Патент на изобретение №2346942
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) 2,4-БИС(ТРИФТОРЭТОКСИ)ПИРИДИНОВОЕ СОЕДИНЕНИЕ И СОДЕРЖАЩЕЕ ЕГО ЛЕКАРСТВЕННОЕ СРЕДСТВО
(57) Реферат:
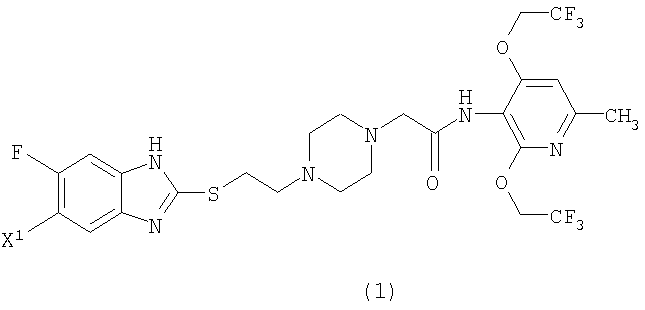
Изобретение относится к 2,4-бис(трифторэтокси)пиридиновому соединению, представленному формулой (1), где X1 представляет атом фтора или водорода, или его соли в качестве ингибитора ацил-кофермента А холестерин-ацилтрансферазы (АСАТ), а также лекарственному средству и лекарственной композиции на их основе, его применению, способу его получения, а также к новым промежуточным соединениям. Соединения могут найти применение для предотвращения или лечения заболеваний, опосредованных активностью АСАТ, таких как гипергликемия и артериосклероз. 9 н. и 1 з.п. ф-лы, 4 табл., 1 ил.
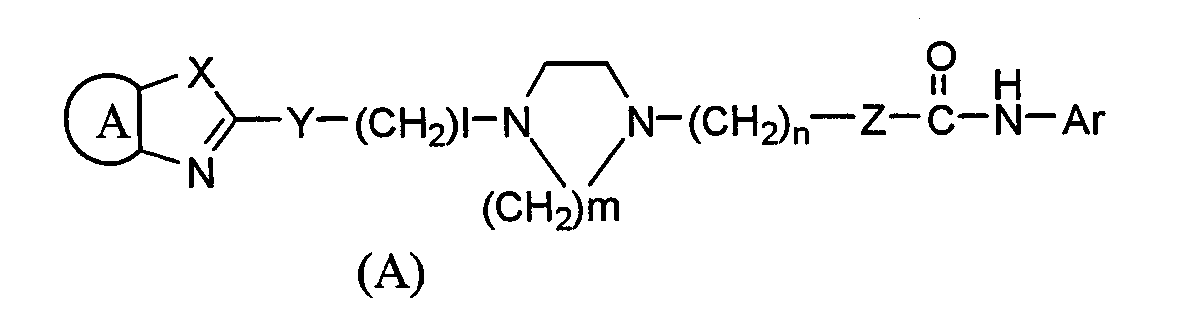
Область техники Настоящее изобретение относится к 2,4-бис(трифторэтокси)пиридиновому соединению, которое проявляет сильное ингибирующее действие против ацил-кофермент А холестерин-ацилтрансферазы (АСАТ) при пероральном введении и таким образом может применяться для предотвращения или лечения гиперлипидемии, артериосклероза или аналогичных расстройств и к промежуточным соединениям, которые можно применять для получения этого соединения. Уровень техники Ацил-кофермент А холестерин-ацилтрансфераза (АСАТ) представляет собой фермент, который катализирует синтез сложного эфира холестерина из холестерина и играет важную роль в метаболизме холестерина и его всасывании в пищеварительных органах. Хотя многие из обычных ингибиторов АСАТ, которые служат в качестве средств против гиперлипидемии или средств против артериосклероза, действуют на АСАТ в тонкой кишке или в печени для уменьшения уровня холестерина в крови, такие средства оказывают неблагоприятные побочные эффекты, такие как кишечное кровотечение, кишечные расстройства, диарея и печеночные расстройства. В соответствии с недавними исследованиями ожидается, что регрессия очагов артериосклероза может быть осуществлена предотвращением образования пенистых клеток из макрофагов, которые играют ключевую роль в формировании очагов артериосклероза. В частности, присутствие происходящих из макрофагов пенистых клеток (которые накапливают в себе сложные эфиры холестерина в виде жировых капелек) наблюдается в очаге атеросклероза. Было выявлено, что образование происходящих из макрофагов пенистых клеток тесно связано с прогрессированием поражения. Кроме того, на участке артериосклеротического поражения активность АСАТ на сосудистой стенке была повышена и на ней накопился сложный эфир холестерина. Таким образом, активность АСАТ на ней может быть тесно связана с артериосклерозом (Exp. Mol. Pathol., 44, 329-339 (1986)). Соответственно, когда ингибитор АСАТ ингибирует этерификацию холестерина на сосудистых стенках, свободный холестерин накапливается в клетках сосудистой стенки. Накопившийся свободный холестерин удаляется липопротеином высокой плотности (HDL, ЛВП) из клеток в печень (обратный транспорт ЛВП) и затем метаболизируется. Таким образом, можно ожидать, что такой ингибитор АСАТ будет ингибировать накопление сложных эфиров холестерина на участках поражения при артериосклерозе (Biochem. Biophys. Acta. 2001 15, 1530 (1): 111-122). Как описано выше, считалось, что ингибитор АСАТ, который ингибирует АСАТ, присутствующую на сосудистых стенках, оказывает прямой эффект против артериосклероза. Ранее, после обширных исследований, сосредоточенных на прогнозе того, что соединение, которое селективно ингибирует АСАТ, присутствующую на сосудистых стенках, и таким образом предотвращает трансформацию макрофагов в пенистые клетки, служит в качестве профилактического или терапевтического средства по поводу артериосклероза, в то же самое время вызывая меньшее количество побочных эффектов, заявители обнаружили, что соединение, представленное следующей формулой (А):
(где Ar представляет арильную группу, которая может быть необязательно замещена,
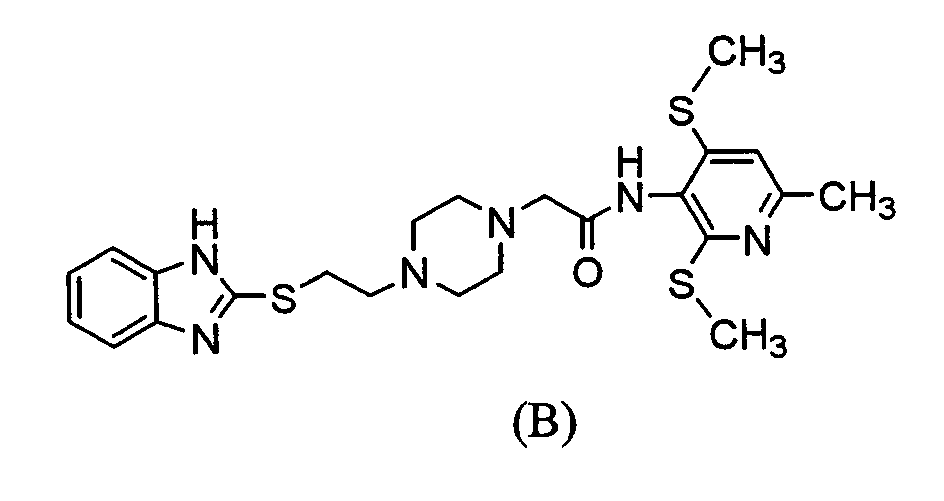
представляет двухвалентный остаток бензола, пиридина, циклогексана или нафталина, который может необязательно быть замещенным, Х представляет NH, атом кислорода или атом серы, Y представляет атом серы или ему подобный, Z представляет простую связь, l представляет собой целое число от 0 до 15, m=2 или 3 и n представляет собой целое число от 1 до 3), его соль или сольват соединения или соли, селективно ингибирует АСАТ, присутствующую в артериальной стенке и таким образом может применяться в качестве профилактического или терапевтического средства по поводу гиперлипидемии или артериосклероза (Международная патентная заявка WO98/54153). Среди соединений, описанных в Международной патентной заявке WO98/54153, было обнаружено, что соединение, представленное ниже формулой (В) его соль имеют высокую растворимость в воде и высокую АСАТ ингибирующую активность и проявляют необычный фармакологический эффект на разнообразных экспериментальных моделях. Хотя соединение (В) и другие соединения, раскрытые в Международной патентной заявке WO98/54153, проявляют отличный фармакологический эффект, относящийся к ингибирующему АСАТ эффекту у животных, эксперименты, выполненные in vitro с использованием микросом печени человека, выявили, что эти соединения быстро метаболизируются и таким образом только небольшой процент неизмененных соединений остается в микросомах печени человека. Поэтому низкая концентрация этих соединений в крови стала вызывать беспокойство. Более того, на основании последних сведений о механизме лекарственного взаимодействия препарат, имеющий более высокую безопасность, продуцируется из соединений, имеющих более высокую метаболическую устойчивость, желательно соединение, имеющее более высокую метаболическую устойчивость в микросомах печени человека. Однако считалось очень трудным повысить устойчивость против метаболизма соединения (В), в то же самое время поддерживая его ингибирующий АСАТ эффект, поскольку соединение (В) имеет множество функциональных групп, которые в целом легко метаболизируются в живых организмах, и считают, что эти функциональные группы существенны для обеспечения фармакологического эффекта.
Сущность изобретения В связи с изложенным выше, заявители провели обширные исследования с целью получения соединения, которое имеет повышенную метаболическую устойчивость в микросомах печени человека, хорошо всасывается при пероральном применении и обеспечивает высокую концентрацию в крови, и неожиданно обнаружили, что 2,4-бис(трифторэтокси)пиридиновое соединение, представленное ниже формулой (1), имеет более высокую концентрацию в крови (Cmax), бóльшую AUC (площадь под кривой зависимости концентрации в крови от времени) и лучше всасывается при пероральном приеме, хотя это пиридиновое соединение имеет более низкую растворимость в воде по сравнению с растворимостью соединения (В). Кроме того, заявители обнаружили, что эти соединения проявляют высокую ингибирующую АСАТ активность и таким образом могут применяться в качестве профилактического или терапевтического средства по поводу гиперлипидемии или артериосклероза. Настоящее изобретение было осуществлено на основании этих данных. Соответственно, настоящее изобретение предоставляет 2,4-бис(трифторэтокси)пиридиновое соединение, представленное формулой (1):
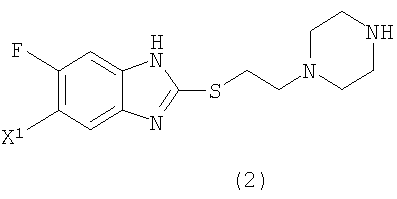
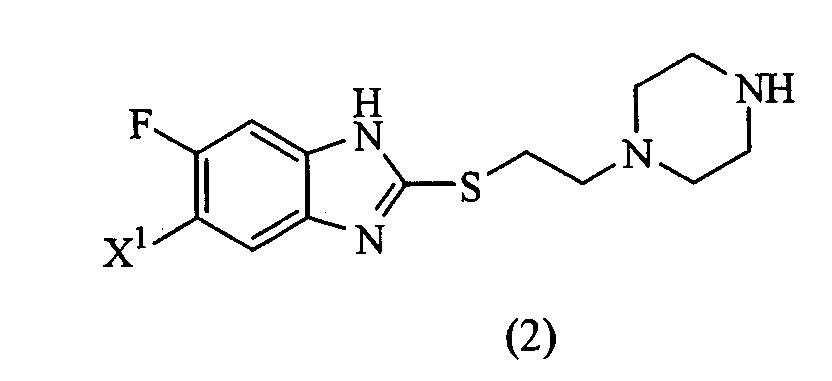
(где Х1 представляет атом фтора или водорода) или его соль и способ получения соединения или соли. Настоящее изобретение также предоставляет пиперазиновое соединение, представленное формулой (2):
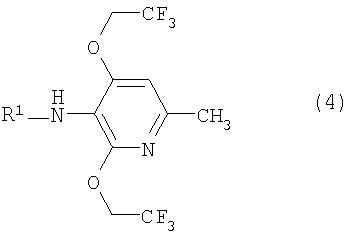
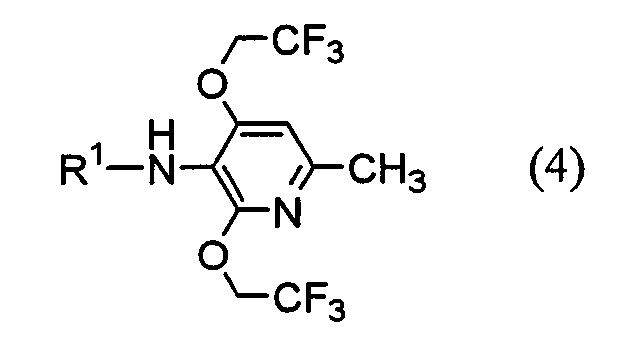
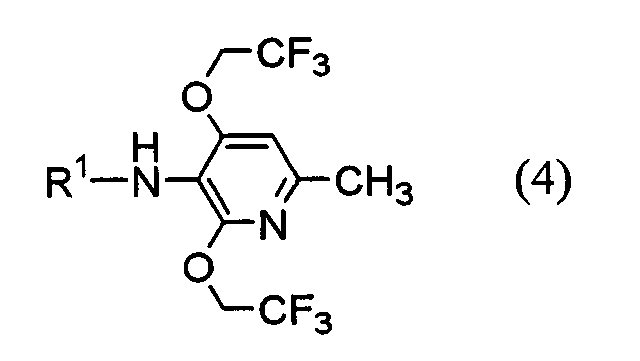
(где Х1 представляет атом водорода или фтора) или его соль. Настоящее изобретение также предоставляет пиридиновое соединение, представленное формулой (4):
(где R1 представляет атом водорода, хлорацетильную группу, бромацетильную группу или йодацетильную группу) или его соль. Настоящее изобретение также предоставляет 2,4-бис(2,2,2-трифторэтокси)-6-метил-3-нитропиридин. Настоящее изобретение также предоставляет N-[2,4-бис(2,2,2-трифторэтокси)-6-метилпиридин-3-ил]-2-[4-(2-гидроксиэтил)пиперазин-1-ил]ацетамид. Настоящее изобретение также предоставляет лекарственное средство, содержащее соединение, представленное указанной выше формулой (1) или его соль, в качестве активного ингредиента. Настоящее изобретение также предоставляет применение соединения, представленного указанной выше формулой (1) или его соль, для изготовления лекарственного средства. Настоящее изобретение также предоставляет способ лечения артериосклероза, включающий введение соединения, представленного указанной выше формулой (1) или его соли в эффективном количестве. Соединение (1) настоящего изобретения селективно ингибирует АСАТ, присутствующую на стенках артерии, обладает высокой устойчивостью против метаболизма в микросомах печени человека, хорошо всасывается при пероральном применении и таким образом может применяться в качестве профилактического или терапевтического средства по поводу гиперлипидемии или артериосклероза. Краткое описание чертежей На чертеже показана устойчивость против метаболизма соединений (1а), (1b) и соединения (В) (гидрохлорида) в микросомах печени человека. Подробное описание предпочтительных вариантов осуществления Соединение (1) настоящего изобретения характеризуется наличием в структуре одного или двух атомов фтора на бензимидазольном кольце и наличием двух 2,2,2-трифторэтокси групп на пиридиновом кольце. В Международной патентной заявке WO 98/54153 не были описаны соединения, имеющие эту необычную химическую структуру. Настоящее изобретение включает следующие два соединения и их соли.
Примеры соли соединения (1) настоящего изобретения включают соли неорганических кислот, такие как гидрохлориды, сульфаты, нитраты, фосфаты; и соли органических кислот, такие как метансульфонаты, малеаты, фумараты, цитраты, бутираты, лактаты, тартраты, аскорбаты, малаты, манделаты, салицилаты, пантотенаты, таннаты, этандисульфонаты, бензолсульфонаты, п-толуолсульфонаты, глутаматы, аспартаты, трифторацетаты, памоаты и глюконаты. Соединение (1) настоящего изобретения или его соль может принимать форму сольвата. Виды сольвата конкретно не ограничиваются, если сольват не оказывает неблагоприятного эффекта на активность по ингибированию АСАТ, и сольват может быть образован посредством добавления растворителя, который используется в способе получения или очистки, такого как вода или спирт. В качестве сольвата предпочтителен гидрат. Соединение (1) настоящего изобретения можно получить посредством, например, следующего способа получения:
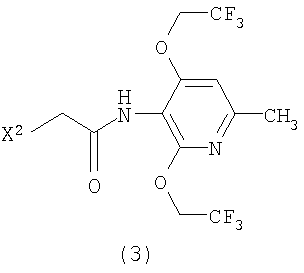
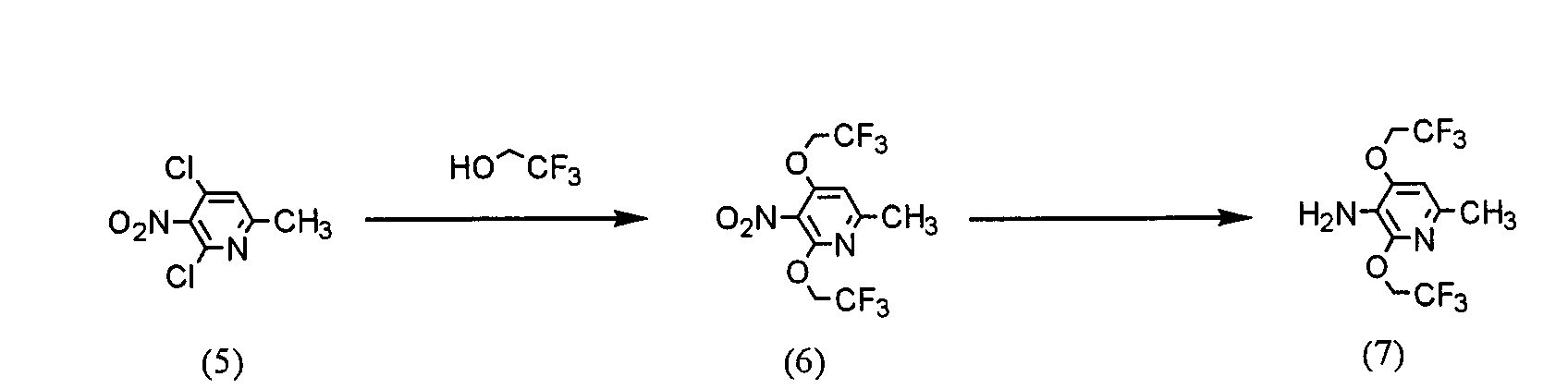
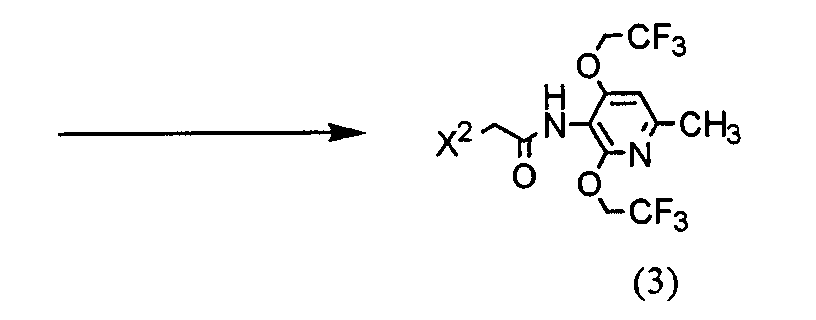
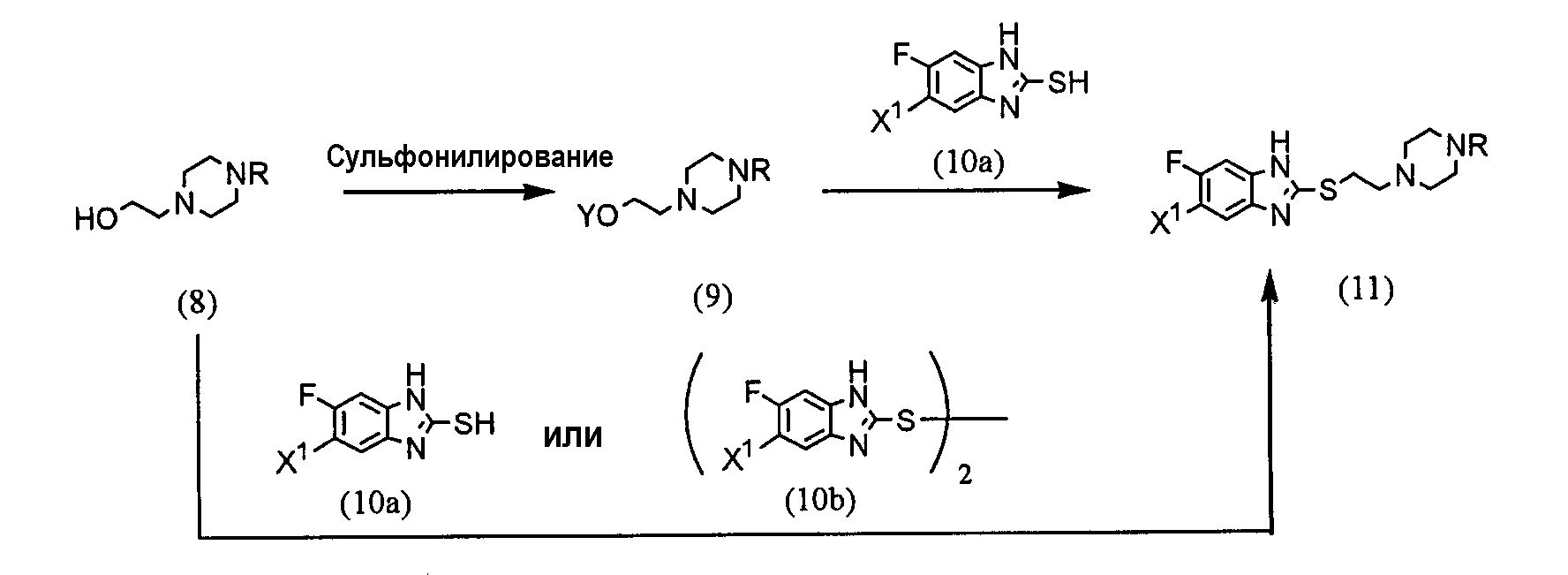
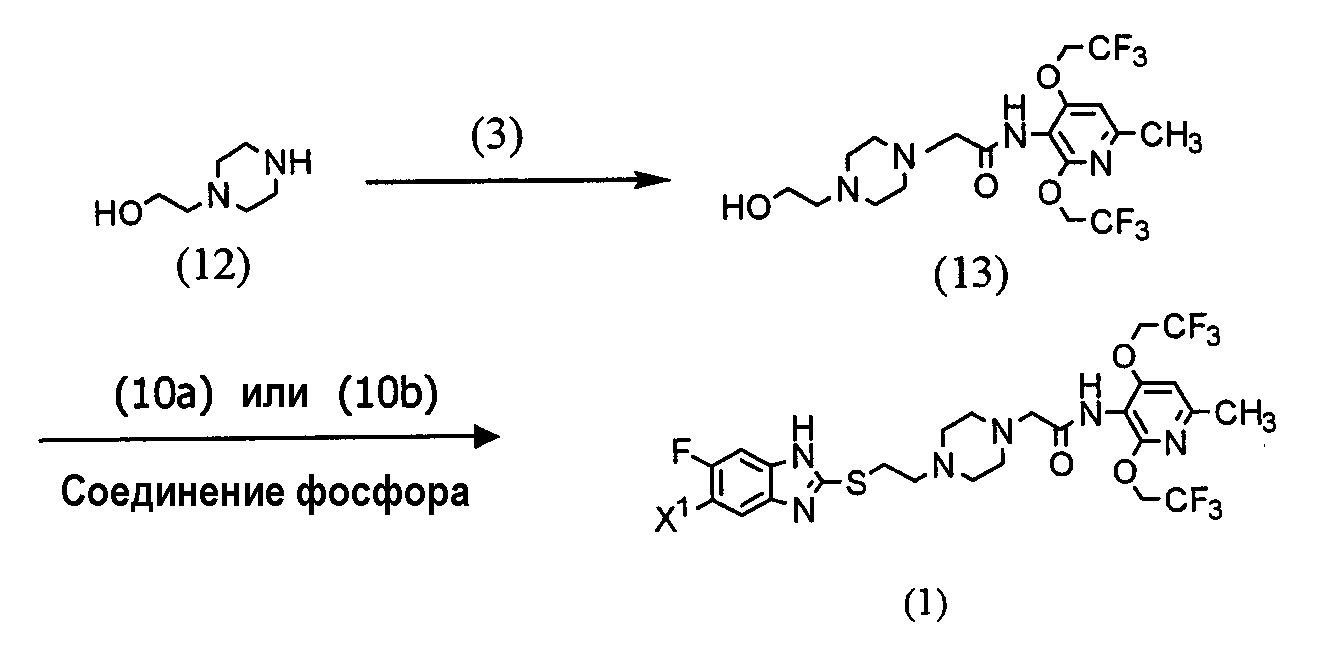
(где R представляет защитную группу, Y представляет алкильную группу или арилсульфонильную группу, Х1 представляет атом водорода или атом фтора, Х2 представляет атом хлора, атом брома или атом йода). В частности, 2,4-дихлор-6-метил-3-нитропиридин (5) взаимодействует с 2,2,2-трифторэтанолом для получения соединения (6). Нитрогруппа соединения (6) восстанавливается для получения соединения (7). Затем соединение (7) взаимодействует с галогенуксусной кислотой или ее реакционноспособным производным для получения соединения (3). Отдельно соединение пиперазинэтанола (8), чья аминогруппа была защищена, сульфонируется для получения соединения (9), и соединение (9) взаимодействует с тиольным производным (10а) для получения соединения (11). Альтернативно, соединение (11) можно получить посредством взаимодействия соединения (8) с тиольным производным (10а) или (10b) в присутствии соединения фосфора. Соединение (2) получают посредством снятия защиты защитной группы (R) соединения (11). Соединение (1) настоящего изобретения получают посредством взаимодействия полученного таким образом соединения (2) с соединением (3). Соответственно, указанное выше соединение (2), соединение (4), представленное ниже формулой (4), и указанное выше соединение (6), которое представляет собой 2,4-бис(2,2,2-трифторэтокси)-6-метил-3-нитропиридин, можно использовать в качестве промежуточных соединений для получения соединения (1) настоящего изобретения.
(где R1 представляет атом водорода, хлорацетильную группу, бромацетильную группу или йодацетильную группу). Далее будет описана каждая стадия указанной выше схемы реакции. Реакция 2,4-дихлор-6-метил-3-нитропиридина (5) с 2,2,2-трифторэтанолом проводится в растворителе (2,2,2-трифторэтаноле или его смеси с растворителями с диметилформамидом (ДМФ), тетрагидрофураном (ТГФ), диметилсульфоксидом (ДМСО) и т.д.) в присутствии основания (например, карбоната щелочного металла, такого как карбонат калия или карбонат натрия; гидроксида щелочного металла, такого как гидроксид калия или гидроксид натрия; или гидрида щелочного металла, такого как гидрид натрия, гидрид калия или гидрид лития) в течение 5-24 ч при комнатной температуре до температуры дефлегмации (предпочтительно, в течение 15-20 ч при температуре дефлегмации). Восстановление соединения (6) предпочтительно проводится посредством одной из следующих реакций восстановления: (i) восстановления посредством применения содержащего серу восстановительного агента, такого как дитионат натрия, сульфид натрия, гидросульфид натрия или сероводород, (ii) восстановления посредством использования содержащего металл восстановительного агента, такого хлорид цинка, железа или олова (II), или (iii) каталитического восстановления в атмосфере водорода. Реакция восстановления (i) выполняется, например, растворением соединения (6) в растворителе, таком как изопропанол, этанол или ТГФ, добавлением при 80°С водного раствора, содержащего серу, восстановительного агента и предоставлением возможности смеси взаимодействовать в течение периода времени от 10 мин до 2 ч. Реакция восстановления (ii) проводится, например, растворением соединения (6) в растворителе, таком как спирт (такой как этанол или изопропанол), уксусная кислота или растворяющая смесь воды и любого из этих растворителей, и предоставлением возможности раствору взаимодействовать в течение периода времени от 30 мин до 24 ч при 0-100°С. При реакции (ii) при необходимости можно добавить кислоту, такую как хлористоводородная кислота или серная кислота. Реакцию каталитического восстановления (iii) проводят растворением соединения (6) в растворителе, таком как диоксан, уксусная кислота, метанол, этанол или изопропанол, или их растворяющая смесь, и предоставлением возможности раствору взаимодействовать в присутствии катализатора, такого как никель Ренея, палладий на угле, гидроксид палладия или палладиевая чернь, в атмосфере водорода в течение периода времени от 30 мин до 12 ч при 0-50°С, предпочтительно, в течение периода времени от 30 мин до 3 ч при комнатной температуре. Примеры галогенуксусной кислоты, которую следует использовать для взаимодействия с соединением (7), включают хлоруксусную кислоту, бромуксусную кислоту и йодуксусную кислоту. Примеры реакционноспособного производного галогенуксусной кислоты включают галогенацетилгалогенид и галогенуксусный ангидрид. Предпочтительно, соединение (7) взаимодействует с галогенацетилгалогенидом. Взаимодействие соединения (7) с галогенацетилгалогенидом проводится, например, в растворителе (таком как метиленхлорид, хлороформ, этилацетат, ацетонитрил или толуол) в присутствии основания (такого как N,N-диметиланилин, триэтиламин, пиридин, 4-диметиламинопиридин или 4-пирролидинпиридин) в течение периода времени от 10 мин до 5 ч при 0-50°С, предпочтительно, в течение периода времени от 10 мин до 60 мин при 0°С. Синтез соединения (11) из соединения пиперазинэтанола (8) может выполняться посредством пути «а» (посредством алкилсульфонирования или арилсульфонирования) или пути «b» (посредством взаимодействия с соединением фосфора). При пути «а» алкилсульфонирование или арилсульфонирование соединения пиперазинэтанола (8) выполняется в растворителе (таком как ДМФ, ТГФ, этилацетат или ацетонитрил) в присутствии основания (такого как триэтиламин, пиридин, N,N-диизопропилэтиламин, N,N-диметиланилин или 4-диметиламинопиридин) посредством использования соединения сульфонилхлорида в качестве агента алкилсульфонирования или агента арилсульфонирования, такого как метансульфонилхлорид, бензолсульфонилхлорид или п-толуолсульфонилхлорид, в течение периода времени от 30 мин до 3 ч при 0-50°С. Защитная группа (R) аминогруппы в соединении пиперазинэтанола (8) может представлять собой группы, используемые в пептидном синтезе. Предпочтительные примеры таких групп включают алкоксикарбонильные группы (такие как бензилоксикарбонил, 2,2,2-трихлорэтилоксикарбонил и трет-бутоксикарбнил) и формильную группу. Взаимодействие соединения (9) с соединением (10а) проводится в растворителе (таком как ДМФ, ДМСО или ацетонитрил) в присутствии основания (таком как карбонат калия или карбонат натрия) и катализатора (такого как 18-крон-6) в течение периода времени от 1 до 5 ч при температуре от комнатной до 100°С, предпочтительно, от 1 до 2 ч при 50-80°С. При пути «b» соединение пиперазинэтанола (8) взаимодействует с тиольным производным (10а) или (10B) в присутствии соединения фосфора. Примеры соединения фосфора включают фосфиновые реагенты, используемые при реакции Мицунобу; фосфорные реагенты, содержащие такие фосфиновые реагенты, и азо реагент или реагент этилендикарбоновой кислоты, такой как диметилмалеат или N,N,N’,N’-тетраметилфумарамид; и реагенты илида фосфония. При пути «b» взаимодействие предпочтительно выполняется посредством любого из следующих способов: (i) взаимодействие соединения (8) с тиольным производным (10а) в присутствии фосфинового реагента и азо реагента или реагента этилендикарбоновой кислоты, такого как диметилмалеат или N,N,N’,N’-тетраметилфумарамид (способ А), (ii) взаимодействие соединения (8) с тиольным производным (10а) в присутствии реагента илида фосфония (способ В) и (iii) взаимодействие соединения (8) с тиольным производным (10b) в присутствии фосфинового реагента (способ С). Способ А Способ А можно выполнить посредством растворения соединения (8), тиольного производного (10а) и фосфинового реагента в реакционном растворителе, добавления к ним азо реагента или реагента этилендикарбоновой кислоты и предоставления возможности смеси взаимодействовать в атмосфере аргона или азота в течение 2-24 ч при 0°С-100°С, предпочтительно, при температуре от комнатной до 80°С. Примеры фосфинового реагента, используемого в способе А, включают триалкилфосфины, такие как триметилфосфин, триэтилфосфин, трипропилфосфин, триизопропилфосфин, трибутилфосфин, триизобутилфосфин и трициклогескилфосфин, и триарилфосфины, такие как трифенилфосфин и дифенилфосфинполистирол. Среди этих соединений, предпочтительны триметилфосфин, трибутилфосфин и трифенилфосфин. Примеры азо реагента включают диэтилазодикарбоксилат (DEAD), 1,1′-азобис(N,N-диметилформамид) (TMAD), 1,1′-(азодикарбонил)дипиперидин (ADDP), 1,1′-азобис(N,N-диизопропилформамид) (TIPA) и 1,6-диметил-1,5,7-гексагидро-1,4,6,7-тетразоцин-2,5-дион (DHTD). Среди них особенно предпочтителен диэтилазодикарбоксилат. Примеры реакционного растворителя, который следует использовать, включают ДМФ, ТГФ, диоксан, ацетонитрил, нитрометан, ацетон, этилацетат, бензол, хлорбензол, толуол, хлороформ и метиленхлорид. Среди них, предпочтительны ДМФ, ТГФ, диоксан и ацетонитрил и особенно предпочтительны ДМФ и ТГФ. Способ В Способ В можно выполнить посредством растворения соединения (8), тиольного производного (10а) и реагента илида фосфония в растворителе для реакции и обеспечения возможности прохождения реакции раствора в атмосфере аргона или азота в течение 2-12 ч при температуре от комнатной до 120°С, предпочтительно, при 80°С-100°С. Примеры реагента илида фосфония, используемого в способе В, включают алканоилметилентриалкилфосфоран, алканоилметилентриарилфосфоран, алкоксикарбонилметилентриалкилфосфоран, алкоксикарбонилметилентриарилфосфоран, цианометилентриалкилфосфоран и цианометилентриарилфосфоран. Примеры триалкила включают триметил, триэтил, трипропил, триизопропил, трибутил, триизобутил и трициклогексил. Примеры триарила включают трифенил и дифенилполистирол. Альтернативно, эту реакцию можно выполнить добавлением в реакционный растворитель соединения (8) и тиольного производного (10а) с реагентом галогенидом фосфония в присутствии основания для получения посредством этого реагента илида фосфония в реакционной системе. Примеры реагента галогенида фосфония, используемые при этой реакции, включают галогенид (цианометил)триалкилфосфония, галогенид (цианометил)триарилфосфония, галогенид (алкилкарбонилметил)триалкилфосфония, галогенид (алкилкарбонилметил)триарилфосфония, галогенид (алкоксикарбонилметил)триалкилфосфония и галогенид (алкоксикарбонилметил)триарилфосфония. Среди указанных выше реагентов галогенида фосфония галогенид (цианометил)триалкилфосфония и галогенид (цианометил)триарилфосфония можно получить посредством взаимодействия соответствующего галогенированного ацетонитрила с соответствующим триалкилфосфином и триарилфосфином (Tetrahedron, Vol.57, pp.5451-5454, 2001). Другие реагенты можно получить посредством взаимодействия соответствующего алканоилгалогенметила или алканоилкарбонилгалогенметила с соответствующим триалкилфосфином или триарилфосфином аналогичным образом. Примеры триалкилфосфина или триарилфосфина включают соединения, перечисленные в связи со способом А. Среди них предпочтительны триметилфосфин, трибутилфосфин и трифенилфосфин, а особенно предпочтителен триметилфосфин. Примеры алканоиловой группы описанного выше алканоилгалогенметила включают формил, ацетил, пропионил и бутирил. Среди них предпочтительны ацетил и пропионил. Примеры алкоксигруппы алкоксикарбонилгалогенметила включают метокси, этокси, пропокси и бутокси. Среди них предпочтительны метокси, этокси и бутокси. Предпочтительные примеры атома галогена включают хлор, бром и йод. Примеры основания включают органические основания, такие как триэтиламин, N,N-диизопропилэтиламин, 1,4-диазабицикло[2,2,2]октан (DABCO), 1,8-диазабицикло[5,4,0]ундека-7-ен (DBU) и 1,5-диазабицикло[4,3,0]нона-5-ен (DBN) и неорганические основания, такие как карбонат калия, карбонат натрия, карбонат цезия, карбонат лития, диизопропиламид лития и гексаметилдисилазид калия. Среди них предпочтительными являются N,N-диизопропилэтиламин, карбонат калия, диизопропиламид лития и гексаметилдисилазид калия, а особенно предпочтительными являются N,N-диизопропилэтиламин и карбонат калия. Предпочтительные примеры растворителя для взаимодействия включают диоксан, ТГФ, толуол, бензол, ДМФ, ДМСО, ацетонитрил и пропионитрил, причем пропионитрил является особенно предпочтительным. Способ С Способ С можно выполнить посредством растворения соединения (8), тиольного производного (10b) и фосфинового реагента в реакционном растворителе, аналогичном растворителю, используемому в связи со способом А, и обеспечения возможности реакции раствора в атмосфере аргона или азота в течение 2-48 ч при температуре от комнатной до 100°С, предпочтительно, при 60°С-100°С. Примеры фосфинового реагента, используемого в способе С, включают триалкилфосфин и триарилфосфин, которые описаны в литературе в связи со способом А. Конкретные примеры включают триметилфосфин, триэтилфосфин, трипропилфосфин, триизопропилфосфин, трибутилфосфин, триизобутилфосфин, трициклогексилфосфин, трифенилфосфин и дифенилфосфинполистирол. Среди них предпочтительны триметилфосфин, трибутилфосфин и трифенилфосфин и особенно предпочтительны триметилфосфин и трифенилфосфин. Тиольное производное (10а) можно получить посредством способа, описанного в указанной выше международной патентной публикации WO98/54153, или посредством способа в соответствии с этой публикацией. Тиольное производное (10b) можно легко получить из тиольного производного (10а). Реакция снятия защиты соединения (11) выполняется посредством известного способа, в зависимости от вида защитной группы, например, посредством гидролиза, восстановления и т.д. Взаимодействие полученного таким образом соединения (2) с соединением (3) проводится в присутствии основания (такого как карбонат калия, карбонат натрия, гидрокарбонат калия или гидрокарбонат натрия) в растворителе (таком как ДМФ, ТГФ или ацетонитрил, или растворяющей смеси воды и любого из этих растворителей) в течение 5-30 ч при температуре от комнатной до 50°С, предпочтительно, в течение 10-20 ч при комнатной температуре. Альтернативно, соединение (1) настоящего изобретения можно получить посредством описанного ниже способа схемы взаимодействия. В частности, 1-(2-гидроксиэтил)пиперазин (12) взаимодействует с соединением галогенацетамида (3) для получения посредством этого соединения (13), и соединение (13) взаимодействует с тиольным производным (10а) или (10b) в присутствии соединения фосфора.
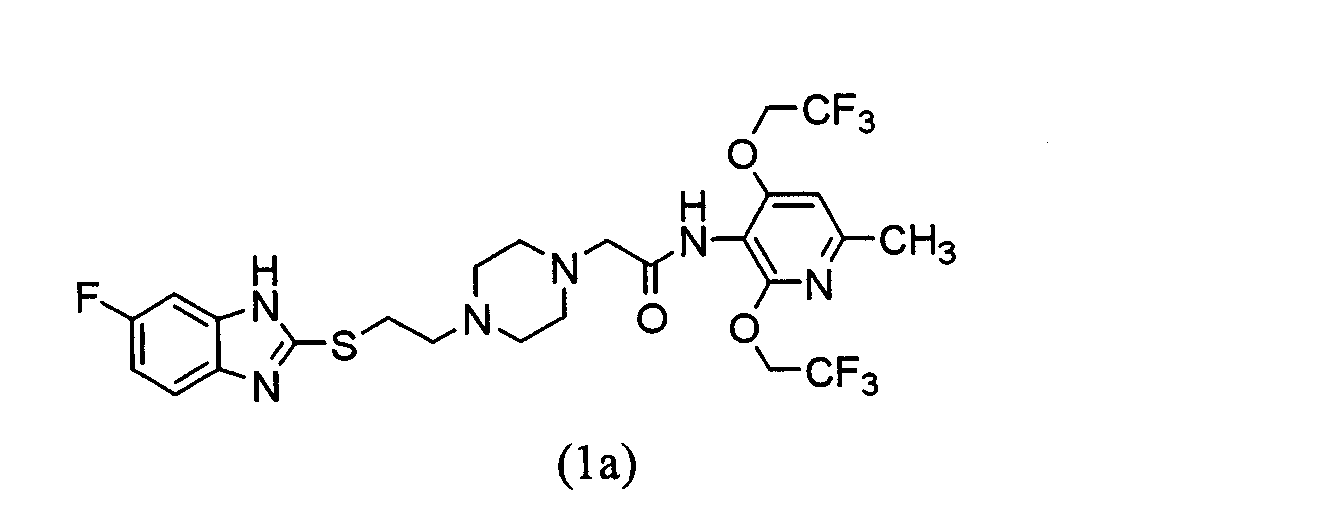
(где Х1 имеет такое же значение, как определено выше). Взаимодействие соединения (12) с соединением (3) проводится в соответствии со способом получения соединения (1) из соединения (2). Взаимодействие соединения (13) с тиольным производным (10а) или (10b) проводится в соответствии с взаимодействием соединения (8) с тиольным производным (10а) или (10b). Это указывает на то, что соединение (13), которое представляет собой N-[2,4-бис(2,2,2-трифторэтокси)-6-метилпиридин-3-ил]-2-[4-(2-гидроксиэтил))пиперадин-1-ил]ацетамид, можно использовать в качестве промежуточного соединения при получении соединения (1) по настоящему изобретению. Выделение и очистка соединения (1) по настоящему изобретению могут выполняться посредством любой подходящей комбинации промывания, экстракции, перекристаллизации, любых типов хроматографии и т.д. Кислотно-аддитивную соль можно получить посредством обычного способа. Устойчивость к метаболизму в микросомах печени человека исследовалась in vitro. На чертеже показана процентная доля неизмененного соединения через 30 мин после инкубации. Как показано на чертеже, было обнаружено, что для соединения (В) (гидрохлорид) остающаяся процентная доля составляет 16%, тогда как было обнаружено, что для соединения (1а) остающаяся процентная доля составляет 27%, а для соединения (1b) выявлена даже более высокая остающаяся процентная доля 62%. То есть соединения настоящего изобретения характеризуются более высокой остающейся процентной долей, чем процентная доля соединения (В) (гидрохлорида). Поэтому было обнаружено, что соединение (1) по настоящему изобретению имеет значительно повышенную метаболическую устойчивость в микросомах печени человека. Кроме того, была исследована растворимость в воде. Как показано в таблице 3, растворимость в воде соединения (1) настоящего изобретения гораздо ниже, чем растворимость соединения (В) (гидрохлорида). Таким образом, предполагалось, что соединение (1) настоящего изобретения имеет низкую всасываемость после перорального введения. Однако данные, полученные посредством теста перорального введения у самцов и самок крыс, выявили совершенно различные результаты. В отличие от ожиданий заявителей было обнаружено, что соединение (1) по настоящему изобретению проявляет в 2-3 раза более высокую концентрацию в крови (Cmax) и в 2-4 раза большую величину AUC по сравнению со случаем, когда использовалось соединение (В) (гидрохлорид). Поэтому было установлено, что соединение (1) по настоящему изобретению имеет более высокое всасывание при пероральном введении по сравнению с соединением (В) (гидрохлоридом). Кроме того, ингибирующую активность АСАТ исследовали in vitro. Как показано в таблице 1, было обнаружено, что соединение (1) по настоящему изобретению проявляет высокую активность ингибирования АСАТ, эквивалентную ингибирующей активности соединения (В) (гидрохлорида). Представленные выше результаты, показывающие, что соединение (1) по настоящему изобретению проявляет высокую активность ингибирования АСАТ, сравнимую с ингибирующей активностью соединения (В), более высокую метаболическую устойчивость в микросомах печени человека, чем метаболическая устойчивость соединения (В), и хорошо всасывается при пероральном введении, указывают на то, что соединении (1) по настоящему изобретению можно применять в качестве профилактического или терапевтического средства по поводу гиперлипидемии или артериосклероза. Соединение (1) по настоящему изобретению оказывает превосходное действие ингибирования АСАТ и таким образом его можно применять в качестве профилактического или терапевтического средства по поводу, например, гиперлипидемии, артериосклероза, артериосклероза шейных и церебральных артерий, цереброваскулярных расстройств, ишемической энтеропатии, коронарного артериосклероза, нефросклероза, артериосклеротического нефросклероза, злокачественного нефросклероза, острой окклюзии сосудов брыжейки, хронического кишечного ишемического синдрома, ишемического колита, аневризмы аорты или облитерирующего артериосклероза (ASO). Когда соединение (1) по настоящему изобретению используется в качестве лекарственного средства, то соединение (1) или его соль могут быть сформированы или отдельно, или в комбинации с одним или несколькими фармакологически приемлемыми носителями (например, наполнителем, связывающим агентом и разбавителем в лекарственную форму, такую как таблетки, капсулы, гранулы, порошки, растворы для инъекций или суппозитории. Такое лекарственное средство может быть изготовлено посредством известных способов. Например, лекарственное средство для перорального введения может быть изготовлено составлением композиции соединения (1) по настоящему изобретения с одним или несколькими подходящими носителями, включая наполнитель, такой как крахмал, манит или лактозу; связывающий агент, такой как карбоксиметилцеллюлоза или гидроксипропилцеллюлоза натрия; разрыхлитель, такой как кристаллическая целлюлоза или карбоксиметилцеллюлоза кальция; смазывающее вещество, такое как тальк или стеарат магния; или агент, повышающий текучесть, такой как легкая безводная кремниевая кислота. Лекарственное средство по настоящему изобретению вводится или перорально, или парентерально, но предпочтительно пероральное введение. Доза лекарственного средства по настоящему изобретению различается в зависимости от, например, массы тела, возраста, пола или симптомов у пациента. Суточная доза соединения (1) по настоящему изобретению для взрослого составляет обычно от 1 до 500 мг, предпочтительно, от 5 до 200 мг. Соединение (1) предпочтительно вводится 1 раз/д или 2 или 3 раза/д разделенным образом. ПРИМЕРЫ Далее настоящее изобретение будет подробнее описано в виде примеров, которые не должны рассматриваться как ограничивающие технический объем изобретения. Пример получения 1 Получение 5,6-дифтор-2-меркаптобензимидазола 4,5-дифтор-2-нитроанилин (5,75 г, 33,03 ммоль) растворяют в уксусной кислоте (100 мл) и концентрированной хлористоводородной кислоте (2,3 мл) и при энергичном перемешивании смеси на ледяной бане к ней в течение 10 мин добавляют порошок цинка (6,91 г, 105,6 ммоль). Полученную смесь перемешивают в течение 20 мин при такой же температуре и затем в течение 130 мин при комнатной температуре. Далее к ней добавляют порошок цинка (1,20 г, 18,35 ммоль) в течение 5 мин при такой же температуре и полученную смесь перемешивают в течение 30 мин при такой же температуре. Реакционную смесь концентрируют при пониженном давлении и остаток нейтрализуют водным насыщенным бикарбонатом с последующей фильтрацией через целит. Фильтрат экстрагируют хлороформом и органический слой промывают насыщенным рассолом. Продукт сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении для получения коричневого масла (4,73 г). Коричневое масло растворяют в этаноле (200 мл) и к нему добавляют О-этилксантат калия (15,75 г, 98,25 ммоль) с последующим кипячением в сосуде с обратным холодильником в течение 14 ч. Реакционную смесь концентрируют при пониженном давлении, остаток экстрагируют этилацетатом – 1-моль/л хлористоводородной кислоты и органический слой промывают насыщенным рассолом. Продукт сушат над безводным сульфатом натрия, затем концентрируют при пониженном давлении и остаток кристаллизуют из хлороформа-гексана для получения 5,6-дифтор-2-меркаптобензимидазола (5,58 г, общий выход 91%) в виде бледно-коричневого порошка. Пример получения 2 Получение 1-трет-бутоксикарбонил-4-[2-(5,6-дифторбензимидазол-2-илтио)этил]пиперазина К раствору 1-трет-бутоксикарбонил-4-[2-гидроксиэтил)пиперазина (7,40 г, 32,13 ммоль) в ТГФ (100 мл) при перемешивании на ледяной бане последовательно добавляют триэтиламин (4,36 г, 43,09 ммоль), 4-диметиламинопиридин (200 мг, 1,64 ммоль) и метансульфонил хлорид (7,40 г, 38,76 ммоль). Температуре смеси дают возможность возвратиться к комнатной температуре, и смесь перемешивают в течение 50 мин. Реакционную смесь фильтруют и фильтрат концентрируют при пониженном давлении. Остаток растворяют в ДМФ (200 мл) и при комнатной температуре к раствору последовательно добавляют 5,6-дифтор-2-меркаптобензимидазол (5,00 г, 26,86 ммоль), карбонат калия (8,64 г, 62,51 ммоль) и 18-крон-6 (500 мг, 1,92 ммоль) с последующим перемешиванием в течение 90 мин при 80°С. Реакционную смесь концентрируют при пониженном давлении и остаток очищают посредством хроматографии на колонке силикагеля (силикагель 200 г, гексан:ацетон=от 8:1 до 1:1). Продукт кристаллизуют из ацетона-эфира-гексана для получения 1-трет-бутоксикарбонил-4-[2-(5,6-дифторбензимидазол-2-илтио)этил]пиперазина (7,26 г, выход 68%) в виде бесцветных кристаллов. Точка плавления: 192,3-193,0°С. ИК (KBr): 3061, 2976, 2836, 1672, 1475, 1427 (см-1). 1H ЯМР (400 МГц, CDCl3) Пример 1 Получение 1-[2-(5,6-дифторбензимидазол-2-илтио)этил]пиперазин трис-трифторацетата В условиях перемешивания на ледяной бане 1-трет-бутоксикарбонил-4-[2-(5,6-дифторбензимидазол-2-илтио)этил]пиперазин (7,26 г, 18,22 ммоль) добавляют к трифторуксусной кислоте (50 мл) в течение 15 мин и растворяют. После перемешивания смеси в течение 10 мин при охлаждении льдом к ней добавляют эфир (100 мл) и гексан (100 мл) и образовавшиеся кристаллы собирают посредством фильтрации. Кристаллы перекристаллизуют из этанола-эфира для получения 1-[2-(5,6-дифторбензимидазол-2-илтио)этил]пиперазин трис-трифтоацетата (9,58 г, выход 82%) в виде бледно-желтого порошка. Точка плавления: 141,2-142,9°С. ИК (KBr): 3417, 3026, 2749, 2483, 1671, 1484 (см-1). 1Н ЯМР (400 МГц, ДМСО-d6) Пример получения 3 Получение 1-[2-(5,6-дифторбензимидазол-2-илтио)этил]-4-формилпиперазина 1-формил-4-(2-гидроксиэтил)пиперазин (1,11 г, 7,0 ммоль), 5,6-дифтор-2-меркаптобензимидазол (1,30 г, 7,0 ммоль) и диизопропилэтиламин (3,62 г, 28,0 ммоль) растворяют в пропионитриле (50 мл) и к ним добавляют йодид цианометилтриметилфосфония (6,80 г, 28,0 ммоль) с последующим перемешиванием в течение 1 ч при 92°С в атмосфере аргона. Реакционной смеси дают возможность охладиться и затем выливают в воду (100 мл) с последующей экстракцией хлороформом (100 мл × 3). Органический слой промывают насыщенным рассолом и затем сушат над безводным сульфатом натрия и продукт концентрируют при пониженном давлении. Неочищенный продукт кристаллизуют из ацетона-эфира для получения 1-[2-(5,6-дифторбензимидазол-2-илтио)этил]-4-формилпиперазина (1,78 г, выход 78%) в виде желтого кристаллического порошка. Точка плавления: 197,0-198,0°С. ИК (KBr)см-1: 3441, 2825, 1648, 1476, 1431, 1363. 1Н ЯМР (ДМСО-d6) МС (m/z): 326 (M+), 140 (100). Пример 2 Получение 1-[2-(5,6-дифторбензимидазол-2-илтио)этил]пиперазина 1-[2-(5,6-дифторбензимидазол-2-илтио)этил]-4-формилпиперазин (1,70 г, 5,2 ммоль) растворяют в метаноле (20 мл) и к раствору добавляют 12N хлористоводородную кислоту (2 мл) с последующим перемешиванием в течение 18 ч при комнатной температуре. Реакционную смесь концентрируют при пониженном давлении и к ней добавляют насыщенный аммиак-метанол с последующим перемешиванием в течение 5 мин при комнатной температуре. Растворитель удаляют при пониженном давлении и остаток очищают посредством хроматографии на колонке силикагеля (хлороформ:насыщенный аммиак-метанол=100:3) для получения 1-[2-(5,6-дифторбензимидазол-2-илтио)этил]пиперазина (1,40 г, выход 90%) в виде коричневого масла. ИК (KBr) см-1: 2925, 2853, 1664, 1602, 1478, 1435, 1364. 1Н ЯМР (400 МГц, CDCl3,) МС (m/z): 298 (M+), 70 (100). Пример получения 4 Получение 2,4-бис(2,2,2-трифторэтокси)-6-метил-3-нитропиридина 2,4-дихлор-6-метил-3-нитропиридин (30 г, 144,9 ммоль) растворяют в 2,2,2-трифторэтаноле (250 мл) и к нему добавляют карбонат калия (50 г, 361,8 ммоль) с последующим кипячением в сосуде с обратным холодильником в течение 21 ч. Реакционную смесь разбавляют водой и затем подвергают экстракции хлороформом. Органический слой промывают насыщенным рассолом и затем сушат над безводным сульфатом натрия с последующей концентрацией при пониженном давлении для получения 2,4-бис(2,2,2-трифторэтокси)-6-метил-3-нитропиридина (45,40 г, выход 94%) в виде бледно-желтого твердого вещества. Точка плавления: 72,8-73,2°С. ИК (KBr): 3432, 3111, 2975, 1610, 1585, 1535 (см-1). 1Н ЯМР (400 МГц, CDCl3) Элементный анализ в виде C10H8F6N2O4. Рассчитано: С, 35,94; Н, 2,41; N, 8,38. Найдено: С, 35,94; Н, 2,45; N, 8,49. Пример 3 Получение 3-амино-2,4-бис(2,2,2-трифторэтокси)-6-метилпиридина 2,4-бис(2,2,2-трифторэтокси)-6-метил-3-нитропиридин (45,00 г, 134,7 ммоль) растворяют в изопропаноле (300 мл) и раствор дитионита натрия (78,00 г, 448,0 ммоль) в воде (300 мл) добавляют к нему при перемешивании при 80°С. Через 15 мин после начала взаимодействия к реакционной смеси добавляют раствор дитионита натрия (16,50 г, 94,8 ммоль) в воде (51 мл). Далее через 25 мин после начала взаимодействия к реакционной смеси добавляют раствор дитионита натрия (11,10 г, 63,8 ммоль) в воде (51 мл) и затем перемешивают в течение 10 мин. После завершения реакции к реакционной смеси добавляют 4 моль/л водной серной кислоты (201 мл) с последующим перемешиванием в течение 30 мин при 90°С. После того, как реакционная смесь остынет, к ней добавляют 28% водный аммиак (360 мл) при охлаждении льдом с последующим перемешиванием в течение 30 мин. Реакционную смесь разводят водой и затем экстрагируют хлороформом. Органический слой промывают насыщенным рассолом и затем сушат над безводным сульфатом натрия с последующей концентрацией при пониженном давлении. Полученные таким образом кристаллы перекристаллизуют из гексана для получения 3-амино-2,4-бис(2,2,2-трифторэтокси)-6-метилпиридина (32,91 г, выход 80%) в виде бледно-желтых иголок. Точка плавления: 53,5-53,8°С. ИК (KBr): 3453, 3314, 2968, 1603, 1505, 1456 (см-1). 1Н ЯМР (400 МГц, CDCl3) Элементный анализ в виде C10H10F6N2O2·0,55Н2О. Рассчитано: С, 38,24; Н, 3,56; N, 8,92. Найдено: С, 37,96; Н, 3,19; N, 8,94. Пример 4 Получение 2-бром-N-[2,4-бис(2,2,2-трифторэтокси)-6-метилпиридин-3-ил]ацетамида N,N-диметиланилин (20,46 г, 168,8 ммоль) добавляют к раствору 3-амино-2,4-бис(2,2,2-трифторэтокси)-6-метилпиридина (42,29 г, 139,0 ммоль) в дихлорметане (600 мл). При перемешивании смеси в условиях охлаждения льдом к ней добавляют раствор бромацетилбромида (28,73 г, 142,3 ммоль) в дихлорметане (100 мл) с последующим перемешиванием в течение 10 мин. Реакционную смесь разводят водой и затем экстрагируют хлороформом. Органический слой промывают насыщенным рассолом и затем сушат над безводным сульфатом натрия с последующей концентрацией при пониженном давлении. Полученные таким образом кристаллы перекристаллизуют из хлороформа-гексана для получения 2-бром-N-[2,4-бис(2,2,2-трифторэтокси)-6-метилпиридин-3-ил]ацетамида (50,25 г, выход 85%) в виде бесцветных иголок. Точка плавления: 152,8-154,0°С. ИК (KBr): 3250, 3053, 1677, 1597, 1541, 1456 (см-1). 1Н ЯМР (400 МГц, CDCl3) Элементный анализ в виде C12H11BrF6N2O3. Рассчитано: С, 33,90; Н, 2,61; N, 6,59. Найдено: С, 34,13; Н, 2,66; N, 6,65. Пример 5 Получение 2-[4-[2-(5,6-дифторбензимидазол-2-илтио)этил]пиперазин-1-ил]-N-[2,4-бис(2,2,2-трифторэтокси)-6-метилпиридин-3-ил]ацетамида (соединения 1b) 1-[2-(5,6-Дифторбензимидазол-2-илтио)этил]пиперазин трис-трифторацетат (4,00 г, 6,25 ммоль) и карбонат калия (31,26 ммоль) растворяют в ацетонитриле (100 мл) и воде (30 мл). При перемешивании раствора в условиях охлаждения льдом к нему в течение 15 мин добавляют 2-бром-N-[2,4-бис(2,2,2-трифторэтокси)-6-метилпиридин-3-ил]ацетамид (2,20 г, 5,22 ммоль). Температуре смеси дают возможность возвратиться до комнатной температуры и смесь перемешивают в течение 15 ч. Затем реакционную смесь разводят водой и экстрагируют хлороформом. Органический слой промывают насыщенным рассолом и сушат над безводным сульфатом натрия с последующей концентрацией при пониженном давлении. Остаток очищают посредством хроматографии на колонке силикагеля (силикагель 150 г, гексан:ацетон=4:1 до 2:1 до 1:1). Полученные таким образом кристаллы перекристаллизуют из хлороформа-гексана для получения 2-[4-[2-(5,6-дифторбензимидазол-2-илтио)этил]пиперазин-1-ил]-N-[2,4-бис(2,2,2-трифторэтокси)-6-метилпиридин-3-ил]ацетамида (3,04 г, выход 91%) в виде бледно-желтого порошка. Точка плавления: 191-192°С. ИК (KBr): 3275, 1686, 1604, 1591, 1509 см-1. 1Н ЯМР (400 МГц, ДМСО-d6) Элементный анализ в виде C25H26F8N6O3S. Рассчитано: С, 46,73; Н, 4,08; N, 13,08. Найдено: С, 46,55; Н, 4,12; N, 12,94. Пример получения 5 Получение 5-фтор-2-меркаптобензимидазола 4-фтор-2-нитроанилина (8,00 г, 51,22 ммоль) растворяют в метаноле (100 мл) и к нему добавляют 10% порошок палладиевой сажи (0,80 г) с последующим перемешиванием в течение 4 ч при комнатной температуре в атмосфере водорода. Реакционную смесь фильтруют и фильтрат концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией (силикагель 150 г, гексан:этилацетат=1:4) для получения посредством этого коричневого масла (5,67 г, выход 88%). Коричневое масло (5,64 г, 44,72 ммоль) растворяют в этаноле (150 мл) и к нему добавляют О-этилксантата калия (8,60 г, 53,65 ммоль) с последующим кипячением в сосуде с обратным холодильником в течение 3 ч. К нему добавляют О-этилксантат калия (1,43 г, 8,92 ммоль) и смесь кипятят в сосуде с обратным холодильником в течение 2 ч. Реакционную смесь концентрируют при пониженном давлении и остаток очищают колоночной хроматографией (силикагель 150 г, гексан:этилацетат=2:1) для получения 5-фтор-2-меркаптобензимидазола (5,93 г, выход 79%) в виде коричневого порошка. Пример получения 6 Получение 1-трет-бутоксикарбонил-4-[2-(5-фторбензимидазол-2-илтио)этил]пиперазина 1-трет-бутоксикарбонил-4-(2-гидроксиэтил)пиперазин (6,00 г, 26,05 ммоль) растворяют в ТГФ (36 мл) и к нему добавляют триэтиламин (3,43 г, 33,90 ммоль) и 4-диметиламинопиридин (159 мг, 1,30 ммоль). При охлаждении льдом к смеси по каплям добавляют раствор метансульфонилхлорида (3,58 г, 31,25 ммоль) в ТГФ (9 мл). Полученную смесь перемешивают в течение 1 ч и затем фильтруют и фильтрат концентрируют при пониженном давлении. Остаток растворяют в ДМФ (90 мл). При перемешивании раствора при комнатной температуре к раствору последовательно добавляют 5-фтор-2-меркаптобензимидазол (4,82 г, 28,66 ммоль), карбонат калия (5,40 г, 39,07 ммоль) и 18-крон-6 (688 мг, 2,60 ммоль) и полученную смесь перемешивают в течение 2 ч при 80°С. Реакционную смесь концентрируют при пониженном давлении и к остатку добавляют воду с последующей экстракцией этилацетатом. Органический слой промывают водой и насыщенным рассолом и затем сушат над безводным сульфатом натрия с последующей концентрацией при пониженном давлении. Остаток очищают посредством хроматографии на колонке силикагеля (силикагель 150 г, гексан:этилацетат=2:1 до 1:1 до 1:2) для получения 1-трет-бутоксикарбонил-4-[2-(5-фторбензимидазол-2-илтио)этил]пиперазина (7,28 г, выход 73%). 1Н ЯМР (CDCl3, 400 МГц) Пример 6 Получение 1-[2-(5-фторбензимидазол-2-илтио)этил]пиперазин трис-трифторацетата При перемешивании трифторуксусной кислоты (17 мл) в условиях охлаждения льдом к кислоте в течение 30 мин добавляют 1-трет-бутоксикарбонил-4-[2-(5-фторбензимидазол-2-илтио)этил]пиперазин (6,50 г, 17,08 ммоль) и растворяют в ней. Температуре смеси дают возможность возвратиться к комнатной температуре и смесь перемешивают в течение 30 мин. Затем к ней добавляют эфир и гексан и образовавшееся твердое вещество собирают посредством фильтрации. Собранный продукт промывают эфиром для получения 1-[2-(5-фторбензимидазол-2-илтио)этил]пиперазин трис-трифторацетата (10,50 г, выход 99%) в виде коричневого порошка. Точка плавления: 127,7-129,3°С. ИК (KBr): 3143, 3032, 2731, 1789, 1747, 1660 (см-1). 1Н ЯМР (400 МГц, ДМСО-d6) Пример получения 7 Получение 1-[2-(5-фторбензимидазол-2-илтио)этил]-4-формилпиперазина 1-формил-4-(2-гидроксиэтил)пиперазин (1,20 г, 7,6 ммоль), 5-фтор-2-меркаптобензимидазол (1,28 г, 7,6 ммоль) и диизопропилэтиламин (3,93 г, 30,4 ммоль) растворяют в пропионитриле (50 мл) и к смеси добавляют йодид цианометилтриметилфосфония (7,39 г, 30,4 ммоль) с последующим перемешиванием в течение 1 ч при 92°С в атмосфере аргона. Реакционной смеси дают возможность охладиться и затем выливают в воду (100 мл) с последующей экстракцией хлороформом (100 мл × 3). Полученный органический слой промывают насыщенным рассолом и затем сушат над безводным сульфатом натрия с последующей концентрацией при пониженном давлении. Неочищенный продукт кристаллизуют из ацетона-эфира для получения 1-[2-(5-фторбензимидазол-2-илтио)этил]-4-формилпиперазина (1,87 г, выход 80%) в виде коричневого кристаллического порошка. Точка плавления: 173,0-175,0°С. ИК (KBr) см-1: 3435, 3051, 2953, 2825, 1648, 1503, 1446. 1Н ЯМР (400 МГц, ДМСО-d6) MC (m/z): 308 (М+), 140 (100). Пример 7 Получение 1-[2-(5-фторбензимидазол-2-илтио)этил]пиперазина 1-[2-(5-фторбензимидазол-2-илтио)этил]-4-формилпиперазин (1,80 г, 5,8 ммоль) растворяют в метаноле (20 мл), к нему добавляют 12N хлористоводородную кислоту с последующим перемешиванием в течение 18 ч при комнатной температуре. Реакционную смесь концентрируют при пониженном давлении и к ней добавляют насыщенный аммиак-метанол с последующим перемешиванием в течение 5 мин при комнатной температуре. Растворитель удаляют при пониженном давлении и остаток очищают посредством хроматографии на колонке силикагеля (хлороформ:насыщенный аммиак-метанол=100:3) для получения 1-[2-(5-фторбензимидазол-2-илтио)этил]пиперазина (1,33 г, выход 81%) в виде коричневого масла. ИК (KBr) см-1: 3059, 2947, 2815, 1626, 1602, 1482, 1444, 1408. 1Н ЯМР (400 МГц, ДМСО-d6) MC m/z): 280 (М+), 70 (100). Пример 8 Получение 2-[4-[2-(5-фторбензимидазол-2-илтио)этил]пиперазин-1-ил]-N-[2,4-бис(2,2,2-трифторэтокси)-6-метилпиридин-3-ил]ацетамида (соединения 1а) 1-[2-(5-фторбензимидазол-2-илтио)этил]пиперазин трис-трифторацетат (6,92 г, 11,12 ммоль) и 2-бром-N-[2,4-бис(2,2,2-трфторэтокси)-6-метилпиридин-3-ил]ацетамид (4,50 г, 10,59 ммоль) суспендируют в ацетонитриле (90 мл) и к суспензии постепенно добавляют карбонат калия (5,85 г, 42,33 ммоль). Смесь перемешивают в течение 5 ч при комнатной температуре и к реакционной смеси добавляют воду (100 мл) с последующей экстракцией этилацетатом. Органический слой промывают насыщенным рассолом и затем сушат над безводным сульфатом натрия с последующей концентрацией при пониженном давлении. Остаток очищают посредством хроматографии на колонке силикагеля (хлороформ:метанол=50:1). Полученные таким образом кристаллы перекристаллизуют из ацетона-эфира для получения 2-[4-[2-(5-фторбензимидазол-2-илтио)этил]пиперазин-1-ил]-N-[2,4-бис(2,2,2-трифторэтокси)-6-метилпиридин-3-ил]ацетамида (4,72 г, выход 71%) в виде бледно-коричневых призм. Точка плавления: 182,0-182,7°С. ИК (KBr): 3282, 2824, 1509, 1413, 1272, 1166 (см-1). 1Н ЯМР (CDCl3, 400 МГц) Элементный анализ в виде C25H27F7N6O3S. Рассчитано: С, 48,08; Н, 4,36; N, 13,46. Найдено: С, 47,98; Н, 4,38; N, 13,31. Пример 9 Получение N-[2,4-бис(2,2,2-трифторэтокси)-6-метилпиридин-3-ил]-2-[4-(2-гидроксиэтил)пиперазин-1-ил]ацетамида 1-(2-гидроксиэтил)пиперазин (1,95 г, 15,0 ммоль) и 2-бром-N-[2,4-бис(2,2,2-трифторэтокси)-6-метилпиридин-3-ил]ацетамид (5,00 г, 12,5 ммоль) растворяют в ацетонитриле (30 мл) и к раствору добавляют карбонат калия (2,25 г, 16,3 ммоль). Смесь перемешивают в течение 5 ч при комнатной температуре и реакционную смесь разводят водой с последующей экстракцией этилацетатом. Органический слой промывают водой и насыщенным рассолом и сушат над безводным сульфатом натрия с последующей концентрацией при пониженном давлении. Полученный остаток очищают посредством хроматографии на колонке силикагеля (проявляющий раствор:аммиак-насыщенный метанол/хлороформ=1/20) для получения N-[2,4-бис(2,2,2-трифторэтокси)-6-метилпиридин-3-ил]-2-[4-(2-гидроксиэтил)пиперазин-1-ил]ацетамида (5,40 г, выход: 91%) в виде бесцветных кристаллов. 1Н ЯМР (400 МГц, CDCl3) Пример 10 Получение 2-[4-[2-(5,6-дифторбензимидазол-2-илтио)этил]пиперазин-1-ил]-N-[2,4-бис(2,2,2-трифторэтокси)-6- метилпиридин-3-ил]ацетамида (соединения 1b) В атмосфере аргона N-[2,4-бис(2,2,2-трифторэтокси)-6-метилпиридин-3-ил]-2-[4-[2-гидроксиэтил]пиперазин-1-ил]ацетамид (4,0 г, 8,43 ммоль), 5,6-дифтор-2-меркаптобензимидазол (5,8 г, 31,2 ммоль) и трифенилфосфин (7,8 г, 29,7 ммоль) растворяют в N,N-диметилформамиде (170 мл) и в условиях охлаждения льдом к смеси по каплям добавляют диэтилазокарбонат (40% мас./об. раствор толуола, 11,0 мл, 25,3 ммоль) с последующим перемешиванием в течение 1,5 ч при той же температуре. К реакционной смеси добавляют этилацетат и 1 моль/л хлористоводородной кислоты и водный слой отделяют. Органический слой далее экстрагируют 1 моль/л хлористоводородной кислоты. Водный слой объединяют и полученную смесь подщелачивают гидроксидом натрия (1 моль/л) с последующей экстракцией этилацетатом. Органический слой промывают водой и насыщенным рассолом и затем сушат над безводным сульфатом натрия с последующей концентрацией при пониженном давлении. Остаток очищают посредством хроматографии на колонке силикагеля (проявляющий раствор:хлороформ:аммиакнасыщенный метанол =100:3) для получения 2-[4-[2-(5,6-дифторбензимидазол-2-илтио)этил]пиперазин-1-ил]-N-[2,4-бис(2,2,2-трифторэтокси)-6-метилпиридин-3-ил]ацетамида (4,9 г, выход: 90,1%) в виде бесцветных кристаллов. Пример испытания 1 Испытание ингибирующей АСАТ активности в клетках J774A Клетки J774 (2×105 клеток/лунку) высевали на 24-луночный планшет и инкубировали в течение 24 ч в DMEM (модифицированной Дульбеко среде Игла) (10% FBS (фетальная телячья сыворотка), 500 мкл). После замещения новой средой к ней добавляли 25-гидроксихолестерин (10 мкг/мл) и ингибитор АСАТ (конечная концентрация: 0, 10-9 до 10-5 моль/л) с последующей инкубацией в течение 18 ч. После промывания 0,9% хлоридом натрия липид экстрагировали гексаном-изопропанолом (3:2) (250 мкл) и затем снова гексаном-изопропанолом (3:2) (250 мкл). Экстракты объединяли и растворитель удаляли. Полученный таким образом сложный эфир холестерина (СЕ) количественно анализировали посредством флюоресцентного ферментного анализа. Клетки, из которых экстрагировали липид, подвергали белковому анализу (микроанализ ВСА) для определения посредством этого количества СЕ на 1 мг белка. По соотношению продукции СЕ между испытуемым соединением и контролем рассчитывали IC50 (концентрацию соединения, ингибирующую 50% продукции СЕ) при N=4. Результаты показаны в таблице 1. Как показано в таблице 1, было подтверждено, что соединения (1а) и (1b) имеют высокую ингибирующую АСАТ активность.
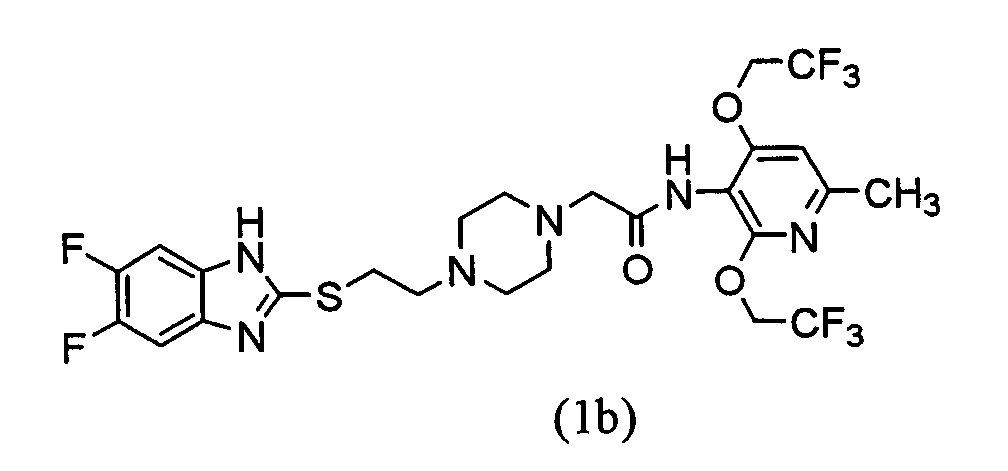
Пример испытания 2 Испытание метаболической устойчивости в микросомах печени человека В соответствии с описанной ниже таблицей 2 раствор NRS (систему, регенерирующую NADPH) и 16% альбумин сыворотки человека добавляли к 0,1 моль/л фосфатного буфера (рН 7,4) и к нему добавляли раствор испытуемого соединения (100 мкМ) в ацетонитриле (0,01 мл). Смесь предварительно инкубировали в течение 5 мин на теплой бане при 37°С и к ней добавляли микросому печени человека (POOLED HUMAN LIVER MICROSOMES, Лот № 20, продукт GENTEST) с последующим предоставлением возможности взаимодействия в течение 30 мин на теплой бане при 37°С. Аликвотное количество (0,25 мл) брали из реакционной смеси через 0 и 30 мин после начала реакции с последующей экстракцией*. Количество испытуемого соединения определяли посредством ВЭЖХ. Остаточную процентную долю неизмененного соединения через 30 мин рассчитывали на основании следующего уравнения: (площадь пика через 30 мин/площадь пика через 0 мин) × 100. Результаты показаны на чертеже. Как показано на чертеже, было подтверждено, что соединения (1а) и (1b) имеют резко улучшенную метаболическую устойчивость в микросомах печени человека по сравнению с соединением (В) (гидрохлоридом).
* Процедура экстракции К каждому образцу добавляли глициновый буфер (рН 10, 1,0 мл), вещество внутреннего стандарта (0,1 мл) и трет-бутилметиловый эфир (5,0 мл). Смесь встряхивали в течение 10 мин и затем центрифугировали при 2500 об/мин в течение 10 мин, и органический слой собирали. Пример испытания 3 Испытание растворимости (раствор I по Фармакопее Японии) Каждое испытуемое соединение растворяли в ацетонитриле для формирования раствора 100 мкМ и раствор добавляют к раствору I по Фармакопее Японии для формирования раствора 1000 нг/мл. Полученный раствор перемешивали в течение 10 мин и аликвотное количество (1 мл) помещали в шприц и затем пропускали через фильтр 0,2 мкм (HLC-DISK 13, вода/растворитель, Kanto Kagaku Kabushiki-kaisya). Фильтрат (0,5 мл) подвергали процедуре экстракции* и количество испытуемого соединения определяли посредством ВЭЖХ. Результаты показаны в таблице 3. Как показано в таблице 3, было обнаружено, что соединение (1а) и соединение (1b) имеют меньшую растворимость по сравнению с растворимостью соединения (В) (гидрохлорида). На основании описанной выше более низкой растворимости ожидалось, что соединения настоящего изобретения имеют низкую всасываемость после перорального введения.
Экстракцию выполняли аналогично способу экстракции испытания метаболической устойчивости в микросомах печени человека. Пример испытания 4 Испытание перорального введения у крыс Каждое испытуемое соединение растворяли в 0,01N растворе хлористоводородной кислоты, и раствор перорально вводили самцам и самкам крыс в дозе 10 мг/5 мл/кг. Образца крови (каждый по 0,25 мл) брали через 30, 60, 120, 180, 240 и 360 мин после введения. Взятые образцы крови центрифугировали в течение 5 мин при 4°С и 9000 g для приготовления посредством этого образцов плазмы. Образцы плазмы хранили при -30°С перед измерением. Образцы подвергали процедуре экстракции*, и уровни в плазме испытуемого соединения определяли посредством ЖХ/МС/МС. Результаты показаны в таблице 4. Как показано в таблице 4, было обнаружено, что соединение (1а) и соединение (1b) проявляют более высокую Cmax и более высокую AUC (площадь под кривой), по сравнению с соединением (В) (гидрохлоридом), подтверждая то, что соединение (1а) и соединение (1b) имеют хорошую биологическую доступность после перорального введения по сравнению с соединением (В) (гидрохлоридом).
(10 мг/кг перорально)
Экстракцию выполняли аналогично способу экстракции испытания метаболической устойчивости в микросомах печени человека. По сравнению с соединением (В), описанным в Международной патентной публикации WO98/54153, было обнаружено, что соединение (1) по настоящему изобретению проявляет отличную устойчивость против метаболизма в микросомах печени человека и высокую ингибирующую АСАТ активность. Хотя соединение (1) по настоящему изобретению имеет более низкую растворимость в воде. По сравнению с соединением (В) (гидрохлоридом) оно проявляет хорошее всасывание при пероральном введении, как показывает испытание перорального введения у крыс. Поэтому ожидается, что соединение (1) по настоящему изобретению имеет превосходную биологическую доступность у людей.
Формула изобретения
1. 2,4-Бис(трифторэтокси)пиридиновое соединение, представленное формулой (I)
где X1 представляет атом фтора или водорода или его соль. 2. Лекарственное средство, обладающее активностью ингибитора ацил-кофермента А холестерин-ацилтрансферазы (АСАТ), содержащее в качестве активного ингредиента соединение или его соль по п.1. 3. Лекарственное средство по п.2, для профилактики или терапии гиперлипидемии и/или артериосклероза. 4. Способ получения соединения или его соли по п.1, включающий взаимодействие пиперазинового соединения, представленного формулой (2)
где X1 представляет атом фтора или водорода с пиридиновым соединением, представленным формулой (3)
где X2 представляет атом хлора, брома или йода. 5. Пиперазиновое соединение, представленное формулой (2)
где X1 представляет атом фтора или водорода или его соль. 6. Пиридиновое соединение, представленное формулой (4)
где R1 представляет атом водорода, хлорацетильную группу, бромацетильную группу или йодацетильную группу или его соль. 7. 2,4-Бис(2,2,2-трифторэтокси)-6-метил-3-нитропиридин. 8. N-[2,4-бис(2,2,2-трифторэтокси)-6-метилпиридин-3-ил]-2-[4-(2-гидроксиэтил)пиперазин-1-ил]ацетамид. 9. Лекарственная композиция, обладающая активностью ингибитора ацил-кофермента А холестерин-ацилтрансферазы (АСАТ), включающая соединение или его соль по п.1 в эффективном количестве и фармакологически приемлемый носитель. 10. Применение соединения по п.1. или его соли для изготовления лекарственного средства, обладающего активностью ингибитора ацилкофермента А холестерин-ацилтрансферазы (АСАТ).
РИСУНКИ
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

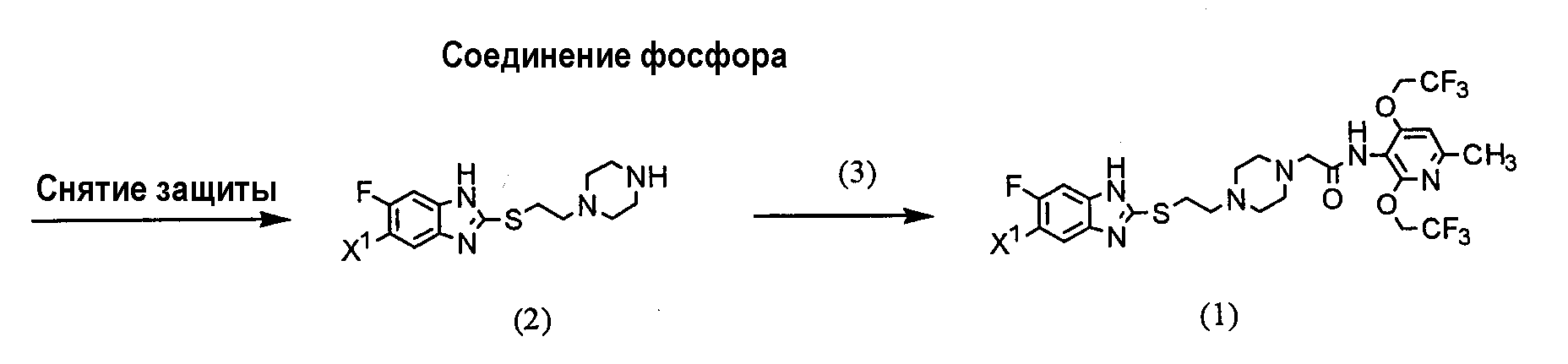
 1,50 (9Н, с), 2,51-2,68 (4Н, м), 2,94 (2Н, т, J=5,4 Гц), 3,28 (2Н, т, J=5,4 Гц), 3,45-3,65 (4Н, м), 6,85-7,62 (2Н, м).
1,50 (9Н, с), 2,51-2,68 (4Н, м), 2,94 (2Н, т, J=5,4 Гц), 3,28 (2Н, т, J=5,4 Гц), 3,45-3,65 (4Н, м), 6,85-7,62 (2Н, м). -никотинамидадениндинуклеотида, в окисленной форме,
-никотинамидадениндинуклеотида, в окисленной форме, Процедура экстракции
Процедура экстракции