Патент на изобретение №2350621
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) ПРОИЗВОДНЫЕ АНТИБИОТИКА ГРУППЫ АУРЕОЛОВОЙ КИСЛОТЫ ОЛИВОМИЦИНА 1, ОБЛАДАЮЩИЕ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ, И СПОСОБ ИХ ПОЛУЧЕНИЯ
(57) Реферат:
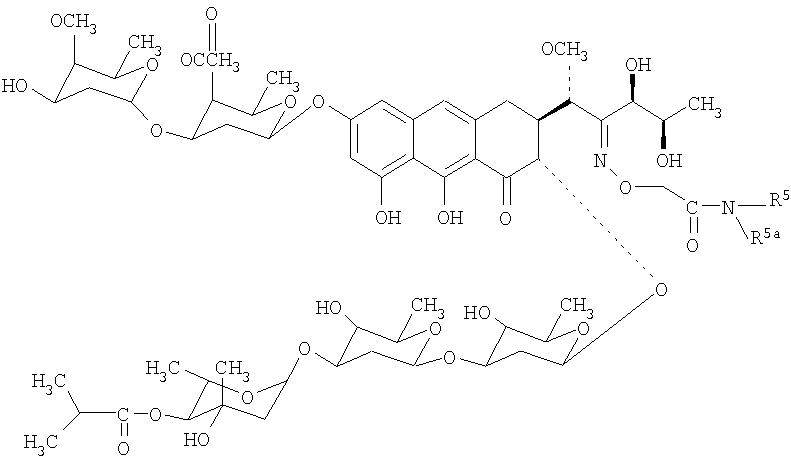
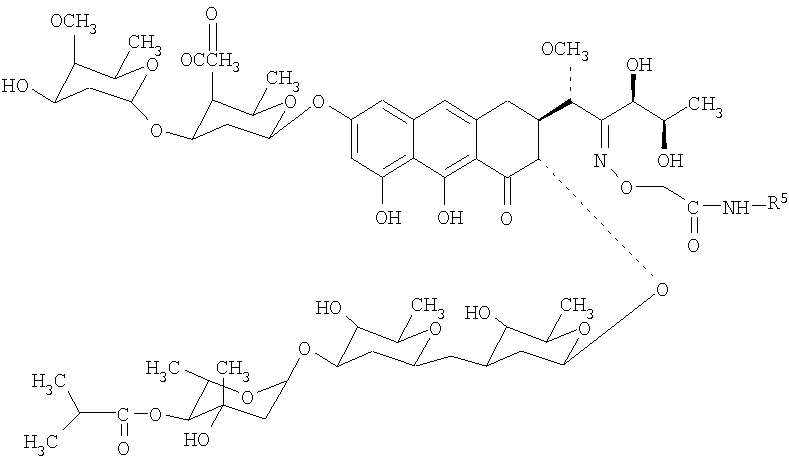
Изобретение относится к обладающим противоопухолевой активностью производным антибиотика группы ауреоловой кислоты оливомицина I, соответствующим структурной формуле, приведенной ниже, в которых R5 представляет собой водород, С3-С10-циклоалкил или C1-C4-алкил с прямой или разветвленной углеводородной цепью, необязательно замещенный одним или несколькими гидроксилами. Изобретение относится также к способу получения указанных производных, заключающемуся в селективной модификации 2′-карбонильной группы оливомицина 1 реакцией с аминооксиуксусной кислотой, с последующим проведением реакции амидирования полученного ключевого интермедиата 2′-(карбоксиметоксим)оливомицина 1 с соответствующими аминами в присутствии конденсирующего агента. 2 н.п. ф-лы, 5 табл.
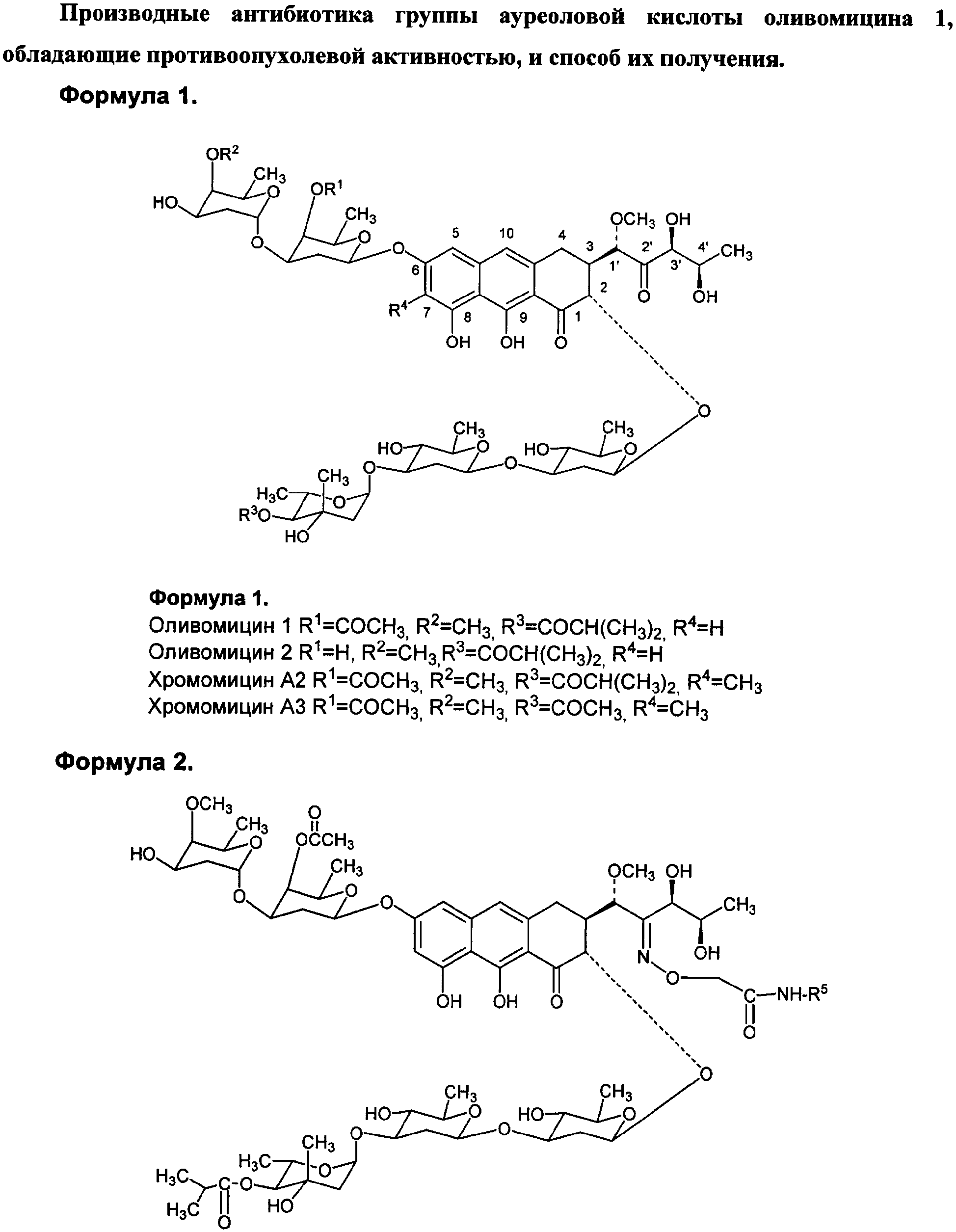
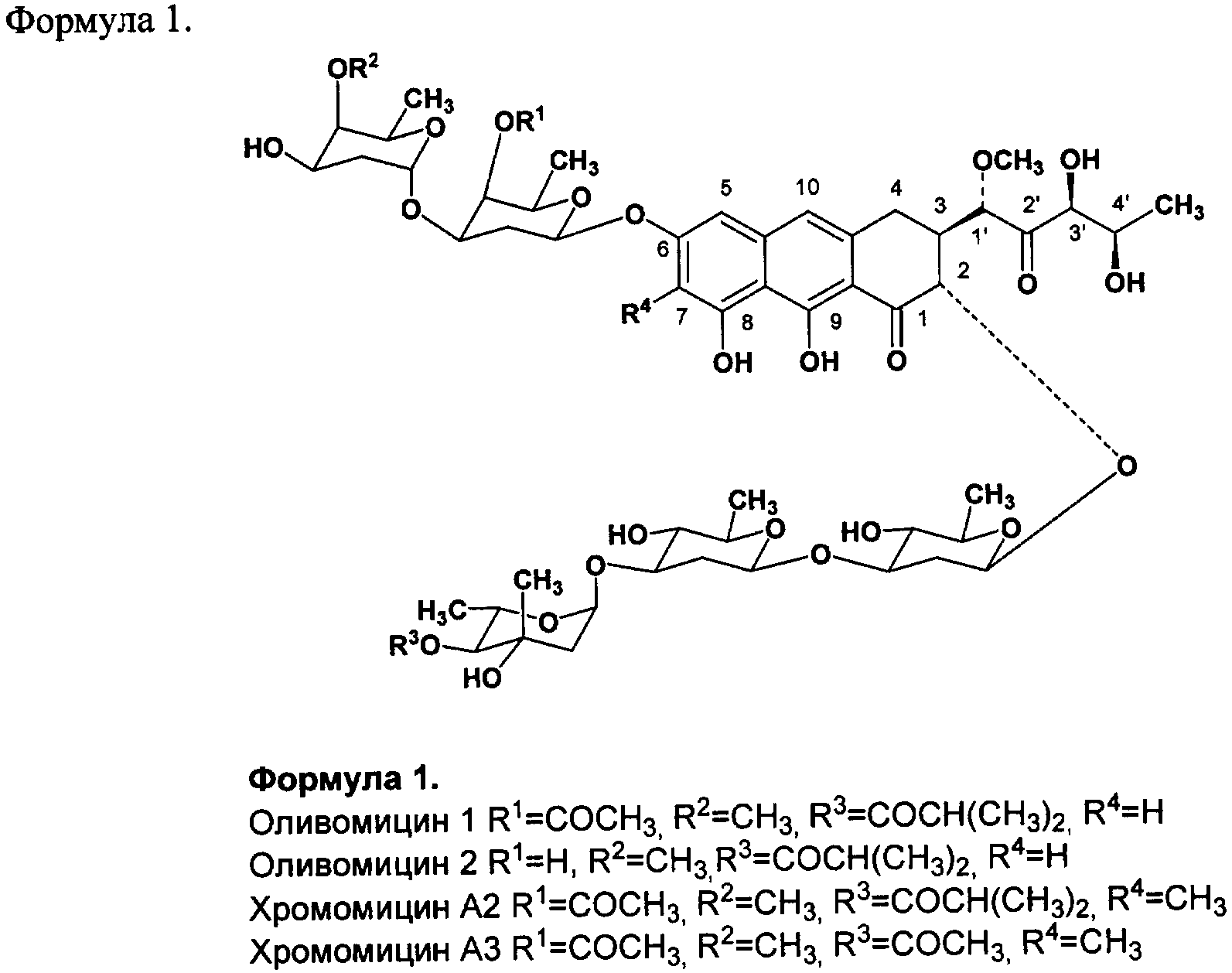
Изобретение относится к фармацевтической промышленности и касается новых производных противоопухолевого антибиотика группы ауреоловой кислоты оливомицина 1 и способа их получения. Антибиотики группы ауреоловой кислоты являются высокоэффективными природными противоопухолевыми препаратами, некоторые из которых разрешены к применению в качестве антинеопластических агентов для лечения ряда опухолевых заболеваний. Важнейшими представителями этой группы являются: оливомицин 1 (оливомицин А) (формула 1, R1=СОСН3, R2=СН3, R3=COCH(CH3)2, R4=H), оливомицин 2 (формула 1, R1=H, R2=СН3, R3=COCH(CH3)2, R4=H), хромомицин А2 (формула 1, R1=СОСН3, R2=СН3, R3=COCH(CH3)2, R4=CH3), хромомицин A3 (формула 1, R1=СОСН3, R2=СН3, R3=COCH3, R4=CH3), митрамицин.
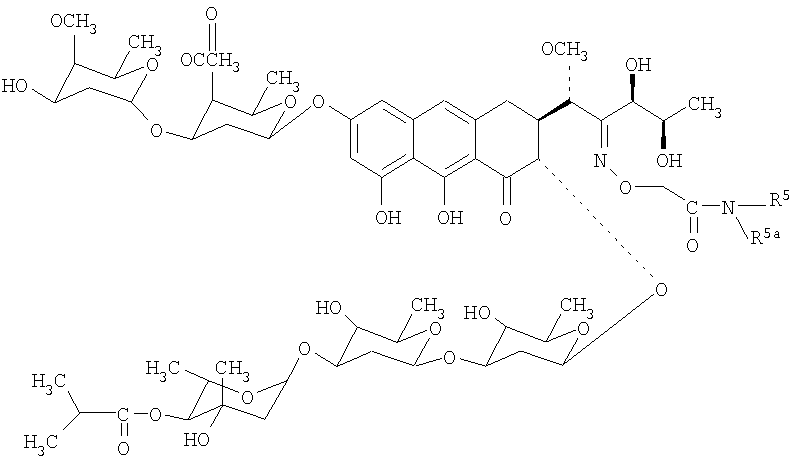
Механизм действия антибиотиков группы ауреоловой кислоты основан на взаимодействии их с GC-парами в малой бороздке ДНК и нарушении структуры и функции нуклеиновых кислот, в том числе генной транскрипции [Симонова B.C., Самусенко А.В., Филиппова Н.А., Тевяшова А.Н., Лынив Л.С., Кулик Г.И., Чехун В.Ф., Штиль А.А. «Оливомицин вызывает апоптоз опухолевых клеток и подавляет р53-индуцированную транскрипцию». Бюллетень экспериментальной биологии и медицины, 2005, том 139, № 4, стр.451-455]. Антибиотики группы ауреоловой кислоты наряду с ценными свойствами имеют ряд недостатков, основными из которых являются высокая токсичность, мутагенность, канцерогенность, миело- и иммунодепрессивное действие. Настоящее изобретение призвано получить новые производные антибиотика группы ауреоловой кислоты оливомицина 1, обладающие противоопухолевой активностью и низкой токсичностью. Изобретение включает соединения, соответствующие формуле 2. Формула 2.
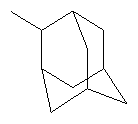
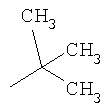
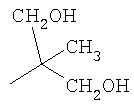
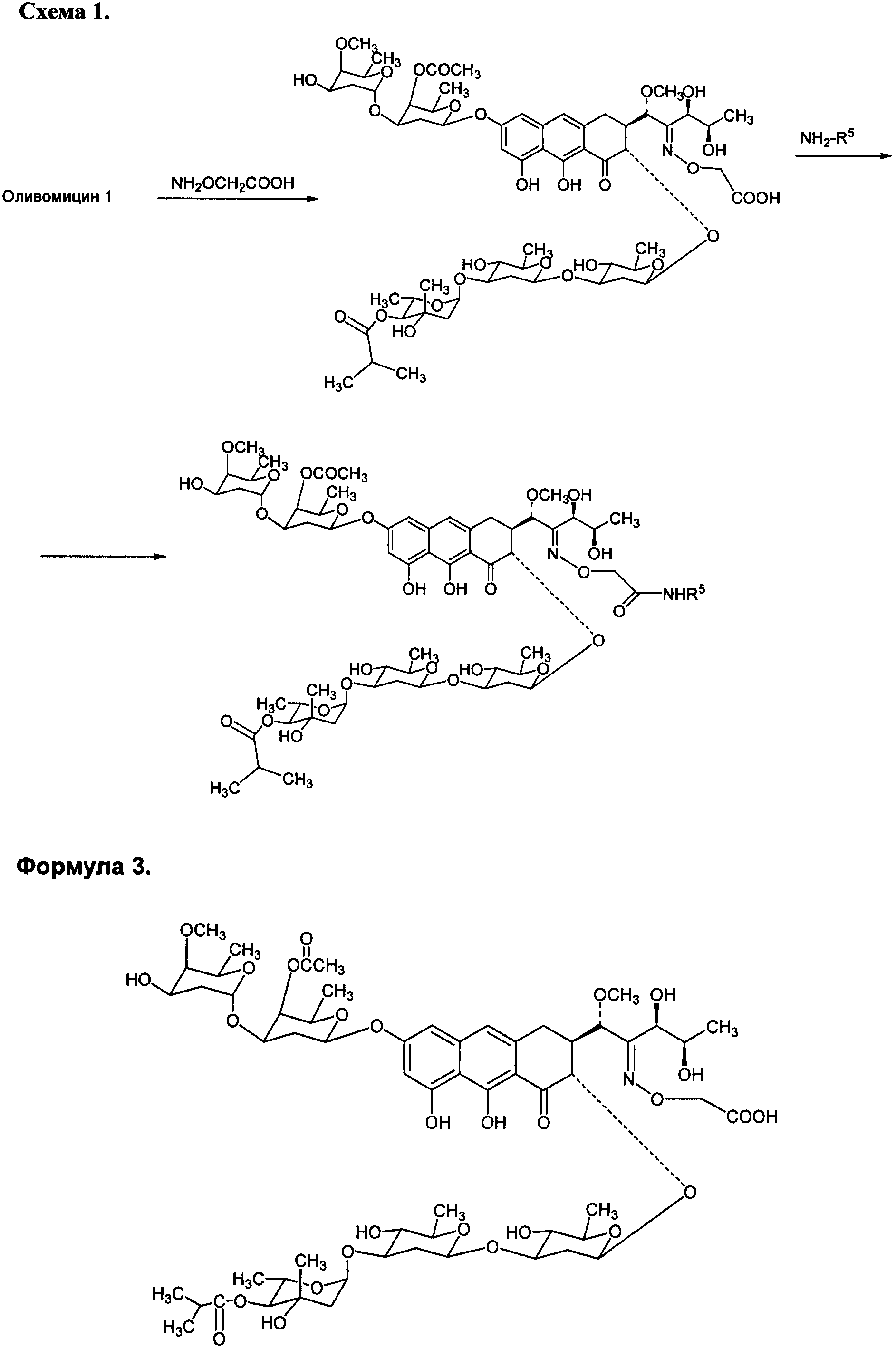
где R5 представляет собой водород, С3-С10-циклоалкил или С1-С4-алкил с прямой или разветвленной углеводородной цепью, необязательно замещенный одним или несколькими гидроксилами. Хотя химическая модификация природных антибиотиков, в том числе противоопухолевых, является важнейшим способом получения новых препаратов, обладающих преимуществами перед исходными антибиотиками, химическая модификация антибиотиков группы ауреоловой кислоты практически не проводилась. Известен полный синтез антибиотика оливомицина 1 [Roush W.R., Hartz R.A., Gustin D.J. «Total synthesis of olivomycin A», Journal of American Chemical Society, 1999, vol. 121, pp.1990-1991]. Описаны подходы к модификации оливомицина 1 по 2′-карбонильной группе и по ароматической части агликона. Авторами были описаны основные типы модификации оливомицина 1 по 2′-карбонильной группе: имин, оксимы, гидразон, семикарбазон. Все известные производные оливомицина 1 по карбонильной группе обладают цитотоксичностью, сравнимой с исходным оливомицином 1 [Kumar V., Remers W.A. “Preparation and antitumor activity of olivomycin A analogues”. Journal of Medicinal chemistry, 1980, vol.23, pp.376-379]. Изобретение также включает в себя способ получения производных антибиотика группы ауреоловой кислоты оливомицина 1 (формула 2), заключающийся в селективной модификации 2′-карбонильной группы оливомицина 1 реакцией с аминооксиуксусной кислотой (Схема 1), с последующим проведением реакции амидирования полученного ключевого интермедиата 2′-(карбоксиметоксим)оливомицина 1 (формула 3) с соотвествующими аминами в присутствии конденсирующего агента. Реакцию амидирования 2′-(карбоксиметоксим)оливомицина 1 (формула 3) амином с получением соединения формулы 2 проводят в присутствии конденсирующих агентов, известных из уровня техники и применяемых для образования амидной связи, например бензотриазол-1-ил-окси-триспирролидинофосфоний гексафторфосфата (РуВОР) или O-(бензотриазол-1-ил)-N,N,N’,N’-бис(тетраметилен)) гексафторфосфат мочевины (HBPyU). Реакцию амидирования 2′-(карбоксиметоксим)оливомицина 1 (формула 3) амином с получением соединения формулы 2 проводят в растворителе, выбираемом из метанола, этанола, N,N-диметилформамида, диметилсульфоксида, толуола, ксилола и хлороформа. Соединения формулы 2 обладают выраженной противоопухолевой активностью и сниженной токсичностью по сравнению с исходным оливомицином 1 (см. Примеры 4-6) и могут быть использованы для лечения онкологических заболеваний. Вспомогательные средства. Полугидрохлорид аминооксиуксусной кислоты, бензотриазол-1-ил-окси-триспирролидинофосфоний гексафторфосфат (РуВОР), O-(бензотриазол-1-ил)-N,N,N’,N’-бис(тетраметилен)) гексафторфосфат мочевины (HBPyU) были коммерческими продуктами фирмы Acros. Гидрохлорид 2-адамантиламина, триэтиламин были коммерческими продуктами фирмы Aldrich (США). Тонкослойную хроматографию осуществляли на пластинках с силикагелем G60 (Merck) в смесях растворителей: хлороформ-метанол-муравьиная кислота: 9:1:0.05 (А). Для препаративной очистки использовали колоночную хроматографию на силикагеле Merck G60 с размером частиц 0.040-0.063 ВЭЖХ проводили на приборе Shimadzu HPLC LC 10 на колонке Diaspher С18 № 1500, элюент: 0.01 н. Н3PO4 – MeCN, pH 2.6, в градиенте ацетонитрила от 40% до 80%, скорость потока 1.1 мл/мин. Регистрация велась на длине волны 274 нм, при температуре 20°С. ИК-спектры снимали в таблетке КВг на спектрофотометре DTGS. 1H ЯМР и 13С ЯМР спектры регистрировали на спектрометрах Varian VXR-400 (США) при частоте 400 МГц. Масс-спектры при ионизации электрораспылением (ESI) получали на приборе Finnigan MAT 900S (Германия), масс-спектры, полученные при ионизации методом MALDI (матричная лазерная десорбционная ионизация), получены на приборе Brucker BIFLEX III. Примеры получения производных антибиотика группы ауреоловой кислоты оливомицина 1 по настоящему изобретению и изучение их противоопухолевой активности. Пример 1. Получение 2′-(карбоксиметоксим)оливомицина 1 (LCTA 1295) (Формула 3). К раствору оливомицина I (0.100 мг, 0.084 ммоль) в МеОН (3 мл) добавляли полугидрохлорид аминооксиуксусной кислоты (4.5 мг, 0.42 ммоль). Реакционную смесь выдерживали при 37°С в течение 50 ч. Реакционную смесь упаривали, наносили на колонку с силикагелем, элюировали смесью CHCl3-МеОН-НСООН (9:1:0.05). Фракции, содержащие целевое вещество объединяли, удаляли растворитель на роторном вакуумном испарителе до минимального объема и добавляли петролейный эфир. Выпавший осадок отфильтровывали, промывали петролейным эфиром, высушивали. Выход: 53 мг (50%). Rf(A) 0.42, Rt 12.38. Тпл 140-142°С (с разложением). 13С-ЯМР (DMSO-d6) спектр 2′-(карбоксиметоксим)оливомицина 1 полностью соответствует 13С-ЯМР спектру исходного оливомицина (DMSO-d6) [отнесение сигналов см. Yoshimura Y., Koenuma M., Matsumoto К., Tori К., Terui Y. “MNR studies of Chromomycins, olivomycins and their derivatives”. The Journal of Antibiotics, 1988, vol. XLI, № 1, pp.53-67], за исключением: отсутствует сигнал 211.81 м.д., соответствующий 2′-карбонильной группе в оливомицине 1; появляются сигналы: 171.203 (-СООН), 158.815 (-C=N-), 70.45 (-ОСН2). MS MALDI: MW вычислено для С60Н87HO28 1269.54, найдено 1292.63 (M+Na)+. Пример 2. Общая методика амидирования 2′-(карбоксиметоксим)оливомицина 1. К раствору 2′-(карбоксиметоксим)оливомицина 1 (0.039 ммоль) в МеОН (3 мл) добавляли гидрохлорид амина (0.078 ммоль). Значение рН реакционной смеси доводили до 8-8.8 добавлением триэтиламина. Несколькими порциями добавляли конденсирующий реагент (0.058 ммоль), контролируя рН реакционной смеси добавлением триэтиламина. Реакционную смесь перемешивали в течение 2 ч, удаляли растворитель на роторном вакуумном испарителе. Остаток растворяли в CHCl3 и наносили на колонку с силикагелем. Вещество элюировали сначала CHCl3, а затем смесью CHCl3-МеОН-НСООН (9:1:0.05). Фракции, содержащие целевое вещество, объединяли, удаляли растворитель на роторном вакуумном испарителе до минимального объема и добавляли петролейный эфир. Выпавший осадок отфильтровывали, промывали петролейным эфиром, высушивали. Чистота полученных соединений изучалась методами ТСХ и ВЭЖХ. Структура полученных соединений была подтверждена данными масс-спектрометрии. Пример 3. Получение 2-адамантиламид 2′-(карбоксиметоксим)оливомицина 1 (LCTA-1297). К раствору 2′-(карбоксиметоксим)оливомицина 1 (50 мг, 0.039 ммоль) в МеОН (3 мл) добавляли гидрохлорид 2-адамантиламина (14.6 мг, 0.078 ммоль). Значение рН реакционной смеси доводили до 8-8.8 добавлением триэтиламина. Несколькими порциями добавляли РуВОР (30 мг, 0.058 ммоль), контролируя рН реакционной смеси добавлением триэтиламина. Реакционную смесь перемешивали в течение 2 ч, удаляли растворитель на роторном вакуумном испарителе. Остаток растворяли в CHCl3 и наносили на колонку с силикагелем. Вещество элюировали сначала CHCl3, а затем смесью CHCl3-МеОН-НСООН (9:1:0.05). Фракции, содержащие целевое вещество, объединяли, удаляли растворитель на роторном вакуумном испарителе до минимального объема и добавляли петролейный эфир. Выпавший осадок отфильтровывали, промывали петролейным эфиром, высушивали. Выход: 30 мг (55%). Rf(A) 0.27, Rt 19.46. Тпл 155-157°С (с разложением). MS MALDI: MW вычислено для C70H102N2O27 1402.67, найдено 1425.47 (M+Na)+. Аналогичным образом были получены в том числе следующие соединения: LCTA 1296 (амид 2′-(карбоксиметоксим)оливомицина 1), LCTA 1298 (этаноламид 2′-(карбоксиметоксим)оливомицина 1), LCTA 1344 (трет-бутиламид 2′-(карбоксиметоксим)оливомицина 1), LCTA 1345 (ди-(гидроксиметил)-метил-метил амид 2′-(карбоксиметоксим)оливомицина 1). Данные соединения соответствуют Формуле 2, где значения R5 представлены в таблице 1.
Пример 4. Изучение антипролиферативной активности заявленных новых производных оливомицина 1 проводилось на культуре опухолевых клеток мышиного лейкоза L1210/0. Данные об антипролиферативной активности изученных производных оливомицина 1 представлены в Таблице 2.
IC50 – концентрация соединения, вызывающая гибель 50% клеток Изучение антипролиферативной активности производных оливомицина 1 проводилось также на культурах опухолевых клеток линий лейкоза человека К562, рака толстой кишки НСТ116 и рака молочной железы MCF-7. Данные этих экспериментов представлены в таблице 3. Таблица 3. Антипролиферативная активность оливомицина 1 и новых производных оливомицина 1 в отношении опухолевых клеток линии MCF-7, К562, НСТ116.
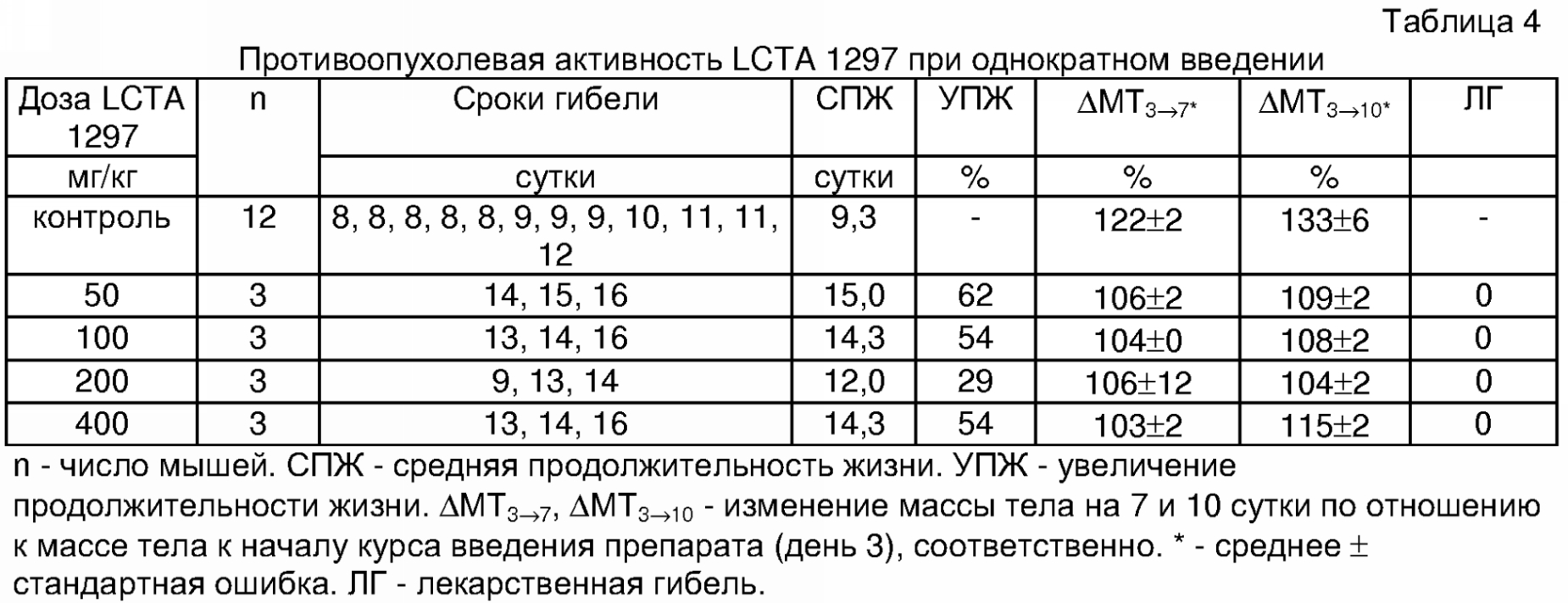
IC50 – концентрация соединения, вызывающая гибель 50% клеток Пример 5. Изучение противоопухолевой активности препарата LCTA 1297 (2-адамантиламид 2′-(карбоксиметоксим)оливомицина 1) при однократном введении проводили на мышах с лимфомой Р388. Самцы мышей B6D2F1 привиты внутрибрюшинно по 106 клеток асцитной лимфомы Р388 в день 0. Препарат LCTA 1297 растворяли в 0,1% ДМСО, разводили физиологическим раствором поваренной соли и вводили однократно внутрибрюшинно в день 3 (Таблица 4).
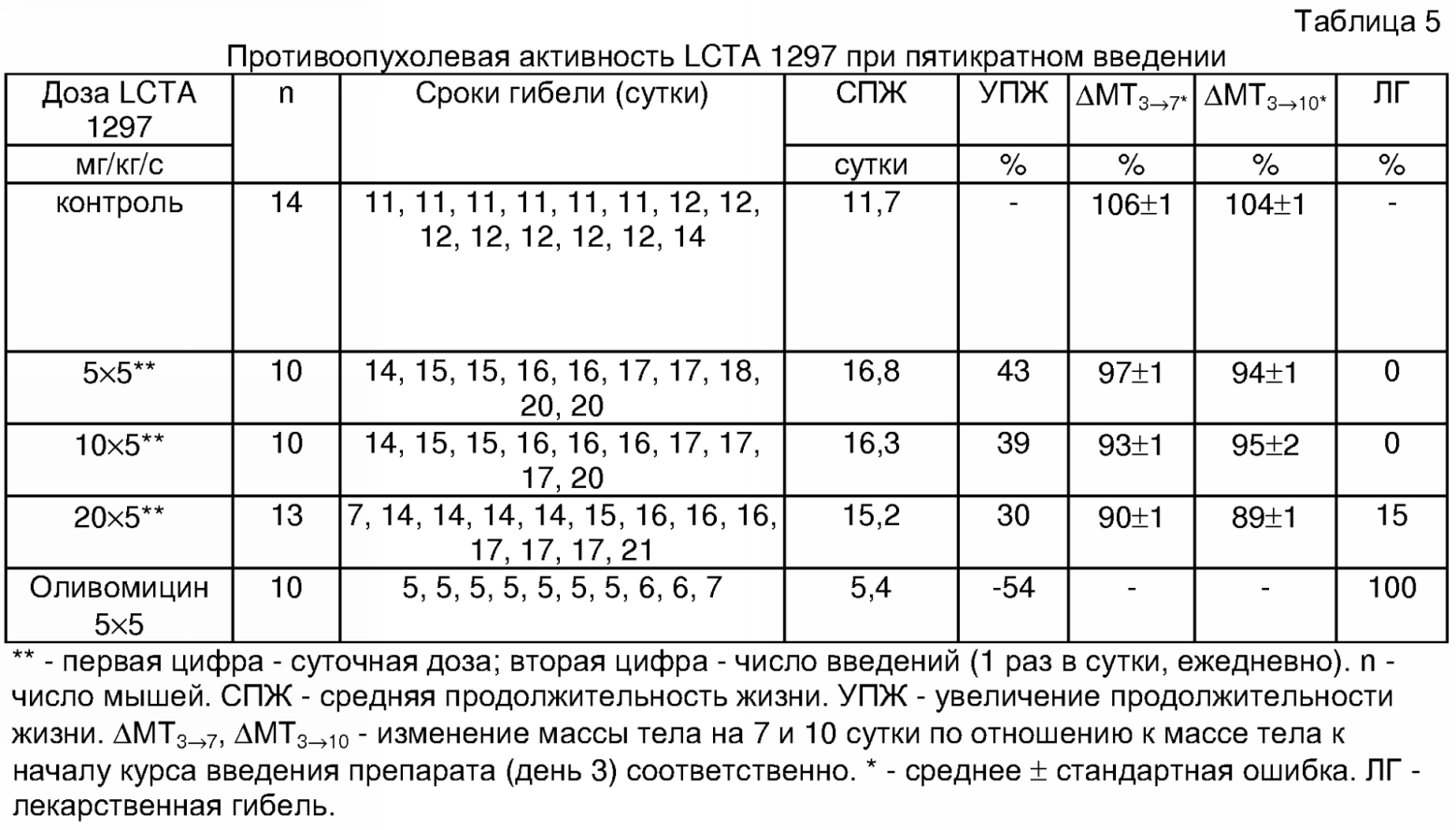
Как видно из представленных данных, при введении соединения в дозах от 50 до 400 мг/кг внутрибрюшинно наблюдалось выраженное противоопухолевое действие препарата при отсутствии токсической гибели мышей. Продолжительность жизни леченых мышей была выше по сравнению с контрольной группой (УПЖ до 62%). Введение препарата LCTA 1297 тормозило накопление асцита, что выражалось в высоко достоверном замедлении увеличения массы тела, масса тела у контрольных мышей увеличилась к 7 суткам до 122% за счет накопления асцита. Пример 6. Изучение противоопухолевой активности препарата LCTA 1297 при многократном введении проводили на мышах с лимфомой Р388. Самцы мышей B6D2F1 привиты внутрибрюшинно по 106 клеток асцитной лимфомы Р388 в день 0. Препарат LCTA 1297 растворяли в 0,1% ДМСО, разводили физиологическим раствором поваренной соли и вводили курсом с 3 по 7 сутки внутрибрюшинно (Таблица 5).
Как видно из представленных данных, при введении соединения LCTA 1297 в дозах от 5 до 10 мг/кг в сутки внутрибрюшинно отмечено выраженное противоопухолевое действие препарата и отсутствие токсической гибели мышей. Продолжительность жизни леченых мышей превышала таковую у контрольных мышей (УПЖ до 43%). Введение LCTA 1297 тормозило накопление асцита, что выражалось в высокодостоверном замедлении увеличения массы тела. Масса тела у контрольных мышей увеличилась к 7 и 10 суткам до 106% и 104% соответственно за счет накопления асцита. При применении препарата в дозе 20 мг/кг в сутки внутрибрюшинно наблюдалась токсическая гибель 2 из 13 мышей на фоне значительного снижения веса животных. При применении оливомицина 1 в дозе до 5 мг/кг в сутки внутрибрюшинно наблюдалась токсическая гибель всех мышей, причем в большинстве случаев после трех инъекций. Таким образом, предложенный новый способ модификации антибиотика группы ауреоловой кислоты оливомицина 1 позволяет получать новые производные оливомицина 1, обладающие преимуществами перед исходным антибиотиком оливомицином 1, а именно: высокой противоопухолевой активностью и низкой токсичностью соединений.
Формула изобретения
1. Производные антибиотика группы ауреоловой кислоты, соответствующие формуле: где R5 представляет собой водород, С3-С10-циклоалкил или С1-С4-алкил с прямой или разветвленной углеводородной цепью, необязательно замещенный одним или несколькими гидроксилами. 2. Способ получения производных антибиотика группы ауреоловой кислоты оливомицина 1 по п.1, заключающийся в селективной модификации 2′-карбонильной группы оливомицина 1 реакцией с аминооксиуксусной кислотой, с последующим проведением реакции амидирования полученного ключевого интермедиата 2′-(карбоксиметоксим)оливомицина 1 с соответствующими аминами в присутствии конденсирующего агента.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 м.
м.