Патент на изобретение №2348643
|
||||||||||||||||||||||||||
(54) СПОСОБ ПОЛУЧЕНИЯ СОЛЕЙ 5′-ТРИФОСФАТОВ ПРИРОДНЫХ И МОДИФИЦИРОВАННЫХ ДЕЗОКСИРИБО- И РИБООЛИГОНУКЛЕОТИДОВ
(57) Реферат:
Изобретение относится к способу получения солей 5′-трифосфатов природных и модифицированных дезокси- и рибоолигонуклеотидов, заключающемуся в том, что исходный реагент – защищенный природный или модифицированный олигонуклеотид, дезокси- или риборяда, в виде 0,1-0,05 М раствора, монофосфорилируют в пиридине 2-3-кратным избытком хлорокиси фосфора в течение 10-15 минут, далее полученное активированное производное олигонуклеотида обрабатывают 10-15-кратным избытком раствора бис-трибутиламмонийной соли пирофосфата в ацетонитриле и 20-кратным избытком третичного амина и выдерживают реакционную смесь в течение 15-30 минут с последующим разложением промежуточного триметафосфатного производного олигонуклеотида триэтиламмонийбикарбонатным буфером, очисткой целевого продукта с помощью обращенно-фазовой хроматографии (ОФХ), удалением защитных групп с функциональных групп олигонуклеотида и повторной очисткой целевого продукта с помощью ОФХ. Выходы целевых продуктов после очистки составляют 45-92%. Чистота полученных соединений более 95% по данным ВЭЖХ и ЯМР-спектроскопии. Данные соединения могут быть использованы в молекулярно-биологических и генно-инженерных исследованиях. 1 н. и 1 з.п. ф-лы, 1 ил., 2 табл.
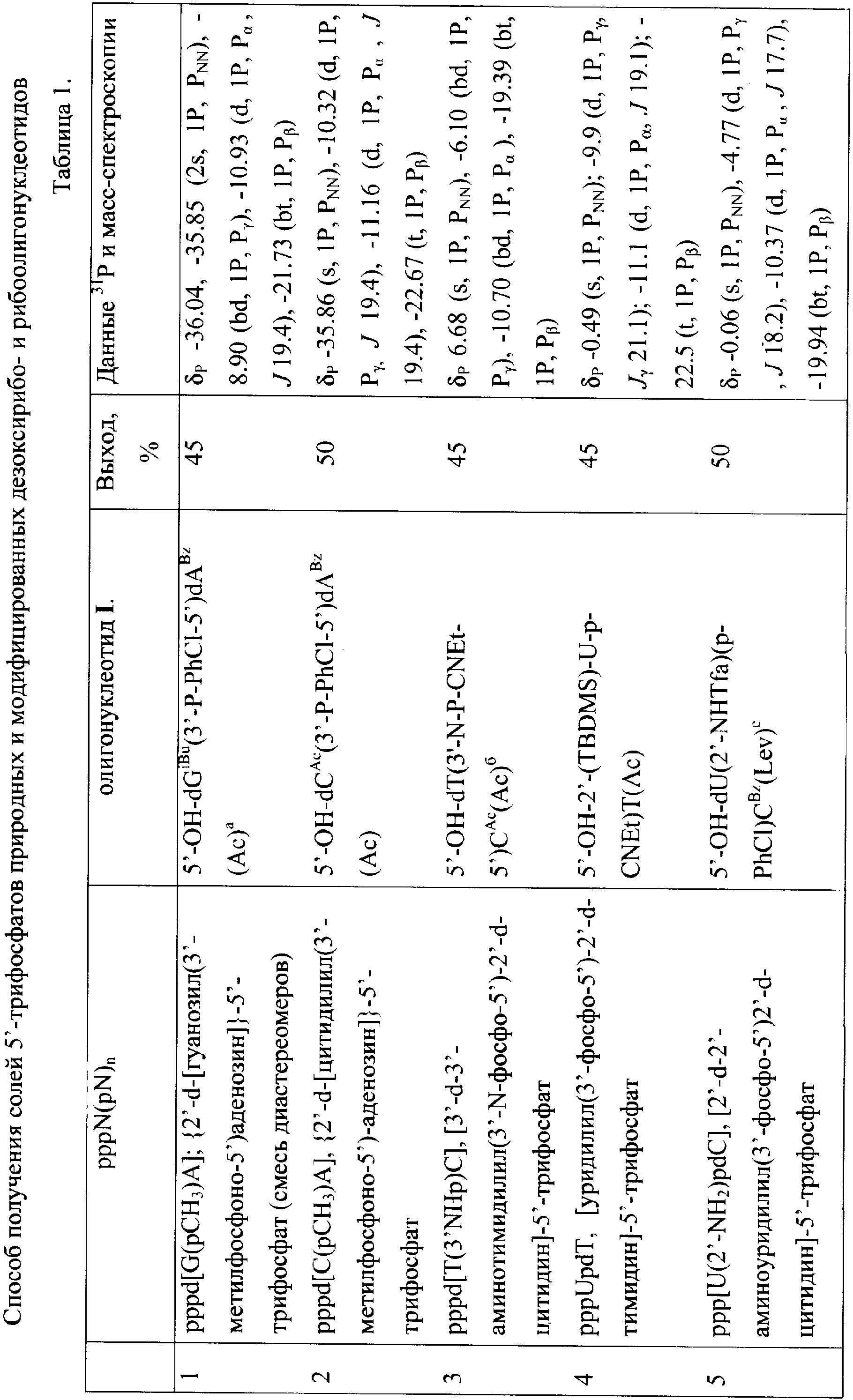
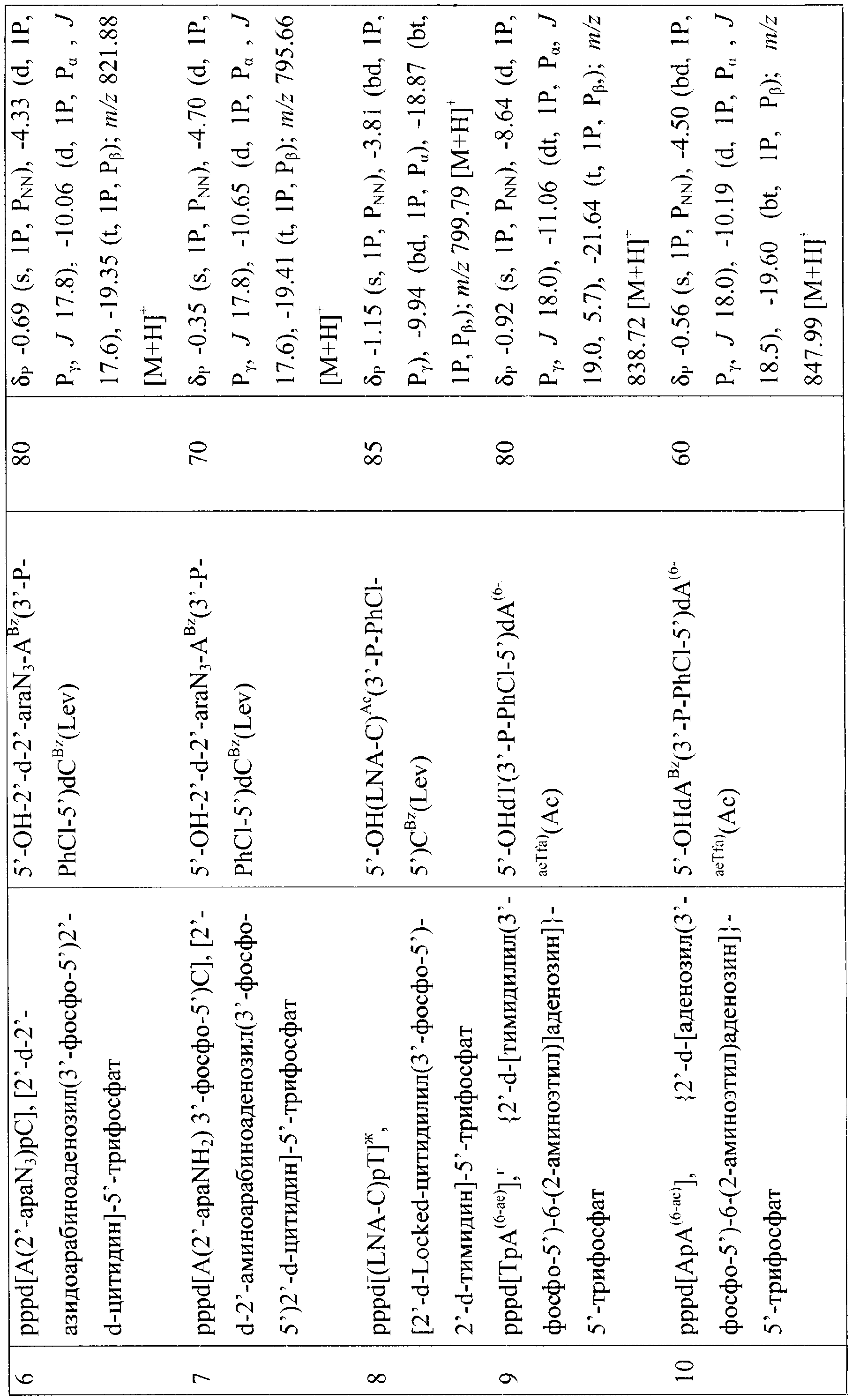
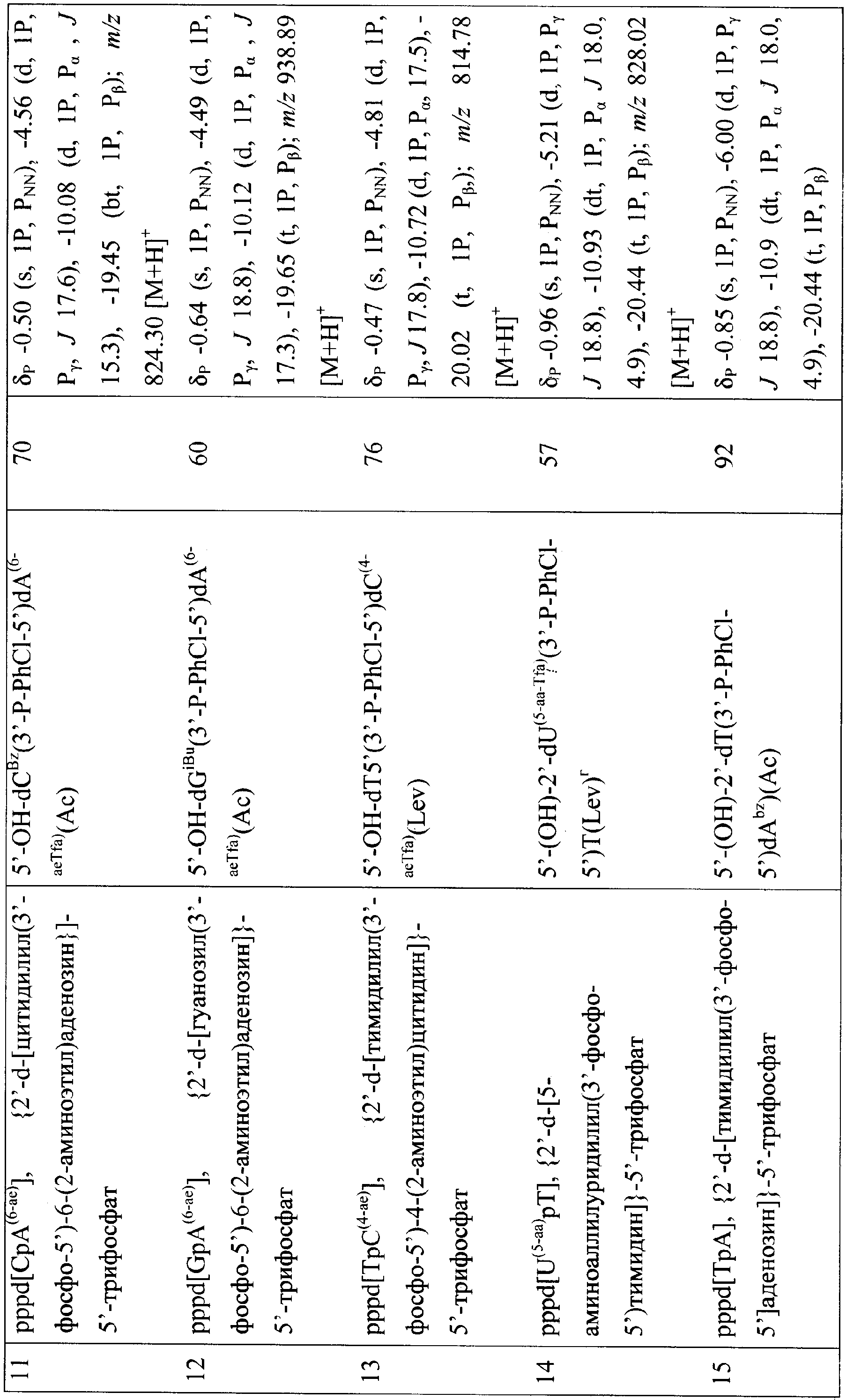
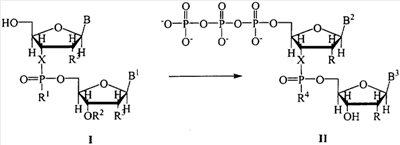
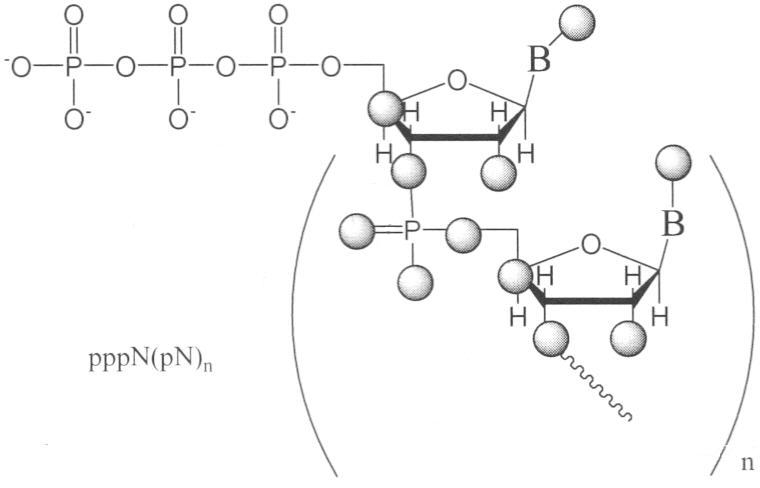
Изобретение относится к области биоорганической химии, в частности к усовершенствованному способу получения солей 5′-трифосфатов природных и модифицированных дезоксирибо- и рибоолигонуклеотидов общей формулы
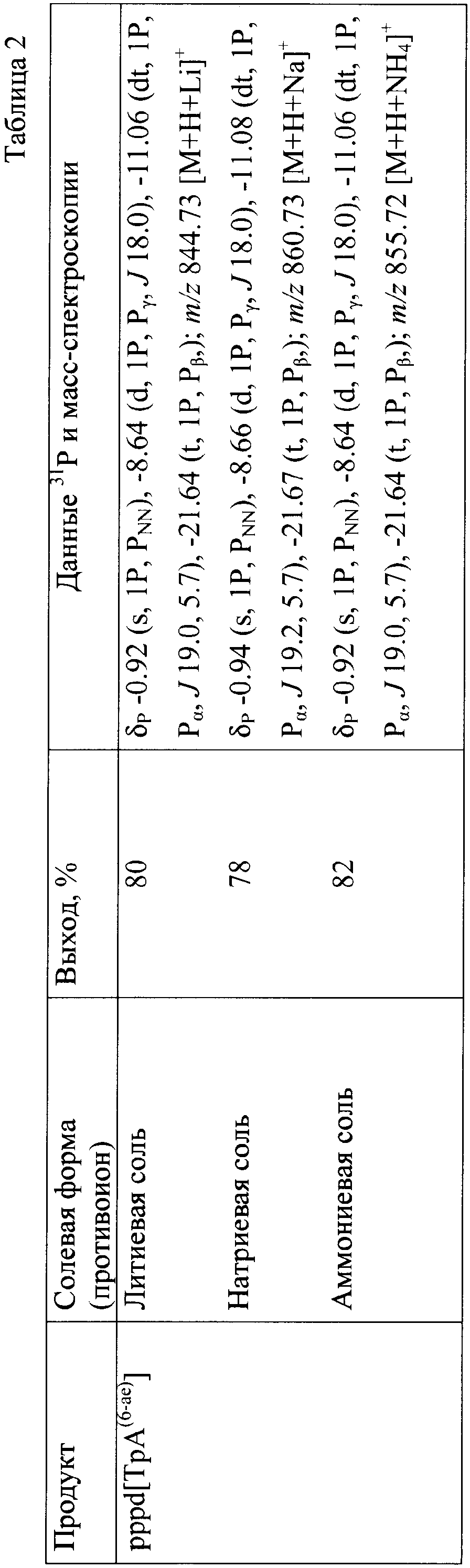
где В – гетероциклическое основание, природное или модифицированное; полупрозрачные сферы обозначают места возможной модификации олигонуклеотида; n 5′-Трифосфаты природных и модифицированных дезоксирибо- и рибоолигонуклеотидов применяются в молекулярно-биологических и генно-инженерных исследованиях, однако коммерчески малодоступны из-за отсутствия универсального и эффективного способа синтеза этих соединений. Доступность 5′-трифосфатов природных и модифицированных дезоксирибо- и рибоолигонуклеотидов позволит широко использовать эти соединения при изучении процессов нуклеинового обмена (например, процессинга пре-мРНК). Недостатком этого способа является то, что при обработке исходного динуклеотида хлорокисью фосфора на первой стадии синтеза происходит полная изомеризация межнуклеотидной фосфодиэфирной связи с образованием смеси 2′(3′)-5′-фосфодиэфирных связей, что снижает выход целевого соединения в 2 раза и усложняет процедуру его очистки. Кроме того, способ не позволяет синтезировать трифосфаты модифицированных олигонуклеотидов, содержащие активные функциональные группы (амино(ди)алкильные, тиоалкильные, оксиалкильные). 2+с последующим получением 5′-трифосфатного производного олигорибонуклеотида ферментативным путем. Недостатком этого способа является то, что он не пригоден для получения 5′-трифосфатов олигорибонуклеотидов с природными 3′-5′-фосфодиэфирными связями. Кроме того, с помощью указанного способа можно синтезировать только производные аденозина. 2, pppdT3, pppdT9, pppdT15, pppd(ACTGT) и pppdC2. Выходы продуктов после очистки составляют 15-30%. Недостатками указанного способа являются следующие: 1) использование огромных избытков реагентов (до 3000-кратного для 2-хлоро-4Н1,3,2-бензодиоксафосфорин-4-она и до 6000-кратного для пирофосфата), а также больших объемов растворителей в расчете на 1 мкмоль исходного соединения; 2) низкие выходы целевых соединений (15-30%); 3) малое количество получаемого продукта, не превышающее 2-5 мкмоль (2-5 мг), и трудности в масштабировании процесса; 4) наличие значительных количеств побочных продуктов; 5) применение дорогостоящей анионообменной ВЭЖХ для очистки продукта. Технической задачей изобретения является повышение выхода целевых продуктов, сокращение избытка используемых реагентов и растворителей, а также расширение функциональных возможностей способа за счет обеспечения возможности получения 5′-трифосфатов олигонуклеотидов не только природного строения дезокси- и риборяда, но также и содержащих различные модификации в сахарофосфатном остове и гетероциклических основаниях. Поставленная техническая задача достигается предлагаемым новым способом получения 5′-трифосфатов природных и модифицированных олигонуклеотидов как дезоксирибо-, так и риборяда с формулой pppN(pN)n, заключающимся в следующем. На чертеже представлена схема синтеза 5′-трифосфатов природных и модифицированных дезоксирибо- и рибоолигонуклеотидов на примере синтеза 5′-трифосфата модифицированного дезоксидинуклеотида {2′-d-[цитидилил(3′-фосфо-5′)-5-аминоаллилуридин]}-5′-трифосфата, где В=N4- бензоилцитозин, В’=5-трифторацетамидоаллилурацил, В2=цитозин, В3=5-(аминоаллил)урацил, R1=O-n-хлорфенил, R2=ацетил, R=R3=Н, R4=О-, Х=О; i), POCl3; ii) [(С4Н9)3N]2Н2Р2O7; iii) NH3/H2O. Синтез осуществляют в три стадии в растворе при комнатной температуре. На первой стадии исходный реагент I – защищенный природный или модифицированный олигонуклеотид, имеющий свободной только одну функциональную группу – 5′-оксигруппу, дезокси- или риборяда, в виде 0,1-0,05 М раствора, монофосфорилируют в пиридине 2-3-кратным избытком хлорокиси фосфора в течение 10-15 мин с получением активированного производного 5′-монофссфата природного или модифицированного защищенного дезокси- или рибоолигонуклеотида. На второй стадии (ii) в раствор 10-15-кратного избытка бис-трибутиламмонийной соли пирофосфата в ацетонитриле и 20-кратного избытка третичного амина (преимущественно трибутиламина) добавляют раствор активированного монофосфорилированного производного в пиридине и выдерживают смесь в течение 15-30 минут. На третьей стадии (iii) производят разложение промежуточного триметафосфатного производного олигонуклеотида триэтиламмонийбикарбонатным буфером (ТЕАБ), рН 7,5, и удаление защитных групп с сахарофосфатного остова, гетероциклических оснований и других функциональных групп олигонуклеотида. Очистку целевого продукта II проводят с помощью обращено-фазовой хроматографии (ОФХ) после разложения реакционной смеси ТЕАБ на сорбенте Porasil С-18 (Waters, США) и повторной ОФХ на том же сорбенте после удаления защитных групп. Чистота полученных соединений более 95% по данным аналитической высокоэффективной жидкостной хроматографии (ВЭЖХ) и ЯМР-спектроскопии. Целевые продукты получают в виде литиевых солей, поскольку они наиболее стабильны при хранении. Для этого после завершения очистки целевой продукт осаждают из водного раствора десятикратным объемом 6% раствора перхлората лития в ацетоне. Осадок промывают ацетоном и серным эфиром и высушивают. Выходы целевых 5′-трифосфатов природных и модифицированных дезокси- и рибоолигонуклеотидов после очистки составляют 45-92% в зависимости от нуклеотидной последовательности и длины олигонуклеотида. Количество получаемых продуктов составляет 10-100 мкмоль (10-100 мг) и выше. Примеры модификаций, введенных в 5′-трифосфаты олигонуклеотидов (см. чертеж, соединения II, таблица 1), включают в себя модификации по гетероциклическому основанию (4-N-(аминоалкил)цитозин, в том числе 5-метилированный, 5-(3-аминоаллил)урацил, 5-(3-аминопропилоксиметил)урацил, 6-N-(аминоалкил)аденин), пример конкретного выполнения 1; модификации углеводного остатка (арабиноза вместо рибозы или дезоксирибозы, 2′-дезокси-2′-амино, 2′-азидо или 2′-метоксирибоза, стерически затрудненные 4′-СН2-О-2′-locked-нуклеозиды, пример конкретного выполнения 2; получение химерных рибо-дезоксирибоолигонуклеотидов, пример конкретного выполнения 3; модификации остатка фосфорной кислоты (метилфосфоно, тиофосфаты), пример конкретного выполнения 4, а также не имеющие модификаций – природные, пример конкретного выполнения 5). 5′-Трифосфаты модифицированных дезокси- и рибоолигонуклеотидов, преимущественно в виде литиевых солей, полученные по заявляемому способу, приведены в табл.1 и табл.2, где а P-PhCl – n-хлорфенилфосфатная группа, б P-CNEt – цианоэтилфосфатная группа, в Tfa – трифторацетил, Г ае – аминоэтил, д аа – аминоаллил, е Lev – левулинил, ж LNA – стерически затрудненные 4′-СН2-О-2′-locked-нуклеозиды. Строение полученных соединений подтверждено с помощью 31Р- и масс-спектроскопии. Данные по некоторым соединениям приведены в табл.1 и табл.2. Определяющими отличиями предлагаемого способа от прототипа, являются следующие. 1. Синтез проводят в растворе, при этом в качестве исходного реагента используют 0.1-0.05 М раствор предварительно очищенного соответствующим образом защищенного природного или модифицированного динуклеозидфосфата, имеющего свободной только одну функциональную группу – 5′-оксигруппу, дезокси- или риборяда, что позволяет избежать высоких избытков реагентов, а также больших объемов растворителей в расчете на 1 мкмоль исходного реагента, а также легко масштабировать процесс, что обеспечивает получение больших (миллимолярных) количеств продукта. В прототипе получение продукта осуществляют в твердофазном варианте, а в качестве исходного реагента используют соответствующим образом защищенный дезоксиолигонуклеотид (защитные группы на гетероциклических основаниях нуклеозидов и межнуклеотидных фосфатных группах, свободная 5′-оксигруппа дезоксиолигонуклеотидной цепи), взятый непосредственно из реакционной смеси в процессе синтеза олигонуклеотида. 2. Исходный реагент активируют в пиридине 2-3-кратным избытком хлорокиси фосфора в течение 10-15 минут, что позволяет эффективно и быстро получить монофосфорилированное производное, так как использование соответствующим образом защищенных исходных реагентов исключает возможные побочные реакции с избытком хлорокиси фосфора, в то же время проведение активации в растворе позволяет избежать необходимости применения больших избытков фосфорилирующего агента. В прототипе в качестве активирующего реагента используют 3000-кратный избыток 2-хлоро-4Н1,3,2-бензодиоксафосфорин-4-она. 3. Активированный монофосфорилированный олигонуклеотид обрабатывают 10-15-кратным избытком раствора бис-трибутиламмонийной соли пирофосфата в ацетонитриле и 20-кратным избытком третичного амина (преимущественно трибутиламина) и выдерживают реакционную смесь в течение 15-30 минут, что позволяет повысить выход целевого продукта и существенно сократить расход пирофосфата. В прототипе используют 6000-кратный избыток пирофосфата. 4. Очистку целевого продукта проводят с помощью двух обращенно-фазовых хроматографий, причем первую обращенно-фазовую ВЭЖХ проводят после разложения промежуточного триметафосфатного производного защищенного олигонуклеотида, а вторую – после удаления защитных групп. Применение двух последовательных обращенно-фазовых ВЭЖХ позволяет обеспечить полное отделение пирофосфата от целевого продукта в случае динуклеотидов, а также избежать применения дорогостоящих анионообменных сорбентов. В прототипе используют анионообменную ВЭЖХ для очистки целевого соединения. Способ иллюстрируется следующими примерами конкретного выполнения. Пример 1. Получение pppd[TpA(6-ae)], {2”-d-[тимидилил(3′-фосфо-5′)-6-(2-аминоэтил)аденозин]}-5′-трифосфата в виде трилитиевой соли. В колбу вместимостью 25 мл загружают 0.340 г (0.4 ммоль) 5′-окси-2′-d-[тимидилил-(3′-n-хлорфенилфосфо-5′)-6-(2-трифторацетиламиноэтил)-3′-O-ацетиладенозин]а и 5 мл сухого пиридина, реакционный сосуд охлаждают в ледяной бане. К раствору оксипроизводного (концентрация 0.08 М) при перемешивании на магнитной мешалке добавляют 73 мкл (0.8 ммоль, двукратный избыток) хлорокиси фосфора. Реакционную смесь перемешивают 10 мин, затем переносят при интенсивном перемешивании в круглодонную колбу объемом 100 мл, в которой находятся 8 мл 0.5 М раствора бис-трибутиламмонийпирофосфата в ацетонитриле (4 ммоль, 10-кратный избыток), и 1.92 мл (8 ммоль, 20-кратный избыток) трибутиламина. Охлаждение убирают, перемешивание продолжают в течение 30 минут. По окончании реакции в реакционную смесь добавляют 40 мл 1 М триэтиламмонийбикарбоната, рН 7.5. Через 1 час реакционную смесь упаривают при пониженном давлении на ротационном испарителе, остаток растворяют в 10 мл воды и наносят на колонку (3×20 см) с обращенно-фазовой смолой Porasil С18 (55-100 мкм, Waters, США). Элюцию проводят в градиенте этанола в воде от 0 до 50%. Фракции, содержащие трифосфатное производное, упаривают. К остатку добавляют 20 мл концентрированного водного аммиака для удаления защитных групп (ацильной, n-хлорфенильной, трифторацетильной). Реакционную смесь перемешивают 2 суток при комнатной температуре, затем упаривают. Остаток растворяют в 5 мл 1 М триэтиламмонийацетата (ТЭА-НОАс), рН 7.0, и наносят на колонку с обращенно-фазовой смолой LiChroprep C18 (15-25 мкм, Merck, Германия). Элюцию проводят в градиенте ацетонитрила от 0 до 10% в 0.05 М ТЭА-НОАс, рН 7.0. Целевые фракции упаривают. Продукт – трилитиевую соль {2′-d-[тимидилил(3′-фосфо-5′)-6-(2-аминоэтил)аденозин]}-5′-трифосфата Li3pppd[TpA(6-ae)], растворяют в воде (5 мл) и осаждают добавлением 50 мл 6% раствора перхлората лития в ацетоне. Осадок отделяют центрифугированим или фильтрованием, промывают ацетоном и высушивают до постоянного веса в вакууме. Выход Li3pppd[TpA(6-ae)] 270.0 мг, 0.32 ммоль, 80%. Пример 2. Получение 5′-трифосфата [2′-d-2′-азидоарабиноаденозил(3′-фосфо-5′)цитидин]а в виде трилитиевой соли Li3pppd[А(2′-apaN3)рС] В колбу вместимостью 25 мл загружают 0.150 г (0.15 ммоль) 5′-окси-[2′-d-2′-азидоарабиноаденозил-(3′-n-хлорфенилфосфо-5′)-2′-d-3′-O-ацетил-4-N-бензоилцитидин]а и 3 мл сухого пиридина, реакционный сосуд охлаждают в ледяной бане. К раствору оксипроизводного (концентрация 0.05 М) при перемешивании на магнитной мешалке добавляют 41 мкл (0.45 ммоль, трехкратный избыток) хлорокиси фосфора. Реакционную смесь перемешивают 15 мин, затем переносят при интенсивном перемешивании в круглодонную колбу объемом 100 мл, в которой находятся 5 мл 0.5 М раствора бис-трибутиламмонийпирофосфата в ацетонитриле (2.25 ммоль, 15-кратный избыток), и 0.72 мл (3 ммоль, 20-кратный избыток) трибутиламина. Охлаждение убирают, перемешивание продолжают в течение 15 минут. По окончании реакции в реакционную смесь добавляют 30 мл 1 М триэтиламмонийбикарбоната, рН 7.5. Далее процесс осуществляют аналогично примеру 1. Из 0.15 г (0.150 ммоль) исходного соединения получают 0.1 г (0.12 ммоль, выход 80%) Li3pppd[A(2′-apaN3)рС]. Пример 3. Получение 5′-трифосфата [уридилил(3′-фосфо-5′)-2′-d-тимидин]а в виде трилитиевой соли проводят аналогично примеру 1 со следующими отличиями. В качестве исходного соединения применяют 5′-окси-[2′-O-трет-бутилдиметилсилилуридин(3′-(2-цианоэтил)фосфо-5′)-2′-d-3′-O-ацетилтимидин]. Для удаления трет-бутилдиметилсилильной защитной группы после обработки аммиаком промежуточного продукта реакционную смесь упаривают, к остатку добавляют 10 мл 1 М раствора тетрабутиламмонийфторида в диоксане. Раствор перемешивают 1 сутки и упаривают. Из 0.05 г (0.050 ммоль) исходного соединения получают 18 мг (0.022 ммоль, выход 45%) Li3pppUpdT]. Пример 4. Получение 5′-трифосфата 2′-d-[цитидилил(3′-метилфосфоно-5′)аденозин]а в виде дилитиевой соли Li2pppd[С(рСН3)А] проводят аналогично примеру 1 со следующими отличиями. В качестве исходного соединения берут 5′-окси-2′-d-[4-N-ацетилцитидилил-(3′-метилфосфоно-5′)-6-(2-трифторацетиламиноэтил)-3′-O-ацетил-6-N-бензоиладенозин]. Удаление основнолабильных защитных групп проводят путем обработки водным аммиаком в течение 1 суток. Из 0.05 г (0.06 ммоль) исходного соединения получают 24 мг (0,03 ммоль, 50%) Li2pppd[C(pCH3)A]. Пример 5. Получение 5′-трифосфата 2′-d-[тимидилил(3′-фосфо-5′)аденозин]а в виде трилитиевой соли Li3pppd(TpA) проводят аналогично примеру 1 со следующими отличиями. В качестве исходного соединения берут 5′-окси-2′-d-[тимидилил-(3′-N-хлорфенилфосфо-5′)-6-N-бензоил-3′-O-ацетиладенозин]. Из 0.1 г (0.1 ммоль) исходного соединения получают 75 мг (0.092 ммоль, 92%) Li3pppd(TpA). Пример 6. Получение Na3pppd[TpA(6-ae)], {2′-d-[тимидилил(3′-фосфо-5′)-6-(2-аминоэтил)аденозин]}-5′-трифосфата в виде тринатриевой соли проводят аналогично примеру 1 со следующими отличиями. Монофосфорилирование защищенного исходного соединения проводят без охлаждения реакционного сосуда в ледяной бане. После очистки целевого соединения на колонке с обращенно-фазовой смолой LiChroprep С18 (15-25 мкм, Merck, Германия) и упаривания целевых фракций продукт – тринатриевую соль {2′-d-[тимидилил(3′-фосфо-5′)-6-(2-аминоэтил)аденозин]}-5′-трифосфата Na3pppd[TpA(6-ae)], растворяют в воде (5 мл) и осаждают добавлением 50 мл 6% раствора перхлората натрия в ацетоне. Осадок отделяют центрифугированим или фильтрованием, промывают ацетоном и высушивают до постоянного веса в вакууме. Выход Na3pppd[TpA(6-ae)] 280.0 мг, 0.31 ммоль, 78%. Пример 7. Получение (NH4)3pppd[TpA(6-ae], {2′-d-[тимидилил(3′-фосфо-5′)-6-(2-аминоэтил)аденозин]}-5′-трифосфата в виде триаммониевой соли проводят аналогично примеру 1 со следующими отличиями. После очистки целевого соединения на колонке с обращенно-фазовой смолой LiChroprep С18 (15-25 мкм, Merck, Германия) и упаривания целевых фракций продукт – триаммониевую соль {2′-d-[тимидилил(3′-фосфо-5′)-6-(2-аминоэтил)аденозин]}-5′-трифосфата (NH4)3pppd[TpA(6-ae)] получают пропусканием раствора трифосфата в воде (15 мл) через колонку (20 мл) со смолой ДЕАЕ-Сефадекс А-25 (Pharmacia, Швеция) в аммониевой форме и промывкой колонки 1.5 М NH4НСО3 20% водным этанолом (40 мл) с последующим упариванием полученного раствора и высушиванием остатка до постоянного веса в вакууме. Выход (NH4)3pppd[TpA(6-ac)] 275.0 мг, 0.33 ммоль, 82%. Таким образом, разработан универсальный способ получения 5′-трифосфатов дезокси- и рибоолигонуклеотидов в милимолярных количествах с высокими выходами, меньшим избытком реагентов и объемов растворителей, необходимых при синтезе.
Формула изобретения
1. Способ получения солей 5′-трифосфатов природных и модифицированных дезокси- и рибоолигонуклеотидов, заключающийся в том, что исходный реагент – защищенный природный или модифицированный олигонуклеотид, дезокси- или риборяда, в виде 0,1-0,05 М раствора, монофосфорилируют в пиридине 2-3-кратным избытком хлорокиси фосфора в течение 10-15 мин, далее полученное активированное производное олигонуклеотида обрабатывают 10-15-кратным избытком раствора бис-трибутиламмонийной соли пирофосфата в ацетонитриле и 20-кратным избытком третичного амина и выдерживают реакционную смесь в течение 15-30 мин с последующим разложением промежуточного триметафосфатного производного олигонуклеотида триэтиламмонийбикарбонатным буфером, очисткой целевого продукта с помощью обращенно-фазовой хроматографии (ОФХ), удалением защитных групп с функциональных групп олигонуклеотида и повторной очисткой целевого продукта с помощью ОФХ. 2. Способ по п.1, отличающийся тем, что в качестве третичного амина используют трибутиламин.
РИСУНКИ
|
||||||||||||||||||||||||||
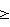 1.
1.