Патент на изобретение №2351605
|
||||||||||||||||||||||||||
(54) СПОСОБ ЭТЕРИФИКАЦИИ ТИОКАРБОНОВОЙ КИСЛОТЫ
(57) Реферат:
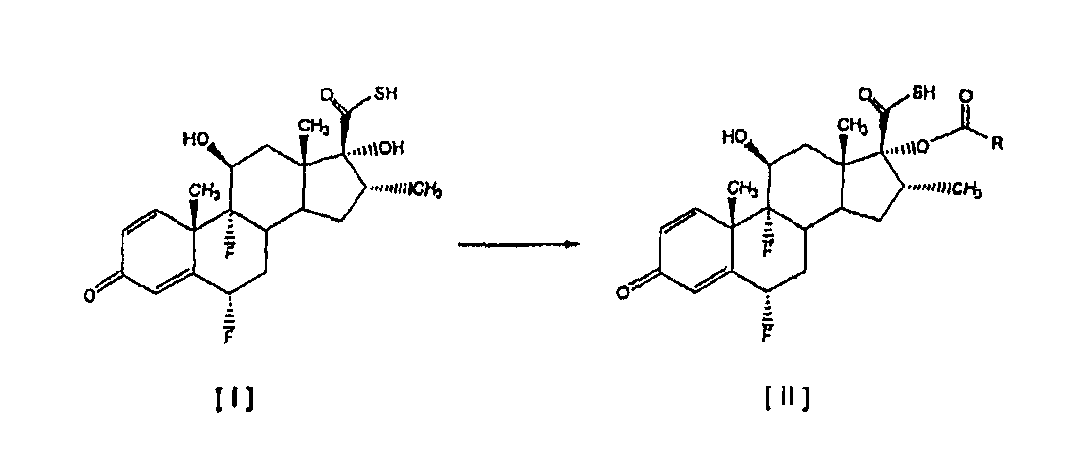
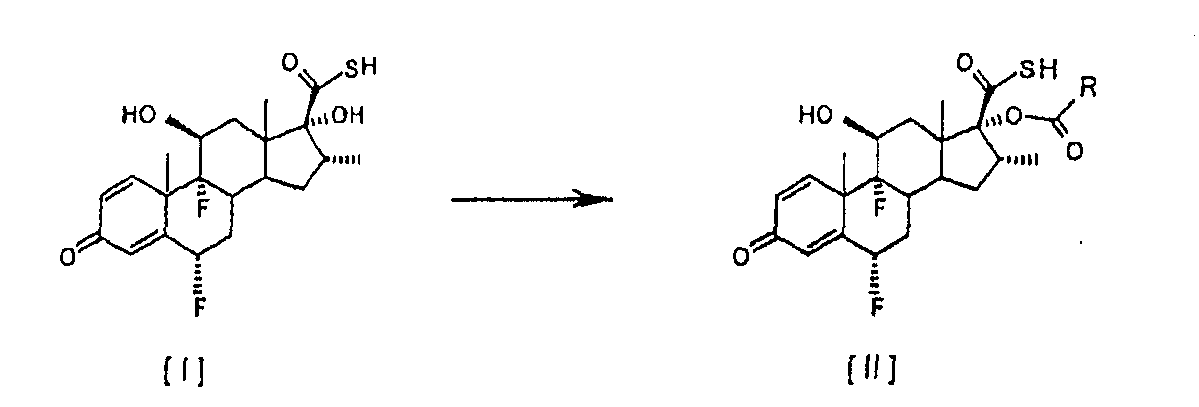
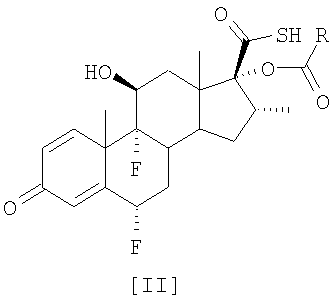
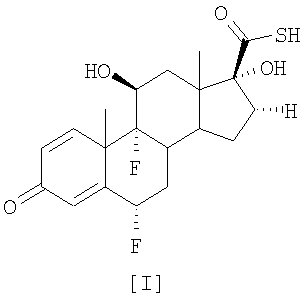
Описан улучшенный способ получения соединения формулы [II], промежуточного продукта в синтезе флутиказона пропионата, путем этерификации С-17 гидроксильной группы 6
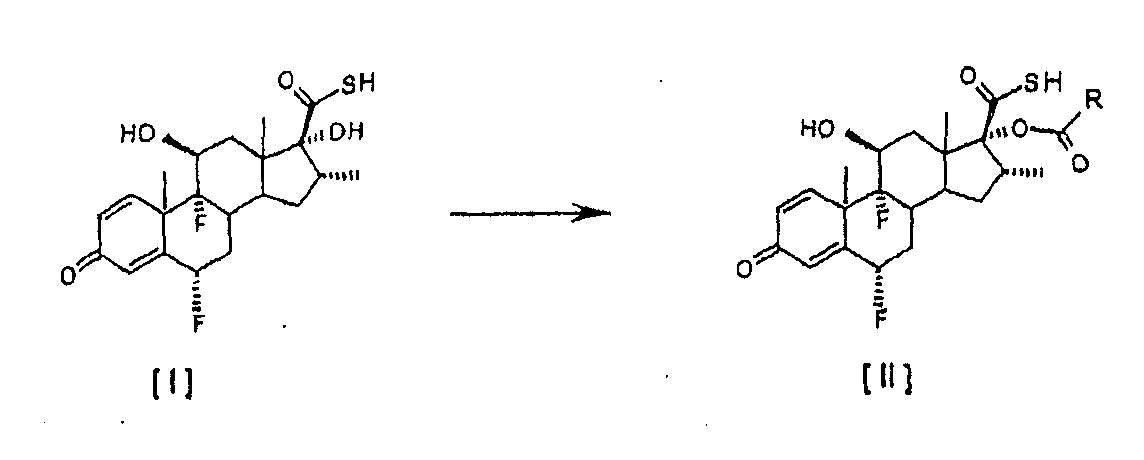
Настоящее изобретение относится, в общем случае, к способу этерификации тиокарбоновой кислоты, в том числе, но не исключительно, к получению флутиказона пропионата, и к применению некоторых промежуточных соединений. В одном аспекте настоящее изобретение относится к усовершенствованному способу этерификации С-17 гидроксильной группы 6
Настоящее изобретение предлагает способ этерификации, который значительно проще и более эффективен экономически, чем способы, известные в этой области, поскольку он подавляет одну из двух химических реакций, описанных в этих способах. Другой отличительной особенностью способа настоящего изобретения является то, что в результате получаются 17 Более конкретно, это изобретение относится к усовершенствованному способу получения 6
Патент US 4335121, британские патенты GB 2088877, GB 2137206, патент US 4578221 и J.Med.Chem., 1994, 37, 3717-3729, описывают 17
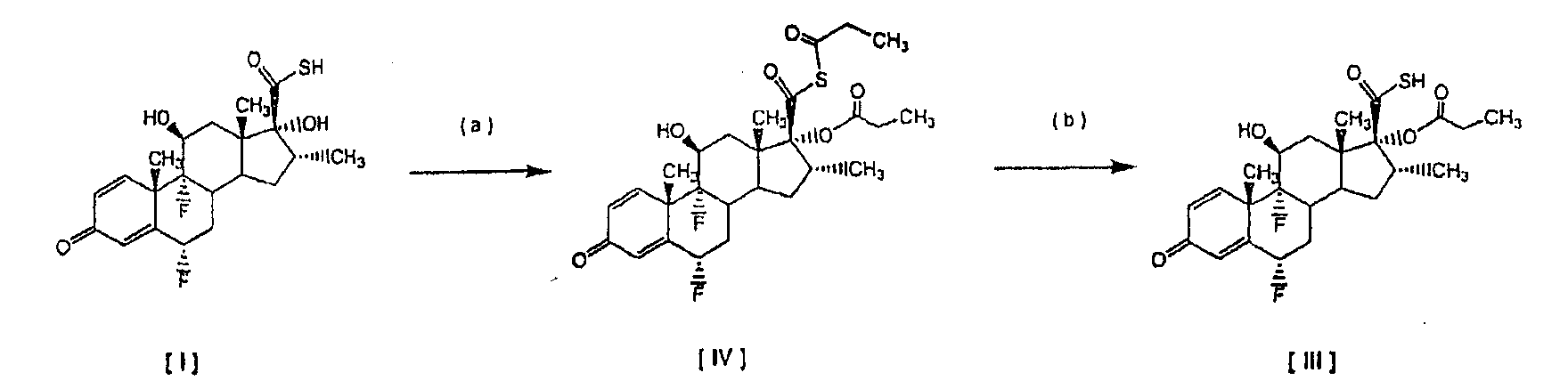
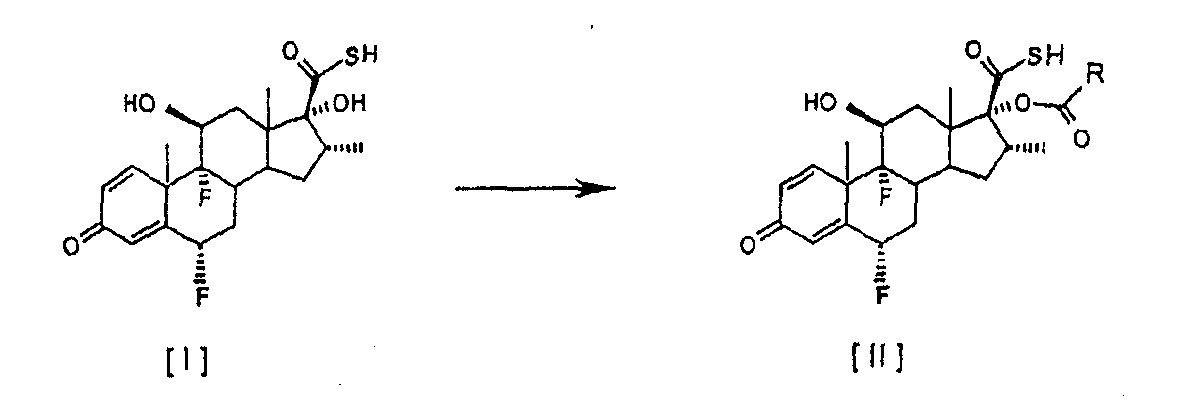
смешанный ангидрид На первой стадии (а) смешанный ангидрид [IV] получают с избытком, по меньшей мере, 2 молей пропионилхлорида на моль соединения [I] в присутствии триэтиламина, используя дихлорметан в качестве растворителя. После завершения реакции пропионилирования реакционную смесь обрабатывают и получают светло-коричневый твердый осадок. На второй стадии (b) этот твердый осадок растворяют в ацетоне и обрабатывают диэтиламином для превращения смешанного ангидрида в соединение [III]. По окончании реакции аминолиза реакционную смесь обрабатывают, чтобы выделить соединение [III]. Международная патентная заявка WO 03/066654 описывает получение промежуточного соединения [III] с помощью (а) взаимодействия соединения [I] с, по меньшей мере, 1,3 молями активированного производного пропионовой кислоты на моль соединения [I] и удаления фрагмента, связанного с серой, из любого соединения формулы [IV] органическим первичным или вторичным амином, таким как диэтаноламин или N-метилпиперазин. Патентная заявка WO 01/62722 раскрывает 17
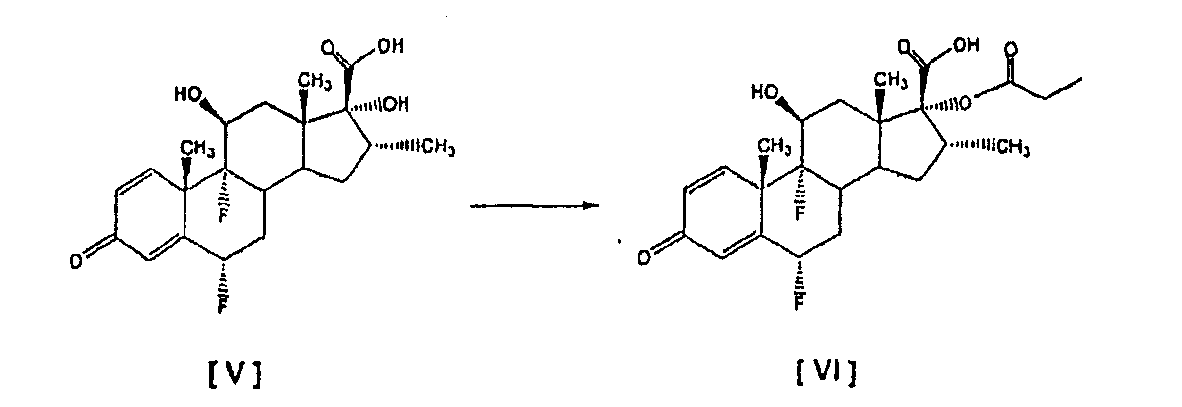
путем (а) взаимодействия оксикислоты формулы [V] с 2,3 моля пропионилхлорида на моль соединения [V], используя триэтиламин в качестве основания, и (b) in situ взаимодействия соединения, полученного на стадии (а), с диэтиламином. Все процедуры 17 В настоящее время авторами было обнаружено, что трансформация соединения тиокарбоновой кислоты [I] в соединения общей формулы [II] избирательно может происходить прямо с незначительным образованием соответствующего смешанного ангидрида при условиях данного изобретения, описанных ниже. Следуя способу настоящего изобретения, промежуточная 6
путем реакции с небольшим избытком соответствующего ацилхлорида в инертном растворителе в присутствии соответствующего третичного амина. Особым преимуществом настоящего изобретения по сравнению с тем, что уже известно из уровня техники, является то, что соединения общей формулы [II] получают непосредственно из соединения [I], без необходимости проведения аминолиза соответствующего смешанного ангидрида. Кроме того, в условиях аминолиза, описанных в уровне техники, химическая стабильность производных тиокарбоновой кислоты, таких как соединение [III], соответственно ограничивается, при этом упрощенный способ настоящего изобретения, путем исключения реакции аминолиза, дает 17 В соответствии с настоящим изобретением предлагается способ получения соединений формулы [II]
путем этерификации С-17 гидроксильной группы 6 Изобретение также предусматривает применение соединений формулы [II], полученных в соответствии со способом настоящего изобретения, для получения используемых в терапевтических целях лекарственных средств. Настоящее изобретение предлагает усовершенствованный и упрощенный способ селективной 17
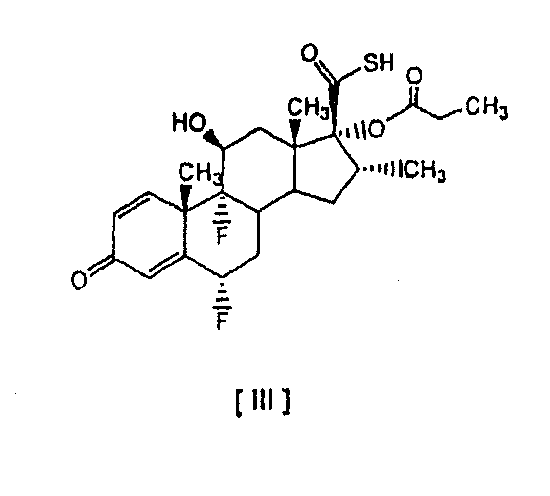
в которой R обозначает -СН2СН3, -СН2СН2СН3 или -СН(СН3)2, без необходимости проведения реакции аминолиза соответствующих смешанных ангидридов. Способ включает реакцию соединения [I] 6
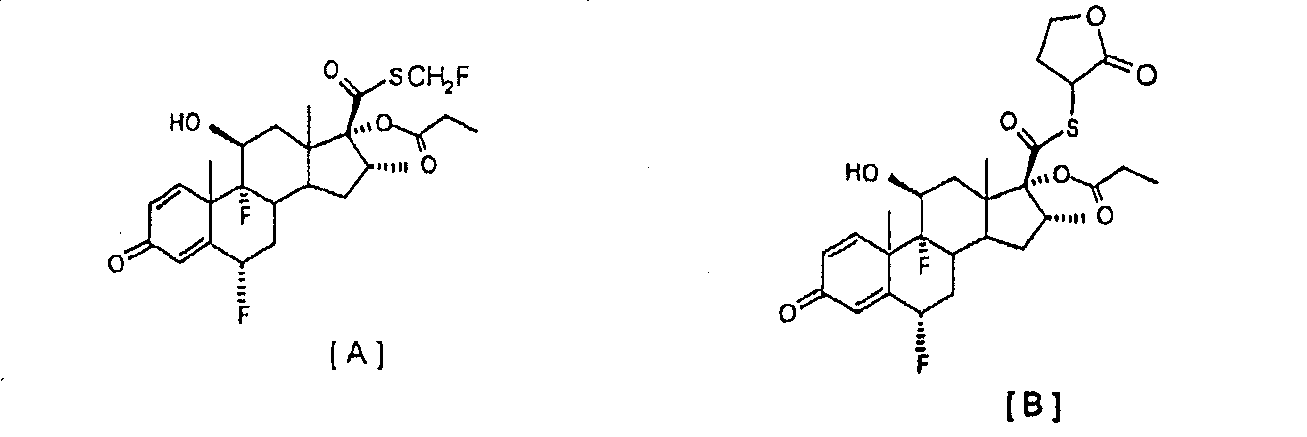
в которой R обозначает -СН2СН3, -СН2СН2СН3 или -СН(СН3)2. Особенно предпочтительным вариантом осуществления настоящего изобретения является предоставление усовершенствованного способа получения 6 Соединение формулы [III] является известным промежуточным соединением, которое используется при получении противовоспалительных стероидов, таких как флутиказона пропионат формулы [А] (описанный в патенте US 4335121), высоко эффективного при лечении воспалительных заболеваний, таких как астма и хроническое обструктивное заболевание легких (ХОЗЛ), и при получении сходного S-(2-оксотетрагидрофуран-3-ил)ового эфира 6
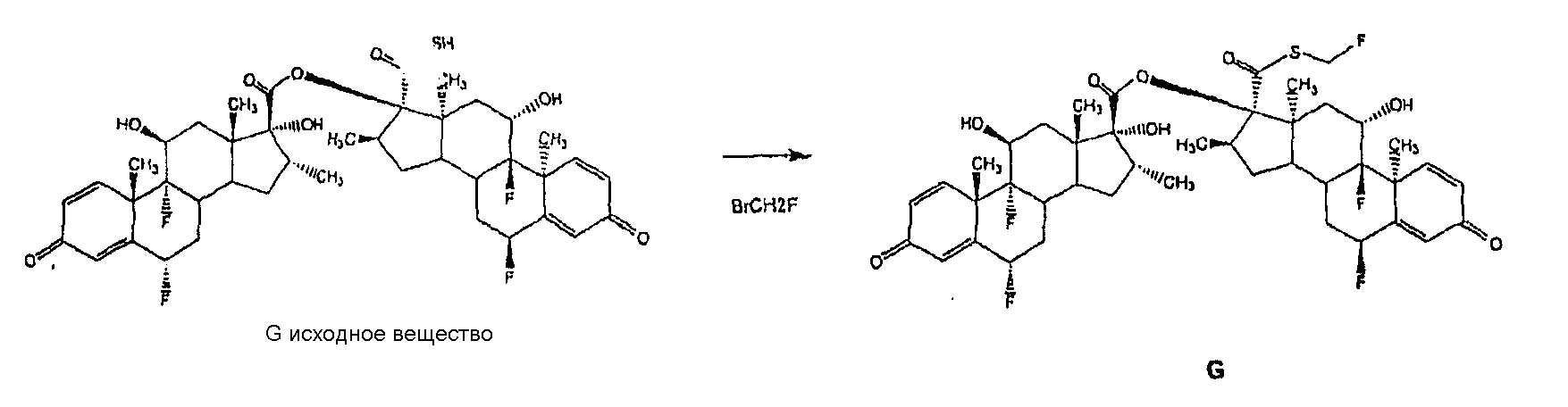
Соответственно, изобретение также предлагает способ получения флутиказона пропионата, включающий получение 6 Также предусматривается способ получения S-(2-оксотетрагидрофуран-3-ил)ового эфира 6 Настоящее изобретение предусматривает эффективный способ получения 6 Предпочтительно использовать хлорангидрид карбоновой кислоты общей формулы R-COCl в способе настоящего изобретения в молярном отношении от 1,0 до 1,2 моля хлорангидрида на моль исходного соединения [I], предпочтительно в количестве, входящим в этот интервал. Инертные растворители для процесса 17 Соответствующие третичные амины для выполнения избирательного 17 Реакция 17 Использование третичных аминов, определенных выше как адекватных для способа по изобретению, представляет преимущества по сравнению с другими основаниями, ранее описанными в этой области, такими как триэтиламин, трипропиламин, этилдиизопропиламин, N,N-диметиланилин, N,N-диметилциклогексиламин, неорганические карбонаты, такие как, например, бикарбонат натрия, карбонат калия и карбонат натрия. При использовании этих оснований предотвращается образование больших количеств соединения смешанного ангидрида [IV] или неполное расходование соединения [I], или образование большого количества примесей, как это имеет место с бикарбонатом натрия. В предпочтительных условиях способа настоящего изобретения 17 При условиях, описанных в известном уровне техники, когда соединение [III] получают через смешанный ангидрид, может наблюдаться частичная деградация соединения [III] во время реакции аминолиза. Эта деградация особенно проблематична, когда имеет место продолжительная реакция, которую следует ожидать в промышленном масштабе. При условиях, раскрытых в настоящем изобретении, эта in situ деградация соединения [III] не происходит. Было обнаружено, что, следуя известному уровню техники, образуется исходное вещество (G исходное вещество) для примеси (примесь G).
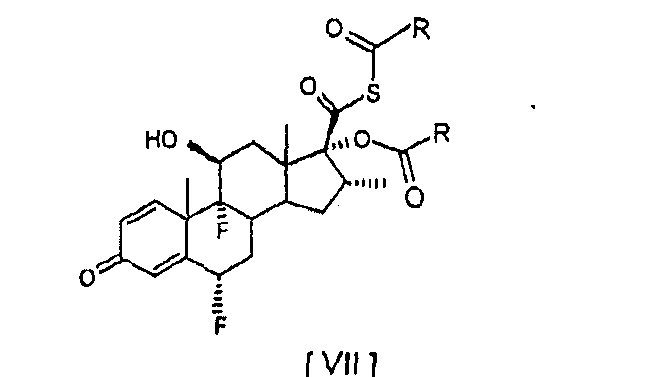
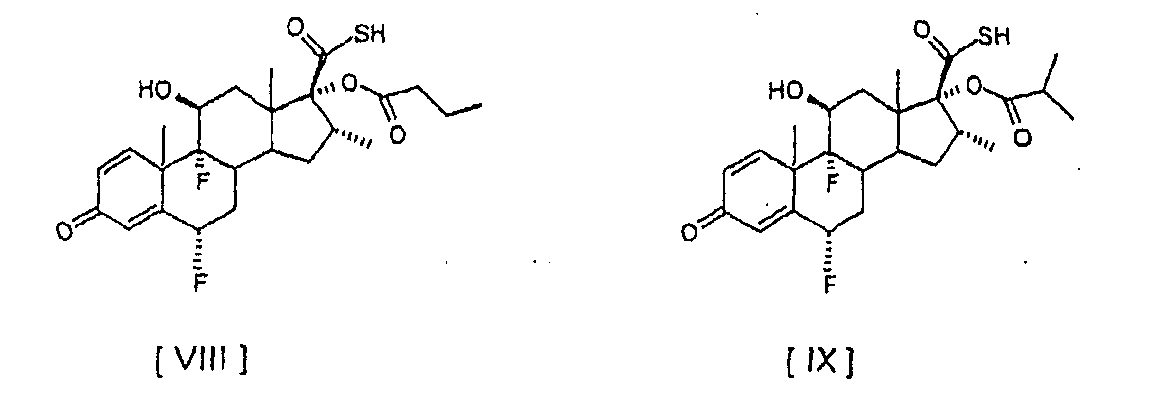
При способе настоящего изобретения димер G, который трудно удалить из конечного продукта, флутиказона пропионата, с помощью перекристаллизации, когда его уровень выше чем 0,5%, образуется на уровне ниже 0,3%. Следовательно, можно избежать повторных перекристаллизаций для получения вещества необходимой чистоты, которые, если выполняются, уменьшают общий выход продукта. Другой вариант осуществления настоящего изобретения предусматривает способ получения соединений: 6
Соединения формул [VIII] и [IX] получают непосредственно из соединения [I] с помощью реакции с небольшим избытком хлорангидрида карбоновой кислоты, без необходимости проведения реакции аминолиза соответствующих смешанных ангидридов. В соответствии с предпочтительным аспектом настоящего изобретения этерификацию соединения [I] для получения соединения [VIII] выполняют с 1,0 – 1,2 молями бутирилхлорида в ацетоне в присутствии пиридина при температуре от 5 до -20°С, предпочтительно от 5 до 0°С. Этерификацию соединения [I] для получения соединения [IX] предпочтительно выполняют с 1,0-1,2 молями изобутирилхлорида в ацетоне в присутствии пиридина при температуре от 5 до -20°С, предпочтительно от 5 до 0°С. Следующие примеры иллюстрируют настоящее изобретение и определенные предпочтительные варианты осуществления и не ограничивают сущность изобретения. Пример 1. Получение 6 Суспензию соединения [I] (10 г, 0,024 моля) в ацетоне (175 мл) охладили до -20°С и обработали пиридином (30 мл) и пропионилхлоридом (2,3 мл, 0,026 моля), поддерживая температуру от -20 до -15°С. Смесь перемешивали при -20/-15°С до полного завершения реакции. Можно вторично добавить пропионилхлорид (0,02 мл), если необходимо, для полного завершения реакции. Соединение [III] осадили путем добавления воды (240 мл) и концентрированной соляной кислоты (30 мл). Суспензию перемешивали в течение 2 часов при температуре от 0 до 5°С, отфильтровали и влажный осадок промыли холодной водой до получения нейтрального значения рН. Твердый осадок высушили при температуре ниже 40°С под вакуумом, чтобы получить названное соединение в виде от белого до грязно-белого твердого осадка (10,04 г; 88%; чистота, площадь пика % ВЭЖХ: 96,1%). Пример 2. Получение 6 Изобутирилхлорид (0,28 мл, 0,0027 моля) медленно добавили к ацетону (17,5 мл), соединению [I] (1 г, 0,0024 моля) и пиридину (3 мл) при температуре от 5 до 0°С и смесь перемешивали в этом температурном интервале до полного израсходования соединения [I]. После завершения реакции названное соединение осаждается при добавлении воды (24 мл) и концентрированной соляной кислоты (3 мл). Суспензию перемешивали в течение 1 – 2 часов при температуре от 0 до 5°С, отфильтровали и влажный осадок промыли холодной водой до нейтрального значения рН. Твердый осадок высушили при температуре ниже 40°С под вакуумом, чтобы получить названное соединение в виде от белого до грязно-белого твердого осадка (1,09 г; 93%; чистота, площадь пика % ВЭЖХ: 94,8%). Пример 3. Получение 6 Суспензию соединения [I] (1 г, 0,0024 моля) в ацетоне (17 мл) охладили до температуры в пределе от -20 до -15°С. N-Метилимидазол (2,9 мл) и пропионилхлорид (0,23 мл, 0,0026 моля) последовательно добавили к раствору, поддерживая температуру от -20 до -15°С. После завершения реакции соединение [III] выпало в осадок при добавлении воды (24 мл) и концентрированной соляной кислоты (3 мл). Суспензию перемешивали в течение 2 часов при температуре приблизительно 0°С, отфильтровали и влажный осадок промыли холодной водой до получения нейтрального значения рН. Влажный осадок высушили при температуре ниже 40°С под вакуумом, чтобы получить названное соединение в виде от белого до грязно-белого твердого осадка (1,0 г; 88%; чистота, площадь пика % ВЭЖХ: 96,8%).
Формула изобретения
1. Способ получения соединения формулы [II] 2. Способ по п.1, который осуществляют при температуре от 5 до -20°С. 3. Способ по п.1 или 2, в котором хлорангидрид карбоновой кислоты общей формулы R-COCl является пропионилхлоридом, где R обозначает -СН2СН3. 4. Способ по п.1, в котором третичный амин является пиридином или N-метилимидазолом. 5. Способ по п.1, в котором третичный амин является пиридином. 6. Способ по п.1, в котором инертный растворитель является ацетоном. 7. Способ по п.1, в котором пиридин используется как третичный амин и инертный растворитель является ацетоном, тетрагидрофураном, диметилацетамидом, дихлорметаном, этилацетатом, 2-пентаноном, 3-пентаноном, 4-метил-2-пентаноном или 2-бутаноном, предпочтительно ацетоном. 8. Способ по п.1, в котором реакция проводится при температуре от -15 до -20°С. 9. Способ по п.1, в котором хлорангидрид карбоновой кислоты общей формулы R-COCl является бутирилхлоридом, где R обозначает -СН2СН2СН3, отличающийся тем, что соединение [I] обрабатывают от 1,0 моля до 1,2 моля бутирилхлорида на моль соединения [I] в присутствии пиридина в ацетоне при температуре от 5 до -20°С, предпочтительно от 5 до 0°С. 10. Способ по п.1, в котором хлорангидрид карбоновой кислоты общей формулы R-COCl является изобутирилхлоридом, где R обозначает – СН(СН3)2, отличающийся тем, что соединение [I] обрабатывается от 1,0 моля до 1,2 моля изобутирилхлорида на моль соединения [I] в присутствии пиридина в ацетоне при температуре от 5 до -20°С, предпочтительно от 5 до 0°С. 11. Способ получения флутиказона пропионата, включающий получение 6 12. Способ получения S-(2-оксотетрагидрофуран-3-ил)ового эфира 6 13. Применение соединений формулы [II], полученных в соответствии со способом по любому из пп.1-11, для получения полезных терапевтических лекарственных средств.
|
||||||||||||||||||||||||||
 22, (1994), p.3724.
22, (1994), p.3724.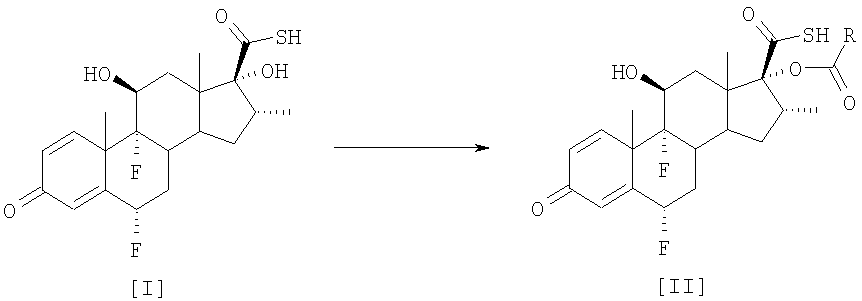
 ,9
,9 ,17
,17