Патент на изобретение №2347786
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) ПРОИЗВОДНЫЕ СЛОЖНОЭФИРНЫХ ЛИПИДОВ НУКЛЕОТИДОВ
(57) Реферат:
Настоящее изобретение относится к сложноэфирным липидам галогенированных адениновых нуклеотидов формулы I, которые могут быть использованы при лечении опухолевых заболеваний.
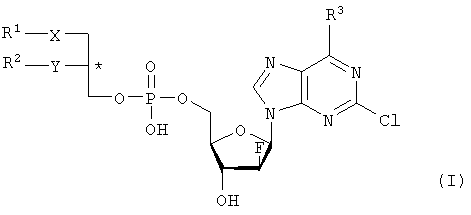
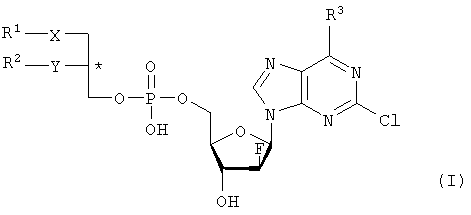
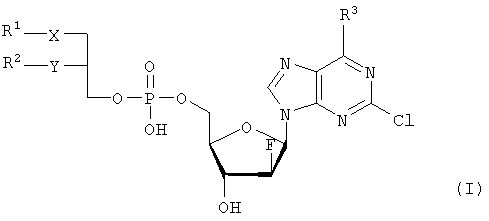
где R1 – С1-С20-алкил, необязательно замещенный C1-С6-алкоксильным радикалом, C1-С6-алкилмеркаптановым радикалом, C1-С6-алкилсульфинильной или C1-С6-алкилсульфонильной группами, R2 – С1-С20-алкил, необязательно замещенный C1-С6-алкоксильным радикалом, C1-С6-алкилмеркаптановым радикалом или C1-С6-алкилсульфонильной группой, R3 – аминогруппа, Х – атом серы, сульфинильная или сульфонильная группы, Y – атом кислорода. Технический результат – получение новых биологически активных соединений. 3 н. и 5 з.п. ф-лы, 1 табл.
(56) (продолжение): CLASS=”b560m”MEDICINAL CHEMISTRY LETTERS, 1995, 2999-3002. MONTGOMERY J.A. ET ALL, JOURNAL OF MEDICINAL CHEMISTRY, 1992, VOL.35, №2, 397-401.
Объектом настоящего изобретения являются специфические сложноэфирные липиды нуклеотидов общей формулы I
где R1 – неразветвленный или разветвленный, насыщенный или ненасыщенный алкильный радикал, имеющий 1-20 углеродных атомов, необязательно моно- или полизамещенный галогеном, алкоксильным радикалом C1-С6, алкилмеркаптановым радикалом C1-С6, алкоксикарбонилом C1-С6, C1-С6-алкилсульфинил – или C1-С6-алкилсулфонилгруппами, R2 – атом водорода, разветвленная или неразветвленная, насыщенная или ненасыщенная алкильная цепь, имеющая 1-20 углеродных атомов, необязательно моно- или полизамещенная галогеном, алкоксильным радикалом C1-С6, алкилмеркаптановым радикалом C1-С6, C1-С6-алкоксикарбонил- или C1-С6-алкилсульфонилгруппами, R3 – аминогруппа или OR4, где R4 – алкил-C1-C8, Х представляет собой серу, сульфинил – или сульфонилгруппу и Y – атом кислорода, их таутомеры и физиологически приемлемые соли органических и неорганических кислот и щелочей, а также способы их получения и медикаменты, содержащие эти соединения в качестве активных ингредиентов. Аминогруппа аденинового остатка общей формулы I может быть также защищена хорошо известными протекторами аминогрупп. Так как соединения общей формулы I содержит асимметричные углеродные атомы, все оптически активные формы и рацемические смеси этих соединений также являются объектом настоящего изобретения. В J.Biol. Chem. 265, 6112 (1990), и ЕР-А-0350287 описано получение и использование липонуклеотидов в качестве противовирусных лекарственных препаратов. Однако в указанных документах раскрываются только остатки димиристоилфосфатидила и дипальмитоилфосфатидила, связанные с хорошо известными нуклеозидами, например, AZT (3′-азидо-3-деокситимидин) и DDC, включая фрагменты их эфиров жирных кислот. J. Med. Chem., 33, 1380, (1990), описывает нуклеозидные конъюгаты простых тиоэфиров липидов с цитидиндифосфатом, которые обладают противоопухолевой активностью и могут найти применение в онкологии. Chem. Pharm. Bull., 36, 209 (1988), описывает 5′-(3-sn-фосфатидил)нуклеозиды, обладающие противолейкозной активностью, а также их ферментативный синтез из соответствующих нуклеозидов и фосфохолинов в присутствии D-фосфолипазы с трансферазной активностью. В заявке WO 92/03462 описаны конъюгаты липидов простых тиоэфиров, обладающие противовирусной активностью, в частности, при лечении ВИЧ-инфекций. Синтез 2-хлор-9-(2′-деокси-2′-фтор- Фармакологическая активность 2-хлор-9-(2′-деокси-2′-фтор- Другие галогенарабиноаденозины, обладающие противораковой активностью, упоминаются в патенте США 5384310 и заявке WO 92/20347. Противовирусная активность таких пуриновых производных показана в ЕР 0314011. 2-Хлор-9-(2′-деокси-2′-фтор- Соединения общей формулы I по настоящему изобретению, которая включает химическую структуру 2-хлор-9-(2′-деокси-2′-фтор- Указанные соединения настоящего изобретения обладают ценными фармакологическими свойствами. В частности, они пригодны для лечения и профилактики злокачественных опухолей, включая карциномы, саркомы или лейкозы. По сравнению с несвязанными производными нуклеозидов, до сих пор применявшихся при лечении злокачественных опухолей, соединения по настоящему изобретению обладают увеличенной эффективностью/активностью при специфических показаниях или меньшей токсичностью и, следовательно, обладают более широким терапевтическим окном. В некоторых вариантах осуществления настоящего изобретения прием внутрь фармацевтических композиций, содержащих указанные соединения, может осуществляться непрерывно, в течение продолжительного периода времени. Заболеваемость при отказе от приема или при дробном введении, которые зачастую случаются при приеме химиотерапевтических агентов по причине их нежелательных побочных эффектов, может быть снижена при помощи соединений настоящего изобретения по сравнению с родственными соединениями. Более того, могут применяться увеличенные дозы ввиду снижения токсических побочных эффектов по причине увеличенной селективности к опухолевой цитотоксичности. Сложные липидэфирные соединения по настоящему изобретению пригодны также для лечения аутоиммунных расстройств, включающих в себя рассеянный склероз, ревматоидный артрит, волчанку, общий васкулит, воспалительное заболевание кишечника, склеродермию и синдром Шегрена. Лецитиноподобная структура липидной группы является желательной для заявляемых улучшенных соединений общей формулы I. Облегчается проникновение сквозь мембраны и резорбционные барьеры, и соответствующие формуле I конъюгаты проявляют депозитарный эффект в различных тканях. Образование липидных конъюгатов может также облегчать преодоление гематоэнцефалитического барьера ввиду улучшенной диффузии или активации процессов переноса. Аналогично соединения по настоящему изобретению и их фармацевтические составы могут быть использованы в свободном виде или в комбинации с другими лекарствами для лечения и профилактики указанных выше заболеваний. Примеры таких лекарств включают в себя такие агенты, как, например, ингибиторы непрямого деления клетки, например колхицины, винбластины, алкилирующие цитостатические агенты, например циклофосфамид, мельфалан, милеран или цисплатин, антиметаболитики, такие как антагонисты фолиевой кислоты (метотрексат) и антагонисты пуриновых и пиримидиновых оснований (меркаптопурин, 5-фторуридин, цитарабин), антибиотики с цитостатической активностью, такие как антрациклины (например, доксорубицин, даунорубицин), гормоны, такие как фосфестрол, тамоксифен, таксаны, например таксол, и другие цитостатические/цитотоксические активные химиотерапевтические и биологические реагенты. Варианты осуществления настоящего изобретения также охватывают соли указанных соединений общей формулы I, включая соли щелочных, щелочноземельных металлов и соли аммония фосфатной группы. Примеры солей щелочных металлов включают соли лития, натрия и калия. Соли щелочноземельных металлов включают соли кальция и магния, и под солями аммония подразумеваются соли, содержащие ион аммония, который может быть замещен алкильными радикалами, содержащими 1-4 углеродных атома, в количестве до 4, и/или арильными остатками, такими как бензильные. В этих случаях заместители могут быть одинаковыми или различными. Соединения общей формулы I могут содержать щелочные группы, в частности аминогруппы, которые могут быть превращены в кислотсодержащие соли при помощи добавления органических или неорганических кислот. Для этого в качестве таких кислот возможно использование, в частности, соляной, бромистоводородной, серной, фосфорной кислот, фумаровой, янтарной, винной, лимонной, молочной, малеиновой или метансульфоновой кислот. В общей формуле I R1 предпочтительно представляет собой неразветвленный алкильный остаток C8-C16, который, в свою очередь, может быть замещен C1-С6-алкокси или C1-C6-алкилмеркаптогруппой. Более предпочтительно R1 представляет собой нонильный, децильный, ундецильный, додецильный, тридецильный, тетрадецильный или пентадецильный радикал. Предпочтительно в качестве заместителей остатка R1 возможны метоксильные, этоксильные, бутоксильные и гексилоксильные группы. В случае, когда R1 представляет собой замещенный C1-С6алкилмеркаптаном остаток, то следует понимать, что это, в частности, метилмеркаптановый, этилмеркаптановый, пропилмеркаптановый, бутилмеркаптановый и гексилмеркаптановый остаток. Предпочтительно R2 представляет собой неразветвленную C8-C15алкильную группу, которая, в свою очередь, может быть замещена C1-С6алкоксильной или C1-С6алкилмеркаптановой группой. Более предпочтительно R2 представляет собой октильную, нонильную, децильную, ундецильную, додецильную, тридецильную или тетрадецильную группу. Предпочтительно в качестве C1-С6алкоксильных заместителей R2 предпочтительны метоксильные, этоксильные, бутоксильные и гексилоксильные группы. В случае, когда R2 замещен C1-С6алкилмеркаптановым остатком, то следует понимать, что он представляет собой, в частности, метилмеркаптановый, этилмеркаптановый, пропилмеркаптановый, бутилмеркаптановый, пентилмеркаптановый и гексилмеркаптановый остаток. Примером предпочтительного липидного остатка является группа
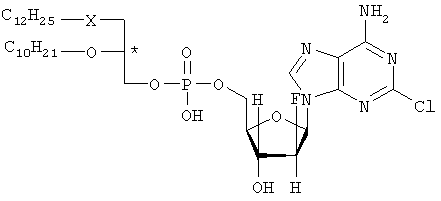
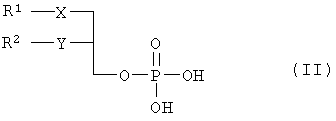
где R1 – C12H25, R2 – C10H21, X – S, SO или SO и Y – O. Наиболее предпочтительными соединениями являются (3-додецилмеркапто-2-децилокси)пропиловыйэфир [2-хлор-9-(2′-деокси-2′-фтор- Указанные соединения общей формулы I могут быть получены посредством 1) взаимодействия соединения общей формулы II или его соли
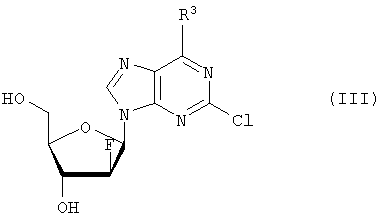
где R1, R2, Х и Y имеют указанные значения, с соединением общей формулы III
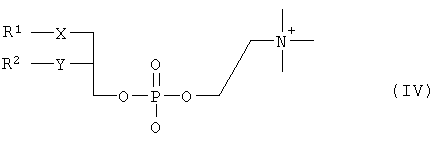
где R3 представляет собой амино или OR4-группу, где R4 – C1-C8алкильная группа, и 3′-гидроксильная группа может быть необязательно защищена блокирующей кислород группой, известной специалисту, и соединение формулы II может быть активировано в присутствии подходящего кислотного хлорида, такого как, например, хлорид 2,4,6-триизопропилбензолсульфония, и третичного азотсодержащего основания, например пиридина или лутидина, в инертном растворителе, например в толуоле, или непосредственно в безводном пиридине, и необязательно, после гидролиза, удаление блокирующих кислород групп, в соответствии с общепринятыми в химии нуклеозидов методиками, и, если R3 представляет собой аминогруппу в соединениях формулы I, необязательно, превращение OR4-группы в положении 6 пуринового кольца в аминогруппу, или взаимодействием липидоспирта (соответствующего формуле II) с нуклеозид-5′-монофосфатом (соответствующим формуле III) способом, аналогичным описанному выше, или 2) взаимодействия соединения общей формулы IV
где R1, R2, Х и Y имеют вышеуказанные значения, с соединением общей формулы III, где R3 представляет собой амино или OR4-группу, где R4 – C1-C8алкильная группа, и 3′-гидроксильная группа может необязательно быть защищена блокирующей кислород группой, известной специалисту, в присутствии D-фосфолипазы стрептомицинов в инертном растворителе, например хлороформе, в присутствии подходящего буфера, и, необязательно, после взаимодействия, удаления блокирующих кислород групп, в соответствии с общепринятыми в химии нуклеозидов методиками, и, если R3 представляет собой аминогруппу в соединениях формулы I, необязательно, превращение OR4-группы в положении 6 пуринового кольца в аминогруппу. Получение указанных соединений общих формул II и IV осуществляется по аналогии с Lipids 22, 947 (1987), и J. Med. Chem. 34, 1377 (1991). Соединения формулы III синтезируются аналогично J. Org. Chem. 34, 2632-2636 (1969), J. Med. Chem. 35, 397-401 (1992), или WO 01/60383, если R3 представляет собой аминогруппу или если R3=OR4 в две стадии. Первый шаг включает в себя получение 2,6-дихлор-9-(3′,5′-O-дибензоил-2′-деокси-2′-фтор- Указанные соединения формулы I, где Х – сульфинил или сульфонил, могут быть получены окислением соответствующих соединений формулы I, где X – сера, при помощи, например, Н2O2/уксусной кислоты или с использованием подходящих исходных соединений формул II или IV. Другие соединения по настоящему изобретению представляют собой дифосфаты формулы V, где n=2 и R1, R2, R3, Х и Y имеют такие же значения, как и в формуле I.
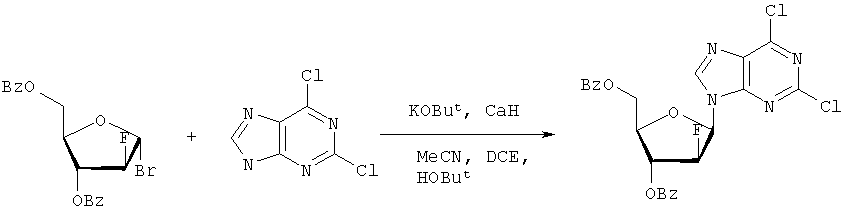
Они могут быть получены реакцией липидофосфата (соответствующего формуле II) с нуклеозид-5′-монофосфатом (полученным из нуклеозидов, соответствующих формуле III). Фосфат липида может быть предварительно активирован с использованием известных специалисту способы. Соли соединений общей формулы I образуются при взаимодействии свободной кислоты с гидроксидами щелочных или щелочноземельных металлов, алкоголятами или ацетатами. “Энантиомеры” липидных фрагментов указанных соединений формулы I могут быть получены разделением через диастереомерные соли или посредством энантиоселективного синтеза липидных остатков, начиная с оптически активного С3-прекурсора формулы II. Лекарства для лечения рака, содержащие соединения формулы I, могут приниматься орально или парентерально, в жидкой или твердой формах. Возможны общепринятые формы использования, например таблетки, капсулы, покрытые таблетки, сиропы, растворы или суспензии. Предпочтительно в качестве среды для инъекций используется вода, содержащая такие добавки, как стабилизаторы, облегчающие растворение вещества, и буферные вещества, которые обычно используются с растворами для инъекций. Такие добавки представляют собой, например, виннокислые или лимоннокислые буферные растворы, этанол, комплексообразующие агенты, например этилендиаминтетрауксусную кислоту и ее нетоксичные соли, высокомолекулярные полимеры для регулирования вязкости, например, полиэтиленоксид. Жидкие переносчики инъекционных растворов должны быть стерильными и предпочтительно помещены в ампулы. Твердые переносчики представлены, например, крахмалом, лактозой, маннитом, метилцеллюлозой, тальком, высокодисперсными кремниевыми кислотами, высокомолекулярными жирными кислотами, например стеариновой кислотой, желатином, агар-агаром, фосфатом кальция, стеаратом магния, животными и растительными жирами, твердыми высокомолекулярными полимерами, например полиэтиленгликолем, и так далее. При желании пригодные для орального приема составы могут включать ароматизаторы или подсластители. Дозировка может зависеть от различных факторов, таких как способы приема, вид, возраст или индивидуальное состояние. Указанные соединения по настоящему изобретению могут надлежащим образом приниматься орально или внутривенно (в.в.) в количествах 0,1-100 мг, предпочтительно в интервале 0,2-80 мг, на килограмм живого веса в сутки. При некоторых режимах дозировки суточная доза делится на 2-5 приемов таблетками, содержащими от 0,5 до 500 мг активного вещества на каждый прием. Аналогично указанные таблетки могут обладать постепенным высвобождением средства, что позволяет снизить число приемов, например, до 1-3 в сутки. Содержание активного ингредиента в таблетках с постепенным высвобождением может находиться с пределах от 2 до 1000 мг. Активный ингредиент может также приниматься путем внутривенного введения пилюли или непрерывного вливания, причем, как правило, достаточные количества составляют от 5 до 1000 мг в сутки. В дополнение к упомянутым в примерах соединениям следующие соединения формулы I и их фармацевтически приемлемые соли также являются примером соединений по настоящему изобретению: 1) (3-додецилмеркапто-2-децилокси)пропиловый эфир [2-хлор-9-(2′-деокси-2′-фтор- 2) (3-додецилсульфинил-2-децилокси)пропиловый эфир [2-хлор-9-(2′-деокси-2′-фтор- 3) (3-додецилсульфонил-2-децилокси)пропиловый эфир [2-хлор-9-(2′-деокси-2′-фтор- 4) (3-ундецилмеркапто-2-децилокси)пропиловый эфир [2-хлор-9-(2′-деокси-2′-фтор- 5) (3-ундецилмеркапто-2-ундецилокси)пропиловый эфир [2-хлор-9-(2′-деокси-2′-фтор- 6) (3-децилмеркапто-2-додецилокси)пропиловый эфир [2-хлор-9-(2′-деокси-2′-фтор- 7) (3-додецилмеркапто-2-додецилокси)пропиловый эфир [2-хлор-9-(2′-деокси-2′-фтор- 8) (3-децилмеркапто-2-децилокси)пропиловый эфир [2-хлор-9-(2′-деокси-2′-фтор- 9) (3-ундецилсульфинил-2-децилокси)пропиловый эфир [2-хлор-9-(2′-деокси-2′-фтор- 10) (3-ундецилсульфонил-2-децилокси)пропиловый эфир [2-хлор-9-(2′-деокси-2′-фтор- 11) (3-ундецилсульфинил-2-ундецилокси)пропиловый эфир [2-хлор-9-(2′-деокси-2′-фтор- 12) (3-ундецилсульфонил-2-ундецилокси)пропиловый эфир [2-хлор-9-(2′-деокси-2′-фтор- 13) (3-тридецилмеркапто-2-ундецилокси)пропиловый эфир [2-хлор-9-(2′-деокси-2′-фтор- 14) (3-тридецилмеркапто-2-децилокси)пропиловый эфир [2-хлор-9-(2′-деокси-2′-фтор- 15) (3-тридецилсульфинил-2-децилокси)пропиловый эфир [2-хлор-9-(2′-деокси-2′-фтор- Далее, настоящее изобретение охватывает аналоги нижеследующих примеров соединений, в которых заместитель в 6 положении пуринового кольца представляет собой C1-C8алкоксил, предпочтительно метоксил. Такие соединения также обладают отличными фармацевтическими свойствами и, кроме того, пригодны в качестве промежуточных соединений при получении указанных нижеследующих примеров соединений. ПРИМЕР 1 Получение 2-хлор-6-метокси-9-(2′-деокси-2′-фтор- Первым шагом является получение 2,6-дихлор-9-
В колбу емкостью 1000 мл загружают 2,6-дихлорпурин (12,65 г, 66,9 ммоль), гидрид кальция (2,43 г, 57,7 ммоль) и ацетонитрил (150 мл) и начинают перемешивание. Через 5 минут добавляют раствор трет-бутоксида калия (60,6 мл, 60,6 ммоль, 1,0 М в трет-бутаноле), получая вязкую, но перемешиваемую суспензию. Через 45 минут, при температуре окружающей среды, добавляют раствор 3,5-O-дибензоил-2-деокси-2-фтор- 1Н ЯМР (ДМСО-d6): ИК (KBr) 3431, 3139, 3063, 2966, 1726, 1596, 1272, 1091, 714 см-1. УФ (H2O/MeCN) Масс-спектр, (с электрораспылением, положительный) m/e [М+Н]+=531. Элементный анализ, рассчитанный для C24H17Cl2FN4O5: С 54,25; Н 3,22; Cl 13,35; F 3,58; N 10,54. Найдено: С 54,19; Н 3,11; Cl 13,20; F 3,49; N 10,52. Вторым шагом является получение 2-хлор-6-метокси-9-(2′-деокси-2′-фтор-
В колбу емкостью 500 мл загружают блокированный 2,6-дихлор-9- 1H ЯМР (ДМСО-d6): ИК (KBr): 3438, 3235, 3113, 2916, 1599, 1471, 1389, 1320, 1238, 1045, 925, 691 см-1. УФ (H2O/MeCN): Масс-спектр (с электрораспылением, положительный) m/e [M+H]+=319. Элементный анализ, рассчитанный для C11H12CIFN4O4: С 41,46; Н 3,80; Cl 11,12; F 5,96; N 17,58. Найдено: С 41,70; Н 3,36; Cl 11,12; F 5,75; N 17,54. ПРИМЕР 2 Получение (3-додецилмеркапто-2-децилокси)пропилового эфира [2-хлор-9-(2′-деокси-2′-фтор- Первым шагом является получение (3-додецилмеркапто-2-децилокси)пропилового эфира [2-хлор-9-(2′-фтор-
3,12 г (3-додецилмеркапто-2-децилокси)пропилового эфира фосфорной кислоты дважды обрабатывают 60 мл безводного пиридина и концентрируют упариванием. Образовавшийся остаток растворяют в 60 мл безводного пиридина при комнатной температуре, обрабатывают 3,80 г 2,4,6-триизопропилбензолсульфонилхлорида (тризилхлорида) в атмосфере азота и перемешивают 2 часа при 20°С. Затем добавляют сначала 2,00 г 2-хлор-9-(2′-деокси-2′-фтор- Образец описанного выше сырого материала очищают при помощи колоночной хроматографии на аппарате Lichrospher 60 RPSelect В с использованием смеси 90:10 метанол/водный 40 мМ раствор ацетата натрия в качестве элюента. Содержащие этот продукт фракции упаривают и остаток распределяют между 50 мл трет-бутилметилового эфира и 10 мл 2 н. соляной кислоты. Органический слой испаряют и остаток растворяют в смеси 5 мл толуола и 5 мл метанола. рН установили на уровне 7 добавлением метилата натрия. Отгоняют растворитель и остаток высушивают в вакууме. Натриевую соль (3-додецилмеркапто-2-децилокси)пропилового эфира [2-хлор-9-(2′-фтор- 1H ЯМР (300 MHz, ДМСО-d6): 8,5 (s, 1H, H8), 6,6, (s (br), 1H, 3′-OH), 6,4 (dd, 1H, H1′), 5,3 (dt, 1H, H2′), 4,4, (dt, 1H, Н3′), 4,1 (s, 3Н, ОСН3), 3,9-4,0, (m, 3H, Н4′, POCH2), 3,6, (m, 1H, Н5’а), 3,6 (m, 1H, Н5’b), 3,3-3,4 (m, 3Н, >СНОСН2-), 2,5-2,6 (m, 4H, CH2SCH2), 1,1-1,5 (m, 36Н, -(СН2)9-, -(CH2)7-), 0,8 (m, 6H, СН2-СН3); 3J1′-H,2′-H 13С ЯМР (75,0 MHz, ДМСО-d6): 160,8, 152,6, 151,7 (С-2, С-4, С-6), 142,9, (С-8), 119,6, (С-5), 94,9, (С-2′), 82,2, (С-4′), 81,6, (С-1′), 78,7, (O-СН<), 73,7, (С-3′), 69,1, (СН2–СН2O-СН<), 64,8, (С-5′), 63,4, (5′-O-Р(O)2OCH2), 55,0, (6-СН3), 32.1, 32,3, (-CH2SCH2-), 20,0-31,2 (-(СН2)9-, -(СН2)7-), 13,9, (2×СН3). 31Р ЯМР (121,5 MHz, ДМСО-d6): -0,46 ppm. 19F ЯМР (282 MHz, ДМСО-d6): -198,7 ppm. УФ (метанол): Масс-спектр (FAB–): m/z=795 [М-Na+]. Вторым шагом является аминолиз сырого (3-додецилмеркапто-2-децилокси)пропилового эфира [2-хлор-9-(2′-фтор-
Стадию аминолиза осуществляют реакторе из нержавеющей стали при 80°С. Вышеуказанный сырой материал (7,38 г) растворяют в 30 мл 7 М раствора NH3 в этаноле (насыщенный при – 5°С). После 20 часов нагревания никаких метоксипроизводных (3-додецилмеркапто-2-децилокси)пропилового эфира [2-хлор-9-(2′-фтор- 1H ЯМР (300 MHz, ДМСО-d6): 8,2 (s, 1H, H8), 7,7, (s (br), 1H, NH2), 6,5, (s (br), 1H, 3′-OH), 6,2 (dd, 1H, H1‘), 5,2 (dt, 1H, H2‘), 4,4, (dt, 1H, Hz, Н3‘), 3,8-4,0, (m, 3Н, Н4‘, POCH2), 3,6, (m, 1H, -H5a’), 3,6 (m, 1H, H5‘b), 3,3-3,5 (m, 3Н, >СНОСН2-), 2,5-2,7 (m, 4H, CH2SCH2), 1,1-1,4 (m, 36H, -(СН2)9-, -(СН2)7-), 0,8 (m, 6H, СН2-СН3); 3J1′-Н,2′-Н 13С ЯМР (75,0 MHz, ДМСО-d6): 156,8, 153,3, 150,1 (С-2, С-4, С-6), 139,8, (С-8), 117,3, (С-5), 95,0, (С-2′), 81,8, (С-4′), 81,2,(С-1′), 78,8, (O-СН<), 72,9, (С-3′), 69,1 (СН2–СН2O-СН<), 64,8, (С-5′), 64,4, (5′-O-Р(O)2OCH2), 32,1, 31,3, (-CH2SCH2-), 22,1-29,7 (-(СН2)9-, -(СН2)7-), 13,9 (2×СН3). 31P ЯМР (121,5 MHz, ДМСО-d6): -0,48 ppm. 19F ЯМР (282 MHz, ДМСО-d6): -198,7 ppm. ПРИМЕР 3 Получение (3-додецилмеркапто-2-децилокси)пропилового эфира [2-хлор-9-(2′-деокси-2′-фтор- 0,91 г 3-додецилмеркапто-2-децилокси)пропилового эфира фосфорной кислоты дважды обрабатывают 20 мл безводного пиридина и концентрируют упариванием. Образовавшийся остаток растворяют в 20 мл безводного пиридина при комнатной температуре, обрабатывают 1,07 г 2,4,6-триизопропилбензолсульфонилхлорида (тризилхлорида) в атмосфере азота и перемешивают 0,5 часа при 25°С. Затем добавляют сначала 0,5 г 2-хлор-9-(2′-деокси-2′-фторарабинофуранозил)аденина и загруженную смесь перемешивают в атмосфере азота в течение 20 часов. Гидролиз проводят при добавлении 5 мл воды, смесь перемешивают еще 0,5 часа при комнатной температуре, освобождают от растворителя под вакуумом и дважды промывают 50 мл толуола. Образовавшийся остаток очищают методом колоночной хроматографии на аппарате Lichrospher 60 RPSelect В с использованием смеси 88:12 метанол/водный 40 мМ раствор ацетата натрия в качестве элюента. Содержащие этот продукт фракции упаривают. Остаток распределяют между 50 мл трет-бутилметилового эфира и 10 мл 2 н. соляной кислоты. Органический слой упаривают и остаток растворяют в смеси 5 мл толуола и 5 мл метанола. Значение рН установили на уровне 7 добавлением метилата натрия. Отгоняют растворитель и остаток высушивают в вакууме. Выход составляет 0,82 г (62%) белого порошка. (3-Додецилмеркапто-2-децилокси)пропиловый эфир фосфорной кислоты получен в соответствии с описанным в WO 92/03462. Пример 4. Получение (3-додецилмеркапто-2-децилокси)пропилового эфира [2-хлор-9-(2′-деокси-2′-фтор- Первым шагом является получение 2-хлор-6-метокси-9-(2′-деокси-2′-фтор-5′-O-фосфат-
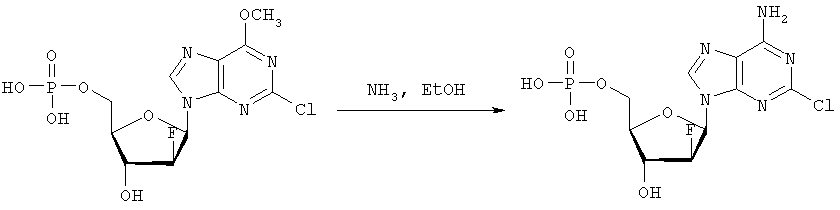
В колбу загружают 2-хлор-6-метокси-9-(2′-деокси-2′-фтор- Затем при перемешивании добавляют лед (1,4 г/ммоль нуклеозида) и воду (8,7 мл/моль нуклеозида) и данную смесь переносят в делительную воронку. Добавляют МТВЕ (4,4 мл/моль нуклеозида) и после перемешивания разделяют фазы. Органическую фазу дважды промывают водой (8,7 мл/ммоль нуклеозида). Объединенные водные экстракты подкисляют до значения рН приблизительно 2 добавлением NaOH (50% водн.), а затем перемешивают с активированным углем (5,7 г/ммоль нуклеозида) в течение достаточного времени (например, 2 часов). Образовавшуюся смесь фильтруют и фильтрат отделяют. Активированный уголь перемешивают со смесью МеОН (4,4 мл/ммоль нуклеозида), гидроксидом аммония (конц., 0,44 мл/ммоль нуклеозида) и водой (например, 3,9 мл/ммоль нуклеозида) в течение достаточного периода времени (30 минут) и фильтруют. Данную процедуру повторяют (например, 5 раз) и фильтраты объединяют. Упаривая объединенные фильтраты, получают сырой 2-хлор-6-метокси-9-(2′-деокси-2′-фтор-5′-O-фосфат- Вторым шагом является аминолиз 2-хлор-6-метокси-9-(2′-деокси-2′-фтор-
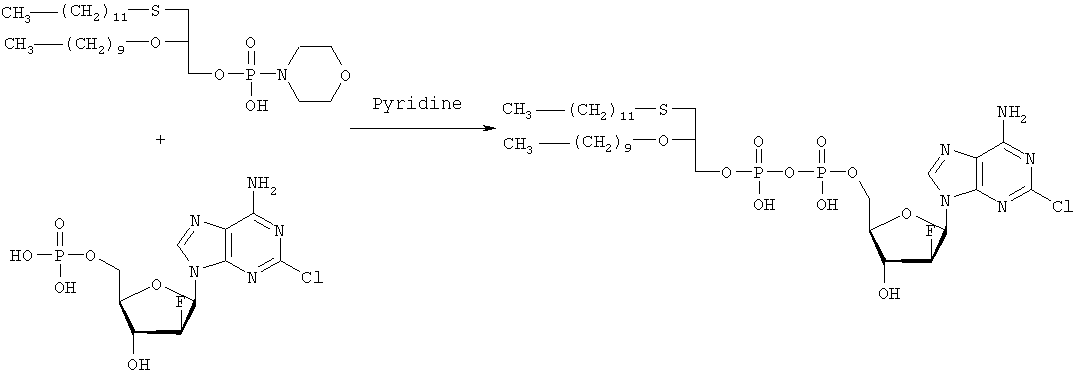
2-Хлор-6-метокси-9-(2′-деокси-2′-фтор- Третьей стадией является получение (3-додецилмеркапто-2-децилокси)пропилового эфира (2-хлор-9-(2′-деокси-2′-фтор-
Морфолидат моно-(3-додецилмеркапто-2-децилокси)-1-пропилового эфира фосфорной кислоты получают аналогично Bioorg. Med. Chem., 7, 1195-1200, (1999), где моно-(3-додецилмеркапто-2-децилокси)-1-пропиловый эфир фосфорной кислоты и морфилин растворяют в смеси воды и трет-бутилового спирта (1: 1, по объему). Дициклогексилкарбодиимид (DCC) в трет-бутиловом спирте добавляют к указанному раствору (примерно 4-молярный избыток DCC по отношению к моно-(3-додецилмеркапто-2-децилокси)-1-пропиловому эфиру фосфорной кислоты), и реакционную смесь нагревают с обратным холодильником (в течение 3,5 часов). Объем уменьшают упариванием и смесь охлаждают, чтобы вызвать осаждение фосфоморфолидата. Фосфоморфолидат (1,13 моль на моль производного аденозина) образуется как безводный пиридин (23 мл/ммоль производного аденозина), и при перемешивании добавляют 2-хлор-9-(2′-деокси-2′-фтор-5′-О-фосфат- ПРИМЕР 5 Получение (3-додецилмеркапто-2R-децилокси)пропилового эфира [2-хлор-9-(2′-деокси-2′-фтор- Целевой продукт получают 6-стадийным синтезом, причем в качестве исходного вещества используют энантиомерно чистый R-(-)-хлор-1,2-пропандиол. Стадия 1: получение R-3-додецилмеркаптопропан-1,2-диола
Раствор 109,9 г (543 ммоль) 1-додекантиола обрабатывают 97,9 мл (543 ммоль) натрия метилата (30% раствор в метаноле) с перемешиванием в течение 30 мин при комнатной температуре, а затем раствором 50,0 г (452 ммоль) R-(-)-хлор-1,2-пропандиола. Полученную смесь перемешивают в течение 3 часов при комнатной температуре. Полный расход хлорида определяют по данным TLC (н-гексан/ацетон, 2:1). Полученный раствор разбавляют 250 мл МТВЕ и экстрагируют 2 н. соляной кислотой (2×200 мл) и водой (200 мл). Органическую фазу упаривают досуха. Полученный осадок кристаллизуют из 1000 мл н-гексана. Получают 120,4 г (96%) бесцветных кристаллов. Т.пл. 55°С, оптическое вращение [ Стадия 2: получение (R)-1-додецилсульфанил-3-тритилоксипропан-2-ола
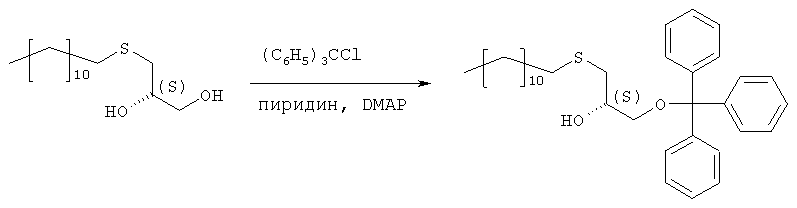
Растворяют 119,7 г (433 ммоль) продукта стадии 1 в 500 мл пиридина при температуре 0°С. Добавляют 132,8 г (476 ммоль) тритилхлорида и 2,57 г (5 мол.%) 4-диметиламинопиридина и полученную смесь перемешивают в течение 48 часов при температуре от 0 до 4°С. Затем гидролизуют с 100 мл воды, пиридин упаривают и осадок разбавляют в 250 мл МТВЕ и 100 мл 2 н. соляной кислоты. Органическую фазу промывают 100 мл соляной кислоты и нейтрализуют путем промывки насыщенным раствором бикарбоната натрия. Сушенную (MgSO4) органическую фазу упаривают в вакууме, полученный осадок растворяют в 500 мл изогексана и фильтруют через слой из 5 г силикагеля. Силикагель промывают 4×25 мл изогексана, и фильтрат и промывную жидкость хранят при температуре 4°С. Полученные кристаллы фильтруют и промывают холодным изогексаном с последующей сушкой в вакууме. Получают 197,8 г (88%) тритилового эфира. Т.пл. 48°С, [ Стадия 3: получение (R)-2-децил-1-додецилмеркапто-3-тритилоксипропана
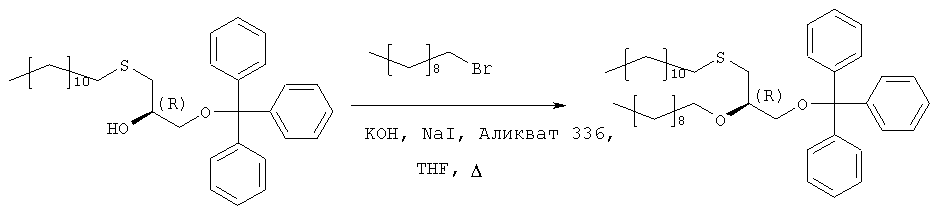
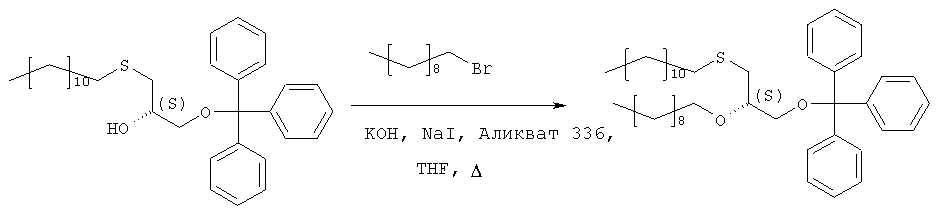
Растворяют 193,3 г (373 ммоль) продукта стадии 2 в 250 мл THF вместе с 132 г (600 ммоль) 1-бромдекана и 37 г Аликвата 336. Добавляют порошок гидроксида калия (190 г) и 38 г иодида натрия и полученную суспензию нагревают с обратным холодильником в течение 5 часов при сильном перемешивании. После полной конверсии исходного материала (TLC-контроль: исходный материал – Rf=0,27 в н-гексане/МТВЕ 9:1, продукт – Rf=0,74) реакционную смесь охлаждают в ванне со льдом (300 г), причем добавляют 300 мл МТВЕ при перемешивании. Полученную органическую фазу промывают 2 н. соляной кислотой (2×200 мл) и насыщенным раствором хлорида натрия (150 мл), фильтруют через целит и растворитель отгоняют в вакууме. Полученный осадок растворяют в 1 л н-гексана, фильтруют через слой из 20 г силикагеля и упаривают досуха. Полученный масляный осадок используют на следующей стадии без дополнительной очистки. Стадия 4: получение (R)-2-децил-1-додецилмеркаптопропан-3-ола
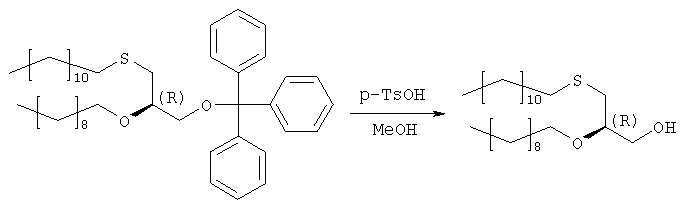
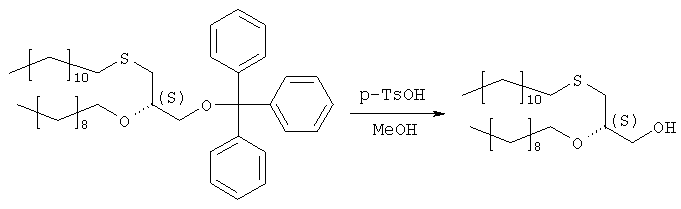
Сырой продукт стадии 3 (284,7 г) растворяют в 1000 мл метанола, добавляют 24 г моногидрата 4-толуолсульфоновой кислоты и 20 мл воды и полученную смесь перемешивают в течение 40 часов. Затем добавляют 200 г ацетата натрия, 100 мл воды и 500 мл н-гексана и перемешивают в течение 30 мин. Двухфазную реакционную смесь подвергают сепарации в делительной воронке, причем нижнюю фазу отделяют. Верхнюю фазу промывают 400 мл 50%-ого ацетата натрия в метаноле, растворитель отгоняют, повторно растворяют в 500 мл н-гексана и охлаждают в холодильнике. Полученные кристаллы отделяют фильтрацией, а полученный маточный раствор сгущают в вакууме до получения 208,8 г масла. Сырой продукт используют на следующей стадии без дополнительной очистки. Образец (5,5 г) сырого продукта очищают на силикагеле в н-гексане/МТВЕ (3:1) в качестве элюента. Получают 3,52 г (85% в расчете на продукт стадии 2) бесцветного масла. Оптическое вращение [ Стадия 5: получение (3-додецилтио-2R-децилокси)пропилового эфира фосфорной кислоты
Растворяют 12,9 мл хлорокиси фосфора в 410 мл толуола и охлаждают в ванне со льдом. Добавляют сырой продукт стадии 4 (52,20 г) и 2,6-лутидин, разбавленный в 410 мл толуола, в течение 1,5 часов. Полученную смесь перемешивают в течение 1,5 часов при 0°С. Данную смесь добавляют в течение 20 мин к 300 мл раствора бикарбоната триэтаноламмония (2 н.), охлажденного льдом. Двухфазную смесь перемешивают в течение ночи при температуре 0-4°С, а затем окисляют 350 мл 2 н. соляной кислоты. После добавления 350 мл МТВЕ водную фазу отделяют, а органическую фазу промывают 250 мл 2 н. соляной кислоты, фильтруют через целит и растворитель удаляют. Полученный масляный осадок (59,2 г) растворяют в 880 мл ацетона, добавляют раствор ацетата кальция (21 г) в 120 мл воды и перемешивают при 0°С. Осажденную соль фильтруют и сушат в вакууме. Полученную кальциевую соль перемешивают в течение ночи в 450 мл ацетона и отделяют путем фильтрации. Получают 46,4 г продукта. Образец (6,73 г) кальциевой соли растворяют в 50 мл 2 н. соляной кислоты и 100 мл МТВЕ, полученный раствор подвергают сепарации в делительной воронке, причем нижнюю фазу отделяют. Верхнюю фазу промывают 50 мл 2 н. соляной кислоты, сгущают до вязкого масла и сушат в высоком вакууме. Получают 5,21 г хроматографически чистого целевого продукта. Стадия 6: получение (3-додецилмеркапто-2R-децилокси)пропилового эфира [2-хлор-9-(2′-деокси-2′-фтор-
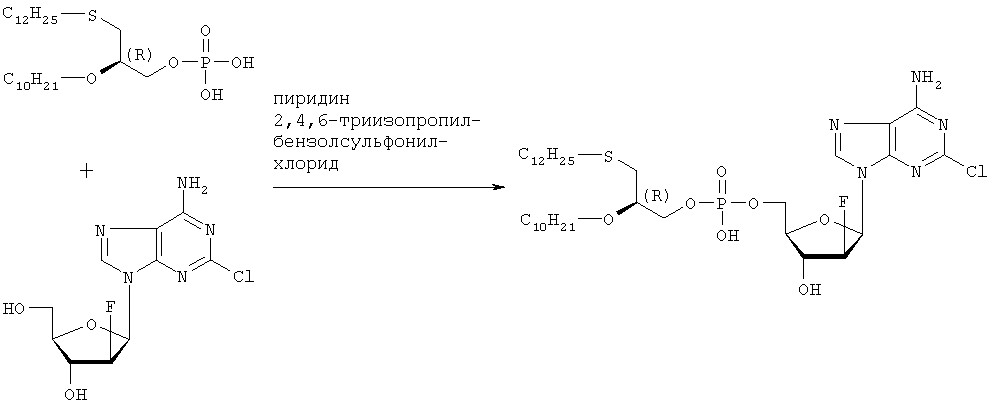
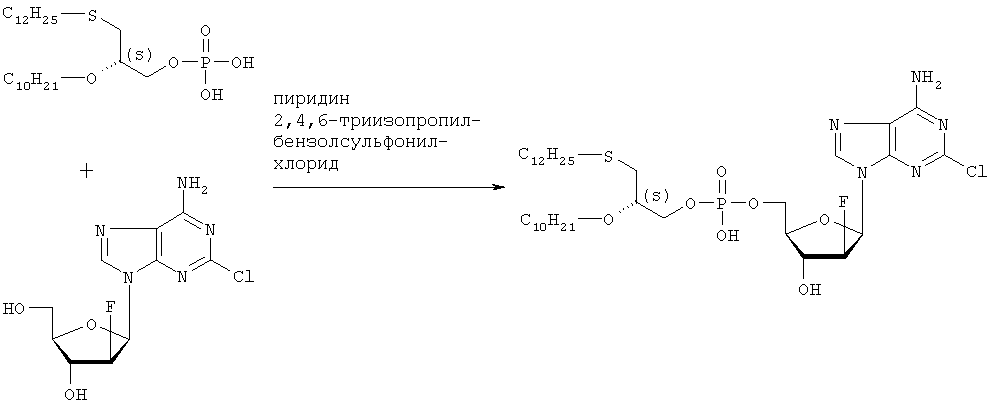
Растворяют 5,21 г (105 ммоль, в расчете на свободную кислоту) продукта стадии 5 в 210 мл пиридина, добавляют 6,35 г (210 ммоль) 2,4,6-триизопропилбензолсульфонилхлорида в атмосфере азота и перемешивают в течение часа при температуре 25°С. Затем добавляют 3,19 г (105 ммоль) [2-хлор-9-(2′-деокси-2′-фтор- Данные по 1H ЯМР, 13С ЯМР и 31P ЯМР соответствуют данным, приведенным в примере 2. Данные по 1Н-развязанному 19F ЯМР проявляют отдельную линию в позиции -198,208 ppm (причем, в случае диастереомерной смеси, согласно примерам 2 и 3, имеются 2 отдельные линии в позициях -198,208 ppm и -198,142 ppm). Оптическое вращение ПРИМЕР 6 Получение (3-додецилмеркапто-2S-децилокси)пропилового эфира [2-хлор-9-(2′-деокси-2′-фтор- Целевой продукт получают указанным выше образом с той лишь разницей, что в качестве исходного вещества используют энантиомерно чистый S-(+)-хлор-1,2-пропандиол. Стадия 1: получение S-3-додецилмеркаптопропан-1,2-диола
Получают 81% бесцветных кристаллов. Т.пл. 57°С, оптическое вращение [ Стадия 2: получение (S)-1-додецилсульфанил-3-тритилоксипропан-2-ола
Получают 82% тритилового эфира. Т.пл. 46°С, [ Стадия 3: получение (S)-2-децил-1-додецилмеркапто-3-тритилоксипропана
Полученный из 103,1 г продукта стадии 2 масляный сырой продукт (134,6 г) используют на следующей стадии без дополнительной очистки. Стадия 4: получение (S)-2-децил-1-додецилмеркаптопропан-3-ола
Получают 60% в расчете на продукт стадии 2) бесцветного масла. Оптическое вращение [ Стадия 5: получение (3-додецилтио-2R-децилокси)пропилового эфира фосфорной кислоты
Продукт обрабатывают указанным выше на стадии 5 образом с получением хроматографически чистого целевого продукта (количественный выход). Стадия 6: получение (3-додецилмеркапто-2S-децилокси)пропилового эфира [2-хлор-9-(2′-деокси-2′-фтор-
Растворяют 5,41 г (109 ммоль, в расчете на свободную кислоту) продукта стадии 5 в 220 мл пиридина, добавляют 6,60 г (218 ммоль) 2,4,6-триизопропилбензолсульфонилхлорида в атмосфере азота и перемешивают в течение часа при температуре 25°С. Затем добавляют 3,31 г (108 ммоль) [2-хлор-9-(2′-деокси-2′-фтор- Данные по 1Н ЯМР, 13С ЯМР и 31P ЯМР соответствуют данным, приведенным в примере 2. Данные по 1Н-развязанному 19F ЯМР проявляют отдельную линию в позиции -198,127 ppm. Оптическое вращение ПРИМЕР 7 Состав таблетки 1,50 кг натриевой соли (3-додецилмеркапто-2-децилокси)пропилового эфира (2-хлор-9-(2′-деокси-2′-фтор- 1,42 кг микрокристаллической целлюлозы, 1,84 кг лактозы, 0,04 кг поливинилпирролидона, 0,20 кг стеарата магния смешивают в сухой форме, увлажняют водой и гранулируют. После высушивания материал прессуют в таблетки весом 500 мг. ПРИМЕР 8 Состав для инъекции 10,0 г натриевой соли (3-додецилмеркапто-2-децилокси)пропилового эфира [2-хлор-9-(2′-деокси-2′-фтор- ПРИМЕР 9 Противоопухолевая активность (3-додецилмеркапто-2-децилокси)пропилового эфира [2-хлор-9-(2′-деокси-2′-фтор- Противоопухолевую активность нуклеотидного конъюгата и соответствующего ему нуклеозида сравнивают на модели ксенотрансплантата карциномы толстой кишки человека НСТ-15 у бестимусных мышей. Пораженных опухолью мышей на 7 день после инокуляции клеток опухоли НСТ-15 перемешивают и разделяют на группы для лечения, по 9 животных в группе. Лечение началось на 8 день. Животным вводили нуклеотидный конъюгат или нуклеозид внутрибрюшинно (ip) один раз в сутки в течение 5 дней подряд. Дозировки включали 50 и 25% от максимальной переносимой дозы (MTD). Контрольным животным вводили соответствующие растворители (переносчик 1 или 2). На 28 день первоначальные опухоли были культивированы и определены их массы. Средние массы опухолей приведены в таблице.
Противоопухолевая эффективность нуклеотидного конъюгата была значительно (р<0.01) выше, чем у соответствующего нуклеозида при всех дозировках.
Формула изобретения
1. Нуклеотидное производное формулы I
где R1 выбирают из группы, состоящей из неразветвленной или разветвленной алкильной цепи, имеющей 1-20 углеродных атомов, которая незамещена или замещена С1-С6-алкоксильным радикалом, С1-С6-алкилмеркаптановым радикалом, С1-С6-алкилсульфинильной или С1-С6-алкилсульфонильной группами, R2 выбирают из группы, состоящей из разветвленной или неразветвленной алкильной цепи, имеющей 1-20 углеродных атомов, которая незамещена или замещена C1-С6-алкоксильным радикалом, C1-С6-алкилмеркаптановым радикалом или C1-С6-алкилсульфонильной группой, R3 – аминогруппа, Х выбирают из группы, состоящей из атома серы, сульфинильной и сульфонильной групп, Y – атом кислорода; его отдельные диастереомерные формы и их смеси, а также его физиологически-приемлемые соли неорганических и органических кислот или щелочей. 2. Нуклеотидное производное по п.1, где R1 представляет собой неразветвленный С8-С15-алкильный радикал, который незамещен или замещен С1-С6 -алкоксильной или C1-С6-алкилмеркаптановой группой. 3. Нуклеотидное производное по п.1, где R2 представляет собой неразветвленную С8-С15-алкильную группу, которая незамещена или замещена C1-С6-алкоксильной или C1-С6-алкилмеркаптановой группой. 4. Нуклеотидное производное по пп.1-3, где указанное соединение имеет вид
где Х – сера, сульфинил или сульфонил, и его отдельные диастереомерные формы и их смеси, а также его физиологически-приемлемые соли неорганических и органических кислот и щелочей. 5. Фармацевтическая композиция для лечения злокачественных опухолей, включающая, по меньшей мере, одно соединение по пп.1-4 в комбинации с фармацевтически приемлемым вспомогательным адьювантом или носителем. 6. Применение соединения по пп.1-4 для приготовления лекарственного средства для лечения злокачественных опухолей, включающего прием пациентом, нуждающимся в таком лечении, эффективного количества соединения по пп.1-4 для лечения указанных опухолей. 7. Применение по п.6, где указанная опухоль выбирается из группы, состоящей из карцином, сарком или лейкозов. 8. Применение по пп.6-7 в фиксированной или произвольной комбинации с другими противораковыми агентами.
TZ4A – Поправки к описаниям изобретений
Часть описания, где обнаружена ошибка: Текст опис., страница 5, строка 38
Напечатано: X-S, SO или SO и
Следует читать: X-S, SO или SO2 и
Номер и год публикации бюллетеня: 6-2009
Извещение опубликовано: 10.03.2010 БИ: 07/2010
TZ4A – Поправки к описаниям изобретений
Часть описания, где обнаружена ошибка: страница 8, строка 35
Напечатано: …-5′-фосфор ной кислоты
Следует читать: …-5′-фосфорной кислоты
Номер и год публикации бюллетеня: 6-2009
Извещение опубликовано: 10.03.2010 БИ: 07/2010
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||
 -D-арабинофуранозил)аденина описывается в J. Org. Chem. 34, 2632- 2636 (1969), заявке WO 01/60383 и патенте США 6680382.
-D-арабинофуранозил)аденина описывается в J. Org. Chem. 34, 2632- 2636 (1969), заявке WO 01/60383 и патенте США 6680382.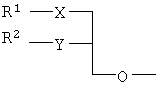
 -D-арабинофуранозила в подходящем растворителе в присутствии затрудненного калиевого основания, предпочтительно трет-бутоксида калия или трет-амилата калия. Подходящие блокирующие группы включают в себя бензоил и ацетил. Подходящие галогениды включают бромид и хлорид. Подходящие инертные растворители включают, но не ограничиваются, трет-бутиловый спирт, ацетонитрил, дихлорметан, дихлорэтан, трет-амиловый спирт, тетрагидрофуран или их смеси. Предпочтительный растворитель содержит смесь ацетонитрила, трет-бутанола и 1,2-дихлорэтана. Необязательно, к реакционной смеси может быть добавлен гидрид кальция. Второй шаг включает воздействие на нуклеозидное производное 2,6-дихлорпурина таких условий, при которых обеспечивается разблокирование и реакция ароматического нуклеофильного замещения, например, в гидроксиде натрия и C1-C8-спирте, или C1-С8-алкоксиде натрия в соответствующем C1-C8 спирте (например, метанол с метоксидом натрия, этанол с этоксидом натрия и т.п.), или другом подходящем неспиртовом растворителе, приводящее к желаемому 6-алкоксипуриновому нуклеозидному C1-C8 соединению формулы III.
-D-арабинофуранозила в подходящем растворителе в присутствии затрудненного калиевого основания, предпочтительно трет-бутоксида калия или трет-амилата калия. Подходящие блокирующие группы включают в себя бензоил и ацетил. Подходящие галогениды включают бромид и хлорид. Подходящие инертные растворители включают, но не ограничиваются, трет-бутиловый спирт, ацетонитрил, дихлорметан, дихлорэтан, трет-амиловый спирт, тетрагидрофуран или их смеси. Предпочтительный растворитель содержит смесь ацетонитрила, трет-бутанола и 1,2-дихлорэтана. Необязательно, к реакционной смеси может быть добавлен гидрид кальция. Второй шаг включает воздействие на нуклеозидное производное 2,6-дихлорпурина таких условий, при которых обеспечивается разблокирование и реакция ароматического нуклеофильного замещения, например, в гидроксиде натрия и C1-C8-спирте, или C1-С8-алкоксиде натрия в соответствующем C1-C8 спирте (например, метанол с метоксидом натрия, этанол с этоксидом натрия и т.п.), или другом подходящем неспиртовом растворителе, приводящее к желаемому 6-алкоксипуриновому нуклеозидному C1-C8 соединению формулы III.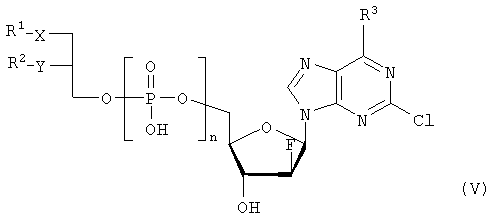
 8,84 (d, 1Н, J=2,82 Hz, H8), 8,14-8,00 (m, 4H, Bz), 7,76-7,50 (m, 6H, Bz), 6,81 (dd, 1H, J=18,2, 3,9 Hz, H1′), 5,95 (m, 2H, Н3′), 5,91 (dm, 1H, J=75,4 Hz, H2′), 4,84-4,79 (m, 3Н, Н4′ и H5′). 13С ЯМР (ДМСО-d6)
8,84 (d, 1Н, J=2,82 Hz, H8), 8,14-8,00 (m, 4H, Bz), 7,76-7,50 (m, 6H, Bz), 6,81 (dd, 1H, J=18,2, 3,9 Hz, H1′), 5,95 (m, 2H, Н3′), 5,91 (dm, 1H, J=75,4 Hz, H2′), 4,84-4,79 (m, 3Н, Н4′ и H5′). 13С ЯМР (ДМСО-d6)  max1 214 nm (0,94 AU),
max1 214 nm (0,94 AU), 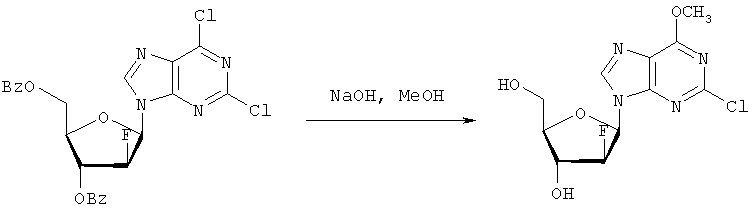

 3J2′-H,3′-H
3J2′-H,3′-H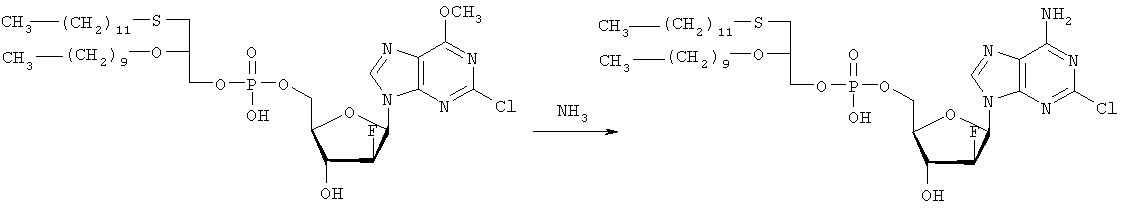
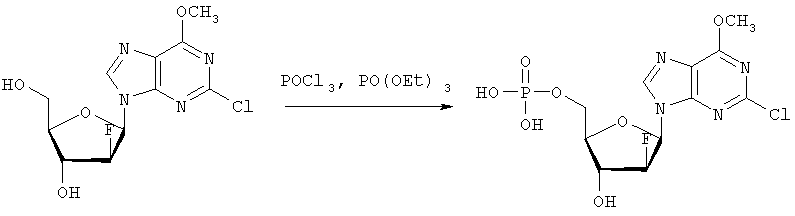
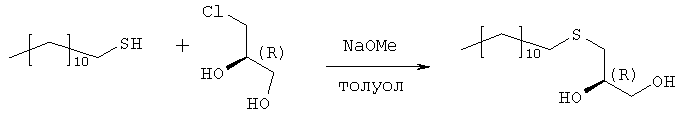
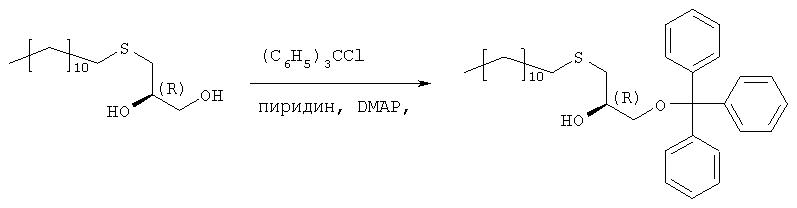
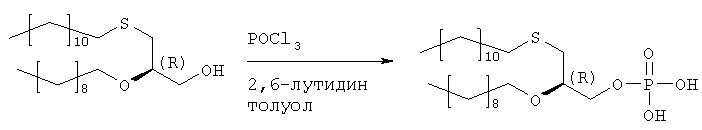
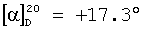 (с=0.25 в метаноле), причем в случае диастереомерной смеси, согласно примерам 2 и 3, оптическое вращение составляет
(с=0.25 в метаноле), причем в случае диастереомерной смеси, согласно примерам 2 и 3, оптическое вращение составляет 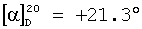 (с=0.23 в метаноле).
(с=0.23 в метаноле).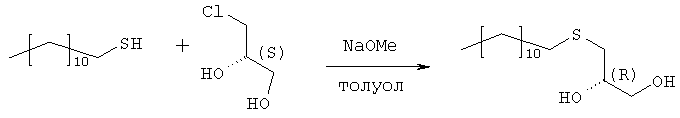
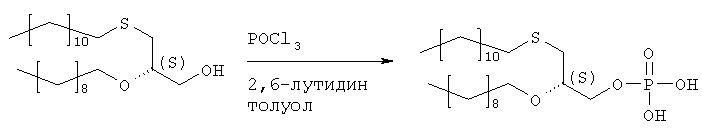
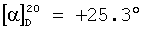 (с=0.25 в метаноле).
(с=0.25 в метаноле).