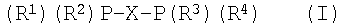Патент на изобретение №2352389
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) КАТАЛИТИЧЕСКАЯ ТРИМЕРИЗАЦИЯ ОЛЕФИНОВЫХ МОНОМЕРОВ
(57) Реферат:
Настоящее изобретение относится к катализатору тримеризации олефиновых мономеров. Описана каталитическая композиция для тримеризации олефиновых мономеров, содержащая а) источник хрома, молибдена или вольфрама; b) лиганд общей формулы (I), в которой Х означает бивалентную органическую мостиковую группу, выбранную из замещенных или незамещенных алкиленовых групп, причем в случае замещенных групп заместители представляют собой углеводородные группы; R1 и R3 представляют собой циклоароматические группы, которые не содержат полярных заместителей ни в одном из орто-положений; R2 и R4 независимо выбирают из необязательно замещенных циклоароматических групп, причем каждый из R2 и R4 имеет полярный заместитель, по меньшей мере, в одном орто-положений; и с) сокатализатор. Также описан способ тримеризации олефиновых мономеров, включающий взаимодействие, по меньшей мере, одного олефинового мономера в условиях реакции тримеризации с указанной выше каталитической композицией. Технический результат – селективное получение 1-гексена из этилена при снижении уровня образования побочных продуктов, особенно С10. 2 н. и 8 з.п. ф-лы, 2 табл.
Область техники, к которой относится изобретение Настоящее изобретение относится к катализатору тримеризации олефиновых мономеров. Настоящее изобретение также относится к способу тримеризации олефиновых мономеров, в частности к получению 1-гексена из этилена в присутствии упомянутого катализатора. Уровень техники изобретения Эффективная каталитическая тримеризация олефиновых мономеров, такая как тримеризация этилена в 1-гексен, представляет собой область повышенного интереса для получения олефиновых тримеров различных степеней коммерческой ценности. В частности, 1-гексен представляет собой ценный сомономер для линейного полиэтилена низкой плотности (ЛПЭНП). 1-гексен можно также получать обычным способом олигомеризации с помощью переходного металла, хотя путь тримеризации более предпочтителен, так как он в большой степени избегает образования нежелательных олефинов. В известном уровне техники описано несколько различных каталитических систем для тримеризации этилена в 1-гексен. Ряд указанных катализаторов основан на хроме. US-А-5198563 (Phillips) описывает катализаторы на основе хрома, содержащие монодентатные аминовые лиганды, пригодные для тримеризации олефинов. US-А-5968866 (Phillips) описывает способ олигомеризации/тримеризации этилена, который использует катализатор, содержащий комплекс хрома, который содержит координирующий асимметричный тридентатный фосфановый, арсановый или стибановый лиганд и алюмоксан, для получения альфа-олефинов, которые обогащены 1-гексеном. Патент США 5523507 (Phillips) описывает катализатор на основе источника хрома, 2,5-диметилпиррольного лиганда и алкилалюминиевого активатора для применения в тримеризации этилена в 1-гексен. Chem. Commun., 2002, 8, 858-859 (BP) описывает комплексы хрома с лигандами типа Ar2PN(Me)PAr2 (Ar=орто-метокси-замещенная арильная группа) в качестве катализаторов тримеризации этилена. WO 02/04119 (BP) описывает катализатор тримеризации олефинов, содержащий источник хрома, молибдена или вольфрама, лиганд, содержащий, по меньшей мере, один атом фосфора, мышьяка или сурьмы, связанный, по меньшей мере, с одной углеводородной или гетероуглеводородной группой, имеющей полярный заместитель, но исключая случай, когда все такие полярные заместители представляют собой фосфановые, арсановые или стибановые группы и, возможно, активатор. Лиганд, используемый в большинстве примеров, представляет собой (2-метоксифенил)2PN(Me)P(2-метоксифенил)2. Хотя катализаторы, описанные в упомянутых документах ВР, имеют хорошую селективность по 1-гексену внутри фракции С6, наблюдается относительно высокий уровень образования побочных продуктов (например, деценов). Поэтому было бы желательно предложить катализатор тримеризации олефиновых мономеров, особенно для тримеризации этилена в 1-гексен, который снижает образование побочных продуктов (например, деценов) при сохранении селективности по 1-гексену. Неожиданно было обнаружено, что композиции катализаторов и способы по настоящему изобретению обеспечивают эффективный путь селективного получения 1-гексена из этилена при снижении уровня образования побочных продуктов, особенно С10. Сущность изобретения Согласно одному аспекту настоящего изобретения предлагается композиция катализатора для тримеризации олефиновых мономеров, которая содержит а) источник хрома, молибдена или вольфрама; b) лиганд общей формулы (I):
в которой Х означает двухвалентную органическую мостиковую группу, содержащую от 1 до 10 атомов углерода в мостике; R1 и R3 независимо выбирают из углеводородных, замещенных углеводородных, гетероуглеводородных и замещенных гетероуглеводородных групп с условием, что, когда R1 и R3 означают циклоароматические группы, они не содержат полярных заместителей в любом из орто-положений; R2 и R4 независимо выбирают из необязательно замещенных циклоароматических групп, причем каждая R2 и R4 имеет полярный заместитель в, по меньшей мере, одном из орто-положений; и с) сокатализатор. Согласно дополнительному аспекту настоящего изобретения предлагается способ тримеризации олефиновых мономеров, включающий взаимодействие, по меньшей мере, одного олефинового мономера в условиях реакции тримеризации с упомянутой композицией катализатора. Композиции катализаторов настоящего изобретения особенно подходят для тримеризации олефиновых мономеров, особенно для тримеризации этилена в 1-гексен. Композиции катализаторов и способ по настоящему изобретению неожиданно дают, по существу, низкие концентрации побочных олефиновых продуктов (например, деценов, преимущественно 1-децена, который получается добавлением двух этиленовых мономеров к полученному 1-гексену) при сохранении высокой селективности по 1-гексену. Кроме того, композиции катализаторов по настоящему изобретению демонстрируют улучшенные профили скорости активности/затухания по сравнению с Cr(III)(2-метоксифенил)2PN(Me)P(2-метоксифенил)2 катализаторами, описанными в WO 02/04119, упомянутом выше. В частности, композиции катализаторов по настоящему изобретению показывают хорошую начальную активность, но менее быстрое затухание, чем Cr(III)(2-метоксифенил)2PN(Me)P(2-метоксифенил)2 катализаторы. Подробное описание изобретения Используемый в данном описании термин “тримеризация” означает каталитическую тримеризацию олефиновых мономеров с получением композиции продукта, обогащенной соединением, полученным по реакции трех упомянутых олефиновых мономеров. Термин тримеризация включает в себя случаи, когда все олефиновые мономеры в потоке сырья идентичны, как и случаи, когда поток сырья содержит два или более разных олефиновых мономеров. В частности, термин “тримеризация”, когда используется в отношении тримеризации этилена, означает тримеризацию этилена с образованием С6алкена, особенно 1-гексена. Термин “селективность тримеризации”, когда используется в отношении тримеризации этилена в 1-гексен, означает количество полученной С6 фракции в композиции продукта. Термин “селективность по 1-гексену”, когда используется в отношении тримеризации этилена в 1-гексен, означает количество полученного 1-гексена в композиции продукта. Общий выход 1-гексена в тримеризации этилена представляет собой результат умножения “селективности тримеризации” на “селективность по 1-гексену”. Композиция катализатора по настоящему изобретению содержит а) источник хрома, молибдена или вольфрама; b) лиганд; с) сокатализатор. Каждый из указанных трех существенных компонентов подробно описан ниже. Источник хрома, молибдена или вольфрама, компонент (а), для композиции катализатора может включать в себя простые неорганические или органические соли хрома, молибдена или вольфрама. Примеры простых неорганических или органических солей представляют собой галогениды, ацетилацетонаты, карбоксилаты, оксиды, нитраты, сульфаты и подобное. Дополнительные источники хрома, молибдена или вольфрама могут также включать в себя координационные или металлоорганические комплексы, например комплекс трихлорида хрома с тетрагидрофураном, (бензол)трикарбонилхром, гексакарбонилхром и подобное. Источник хрома, молибдена или вольфрама может также включать в себя смесь простых неорганических солей, простых органических солей, координационных комплексов и металлорганических комплексов. В предпочтительных вариантах осуществления данного изобретения компонент (а) представляет собой хром, особенно хром (III). Предпочтительные источники хрома для использования в данном изобретении представляют собой неорганические и органические соли хрома. Более предпочтительные источники хрома для использования в данном изобретении представляют собой галогениды хрома, такие как хлорид хрома, бромид хрома, фторид хрома и иодид хрома. Особенно предпочтительный источник хрома для использования в данном изобретении представляет собой хлорид хрома, CrCl3. Лиганд композиции катализатора по настоящему изобретению, компонент (b), имеет общую формулу (I):
в которой Х означает двухвалентную органическую мостиковую группу, содержащую от 1 до 10 атомов углерода в мостике; R1 и R3 независимо выбирают из углеводородных, замещенных углеводородных, гетероуглеводородных и замещенных гетероуглеводородых групп с условием, что, когда R1 и R3 представляют собой циклоароматические группы, они не содержат полярных заместителей в любом из орто-положений; R2 и R4 независимо выбирают из необязательно замещенных циклоароматических групп, причем каждая R2 и R4 имеет полярный заместитель в, по меньшей мере, одном из орто-положений. В общей формуле (I) Х обозначает двухвалентную органическую мостиковую группу, содержащую от 1 до 10, предпочтительно от 2 до 6, более предпочтительно от 2 до 4 и особенно от 2 до 3 атомов углерода в мостике. Предпочтительный вариант осуществления имеет 2 атома углерода в мостике. Под выражением “в мостике” понимают наиболее короткое соединение между двумя атомами фосфора. Подходящие мостиковые группы включают в себя замещенные и незамещенные алкиленовые группы. Алкиленовые группы могут возможно содержать один или несколько гетероатомов в мостике, таких как N, S, Si или О. Предпочтительно, если алкиленовая группа содержит только атомы углерода в мостике. Алкиленовые группы могут быть замещены одним или несколькими заместителями. Заместители могут быть присоединены к любой части соединения. Заместители алкиленовой мостиковой группы могут содержать атомы углерода и/или гетероатомы. Подходящие заместители включают в себя углеводородные группы, которые могут быть линейными или разветвленными, насыщенными или ненасыщенными, ароматическими или неароматическими. Углеводородные заместители могут содержать гетероатомы, такие как Si, S, N или О. Подходящие ароматические углеводородные заместители включают в себя циклоароматические группы, предпочтительно имеющие от 5 до 10 атомов углерода в цикле, такие как фенильная и С1-С4алкилфенильная группы. Подходящие неароматические углеводородные заместители включают в себя линейные или разветвленные, алкильные или циклоалкильные группы, предпочтительно имеющие от 1 до 10 атомов углерода, более предпочтительно от 1 до 4 атомов углерода. Другие подходящие заместители алкиленовой мостиковой группы включают в себя галогениды, такие как хлорид, бромид и иодид, тиол, -ОН, А1-О-, -S-A1, -CO-A1, -NH2, -NHA1, -NA1A2, -CO-NA1A2, -PO4, -NO2, -CO, -SO2, в которых А1 и А2 независимо представляют собой неароматические группы, предпочтительно имеющие от 1 до 10 атомов углерода, более предпочтительно от 1 до 4 атомов углерода, например метил, этил, пропил и изопропил. Когда алкиленовая мостиковая группа замещена, предпочтительные заместители представляют собой углеводородные группы. Особенно предпочтительные углеводородные заместители представляют собой С1-С4алкильные группы, предпочтительно метил, этил, пропил, изопропил, бутил, изобутил, наиболее предпочтительно метил. Примеры незамещенных алкиленовых мостиковых групп включают в себя метиленовую, этиленовую и триметиленовую группы. Примеры замещенных алкиленовых мостиковых групп включают в себя 2,2-диметил-триметилен, 2,2-диэтил-триметилен, 2,2-диметил-тетраметилен, 2-метил, 2-гидроксиметил-триметилен и 2,2-ди-гидроксиметил-триметилен. Особенно предпочтительные органические алкиленовые мостиковые группы для использования в данном изобретении представляют собой незамещенные алкиленовые мостиковые группы. Особенно предпочтительная органическая мостиковая группа представляет собой этилен, то есть -СН2-СН2-. Другие подходящие мостиковые группы представляют собой те, где соединение образует часть неароматической или ароматической кольцевой структуры. Такие мостиковые группы содержат одну или несколько замещенных или незамещенных, насыщенных или ненасыщенных неароматических кольцевых структур и/или одну или несколько замещенных или незамещенных циклоароматических (включая гетероароматические) кольцевых структур. Неароматическая кольцевая структура может прерываться одним или несколькими гетероатомами, такими как N, S, Si или О. Предпочтительно такая мостиковая группа также содержит от 2 до 6 атомов углерода в мостике. Подходящие неароматические кольцевые структуры включают в себя циклопентан, циклогексан, циклогексен, циклопентен, 3,4-фуран и 3,4-тиофен. Подходящие ароматические кольцевые структуры включают в себя фенилен, особенно 1,2-фенилены и нафтилен, особенно 1,8- или 1,2-нафтилены. Указанные кольцевые структуры могут быть замещены любым типом заместителя, включая гетероатомы, алкильные группы, циклоалкильные группы и циклоароматические группы. Подходящие заместители включают в себя заместители, упомянутые выше в отношении алкиленовых мостиковых групп. Предпочтительно, когда два атома фосфора добавлены к циклической системе в соседних положениях, то есть положениях 1 и 2. R1 и R3 независимо выбирают из углеводородных, замещенных углеводородных, гетероуглеводородных и замещенных гетероуглеводородных групп при условии, что, когда R1 и R3 представляют собой циклоароматические группы, они не содержат полярных заместителей в любом из орто-положений. Используемый в данном описании термин “углеводородный” относится к группе, содержащей атомы только углерода и водорода. Углеводородная группа может быть насыщенной или ненасыщенной, линейной или разветвленной, неароматическим кольцом или циклоароматическим кольцом. Предпочтительные для использования в данном изобретении углеводородные группы содержат от 1 до 20 атомов углерода. Используемый в данном описании термин “замещенный углеводородный” относится к углеводородным группам, которые содержат одну или несколько функциональных групп, содержащих инертные гетероатомы. Под “функциональными группами, содержащими инертные гетероатомы”, понимают, что данные функциональные группы не мешают в какой-либо существенной степени процессу тримеризации. Используемый в данном описании термин “гетероуглеводородный” относится к углеводородной группе, в которой один или несколько из атомов углерода замещен гетероатомом, таким как S, N или О. Используемый в данном описании термин “замещенный гетероуглеводородный” относится к гетероуглеводородным группам, которые содержат одну или несколько функциональных групп, содержащих инертные гетероатомы. Используемый в данном описании термин “циклоароматический” относится к моноциклическому или полициклическому, ароматическому или гетероароматическому кольцу, имеющему от 5 до 14 кольцевых атомов, необязательно содержащему от 1 до 3 гетероатомов, выбранных среди N, O и S. Циклоароматические группы предпочтительно являются моноциклическими или полициклическими ароматическими кольцами, такими как циклопентадиенил, фенил, нафтил или антраценил. Еще более предпочтительные циклоароматические группы представляют собой моноциклические или полициклические ароматические кольца, имеющие от 5 до 10 кольцевых атомов. Особенно предпочтительные циклоароматические группы представляют собой моноциклические ароматические кольца, содержащие от 5 до 6 атомов углерода, таких как фенил и циклопентадиенил, и наиболее предпочтительная циклоароматическая группа представляет собой фенильную группу. Используемый в данном описании термин “замещенный циклоароматический” означает, что циклоароматическая группа может быть замещена одним или несколькими заместителями. Подходящие заместители включают в себя заместители, упомянутые выше в отношении алкиленовых мостиковых групп. В одном предпочтительном варианте осуществления R1 и R3 независимо выбирают из замещенных или незамещенных циклоароматических групп, которые не содержат полярного заместителя в любом из орто-положений. В еще более предпочтительном варианте осуществления R1 и R3 независимо выбирают из необязательно замещенных фенильных групп, которые не содержат полярного заместителя в любом из орто-положений. В особенно предпочтительном варианте осуществления R1 и R3 представляют собой незамещенные фенильные группы. Предпочтительно, чтобы R1 и R3 группы были одинаковыми. R2 и R4 независимо выбирают из необязательно замещенных циклоароматических групп, причем каждая R2 и R4 группа имеет полярный заместитель, по меньшей мере, в одном из орто-положений. Во избежание неопределенности фраза “каждая R2 и R4 группа имеет полярный заместитель, по меньшей мере, в одном из орто-положений” означает, что в том же лиганде R2 замещена полярным заместителем в одном или обоих из ее орто-положений и R4 замещена полярным заместителем в одном или обоих из ее орто-положений. Термин “возможно замещенный” в отношении R2 и R4 означает, что, в добавление к полярному заместителю, по меньшей мере, в одном из орто-положений, R2 и R4 группы могут содержать один или несколько заместителей. Подходящие заместители включают в себя заместители, упомянутые выше в отношении алкиленовых мостиковых групп. Предпочтительно, если R2 и R4 независимо выбирают из необязательно замещенных циклоароматических групп, имеющих от 5 до 14 кольцевых атомов, предпочтительно от 5 до 10 кольцевых атомов, причем каждая из R2 и R4 имеет полярный заместитель, по меньшей мере, в одном из орто-положений. В одном предпочтительном варианте осуществления R2 и R4 независимо выбирают из возможно замещенных фенильных групп, причем каждая из R2 и R4 имеет полярный заместитель, по меньшей мере, в одном из орто-положений. Предпочтительно, если каждая из R2 и R4 имеет полярный заместитель, по меньшей мере, в одном из двух орто-положений. Используемый в данном описании термин “полярные заместители” означает заместитель, который включает в себя электроотрицательный центр. Подходящие для использования в данном изобретении полярные заместители включают в себя, но не ограничены ими, необязательно разветвленные С1-С20 алкоксигруппы, т.е. углеводородные группы, связанные с R2 и R4 циклоароматическим кольцом через мостиковый атом кислорода; необязательно замещенные С5-С14арилоксигруппы, т.е. необязательно замещенные циклоароматические группы, связанные с R2 и R4 циклоароматическим кольцом через мостиковый атом кислорода; необязательно разветвленные С1-С20алкил(С1-С20)алкоксигруппы, т.е. С1-С20углеводородные группы, имеющие С1-С20алкоксигруппу; гидроксил; амино; (ди-)С1-С6алкиламино; нитро; С1-С6алкилсульфанил; С1-С6алкилтиоС1-С6алкильные группы; и тозильные группы. Примеры подходящих полярных заместителей включают в себя метокси, этокси, изопропокси, фенокси, пентафторфенокси, триметилсилокси, диметиламино, метилсульфанил, тозил, метоксиметил, метилтиометил, 1,3-оксазолил, гидроксил, амино, сульфат, нитро и подобное. Предпочтительно, если полярные заместители для R2 и R4 независимо выбирают из необязательно разветвленных С1-С20алкоксигрупп, необязательно замещенных С5-С14арилоксигрупп и необязательно разветвленных С1-С20алкил(С1-С20)алкоксигрупп. Более предпочтительно, если полярные заместители для R2 и R4 независимо выбирают из необязательно разветвленных С1-С20алкоксигрупп, особенно необязательно разветвленных С1-С6алкоксигрупп, таких как, например, метокси, этокси или изопропокси. Особенно предпочтительным полярным заместителем для R2 и R4 является метокси. Предпочтительно, когда R2 и R4 группы одинаковы и имеют одинаковое число и тип полярных заместителей. Особенно предпочтительно, если R2 имеет только один полярный заместитель в одном из двух ее орто-положений и если R4 имеет только один полярный заместитель в одном из двух ее орто-положений. Лиганды согласно формуле (I) можно приготовить, используя методики, известные специалистам в данной области техники или описанные в опубликованной литературе. Примеры таких соединений представляют собой (2-метоксифенил)(фенил)РСН2СН2Р(2-метоксифенил)(фенил), (2-метоксифенил)(фенил)РСН2Р(2-метоксифенил)(фенил), (2-метоксифенил)(фенил)РСН2СН2СН2Р(2-метоксифенил)(фенил), (2-этоксифенил)(фенил)РСН2СН2Р(2-этоксифенил)(фенил), (2-этоксифенил)(фенил)РСН2Р(2-этоксифенил)(фенил), (2-этоксифенил)(фенил)РСН2СН2СН2Р(2-этоксифенил)(фенил), (2-изопропоксифенил)(фенил)РСН2СН2Р(2-изопропоксифенил)(фенил), (2-изопропоксифенил)(фенил)РСН2Р(2-изопропоксифенил)(фенил), (2-изопропоксифенил)(фенил)РСН2СН2СН2Р(2-изопропоксифенил)(фенил). Особенно предпочтительный лиганд для использования в данном изобретении представляет собой (2-метоксифенил)(фенил)РСН2СН2Р(2-метоксифенил)(фенил). Источник хрома, молибдена или вольфрама, компонент (а), и лиганд, компонент (b), могут присутствовать в композиции катализатора по настоящему изобретению в соотношении в диапазоне от 10000:1 до 1:10000, предпочтительно от 100:1 до 1:100, более предпочтительно от 10:1 до 1:10. Наиболее предпочтительно компоненты (а) и (b) присутствуют в соотношении в диапазоне от 3:1 до 1:3. Обычно количества (а) и (b) приблизительно равны, т.е. соотношение в диапазоне от 1,5:1 до 1:1,5. Сокатализатор, компонент (с), может в принципе быть любым соединением или смесью соединений, которая образует активный катализатор с источником хрома, молибдена или вольфрама, компонентом (а) и лигандом, компонентом (b). Соединения, которые подходят в качестве сокатализатора, включают в себя алюмоорганические соединения, борорганические соединения и неорганические кислоты и соли, такие как эфират тетрафторборной кислоты, тетрафторборат серебра, гексафторантимонат натрия и подобное. Особенно предпочтительные сокатализаторы представляют собой алюмоорганические соединения. Подходящие для использования в данном изобретении алюмоорганические соединения представляют собой соединения, имеющие формулу AlR3, в которой каждую R группу независимо выбирают из С1-С30алкила, кислорода или галогенидов, или соединений, таких как LiAlH4 и подобного. Неограничивающие примеры подходящих алюмоорганических соединений включают в себя триметилалюминий (ТМА), триэтилалюминий (ТЭА), триизобутилалюминий (ТИБА), три-н-октилалюминий, дихлорид метилалюминия, дихлорид этилалюминия, хлорид диметилалюминия, хлорид диэтилалюминия и алюмоксаны. Смеси алюмоорганических соединений также пригодны для использования в данном изобретении. В предпочтительном варианте осуществления данного изобретения сокатализатор представляет собой алюмоксановый сокатализатор. Указанные алюмоксановые сокатализаторы могут содержать любое алюмоксановое соединение или смесь алюмоксановых соединений. Алюмоксаны можно приготовить регулируемым добавлением воды к алкилалюминиевому соединению, такому как соединения, упомянутые выше или коммерчески доступные. В данном контексте следует отметить, что термин “алюмоксан”, используемый в данном описании, включает в себя коммерчески доступные алюмоксаны, которые могут содержать долю, обычно примерно 10 мас.%, но возможно до 50 мас.%, соответствующего триалкилалюминия. Например, коммерческий метилалюмоксан (МАО) обычно содержит приблизительно 10 мас.% триметилалюминия (ТМА), тогда как модифицированный метилалюмоксан (ММАО) содержит как ТМА, так и триизобутиллюминий (ТИБА). Молярное отношение воды к соединению алюминия при приготовлении алюмоксанов предпочтительно лежит в диапазоне от 0,01:1 до 2,0:1, более предпочтительно от 0,02:1 до 1,2:1, еще более предпочтительно от 0,4:1 до 1:1, особенно 0,5:1. Указанные алюмоксановые соединения могут быть линейными, циклическими каркасами или их смесями. Предпочтительные алюмоксаны представляют собой линейные алюмоксаны формулы R5(R6AlO)n, в которой n обозначает число от примерно 2 до 50 и R5 и R6 обозначают алкильные группы от С1 до С6. Наиболее предпочтительные алюмоксаны представляют собой метилалюмоксан (МАО) или модифицированный метилалюмоксан (ММАО), который содержит как ТМА, так и ТИБА. Другие подходящие сокатализаторы включают в себя сокатализаторы, описанные в WO 02/04119. Количество сокатализатора, используемого в настоящем изобретении, обычно достаточно для обеспечения соотношения в диапазоне от 0,1 до 20000, предпочтительно от 1 до 2000, атомов алюминия или бора на атом хрома, молибдена или вольфрама. Композиция катализатора по настоящему изобретению может также быть смешана, по меньшей мере, с одним другим катализатором тримеризации. Три существенных компонента катализатора (а), (b) и (с) могут добавляться вместе одновременно или последовательно в любом порядке так, чтобы предоставить активный катализатор. Три существенных компонента катализатора могут взаимодействовать в присутствии любого подходящего растворителя. Подходящие растворители известны специалистам в данной области техники. Примерами подходящих растворителей являются растворители, описанные в WO 02/04119. Композицию катализатора по настоящему изобретению можно готовить либо в присутствии (т.е. “in-situ”), либо в отсутствие олефинового мономера. Три существенных компонента композиции катализатора можно объединять полностью в отсутствие олефинового мономера, или олефиновый мономер можно добавлять до взаимодействия компонентов катализатора, одновременно с компонентами катализатора или в любой момент процесса взаимодействия компонентов катализатора. Три существенных компонента катализатора могут быть не нанесенными или нанесенными на материал носителя. Примеры подходящих материалов носителя можно найти в WO 02/04119. Олефиновые мономеры, подходящие для использования в способе тримеризации по настоящему изобретению, могут быть любыми олефиновыми мономерами, которые можно превращать в тример. Подходящие олефиновые мономеры включают в себя, но не ограничены, этилен, пропилен, необязательно разветвленные С4-С20 Смеси олефиновых мономеров также можно использовать в способе по настоящему изобретению. Предпочтительные олефиновые мономеры для использования в способе тримеризации по настоящему изобретению представляют собой пропилен и этилен. Особенно предпочтителен этилен. Композиции катализатора и способ по настоящему изобретению особенно пригодны для тримеризации этилена в 1-гексен. Способ тримеризации по настоящему изобретению можно осуществлять в широком диапазоне условий способа, известных специалистам в данной области техники или описанных в опубликованной литературе, таких как, например, условия, описанные в WO 02/04119. Реакцию тримеризации можно осуществлять в растворе, суспензии, газовой фазе или в массе. Когда тримеризацию проводят в растворе или суспензии, можно применять разбавитель или растворитель, который, по существу, инертен в условиях тримеризации. Подходящие разбавители или растворители представляют собой алифатические и ароматические углеводороды, галогенированные углеводороды и олефины, которые, по существу, инертны в условиях тримеризации, такие как растворители, описанные в WO 02/04119. Способ тримеризации по настоящему изобретению можно осуществлять в широком диапазоне условий способа, которые хорошо известны специалистам в данной области техники. Обычно температура лежит в диапазоне от -100°С до 200°С, предпочтительно от 0°С до 150°С и более предпочтительно от 25°С до 100°С. Обычно давление находится в диапазоне от 0 до 100 бар, предпочтительно от 1 до 50 бар. Способ тримеризации по настоящему изобретению можно осуществлять в любом из ряда подходящих реакторов, которые хорошо известны специалистам в данной области техники. Обычно способ тримеризации по настоящему изобретению проводят в периодическом, полупериодическом или непрерывном режиме. Разделение продукта, реагента и катализатора можно выполнять с помощью любой технологии, известной специалистам в данной области техники, такой как дистилляция, фильтрация, центрифугирование, разделение жидкость/жидкость, экстракция и т.д. Дополнительные детали, касающиеся подходящих условий реакции тримеризации, включая дополнительные детали по реакторам, растворителям, технологиям разделения и подобному, можно найти в WO 02/04119. Применение композиции катализатора и способа по настоящему изобретению для каталитической тримеризации этилена в 1-гексен обеспечивает очень высокую селективность по 1-гексену над всеми другими продуктами, образующимися в данной реакции. Композиция катализатора по настоящему изобретению обеспечивает полный выход 1-гексена за счет тримеризации этилена, который больше чем полный выход 1-гексена за счет тримеризации этилена при использовании эквивалентной композиции катализатора, которая не содержит лиганда такого типа, как описано в настоящем изобретении (но которая, например, содержит лиганд формулы -PN(CH3)P-, как описано в WO 02/04119), в идентичных условиях реакции. Предпочтительно, если композиция катализатора по настоящему изобретению обеспечивает полный выход 1-гексена за счет тримеризации этилена, который до 35% больше чем полный выход 1-гексена за счет тримеризации этилена с использованием эквивалентной композиции катализатора, которая не содержит лиганда такого типа, как описано в настоящем изобретении, в идентичных условиях реакции. Более предпочтительно, если композиция катализатора по настоящему изобретению обеспечит полный выход 1-гексена за счет тримеризации этилена, который, по меньшей мере, на 5% больше чем полный выход 1-гексена за счет тримеризации этилена с использованием эквивалентной композиции катализатора, которая не содержит лиганда такого типа, как описано в настоящем изобретении, в идентичных условиях реакции. Количество 1-гексена, получаемого за счет тримеризации этилена с использованием композиции катализатора по настоящему изобретению, составляет, по меньшей мере, 80 мас.%, предпочтительно, по меньшей мере, 85 мас.%, более предпочтительно, по меньшей мере, 90 мас.% и особенно, по меньшей мере, 95 мас.%, от композиции конечного продукта. Селективность тримеризации (т.е. количество фракции С6 в композиции продукта) для тримеризации этилена с использованием композиции катализатора по настоящему изобретению составляет, по меньшей мере, 80 мас.%. Предпочтительно, если селективность тримеризации для тримеризации этилена с использованием композиции катализатора по настоящему изобретению больше чем селективность тримеризации для получения С6 соединений тримеризацией этилена с использованием эквивалентной композиции катализатора, которая не содержит лиганда такого типа, как описано в настоящем изобретении (но которая, например, содержит лиганд формулы -PN(CH3)P-, как описано в WO 02/04119), в идентичных условиях реакции. Предпочтительно, если селективность тримеризации для тримеризации этилена с использованием композиции катализатора по настоящему изобретению до 40% больше чем селективность тримеризации для тримеризации этилена с использованием эквивалентной композиции катализатора, которая не содержит лиганда такого типа, как описано в настоящем изобретении, в идентичных условиях реакции. Также предпочтительно, что композиция катализатора по настоящему изобретению имеет селективность тримеризации для тримеризации этилена, которая, по меньшей мере, на 5% больше чем селективность тримеризации для тримеризации этилена с использованием эквивалентной композиции катализатора, которая не содержит лиганда такого типа, как описано в настоящем изобретении, в идентичных условиях реакции. Образование побочных С10 соединений при тримеризации этилена с использованием композиции катализатора по настоящему изобретению составляет предпочтительно не больше чем 60% от уровня побочных С10 соединений, образующихся при тримеризации этилена с использованием эквивалентной композиции катализатора, которая не содержит лиганда такого типа, как описано в настоящем изобретении (например, Cr(III)(2-метоксифенил)2)PN(CH3)P(2-метоксифенил)2), при идентичных условиях реакции. Более предпочтительно, когда получение побочных соединений С10 при тримеризации этилена с использованием катализатора по настоящему изобретению не больше чем 50% от уровня побочных соединений С10, полученных при тримеризации этилена с использованием эквивалентной композиции катализатора, которая не содержит лиганда такого типа, как описано в настоящем изобретении, при идентичных условиях реакции. Еще более предпочтительно, когда получение побочных продуктов соединений С10 при тримеризации этилена с использованием катализатора по настоящему изобретению не больше чем 30% от уровня побочных соединений С10, полученных при тримеризации этилена с использованием эквивалентной композиции катализатора, которая не содержит лиганда такого типа, как описано в настоящем изобретении, при идентичных условиях реакции. В особенно предпочтительном варианте осуществления получение побочных соединений С10 при тримеризации этилена с использованием катализатора по настоящему изобретению не больше чем 20% от уровня побочных соединений С10, полученных при тримеризации этилена с использованием эквивалентной композиции катализатора, которая не содержит лиганда такого типа, как описано в настоящем изобретении, при идентичных условиях реакции. Каталитическая композиция и способ по настоящему изобретению иллюстрируются следующими неограничивающими примерами. ПРИМЕРЫ Ряд композиций (композиции 1, 2 и 3), содержащие компонент лиганда и источник хрома, получали для использования в реакциях тримеризации, описанных ниже. Композиция 1 (2-Метоксифенил)(фенил)PCH2CH2P(2-метоксифенил)(фенил) в соотношении 1:1 с CrCl3 (2-метоксифенил)(фенил)PCH2CH2P(2-метоксифенил)(фенил) лиганд получали по следующему способу. В атмосфере азота к раствору о-броманизола (0,54 моль) в пентане (150 мл) медленно прибавляли при постоянном перемешивании раствор н-бутиллития (337 мл, 0,54 моль). Смесь перемешивали в течение ночи, после чего перемешивание прекращали и суспензии давали возможность распределиться. Жидкость декантировали и твердый осадок о-анизиллития промывали пентаном и сушили в высоком вакууме. 0,20 моль о-анизиллития растворяли в диэтиловом эфире (400 мл) и охлаждали до -20°С. К полученному раствору медленно прибавляли при постоянном перемешивании 0,1 моль этилфенилфосфината. Затем раствору давали возможность достичь 25°С, после чего раствор кипятили с обратным холодильником в течение 2 часов. Затем раствору давали возможность охладиться, после чего прибавляли 0,1М соляную кислоту (150 мл). Продукт экстрагировали тремя порциями по 50 мл дихлорметана. Объединенные органические слои сушили с использованием сульфата магния. Растворители затем удаляли с получением масла и затем избыток анизола удаляли при нагревании (70°С) в вакууме. Последние следы анизола удаляли промыванием полученного белого осадка ((2-метоксифенил)(фенил)фосфиноксид) диэтиловым эфиром с последующей кристаллизацией из смеси хлороформ/диэтиловый эфир. 40 ммоль (2-метоксифенил)(фенил)фосфиноксида прибавляли к тетрагидрофурану (600 мл) и к полученному раствору прибавляли раствор н-бутиллития (25 мл, 40 ммоль) при 0°С. Образовавшийся оранжевый гомогенный раствор соли лития затем перемешивали в течение 1 часа при комнатной температуре и затем охлаждали до 0°С. К данному раствору добавляли 1,2-этандиилбистозилат (20 мл). Температуре раствора далее давали возможность увеличиться до комнатной температуры. Суспензия образовывалась при нагревании раствора и кипячении с обратным холодильником в течение ночи. Затем смесь охлаждали и реакцию гасили добавлением воды (150 мл). Продукт затем экстрагировали дихлорметаном (3×100 мл) с последующим высушиванием сульфатом магния. Концентрирование раствора приводит к продукту 1,2-этандиил(2-метоксифенил)(фенил)фосфиноксиду, как белому твердому веществу. К 2 ммоль раствора продукта 1,2-этандиил(2-метоксифенил)(фенил)фосфиноксида в тетрагидрофуране (250 мл) прибавляли по каплям алюмогидрид (AlH3×1/3(C2H5)2O, 20 ммоль). Раствор затем кипятили с обратным холодильником до завершения (обычно в течение ночи), после чего реакцию гасили добавлением метанола (10 мл) с последующей фильтрацией осадка алюминиевой соли. Фильтрат затем концентрировали. Добавление метанола приводит к кристаллическому продукту (2-метоксифенил)(фенил)PCH2CH2P(2-метоксифенил)(фенил). Композиция 2 (сравнительная) (2-Метоксифенил)2PN(CH3)P(2-метоксифенил)2 в соотношении 1:1 с CrCl3 (2-метоксифенил)2PN(CH3)P(2-метоксифенил)2 лиганд готовили первичным образованием раствора 1,59 г (5 ммоль) (2-метоксифенил)2PNEt2 в 20 мл диэтилового эфира. К данному раствору прибавляли раствор 10 мл 1М HCl в диэтиловом эфире (10 ммоль HCl) в инертной атмосфере при комнатной температуре. Полученную таким образом суспензию перемешивали в течение ночи. Диэтиловый эфир удаляли из продукта в вакууме и прибавляли 20 мл сухого толуола. Полученный раствор фильтровали и толуол удаляли из фильтрата в вакууме с получением продукта в виде белого осадка (2-метоксифенил)2PCl. Раствор 0,51 г (5 ммоль) триэтиламина в 20 мл сухого дихлорметана прибавляли к (2-метоксифенил)2PCl продукту. К полученной смеси прибавляли раствор 1,25 мл 2М H2NMe в ТГФ (2,5 ммоль) и оставляли с перемешиванием в течение ночи. Растворители удаляли из полученного раствора в вакууме и прибавляли 20 мл сухого толуола. Смесь затем фильтровали. Толуол удаляли из фильтрата в вакууме и прибавляли 10 мл метанола. Суспензию фильтровали еще раз и выделяли белое твердое вещество продукта (2-метоксифенил)2PN(CH3)(2-метоксифенил)2. Композиция 3 (сравнительная) (2-Метоксифенил)(фенил)PN(CH3)P(2-метоксифенил)(фенил) в соотношении 1:1 с CrCl3 (2-метоксифенил)(фенил)PN(CH3)P(2-метоксифенил)(фенил) лиганд готовили первичным образованием суспензии 0,42 г лития (60 ммоль) в 80 мл ТГФ, к которой прибавляли 9,66 г лиганд (2-метоксифенил)2P(фенил) (30 ммоль) при 0°С в атмосфере аргона. Смесь перемешивали в течение 4 часов, после чего прибавляли аликвоту метанола 5 мл. 60 мл толуола прибавляли к смеси, после чего раствор экстрагировали двумя порциями по 40 мл воды. Экстрагированный толуольный раствор затем концентрировали до объема приблизительно 20 мл, что приводило к образованию суспензии. Концентрированный толуольный раствор фильтровали и к толуольному фильтрату прибавляли 4,6 г С2Cl6 (24 ммоль), который затем перемешивали в течение 2 часов при 90°С. Газообразный HCl, выделяющийся из реакционной смеси, «улавливали» щелочной баней. Смесь затем охлаждали до комнатной температуры и продували азотом для удаления всего остаточного HCl, присутствующего в растворе. При комнатной температуре прибавляли 5-мл аликвоту триэтиламина к концентрированному толуольному раствору и оставляли на несколько минут, после чего прибавляли 6 мл 2М H2NMe (12 ммоль) по несколько капель за один раз. Суспензию фильтровали и промывали 20 мл толуола. Толуольный фильтрат и промытую толуолом фракцию объединяли. Объединенные толуольные фракции упаривали досуха и прибавляли 30 мл метанола. Метанольный раствор оставляли на ночь при -35°С, при этом в растворе образовывался белый осадок (2-метоксифенил)(фенил)PN(CH3)P(2-метоксифенил)(фенил). Осажденный лиганд затем выделяли. Осажденный лиганд состоял из двух изомеров, рацемического изомера (RR и/или SS энантиомеры лиганда) и мезоизомера (RS энантиомер лиганда). Соотношение данных двух изомеров определяли с помощью 31P ЯМР с пиками при 63,18 м.д. и 64,8 м.д., относящимися к двум различным изомерам соответственно. Два образца (2-метоксифенил)(фенил)PN(CH3)P(2-метоксифенил)(фенил) использовали в примерах. Данные два образца состояли из смеси как рацемического изомера, так и мезоизомера с массовым соотношением 57/43 и 92/8 соответственно. Методика работы на установке Примеры 1-8 проводили с использованием следующей установки и методики. «Установка» (торговая марка Argonaut Technologies, Inc.) представляет собой мультиреакторную установку, содержащую восемь облицованных стеклом реакторов по 15 мл, используемую для проведения реакций под давлением (до 30 бар). Настоящие реакции проводили в объеме от 5 до 10 мл. Методику тримеризации этилена до 1-гексена выполняли следующим образом. Реакторы продували три раза этиленом при 100°С и давлении 30 бар. Затем реакторы оставляли охлаждаться до комнатной температуры, поддерживая в это время давление этилена 20-30 бар. Входной клапан этилена закрывали и реакторы оставляли на ночь. К тому же контролем за давлением этилена внутри реакторов в течение ночи реакторы проверяли на утечку. После этого реакторы были готовы для проведения реакций на следующий день. Предварительно смешанный каталитический раствор готовили для соответствующего катализатора, который должен быть использован. Предварительно смешанный каталитический раствор готовили взвешиванием 10 мкмоль композиции 1, 2 или 3, прибавлением 7,4 г сухого толуола и прибавлением 1,26 г (3 ммоль) модифицированного раствора метилалюмоксана (обозначенного здесь как ММАО) (6,4 мас.% Al в гептане, полученного у Witco Co.). Таким образом, приготовленный предварительно смешанный раствор (10 мл) содержал суммарно 10 мкмоль Cr и 3 ммоль Al (1 мМ Cr, 0,3М Al), и, следовательно, соотношение Al:Cr составляло 300:1. Предварительно смешанный раствор перемешивали в течение ночи в атмосфере азота при комнатной температуре и атмосферном давлении. 0,2М ММАО поглотительный раствор (5 мл) готовили прибавлением 1 ммоль (422 мг) количества раствора ММАО (6,4 мас.% Al в гептане) к 3,9 г толуола. Реакторы затем загружали соответствующим количеством 0,2М поглотительного раствора ММАО и 2,5 мл дополнительного количества толуола. Реакторы затем нагревали до 80°С и заполняли этиленом до желаемого реакционного давления. Чтобы начать реакцию тримеризации, аликвоту предварительно смешанного раствора вводили в реакторы. Дополнительные 0,5 мл толуола затем вводили, чтобы очистить линию ввода от оставшегося предварительно смешанного каталитического раствора. Реакцию прекращали или при достижении максимального поглощения этилена, или после установленного времени, закрывая входной клапан этилена, охлаждая до комнатной температуры, сбрасывая давление и открывая реактор. Выражение «прекращали при достижении максимального поглощения этилена», при использовании в данном описании, означает, что количество этилена, поглощенное в соответствующей реакции, соответствует количеству этилена, требуемому для получения желаемого объема 1-гексена. Например, если желательный конечный объем 1-гексена равен 5 мл (0,04 моль), количество молей этилена, требуемого для получения 5 мл конечного объема 1-гексена, должно быть 0,12 моль, таким образом, подача этилена в реактор должна быть прекращена, когда подано 0,12 моль этилена. Важно, чтобы объем реактора на «установке» составлял приблизительно 15 мл и таким образом желаемый конечный объем продукта и любого из оставшихся исходных материалов составлял меньше чем 15 мл. Обычно желателен конечный объем, равный 5-10 мл. Смесь продуктов собирали и взвешивали. Взвешенное количество анализировали с помощью газовой хроматографии (ГХ) (50 м CPSIL 5 CB y 0,25 колонка, газ-носитель гелий, ПИД (пламенно-ионизационный) детектор) с известным количеством гексилбензола в качестве внутреннего стандарта. Пример 1 В данном эксперименте реактор, содержащий 0,5 мл 0,2М ММАО поглотительного раствора и нагретый до 80°С, заполняли этиленом до давления, равного 8 бар. Аликвоту 0,5 мл предварительно смешанного каталитического раствора, содержащего композицию 1, вводили в реактор, чтобы начать реакцию (соотношение Al:Cr 500:1). Реакцию прекращали, когда было достигнуто максимальное поглощение этилена (161 минута). Смесь продуктов анализировали с помощью ГХ. Результаты представлены в табл.1. Пример 2 В данном эксперименте реактор, содержащий 0,5 мл 0,2М ММАО поглотительного раствора и нагретый до 80°С, заполняли этиленом до давления, равного 20 бар. Аликвоту 0,5 мл предварительно смешанного каталитического раствора, содержащего композицию 1, вводили в реактор, чтобы начать реакцию (соотношение Al:Cr 500:1). Реакцию прекращали, когда было достигнуто максимальное поглощение этилена (96 мин). Смесь продуктов анализировали с помощью ГХ. Результаты представлены в табл.1. Пример 3 В данном эксперименте реактор, содержащий 0,5 мл 0,2М ММАО поглотительного раствора и нагретый до 80°С, заполняли этиленом до давления, равного 8 бар. Аликвоту 0,5 мл предварительно смешанного каталитического раствора, содержащего композицию 1, вводили в реактор, чтобы начать реакцию (соотношение Al:Cr 500:1). Реакцию прекращали через 1 час. Смесь продуктов анализировали с помощью ГХ. Результаты представлены в табл.1. Пример 4 В данном эксперименте реактор, содержащий 0,35 мл 0,2М ММАО поглотительного раствора и нагретый до 80°С, заполняли этиленом до давления, равного 15 бар. Аликвоту 0,35 мл предварительно смешанного каталитического раствора, содержащего композицию 1, вводили в реактор, чтобы начать реакцию (соотношение Al:Cr 500:1). Реакцию прекращали через 1 час. Смесь продуктов анализировали с помощью ГХ. Результаты представлены в табл.1. Пример 5 (сравнительный) В данном эксперименте реактор, содержащий 0,5 мл 0,2М ММАО поглотительного раствора и нагретый до 80°С, заполняли этиленом до давления, равного 8 бар. Аликвоту 0,5 мл предварительно смешанного каталитического раствора, содержащего композицию 2, вводили в реактор, чтобы начать реакцию (соотношение Al:Cr 500:1). Реакцию прекращали, когда было достигнуто максимальное поглощение этилена, 105 минут. Смесь продуктов анализировали с помощью ГХ. Результаты представлены в табл.1. Пример 6 (сравнительный) В данном эксперименте реактор, содержащий 0,5 мл 0,2М ММАО поглотительного раствора и нагретый до 80°С, заполняли этиленом до давления, равного 20 бар. Однако благодаря высокой скорости реакции, превышающей скорость подачи исходного сырья в реактор, давление во время реакции составляло только 7-10 бар. Аликвоту 0,5 мл предварительно смешанного каталитического раствора, содержащего композицию 2, вводили в реактор, чтобы начать реакцию (соотношение Al:Cr 500:1). Реакцию прекращали, когда было достигнуто максимальное поглощение этилена, 96 минут. Смесь продуктов анализировали с помощью ГХ. Результаты представлены в табл.1. Пример 7 (сравнительный) В данном эксперименте реактор, содержащий 0,5 мл 0,2М ММАО поглотительного раствора и нагретый до 80°С, заполняли этиленом до давления, равного 8 бар. Аликвоту 0,5 мл предварительно смешанного каталитического раствора, содержащего композицию 2, вводили в реактор, чтобы начать реакцию (соотношение Al:Cr 500:1). Реакцию прекращали через 1 час. Смесь продуктов анализировали с помощью ГХ. Результаты представлены в табл.1. Пример 8 (сравнительный) В данном эксперименте реактор, содержащий 0,35 мл 0,2М ММАО поглотительного раствора и нагретый до 80°С, заполняли этиленом до давления, равного 15 бар. Аликвоту 0,35 мл предварительно смешанного каталитического раствора, содержащего композицию 2, вводили в реактор, чтобы начать реакцию (соотношение Al:Cr 500:1). Реакцию прекращали через 1 час. Смесь продуктов анализировали с помощью ГХ. Результаты представлены в табл.1.
Методика с 1-литровым реактором периодического действия 1-литровый реактор периодического действия нагревали в атмосфере азота до 70°С, промывали N2 три раза и вакуумировали в течение 5 минут. В реактор помещали раствор 250 мл сухого толуола и 1 г раствора МАО (5,11% Al в толуоле) для того, чтобы «протравить» реактор в течение, по меньшей мере, 2 часов при 70°С. Толуол и МАО раствор для «протравливания» удаляли и реактор вакуумировали в течение 5 минут, поддерживая реактор при температуре 70°С. Реактор затем снова заполняли 250 мл сухого толуола, нагнетали этилен до реакционного давления и вводили соответствующее количество поглотителя МАО. Раствор затем перемешивали в течение, по меньшей мере, 5 минут при 70°С. Предварительно смешанный каталитический раствор готовили взвешиванием 10 мкмоль композиции 1, 2 или 3, прибавлением 7,1 г сухого толуола и прибавлением 1,59 г (3 ммоль) раствора МАО (5,11% Al в толуоле). Таким образом, полученный предварительно смешанный раствор (10 мл) содержал суммарно 10 мкмоль Cr и 3 мкмоль Al (1 мМ Cr, 0,3М Al) и имел соотношение Al:Cr, равное 300:1. После перемешивания начинали реакцию тримеризации введением аликвоты каталитического предварительно смешанного раствора в находящийся под давлением реактор. Реактор затем нагревали до температуры реакции, равной 80°С. Реакцию проводили в течение известного отрезка времени, пока поддерживалось реакционное давление, и затем прекращали, быстро охлаждая реактор до примерно 30°С (приблизительно 5 минут). Содержимое реактора удаляли со дна 1-литрового реактора периодического действия. Полученную смесь продукта собирали и взвешивали. Взвешенное количество использовали для ГХ анализа с гексилбензолом в качестве внутреннего стандарта. Пример 9 Аликвоту 10 мл предварительно смешанного каталитического раствора, приготовленного с использованием композиции 1, вводили в 1-литровый реактор, содержащий 3 ммоль МАО в качестве поглотителя (1,59 г раствора МАО). Реакцию проводили при 80°С и давлении 15 бар атмосферы этилена. Реакцию прекращали после 5 часов. Суммарно было поглощено 275 литров этилена. Смесь продукта анализировали с помощью ГХ. Результаты представлены в табл.2. Пример 10 (сравнительный) Аликвоту 2 мл предварительно смешанного каталитического раствора, приготовленного с использованием композиции 1, вводили в 1-литровый реактор, содержащий 0,6 ммоль МАО в качестве поглотителя (317 мг раствора МАО). Реакцию проводили при 80°С и давлении 15 бар атмосферы этилена. После 205 минут вводили дополнительные 2 мл предварительно смешанного каталитического раствора. Реакцию прекращали после 4,5 часов. Суммарно было поглощено 325 литров этилена. Смесь продукта анализировали с помощью ГХ. Результаты представлены в табл.2. Пример 11 (сравнительный) Аликвоту 2 мл предварительно смешанного каталитического раствора, приготовленного с использованием композиции 3 в соотношении 57/43, вводили в 1-литровый реактор, содержащий 0,6 ммоль МАО как поглотитель (317 мг раствора МАО). Реакцию проводили при 80°С и давлении 15 бар атмосферы этилена. Реакцию прекращали после 3 часов. Суммарно было поглощено 250 литров этилена. Смесь продуктов анализировали с помощью ГХ. Результаты представлены в табл.2. Пример 12 (сравнительный) Аликвоту 2 мл предварительно смешанного каталитического раствора, приготовленного с использованием композиции 3 в соотношении 92/8, вводили в 1-литровый реактор, содержащий 0,6 ммоль МАО в качестве поглотителя (317 мг раствора МАО). Реакцию проводили при 80°С и давлении 15 бар атмосферы этилена. Реакцию прекращали после 4,5 часов. Суммарно было поглощено 308 литров этилена. Смесь продуктов анализировали с помощью ГХ. Результаты представлены в табл.2.
Из представленных выше результатов в табл.1 и 2 очевидно, что использование каталитической композиции по настоящему изобретению, содержащей лиганд формулы (I) как определено выше, а именно (метоксифенил)(фенил)PCH2CH2P(метоксифенил)(фенил), приводит к уменьшению выхода побочных продуктов С10 по сравнению с использованием при эквивалентных условиях реакции эквивалентной каталитической композиции, содержащей лиганд, имеющий формулу (2-метоксифенил)2PN(CH3)P(2-метоксифенил)2 (раскрытый в примерах WO 02/04119) или лиганд, имеющий формулу (2-метоксифенил)(фенил)PN(CH3)P(метоксифенил)(фенил), ни один из которых не попадает в формулу (I), как определено выше.
Формула изобретения
1. Каталитическая композиция для тримеризации олефиновых мономеров, содержащая 2. Каталитическая композиция по п.1, в которой бивалентная органическая мостиковая группа Х означает алкиленовую группу, которая содержит от 2 до 6 атомов углерода в мостике. 3. Каталитическая композиция по п.1 или 2, в которой бивалентная органическая мостиковая группа Х означает -СН2-СН2-. 4. Каталитическая композиция по п.1 или 2, в которой R1 и R3 независимо выбирают из необязательно замещенных фенильных групп, которые не содержат полярных заместителей ни в одном из ортоположений. 5. Каталитическая композиция по п.1 или 2, в которой R2 и R4 независимо выбирают из необязательно замещенных фенильных групп, при этом полярные заместители представляют собой необязательно разветвленные С1-С20алкоксигруппы. 6. Каталитическая композиция по п.1 или 2, в которой R2 и R4 означают 2-метоксифенильные группы. 7. Каталитическая композиция по п.1 или 2, в которой сокатализатор выбирают из метилалюмоксана или модифицированного метилалюмоксана. 8. Каталитическая композиция по п.1 или 2, в которой компонент а) представляет собой источник хрома. 9. Каталитическая композиция по п.8, в которой источник хрома представляет собой CrCl3. 10. Способ тримеризации олефиновых мономеров, включающий взаимодействие, по меньшей мере, одного олефинового мономера в условиях реакции тримеризации с каталитической композицией по любому из пп.1-9.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 -олефины, необязательно разветвленные С4-С20 внутренние олефины, необязательно разветвленные С4-С20 винилиденовые олефины, необязательно разветвленные С4-С20 циклические олефины и необязательно разветвленные С4-С20 диены, так же как и необязательно разветвленные С4-С20 функционализированные олефины. Примеры подходящих олефиновых мономеров включают в себя, но не ограничены ими, этилен, пропилен, 1-бутен, 1-пентен, 1-гексен, 4-метилпент-1-ен, 1-гептен, 1-октен, 1-нонен, 1-децен, 1-ундецен, 1-додецен, 1-тридецен, 1-тетрадецен, 1-пентадецен, 1-гексадецен, 1-гептадецен, 1-октадецен, 1-нонадецен, 1-эйкозен, стирол, 2-бутен, 1-этил-1-гексен, циклогексен, норборнен и подобные.
-олефины, необязательно разветвленные С4-С20 внутренние олефины, необязательно разветвленные С4-С20 винилиденовые олефины, необязательно разветвленные С4-С20 циклические олефины и необязательно разветвленные С4-С20 диены, так же как и необязательно разветвленные С4-С20 функционализированные олефины. Примеры подходящих олефиновых мономеров включают в себя, но не ограничены ими, этилен, пропилен, 1-бутен, 1-пентен, 1-гексен, 4-метилпент-1-ен, 1-гептен, 1-октен, 1-нонен, 1-децен, 1-ундецен, 1-додецен, 1-тридецен, 1-тетрадецен, 1-пентадецен, 1-гексадецен, 1-гептадецен, 1-октадецен, 1-нонадецен, 1-эйкозен, стирол, 2-бутен, 1-этил-1-гексен, циклогексен, норборнен и подобные.