, такие как тетрагидрат гептамолибдата аммония и молибденгалогениды, такие как молибденхлорид. Пригодными, применяемыми согласно изобретению исходными соединениями для элемента ванадия V являются оксисульфатгидрат ванадия, ванадил-ацетилацетонат, ванадаты, такие, как метаванадат аммония, оксиды ванадия, такие как пентоксид ванадия (V2O5), галогениды ванадия, такие как тетрахлорид ванадия (VCl4) и оксигалогениды ванадия, такие как VOCl3. При этом в качестве исходных ванадиевых соединений могут применяться также и такие, которые содержат ванадий степени окисления +4. В качестве источника элемента теллура пригодны согласно изобретению оксиды теллура, такие как диоксид теллура, металлический теллур, галогениды теллура, такие как TeCl2, а также теллуровые кислоты, такие как ортотеллуровая кислота Н6ТеО6. Предпочтительными исходными соединениями сурьмы являются галогениды сурьмы, такие как SbCl3, оксиды сурьмы, такие, как триоксид сурьмы (Sb2O3), сурьмяные кислоты, такие, как HSb(ОН)6, а также соли оксидов сурьмы, такие как сульфат оксида сурьмы (SbO)2SO4. Пригодными согласно изобретению источниками ниобия являются, например, оксиды ниобия, такие как пентоксид ниобия (Nb2O5), галогениды оксида ниобия, такие как NbOCl3, галогениды ниобия, такие как NbCl5, а также комплексные соединения из ниобия и органических карбоновых кислот и/или дикарбоновых кислот, например, оксалаты и алкоголяты. Само собой разумеется, в качестве источников ниобия подходят также и применяемые в ЕР-А 895 809 содержащие Nb растворы. Относительно всех других возможных элементов (в особенности Pb, Ni, Cu, Со, Bi и Pd) в качестве пригодных исходных соединений возможны прежде всего галогениды, нитраты, формиаты, оксалаты, ацетаты, карбонаты и/или гидроксиды. Пригодными исходными соединениями являются оксосоединения, например вольфраматы, соответственно, производные от них кислоты. Часто в качестве исходных соединений применяются также и аммониевые соли. Далее в качестве исходных соединений пригодны также и полианионы типа Андерсона, как описано, например, в публикации Polyhedron Vol.6, No.2, pp.213-218, 1987. Еще одним пригодным литературным источником для полианионов Андерсена является Kinetics and Catalysis, Vol.40, No.3, 1999, стр. от 401 до 404. Другими пригодными в качестве исходных соединений полианионов являются полианионы типа Dawson или Keggin. Предпочтительно применяются такие исходные соединения, которые при высоких температурах или в присутствии или при исключении кислорода, в случае необходимости, при высвобождении газообразных соединений могут превращаться в свои оксиды. Получаемые, как описано, массы оксидов металлов смешанных i-/k-фазных кристаллов (чистые i-фазные оксиды металлов получают описанными способами только случайно) могут потом переводиться посредством подходящей промывки в оксиды металлов (I) согласно изобретению. Повышенная доля i-фазы (и в благоприятных случаях в основном чистая i-фаза) устанавливается при получении форконтактов оксидов металлов (которые описанным промыванием могут быть переведены в оксиды металлов (I) согласно изобретению) тогда, когда их получение происходит гидротермальным путем, как описано, например, в DE-A 10029338 и в JP-A 2000-143244. Получение масс оксидов металлов (I) согласно изобретению может осуществляться также и таким образом, что сначала получают массу оксидов металлов I Подобная, предпочтительно тонкодисперсная масса оксидов металлов I Получаемые, как описано, оксиды металлов (I) согласно изобретению могут применяться для способа согласно изобретению как таковые [например, как порошок или после таблетирования порошка (часто при добавке от 0,5 до 2 вес.% тонкодисперсного графита) и измельченными последующим дроблением в мелкий щебень] или отформованными в формованные изделия. При этом катализаторный слой может быть неподвижным, подвижным или псевдоожиженным слоем. Формование формованных изделий может осуществляться, например, нанесением на носитель, как это описано, например, в документах DE-A 10118814, соответственно, РСТ/ЕР/02/04073, соответственно DE-A 10051419. Применяемые для масс оксидов металлов (I) носители являются предпочтительно химически инертными. Это означает то, что они в основном не участвуют в процессе частичного каталитического газофазного окисления, соответственно аммокисления углеводорода (например, пропана и/или пропена в акриловую кислоту), который катализируется применяемыми согласно изобретению массами оксидов металлов (I). В качестве материала для носителя пригодны согласно изобретению в особенности оксид алюминия, диоксид кремния, силикаты, такие как глина, каолин, стеатит (предпочтительно с небольшим содержанием растворимой в воде щелочи), пемза, силикат алюминия и силикат магния, карбид кремния, диоксид циркония и диоксид тория. Поверхность носителя может быть как гладкой, так и шероховатой. С преимуществом поверхность носителя выполняется шероховатой, так как повышенная шероховатость поверхности, как правило, обуславливает повышенную адгезионную прочность нанесенного слоя активной массы. Часто шероховатость поверхности Rz носителя составляет от 5 до 200 мкм, зачастую от 20 до 100 мкм (определено по стандарту Германии ДИН DIN 4768 Blatt 1 инструментом “Hommel Tester für DIN-ISO Oberflächenmeßgrößen” фирмы Hommelwerke, DE). Далее материал носителя может быть пористым или непористым. Целесообразным образом материал носителя является непористым (общий объем пор, в пересчете на объем носителя, составляет Толщина нанесенной на носитель катализатора (оболочкового катализатора) согласно изобретению активной оболочки оксидов металлов составляет обычно от 10 до 1000 мкм. Она может также составлять от 50 до 700 мкм, от 100 до 600 мкм или от 150 до 400 мкм. Возможная толщина оболочки составляет также от 10 до 500 мкм, от 100 до 500 мкм или от 150 до 300 мкм. В принципе для способа согласно изобретению пригодны любые геометрические формы носителя. Их продольный размер, как правило, составляет от 1 до 10 мм. Предпочтительно применяется шаровая форма или цилиндрическая форма, в особенности форма полых цилиндров. Диаметр носителя оставляет от 1,5 до 4 мм. Если в качестве носителя используются цилиндры, то их длина составляет предпочтительно от 2 до 10 мм и их внешний диаметр предпочтительно от 4 до 10 мм. В случае колец толщина стенки обычно составляет от 1 до 4 мм. Пригодные согласно изобретению кольцеобразные носители могут также иметь длину от 3 до 6 мм, внешний диаметр от 4 до 8 мм и толщину стенки от 1 до 2 мм. Возможно также и такая кольцевая форма, при которой имеются размеры 7 мм × 3 мм × 4 мм или 5 мм × 3 мм × 2 мм (внешний диаметр × длина × внутренний диаметр). Получение применяемых согласно изобретению нанесенных (оболочковых) катализаторов простым образом осуществляется так, что предварительно изготавливают применяемую согласно изобретению массу оксидов общей формулы (I), ее переводят в тонкодисперсную форму и затем с помощью жидкого связующего наносят на поверхность носителя. Для этого поверхность носителя увлажняют простым образом жидким связующим и приведением в контакт с тонкодисперсной активной массой оксидов общей формулы (I) прикрепляют слой активной массы на увлажненную поверхность. В заключение покрытый слоем носитель сушат. Само собой разумеется для обеспечения более толстого слоя процесс периодически повторяют. В этом случае снабженная слоем основа становится новым Тонкость наносимой на поверхность носителя каталитически активной массы оксидов металлов общей формулы (I) подгоняется к желаемой толщине оболочки. Для диапазона толщин оболочки от 100 до 500 мкм пригодны, например, такие порошки активной массы, при которых, по меньшей мере, 50% общего числа порошковых частиц проходит через сито с шириной отверстий от 1 до 20 мкм и численная доля частиц которых с продольным размером выше 50 мкм составляет менее 10%. Как правило, распределение продольных размеров порошковых частиц соответствует гауссовому распределению. Часто распределение размеров частиц соответствует следующим величинам:
При этом: D = диаметр частицы, х = процентная доля частиц, диаметр которых и у = процентная доля частиц, диаметр которых < D. Для проведения способа нанесения слоя в техническом масштабе рекомендуется, например, применение раскрытого в DE-A 2909671, а также в DE-A 10051419 метода. При этом подлежащие покрытию слоем носители помещают в предпочтительно наклонную (угол наклона составляет, как правило, При потребности покрытый таким образом основным слоем носитель в ходе следующего оборота снова проходит под соплом, снова контролировано увлажняется. чтобы в ходе дальнейшего движения воспринять еще один слой тонкодисперсной оксидной активной массы и т.д. (промежуточной сушки, как правило, не требуется). Тонкодисперсная (порошковая) оксидная активная масса и жидкое связующее подаются, как правило, непрерывно и синхронно. Удаление жидкого связующего может происходить после окончания покрытия, например, воздействием горячих газов, таких, N2 или воздух. Следует заметить, что описанный способ нанесения покрытия способствует удовлетворительной адгезии как следующих друг за другом слоев, так и основного слоя на поверхности носителя. Существенным для вышеописанного способа нанесения слоев является то, что увлажнение подлежащей покрытию поверхности носителя производится контролированным образом. Короче говоря, это означает то, что поверхность носителя увлажняют таким образом, что она правда имеет абсорбированное связующее, однако на поверхности носителя визуально не просматривается жидкая фаза. Если поверхность носителя слишком влажная, тонкодисперсная каталитическая активная оксидная масса агломерируется в отдельные агломераты, вместо того чтобы распределяться по поверхности. Подробные данные об этом можно найти в заявках DE-A 2909671 и DE-A 10051419. Заключительное удаление примененного жидкого связующего может осуществляться контролированным образом, например, посредством отпаривания и/или сублимации. В самом простом случае это может осуществляться воздействием горячих газов соответствующей температуры (часто от 50 до 300, часто 150°С). Воздействием горячих газов можно способствовать только предварительной сушке. Окончательная сушка может осуществляться затем, например, в сушильной печи любой конструкции (например, в ленточной сушилке) или в реакторе. Температура при этом не должна лежать выше применяемой для получения оксидной активной массы температуры кальцинирования. Само собой разумеется сушка может проводиться исключительно в сушильной печи. В качестве связующего для процесса нанесения слоя могут применяться независимо от вида и геометрической формы носителя следующие: вода, одноатомные спирты, такие как этанол, метанол, пропанол и бутанол и многоатомные спирты, такие как этиленгликоль, 1,4-бутандиол, 1,6-гександиол или глицерин, одно- или многовалентные карбоновые кислоты, такие как пропионовая кислота, щавелевая кислота, малоновая кислота, глутаровая кислота и малеиновая кислота, аминоспирты, такие как этаноламин или диэтаноламин, а также одно или многовалентные органические амиды. Пригодными связующими являются также растворы, состоящие из 20 до 90 вес.% воды и 10 до 80 вес.% растворенного воде органического соединения, точка кипения которого или температура сублимации при нормальном давлении (1 атм) составляет >100°С, предпочтительно >150°С. С преимуществом выбирают органическое соединение из вышеприведенного перечня возможных органических связующих. Предпочтительно органическая доля вышеприведенных водных растворов связующих составляет от 10 до 50 и особенно предпочтительно от 20 до 30 вес.%. В качестве органических компонентов при этом пригодны также и моносахариды и олигосахариды, такие как глюкоза, фруктоза, сахароза или лактоза, а также полиэтиленоксиды и полиакрилаты. Важным является то, что получение пригодных согласно изобретению оболочковых (нанесенных) катализаторов может происходить не только посредством нанесения подготовленной, тонко измельченной активной оксидной массы общей формулы (I) на увлажненную поверхность носителя. Вместо активной оксидной массы на увлажненную поверхность носителя может также наноситься ее предварительная масса (при применении одного и того же способа нанесения и связующего) и производиться кальцинирование после сушки покрытых носителей (носители могут также пропитываться раствором предварительной массы, затем сушиться и в заключение кальцинироваться). Под конец могут, в случае необходимости, вымываться фазы, отличные от i-фазы. В качестве такой тонкодисперсной предварительной массы пригодны, например, такие массы, которые получают таким образом, что из источников элементарных компонентов желаемой активной оксидной массы общей формулы (I) производят по возможности хорошо перемешанную, тонкодисперсную сухую смесь (например, распылительной сушкой водной суспензии или раствора источника) и эту сухую смесь (в случае необходимости, после таблетирования при добавке от 0,5 до 2 вес.% тонкодисперсного графита) термически обрабатывают (несколько часов) при температуре от 150 до 350°С, предпочтительно от 250 до 350°С под окисляющей (содержащей кислород) атмосфере (например, под воздухом) и затем подвергают при необходимости измельчению. После нанесения на носитель предварительной массы производят кальцинирование, предпочтительно под инертной атмосферой (все другие атмосферы также пригодны) при температуре от 360 до 700°С, соответственно от 400 до 650°С или от 400 до 600°С. Само собой разумеется, формование применяемых согласно изобретению масс оксидов металлов (I) может осуществляться также и экструзией и/или таблетированием как тонкодисперсных масс оксидов металлов (I), так и тонкодисперсных предварительных масс оксидов металлов (I) (в случае необходимости в заключение можно проводить вымывание фаз, отличных от i-фаз). В качестве геометрической формы при этом пригодны как шаровидные, цилиндрические сплошные и цилиндрические полые (кольцевые) формы. Продольный размер вышеуказанных геометрических форм составляет при этом, как правило, от 1 до 10 мм. В случае цилиндров их длина составляет предпочтительно от 2 до 10 мм и их внешний диаметр предпочтительно от 4 до 10 мм. В случае колец их толщина стенки обычно составляет от 1 до 4 мм. Пригодные согласно изобретению кольцеобразные цельные катализаторы могут иметь длину от 3 до 6 мм, внешний диаметр от 4 до 8 мм и толщину стенки от 1 до 2 мм. Возможна также такая геометрическая форма цельных кольцеобразных катализаторов, при которой имеются следующие размеры: 7 мм × 3 мм × 4 мм или 5 мм × 3 мм × 2 мм (внешний диаметр × длина × внутренний диаметр). Само собой разумеется для способа согласно изобретению пригодны также геометрические формы применяемых масс оксидов металлов (I), раскрытые в DE-A 10101695. Согласно изобретению существенным является то, что, как уже было упомянуто, применяемые согласно изобретению массы оксидов металлов (I) имеют рентгеновскую дифрактограмму (в настоящем описании всегда в отношении Cu-К – дифракционный рефлекс h является самым интенсивным внутри рентгеновской дифрактограммы, а также имеет полуширину пика максимально 0,5°, – интенсивность Pi дифракционного рефлекса i и интенсивность Pk дифракционного рефлекса k выполняют соотношение 0,65 R=Pi/(Pi+Pk) и – полуширина пика дифракционного рефлекса i и дифракционного рефлекса k составляет каждая Одновременно рентгеновская дифрактограмма не должна иметь дифракционного рефлекса с пиком 2 Определение интенсивности дифракционного рефлекса на рентгеновской дифрактограмме относится в этом описании, как уже было упомянуто, к установленному в DE-A 19835247, а также в DE-A 10051419 и DE-A 10046672 определению. Это означает то, что если А1 обозначает пик (максимум кривой) рефлекса 1 и В1 обозначает на линии рентгеновской дифрактограммы, если смотреть вдоль оси интенсивности, вертикальной к оси 2 Полуширина пика представляет собой в настоящем описании соответствующим образом длину прямого участка, между двумя точками пересечения Н1 и Н2, получаемыми, если посередине прямого участка А1C1 проходит параллель к 2 Проведение определения полуширины пика и интенсивности показано как пример на фиг.6 в заявке DE-A 10046672. Само собой разумеется, применяемые согласно изобретению массы оксидов металлов (I) могут также применяться в разбавленной тонкодисперсными, например, коллоидальными материалами, такими, как диоксид кремния, диоксид титана, оксид алюминия, оксид циркония, оксид ниобия форме в качестве каталитической активной массы. Весовое соотношение разбавления может при этом составлять до 9 (разбавитель): 1 (активная масса). Это означает то, что возможные весовые соотношения разбавления составляют, например, 6 (разбавитель): 1 (активная масса) и 3 (разбавитель): 1 (активная масса). Ввод разбавителя может осуществляться до и/или после кальцинирования, как правило, даже перед сушкой. Если ввод происходит перед сушкой, соответственно перед кальцинированием, разбавитель должен выбираться таким образом, чтобы он в основном сохранялся в жидкой среде соответственно при кальцинировании. Это имеется, например, в случае обожженных при соответствующим образом высокой температуре оксидов. Массы оксидов металлов (I) согласно изобретению как таковые или в разбавленной, как описано, выше форме пригодны в качестве активных масс для гетерогенно-катализируемого частичного газофазного окисления (включая оксидегидрирование) и/или аммокисления насыщенных и/или ненасыщенных углеводородов. Подобными насыщенными и/или ненасыщенными углеводородами являются, в частности, этан, этилен, пропан, пропилен, н-бутан, изобутан и изобутен. Целевыми продуктами при этом являются прежде всего акролеин, акриловая кислота, метакролеин, метакриловая кислота, акрилнитрил и метакрилнитрил. Они пригодны также и для гетерогенно-катализируемого частичного газофазного окисления и/или аммокисления таких соединений, как акролеин и метакролеин. Целевыми продуктами могут также быть этилен, пропилен и уксусная кислота. Под полным окислением углеводородов в настоящем описании понимается то, что содержащийся в общем в углеводороде углерод превращается в оксиды углерода (СО, СО2). Все отличные от этого превращения углеводорода при активном воздействии молекулярного кислорода называются в настоящем описании частичным окислением. Дополнительное реакционноспособное воздействие аммиака обозначается частичным аммокислением. Массы оксидов металлов (I) согласно настоящему изобретению пригодны в качестве каталитически активных масс для превращения пропана в акролеин и/или акриловую кислоту, пропана в акриловую кислоту и/или акрилнитрил, пропилена в акролеин и/или акриловую кислоту, пропилена в акрилнитрил, изобутана в метакролеин и/или метакриловую кислоту, изобутана в метакриловую кислоту и/или метакрилнитрил, этана в этилен, этана в уксусную кислоту и этилена в уксусную кислоту. Проведение подобного частичного окисления и/или аммокисления (регулируемое известным образом выбором содержания аммиака в реакционной газовой смеси реакцию можно в основном проводить исключительно как частичное окисление или исключительно как частичное аммокисление или наложением обеих реакций, например, см., WO 98/22421) известно из систем смешанных кристаллов i-/k-фаз известного уровня техники и может осуществляться полностью аналогичным образом. Если в качестве углеводорода применяется сырой пропан или сырой пропилен, то он имеет состав предпочтительно, как описано в документах DE-A 10246119, соответственно DE-A 10118814, соответственно РСТ/ЕР/02/04073. Также предпочтительно способ проводят, как описано в этих документах. Проводимое на катализаторах с активной массой оксидов металлов (I) частичное окисление пропана в акриловую кислоту может проводиться, как описано в ЕР-А 608838, WO 0029106, JP-A 10-36311 и ЕР-А 1192987. В качестве источника требуемого молекулярного кислорода может применяться, например, воздух, обогащенный кислородом и обедненный кислородом воздух или чистый кислород. Такой способ дает тогда преимущества, когда реакционная газовая исходная смесь не содержит в качестве инертного разбавляющего газа благородный газ, в частности гелий. Впрочем, реакционная газовая смесь наряду с пропаном и молекулярным кислородом может включать инертные разбавляющие газы, такие как N2, CO и СО2. Водяной пар в качестве компонента реакционной газовой смеси является предпочтительным. Таким образом реакционная газовая исходная смесь, нагрузке которой подлежат массы оксидов металлов согласно изобретению при температуре, например, от 200 до 550°С или от 230 до 480°С, соответственно от 300 до 440°С и давлении от 1 до 10 бар, соответственно от 2 до 5 бар, может иметь следующий состав: от 1 до 15, предпочтительно от 1 до 7 об.% пропана, от 44 до 99 об.% воздуха и от 0 до 55 об.% водяного пара. Предпочтительны содержащие водяной пар реакционные газовые исходные смеси. В качестве еще одного возможного состава реакционной газовой исходной смеси пригоден следующий: от 70 до 95 об.% пропана, от 5 до 30 об.% молекулярного кислорода и от 0 до 25 об.% водяного пара. Само собой разумеется, таким способом получают газовую смесь продукта, которая не состоит исключительно из акриловой кислоты. Газовая смесь продукта содержит наряду с непрореагировавшим пропаном такие побочные компоненты, как пропен, акролеин, CO2, СО, H2O, уксусную кислоту, пропионовую кислоту и т.п., от которых нужно отделять акриловую кислоту. Это может осуществляться так, как это известно для гетерогенно-катализируемого газофазного окисления пропена в акриловую кислоту. Это означает то, что из газовой смеси продукта полученная акриловая кислота может абсорбироваться водой или высококипящим инертным гидрофобным органическим растворителем (например, смесью простого дифенилового эфира и дифила (Diphyl®, представляющий собой смесь бифенила с простым дифениловым эфиром), которая, в случае необходимости, может содержать еще добавки, такие как диметилфталат). Получаемая при этом смесь из абсорбента и акриловой кислоты может затем перерабатываться известным образом ректификацией, экстракцией и/или кристаллизацией до чистой акриловой кислоты. Альтернативно основное отделение акриловой кислоты из газовой смеси продукта может происходить также и фракционированной конденсацией, как это описано в DE-A 19924532. Получаемый при этом водный конденсат акриловой кислоты может потом еще очищаться, например, фракционированной конденсацией (например, суспензионной кристаллизацией и/или слоистой кристаллизацией). Остающаяся при основном отделении акриловой кислоты остаточная газовая смесь содержит, в частности, непрореагировавший пропан, который предпочтительно рециркулируется на газофазное окисление. Для этого он может частично или полностью отделяться из остаточной газовой смеси, например, фракционированной ректификацией под давлением и затем рециркулироваться на газофазное окисление. Однако благоприятнее приводить остаточный газ в экстракционном устройстве в контакт с гидрофобным, органическим растворителем (например, пропусканием через него), который предпочтительно в состоянии абсорбировать пропан. Посредством последующей десорбции и/или стриппинга воздухом абсорбированный пропан может снова высвобождаться и рециркулироваться в процесс согласно изобретению. Таким образом, могут быть получены экономичные конверсии всего пропана. Образовавшийся как побочный компонент пропен при этом, так же как и при других способах отделения, как правило, не отделяется или не полностью отделяется от пропана и вместе с ним циркулируется. Это действительно также и в случае других гомологических насыщенных или олефиновых углеводородов. Прежде всего это действительно в общем для гетерогенно-катализируемого частичного окисления и/или аммокисления насыщенных углеводородов. При этом следует отметить, что массы оксидов металлов согласно изобретению могут катализировать также и частичное окисление и/или аммокисление гомологического олефинового углеводорода до такого же целевого продукта. Таким образом массами оксидов металлов (I) согласно изобретению в качестве активных масс можно получать акриловую кислоту посредством гетерогенно-катализируемого частичного газофазного окисления пропена молекулярным кислородом, как это описано в DE-A 10118814, соответственно РСТ/ЕР/02/04073 или JP-A 7-53448. То есть для проведения способа согласно изобретению достаточна одна-единственная реакционная зона А. В этой реакционной зоне находятся в качестве каталитически активной массы исключительно катализаторы на основе масс оксидов металлов (I). Это является необычным, т.к. в общем гетерогенно-катализируемое газофазное окисление пропена в акриловую кислоту осуществляется в две следующие по времени друг за другом стадии. На первой стадии обычно пропен окисляется в значительной степени в акролеин, и на второй стадии обычно полученный акролеин окисляется в акриловую кислоту. Обычные способы гетерогенно-катализируемого газофазного окисления пропена в акриловую кислоту для каждой из обеих названных стадий окисления применяют специальный, точно подогнанный для стадии окисления тип катализатора. То есть обычные способы гетерогенно-катализируемого газофазного окисления пропена в акриловую кислоту работают в отличие от способа согласно изобретению с двумя реакционными зонами. Само собой разумеется, при предлагаемом способе частичного окисления пропена в реакционной зоне А может находиться только один катализатор или более одного катализатора из масс оксидов металлов (I). Конечно, применяемые согласно изобретению катализаторы могут быть разбавлены инертным материалом, который в настоящем описании рекомендуется также как материал носителя. Вдоль одной реакционной зоны А при способе частичного окисления пропена согласно изобретению может преобладать только одна или же изменяющаяся вдоль реакционной зоны А температура теплоносителя для термостатирования реакционной зоны А. Это изменение температуры может быть повышающимся или понижающимся. Если способ частичного окисления пропена согласно изобретению проводится как окисление на неподвижном слое, то он осуществляется целесообразным образом в кожухотрубном реакторе, контактные трубы которого загружены катализатором. Вокруг контактных труб в обычном случае направляется в качестве теплоносителя жидкость, как правило, солевая баня. Несколько температурных зон вдоль реакционной зоны А могут быть реализованы простым образом за счет того, что участками по длине контактных труб вокруг контактных труб направляется более одной солевой бани. Реакционная газовая смесь в контактных трубах, если смотреть вдоль реактора, направляется или в параллельном потоке или в противотоке к солевой бане. Солевая баня сама может двигаться относительно контактных труб в чисто параллельном потоке. Само собой разумеется, на этот поток может накладываться поперечный поток. В общем солевая баня может выполнять также и меандрообразное движение, которое, если смотреть только вдоль реактора, направляет в параллельном потоке или противотоке в реакционной газовой смеси. Температура реакции при способе частичного окисления пропена согласно изобретению вдоль всей реакционной зоны А может составлять от 200° до 500°С. Обычно она составляет от 250 до 450°С. Предпочтительно температура реакции составляет от 330 до 420°С, особенно предпочтительно от 350 до 400°С. Рабочее давление при способе частичного окисления пропена согласно изобретению может составлять как 1 бар, так и менее 1 бар или более 1 бар. Типичным давлением согласно изобретению являются от 1,5 до 10 бар, часто от 1,5 до 5 бар. К применяемому в способе частичного окисления пропена согласно изобретению пропену не предъявляется никаких особых требований относительно его чистоты. В качестве пропена для такого способа, как уже было упомянуто и как это обычно для всех одно- или двухстадийных способов гетерогенно-катализируемого газофазного окисления пропена в акролеин и/или акриловую кислоту, может полностью без проблем применяться пропен (также называемый сырым пропеном) следующих характеристик: а) полимерно чистый пропилен:
b) химически чистый пропилен:
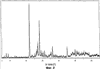
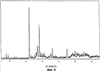
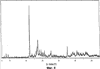
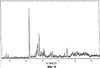
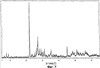
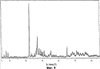
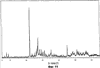
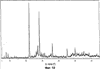
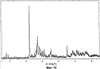
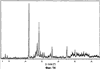
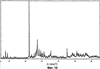
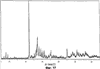
Само собой разумеется, все вышеприведенные возможные сопутствующие соединения пропена могут содержаться в сыром пропене в двойном или десятикратном количестве приведенных индивидуальных количеств, в общем не оказывая отрицательного воздействия на применимость сырого пропена для способа согласно изобретению, соответственно для известных способов одно- или двухстадийного гетерогенно-катализируемого газофазного окисления пропена в акролеин и/или акриловую кислоту. Это действительно особенно тогда, когда, как и при насыщенных углеводородах, водяном паре, оксидах углерода или молекулярном кислороде, речь идет о соединениях, которые и так принимают участие в прохождении реакции при вышеприведенных способах в повышенных количествах как инертные разбавляющие газы или как реагенты. Обычно сырой пропен смешивается, как таковой, с циркулирующим газом, воздухом и/или молекулярным кислородом для способа согласно изобретению и всех прочих способов гетерогенно-катализируемого газофазного окисления пропена в акролеин и/или акриловую кислоту. В качестве источника пропена при способе согласно изобретению пригоден также и пропен, который в рамках отличного от способа согласно изобретению способа образуется как побочный продукт и содержит, например, до 40% своего веса пропан. При этом этот пропен может сопровождаться другими, не мешающими способу согласно изобретению сопутствующими компонентами. В качестве источника кислорода для способа частичного окисления пропена согласно изобретению может применяться как чистый кислород, так и обогащенный, соответственно обедненный кислородом воздух. Наряду с молекулярным кислородом и пропеном применяемая в способе согласно изобретению реакционная газовая смесь содержит обычно еще, по меньшей мере, разбавляющий газ. В качестве такого газа пригодны азот, оксиды углерода, благородные газы и низшие углеводороды, такие как метан, этан и пропан (высших, например, С4-углеводородов следует избегать). В качестве разбавляющего газа часто применяется также и водяной пар. Часто смеси из вышеприведенных газов представляют разбавляющий газ для способа частичного окисления пропена согласно изобретению. Согласно изобретению гетерогенно-катализируемое окисление пропена производится в присутствии пропана. Типичным образом реакционная газовая смесь для способа согласно изобретению имеет следующий состав (молярные соотношения): пропен: кислород: Н2O: прочие разбавляющие газы = 1:(0,1-10):(0-70):(0-20). Предпочтительно это соотношение составляет 1:(1-5):(1-40):(0-10). Если в качестве разбавляющего газа применяется пропан, то он может частично окисляться также до акриловой кислоты. Предпочтительно реакционная газовая смесь содержит согласно изобретению молекулярный азот, СО, CO2, водяной пар и пропан в качестве разбавляющего газа. Молярное соотношение пропана к пропену может принимать при способе согласно изобретению следующие значения: от 0 до 15, часто от 0 до 10, многократно от 0 до 5, целесообразно от 0,01 до 3. Нагрузка катализаторного слоя пропеном может при способе частичного окисления пропена согласно изобретению составлять, например, от 40 до 250 нл/л·ч. Нагрузка реакционной газовой смесью часто составляет от 500 до 15000 нл/л·ч, многократно от 600 до 10000 нл/л·ч, зачастую от 700 до 5000 нл/л·ч. Само собой разумеется, при способе частичного окисления пропена в акриловую кислоту согласно изобретению получают газовую смесь продукта, которая не состоит исключительно из акриловой кислоты. Эта газовая смесь продукта наряду с непрореагировавшим пропеном содержит такие побочные компоненты, как пропан, акролеин, CO2, CO, H2O, уксусную кислоту, пропионовую кислоту и т.п., от которых нужно отделять акриловую кислоту. Это может осуществляться так, как это известно из гетерогенно-катализируемого, двухстадийного (проводимого в двух реакционных зонах), газофазного окисления пропена в акриловую кислоту. Это означает то, что из газовой смеси продукта полученная акриловая кислота может поглощаться абсорбцией водой или абсорбцией высококипящим инертным гидрофобным органическим растворителем (например, смесью дифенилового эфира с дифилом (Diphyl® представляет собой смесь бифенила с простым дифениловым эфиром), которая, в случае необходимости, может еще содержать такие добавки, как диметилфталат). Получаемая при этом смесь из абсорбера и акриловой кислоты может затем перерабатываться известным образом, например, ректификацией, экстракцией и/или кристаллизацией до чистой акриловой кислоты. Альтернативно основное отделение акриловой кислоты от газовой смеси продукта может происходить также и фракционированной конденсацией, как это описано в DE-A 19924532. Получаемый при этом водный концентрат акриловой кислоты может затем далее очищаться, например, фракционированной кристаллизацией (например, суспензионной кристаллизацией и/или слоистой кристаллизацией). Остающаяся при основном отделении акриловой кислоты остаточная смесь содержит, в частности, непрореагировавший пропен (и, в случае необходимости, пропан). Он может отделяться от остаточной газовой смеси, например, фракционированной ректификацией под давлением и затем рециркулироваться в процесс газофазного окисления согласно изобретению. Однако более предпочтительно приводить остаточный газ в экстракционном устройстве в контакт с гидрофобным органическим растворителем (например, пропусканием через него), который может преимущественно абсорбировать пропен (и, в случае необходимости, пропан). Посредством последующей десорбции и/или стриппинга воздухом абсорбированный пропен (и, в случае необходимости, пропан) может снова высвобождаться и рециркулироваться в процесс согласно изобретению. Таким образом, обеспечивается экономичность способа. Если пропен частично окисляется в присутствии пропана, то пропен и пропан предпочтительно отделяются и рециркулируются совместно. Совершенно таким же образом могут применяться оксиды металлов (I) согласно изобретению в качестве катализаторов для частичного окисления изобутана и/или изобутена в метакриловую кислоту. Их применение для аммокисления пропана и/или пропена может осуществляться, как описано в ЕР-А 529853, DE-A2351151, JP-A 6-166668 и JP-A 7-232071. Их применение для аммокисления н-бутана и/или н-бутена может происходить, как описано в JP-A 6-211767. Их применение для оксидегидрирования этана в этилен соответственно в дальнейшей реакции до уксусной кислоты можно осуществлять, как описано в US-A 4250346 или в ЕР-В 261264. Массы оксидов металлов (I) согласно изобретению могут быть интегрированы в другие массы оксидов металлов (например, их тонкодисперсные массы смешиваются, в случае необходимости, спрессовываются, кальцинируются или в качестве суспензий (предпочтительно водных) смешиваются, сушатся и кальцинируются (например, как описано в ЕР-А 529853 для масс оксидов металлов (I), где d=0)). Предпочтительно кальцинирование проводят под инертным газом. Получаемые при этом масс оксидов металлов (далее называемые общими массами) содержат в пересчете на Также и общие массы предпочтительно не содержат при 2 Если общая масса при 2 Придание геометрических форм осуществляется для общих масс таким же образом, как это описано для масс оксидов металлов (I). Преимущество масс оксидов металлов (I) согласно изобретению основано на их прекрасной селективности целевого продукта. Неожиданным является то, что промоторы М3 активны также и в чистой i-фазе, а именно как относительно приведенного в описании частичного окисления, так и в отношении частичного аммокисления. Для гетерогенно-катализируемого частичного газофазного окисления пропана в акриловую кислоту массы оксидов металлов (I) согласно изобретению соответственно катализаторы используются как предпочтительно описывается в немецкой заявке DE-A 10122027. Примеры А) Получение оболочкового (нанесенного) катализатора на основе масс оксидов металлов Сравнительный пример 1 (получение катализатора на основе масс оксидов металлов с активной массой Mo1,0V0,33Te0,19Nb0,11Ni0,01Ox, содержащей i- и k-фазу) 87,61 г метаванадата аммония (78,55 вес.% V2O5, фирмы G.f.E. Nürnbern, DE) при перемешивании растворяют при 80°С в 3040 мл воды (трехгорлая колба с мешалкой, термометр, обратный холодильник и нагревающее устройство). Получают прозрачный желтоватый раствор. Этот раствор охлаждают до 60°С и потом при сохранении температуры 60°С в раствор последовательно вводят 117,03 г теллуровой кислоты (99 вес.% Н6ТеО6, фирмы Aldrich) и 400,00 г гептамолибдата аммония (82,52 вес.% МоО3, фирмы Starck/Goslar). Получаемый темно-красный раствор охлаждают до 30°С и потом при поддержании 30°С смешивают с 25,60 г водного раствора 6,80 г гексагидрата нитрата никеля (II) (98 вес.%, фирмы Fluka) в 20 г воды (растворение происходит при 25°С). Таким образом получают раствор А, который имеет температуру 30°С. В химическом стакане отдельно от этого растворяют при 60°С 112,67 г аммоний-ниобий оксалата (20,8 вес.% Nb, фирмы Starck/Goslar) в 500 мл воды при получении раствора В. Раствор В охлаждают до 30°С и при этой температуре объединяют с имеющим ту же температуру раствором А, причем раствор В подают к раствору А. Эту подачу осуществляют в течение 5 минут. Получают оранжевую суспензию. После этого суспензию сушат распылительной сушкой в распылительной сушилке фирмы Niro (распылительная сушилка фирмы Niro A/S, DK). Температура суспензии составляет 30°С. Входная температура газа Твх составляет 320°С, выходная температура газа Твых составляет 110°С. Получаемый порошок имеет также оранжевый цвет. 100 г полученного распылительной сушкой порошка нагревают в ротационной шаровой печи согласно фиг.1 (кварцевый шар с 1 литром внутреннего объема; 1 = корпус печи, 2 = вращающаяся колба, 3 = нагреваемое пространство, 4 = поток азота/воздуха) при потоке воздуха 50 нл/в в течение 27,5 мин сначала линейно от 25°С до 275°С и сохраняют эту температуру и этот поток воздуха в течение 1 часа. Непосредственно после этого поток воздуха заменяют потоком азота в 50 нл/ч и нагревают в течение 32,5 минут линейно от 275°С до 600°С. Температуру и поток азота сохраняют в течение 2 часов. В заключение при сохранении потока азота охлаждают всю ротационную шаровую печь до 25°С. Получают черный порошок с составом Mo1,0V0,33Te0,19Nb0,11Ni0,01Ox (стехиометрия навески: Mo1,0V0,33Te0,22Nb0,11Ni0,01Ox). Соответствующая рентгеновская дифрактограмма приведена на фиг.2 (R=0,26). БЭТ=8,0 м2/г. Порошок активной массы затем измельчают в мельнице (центробежная мельница типа ZM 100, фирмы Retsch, DE) (величина частиц 38 г имеющегося после измельчения порошка наносят на 150 г шаровых носителей с диаметром от 2,2 до 3,2 мм (Rz=45 мм, материал носителя = стеатит фирмы Ceramtec, DE, общий объем пор носителя После окончания нанесения покрытый носитель сушат в атмосфере воздуха в течение 16 часов при 150°С в муфельной печи. Получают оболочковый (нанесенный) катализатор VB1 с долей активной массы 20 вес.%. Пример 1 Как и сравнительный пример 1. Однако полученный после измельчения в мельнице фирмы Retsch порошок перемешивают в 1000 мл 10 вес.%-го водного раствора HNO3 в течение 7 часов при 70°С при нагреве с обратным холодильником. Остающееся при этом твердое вещество отфильтровывают из получаемой суспензии, промывают водой до свободного от нитрата состояния. После этого фильтрат сушат в течение ночи в атмосфере воздуха при 110°С в муфельной печи. Получаемая активная масса имеет следующий состав: Mo1,0V0,29Te0,14Nb0,13Ni0,007Оx. Соответствующая рентгеновская дифрактограмма показана на фиг.3 (R=0,71). Удельная поверхность по БЭТ=20,2 м2/г. Эту массу наносят на тот же носитель, что и в сравнительном примере 1, так что получают оболочковый катализатор В1 с долей активной массы 20 вес.%. Сравнительный пример 2 Как и сравнительный пример 1, только вместо 6,80 г гексагидрата нитрата никеля (II) применяют 6,17 г дигидрата нитрата палладия (II) (98%, фирмы Fluka). Получаемая активная масса имеет следующий состав Mo1,0V0,33Te0,19Nb0,11Pd0,01Ox. Соответствующая рентгеновская дифрактограмма показана на фиг.4 (R=0,25). БЭТ=9,3 м2/г. Ее наносят таким же образом на тот же носитель, что и в сравнительном примере 1, так что получают оболочковый катализатор VB2 с долей активной массы 20 вес.%. Пример 2 Как пример 1, только активную массу из сравнительного примера 2 промывают водной азотной кислотой. Получаемая активная масса имеет следующий состав: Mo1,0V0,28Te0,13Nb0,13Pd0,001Ox. Соответствующая рентгеновская дифрактограмма показана на фиг.5 (R=0,73). БЭТ=22,5 м2/г. Ее наносят таким же образом на тот же носитель, что и в сравнительном примере 1, так что получают оболочковый катализатор В2 с долей активной массы 20 вес.%. Сравнительный пример 3 Как сравнительный пример 1, только количество реакционной смеси снижают на половину и вместо 3,40 г гексагидрата нитрата никеля (II) применяют 2,34 г дигидрата нитрата палладия (II) (98%, фирмы Fluka). Получаемая активная масса имеет следующий состав: Mo1,0V0,33Te0,22Nb0,11Pd0,04Оx. Соответствующая рентгеновская дифрактограмма показана на фиг.6 (R=0,35). BET=9,3 m2/g. Ее наносят таким же образом на тот же носитель, что и в сравнительном примере 1, так что получают оболочковый катализатор VB3 с долей активной массы 20 вес.%. Пример 3 Как пример 1, однако активную массу из сравнительного примера 3 промывают водной азотной кислотой. Получаемая активная масса имеет следующий состав: Mo1,0V0,29Te0,13Nb0,13Pd0,001Ox. Соответствующая рентгеновская дифрактограмма показана на фиг.7 (R=0,74). БЭТ=17,4 м2/г. Ее наносят таким же образом на тот же носитель, что и в сравнительном примере 1, так что получают оболочковый катализатор В2 с долей активной массы 20 вес.%. Сравнительный пример 4 Как сравнительный пример 1, однако вместо 6,80 г гексагидрата нитрата никеля (II) применяют 3,41 г гексагидрата нитрата кобальта (II) (98%, фирмы Riedel-de-Haen). Получаемая активная масса имеет следующий состав: Mo1,0V0,33Te0,19Nb0,11Co0,005Оx. Соответствующая рентгеновская дифрактограмма показана на фиг.8 (R=0,24). БЭТ=8,9 м2/г. Ее наносят таким же образом на тот же носитель, что и в сравнительном примере 1, так что получают оболочковый катализатор VB4 с долей активной массы 20 вес.%. Пример 4 Как пример 1, однако активную массу из сравнительного примера 4 промывают азотной кислотой. Получаемая активная масса имеет следующий состав: Мо0,1V0,29Те0,13Nb0,13Со0,004Ох. Соответствующая рентгеновская дифрактограмма показана на фиг.9 (R=0,73). БЭТ=24,6 м2/г. Ее наносят таким же образом на тот же носитель, что и в сравнительном примере 1, так что получают оболочковый катализатор В4 с долей активной массы 20 вес.%. Сравнительный пример 5 Как сравнительный пример 1, однако вместо 6,80 г гексагидрата нитрата никеля (II) применяют 5,65 г тригидрата нитрата меди (II) (99%, фирмы Acres Organics). Получаемая активная масса имеет следующий состав: Mo1,0V0,33Te0,19Nb0,11Cu0,01Ox. Соответствующая рентгеновская дифрактограмма показана на фиг.10 (R=0,27). БЭТ=6,7 м2/г. Ее наносят таким же образом на тот же носитель, что и в сравнительном примере 1, так что получают оболочковый катализатор VB5 с долей активной массы 20 вес.%. Пример 5 Как пример 1, только активную массу из сравнительного примера 5 промывают водной азотной кислотой. Получаемая активная масса имеет следующий состав: Мо1,0V0,28Те0,13Nb0,13Cu0,003Ох. Соответствующая рентгеновская дифрактограмма показана на фиг.11 (R=0,74). БЭТ=23,1 м2/г. Ее наносят таким же образом на тот же носитель, что и в сравнительном примере 1, так что получают оболочковый катализатор В5 с долей активной массы 20 вес.%. Сравнительный пример 6 Как сравнительный пример 1, только вместо 6,80 г гексагидрата нитрата никеля (II) применяют 5,68 г пентагидрата нитрата висмута (III) (98,5%, фирмы Merck). Получаемая активная масса имеет следующий состав: Mo1,0V0,33Te0,19Nb0,11Bi0,004Оx. Соответствующая рентгеновская дифрактограмма показана на фиг.12 (R=0,18). БЭТ=9,0 м2/г. Ее наносят таким же образом на тот же носитель, что и в сравнительном примере 1, так что получают оболочковый катализатор VB6 с доли активной массы 20 вес.%. Пример 6 Как пример 1, однако активную массу из сравнительного примера 6 промывают водной азотной кислотой. Получаемая активная масса имеет следующий состав: Mo1,0V0,28Te0,15Nb0,14Bi0,005Оx. Соответствующая рентгеновская дифрактограмма показана на фиг.13 (R=0,70). BET=22,0 m2/g. Ее наносят таким же образом на тот же носитель, что и в сравнительном примере 1, так что получают оболочковый катализатор В6 с долей активной массы 20 вес.%. Сравнительный пример 7 Как сравнительный пример 1, только вместо 6,80 г гексагидрата нитрата никеля применяют 3,84 г нитрата свинца (II) (фирмы Riedel-de-Haen, 99%). Получаемая активная масса имеет следующий состав: Mo1,0V0,34Te0,18Nb0,11Pb0,004. Соответствующая рентгеновская дифрактограмма показана на фиг.14 (R=0,30). БЭТ=2,2 м2/г. Ее наносят таким же образом на тот же носитель, что и в сравнительном примере 1, так что получают оболочковый катализатор VB7 с долей активной массы 20 вес.%. Пример 7 Как пример 1, только активную массу из сравнительного примера 7 промывают водной азотной кислотой. Получаемая активная масса имеет следующий состав: Mo1,0V0,28Te0,13Nb0,13Pb0,001Ox. Соответствующая рентгеновская дифрактограмма показана на 15 (R=0,67). БЭТ=27,1 м2/г. Ее наносят таким же образом на тот же носитель, что и в сравнительном примере 1, так что получают оболочковый катализатор В7 с долей активной массы 20 вес.%. Сравнительный пример 8 Как сравнительный пример 1, однако с той разницей, что не производят добавки 5,60 г гексагидрата нитрата никеля (II). Получаемая активная масса имеет следующий состав: Mo1,0V0,33Te0,16Nb0,11Ox. Соответствующая рентгеновская дифрактограмма показана на фиг.16 (R=0,26). БЭТ=6,7 м2/г. Ее наносят таким же образом на тот же носитель, что и в сравнительном примере 1, так что получают оболочковый катализатор VB8 с долей активной массы 20 вес.%. Сравнительный пример 9 Как пример 1, только активную массу из сравнительного примера 7 промывают водной азотной кислотой. Получаемая активная масса имеет следующий состав: Мо1,0V0,29Те0,13Nb0,13Ох. Соответствующая рентгеновская дифрактограмма показана на фиг.17 (R=0,68). Удельная поверхность про БЭТ=26,0 м2/г. Ее наносят таким же образом на тот же носитель, что и в сравнительном примере 1, так что получают оболочковый катализатор VB9 с долей активной массы 20 вес.%. В) Испытание полученных в А) оболочковых катализаторов на основе масс оксидов металлов По 35,0 г каждого оболочкового катализатора из А) загружают в изготовленный из стали трубчатый реактор (внутренний диаметр: 8,5 мм, длина: 140 см, толщина стенки: 2,5 см) (длина катализаторной засыпки во всех случаях прибл. 53 см). Перед катализаторной засыпкой (слоем) размещают засыпку в 30 см из шариков стеатита (диаметр: от 2,2 до 3,2 мм, изготовитель фирма Ceramtec) и после катализаторной засыпки по остаточной длине трубчатого катализатора размещают дополнительную засыпку из таких же стеатитных шариков. С помощью электрически нагреваемых матов внешнюю температуру загруженной реакционной трубы устанавливают по всей длине на 350°С. Потом реакционную трубу загружают реакционной газовой смесью молярного состава пропан:воздух:Н2О = 1:15:14 (место ввода смеси со стороны дополнительной засыпки). Время пребывания (в пересчете на объем катализаторной засыпки) устанавливают на 2,4 сек. Входное давление составляет 2 абс. бар. Загрузку реакционной трубы производят сначала при вышеприведенной внешней температуре загруженной реакционной трубы в течение 24 часов, прежде чем внешнюю температуру повышают настолько, что в пересчете на один проход реакционной трубы во всех случаях получают конверсию пропана (UPAN) прибл. 78 мол.%. Нижеприведенная таблица показывает в зависимости от примененного оболочкового (нанесенного) катализатора необходимую для этой конверсии внешнюю температуру Т (°С), а также получаемую при этом селективность образования акриловой кислоты (SACS (мол.%)) и селективность образования побочного продукта пропена (SPEN (мол.%)). Дополнительно таблица показывает соотношение интенсивности R находящейся на оболочковом катализаторе активной массы и состав этой активной массы.
Формула изобретения
1. Масса оксидов металлов, предназначенная как катализатор для гетерогенно-катализируемого частичного окисления и/или аммокисления, по меньшей мере, одного насыщенного и/или ненасыщенного углеводорода, общей стехиометрии I 2. Масса оксидов металлов по п.1, отличающаяся тем, что 0,67 3. Масса оксидов металлов по п.1, отличающаяся тем, что 0,69 4. Масса оксидов металлов по п.1, отличающаяся тем, что 0,71 5. Масса оксидов металлов по п.1, отличающаяся тем, что R=0,72. 6. Масса оксидов металлов по п.1, отличающаяся тем, что ее удельная поверхность составляет от 11 до 40 м2/г. 7. Масса оксидов металлов по п.1, отличающаяся тем, что ее рентгеновская дифрактограмма имеет еще другие дифракционные рефлексы с пиком под следующими углами дифракции 2 8. Масса оксидов металлов по п.7, отличающаяся тем, что ее рентгеновская дифрактограмма имеет еще другие дифракционные рефлексы с пиком под следующими углами дифракции 2 9. Масса оксидов металлов по п.8, отличающаяся тем, что интенсивность дифракционных рефлексов h, i, l, m, n, о, р, q по одинаковой шкале интенсивности имеет следующие значения: 10. Масса оксидов металлов по п.1, отличающаяся тем, что а=0,1 до 0,6; 11. Масса оксидов металлов по п.1, отличающаяся тем, что М1=Те, М2=Nb и М3 означает, по меньшей мере, один элемент из группы, включающей Ni, Co и Pd. 12. Масса оксидов металлов, которая содержит равно или больше 80 вес.%, по меньшей мере, одной массы оксидов металлов по одному из пп.1-11 и рентгеновская дифрактограмма которой имеет дифракционный рефлекс с пиком 2 13. Способ гетерогенно-катализируемого частичного газофазного окисления, по меньшей мере, одного насыщенного или ненасыщенного углеводорода, отличающийся тем, что в качестве каталитической активной массы применяют, по меньшей мере, одну массу оксидов металлов по одному из пп.1-12. 14. Способ по п.13, отличающийся тем, что углеводородом является пропан, пропен или смесь пропана и пропена. 15. Способ гетерогенно-катализируемого частичного газофазного аммокисления, по меньшей мере, одного насыщенного или ненасыщенного углеводорода, отличающийся тем, что в качестве каталитической активной массы применяют, по меньшей мере, одну массу оксидов металлов по одному из пп.1-12. 16. Способ по п.15, отличающийся тем, что углеводородом является пропан, пропен или смесь пропана и пропена. 17. Способ получения массы оксидов металлов по одному из пп.1-11, отличающийся тем, что из источников ее элементарных компонентов получают хорошо перемешанную сухую смесь, эту смесь кальцинируют при температуре от 350 до 700°С и получающийся при этом продукт промывают водным раствором органической и/или неорганической кислоты.
РИСУНКИ
TH4A – Переиздание описания изобретения к патенту Российской Федерации
Причина переиздания: Коррекция в текстах реферата и описания изобретения
Извещение опубликовано: 20.05.2010 БИ: 14/2010
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
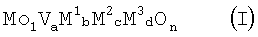 ,
, ) 22,2±0,5° (h), 27,3±0,5° (i) и 28,2±0,5° (k), причем
) 22,2±0,5° (h), 27,3±0,5° (i) и 28,2±0,5° (k), причем R
R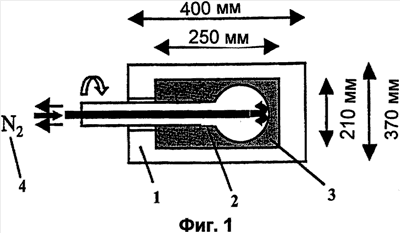
 Ammoxidation of propane over Mo-V-Nb-Te mixed oxide catalysts” из
Ammoxidation of propane over Mo-V-Nb-Te mixed oxide catalysts” из  -излучения в качестве рентгеновского излучения рентгеновской дифрактограммы (дифрактометр фирмы Сименс Siemens Theta-Theta D-5000, напряжение трубки: 40 кВ, ток в трубке: 40 мА, апертурная диафрагма V20 (варьируемая), диафрагма рассеянного излучения V20 (варьируемая), вторичного монохроматора (0,1 мм), диафрагма детектора (0,6 мм), измерительный интервал (2
-излучения в качестве рентгеновского излучения рентгеновской дифрактограммы (дифрактометр фирмы Сименс Siemens Theta-Theta D-5000, напряжение трубки: 40 кВ, ток в трубке: 40 мА, апертурная диафрагма V20 (варьируемая), диафрагма рассеянного излучения V20 (варьируемая), вторичного монохроматора (0,1 мм), диафрагма детектора (0,6 мм), измерительный интервал (2 , которая отличается от масс оксидов металлов (I) только тем, что в ней d=0.
, которая отличается от масс оксидов металлов (I) только тем, что в ней d=0.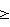 D;
D;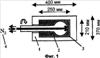



{kind=link}