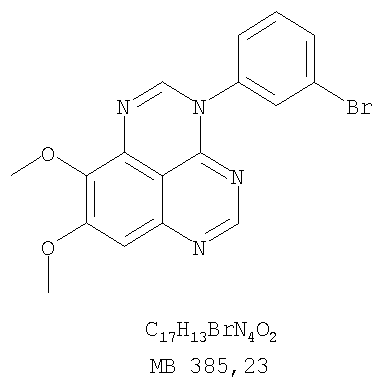
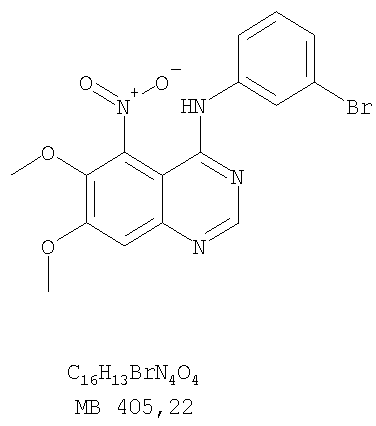
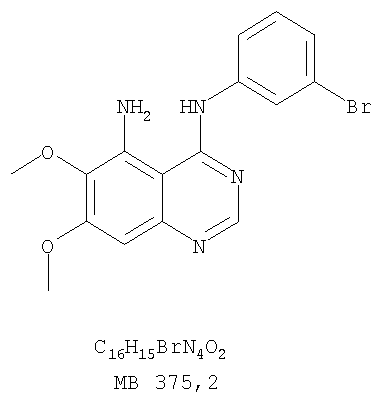
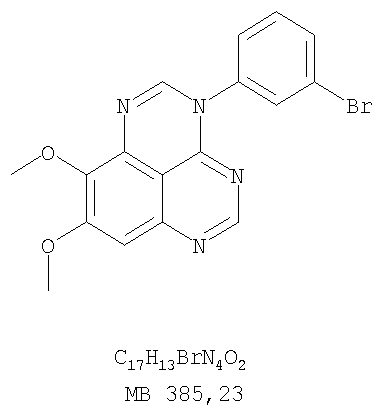
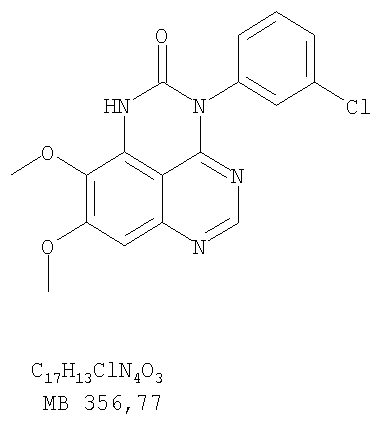
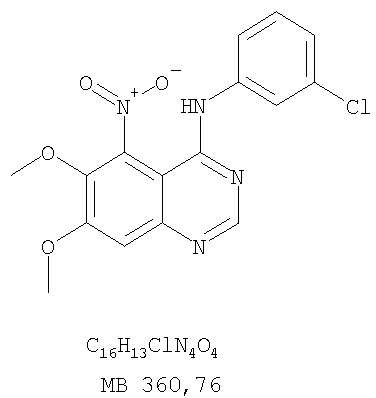
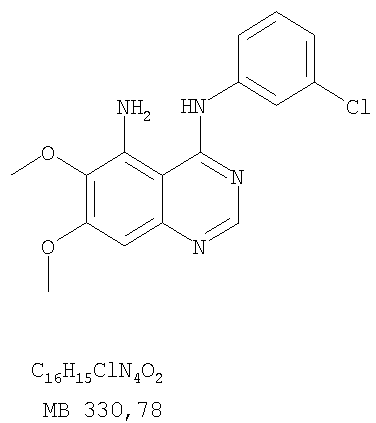
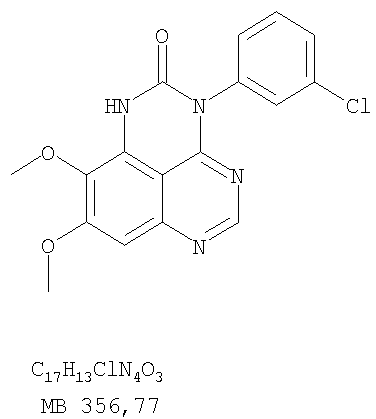
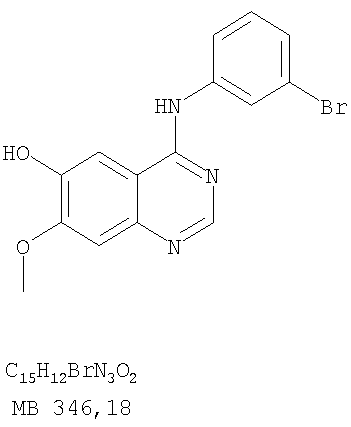
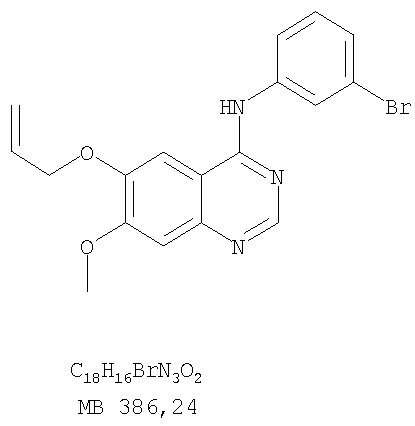
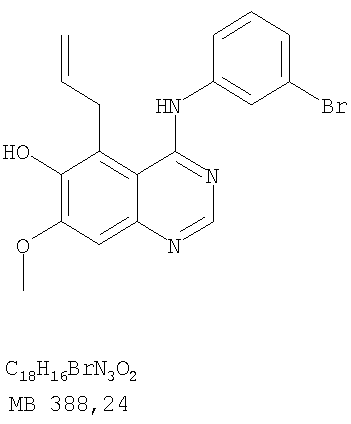
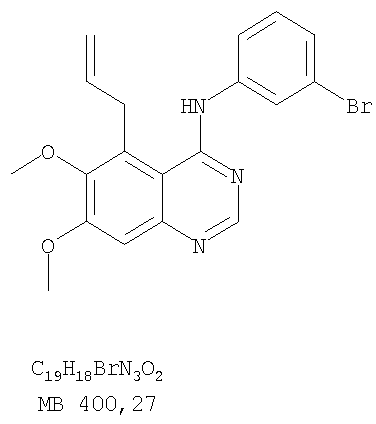
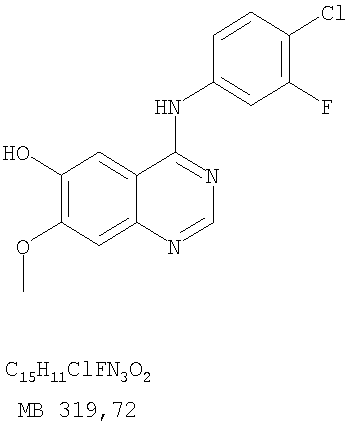
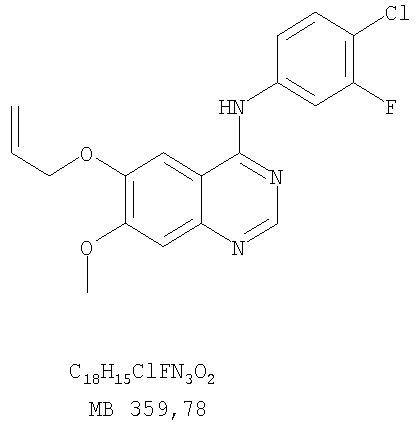
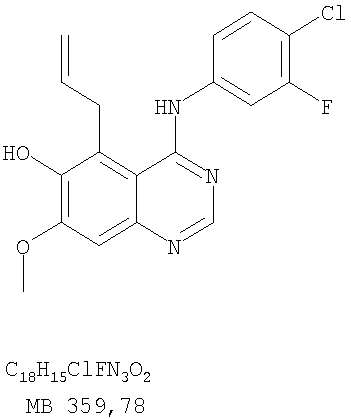
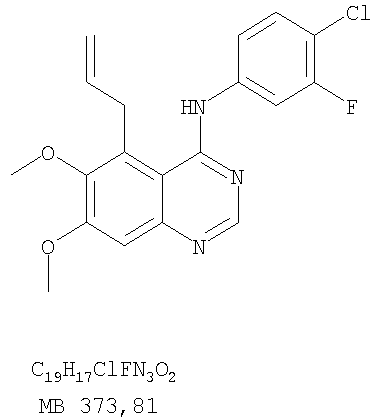
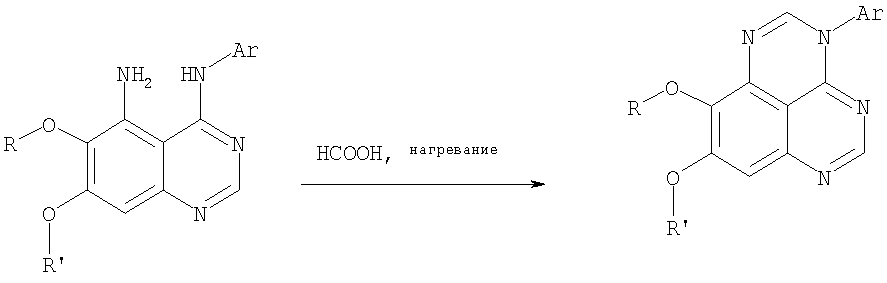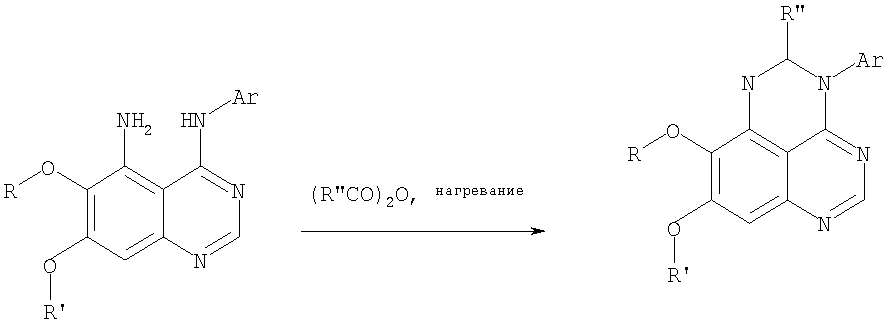Патент на изобретение №2346946
|
||||||||||||||||||||||||||
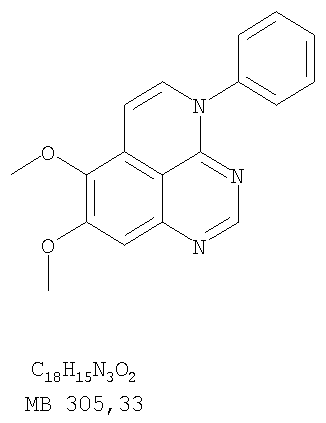
(54) ПРОИЗВОДНЫЕ 1, 3, 4-ТРИАЗАФЕНАЛЕНА И 1, 3, 4, 6-ТЕТРААЗАФЕНАЛЕНА, ОБЛАДАЮЩИЕ СВОЙСТВАМИ ИНГИБИТОРА ЭПИДЕРМАЛЬНОГО ФАКТОРА РОСТА ТИРОЗИНКИНАЗЫ
(57) Реферат:
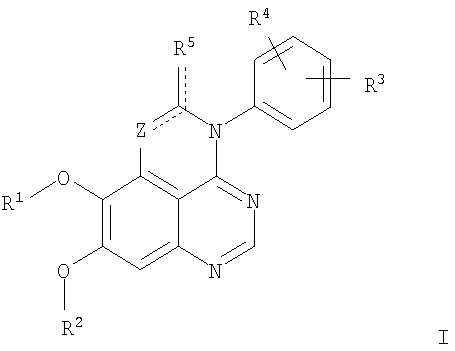
Изобретение относится к новым производным 1,3,4-триазафеналена и 1,3,4,6-тетраазафеналена формулы I и их фармацевтически приемлемым солям, обладающим свойствами ингибировать эпидермальный фактор роста (РЭФР) тирозинкиназы и обладающим антипролиферативной активностью. Соединения могут быть использованы при лечении и профилактике, например, рака, в частности твердых опухолей. В формуле I
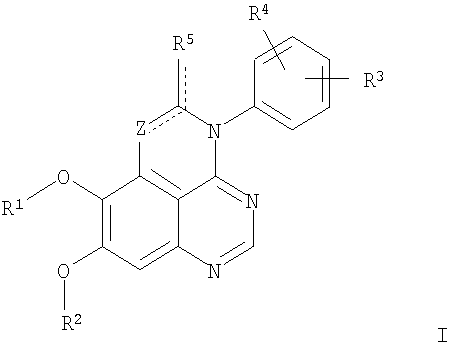
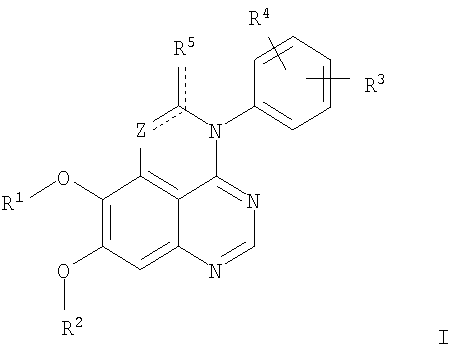
Z означает С или N; R1 и R2 независимо выбраны из Н, низшего алкила; R3 и R4 независимо друг от друга выбраны из Н, F, Cl или Br; R5 выбран из Н, оксогруппы, тиона, C1-С3алкила. Изобретение также относится к фармацевтическим композициям, содержащим указанные соединения. 2 н. и 20 з.п. ф-лы.
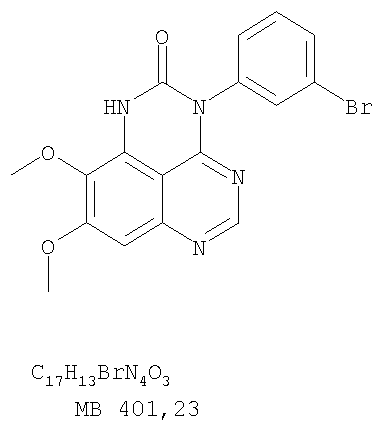
(56) (продолжение): CLASS=”b560m”ONCOLYTIC EGF RECEPTOR TYROSINE KINASE INHIBITOR. DRUGS OF THE FUTURE, 2002, vo.l.27, №4, p.339-345. RUSNAK D.W. et al. THE CHARACTERIZATION OF NOVEL, DUAL ERB-2/EGFR, TYROSINE KINASE INHIBITORS: POTENTIAL THERAPY FOR CANCER-CANCER RESEARCH, AMERICAN ASSOCIATION FOR CANCER RESEARCH, 2001, vol.61, №19, p.7196-7203. PETER M.TRAXLER et al. Protein tyrosine kinase inhibitors in cancer treatment EXPERT OPINION ON THERAPEUTIC PATENTS, ASHLEY PUBLICATIONS, GB, 1997, vol.7, №6, p.571-588.
Данное изобретение предлагает производные 1,3,4-триазафеналена и 1,3,4,6-тетраазафеналена, которые ингибируют рецептор эпидермального фактора роста (РЭФР) тирозинкиназы. Эти соединения и их фармацевтически приемлемые соли, и эфиры обладают антипролиферативной активностью и применимы, в числе прочего, для лечения или профилактики рака, в частности твердых опухолей. Изобретение предлагает также фармацевтические композиции, включающие такие соединения, и методы лечения или профилактики рака, наиболее часто опухолей груди, легкого, толстой кишки или простаты. Кроме того, изобретение предлагает промежуточные соединения, используемые в получении новых производных феналена, описанных в нем. Рецепторы тирозинкиназ являются межмембранными белками, участвующими в передаче клеточного сигнала, а именно, они проводят сигналы фактора роста от клеточной поверхности до определенных внутриклеточных процессов, которые контролируют такие важные функции, как рост, дифференциацию и ангиогенез. (A.A.Adjei, Drugs of the Future 2001, 26, №11, сс.1087-1092). Многие виды рака связаны с аберрантными сигналами. В частности, нерегулируемая передача сигнала через тирозинкиназы играет ключевую роль в росте и распространении рака (A.D.Laird и J.M.Cherrington, Expert Opin. Invetig. Drugs, 2003, 12, №1, сс.51-64). Одно из семейств рецептора тирозинкиназ представлено рецептором эпидермального фактора роста (РЭФР) тирозинкиназы. Было найдено, что эти рецепторы производятся в чрезмерном количестве в ряде эпидермальных видов рака и участвуют в развитии ракового заболевания. Роль рецептора тирозинкиназ (РТК) и, в частности, РЭФР в росте и распространении рака твердо установлена (Laird и Cherrington, выше; и Adjey, выше). Интенсивно развиваются исследования по созданию малых молекул-ингибиторов РТК и, в частности, РЭФР. Обзор соединений, ингибирующих РЭФР, и их терапевтическое применение содержится в работах: Laird и Cherrington, выше; Adjey, выше; Drugs of the Future 2002, 27, №4, сс.339-345; M.R.Myers и др., Bioorganic & Medicinal Chemistry Letters, 1997, 7, №4, сс.421-424; А.J.Bridges и др., J. Med. Chem., 1996, 39, сс.267-276; G.W.Rewcastle и др., J. Med. Chem., 1995, 38, сс.3482-3487. Данное изобретение предлагает новые производные 1,3,4-триазафеналена и 1,3,4,6-тетраазафеналена формулы
в которой Z означает С или N; R1 и R2 независимо выбраны из Н, низшего алкила, низшего алкила, замещенного OR6, NR6R7, гетероцикла или гетероарила; R3 и R4 независимо друг от друга выбраны из Н, F, Cl или Br; R5 выбран из Н, ОН, SH, оксогруппы, тиона, C1-С3алкила и R6 и R7 независимо друг от друга выбраны из Н и низшего алкила или NR6R7 могут вместе образовывать кольцо с 3-7 атомами, и это кольцо может необязательно включать до трех дополнительных гетероатомов и может необязательно быть замещенным одним или более низшим алкилом; или их фармацевтически приемлемые соли или эфиры. Двойные линии в вышеприведенной формуле означают, что двойная связь существует или между Z и атомом углерода, к которому присоединен R5, или между атомом углерода и R5. Использование двойных линий для обозначения таутомерных форм хорошо известно профессионалам. Эти соединения ингибируют рецептор эпидермального фактора роста (РЭФР) тирозинкиназы. Эти соединения и их фармацевтически приемлемые соли, и эфиры обладают антипролиферативной активностью и, среди прочего, используются при лечении и профилактике рака, в частности твердых опухолей. Настоящее изобретение также представляет фармацевтические композиции, содержащие одно или более соединений настоящего изобретения, или фармацевтически приемлемую соль или эфир из них и фармацевтически приемлемый носитель или наполнитель. Настоящее изобретение также представляет способ лечения или профилактики рака, более узко – способ лечения или профилактики твердых опухолей, наиболее узко – способ лечения или профилактики рака груди, легкого, толстой кишки и простаты введением больному при необходимости терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли или эфира. Наконец, предлагаемое изобретение представляет новые промежуточные соединения, используемые при получении соединения формулы I. Используемые здесь следующие термины будут иметь следующие обозначения. “Алкил”, один или в сочетании с другим термином, например, алкилгетероцикл означает прямоцепочечный или разветвленный насыщенный алифатический углеводород, имеющий 1-12, предпочтительно 1-10 атомов углерода. Предпочтительные алкильные группы означают “низшие алкильные” группы, имеющие 1-6, предпочтительно 1-4 атома углерода. Типичные низшие алкильные группы включают метил, этил, пропил, изопропил, бутил, трет-бутил, 2-бутил, пентил, гексил и подобные. Как здесь используется, С1-С3 алкил означает алкильную группу, имеющую от 1 до 3 атомов углерода. “Арил”, один или в сочетании с другим термином, означает моновалентный, моноциклический или бициклический, ароматический карбоциклический углеводородный радикал, предпочтительно 6-10-членную ароматическую циклическую систему. Предпочтительные арильные группы включают, но не ограничиваются фенилом, нафтилом, толилом и ксилилом. “Эффективное количество” означает количество, достаточно эффективное для того, чтобы прервать, смягчить или облегчить симптомы заболевания или пролонгировать выживание субъекта, подвергшегося лечению. “Галоген” означает фтор, хлор, бром и иод, предпочтительно фтор, хлор или бром. “Гетероатом” означает атом, выбранный из N, О и S. “Гетероарил” означает ароматическую гетероциклическую циклическую систему, содержащую до двух циклов. Предпочтительные гетероарильные группы включают, но не ограничиваются тиенилом, фурилом, индолилом, пирролилом, пиридинилом, пиридином, пиразинилом, оксазолилом, тиаксолилом, хинолинилом, пиримидинилом, имидазолом и тетразолилом. “Гетероцикл” или “гетероциклил” означает насыщенный или частично ненасыщенный, неароматический циклический радикал с 3-8 атомами в цикле, из которых 1-4 атома являются гетероатомами, выбранными из азота, кислорода, серы, или их комбинации, а остающиеся атомы цикла являются атомами углерода. Примерами предпочтительных гетероциклов являются пиперидин, пиперазин, пирролидин, морфолин, индолил, тетрагидропиранил, тиоморфолин, пентаметиленсульфид и пентаметиленсульфон. “IC50” означает концентрацию определенного соединения, согласно предлагаемому изобретению, которая ингибирует 50% специфической измеряемой активности. IC50 может быть измерена, среди прочего, так, как описано в Примере 23 ниже. “Оксо” означает =O. “Фармацевтически приемлемый эфир” означает этерифицированное соединение формулы I, имеющее спиртовую или карбоксильную группу, которое сохраняет биологическую эффективность и свойства соединений формулы I, и раскрывается in vivo (в организме) в соответствующий активный спирт или карбоновую кислоту. Примерами эфирных групп, которые раскрываются (в данном случае гидролизуются) in vivo в соответствующие карбоновые кислоты (R40С(=O)ОН), являются низшие алкиловые эфиры, которые могут быть замещены NR41R42, где R41 и R42 означают низшие алкилы или где NR41R42, объединенные вместе, образуют моноциклический алифатический гетероцикл, такой как пирролидин, пиперидин, морфолин, N-метилпиперазин и т.д.; и карбоэфиры формулы R40C(=O)OCYR43OC(=O), где R43 означает водород или метил. R40 означает соединение согласно настоящему изобретению. Оно показано в форме его карбоновой кислоты (после введения больному в виде эфира, расщепившегося до соответствующей кислоты). Примерами низших алкиловых эфиров являются ацетильные, пропионильные эфиры и подобные. Примерами низших алкиловых эфиров, замещенных NR41R42, являются диэтиламиноацетил, 2-(4-морфолинил)ацетил, 2-(4-метилпиперазин-1-ил)ацетильные эфиры и подобные. Примерами карбоэфиров являются ацетоксиметоксиформил и 2-(диметиламино)ацетоксиметоксиформильный эфиры. Дальнейшая информация, касающаяся примеров и использования эфиров для получения фармацевтически полезных соединений, представлена в Design of Prodrugs. Bundgaard H ed. (Elsevier, 1985), а также: Н.Ansel и др., Pharmaceutical Dosage Forms and Drug Delivery Systems (6-е изд., 1995), сс.108-109; Krogsgaard-Larsen, и др., Textbook of Drug Design and Development (2-е изд., 1996), сс.152-191; H.Gao и др., Synthesis, 2000, сс.329-351; J.Alexander и др., J. Med. Chem. 1988, 31, cc.318-322. “Фармацевтически приемлемые соли” означают подходящие кислотно-аддитивные соли или основно-аддитивные соли, которые обладают биологической эффективностью и свойствами соединений формулы I и получаются из подходящих нетоксичных органических или неорганических кислот или органических или неорганических оснований. Примеры кислотно-аддитивных солей включают соли, образованные из неорганических кислот, таких как хлористо-водородная кислота, бромисто-водородная кислота, иодистоводородная кислота, серная кислота, сульфаминовая кислота, фосфорная кислота и азотная кислота, и соли, образованные из органических кислот, таких как п-толуолсульфоновая кислота, салициловая кислота, метансульфоновая кислота, щавелевая кислота, янтарная кислота, лимонная кислота, малоновая кислота, молочная кислота, фумаровая кислота и подобные. Примеры основно-аддитивных солей включают соли, образованные аммонием, натрием, калием и четвертичными гидроксидами аммония, как, например, гидроксид тетраметиламмония. Химическая модификация фармацевтически полезных соединений (т.е. лекарств) в соли технически хорошо известна химикам-фармацевтам для получения улучшенной физической и химической стабильности, гигроскопичности, текучести и растворимости соединений. Смотри, например, Н.Ansel и др. Pharmaceutical Dosage Forms and Drug Delivery Systems (6-е изд,. 1995), сс.196 и 1456-1457. “Фармацевтически приемлемый”, например, фармацевтически приемлемый носитель, наполнитель и т.д., означает фармакологически приемлемый и существенно нетоксичный для субъекта, которому вводится определенное соединение. “Замещенный”, например, алкил-замещенный, означает, что замещение может иметь место в одном или нескольких положениях и, если не указано обратное, заместители в каждом положении замещения могут быть выбраны независимо из специфических групп. “Терапевтически эффективное количество” означает количество по крайней мере одного соединения формулы I или его фармацевтически приемлемой соли или эфира, которое существенно ингибирует пролиферацию и/или прерывает дифференциацию человеческой раковой клетки, включая линии человеческих раковых клеток. “Тион” означает =S при неконцевом атоме углерода. Предлагаемое изобретение представляет соединения формулы I
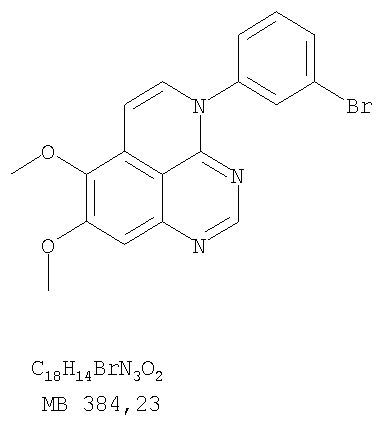
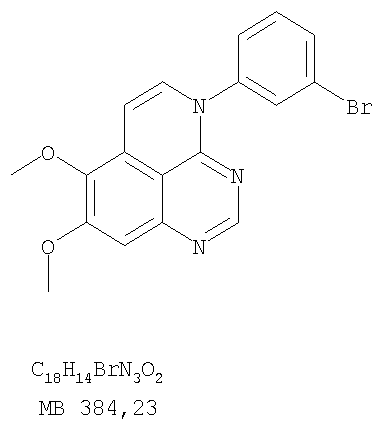
или их фармацевтически приемлемые соли или эфиры, в которых Z и заместители от R1 до R5 определены выше. Предпочтительно в некоторых соединениях формулы I Z означает N, в других Z означает С. В других предпочтительных примерах соединений формулы I R1 и R2 независимо друг от друга означают низший алкил, предпочтительно метил. В наиболее предпочтительных примерах оба радикала R1 и R2 означают метил. В других предпочтительных примерах соединений формулы I R1 и R2 независимо друг от друга выбраны из Н, Br, Cl и F, наиболее предпочтителен Н. В наиболее предпочтительных примерах оба радикала R3 и R4 означают Н. В других предпочтительных примерах соединений формулы I R5 выбран из =O, =S, Н и низшего алкила, наиболее предпочтителен Н и низший алкил. Следующие соединения являются предпочтительными согласно предлагаемому изобретению: 8,9-диметокси-3-фенил-1H,3H-1,3,4,6-тетраазафенален-2-он (Пример 1D); 8,9-диметокси-3-фенил-1H,3H-1,3,4,6-тетраазафенален-2-тион (Пример 2); 8,9-диметокси-3-фенил-3H-1,3,4,6-тетраазафенален (Пример 3); 8,9-диметокси-2-метил-3-фенил-3H-1,3,4,6-тетраазафенален (Пример 4); 2-этил-8,9-диметокси-3-фенил-3H-1,3,4,6-тетраазафенален (Пример 4); 3-(3-бромфенил)-8,9-диметокси-3H-1,3,4,6-тетраазафенален (Пример 6C); 3-(3-бромфенил)-8,9-диметокси-1H,3H-1,3,4,6-тетраазафенален-2-он (Пример 7); 3-(3-бромфенил)-8,9-диметокси-2-метил-3H-1,3,4,6-тетраазафенален (Пример 8); 3-(3-бромфенил)-8,9-диметокси-1H,3H-1,3,4,6-тетраазафенален-2-тион (Пример 9); 3-(3-бромфенил)-2-этил-8,9-диметокси-3H-1,3,4,6-тетраазафенален (Пример 10); 3-(3-хлорфенил)-8,9-диметокси-1H,3H-1,3,4,6-тетраазафенален-2-он (Пример 11C); 3-(3-хлорфенил)-8,9-диметокси-2-метил-3H-1,3,4,6-тетраазафенален (Пример 12); 3-(4-хлорфенил)-8,9-диметокси-3H-1,3,4,6-тетраазафенален (Пример 13C); 3-(4-хлорфенил)-8,9-диметокси-1H,3H-1,3,4,6-тетраазафенален-2-тион (Пример 14); 7,8-диметокси-5-метил-4-фенил-4H-1,3,4-триазафенален (Пример 15G); 4-(3-хлорфенил)-7,8-диметокси-5-метил-4H-1,3,4-триазафенален (Пример 16G); 4-(3-бромфенил)-7,8-диметокси-5-метил-4H-1,3,4-триазафенален (Пример 17G); 4-(3-бромфенил)-7,8-диметокси-5-метил-4H-1,3,4-триазафенален (Пример 18G); 4-(4-бром-2-фторфенил)-7,8-диметокси-5-метил-4H-1,3,4-триазафенален (Пример 19G); 7,8-диметокси-4-фенил-4H-1,3,4-триазафенален (Пример 20В); 4-(4-хлорфенил)-7,8-диметокси-4H-1,3,4-триазафенален (Пример 21В); и 4-(3-бромфенил)-7,8-диметокси-4H-1,3,4-триазафенален (Пример 22В). Соединения, приведенные здесь и охваченные формулой I, могут существовать в виде таутомеров или структурных изомеров. Предполагается, что настоящее изобретение включает любую таутомерную или структурно-изомерную форму этих соединений или смесей таких форм и не ограничивается любой одной таутомерией или структурно-изомерной формой, указанной в вышеприведенной формуле. Соединения предлагаемого изобретения могут быть получены любыми подходящими способами. Подходящие способы синтеза этих соединений приведены в примерах. В общем, соединения формулы I могут быть получены согласно одному из нижеописанных синтетических путей. Схема 1
Схема 2
Схема 3
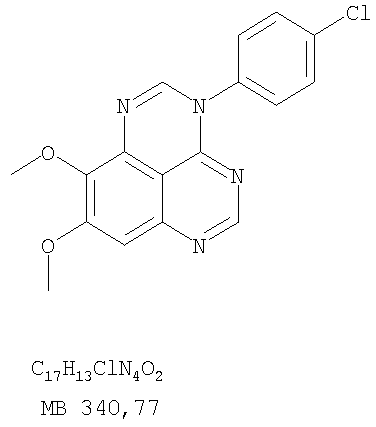
Схема 4
Схема 5
Схема 6
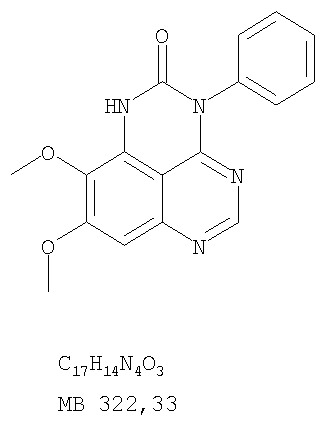
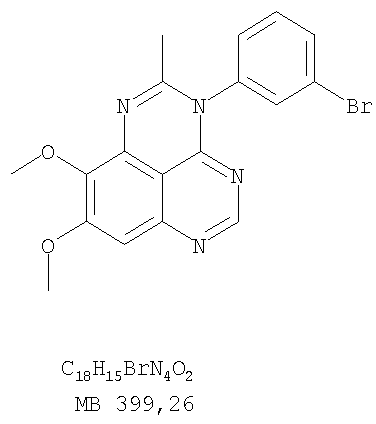
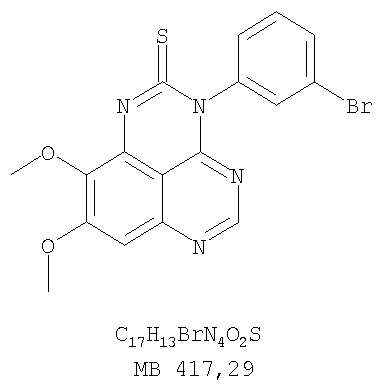
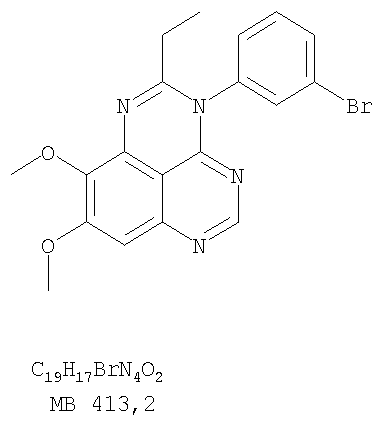
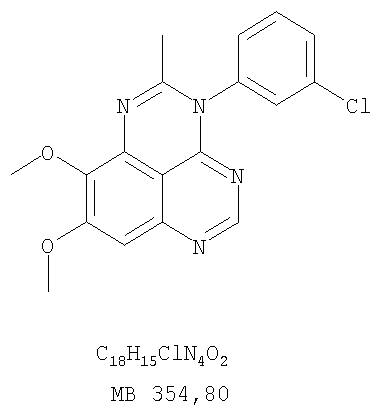
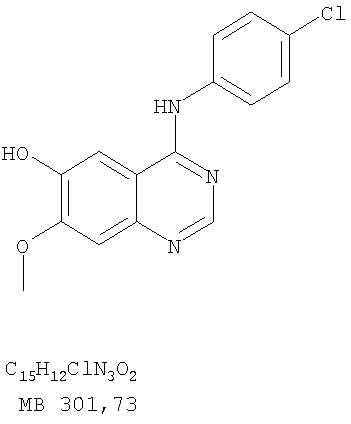
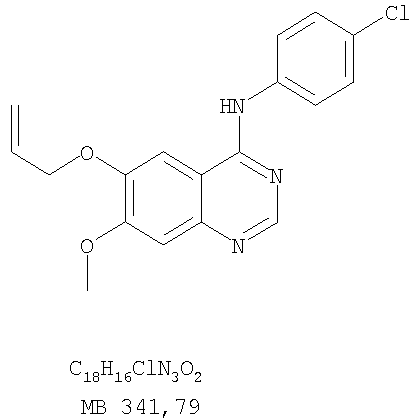
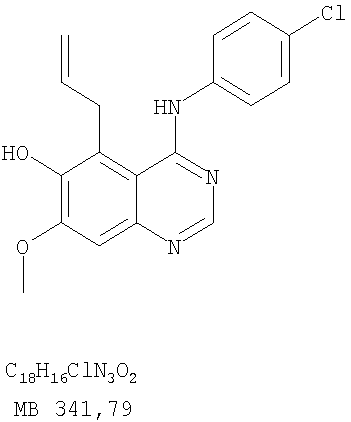
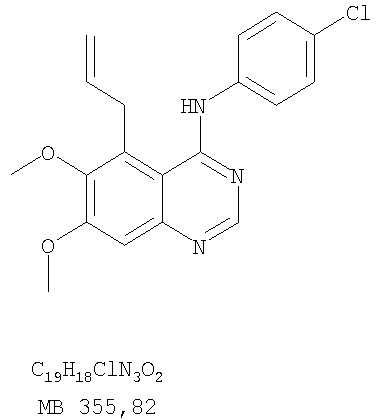
Разделение смеси стереоизомеров на оптически чистые стереоизомеры (когда соединение формулы I является хиральным). Разделение изомерных структур соединений формулы I может быть проведено согласно известным методам, таким как, например, механическое разделение или хиральная жидкостная хроматография высокого давления (также известная как хиральная ВДЖХ). Методы разделения хорошо известны и суммированы в “Enantiomers, Racemates, and Resolutions” (Jacques, J. и др. John Wiley and Sons, Нью-Йорк, 1981). Методы хиральной ВДЖХ также хорошо известны и суммированы в “Separation of Enantiomers by Liquid Chromatographic Methods” (Pirkle, W.H. и Finn, J. в “Asymmetric Synthesis”, T.1, Morrison, J.D., Ed., Academic Press, Inc., Нью-Йорк 1983, сс.87-124). Превращение соединения формулы I, имеющего основный атом азота, в фармацевтически приемлемую кислотно-аддитивную соль. Выборочное превращение соединения формулы I, имеющего основный атом азота, в фармацевтически приемлемую кислотно-аддитивную соль может быть осуществлено обычными методами. Так, например, соединение может быть обработано неорганической кислотой, например, хлористо-водородной, бромисто-водородной, серной, азотной, фосфорной кислотой, или подходящей органической кислотой, например, уксусной, лимонной, винной, метансульфоновой, п-толуолсульфоновой кислотой и так далее. Превращение соединения формулы I, имеющего карбоксильную группу, в фармацевтически приемлемую соль щелочного металла. Выборочное превращение соединения формулы I, имеющего карбоксильную группу, в фармацевтически приемлемую соль щелочного металла может быть осуществлено обычными методами, например соединение может быть обработано неорганическим основанием, таким как гидроксид лития, гидроксид натрия, гидроксид калия и так далее. Превращение соединения формулы I, имеющего спиртовую или карбоксильную группу, в фармацевтически приемлемый эфир. Выборочное превращение соединения формулы I, имеющего спиртовую или карбоксильную группу, в фармацевтически приемлемый эфир может быть осуществлено обычными методами. Условия образования эфира зависят от стабильности других групп в молекуле по отношению к условиям реакции. Если другие группы в молекуле стабильны в кислых условиях, эфир может быть получен нагреванием в растворе минеральной кислоты (например, серной) в спирте. Альтернативно, эфир может быть получен конденсацией спирта с ангидридом или галоид ангидридом кислоты в условиях, известных специалисту. Другие способы получения эфира, применяемые в тех случаях, когда молекула неустойчива в кислых условиях, включают обработку соединения спиртом и кислотой в присутствии сочетающего реагента и необязательно в присутствии реагента, ускоряющего реакцию. Специалистам в области органической химии известно много таких сочетающих реагентов. Двумя такими примерами являются дициклогексилкарбодиимид и трифенилфосфин/диэтилазодикарбоксилат. При использовании дициклогексилкарбодиимида в качестве сочетающего реагента реакцию выборочно проводят, обрабатывая кислоту спиртом, дициклогексилкарбодиимидом и необязательно в присутствии каталитического количества (0-10 мольных %) N,N-диметиламинопиридина в инертном растворителе, таком как галоидированные углеводороды (например, дихлорметан) при температуре между 0°С и примерно комнатной температурой. При использовании в качестве сочетающего реагента системы трифенилфосфин/диэтилазодикарбоксилат реакцию обычно проводят, обрабатывая кислоту спиртом, трифенилфосфином и диэтилазодикарбоксилатом в инертном растворителе, таком как эфир (например, тетрагидрофуран) или ароматический углеводород (например, бензол) при температуре между 0°С и примерно комнатной температурой, предпочтительно при 0°С. В другой своей части изобретение предлагает фармацевтические композиции, включающие по крайней мере одно соединение формулы I или его приемлемую соль или эфир и фармацевтически приемлемый наполнитель и/или разбавитель. Предлагаемые фармацевтические композиции могут быть введены орально, например, в форме таблеток, таблеток с покрытием, драже, твердых или мягких желатиновых капсул, растворов, эмульсий и суспензий. Они могут также вводиться ректально, например, в форме суппозиториев, или парентерально, например, в форме инъектируемых растворов. Фармацевтические композиции предлагаемого изобретения, включающие соединения формулы I и/или их соли или эфиры, могут быть получены по методам, известным специалистам, например, обычным смешиванием, энкапсулированием, растворением, гранулированием, эмульгированием, наполнением, формированием драже или в процессе лиофилизации. Эти фармацевтические формы могут сочетаться с терапевтически инертными неорганическими или органическими носителями. В качестве носителей для получения таблеток, драже, твердых желатиновых капсул могут быть использованы лактоза, кукурузный крахмал или его производные, тальк, steric кислота или ее соли. Подходящими носителями для мягких желатиновых капсул служат растительное масло, воски и жиры. В зависимости от природы активного компонента для мягких желатиновых капсул обычно не требуется никаких носителей. Подходящими носителями для приготовления растворов и сиропов являются вода, полиолы, сахароза, инвертный сахар и глюкоза. Подходящими носителями для вливаний являются вода, спирты, полиолы, глицерин, растительные масла, фосфолипиды и поверхностно-активные вещества. Подходящими носителями для суппозиториев являются натуральные или отвержденные масла, воски, жиры и полужидкие полиолы. Фармацевтические композиции могут также содержать консервирующие агенты, агенты, повышающие растворимость, стабилизирующие агенты, увлажняющие агенты, эмульгирующие агенты, умягчители, агенты цвета и запаха, соли для регулирования осмотического давления, буферы, обволакивающие агенты или антиоксиданты. Они могут также содержать другие терапевтически применяемые вещества, включая дополнительные активные ингредиенты, другие, нежели охватываемые формулой I. Дозировки. Как упоминалось выше, соединения предлагаемого изобретения, включая соединения формулы I, применяются при лечении или профилактике клеточных пролиферативных заболеваний, включая хемопрофилактику рака. Хемопрофилактика определяется как ингибирование развития инвазивного рака, или блокирование начального мутагенеза, или блокирование развития пре-злокачественных клеток, которые уже пострадали от удара, или ингибирование рецидива опухоли. Эти соединения и составы, содержащие указанные соединения, особенно применимы при лечении или профилактике твердых опухолей, таких как, например, опухоли груди, толстой кишки, легких и простаты. Терапевтически эффективное количество соединения в соответствии с предлагаемым изобретением означает количество соединения, эффективное для прерывания, смягчения или облегчения симптомов заболевания или пролонгирования выживания субъекта, подвергшегося лечению. Определение терапевтически эффективного количества находится в сфере деятельности специалистов в этой области. Терапевтически эффективное количество или доза соединения согласно предлагаемому изобретению может варьироваться в широких пределах и может быть определено способами, известными специалистам. Такая доза должна быть приспособлена к индивидуальным требованиям в каждом отдельном случае, включая специфику вводимого соединениями), путь введения, условия лечения, равно как и специфику больного, подвергаемого лечению. В общем случае, в случае орального или парентерального введения взрослым людям с весом примерно 70 кг дневная доза должна составлять примерно от 10 мг до примерно 10000 мг, предпочтительно от примерно 200 мг до примерно 1000 мг, хотя нижний предел может быть увеличен, если это показано. Дневная доза может быть введена за один раз или раздельными дозами, или в случае парентерального введения она может даваться как продолжительное вливание. Комбинации Соединения этого изобретения могут применяться в комбинации (вводятся в комбинации или последовательно) с известными антираковыми средствами, такими как радиационная терапия или с цитостатическими или цитотоксическими агентами, такими как, например, но не ограничиваясь, ДНК интерактивными агентами, такими как цисплатин или доксорубицин; ингибиторами топоизомеразы II, такими как этопозид; ингибиторами топоизомеразы I, такими как СПТ-11 или топотекан; агентами, взаимодействующими с тубулином, такими как паклитаксэл, доцетаксэл или эпотилонс; гормональными агентами, такими как тамоксифен; ингибиторами тимидилатсинтазы, такими как 5-фторурацил, и антиметаболитами, такими как метотрексат. Соединения формулы I могут также применяться в комбинации с модуляторами п53 трансактивации. В виде смешанных доз вышеописанные продукты комбинации включают соединения данного изобретения в дозовом интервале, описанном выше, и другие фармацевтически активные агенты или препараты в их санкционированном дозовом интервале. Соединения формулы I могут также вводиться последовательно с известными антираковыми или цитотоксическими агентами в том случае, когда совместное введение или комбинация являются неподходящими. Настоящее изобретение не ограничивает последовательность введения: соединения формулы I могут вводиться или до, или после введения известных антираковых или цитотоксических агентов. Промежуточные соединения В другой части изобретения предлагаются новые промежуточные соединения, используемые в получении соединений формулы I. Эти новые промежуточные соединения включают следующий ряд: (5-Аллил-6,7-диметоксихиназолин-4-ил)-фениламин (Пример 15Е); 5-Йодметил-7,8-диметокси-4-фенил-5,6-дигидро-4Н-1,3,4-триазафенален (Пример 15F); 5-Аллил-4-(4-хлорфениламино)-7-метоксихиназолин-6-ол (Пример 16D); (5-Аллил-6,7-диметоксихиназолин-4-ил)-(4-хлорфенил)амин (Пример 16Е); 4-(4-Хлорфенил)-5-йодметил-7,8-диметокси-5,6-дигидро-4Н-1,3,4-триазафенален (Пример 16F); 5-(Аллил-4-(3-бромфениламино)-7-метоксихиназолин-6-ол (Пример 17D); (5-Аллил-6,7-диметоксихиназолин-4-ил)-(3-бромфенил)амин (Пример 17Е); 4-(3-Бромфенил)-5-йодметил-7,8-диметокси-5,6-дигидро-4Н-1,3,4-триазафенален (Пример 17F); 5-Аллил-4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ол (Пример 18D); (5-Аллил-6,7-диметоксихиназолин-4-ил)-(3-хлор-4-фторфенил)амин (Пример 18Е); 4-(3-Хлор-4-фторфенил)-5-йодметил-7,8-диметокси-5,6-дигидро-4Н-1,3,4-триазафенален (Пример 18F); 5-Аллил-4-(4-бром-2-фторфениламино)-7-метоксихиназолин-6-ол (Пример 19D); (5-Аллил-6,7-диметоксихиназолин-4-ил)-(4-бром-2-фторфенил)амин (Пример 19Е); 4-(4-Бром-2-фторфенил)-5-йодметил-7,8-диметокси-5,6-дигидро-4Н-1,3,4-триазафенален (Пример 19F); 7,8-Диметокси-4-фенил-5,6-дигидро-4Н-1,3,4-триазафенален-5-ол (Пример 20А); 7,8-Диметокси-4-хлорфенил-5,6-дигидро-4Н-1,3,4-триазафенален-5-ол (Пример 21А); 7,8-Диметокси-4-(3-бромфенил)-5,6-дигидро-4Н-1,3,4-триазафенален-5-ол (Пример 22А). Примеры Следующие примеры иллюстрируют предпочтительные методы синтеза, применения соединений и получения лекарственных форм, предлагаемых в настоящем изобретении. Эти примеры и препараты носят иллюстративный характер и не ограничивают весь объем изобретения. Следует иметь в виду, что могут быть другие составные части предлагаемого изобретения, которые определены в формуле изобретения. Сокращения, используемые в изобретении: ДМФ – N,N-диметилформамид ТСХ – тонкослойная хроматография ДБУ – 1,8-диазабицикло[5.4.0]ундец-7-ен Пример 1 8,9-Диметокси-3-фенил-1Н,3Н-1,3,4,6-тетраазафенален-2-он
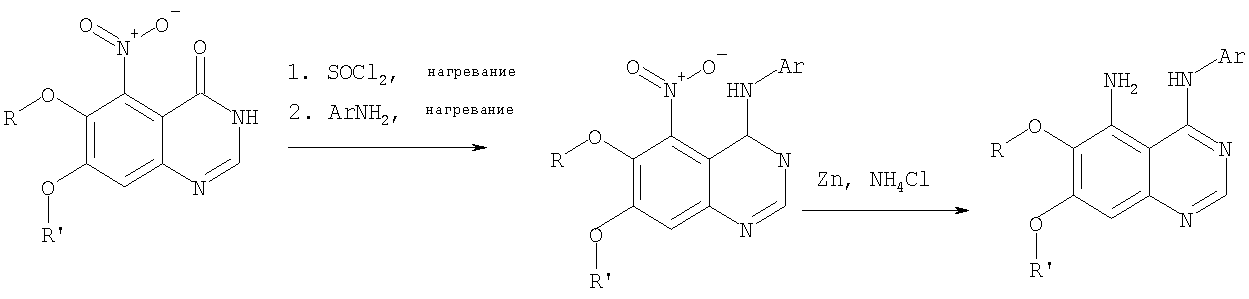
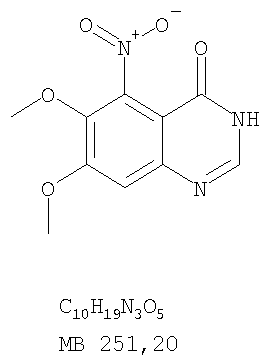
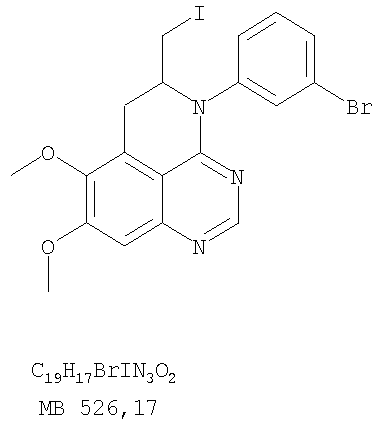
Стадия А: 6,7-диметокси-5-нитро-3H-хиназолин-4-он
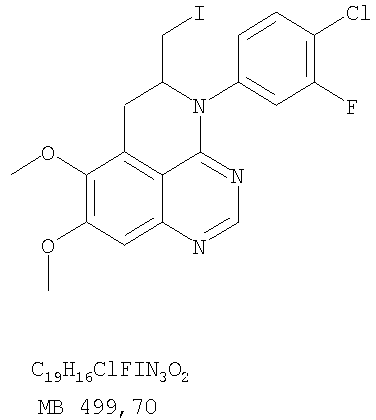
6,7-Диметокси-5-нитро-3-Н-хиназолин-4-он был получен в виде бледно-желтого твердого вещества согласно процессу, описанному Chao, Qi; Deng, Lynn; Shih, Hsiencheng; Leoni, Lorenzo M.; Genini, Davide; Carson, Dennis A.; Cottam, Howard В.; J. Med. Chem, 1999, 42, сс.3860-3873. Стадия В: (6,7-диметокси-5-нитрохиназолин-4-ил)фениламин
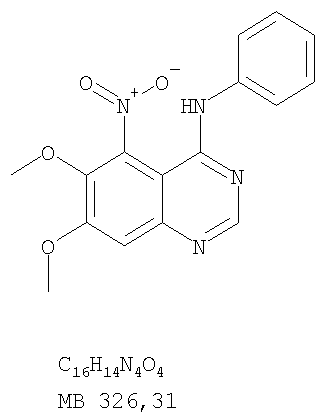
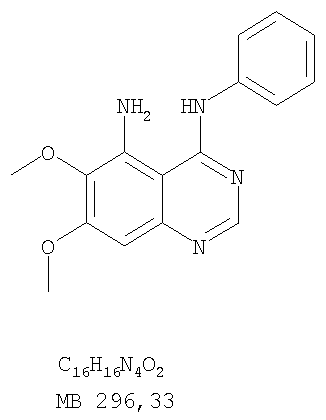
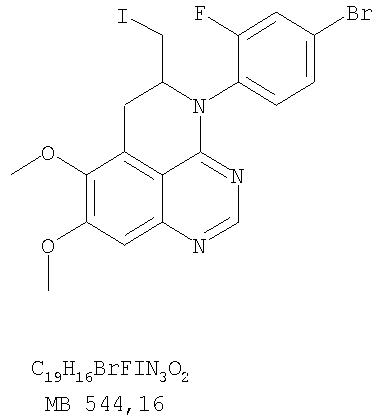
К раствору 6,7-диметокси-5-нитро-3H-хиназолин-4-она (1,0 г, 3,98 ммоль) (из Примера 1, стадия А) в SOCl2 (20 мл) (Aldrich) добавляли несколько капель ДМФ. Затем реакционную смесь нагревали при перемешивании при 90°С в течение 3 час. Растворители удаляли, и остаток высушивали в вакууме. Остаток растворяли в 2-пропаноле (30 мл), затем добавляли анилин (0,36 мл, 3,98 ммоль) (Aldrich). Реакционную смесь нагревали при 110°С в течение 3 час, затем концентрировали при уменьшенном давлении, и остаток очищали хроматографически, используя в качестве элюента EtOAc/CH2Cl2/NEt3 (1:1:0,05), получая желаемый (6,7-диметокси-5-нитрохиназолин-4-ил)фениламин в виде желтого твердого вещества. (Выход 0,9 г, 70%). Стадия С: 6,7-диметокси-N-4-фенилхиназолин-4,5-диамин
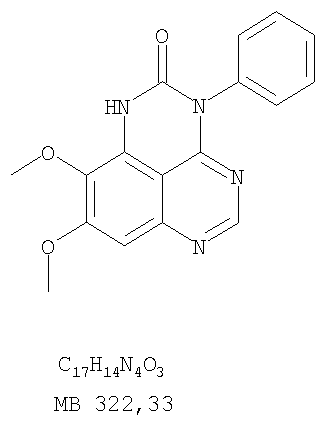
К раствору (6,7-диметокси-5-нитрохиназолин-4-ил)фениламина (1,0 г, 3,14 ммоль) (из Примера 1, стадия В), NH4Cl (1,7 г, 31,4 ммоль) в МеОН, Н2О и CHCl3 (40 мл, 2:1:1) добавляли порошкообразный Zn (2,0 г, 31,4 ммоль) (Aldrich). Реакционную смесь перемешивали при комнатной температуре в течение 1 час, затем фильтровали, фильтрат концентрировали и экстрагировали хлороформом (100 мл). Органический слой отделяли, сушили над Na2SO4 и концентрировали. Остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/CH2Cl2 (1:1) и получая желаемый 6,7-диметокси-N-4-фенилхиназолин-4,5-диамин в виде желтой смолы. (Выход 0,5 г, 54%). Стадия D: 8,9-Диметокси-3-фенил-1H,3H-1,3,4,6-тетраазафенален-2-он
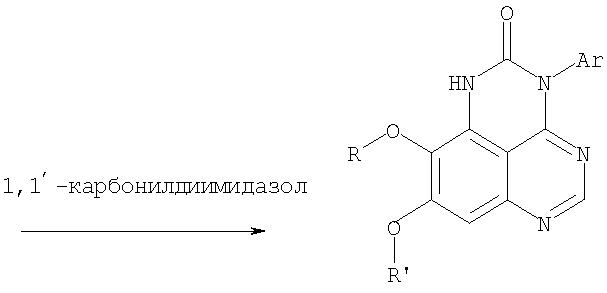
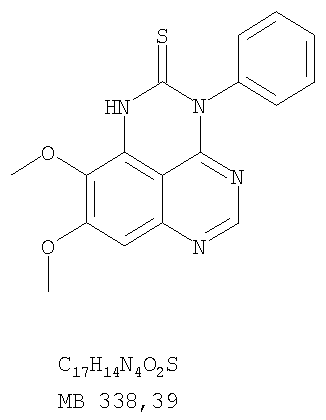
К раствору 6,7-диметокси-N-4-фенилхиназолин-4,5-диамина (0,2 г, 0,68 ммоль) (Пример 1, стадия С, выше) в 1,2-дихлорэтане (50 мл) добавляли 1,1′-карбонилдиимидазол (0.55 г, 3,4 ммоль) (Aldrich). Реакционную смесь нагревали при перемешивании при 90°С в течение 4 час, затем растворитель выпаривали, остаток очищали хроматографически, используя EtOAc/CH2Cl2/Et3N (1:1:0,04) в качестве элюента, и получали желаемый 8,9-диметокси-3-фенил-1H,3H-1,3,4,6-тетраазафенален-2-он в виде твердого желтого вещества (Выход 0,13 г, 59%). Пример 2 8,9-Диметокси-3-фенил-1H,3H-1,3,4,6-тетраазафенален-2-тион
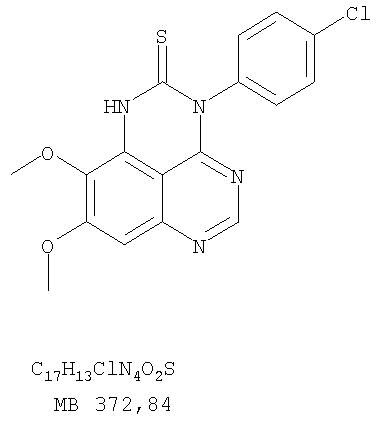
К раствору 6,7-диметокси-N-4-фенилхиназолин-4,5-диамина (80 мг, 0,27 ммоль) (Пример 1, стадия С, выше) в 1,2-дихлорэтане (30 мл) добавляли 1,1′-тиокарбонилдиимидазол (0.58 г, 3,24 ммоль) (Fluka). Реакционную смесь нагревали при перемешивании при 80°С в течение 4 час, затем растворитель выпаривали, остаток очищали хроматографически, используя EtOAc/CH2Cl2/Et3N (1:1:0,04) в качестве элюента, и получали желаемый 8,9-диметокси-3-фенил-1H,3H-1,3,4,6-тетраазафенален-2-тион в виде твердого серого вещества. (Выход 50 мг, 55%). Пример 3 8,9-Диметокси-3-фенил-1H,3H-1,3,4,6-тетраазафенален
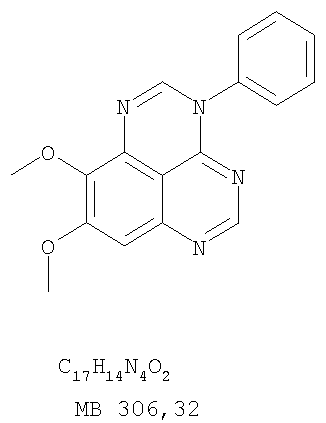
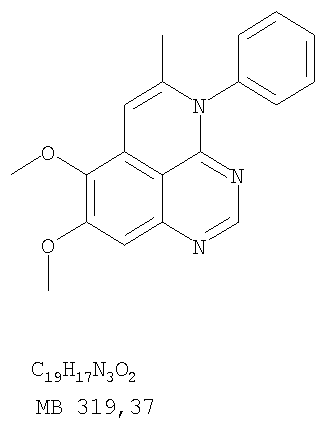
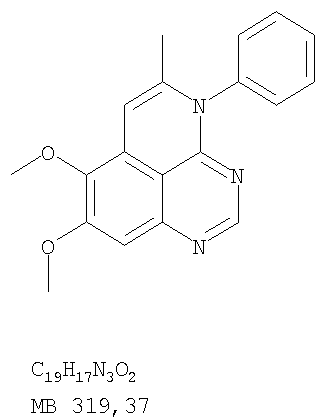
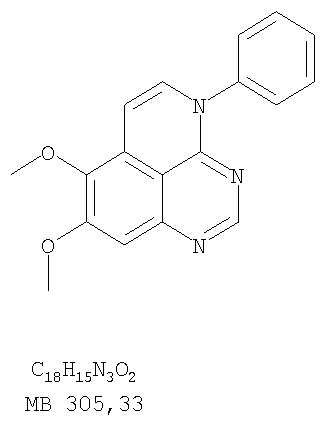
Раствор 6,7-диметокси-N-4-фенилхиназолин-4,5-диамина (80 мг, 0,27 ммоль) (Пример 1, стадия С, выше) в муравьиной кислоте (5 мл) нагревали при 110°С 2 час, затем к реакционной смеси добавляли водный раствор NaOH до рН 10-12. Раствор разбавляли хлороформом (50 мл), отделяли органический слой, сушили его над Na2SO4 и концентрировали. Остаток очищали хроматографически, используя EtOAc/Et3N (1:0,04) в качестве элюента, и получали желаемый 8,9-диметокси-3-фенил-1H,3H-1,3,4,6-тетраазафенален в виде твердого белого вещества. (Выход 45 мг, 54%) Пример 4 8,9-Диметокси-2-метил-3-фенил-3H-1,3,4,6-тетраазафенален
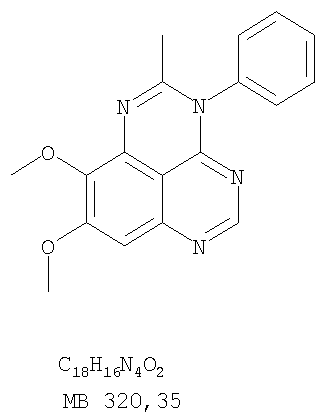
Раствор 6,7-диметокси-N-4-фенилхиназолин-4,5-диамина (60 мг, 0,20 ммоль) (из Примера 1, стадия С) в уксусном ангидриде (2 мл) нагревали при 150°С в течение 2 час. Затем в реакционную смесь добавляли водный раствор NaOH до рН 10-12, после чего смесь разбавляли хлороформом (50 мл). Органический слой отделяли, сушили над Na2SO4 и концентрировали. Остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/Et3N (1:0,04), и получали желаемый 8,9-диметокси-2-метил-3-фенил-3H-1,3,4,6-тетраазафенален в виде желтого твердого вещества. (Выход 20 мг, 62%). Пример 5 2-Этил-8,9-диметокси-3-фенил-3H-1,3,4,6-тетраазафенален
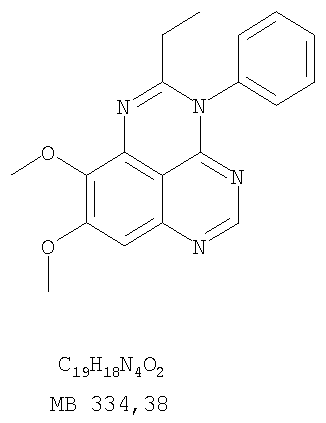
Раствор 6,7-диметокси-N-4-фенилхиназолин-4,5-диамина (130 мг, 0,44 ммоль) (Пример 1, стадия С, выше) в пропионовом ангидриде (3 мл) (Aldrich) нагревали 2 час при 170°С. К реакционной смеси добавляли водный раствор NaOH до рН 10-12, и раствор разбавляли хлороформом (50 мл). Отделяли органический слой. Сушили его над Na2SO4 и концентрировали. Остаток очищали хроматографически, используя EtOAc/Et3N (1:0,05) в качестве элюента, и получали желаемый 2-этил-8,9-диметокси-3-фенил-3H-1,3,4,6-тетраазафенален в виде твердого оранжевого вещества. (Выход 21 мг, 14%). Пример 6 3-(3-Бромфенил)-8,9-диметокси-3H-1,3,4,6-тетраазафенален
Стадия А: (3-Бромфенил)(6,7-диметокси-5-нитрохиназолин-4-ил)амин
К раствору 6,7-диметокси-5-нитро-3H-хиназолин-4-она (1,0 г, 3,98 ммоль) (Пример 1, стадия А, выше) в SO2Cl2 (20 мл) (Aldrich) добавляли несколько капель ДМФ (0,05 мл). Реакционную смесь 3 час нагревали при перемешивании при 90°С, затем растворители отгоняли, и остаток сушили в вакууме. Остаток растворяли в 2-пропаноле (30 мл) и добавляли 3-броманилин (0,69 г, 3,98 ммоль) (Aldrich). Реакционную смесь нагревали при 110°С 3 час, затем удаляли растворители, остаток очищали хроматографически, используя EtOAc/CH2Cl2/Et3N (1:1:0,05) в качестве элюента, и получали желаемый (3-бромфенил)(6,7-диметокси-5-нитрохиназолин-4-ил)амин в виде твердого желтого вещества. (Выход 1,2 г, 74%). Стадия В: N-4-(3-Бромфенил)-6,7-диметоксихиназолин-4,5-диамин
К раствору (3-бромфенил)(6,7-диметокси-5-нитрохиназолин-4-ил)амина (0,2 г, 0,49 ммоль) (из Примера 6, стадия А) и NH4Cl (0,26 г, 4,94 ммоль) в смеси МеОН, Н2О и CHCl3 (30 мл, 6:1:1) добавляли порошкообразный Zn (0,64 г, 9,87 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 час, затем фильтровали, фильтрат концентрировали и затем экстрагировали хлороформом (100 мл). Органический слой отделяли, сушили над Na2SO4 и концентрировали. Остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/CH2Cl2/Net3 (1:1:0,05), и получали желаемый N-4-(3-бромфенил)-6,7-диметоксихиназолин-4,5-диамин в виде желтой смолы. (Выход 0,1 г, 54%). Стадия С: 3-(3-Бромфенил)-8,9-диметокси-3H-1,3,4,6-тетраазафенален
Раствор N-4-(3-бромфенил)-6,7-диметоксихиназолин-4,5-диамина (100 мг, 0,27 ммоль) (Пример 6, стадия В, выше) в муравьиной кислоте (5 мл) нагревали при 110°С 2 час, затем к реакционной смеси добавляли водный раствор NaOH до рН 10-12. Раствор разбавляли хлороформом (100 мл), отделяли органический слой, сушили его над Na2SO4 и концентрировали. Остаток очищали хроматографически, используя EtOAc/Et3N (1:0,04) в качестве элюента, и получали желаемый 3-(3-бромфенил)-8,9-диметокси-3H-1,3,4,6-тетраазафенален в виде твердого белого вещества. (Выход 86 мг, 83%). Пример 7 3-(3-Бромфенил)-8,9-диметокси-1H,3H-1,3,4,6-тетраазафенален-2-он
К раствору N-4-(3-бромфенил)-6,7-диметоксихиназолин-4,5-диамина (0,15 мг, 0,40 ммоль) (Пример 6, стадия В, выше) в 1,2-дихлорэтане (50 мл) добавляли 1,1′-карбонилдиимидазол (0,65 г, 4,0 ммоль) (Aldrich). Реакционную смесь нагревали 4 час при перемешивании при 90°С, отгоняли растворители, остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/CH2Cl2/Net3 (1:1:0,05), и получали желаемый 3-(3-бромфенил)-8,9-диметокси-1H,3H-1,3,4,6-тетраазафенален-2-он в виде твердого коричневого вещества. (Выход 0,12 г, 75%). Пример 8 3-(3-Бромфенил)-8,9-диметокси-2-метил-3H-1,3,4,6-тетраазафенален
Раствор N-4-(3-бромфенил)-6,7-диметоксихиназолин-4,5-диамина (120 мг, 0,32 ммоль) (из Примера 6,стадия В) в уксусном ангидриде (3 мл) нагревали при 150°С в течение 2 час. К реакционной смеси добавляли водный раствор NaOH до рН 10-12. Раствор экстрагировали хлороформом (50 мл). Органический слой отделяли, сушили над Na2SO4 и концентрировали. Остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/Et3N (1:0,05), и получали желаемый 3-(3-бромфенил)-8,9-диметокси-2-метил-3H-1,3,4,6-тетраазафенален в виде коричневого твердого вещества. (Выход 60 мг, 47%). Пример 9 3-(3-Бромфенил)-8,9-диметокси-1H,3H-1,3,4,6-тетраазафенален-2-тион
К раствору N-4-(3-бромфенил)-6,7-диметоксихиназолин-4,5-диамина (80 мг, 0,21 ммоль) (из Примера 6, стадия В) в 1,2-дихлорэтане (50 мл) добавляли 1,1′-тиокарбонилдиимидазол (0,38 г, 2,13 ммоль) (Fluka). Реакционную смесь нагревали при перемешивании при 90°С в течение 4 час. Растворители удаляли, и остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/CH2Cl2/Et3N (1:2:0,05), и получая желаемый 3-(3-бромфенил)-8,9-диметокси-1H,3H-1,3,4,6-тетраазафенален-2-тион в виде желтого твердого вещества. (Выход 80 мг, 91%). Пример 10 3-(3-Бромфенил)-2-этил-8,9-диметокси-3H-1,3,4,6-тетраазафенален
Раствор N-4-(3-бромфенил)-6,7-диметоксихиназолин-4,5-диамина (120 мг, 0,32 ммоль) (из Примера 6, стадия В) в пропионовом ангидриде (3 мл) (Aldrich) нагревали при 170°С в течение 2 час, после чего к реакционной смеси добавляли водный раствор NaOH до рН 10-12. Раствор экстрагировали хлороформом (50 мл). Органический слой отделяли, сушили над Na2SO4 и концентрировали. Остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/Et3N (1:0,05), и получали желаемый 3-(3-бромфенил)-2-этил-8,9-диметокси-3H-1,3,4,6-тетраазафенален в виде коричневого твердого вещества. (Выход 50 мг, 38%). Пример 11 3-(3-Хлорфенил)-8,9-диметокси-1H, 3H-1,3,4,6-тетраазафенален-2-он
Стадия А:(3-Хлорфенил)(6,7-диметокси-5-нитрохиназолин-4-ил)амин
Раствор 6,7-диметокси-5-нитро-3H-хиназолин-4-она (0,5 г, 1,99 ммоль) (из Примера 1,стадия А) в SOCl2 (20 мл) (Aldrich) и нескольких каплях ДМФ (0,05 мл) нагревали при перемешивании при 90°С в течение 3 час. Затем растворители удаляли, и остаток высушивали в вакууме. Остаток растворяли в 2-пропаноле (30 мл), затем добавляли 3-хлоранилин (0,25 г, 1,99 ммоль) (Aldrich). Реакционную смесь нагревали при 110°С в течение 3 час. Растворители удаляли, и остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/CH2Cl2/Et3N (1:2:0,05) и получая желаемый (3-хлорфенил)(6,7-диметокси-5-нитрохиназолин-4-ил)амин в виде желтого твердого вещества. (Выход 0,4 г, 56%). Стадия В: N-4-(3-хлорфенил)-6,7-диметоксихиназолин-4,5-диамин
К раствору (3-хлорфенил)(6,7-диметокси-5-нитрохиназолин-4-ил)амина (0,2 г, 0,55 ммоль) (из Примера 11, стадия А) и NH4Cl (0,24 г, 4,43 ммоль) в смеси МеОН, Н2О и CHCl3 (50 мл, 2:1:12) добавляли порошкообразный Zn (0,72 г, 11 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 час и затем фильтровали. Фильтрат концентрировали и затем экстрагировали хлороформом (100 мл). Органический слой отделяли, сушили над Na2SO4 и концентрировали. Остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/CH2Cl2/Et3N (1:1:0,05), и получали желаемый N-4-(3-хлорфенил)-6,7-диметоксихиназолин-4,5-диамин в виде оранжевой смолы. (Выход 0,1 г, 55%). Стадия С: 3-(3-хлорфенил)-8,9-диметокси-1H,3H-1,3,4,6-тетраазафенален-2-он
К раствору N-4-(3-хлорфенил)-6,7-диметоксихиназолин-4,5-диамина (80 мг, 0,24 ммоль) (из Примера 11, стадия В) в 1,2-дихлорэтане (30 мл) добавляли 1,1′-карбонилдиимидазол (0,2 г, 1,21 ммоль) (Aldrich). Реакционную смесь нагревали при перемешивании при 90°С в течение 4 час. Растворители удаляли, и остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/CH2Cl2/Et3N (1:1:0,05), и получая желаемый 3-(3-хлорфенил)-8,9-диметокси-1H,3H-1,3,4,6-тетраазафенален-2-он в виде коричневого твердого вещества. (Выход 50 мг, 58%). Пример 12 3-(3-Хлорфенил)-8,9-диметокси-2-метил-3H-1,3,4,6-тетраазафенален
Раствор N-4-(3-хлорфенил)-6,7-диметоксихиназолин-4,5-диамина (0,2 г, 0,60 ммоль) (Пример 11, стадия В) в уксусном ангидриде (3 мл) (Aldrich) нагревали при 150°С в течение 2 час, после чего к реакционной смеси добавляли водный раствор NaOH до рН 10-12. Раствор экстрагировали хлороформом (50 мл). Органический слой отделяли, сушили над Na2SO4 и концентрировали. Остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/CH2Cl2/Et3N (1:1:0,05), и получали желаемый 3-(3-хлорфенил)-8,9-диметокси-2-метил-3H-1,3,4,6-тетраазафенален в виде твердого желтого вещества. (Выход 90 мг, 42%). Пример 13 3-(4-Хлорфенил)-8,9-диметокси-3H-1,3,4,6-тетраазафенален
Стадия А: (4-Хлорфенил)-(6,7-диметокси-5-нитрохиназолин-4-ил)амин
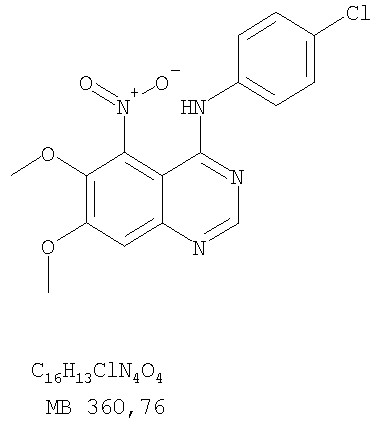
К раствору 6,7-диметокси-5-нитро-3H-хиназолин-4-она (0,5 г, 1,99 ммоль) (Пример 1, стадия А, выше) в SOCl2 (20 мл) (Aldrich) добавляли несколько капель ДМФ (0,05 мл). Реакционную смесь 3 час нагревали при перемешивании при 90°С, затем растворители отгоняли, и остаток сушили в вакууме. Остаток растворяли в 2-пропаноле (30 мл) и добавляли 4-хлоранилин (0,25 г, 1,99 ммоль) (Aldrich). Реакционную смесь нагревали при 110°С 3 час, затем удаляли растворители, остаток очищали хроматографически, используя смесь EtOAc/CH2Cl2/Et3N (1:2:0,05) в качестве элюента, и получали желаемый (4-хлорфенил)-(6,7-диметокси-5-нитрохиназолин-4-ил)амин в виде твердого желтого вещества. (Выход 0,5 г, 70%). Стадия В: N-4-(4-хлорфенил)-6,7-диметоксихиназолин-4,5-диамин
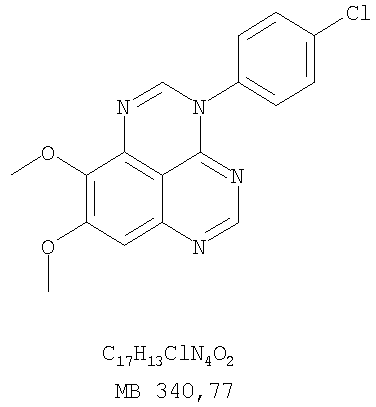
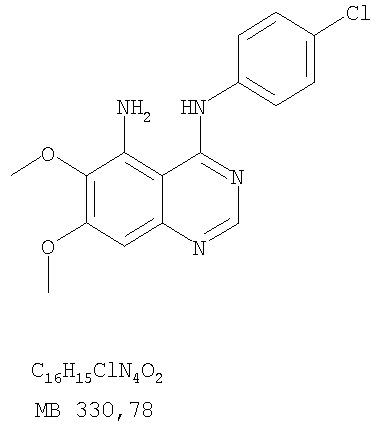
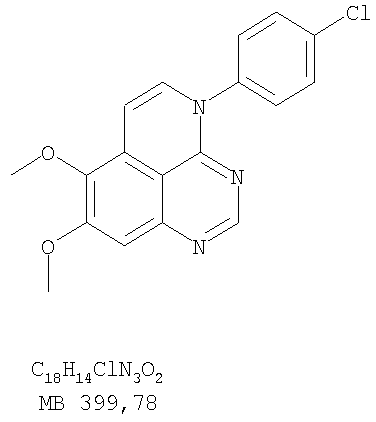
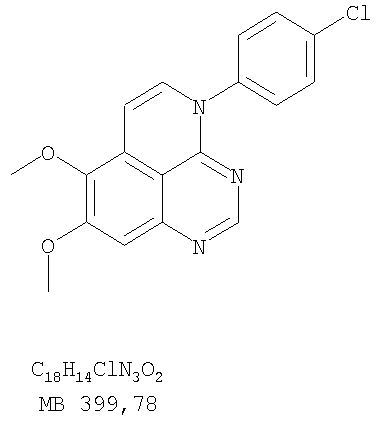
К раствору (4-хлорфенил)(6,7-диметокси-5-нитрохиназолин-4-ил)амина (0,2 г, 0,55 ммоль) (из Примера 11, стадия А) и NH4Cl (0,24 г, 4,43 ммоль) и NH4Cl (0,24 г, 4,43 ммоль) в смеси МеОН, H2O и CHCl3 (50 мл, 2:1:12) добавляли порошкообразный Zn (0,72 г, 11 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 час и затем фильтровали. Фильтрат концентрировали и затем экстрагировали хлороформом (100 мл). Органический слой отделяли, сушили над Na2SO4 и концентрировали. Остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/CH2Cl2/Et3N (1:1:0,05), и получали желаемый N-4-(4-хлорфенил)-6,7-диметоксихиназолин-4,5-диамин в виде бледно-желтой смолы. (Выход 0,16 г, 88%). Стадия С: 3-(4-Хлорфенил)-8,9-диметокси-3H-1,3,4,6-тетраазафенален
Раствор N-4-(4-хлорфенил)-6,7-диметоксихиназолин-4,5-диамина (160 мг, 0,48 ммоль) (Пример 13, стадия В) в муравьиной кислоте (2 мл) нагревали 2 час при 110°С, затем добавляли водный раствор NaOH до рН 10-12, раствор разбавляли хлороформом (100 мл), отделяли органическую фазу, сушили над Na2SO4 и концентрировали. Остаток очищали хроматографически в системе EtOAc/Et3N (1:0,05) в качестве элюента и получали желаемый 3-(4-хлорфенил)-8,9-диметокси-3H-1,3,4,6-тетраазафенален в виде почти белого твердого вещества. (Выход 96 мг, 59%). Пример 14 3-(4-Хлорфенил)-8,9-диметокси-1H,3H-1,3,4,6-тетраазафенален-2-тион
К раствору N-4-(4-хлорфенил)-6,7-диметоксихиназолин-4,5-диамина (120 мг, 0,36 ммоль) (Пример 13, стадия В) в 1,2-дихлорэтане (30 мл) добавляли 1,1′-тиокарбонилдиимидазол (0,65 г, 3,63 ммоль) (Aldrich). Реакционную смесь нагревали при перемешивании при 90°С в течение 4 час. Растворители удаляли, и остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/CH2Cl2/Et3N (1:3:0,05) и получая желаемый 3-(4-хлорфенил)-8,9-диметокси-1H,3H-1,3,4,6-тетраазафенален-2-тион в виде коричневого твердого вещества. (Выход 110 мг, 82%). Пример 15 7,8-Диметокси-5-метил-4-фенил-4H-1,3,4-триазафенален
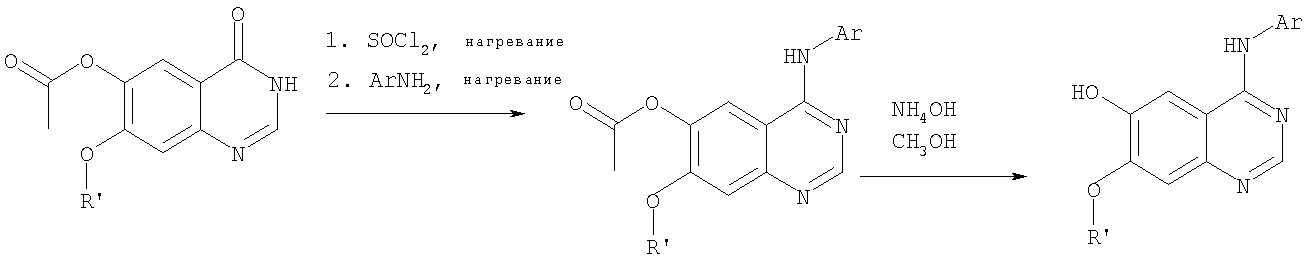
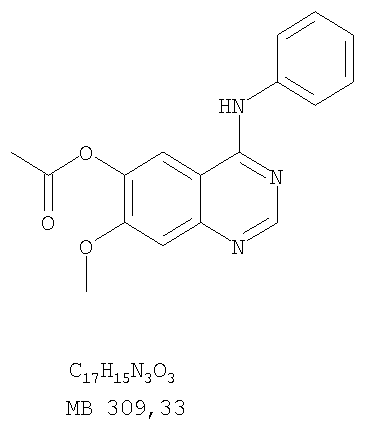
Стадия А: 6-ацетокси-7-метокси-4-фениламинохиназолин
6-Ацетокси-7-метоксихиназолин-4-он был получен согласно процессу, описанному в литературе Gibson, К.Н. и др. Bioorganic & Medicinal Chemistry Letters, 1997, 7, №21, c.c.2723-2728. К раствору 6-ацетокси-7-метоксихиназолин-4-она (1,2 г, 5,13 ммоль) в SOCl2 (30 мл) (Aldrich) добавляли несколько капель ДМФ (0,1 мл). Реакционную смесь затем нагревали при перемешивании при 100°С в течение 3 час, после чего растворители удаляли, и остаток высушивали в вакууме. Остаток растворяли в 2-пропаноле (30 мл), затем добавляли анилин (0,47 мл, 5,13 ммоль) (Aldrich) и нагревали реакционную смесь при 110°С в течение 3 час, после чего охлаждали до комнатной температуры и фильтровали. Осадок собирали, высушивали в вакууме, получая 6-ацетокси-7-метокси-4-фениламинохиназолин в виде белого твердого вещества. (Выход 1,0 г, 63%). Стадия В: 7-метокси-4-фениламинохиназолин-6-ол
К раствору 6-ацетокси-7-метокси-4-фениламинохиназолина (1,01 г, 3,27 ммоль) (из Примера 15, стадия А) в МеОН (30 мл) добавляли водный раствор NH4OH (29%, 2,0 г, 31,4 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 час, затем нагревали при 100°С в течение 1,5 час, затем охлаждали до комнатной температуры и фильтровали. Осадок собирали, высушивали в вакууме, получая желаемый 7-метокси-4-фениламинохиназолин-6-ол в виде твердого белого вещества. (Выход 0,6 г, 69%). Стадия С: (6-аллилокси-7-метоксихиназолин-4-ил)фениламин
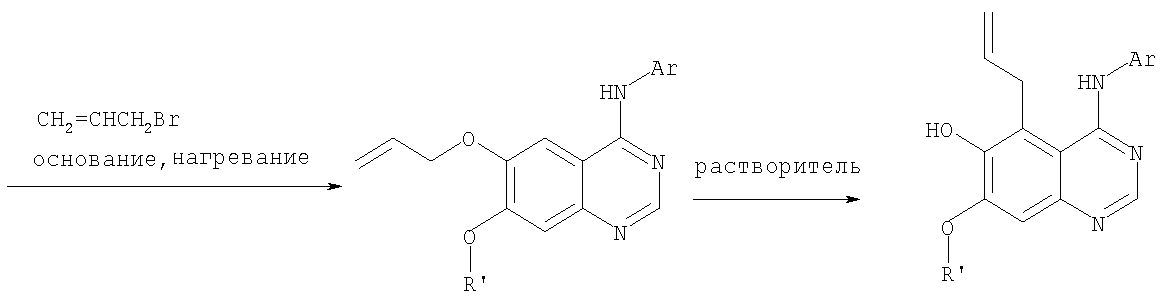
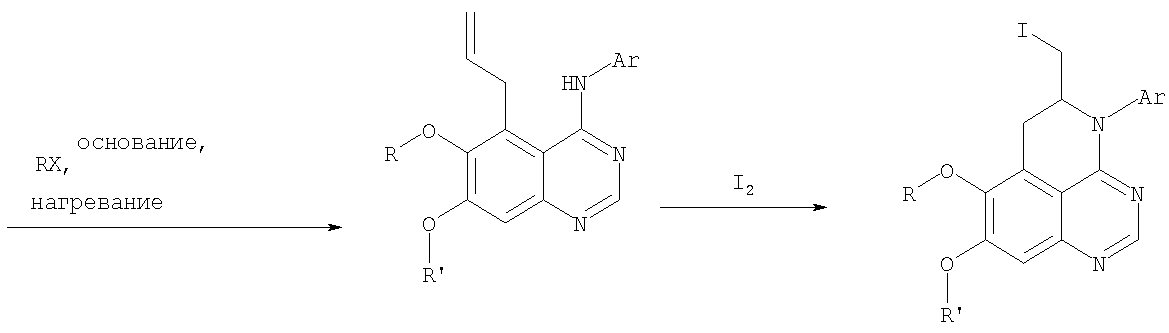
К раствору 7-метокси-4-фениламинохиназолин-6-ола (0,56 г, 2,1 ммоль) (из Примера 15, стадия В) в ацетоне (50 мл) добавляли К2СО3 (1,45 г, 10 ммоль) и аллилбромид (0,2 мл, 2,3 ммоль) (Aldrich). Реакционную смесь нагревали при перемешивании в течение 4 час при 90°С, затем охлаждали до комнатной температуры, фильтровали, фильтрат концентрировали. Остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/CH2Cl2/Et3N (1:4:0,04), и получали желаемый (6-аллилокси-7-метоксихиназолин-4-ил)фениламин в виде твердого желтого вещества. (Выход 0,6 г, 93%). Стадия Д: 5-аллил-7-метокси-4-фениламинохиназолин-6-ол
Раствор (6-аллилокси-7-метоксихиназолин-4-ил)фениламина (0,30 г, 0,98 ммоль) (из Примера 15, стадия С) в о-ксилоле (50 мл) нагревали при 150°С в течение 7 час. В конце этого периода анализ смеси методом ТСХ показал почти полный расход исходного вещества и образование желаемого продукта как главного составляющего смеси. Растворитель удаляли, и остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/Et3N (1:0,04), и получали желаемый 5-аллил-7-метокси-4-фениламинохиназолин-6-ол в виде не очень белого твердого вещества. (Выход 0,26 г, 87%). Стадия Е: (5-аллил-6,7-диметоксихиназолин-4-ил)фениламин
К раствору 5-аллил-7-метокси-4-фениламинохиназолин-6-ола (0,16 г, 0.524 ммоль) (Пример 15, стадия D) в ацетоне (30 мл) добавляли К2СО3 (0,36 г, 2,62 ммоль) и йодистый метил (0,15 г, 1,05 ммоль) (Aldrich). Реакционную смесь нагревали при перемешивании при 90°С в течение 4 час, затем охлаждали до комнатной температуры, фильтровали и концентрировали. Остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/СН2Cl2/Et3N (1:5:0,05), и получали желаемый (5-аллил-6,7-диметоксихиназолин-4-ил)фениламин в виде бледно-желтого твердого вещества. (Выход 0,13 г, 77%). Стадия F: 5-Йодметил-7,8-диметокси-4-фенил-5,6-дигидро-4H-1,3,4-триазафенален
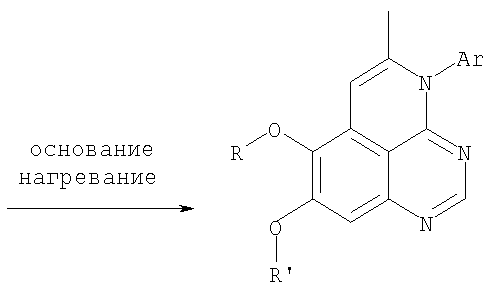
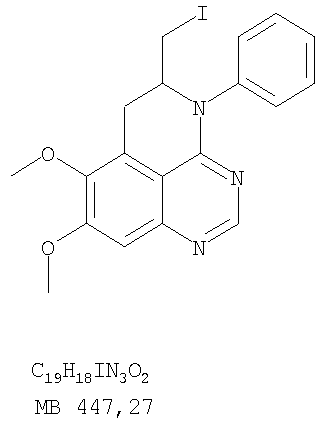
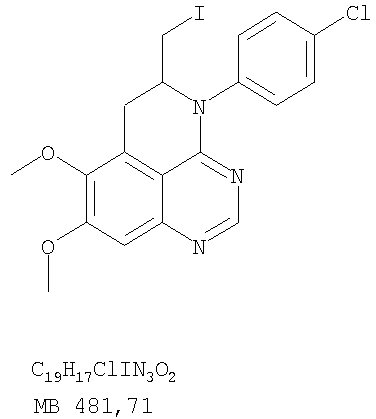
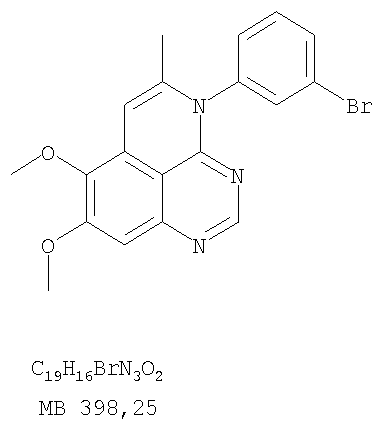
К раствору (5-аллил-6,7-диметоксихиназолин-4-ил)фениламина (0,12 г, 0,37 ммоль) (из Примера 15, стадия Е) в дихлорметане (20 мл) добавляли 1 г (0,24 г, 1,87 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 час, разбавляли хлороформом (100 мл) и промывали насыщенным водным раствором Na2SO3. Органический слой отделяли, высушивали над Na2SO4 и концентрировали. Остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/СН2Cl2/Et3Н (1:2:0,05), и получали желаемый 5-иодметил-7,8-диметокси-4-фенил-5,6-дигидро-4H-1,3,4-триазафенален в виде бледно-желтого твердого вещества. (Выход 0,11 г, 66%). Стадия G: 7,8-Диметокси-5-метил-4-фенил-4H-1,3,4-триазафенален
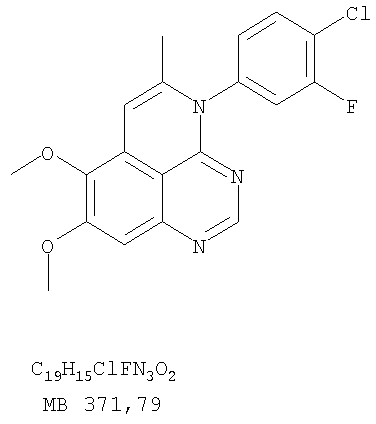
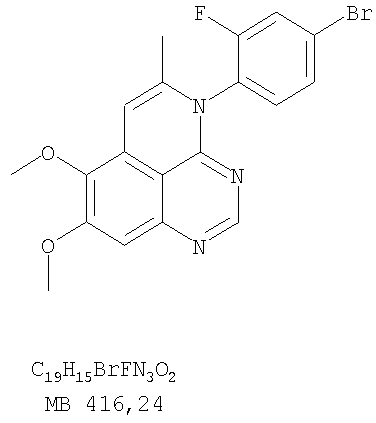
К раствору 5-иодметил-7,8-диметокси-4-фенил-5,6-дигидро-4H-1,3,4-триазафеналена (0,1 г, 0,22 ммоль) (из Примера 15, стадия F) в толуоле (20 мл) добавляли ДБУ (73 мкл, 0,49 ммоль) (Fluka). Реакционную смесь нагревали при 120°С в течение 1 час, затем разбавляли хлороформом (100 мл) и промывали Н2О. Органический слой отделяли, высушивали над Na2SO4 и концентрировали. Остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/CH2Cl2/Et3N (1:1:0,05), и получали желаемый 7,8-диметокси-5-метил-4-фенил-4H-1,3,4-триазафенален в виде бледно-желтого твердого вещества. (Выход 50 мг, 71%). Пример 16 4-(4-Хлорфенил)-7,8-диметокси-5-метил-4H-1,3,4-триазафенален
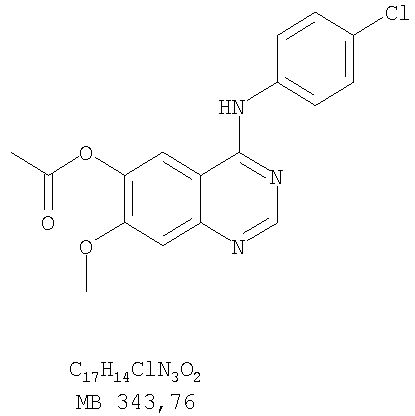
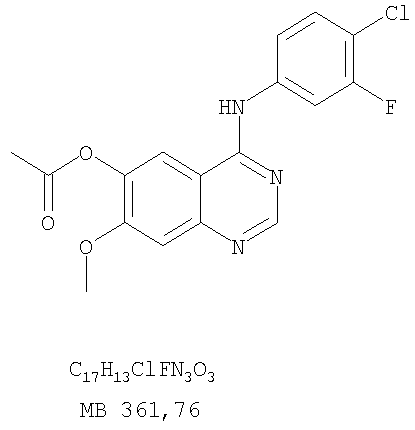
Стадия А: 6-ацетокси-4-(4-хлорфениламино)-7-метоксихиназолин
К раствору 6-ацетокси-7-метоксихиназолин-4-она (1,0 г, 4,26 ммоль) (из Примера 15, стадия А) в SOCl2 (12,5 мл) (Aldrich) добавляли несколько капель ДМФ (0,1 мл). Реакционную смесь затем нагревали при перемешивании при 100°С в течение 3 час. Растворители затем удаляли, и остаток высушивали в вакууме. Остаток растворяли в 2-пропаноле (20 мл), затем добавляли 4-хлоранилин (0,6 г, 4,69 мл) (Fluka). Реакционную смесь нагревали при 110°С в течение 3 час, затем охлаждали до комнатной температуры и фильтровали. Осадок собирали и высушивали в вакууме, получая 6-ацетокси-4-(4-хлорфениламино)-7-метоксихиназолин в виде серого твердого вещества. (Выход 1,45 г, 99%). Стадия В: 4-(4-Хлорфениламино)-7-метоксихиназолин-6-ол
К раствору 6-ацетокси-4-(4-хлорфениламино)-7-метоксихиназолина (1.4 г, 4,07 ммоль) (Пример 16, стадия А) в МеОН (35 мл) добавляли водный раствор NH4OH (29%, 0,82 мл, 12,2 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 час, затем нагревали при 100°С в течение 1,5 час, после чего охлаждали до комнатной температуры и фильтровали. Осадок собирали и высушивали в вакууме, получая желаемый 4-(4-хлорфениламино)-7-метоксихиназолин-6-ол в виде серого твердого вещества. (Выход 0,67 г, 55%). Стадия С: (6-Аллилокси-7-метоксихиназолин-4-ил)(4-хлорфенил)амин
К раствору 4-(4-хлорфениламино)-7-метоксихиназолин-6-ола (0,61 г, 2,02 ммоль) (из Примера 16, стадия В) в ацетоне (50 мл) добавляли К2СО3 (0,84 г, 6,06 ммоль) и аллилбромид (0,52 мл, 6,06 ммоль) (Aldrich). Реакционную смесь нагревали при перемешивании при 90°С в течение 4 час. Смесь охлаждали до комнатной температуры, фильтровали, и фильтрат концентрировали. Остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/CH2Cl2/Et3N (1:1:0,01), и получали желаемый (6-аллилокси-7-метоксихиназолин-4-ил)(4-хлорфенил)амин в виде белого твердого вещества. (Выход 0,5 г, 72%). Стадия D: 5-Аллил-4-(4-хлорфениламино)-7-метоксихиназолин-6-ол
Раствор (6-аллилокси-7-метоксихиназолин-4-ил)(4-хлорфенил)амина(0,50 г, 1,46 ммоль) (из Примера 16, стадия С) в о-ксилоле (50 мл) нагревали при 150°С в течение 7 час. В конце этого периода анализ смеси методом ТСХ показал почти полное использование исходного вещества и образование желаемого продукта как главного составляющего смеси. Растворитель удаляли, и остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/Et3N (1:0,04), и получали желаемый 5-аллил-4-(4-хлорфениламино)-7-метоксихиназолин-6-ол в виде не очень белого твердого вещества. (Выход 0,4 г, 80%). Стадия Е: (5-Аллил-6,7-диметоксихиназолин-4-ил)(4-хлорфенил)амин
К раствору 5-аллил-4-(4-хлорфениламино)-7-метоксихиназолин-6-ола (0,45 г, 1,32 ммоль) (из Примера 16, стадия D) в ацетоне (30 мл) добавляли К2СО3 (0,36 г, 2,6 ммоль) и йодистый метил (0,75 г, 5,3 ммоль) (Aldrich). Реакционную смесь нагревали при перемешивании при 90°С в течение 4 час, затем охлаждали до комнатной температуры, фильтровали, и фильтрат концентрировали. Остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/CH2Cl2/Et3N (1:2:0,01), и получали желаемый (5-аллил-6,7-диметоксихиназолин-4-ил)(4-хлорфенил)амин в виде белого твердого вещества. (Выход 0,3 г, 84%). Стадия F: 4-(4-Хлорфенил)-5-йодметил-7,8-диметокси-5,6-дигидро-4H-1,3,4-триазафенален
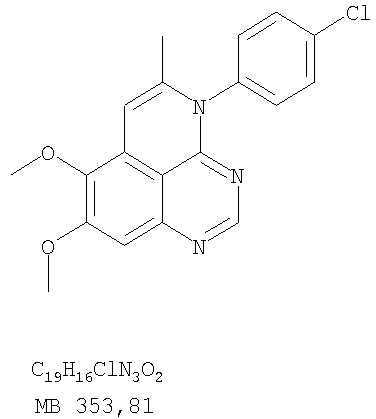
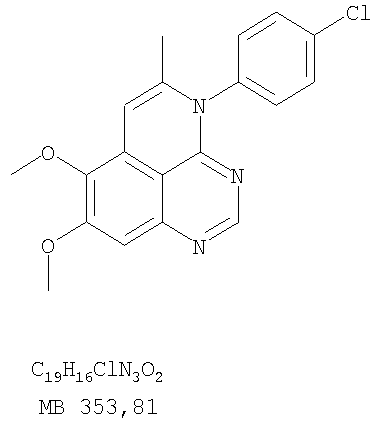
К раствору (5-аллил-6,7-диметоксихиназолин-4-ил)(4-хлорфенил)амина (0,3 г, 0,85 ммоль) (Пример 16, стадия Е) в дихлорметане (50 мл) добавляли I2 (0,53 г, 4,2 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 час, затем разбавляли хлороформом (100 мл) и промывали насыщенным раствором Na2SO3. Отделяли органический слой, сушили его над Na2SO4 и концентрировали. Остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/CH2Cl2/Et3N (2:1:0,01), и получали желаемый 4-(4-хлорфенил)-5-йодметил-7,8-диметокси-5,6-дигидро-4H-1,3,4-триазафенален в виде твердого белого вещества. (Выход 0,15 г, 37%). Стадия G: 4-(4-Хлорфенил)-7,8-диметокси-5-метил-4H-1,3,4-триазафенален
К раствору 4-(4-хлорфенил)-5-йодметил-7,8-диметокси-5,6-дигидро-4H-1,3,4-триазафеналена (0.15 г, 0,31 ммоль) (Пример 16, Стадия F) в толуоле (50 мл) добавляли ДБУ (0,46 мл, 3,1 ммоль) (Fluka). Реакционную смесь нагревали 1 час при 120°С, затем разбавляли хлороформом (100 мл) и промывали Н2О. Органический слой отделяли, сушили над Na2SO4 и концентрировали. Остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/CH2Cl2/Et3N (1:1:0,05), и получали желаемый 4-(4-хлорфенил)-7,8-диметокси-5-метил-4H-1,3,4-триазафенален в виде бледно-желтого твердого вещества. (Выход 0,1 г, 91%). Пример 17 4-(3-Бромфенил)-7,8-диметокси-5-метил-4Н-1,3,4-триазафенален
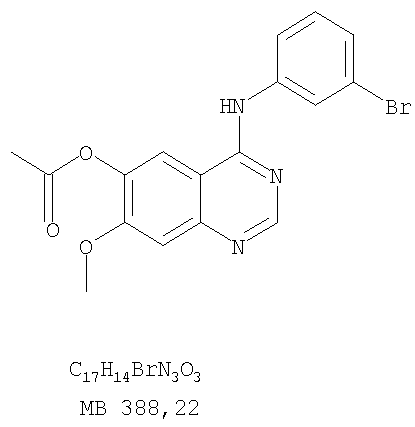
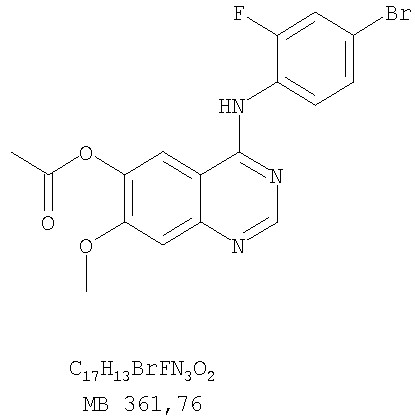
Стадия А: 6-Ацетокси-4-(3-бромфениламино)-7-метоксихиназолин
К раствору 6-ацетокси-7-метоксихиназолин-4-она (1,0 г, 4,26 ммоль) (Пример 15, стадия А) в SOCl2 (12,5 мл) (Aldrich) добавляли несколько капель ДМФ (0,1 мл). Реакционную смесь нагревали 3 час при 100°С, затем растворители отгоняли, и остаток сушили в вакууме. Остаток растворяли в 2-пропаноле (20 мл) и добавляли 3-броманилин (0,806 г, 4,69 ммоль). Реакционную смесь нагревали 3 час при 110°С, затем охлаждали до комнатной температуры и фильтровали. Осадок собирали и сушили в вакууме, получая 6-ацетокси-4-(3-бромфениламино)-7-метоксихиназолин в виде твердого серого вещества. (Выход 1,65 г, 100%). Стадия В: 4-(3-Бромфениламино)-7-метоксихиназолин-6-ол
К раствору 6-ацетокси-4-(3-бромфениламино)-7-метоксихиназолина (1,59 г, 4,11 ммоль) (из Примера 17, стадия А) в МеОН (30 мл) добавляли водный раствор NH4OH (29%, 0,83 мл, 12,3 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 час, затем нагревали при 100°С в течение 1,5 час, после чего охлаждали до комнатной температуры и фильтровали. Осадок собирали и высушивали в вакууме, получая желаемый 4-(3-бромфениламино)-7-метоксихиназолин-6-ол в виде серого твердого вещества. (Выход 1,25 г, 88%). Стадия С: (6-Аллилокси-7-метоксихиназолин-4-ил)(3-бромфенил)амин
К раствору 4-(3-бромфениламино)-7-метоксихиназолин-6-ола (1,24 г, 3,58 ммоль) (из Примера 17, стадия В) в ацетоне (250 мл) добавляли К2СО3 (0,99 г, 7,16 ммоль) и аллилбромид (1,55 мл, 17,9 ммоль) (Aldrich). Реакционную смесь нагревали при перемешивании при 90°С в течение 4 час. Смесь охлаждали до комнатной температуры, фильтровали, и фильтрат концентрировали. Остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/CH2Cl2/Et3N (2:3:0,02), и получали желаемый (6-аллилокси-7-метоксихиназолин-4-ил)(3-бромфенил)амин в виде желтого масла. (Выход 0,55 г, 40%). Стадия D: 5-Аллил-4-(3-бромфениламино)-7-метоксихиназолин-6-ол
Раствор (6-аллилокси-7-метоксихиназолин-4-ил)(3-бромфенил)амина (0,54 г, 1,39 ммоль) (из Примера 17, стадия С) в о-ксилоле (75 мл) нагревали при 150°С в течение 4,5 час. В конце этого периода анализ смеси методом ТСХ показал почти полное использование исходного вещества и образование желаемого продукта как главного составляющего смеси. Раствор концентрировали и фильтровали. Осадок собирали, высушивали в вакууме, получая желаемый 5-аллил-4-(3-бромфениламино)-7-метоксихиназолин-6-ол в виде не очень белого твердого вещества. (Выход 0,33 г, 61%). Стадия Е: (5-Аллил-6,7-диметоксихиназолин-4-ил)(3-бромфенил)амин
К раствору 5-аллил-4-(3-бромфениламино)-7-метоксихиназолин-6-ола (0,27 г, 0,7 ммоль) (из Примера 17, стадия D) в ацетоне (30 мл) добавляли К2СО3 (0,48 г, 3,5 ммоль) и йодистый метил (0,4 г, 2,8 ммоль) (Aldrich). Реакционную смесь нагревали при перемешивании при 90°С в течение 4 час, затем охлаждали до комнатной температуры, фильтровали, и фильтрат концентрировали. Остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/CH2Cl2/Et3N (1:3:0,01), и получали желаемый (5-аллил-6,7-диметоксихиназолин-4-ил)(3-бромфенил)амин в виде желтого твердого вещества. (Выход 0,14 г, 50%). Стадия F: 4-(3-Бромфенил)-5-иодметил-7,8-диметокси-5,6-дигидро-4H-1,3,4-триазафенален
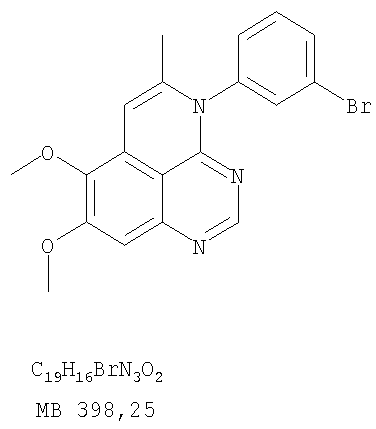
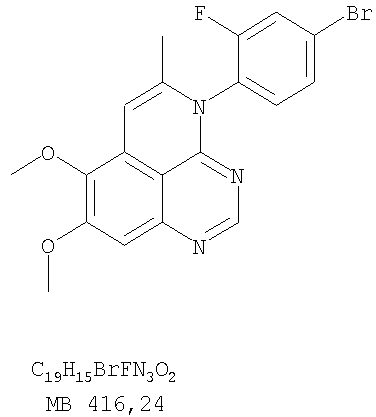
К раствору (5-аллил-6,7-диметоксихиназолин-4-ил)(3-бромфенил)амина (0,14 г, 0,35 ммоль) (из Примера 17, стадия Е) в дихлорметане (20 мл) добавляли I2 (0,23 г, 1,75 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 час, затем разбавляли хлороформом (100 мл) и промывали насыщенным раствором Na2SO3 Отделяли органический слой, сушили его над Na2SO4 и концентрировали. Остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/CH2Cl2/Et3N (1:2:0,01), и получали желаемый 4-(3-бромфенил)-5-иодметил-7,8-диметокси-5,6-дигидро-4H-1,3,4-триазафенален в виде бледно-желтой пены. (Выход 0,12 г, 66%). Стадия G: 4-(3-Бромфенил)-7,8-диметокси-5-метил-4H-1,3,4-триазафенален
К раствору 4-(3-бромфенил)-5-иодметил-7,8-диметокси-5,6-дигидро-4H-1,3,4-триазафеналена (0,12 г, 0,23 ммоль) (из Примера 17, стадия F) в толуоле (20 мл) добавляли ДБУ (0,075 мл, 0,5 ммоль) (Fluka). Реакционную смесь нагревали 1 час при 120°С, затем разбавляли хлороформом (100 мл) и промывали Н2О. Органический слой отделяли, сушили над Na2SO4 и концентрировали. Остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/CH2Cl2/Et3N (1:2:0,05), и получали желаемый 4-(3-бромфенил)-7,8-диметокси-5-метил-4H-1,3,4-триазафенален в виде бледно-желтого твердого вещества. (Выход 0,09 г, 100%). Пример 18 4-(3-Хлор-4-фторфенил)-7,8-диметокси-5-метил-4H-1,3,4-триазафенален
Стадия А: 6-Ацетокси-4-(3-хлор-4-фторфениламино)-7-метоксихиназолин
К раствору 6-ацетокси-7-метоксихиназолин-4-она (1,0 г, 4,26 ммоль) (из Примера 15, стадия А) в SOCl2 (12,5 мл) (Aldrich) добавляли несколько капель ДМФ (0,1 мл). Реакционную смесь нагревали 3 час при 100°С, затем растворители отгоняли, и остаток сушили в вакууме. Остаток растворяли в 2-пропаноле (20 мл) и добавляли 4-хлор-3-фторанилин (0,682 г, 4,69 ммоль). Реакционную смесь нагревали 3 час при 110°С, затем охлаждали до комнатной температуры и фильтровали. Осадок собирали и сушили в вакууме, получая 6-ацетокси-4-(3-хлор-4-фторфениламино)-7-метоксихиназолин в виде серого твердого вещества. (Выход 1,54 г, 100%). Стадия В: 4-(4-Хлор-4-фторфениламино)-7-метоксихиназолин-6-ол
К раствору 6-ацетокси-4-(3-хлор-4-фторфениламино)-7-метоксихиназолина (1,54 г, 4,26 ммоль) (из Примера 18, стадия А) в МеОН (30 мл) добавляли водный раствор NH4OH (29%, 0,86 мл, 12,7 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 час, затем нагревали при 100°С в течение 1,5 час, после чего охлаждали до комнатной температуры и фильтровали. Осадок собирали и высушивали в вакууме, получая желаемый 4-(4-хлор-4-фторфениламино)-7-метоксихиназолин-6-ол в виде твердого серого вещества. (Выход 1,21 г, 89%). Стадия С: (6-Аллилокси-7-метоксихиназолин-4-ил)(3-хлор-4-фторфенил)амин
К раствору 4-(4-хлор-4-фторфениламино)-7-метоксихиназолин-6-ола (0,70 г, 2,18 ммоль) (из Примера 18, стадия В) в ацетоне (140 мл) добавляли К2СО3 (0,61 г, 10,9 ммоль) и аллилбромид (0,94 мл, 10,9 ммоль) (Aldrich). Реакционную смесь нагревали при перемешивании при 90°С в течение 4 час. Смесь охлаждали до комнатной температуры, фильтровали, и фильтрат концентрировали. Остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/CH2Cl2/Et3N (1:1:0,02), и получали желаемый (6-аллилокси-7-метоксихиназолин-4-ил)(3-хлор-4-фторфенил)амин в виде не очень белого твердого вещества. (Выход 0,64 г, 82%). Стадия D: 5-Аллил-4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ол
Раствор (6-аллилокси-7-метоксихиназолин-4-ил)(3-хлор-4-фторфенил)амина (0,92 г, 2,55 ммоль) (из Примера 18, стадия С) в о-ксилоле (150 мл) нагревали при 150°С в течение 7 час. В конце этого периода анализ смеси методом ТСХ показал почти полное использование исходного вещества и образование желаемого продукта как главного составляющего смеси. Раствор концентрировали и фильтровали. Осадок собирали и высушивали в вакууме, получая желаемый 5-аллил-4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ол в виде не очень белого твердого вещества. (Выход 0,65 г, 71%). Стадия Е: (5-Аллил-6,7-диметоксихиназолин-4-ил)(3-хлор-4-фторфенил)амин
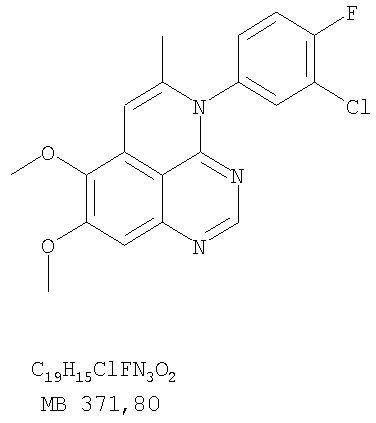
К раствору 5-аллил-4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ола (0,59 г, 1,63 ммоль) (из Примера 18, стадия D) в ацетоне (175 мл) добавляли К2СО3 (0,68 г, 4,91 ммоль) и йодистый метил (2,32 г, 16,4 ммоль) (Aldrich). Реакционную смесь нагревали при перемешивании при 90°С в течение 3 час, затем охлаждали до комнатной температуры, фильтровали, и фильтрат концентрировали. Остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/CH2Cl2/Et3N (1:3:0,01), и получали желаемый (5-аллил-6,7-диметоксихиназолин-4-ил)(3-хлор-4-фторфенил)амин в виде не очень белого твердого вещества. (Выход 0,29 г, 48%). Стадия F: 4-(3-Хлор-4-фторфенил)-5-иодметил-7,8-диметокси-5,6-дигидро-4H-1,3,4-триазафенален
К раствору (5-аллил-6,7-диметоксихиназолин-4-ил)(3-хлор-4-фторфенил)амина (0,102 г, 0,27 ммоль) (из Примера 18, стадия Е) в дихлорметане (20 мл) добавляли I2 (0,69 г, 2,7 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 час, затем разбавляли хлороформом (100 мл) и промывали насыщенным раствором Na2SO4. Отделяли органический слой, сушили его над Na2SO4 и концентрировали, получая желаемый 4-(3-хлор-4-фторфенил)-5-иодметил-7,8-диметокси-5,6-дигидро-4H-1,3,4-триазафенален в виде желтой пены. (Выход 0,134 г, 99%). Стадия G: 4-(3-Бромфенил)-7,8-диметокси-5-метил-4H-1,3,4-триазафенален
К раствору 4-(3-хлор-4-фторфенил)-5-иодметил-7,8-диметокси-5,6-дигидро-4H-1,3,4-триазафеналена (0,125 г, 0,25 ммоль) (из Примера 18, стадия F) в толуоле (25 мл) добавляли ДБУ (0,37 мл, 2,5 ммоль) (Fluka). Реакционную смесь нагревали 1 час при 120°С, затем разбавляли хлороформом (100 мл) и промывали Н2О. Органический слой отделяли, сушили над Na2SO4 и концентрировали. Остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/CH2Cl2/Et3N (3:1:0,05), и получали желаемый 4-(3-бромфенил)-7,8-диметокси-5-метил-4H-1,3,4-триазафенален в виде белого твердого вещества. (Выход 0,093 г, 100%). Пример 19 4-(4-Бром-2-фторфенил)-7,8-диметокси-5-метил-4H-1,3,4-триазафенален
Стадия А: 6-Ацетокси-4-(4-бром-2-фторфениламино)-7-метоксихиназолин
К раствору 6-ацетокси-7-метоксихиназолин-4-она (1,0 г, 4,26 ммоль) (Пример 15, стадия А) в SOCl2 (12,5 мл) (Aldrich) добавляли несколько капель ДМФ (0,1 мл). Реакционную смесь нагревали 3 час при 100°С, затем растворители отгоняли и остаток сушили в вакууме. Остаток растворяли в 2-пропаноле (20 мл) и добавляли 4-бром-2-фторанилин (0,891 г, 4,69 ммоль) (Aldrich). Реакционную смесь нагревали 3 час при 110°С, затем охлаждали до комнатной температуры и фильтровали. Осадок собирали и сушили в вакууме, получая 6-ацетокси-4-(4-бром-2-фторфениламино)-7-метоксихиназолин в виде твердого серого вещества. (Выход 1,42 г, 82%). Стадия В: 4-(4-Бром-2-фторфениламино)-7-метоксихиназолин-6-ол
К раствору 6-ацетокси-4-(4-бром-2-фторфениламино)-7-метоксихиназолина (1,36 г, 3,34 ммоль) (Пример 19, стадия А) в МеОН (30 мл) добавляли водный раствор NH4OH (29%, 0,68 мл, 10 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 час, затем нагревали при 100°С в течение 1,5 час, после чего охлаждали до комнатной температуры и фильтровали. Осадок собирали и высушивали в вакууме, получая желаемый 4-(4-бром-2-фторфениламино)-7-метоксихиназолин-6-ол в виде твердого серого вещества. (Выход 0,93 г, 77%). Стадия С: (6-Аллилокси-7-метоксихиназолин-4-ил)(4-бром-2-фторфенил)амин
К раствору 4-(4-бром-2-фторфениламино)-7-метоксихиназолин-6-ола (0,50 г, 1,37 ммоль) (из Примера 19, стадия В) в ацетоне (100 мл) добавляли К2СО3 (0,38 г, 2,74 ммоль) и аллилбромид (0,59 мл, 6,86 ммоль) (Aldrich). Реакционную смесь нагревали при перемешивании при 90°С в течение 2 час. Смесь охлаждали до комнатной температуры, фильтровали, и фильтрат концентрировали. Остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/CH2Cl2/Et3N (1:1:0,02), и получали желаемый (6-аллилокси-7-метоксихиназолин-4-ил)(4-бром-2-фторфенил)амин в виде бледно-желтого твердого вещества. (Выход 0,52 г, 95%). Стадия D: 5-Аллил-4-(4-бром-2-фторфениламино)-7-метоксихиназолин-6-ол
Раствор (6-аллилокси-7-метоксихиназолин-4-ил)(4-бром-2-фторфенил)амина (0,46 г, 1,13 ммоль) (из Примера 19, стадия С) в о-ксилоле (75 мл) нагревали при 150°С в течение 4 час. В конце этого периода анализ смеси методом ТСХ показал почти полное использование исходного вещества и образование желаемого продукта как главного составляющего смеси. Раствор концентрировали до небольшого объема и фильтровали. Осадок собирали, высушивали в вакууме, получая желаемый 5-аллил-4-(4-бром-2-фторфениламино)-7-метоксихиназолин-6-ол в виде не очень белого твердого вещества. (Выход 0,35 г, 76%). Стадия Е: (5-Аллил-6,7-диметоксихиназолин-4-ил)(4-бром-2-фторфенил)амин
К раствору 5-аллил-4-(4-бром-2-фторфениламино)-7-метоксихиназолин-6-ола (0,34 г, 0,84 ммоль) (из Примера 19, стадия D) в ацетоне (100 мл) добавляли К2СО3 (0,35 г, 2,52 ммоль) и йодистый метил (1,19 г, 8,41 ммоль) (Aldrich). Реакционную смесь нагревали при перемешивании при 90°С в течение 4,5 час, затем охлаждали до комнатной температуры, фильтровали, и фильтрат концентрировали. Остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/CH2Cl2/Et3N (1:4:0,01), и получали желаемый (5-аллил-6,7-диметоксихиназолин-4-ил)(4-бром-2-фторфенил)амин в виде желтого твердого вещества. (Выход 0,33 г, 94%). Стадия F: 4-(4-Бром-2-фторфенил)5-иодметил-7,8-диметокси-5,6-дигидро-4H-1,3,4-триазафенален
К раствору (5-аллил-6,7-диметоксихиназолин-4-ил)(4-бром-2-фторфенил)амина (0,236 г, 0,564 ммоль) (из Примера 19, стадия Е) в дихлорметане (30 мл) добавляли I2 (1,43 г, 5,64 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 час, затем разбавляли хлороформом (100 мл) и промывали насыщенным раствором Na2SO3. Отделяли органический слой, сушили его над Na2SO4 и концентрировали, получая желаемый 4-(4-бром-2-фторфенил)-5-иодметил-7,8-диметокси-5,6-дигидро-4H-1,3,4-триазафенален в виде не очень белого твердого вещества. (Выход 0,034 г, 11%). Стадия G: 4-(4-Бром-2-фторфенил)7,8-диметокси-5-метил-4H-1,3,4-триазафенален
К раствору 4-(4-бром-2-фторфенил)-5-иодметил-7,8-диметокси-5,6-дигидро-4H-1,3,4-триазафеналена (33 мг, 0,06 ммоль) (из Примера 19, стадия F) в толуоле (7 мл) добавляли ДБУ (0,09 мл, 0,6 ммоль) (Fluka). Реакционную смесь нагревали 1 час при 120°С, затем разбавляли хлороформом (100 мл) и промывали Н2О. Органический слой отделяли, сушили над Na2SO4 и концентрировали. Остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/CH2Cl2/Et3N (1:1:0,05), и получали желаемый 4-(4-бром-2-фторфенил)-7,8-диметокси-5-метил-4H-1,3,4-триазафенален в виде белого твердого вещества. (Выход 0,027 г, 100%). Пример 20 7,8-Диметокси-4-фенил-4H-1,3,4-триазафенален
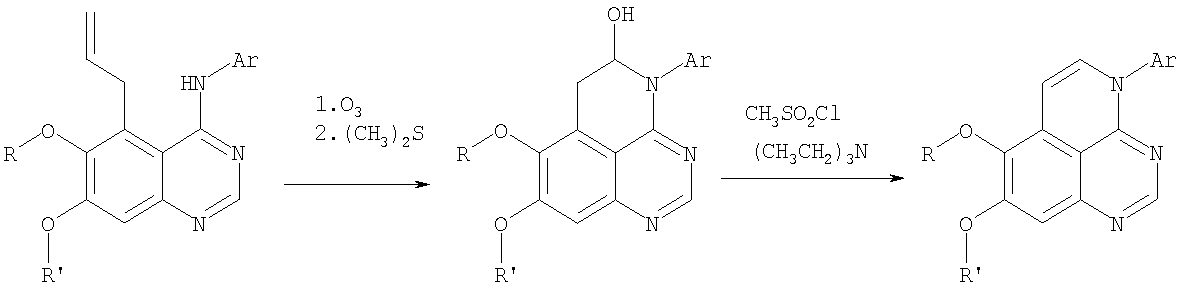
Стадия А: 7,8-Диметокси-4-фенил-5,6-дигидро-4H-1,3,4-триазафенален-5-ол
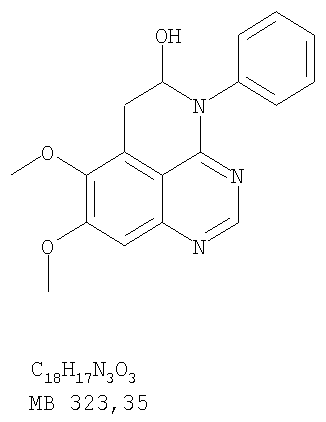
Через раствор (5-аллил-6,7-диметоксихиназолин-4-ил)фениламина (0,35 г, 1,09 ммоль) (Пример 15, стадия Е) в метаноле (50 мл) при -78°С пропускали струю О3 до тех пор, пока цвет раствора не стал голубым. Затем реакционную колбу заполнили аргоном и добавили раствор метилсульфида (5 мл) (Aldrich). Доводили реакционную смесь до комнатной температуры и перемешивали в течение ночи. Раствор концентрировали, и остаток сушили в вакууме, получая желаемый 7,8-диметокси-4-фенил-5,6-дигидро-4H-1,3,4-триазафенален-5-ол в виде сырого желтого твердого вещества. (Выход 0,17 г, 48%). Стадия В: 7,8-Диметокси-4-фенил-4H-1,3,4-триазафенален
К раствору 7,8-диметокси-4-фенил-5,6-дигидро-4H-1,3,4-триазафенален-5-ола (91 мг, 0,28 ммоль) (Пример 20, стадия А) и триэтиламина (0,23 мл, 1,69 ммоль) в дихлорметане (50 мл) при 0°С добавляли метансульфонилхлорид (0,087 мл, 1,12 ммоль) (Aldrich). Реакционный раствор перемешивали 2 час при комнатной температуре, затем концентрировали, и остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/CH2Cl2/Et3N (1:4:0,05), и получали желаемый 7,8-диметокси-4-фенил-4H-1,3,4-триазафенален в виде твердого белого вещества. (Выход 47 мг, 55%). Пример 21 4-(4-Хлорфенил)-7,8-диметокси-4H-1,3,4-триазафенален
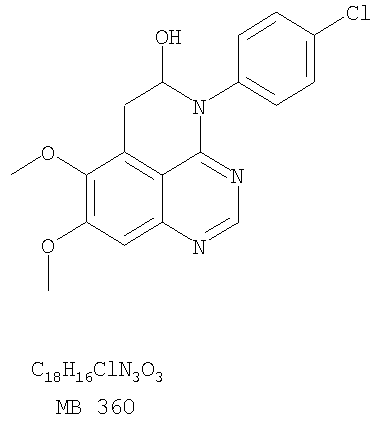
Стадия А: 7,8-Диметокси-4-(4-хлорфенил)-5,6-дигидро-4H-1,3,4-триазафенален-5-ол
Через раствор (5-аллил-6,7-диметоксихиназолин-4-ил)-(4-хлорфенил)амина (0,14 г, 0,39 ммоль) (Пример 16, стадия Е) в метаноле (50 мл) при -78°С пропускали струю О3 до тех пор, пока цвет раствора не стал голубым. Затем реакционную колбу заполняли аргоном и добавляли раствор метилсульфида (5 мл) (Aldrich). Доводили реакционную смесь до комнатной температуры и перемешивали в течение ночи. Раствор концентрировали, и остаток сушили в вакууме, получая желаемый 7,8-диметокси-4-(4-хлорфенил)-5,6-дигидро-4H-1,3,4-триазафенален-5-ол в виде сырого желтого твердого вещества. (Выход 0,14 г, 100%). Стадия В: 4-(4-Хлорфенил)-7,8 диметокси-4H-1,3,4-триазафенален
К раствору 7,8-диметокси-4-(4-хлорфенил)-5,6-дигидро-4H-1,3,4-триазафенален-5-ола (140 мг, 0,39 ммоль) (Пример 21, стадия А) и триэтиламина (0,32 мл, 2,33 ммоль) в дихлорметане (50 мл) при 0°С добавляли метансульфонилхлорид (0,12 мл, 1,55 ммоль) (Aldrich). Реакционную смесь перемешивали 2 час при комнатной температуре, затем концентрировали, и остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/CH2Cl2/Et3N (1:1:0,05), и получали желаемый 4-(4-хлорфенил)-7,8-диметокси-4H-1,3,4-триазафенален в виде твердого желтого вещества. (Выход 47 мг, 55%). Пример 22 4-(3-Бромфенил)-7,8-диметокси-4H-1,3,4-триазафенален
Стадия А: 7,8-Диметокси-4-(3-бромфенил)-5,6-дигидро-4H-1,3,4-триазафенален-5-ол
Через раствор (5-аллил-6,7-диметоксихиназолин-4-ил)-(3-бромфенил)амина (0,17 г, 0,42 ммоль) (Пример 17, стадия Е) в метаноле (50 мл) при -78°С пропускали струю О3 до тех пор, пока цвет раствора не стал голубым. Затем реакционную колбу заполняли аргоном и добавляли раствор метилсульфида (5 мл) (Aldrich). Доводили реакционную смесь до комнатной температуры и перемешивали в течение ночи. Раствор концентрировали, и остаток сушили в вакууме, получая желаемый 7,8-диметокси-4-(3-бромфенил)-5,6-дигидро-4H-1,3,4-триазафенален-5-ол в виде сырого коричневого твердого вещества. (Выход 0,16 г, 100%). Стадия В: 4-(3-Бромфенил)-7,8-диметокси-4H-1,3,4-триазафенален
К раствору 7,8-диметокси-4-(3-бромфенил)-5,6-дигидро-4H-1,3,4-триазафенален-5-ола (0,17 г, 0,42 ммоль) (Пример 22, стадия А) и триэтиламина (0,35 мл, 2,53 ммоль) в дихлорметане (30 мл) при 0°С добавляли метансульфонилхлорид (0,13 мл, 1,68 ммоль) (Aldrich). Реакционную смесь перемешивали 2 час при комнатной температуре, затем концентрировали, и остаток очищали хроматографически, используя в качестве элюента смесь EtOAc/СН2Cl2/Et3N (1:1:0,01), и получали желаемый 4-(3-бромфенил)-7,8-диметокси-4H-1,3,4-триазафенален в виде твердого белого вещества. (Выход 81 мг, 50%). Фармакологические свойства соединений предлагаемого изобретения могут быть подтверждены фармакологическими опытами. Представленный далее фармакологический опыт проводили с соединениями предлагаемого изобретения и их солями. Пример 23: Проверка ингибирования киназы. Для определения способности тестируемых соединений, относящихся к предлагаемому изобретению, ингибировать активность РЭФР проводили опыты, используя разрешенный во времени флуоресцентный метод (Homogeneous Time Resolved Fluorescence). Этот метод описан в работе A.J.Kolb и др., Drug Discovery Today, 1998, 3, №7, с.333. Опыты по определению активности киназы проводили на полипропиленовых пластинах с 96 ячейками (Falcon) с общим объемом в каждой ячейке 90 мкл. Каждая ячейка содержала 1 мкМ субстрата ФЭРФ (Biotin-EEEEYFELV), 1,5 нМ ФЭРФ и тестируемое соединение с одной из 8 концентраций опыта в интервале 100 мкМ-128 пМ (разбавление в серии 1:5). Опыт по определению активности киназы проводили в присутствии 100 мМ ГЭПЭС, рН 7,4, 1 мМ ДТТ, 5 мМ MgCl2, 2 мМ MnCl2, 1% ДМСО, 0,5 мкМ АТФ (Кm для ФЭРФ), 0,1 мМ Na2VO4 и 0,02% БСА. Реакционную смесь инкубировали 30 мин при 37°С. Для того чтобы остановить реакцию с ФЭРФ, 72 мкл смеси переносили на СТОП пластину, содержащую 18 мкл буфера (20 мМ ЭДТА, 50 мМ ГЭПЭС, рН 7,4, 0,02% БСА, 10 нМ меченного Ей анти-pY антитела (конечная концентрация 2 нМ) и 100 нМ стрептавидина (конечная концентрация 20 нМ). После перемешивания 35 мкл этого раствора переносили в дуплицированные ячейки черной пластины (Costar) с 384 ячейками и считывали показания при 615/665 нм на приборе Wallac Victor 5. Значения IC50 тестируемого соединения определяли, получая двойной набор данных, и рассчитывали, используя Exel и фиттируя данные по уравнению Y=[(a-b)/1+(X/c)d]+b, в котором а и b означают активность фермента в отсутствие тестируемого соединения и в присутствии какого-то количества тестируемого соединения соответственно, с означает IC50, и d означает константу Хилла отклика соединения. Величина IC50 означает концентрацию тестируемого соединения, которая понижает активность фермента на 50% в условиях, описанных для проведения теста. Соединения предлагаемого изобретения характеризуются значениями IC50, меньшими, чем 1 мкМ, при испытании в описанных выше условиях. Таким образом, эти соединения обладают способностью ингибировать киназную активность ФЭРФ.
Формула изобретения
1. Соединение формулы
в которой Z означает С или N; R1 и R2 независимо выбраны из Н, низшего алкила; R3 и R4 независимо друг от друга выбраны из Н, F, Cl или Br; R5 выбран из Н, оксогруппы, тиона, C1-С3алкила и или их фармацевтически приемлемые соли. 2. Соединение по п.1, в котором Z означает С. 3. Соединение по п.1, в котором Z означает N. 4. Соединение формулы I, по п.1, в котором R1 и R2 независимо друг от друга выбраны из низшего алкила. 5. Соединение по п.4, в котором либо R1 либо R2 означает метил. 6. Соединение по п.4, в котором оба R1 и R2 означают метил. 7. Соединение формулы I, по п.1, в котором R3 и R4 независимо друг от друга выбраны из Н, Br, Cl и F. 8. Соединение по п.7, в котором или R3, или R4 означает Н. 9. Соединение по п.7, в котором оба R3 и R4 означают Н. 10. Соединение формулы I, по п.1, в котором R5 выбран из =O. =S, Н и низшего алкила. 11. Соединение по п.10, в котором R5 выбран из Н и низшего алкила. 12. Соединение формулы I, по п.1 в котором Z означает N, R1 и R2 означают метил и R3 и R4 означают Н. 13. Соединение формулы I по п.1, в котором Z означает N, R1 и R2 означают метил, один из R3 и R4 означает Н, и другой из R3 и R4 означает Cl или Br. 14. Соединение формулы I по п.1, в котором Z означает С, R1 и R2 означают метил, один из R3 и R4 означает Н, и другой из R3 и R4 означает Cl или Br. 15. Соединение формулы I по п.1, в котором Z означает С, R1 и R2 означают метил, один из R3 и R4 означает F, и другой из R3 и R4 означает Cl или Br. 16. Соединение, выбранное из группы 8,9-диметокси-3-фенил-1Н,3Н-1,3,4,6-тетраазафенален-2-он; 8,9-диметокси-3-фенил-1Н,3Н-1,3,4,6-тетраазафенален-2-тион; 8,9-диметокси-3-фенил-3Н-1,3,4,6-тетраазафенален; 8,9-диметокси-2-метил-3-фенил-3Н-1,3,4,6-тетраазафенален и 2-этил-8,9-диметокси-3-фенил-3Н-1,3,4,6-тетраазафенален. 17. Соединение, выбранное из группы 3-(3-бромфенил)-8,9-диметокси-3Н-1,3,4,6-тетраазафенален, 3-(3-бромфенил)-8,9-диметокси-1Н,3Н-1,3,4,6-тетраазафенален-2-он; 3-(3-бромфенил)-8,9-диметокси-2-метил-3Н-1,3,4,6-тетраазафенален, 3-(3-бромфенил)-8,9-диметокси-1Н,3Н-1,3,4,6-тетраазафенален-2-тион; 3-(3-бромфенил)-2-этил-8,9-диметокси-3Н-1,3,4,6-тетраазафенален и 3-(3-хлорфенил)-8,9-диметокси-1Н,3Н-1,3,4,6-тетраазафенален-2-он. 18. Соединение, выбранное из группы 3-(3-хлорфенил)-8,9-диметокси-2-метил-3Н-1,3,4,6-тетраазафенален; 3-(4-хлорфенил)-8,9-диметокси-3Н-1,3,4,6-тетраазафенален; 3-(4-хлорфенил)-8,9-диметокси-1Н,3Н-1,3,4,6-тетраазафенален-2-тион: 7,8-диметокси-5-метил-4-фенил-4Н-1,3,4-триазафенален; 4-(3-хлорфенил)-7,8-диметокси-5-метил-4Н-1,3,4-триазафенален; 4-(3-бромфенил)-7,8-диметокси-5-метил-4Н-1,3,4-триазафенален. 19. Соединение, выбранное из группы 4-(4-бром-2-фторфенил)-7,8-диметокси-5-метил-4Н-1,3,4-триазафенален; 7,8-диметокси-4-фенил-4Н-1,3,4-триазафенален; 4-(4-хлорфенил)-7,8-диметокси-4Н-1,3,4-триазафенален и 4-(3-бромфенил)-7,8-диметокси-4Н-1,3,4-триазафенален. 20. Фармацевтическая композиция, обладающая свойствами ингибитора эпидермального фактора роста тирозинкиназы, и имеющая антипролиферативную активность, содержащая в качестве активного ингредиента эффективное количество соединения по любому из пп.1-19, и фармацевтически приемлемый носитель или наполнитель. 21. Фармацевтическая композиция по п.20, подходящая для парентерального введения. 22. Соединения по любому из пп.1-19 в качестве активного ингредиента для получения лекарственного средства для лечения пролиферативных заболеваний.
|
||||||||||||||||||||||||||