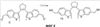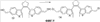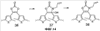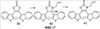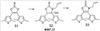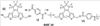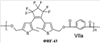Патент на изобретение №2345997
|
||||||||||||||||||||||||||
(54) ФОТОХРОМНЫЕ ПОЛИМЕРЫ ДЛЯ ТРЕХМЕРНОЙ ОПЕРАТИВНОЙ ОПТИЧЕСКОЙ ПАМЯТИ
(57) Реферат:
Изобретение относится к новым фотохромным мономерам и новым полимерам на их основе, предназначенным для создания двухфотонных фотохпромных регистрирующих сред для трехмерной оптической памяти и фотопереключателей оптических сигналов. Описываются мономеры
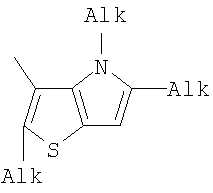
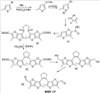
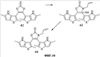
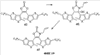
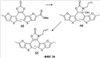
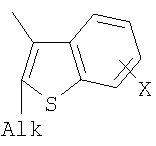
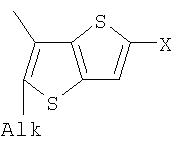
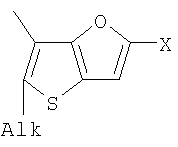
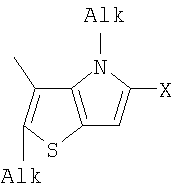
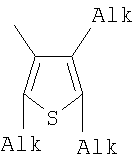
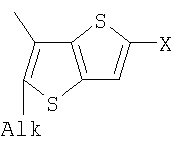
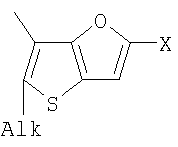
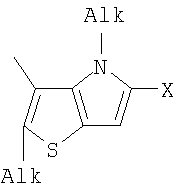
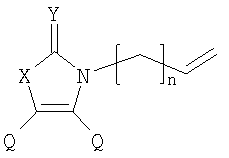
Q= Alk=CH3-С10H21 X=Cl, Br, I, F, NH2, CH2OH, CH2Cl, CH2Br, CHO, CO2H
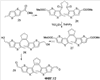
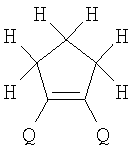
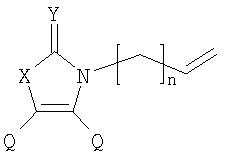
X=CH2, O, S, NAlk; Y=O, S, NAlk; n=0-6; Q= Alk=CH3-C10H21 способы их получения, фотохромные полимеры на их основе, способы получения фотохромных полимеров и их применение. Предложенные материалы обладают термической необратимостью фотохромных превращений и свойствами, обеспечивающими возможность применения фотохромных полимеров в двухфотонной оперативной оптической памяти. 15 н.п. ф-лы, 46 ил.
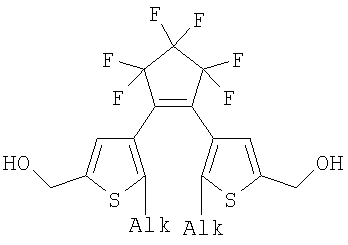
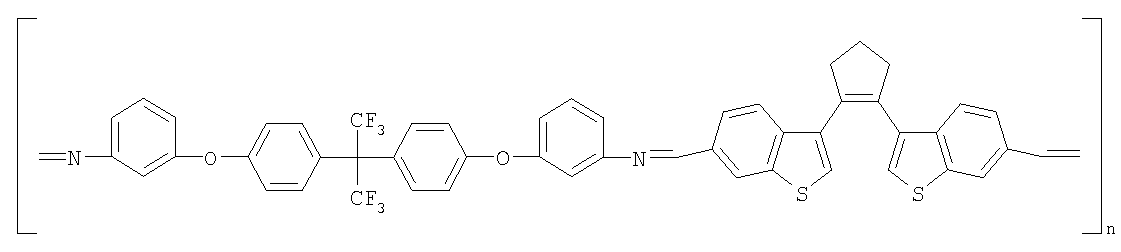
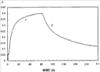
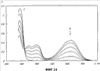
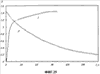
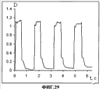
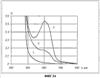
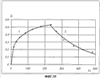
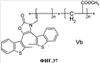
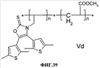
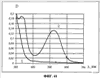
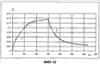
(1) Область техники Настоящее изобретение относится к новым фотохромным полимерам и способам их получения с целью создания двухфотонных фотохромных регистрирующих сред для трехмерной оперативной оптической памяти с побитовой регистрацией оптической информации. (2) Уровень техники В настоящее время актуальной задачей в области информационных технологий является создание баз данных для телекоммуникационных систем связи. В связи с этим активно ведется разработка оптической памяти сверхбольшой информационной емкости за счет перехода от двумерных носителей информации к трехмерным регистрирующим средам, позволяющим достичь максимально возможной плотности записи информации (до 1 Тбит/см3). Создание оперативной трехмерной оптической памяти требует создания двухфотонных регистрирующих сред (I.Cokgor, F.B.McCormick, A.S.Dvornikov, M.Wang, N.Kim, К.Koblentz, S.C.Esener, P.M.Rentzepis. Proc. SPIE, vol.3109, pp.182-186, 1997; S.Kawata, Y.Kawata. Chem. Rev., vol.100, pp.1777-1791, 2000). Такие среды разрабатываются в основном в США (фирма Call/Recall Corporation, Irvine and San Diego Universities of California) и Японии (Japan Science and Technology Corporation, Kyushu, Osaka, and Shizuoka Universities). В этих разработках используются полимерные растворы термически необратимых фотохромных соединений различных классов: диарилэтенов, фульгидов, фульгимидов, феноксипроизводных феноксинафтаценхинов и др. (A.S.Dvornikov, I.Cokgor, M.Wang, F.B.McCormick, S.C.Esener, P.M.Rentzepis. IEEE Transaction. Part A, vol.20, N2, pp.203-212, 1997). Известно, что соединения этого типа обеспечивают двухфотонное возбуждение и, следовательно, запись оптической информации в объеме среды. Недостатком таких регистрирующих сред является то, что фотохромный слой готовится из мономерного фотохромного соединения одного из вышеперечисленных классов и полимерного связующего. По существу, такая среда представляет собой молекулярный раствор фотохромного соединения в полимерной матрице. Это приводит к тому, что концентрация фотохромных молекул, а следовательно, и число светочувствительных центров определяется предельной растворимостью соединений в полимере, которая обычно не превышает 10% от массы сухого полимерного связующего. В таких средах наблюдаются процессы кристаллизации фотохромных веществ, фазовое разделение, агрегация фотохромных молекул и образование градиентов концентрации фотохромного соединения в объеме и на поверхности слоя (D.M.Buland, R.D.Miller, C.A.Walsh. Chem. Rev., vol.94, p.31, 1994). В результате этого информационная емкость и цикличность записи-перезаписи информации в такой фотохромной среде резко снижаются. Кроме того, квантовый выход фотохромных превращений ряда наиболее приемлемых фотохромных соединений, а именно мономерных диарилэтенов, в силу хаотичности их распределения в объеме полимера не превышает В связи с этим наибольший интерес для использования в качестве двухфотонных регистрирующих сред представляют фотохромные полимеры, содержащие валентно связанные фотохромные молекулы либо в основной полимерной цепи, или в виде боковых фрагментов. В фотохромных полимерах ковалентно связанные фотохромные молекулы более стабильны во времени при хранении за счет затруднения их перемещения в объеме среды. К числу таких фотохромных регистрирующих сред относятся, например, фотохромные полимеры с диарилэтеновыми фрагментами на основе мономеров стирола и бутилметакрилата (Е.Kim, Y.-K.Choi, M.-H.Lee, Macromolecules, vol.32, pp.4855-4860, 1999; S.Y.Cho, H.-W.Shin, K.H.Ahn, Y.R.Kim, E.Kim. Optical Materials, vol.21, pp.279-284, 2002). Среды этого типа по сравнению с полимерными растворами позволяют получать слои толщиной 0,05-0,1 мкм высокого качества. Разработанная среда обеспечивает запись оптической информации под действием излучения гелий-кадмиевого лазера (325 нм), а стирание – с использованием лазеров, излучающих в видимой области спектра (663 и 532 нм). Недеструктивное считывание записанной информации достигается за счет фотоиндуцированного изменения показателя преломления ( Наиболее близким аналогом настоящего изобретения являются фотохромные полимеры на основе 1,2-бис(3-тиенил)циклопентенового мономера (публикация заявки на патент США №20040030078 от 12 февраля 2004 г.). Недостаток таких фотохромных полимеров состоит в том, что в них используется вполне определенная структура фотохромного мономера и гомополимеров на их основе. Этот недостаток обусловлен ограниченностью выбора исходных веществ для синтеза фотохромных полимеров. (3) Раскрытие изобретения Целью настоящего изобретения является синтез новых фотохромных сополимеров и полимеров на основе новых фотохромных функциональных соединений из класса диарилэтенов с отличной от ближайшего аналога структурой, но обладающих термической необратимостью фотохромных превращений и другими свойствами, обеспечивающими возможность применения фотохромных полимеров в двухфотонной оперативной оптической памяти. Поставленная цель достигается тем, что в качестве фотохромных мономеров используются следующие функциональные соединения из класса дигетарилэтенов I-III (фиг.1). Фотохромизм диарилэтенов (ДАЭ) состоит в обратимой фотоциклизации, т.е. в фотоиндуцированном переходе из открытой формы А в циклическую форму В (фиг.2). Способ получения фотохромных мономеров класса I включает ацилирование производных бензотиофена, а также пироллотиофена, тиенотиофена или фуранотиофена дихлорангидридом глутаровой кислоты в хлористом метилене в присутствии хлористого алюминия и последующую восстановительную циклизацию полученных дикетонов под действием TiCl4, Zn в ТГФ в присутствии пиридина, с дальнейшим формилированием продуктов реакции дихлорметиловым эфиром в нитробензоле в присутствии хлористого алюминия. Способ получения фотохромных мономеров класса II включает обработку производных 1,3-оксазолан-2-она и тиофена, а также бензотиофена, пироллотиофена, тиенотиофена или фуранотиофена аллиламином и дальнейшую обработку продуктов реакции трифторуксусной кислотой. Способ получения фотохромных мономеров класса III включает обработку тетрагидропиранильного производного бромтиофена бутиллитием и последующее взаимодействие с перфторциклопентеном с дальнейшим гидролизам продукта реакции при действии соляной кислоты. Способ получения фотохромного олигомера из мономеров класса I включает взаимодействие 2,2-бис[4-(3-аминофенокси)фенил]гексафторпропана с диальдегидным производным 1,2-циклопентена при повышенной температуре с одновременной отгонкой воды в виде азеотропа. Способ получения фотохромных полимеров из мономеров класса II включает взаимодействие аллильного производного 1,3-оксазалон-2-она с производным акриловой кислоты при повышенной температуре в присутствии инициатора. Способ получения фотохромных полимеров из мономера класса III включает взаимодействие 1,2-бис(5-гидроксиметил-2-метилтиофен-3-ил)гексафторциклопентана с дихлорангидридом кислоты при повышенной температуре в присутствии пиридина. Новизна заявленных признаков состоит в использовании в качестве фотохромных фрагментов функциональных дигетарилэтенов I-III, отличных от ближайшего аналога. Использование этих фотохромных соединений позволяет фотохромным полимерам на их основе осуществлять двухфотонную регистрацию оптической информации в устройствах трехмерной оперативной оптической памяти. Изучение и анализ известной научно-технической и патентной литературы показал, что полной совокупности признаков, характеризующих данные технические решения, ранее известно не было, т.е. заявляемые решения отвечают критерию патентоспособности “новизна”. (4) Краткое описание чертежей Сущность настоящего изобретения поясняется далее с помощью примеров и чертежей. На фиг.1 приведены структурные формулы фотохромных мономеров I-III, использованных для получения фотохромных сополимеров и полимеров. На фиг.2 приведена обобщенная схема фотохромных превращений дигетарилэтенов. На фиг.3 приведена схема способа получения фотохромного мономера 5 из класса I. На фиг.4 приведены спектры поглощения исходной открытой формы А (кривая 1) и фотоиндуцированной формы В через стеклянный светофильтр УФС-2 (кривые 2-7) при возрастающей экспозиции УФ светом фотохромного соединения 5 в растворе толуола (С=2·10-4М). На фиг.5 приведены кинетические кривые фотоокрашивания УФ светом через светофильтр УФС-6 (кривая 1) и фотообесцвечивания видимым светом через светофильтр ЖС-12 (кривая 2) раствора фотохромного соединения 5 в толуоле на длине волны максимума полосы поглощения циклической формы В. На фиг.6 приведена схема получения фотохромного мономера 10 из класса I. На фиг.7 приведена схема получения фотохромного мономера 12 из класса I. На фиг.8 приведена схема получения фотохромного мономера 13 из класса I. На фиг.9 приведена схема получения фотохромного мономера 14 из класса II. На фиг.10 приведена схема получения фотохромного мономера 21 из класса I. На фиг.11 приведена схема получения фотохромного мономера 24 из класса I. На фиг.12 приведена схема получения фотохромного мономера 30 из класса I. На фиг.13 приведена схема получения фотохромного мономера 35 из класса I. На фиг.14 приведена схема получения фотохромного мономера 38 из класса II. На фиг.15 приведены спектры поглощения исходной открытой формы А (кривая 1) и фотоиндуцированной формы В при возрастающей экспозиции УФ света через светофильтр УФС-2 (кривые 2-6) для фотохромного соединения 38 из класса II в толуоле. На фиг.16 приведены кинетические кривые фотоокрашивания УФ светом через стеклянный светофильтр УФС-2 (кривая 1) и фотообесцвечивания видимым светом через светофильтр ЖС-12 (кривая 2) раствора фотохромного соединения 38 из класса II в толуоле на длине волны максимума полосы поглощения циклической формы В. На фиг.17 приведена схема получения фотохромного мономера 41 из класса II. На фиг.18 приведена схема получения фотохромного мономера 44 из класса II. На фиг.19 приведена схема получения фотохромного мономера 47 из класса II. На фиг.20 приведена схема получения фотохромного мономера 50 из класса II. На фиг.21 приведена схема получения фотохромного мономера 53 из класса II. На фиг.22 приведена схема получения фотохромного мономера 56 из класса II. На фиг.23 приведена схема получения фотохромного соединения 60 из класса III. На фиг.24 приведены спектры поглощения исходной открытой формы А (кривая 1) и фотоиндуцированной формы В при возрастающей экспозиции УФ света через светофильтр УФС-2 (кривые 2-5) для фотохромного соединения 60 из класса III в толуоле. На фиг.25 приведены кинетические кривые фотоокрашивания УФ светом через стеклянный светофильтр УФС-2 (кривая 1) и фотообесцвечивания видимым светом через светофильтр ЖС-12 (кривая 2) раствора фотохромного соединения 60 из класса III в толуоле на длине волны максимума полосы поглощения циклической формы В. На фиг.26 приведена схема получения фотохромного олигомера IV. На фиг.27 приведены спектры поглощения исходной открытой формы А (кривая 1) и фотоиндуцированной В формы (кривая 2) для пленки фотохромного олигомера IV в полиметилметакрилате (10 мас.% от массы сухого полимера) до и после облучения УФ светом соответственно. На фиг.28 приведены кинетические кривые фотоокрашивания УФ светом (кривая 1) и фотообесцвечивания видимым светом (кривая 2) пленки фотохромного олигомера IV в полиметилметакрилате (10 мас.% от массы сухого полимера) на длине волны максимума полосы поглощения циклической формы В. На фиг.29 приведены кривые последовательного фотоокрашивания и фотообесцвечивания при периодической смене светофильтров УФС-8 и ПС-7 + ЖС-18 для пленки фотохромного олигомера IV в полиметилметакрилате (10 мас.% от массы сухого полимера) на длине волны максимума полосы поглощения циклической формы В. На фиг.30 приведена схема получения фотохромного олигомера IVa. На фиг.31 приведена схема получения фотохромного олигомера IVb. На фиг.32 приведена схема получения фотохромного олигомера IVc. На фиг.33 приведена схема синтеза фотохромного полимера V. На фиг.34 приведены спектры поглощения исходной открытой формы А (кривая 1) и фотоиндуцированной формы В (кривая 2) пленки фотохромного полимера V в поликарбонате (10% от массы сухого полимера) до и после облучения УФ светом соответственно. На фиг.35 приведены кинетические кривые фотоокрашивания УФ светом (кривая 1) и фотообесцвечивания видимым светом (кривая 2) пленки фотохромного полимера V в поликарбонате (10% от массы сухого полимера) на длине волны максимума полосы поглощения циклической формы В. На фиг.36 приведена структура фотохромного полимера Va. На фиг.37 приведена структура фотохромного полимера Vb. На фиг.38 приведена структура фотохромного полимера Vc. На фиг.39 приведена структура фотохромного полимера Vd. На фиг.40 приведена схема синтеза фотохромного полимера VII. На фиг.41 приведены спектры поглощения исходной открытой формы А (кривая 1) и фотоиндуцированной формы В после облучения УФ светом через светофильтр УФС-2 (кривая 2) и последующего облучения видимым светом через светофильтр ЖС-12 (кривая 3) для пленки фотохромного полимера VII в поликарбонате (4 мас.% от массы сухого полимера) до и после облучения УФ светом соответственно. На фиг.42 приведены кинетические кривые фотоокрашивания УФ светом (кривая 1) и фотообесцвечивания видимым светом (кривая 2) пленки фотохромного полимера VII в поликарбонате (4 мас.% от массы сухого полимера) на длине волны максимума полосы поглощения циклической формы В. На фиг.43 приведена структура фотохромного полимера VIIa. На фиг.44 приведена структура фотохромного полимера VIIb. На фиг.45 приведена структура фотохромного полимера VIIc. На фиг.46 приведена структура фотохромного полимера VIId. (5) Сведения, подтверждающие возможность осуществления изобретения Пример 1 Фотохромные мономеры 1,2-бис(2-метил-6-формил-1-бензотиофен-3-ил)циклопентены 5а-е из класса использованных фотохромных мономеров, содержащие бензотиофеновые фрагменты, связанные циклопентеновым мостиком, синтезировали на основе бензотиофенов 2а-е по схеме, представленной на фиг.3. Исходные бензотиофены 2а-е были получены по описанным методикам: 2-метилбензотофен (2а) (Синтез сульфидов, тиофенов и тиолов типа соединений, встречающихся в нефти. Под ред. Е.Н.Карауловой, Изд-во «Наука», 1988 г, с.180), 2,4-диметилбензотиофен и 2,7-диметилбензотиофен (2b и 2с) (Monatsh. Chem. 1960, 91, 1070), 2,4-диметилбензотиофен и 2,6-диметилбензотиофен (2d и 2е) (J. Chem. Soc, Chem. Com. 1974, 5, 174). Синтез 1,2-бис(2-метил-6-формил-1-бензотиофен-3-ил)циклопентенов 5а-е включал в себя ацилирование бензотиофенов 2а-е дихлорангидридом глутаровой кислоты в хлористом метилене в присутствии хлористого алюминия и последующую восстановительную циклизацию полученных дикетонов 3а-е, которая отличалась тем, что соединения 4а-е было получены с 60-73% выходом при циклизации дикетонов 3а-е под действием TiCl4, Zn в ТГФ в присутствии пиридина, тогда как в литературе описано получение соединения 4а циклизацией дикетона 3а под действием TiCl4, Zn в ТГФ с выходом 54% (Synthesis 1998, 1092-1094). Последующее формилирование продуктов 4а-е дихлорметиловым эфиром в нитробензоле в присутствии хлористого алюминия привело к образованию 1,2-бис(2-метил-6-формил-1-бензотиофен-3-ил)циклопентенов 5а-е. Общая схема синтеза 1,5-бис-(метил-1-бензотиофен-3-ил)пентан-1,5-дионов 3а-е К хорошо перемешиваемой смеси 20,2 ммоль метилбензотиофена 2а-е и 10,1 ммоль дихлорангидрида глутаровой кислоты в 50 мл хлористого метилена прибавляли при 0°С 42,5 ммоль безводного хлористого алюминия. Реакционную массу перемешивали при комнатной температуре 3-6 часов. Выливали на лед с водой, экстрагировали хлористым метиленом (3×100 мл), собранные органические слои промывали водным раствором NaHCO3, сушили над сульфатом магния, растворитель упаривали под вакуумом. Остаток перекристаллизовывали из гексана. 1,5-Бис-(2-метил-1-бензотиофен-3-ил)пентан-1,5-дион 3а: Получали 3,25 г продукта 3а (82%), Тпл=165-167°С (гексан: хлороформ, 6:1, об.). Масс-спектр, m/z: 392, [М]+. Спектр ЯМР 1Н (ДМСО-d6, 1,5-Бис-(2,5-диметил-1-бензотиофен-3-ил)пентан-1,5-дион 3b: Получали 3,57 г продукта (3b) (84%), Тпл=171-173°С (гексан: хлороформ, 6:1, об.). Масс-спектр, m/z: 420, [М]+. Спектр ЯМР 1Н (ДМСО-d6, 1,5-Бис-(2,7-диметил-1-бензотиофен-3-ил)пентан-1,5-дион 3с: Получали 3,61 г продукта (3с) (85%), Тпл=159-161°С (гексан: хлороформ, 6:1, об.). Масс-спектр, m/z: 420, [М]+. Спектр ЯМР 1Н (ДМСО-d6, 1,5-Бис-(2,4-диметил-1-бензотиофен-3-ил)пентан-1,5-дион 3d: Получали 3,32 г продукта (3d) (78%), Тпл=156-158°С (гексан: хлороформ, 6:1, об.). Масс-спектр, m/z: 420, [М]+. Спектр ЯМР 1Н ДМСО-d6, 1,5-Бис-(2,6-диметил-1-бензотиофен-3-ш)пентан-1,5-дион 3е: Получали 3,66 г продукта (3е) (86%), Тпл=171-173°С (гексан: хлороформ, 6:1, об.). Масс-спектр, m/z: 420, [М]+. Спектр ЯМР 1Н (ДМСО-d6, Общая методика синтеза 1,2-бис(2-метил-1-бензотиофен-3-ш)циклопентена 4 К хорошо перемешиваемой суспензии 7,93 ммоль цинка в свежеперегнанном безводном THF (50 мл) при -10°С под аргоном добавляли по каплям 2,8 мл TiCl4. После окончания прибавления реакционную смесь нагревали под аргоном 1 час. Охлаждали до 20°С и прибавляли 12,8 ммоль дикетона 3а-е и безводный пиридин (5 мл). Кипятили под аргоном еще 20 часов. Выливали в 10% К2СО3 (150 мл) и водный слой экстрагировали Et2O (5×100 мл). Собранные органические экстракты сушили MgSO4 и отгоняли под вакуумом. Остаток чистили с помощью флэш-хроматографии на силикагеле (Merck, 0,063-0,1), элюент – петролейный эфир (40/70) – AcOEt (10:1, об.). 1,2-бис(2-метил-1-бензотиофен-3-ил)циклопентен 4а: Получали 2,76 г продукта 4а (60%) с Тпл=186,5-187,5°С, Тпл(литер.)=187-188°С (Synthesis 1998, 1092-1094). 1,2-бис(2,5-диметил-1-бензотиофен-3-ил)циклопентен 4b: Получали 3,19 г продукта (4b) (69%), Тпл=165-167°С (гексан: хлороформ, 6:1, об.). Масс-спектр, m/z: 388, [М]+. Спектр ЯМР 1Н (ДМСО-d6, 1,2-бис(2,7-диметил-1-бензотиофен-3-ил)циклопентена 4с: Получали 3,37 г продукта (4с) (73%), Тпл=174-176°С (гексан: хлороформ, 6:1, об.). Масс-спектр, m/z: 388, [М]+. Спектр ЯМР 1Н (ДМСО-d6, 1,2-бис(2,4-диметил-1-бензотиофен-3-ил)циклопентена 4d: Получали 2,82 г продукта (4d) (61%), Тпл=171-173°С (гексан: хлороформ, 6:1, об.). Масс-спектр, m/z: 388, [М]+. Спектр ЯМР 1Н (ДМСО-d6, 1,2-бис(2,6-диметил-1-бензотиофен-3-ил)циклопентена 4е: Получали 3,0 г продукта (4е) (65%), Тпл=184-186°С (Масс-спектр, m/z: 388, [М]+. Спектр ЯМР 1Н (ДМСО-d6, Общая методика синтеза 1,2-бис(формил-1-бензотиофен-3-ил)циклопентенов 5а-е К перемешиваемому раствору 3,33 ммоль 1,2-бис(диметил-1-бензотиофен-3-ил)циклопентена 4а-е в нитробензоле (25 мл) при 0°С добавляли 50 ммоль дихлорметилового эфира и 13,35 ммоль безводного хлористого алюминия, перемешивали 30 мин при 0°С и 20 часов при комнатной температуре. Реакционную смесь выливали в лед с водой и продукт экстрагировали этилацетатом, промывали водой, сушили сульфатом магния. После отгонки нитробензола под вакуумом продукт очищали с помощью колоночной хроматографии (Silica Gel, 0,063-0,1), элюент – петролейный эфир (40/70): этилацетат (6:1, об.). 1,2-бис(2-метил-6-формил-1-бензотиофен-3-ил)циклопентен 5а: Получали 0,56 г диальдегида (5а) (40%), Тпл=196-197°С (гексан: хлороформ, 6:1, об.). Масс-спектр, m/z: 416, [М]+. Спектр ЯМР 1Н (ДМСО-d6, Полученное соединение 5а растворяли в толуоле в концентрации С=2·10-4 М. Спектры поглощения форм А и В измеряли на спектрофотометре Сагу 50 (Varian) сразу после растворения (фиг.4, кривая 1) и после облучения УФ светом ртутной лампы ДРШ-250 через стеклянный светофильтр УФС-2 из стандартного набора стеклянных оптических светофильтров (фиг.4, кривые 2-7). Фотохромный мономер характеризуется высокой обратимостью (цикличностью) фотохромных превращений, о чем свидетельствует наличие изобестической точки на кривых поглощения (фиг.4). Затем измеряли изменение оптической плотности (кинетику) раствора в максимуме полосы поглощения циклической формы В под действием того же самого УФ света (процесс фотоокрашивания) (фиг.5, кривая 1) и после достижения состояния равновесия под действием видимого излучения того же источника света, пропущенного через стеклянный светофильтр ЖС-12 (процесс фотообесцвечивания) (фиг.5, кривая 2). Из спектрально-кинетических данных видно, что соединение обладает приемлемыми для практического использования фотохромными свойствами. 1,2-бис(2,5-диметил-6-формил-1-бензотиофен-3-ил)циклопентен 5b: Получали 0,71 г диальдегида (5b) (48%), Тпл=181-183°С (гексан: хлороформ, 6:1, об.). Масс-спектр, m/z: AAA, [М]+. Спектр ЯМР 1Н (ДМСО-d6, 1,2-бис(2,7-диметил-6-формил-1-бензотиофен-3-ил)циклопентен 5с: Получали 0,96 г диальдегида (5с) (65%), Тпл=163-165°С (гексан: хлороформ, 6:1, об.). Масс-спектр, m/z: AAA, [М]+. Спектр ЯМР 1Н (ДМСО-d6, 1,2-бис(2,4-диметил-5-формил-1-бензотиофен-3-ил)циклопентен 5d: Получали 1,05 г диальдегида (5d) (68%), Тпл=187-189°С (гексан: хлороформ, 6:1, об.). Масс-спектр, m/z: AAA, [М]+. Спектр ЯМР 1Н (ДМСО-d6, 1,2-бис(2,6-диметил-5-формил-1-бензотиофен-3-ил)циклопентен 5е: Получали 0,77 г диальдегида (5е) (52%), Тпл=201-203°С (гексан: хлороформ, 6:1, об.). Масс-спектр, m/z: AAA, [М]+. Спектр ЯМР 1Н (ДМСО-d6, Пример 2 1,2-бис(2-децил-6-формил-1-бензотиофен-3-ил)циклопентен 10 получали аналогично 1,2-бис(2-метил-6-формил-1-бензотиофен-3-ил)циклопентену 5а (см. Пример 1 в основных материалах заявки) по схеме, представленной на фиг.6, с использованием в качестве исходного соединения 2-децилбензотиофен 6. Целевой 1,2-бис(2-децил-6-формил-1-бензотиофен-3-ил)циклопентен 10 получали в виде аморфного порошка. Масс-спектр, m/z: 668, [М]+. Спектр ЯМР 1Н (ДМСО-d6, Пример 3 Синтез 1,2-бис(2-метил-6-карбокси-1-бензотиофен-3-ил)циклопентена 12 осуществляли по схеме, представленной на фиг.7. Синтез дикетоэфира 11 К раствору 0,75 г (2,1 ммоль) соединения 4 в 15 мл CH2Cl2 добавили 0,75 г (5,6 ммоль) AlCl3, реакционную смесь охладили до температуре 5°С и затем прикапали 5,5 ммоль хлорангидрида VI. Перемешивали смесь при температуре 5-15°С в течение 3 часов, затем прилили 50 мл воды и экстрагировали 3×50 мл этилацетата. Органический слой сушили над MgSO4 и затем упарили. Остаток чистили на хроматографической колонке (4:1 петролейный эфир – этилацетат). Выход соединения 11 0,45 г (39%). Тпл=158-162°С. Синтез дикислоты 12 К раствору 0,09 г (0,16 ммоль) соединения 11 в 3 мл диоксана добавил 0,035 г (0,8 ммоль) NaOH в 3 мл воды и 0,5 мл Н2О2 (30% раствор в Н2О). Реакционную смесь перемешивал в течение 10 часов, затем прилил 10 мл воды и подкислил раствор до рН 4 разбавленной серной кислотой. Выпавший аморфный осадок отфильтровал и посушил. Выход соединения 12 72%. Масс-спектр, m/z: 448, [М]+. Спектр ЯМР 1H (ДМСО-d6, Пример 4 Синтез 1,2-бис(2-метил-6-метилокси-1-бензотиофен-3-ил)циклопентена 13 осуществляли по схеме, представленной на фиг.8. Восстановление диальдегида 5 проводили по стандартной методике NaBH4 в метаноле. Выход соединения 13 52%. Масс-спектр, m/z: 420, [М]+. Спектр ЯМР 1Н (ДМСО-d6, Пример 5 Синтез 1,2-бис(2-метил-6-хлорметил-1-бензотиофен-3-ил)циклопентена 14 осуществляли по схеме, представленной на фиг.9. Получение дихлорида 14 проводили по стандартной методике в двухфазной системе хлороформ – 32% соляная кислота. Выход 67%. Спектр ЯМР 1Н (ДМСО-d6, Пример 6 Фотохромный 1,2-бис(2-метил-5-формил-4Н-тиено[3,2-b]пиррол-3-ил)циклопентен 21 из класса использованных фотохромных мономеров синтезировали по схеме, представленной на фиг.10. Метиловый эфир 2-азидо-3-(5′-метил-2′-тиенил)акриловой кислоты 16. К смеси метилата натрия, приготовленного из 1,8 г (78,3 ммоль) Na и 30 мл абс. метанола и 22,8 г (0,2 моль) метилового эфира азидоуксусной кислоты при перемешивании и температуре -5-0°С прибавляли 30 ммоль метилтиофенкарбоксальдегида 15 [Метилтиофенкарбоксальдегид 15 синтезировали по методике, описанной в Organic Syntheses, Coll. Vol.4, p.915 (1963); Vol.31, p.108 (1951)]. Смесь перемешивали при 0°С 30 минут и 2 часа при комнатной температуре. Добавляли водный раствор насыщенного NH4CI и перемешивали 10 минут. Выпавший осадок отфильтровывают, сушат. Выход соединения 16 76%, Тпл=51°С (с разл.). Масс-спектр, m/z: 223, [М]+. Спектр ЯМР 1Н (CDCl3, Метиловый эфир 2-метт-4Н-тиено[3,2-b]пиррол-5-карбоновой кислоты 17. Раствор 30 ммоль метилового эфира 2-азидо-3-(тиенил-2′)-акриловой кислоты 16 в 50 мл толуола кипятили в течение 3 часов. Выпавший осадок отфильтровывают, маточник упаривали под вакуумом и перекристаллизовывали из толуола. Осадки объединяют. Выход соединения 17 95%, Тпл=189-190°С (из толуола). Масс-спектр, m/z: 195, [М]+. Спектр ЯМР 1Н (CDCl3, 1,5-бис-(2-метил-5-метоксикарбонил-4Н-тиено[3,2-b]пиррол-3-ил)пентан-1,5-дион 18. К хорошо перемешиваемой смеси 20,2 ммоль тиенопиррола 17 и 10,1 ммоль дихлорангидрида глутаровой кислоты в 50 мл хлористого метилена прибавляли при 0°С 42,5 ммоль безводного хлористого алюминия. Реакционную массу перемешивали при комнатной температуре 6 часов. Выливали на лед с водой, экстрагировали хлористым метиленом (3×100 мл), собранные органические слои промывали водным раствором NaHCO3, сушили над сульфатом магния, растворитель упаривали под вакуумом. Остаток перекристаллизовывали из гексана. Выход соединения 18 82%, Тпл=175-177°С (гексан: хлороформ, 6:1, об.). Масс-спектр, m/z: 486, [М]+. Спектр ЯМР 1Н (ДМСО-d6, 1,2-бис(2-метил-5-метоксикарбонил-4Н-тиено[3,2-b]пиррол-3-ил)циклопентен 19. К хорошо перемешиваемой суспензии 7,93 ммоль цинка в свежеперегнанном безводном THF (50 мл) при -10°С под аргоном добавляли по каплям 2,8 мл TiCl4. После окончания прибавления реакционную смесь нагревали под аргоном 1 час. Охлаждали до 20°С и прибавляли 12,8 ммоль дикетона 18 и безводный пиридин (5 мл). Кипятили под аргоном еще 20 часов. Выливали в 10% К2CO3 (150 мл) и водный слой экстрагировали Et2О (5×100 мл). Собранные органические экстракты сушили MgSO4 и отгоняли под вакуумом. Остаток чистили с помощью флэш-хроматографии на силикагеле (Merck, 0,063-0,1), элюент – петролейный эфир (40/70) – AcOEt (10:1, об.). Выход соединения 19 60%, Тпл=196,5-197,5°С. Масс-спектр, m/z: 454, [М]+. Спектр ЯМР 1Н (ДМСО-d6, 1,2-бис(2-метил-5-оксиметил-4Н-тиено[3,2-b]пиррол-3-ил)циклопентен 20. К хорошо перемешиваемому раствору соединения 19 (12 ммоль) в 5 мл эфира добавили LiAlH4 (10 ммоль) и реакционную смесь перемешивали в течение 1 часа. При охлаждении добавили воды и отделили эфирный слой. Растворитель отогнали, полученный продукт чистили с помощью флэш-хроматографии на силикагеле (Merck, 0,063-0,1), элюент – петролейный эфир (40/70) – AcOEt (10:1, об.). Выход соединения 19 59%, Тпл=166,5-167,5°С. Масс-спектр, m/z: 398, [М]+. Спектр ЯМР 1Н (ДМСО-d6, 1,2-бис(2-метил-5-формил-4Н-тиено[3,2-b]пиррол-3-ил)циклопентен 21. К хорошо перемешиваемому раствору соединения 20 (12 ммоль) в 20 мл хлористого метилена добавили комплекс хромового ангидрида с двумя молекулами пиридина (72 ммоль) и реакционную смесь перемешивали в течение 30 минут при комнатной температуре. Растворитель отогнали, полученный продукт чистили с помощью флэш-хроматографии на силикагеле (Merck, 0,063-0,1), элюент – петролейный эфир (40/70) – AcOEt (10:1, об.). Выход соединения 21 28% в виде вязкого масла. Масс-спектр, m/z: 394, [М]+. Спектр ЯМР 1Н (ДМСО-d6, Пример 7 1,2-бис[N-метил(2-метил-5-формил-4Н-тиено[3,2-b]пиррол-3-ил)]циклопентен 24 синтезировали по схеме, приведенной на фиг.11. 1,2-бис[N-метил(2-метил-5-метоксикарбонил-4Н-тиено[3,2-b]пиррол-3-ил)]циклопентен 22 получили по стандартной методике метилированием соединения 19 йодистым метилом. Масс-спектр, m/z: 482, [М]+. Спектр ЯМР 1Н (ДМСО-d6, 1,2-бис(2-метил-5-оксиметил-4Н-тиено[3,2-b]пиррол-3-ил)циклопентен 23. К хорошо перемешиваемому раствору соединения 22 (12 ммоль) в 5 мл эфира добавили LiAlH4 (10 ммоль) и реакционную смесь перемешивали в течение 1 часа. При охлаждении добавили воды и отделили эфирный слой. Растворитель отогнали, полученный продукт чистили с помощью флэш-хроматографии на силикагеле (Merck, 0,063-0,1), элюент – петролейный эфир (40/70) – AcOEt (10:1, об.). Выход соединения 23 43% с Тпл=155-157,5°С. Масс-спектр, m/z: 426, [М]+. Спектр ЯМР 1Н (ДМСО-d6, 1,2-бис(2-метил-5-формил-4Н-тиено[3,2-b]пиррол-3-ил)циклопентен 24. К хорошо перемешиваемому раствору соединения 23 (12 ммоль) в 20 мл хлористого метилена добавили комплекс хромового ангидрида с двумя молекулами пиридина (72 ммоль) и реакционную смесь перемешивали в течение 30 минут при комнатной температуре. Растворитель отогнали, полученный продукт чистили с помощью флэш-хроматографии на силикагеле (Merck, 0,063-0,1), элюент – петролейный эфир (40/70) – AcOEt (10:1, об.). Выход соединения 24 32% в виде вязкого масла. Масс-спектр, m/z, 422, [М]+. Спектр ЯМР 1Н (ДМСО-d6, Пример 8 Фотохромный 1,2-бис(2-метил-5-формилтиено[3,2-b]тиофен-3-ил)циклопентен 29 из класса I использованных фотохромных мономеров синтезировали по схеме, представленной на фиг.12. 1,5-бис-(2-метил-5-метоксикарбонил-4Н-тиено[3,2-b]тиофен-3-ил)пентан-1,5-дион 26 К хорошо перемешиваемой смеси 20,2 ммоль 2-метил-5-метоксикарбонил-4Н-тиено[3,2-b]тиофена 25 и 10,1 ммоль дихлорангидрида глутаровой кислоты в 50 мл хлористого метилена прибавляли при 0°С 42,5 ммоль безводного хлористого алюминия. Реакционную массу перемешивали при комнатной температуре 6 часов. Выливали на лед с водой, экстрагировали хлористым метиленом (3×100 мл), собранные органические слои промывали водным раствором NaHCO3, сушили над сульфатом магния, растворитель упаривали под вакуумом. Остаток перекристаллизовывали из гексана. Выход соединения 26 82%, Тпл=165-167°С (гексан: хлороформ, 6:1, об.). Масс-спектр, m/z, 520, [М]+. Спектр ЯМР 1Н (ДМСО-d6, 1,2-бис(2-метил-5-метоксикарбонил-4Н-тиено[3,2-b]тиофен-3-ил)циклопентен 27 К хорошо перемешиваемой суспензии 7,93 ммоль цинка в свежеперегнанном безводном THF (50 мл) при -10°С под аргоном добавляли по каплям 2,8 мл TiCl4. После окончания прибавления реакционную смесь нагревали под аргоном 1 час. Охлаждали до 20°С и прибавляли 12,8 ммоль дикетона 26 и безводный пиридин (5 мл). Кипятили под аргоном еще 20 часов. Выливали в 10% К2CO3 (150 мл) и водный слой экстрагировали Et2O (5×100 мл). Собранные органические экстракты сушили MgSO4 и отгоняли под вакуумом. Остаток чистили с помощью флэш-хроматографии на силикагеле (Merck, 0,063-0,1), элюент – петролейный эфир (40/70) – AcOEt (10:1, об.). Выход соединения 27 60%, Тпл=177,5-176,5°С. Масс-спектр, m/z, 488, [М]+. Спектр ЯМР 1Н (ДМСО-d6, 1,2-бис(2-метил-5-оксиметил-4Н-тиено[3,2-b]тиофен-3-ил)циклопентен 28 К хорошо перемешиваемому раствору соединения 27 (12 ммоль) в 5 мл эфира добавили LiAlH4 (10 ммоль) и реакционную смесь перемешивали в течение 1 часа. При охлаждении добавили воды и отделили эфирный слой. Растворитель отогнали, полученный продукт чистили с помощью флэш-хроматографии на силикагеле (Merck, 0,063-0,1), элюент – петролейный эфир (40/70) – AcOEt (10:1, об.). Выход соединения 28 59% в виде вязкого масла. Масс-спектр, m/z, 432, [М]+. Спектр ЯМР 1Н (ДМСО-d6, 1,2-бис(2-метил-5-формил-4Н-тиено[3,2-b]тиофен-3-ил)циклопентен 29 К хорошо перемешиваемому раствору соединения 28 (12 ммоль) в 20 мл хлористого метилена добавили комплекс хромового ангидрида с двумя молекулами пиридина (72 ммоль) и реакционную смесь перемешивали в течение 30 минут при комнатной температуре. Растворитель отогнали, полученный продукт чистили с помощью флэш-хроматографии на силикагеле (Merck, 0,063-0,1), элюент – петролейный эфир (40/70) – AcOEt (10:1, об.). Выход соединения 29 28% в виде вязкого масла. Масс-спектр, m/z, 428, [М]+. Спектр ЯМР 1Н (ДМСО-d6, Пример 9 Фотохромный 1,2-бис(3,5-диметил-2-формилтиено[3,2-b]фуран-6-ил)циклопентен 35 из класса использованных фотохромных мономеров синтезировали по схеме, представленной на фиг.13. Этил 3,5-диметилтиено[3,2-b]фуран-2-карбоксилат 31 3-Гидроокси-5-метилтиофен 30 был получен из 2-метил-4-бром-тиофена по методике, описанной в Organic Syntheses, Coll. Vol.5, p.642 (1973); Vol.43, p.55 (1963). К раствору гидрокситиофена 18 (1,5 ммоль) в 5 мл бензола добавили 1 ммоль NaH и к нагретой до кипения реакционной смеси добавили 1,5 ммоль этилхлорацетоацетата. Смесь кипятили в течение 4 часов, охладили, промыли водой, органический слой сушили над MgSO4. Растворитель отогнали, полученное вещество добавили при перемешивании в течение 1 часа к 3 мл концентрированной серной кислоте при температуре 5°С. Смесь перемешивали еще 1 час. Затем добавили смесь воды и льда и экстрагировали 2 раза по 5 мл бензола. Органический слой промыли водой, раствором карбоната натрия, сушили над MgSO4 и удалили, продукт выделяли на колонке с силикагелем (элюент CHCl3). Выход соединения 31 48%, Тпл=120-121°С. Масс-спектр, m/z: 224, [М]+. Спектр ЯМР 1Н (ДМСО-d6, 1,5-бис(3,5-диметил-2-этоксикарбонилтиено[3,2-b]фуран-6-ил)пентан-1,5-дион 32 К хорошо перемешиваемой смеси 20,2 ммоль тиенофурана 31 и 10,1 ммоль дихлорангидрида глутаровой кислоты в 50 мл хлористого метилена прибавляли при 0°С 42,5 ммоль безводного хлористого алюминия. Реакционную массу перемешивали при комнатной температуре 6 часов. Выливали на лед с водой, экстрагировали хлористым метиленом (3×100 мл), собранные органические слои промывали водным раствором NaHCO3, сушили над сульфатом магния, растворитель упаривали под вакуумом. Остаток перекристаллизовывали из гексана. Выход соединения 32 82%, Тпл=169-170°С (гексан: хлороформ, 6:1, об.). Масс-спектр, m/z: 544, [М]+. Спектр ЯМР 1Н (ДМСО-d6, 1,2-бис(3,5-диметил-2-этоксикарбонилтиено[3,2-b]фуран-6-ил)циклопентен 33 К хорошо перемешиваемой суспензии 7,93 ммоль цинка в свежеперегнанном безводном THF (50 мл) при -10°С под аргоном добавляли по каплям 2,8 мл TiCl4. После окончания прибавления реакционную смесь нагревали под аргоном 1 час. Охлаждали до 20°С и прибавляли 12,8 ммоль дикетона 32 и безводный пиридин (5 мл). Кипятили под аргоном еще 20 часов. Выливали в 10% К2СО3 (150 мл) и водный слой экстрагировали Et2O (5×100 мл). Собранные органические экстракты сушили MgSO4 и отгоняли под вакуумом. Остаток чистили с помощью флэш-хроматографии на силикагеле (Merck, 0,063-0,1), элюент – петролейный эфир (40/70) – AcOEt (10:1, об.). Выход соединения 33 62%, Тпл=198,5-199,5°С. Масс-спектр, m/z: 512, [М]+. Спектр ЯМР 1Н (ДМСО-d6, 1,2-бис(3,5-диметил-2-оксиметилтиено[3,2-b] фуран-6-ил)циклопентен 34 К хорошо перемешиваемому раствору соединения 33 (12 ммоль) в 5 мл эфира добавили LiAlH4 (10 ммоль) и реакционную смесь перемешивали в течение 1 часа. При охлаждении добавили воды и отделили эфирный слой. Растворитель отогнали, полученный продукт чистили с помощью флэш-хроматографии на силикагеле (Merck, 0,063-0,1), элюент – петролейный эфир (40/70) – AcOEt (10:1, об.). Выход соединения 34 59%, Тпл=168,5-169,5°С. Масс-спектр, m/z: 428, [М]+. Спектр ЯМР 1Н (DMSO-d6, 1,2-бис(3,5-диметил-2-формилтиено[3,2-b]фуран-6-ил)циклопентен 35 К хорошо перемешиваемому раствору соединения 34 (12 ммоль) в 20 мл хлористого метилена добавили комплекс хромового ангидрида с двумя молекулами пиридина (72 ммоль) и реакционную смесь перемешивали в течение 30 минут при комнатной температуре. Растворитель отогнали, полученный продукт чистили с помощью флэш-хроматографии на силикагеле (Merck, 0,063-0,1), элюент – петролейный эфир (40/70) – AcOEt (10:1, об.). Выход соединения 35 28% в виде вязкого масла. Масс-спектр, m/z: 424, [М]+. Спектр ЯМР 1Н (ДМСО-d6, Пример 10 Фотохромный мономер 3-аллил-4,5-бис(2,5-диметил-3-тиенил)-1,3-оксазол-2-она 38 из класса II получали по схеме, представленной на фиг.14. Методика синтеза 3-аллил-4,5-бис(2,5-диметил-3-тиенил)-4-гидрокси-1,3-оксазолан-2-она 37 К раствору 1,0 г (3,27 ммоль) карбоната 10 в 10 мл этанола добавили 0,3 г (5,00 ммоль) аллиламина и перемешивали при 25°С 1-1,5 часа. Выпавший осадок отфильтровали и кристаллизовали продукт из этанола. Получали 1,1 г оксазолана 37 (93%), Тпл=181-182°С. Найдено (%): С – 59,54; Н – 5,71; S – 17,55. C18H21NO3S2. Вычислено (%): С – 59,48; Н – 5,82; S – 17,64. Спектр ЯМР 1Н (ДМСО-d6, Методика синтеза 3-аллил-4,5-бис(2,5-диметил-3-тиенил)-1,3-оксазол-2-она 38 Раствор 0,9 г (2,5 ммоль) 1,3-оксазолан-2-она 37 в 25 мл (0,325 моль) CF3COOH перемешивали при 25°С 3-3,5 часов. Упарили растворитель в вакууме и кристаллизовали остаток из этанола. Получили 0,63 г оксазола 38 (73%), Тпл=103-104°С. Найдено (%): С – 62,43; Н – 5,62; S – 18,78. C18H19NO2S2. Вычислено (%): С – 62,58; Н – 5,54; S – 18,56. Спектр ЯМР 1Н (ДМСО-d6, По методике, описанной в примере 1, измеряли аналогичные спектральные (фиг.15) и кинетические (фиг.16) характеристики, свидетельствующие о фотохромных свойствах фотохромного мономера. Пример 11 По методике, описанной в примере 10, из карбоната 39 получили оксазол 41 (Фиг.17). Пример 12 По методике, описанной в примере 10, из карбоната 42 получили оксазол 44 (Фиг.18). Пример 13 По методике, описанной в примере 10, из карбоната 45 получили оксазол 47 (Фиг.19). Пример 14 По методике, описанной в примере 10, из карбоната 48 получили оксазол 50 (Фиг.20). Пример 15 По методике, описанной в примере 10, из тиокарбоната 51 получили оксазол 53 (Фиг.21). Пример 16 По методике, описанной в примере 10, из тиокарбоната 54 получили оксазол 56 (Фиг.22). Пример 17 Фотохромный мономер 1,2-бис(5-гидроксиметил-2-метилтиен-3-ил)гексафторпентена 60 синтезировали по схеме, приведенной на фиг.23. Данный способ отличается тем, что в синтезе 1,2-бис(5-гидроксиметил-2-алкилтиофен-3-ил)гексафторпентена 60 в качестве защитной группы использовался более доступный 1,2-дигидропиран, тогда как в литературе применялась труднодоступная трет-бутилдиметилсилильная защита (Chem. Letters, 1998, p.1093-1094). Методика синтеза 2-[(4-бром-5-метилтиофен-2-ил)метокси]тетрагидро-2Н-пирана 58 К 5,4 г 4-бром-2-гидроксиметил-5-метилтиофена 57 в 40 мл метанола прибавляли 2,6 мл 1,2-дигидропирана и 0,2 г п-толуолсульфокислоты. Реакционную смесь перемешивали при комнатной температуре 24 часа. Метанол отгоняли под вакуумом, остаток перегоняли на масляном насосе. Получали 6,82 г продукта 58 (90%) с Ткип=125-130°С (5 мм рт.ст.), nd 16‘5=1,5610. Масс-спектр, m/z: 291, [М]+. Спектр ЯМР 1Н (CDCl3, Методика синтеза 1,2-бис{5-метокси(тетрагидро-2Н-пирил-2)-2-метилтиен-3-ил}гексафторпентена 59 К 5,3 г 2-[(4-бром-5-метилтиофен-2-ил)метокси]тетрагидро-2Н-пирана в 30 мл безводного ТГФ при -78°С и под аргоном прибавляли 14 мл 1,6 М раствора бутиллития в гексане, реакционную смесь перемешивали при -78°С 10 минут и приливали 1,93 г октафторциклопентена. Реакционную смесь выдерживали при температуре -78°С 1 час и оставляли на ночь при комнатной температуре. Выливали в воду, экстрагировали эфиром, сушили над сульфатом магния. Растворитель отгоняли под вакуумом, образовавшееся масло хроматографировали. Получали 3,37 г маслообразного продукта 59 (62%). Масс-спектр, m/z: 596, [М]+. Спектр ЯМР 1Н (CDCl3, Методика синтеза 1,2-бис(5-гидроксиметил-2-метилтиен-3-ил)гексафторпентена 60 К 3 г 1,2-бис{5-метокси(тетрагидро-2Н-пирил-2)-2-метилтиен-3-ил}гексафторпентена 59 в 30 мл метанола прибавляли 3 мл концентрированной соляной кислоты. Реакционную смесь перемешивали при комнатной температуре 20 часов. Избыток метанола отгоняли под вакуумом, остаток перекристаллизовывали из гексана. Получали 1,62 г продукта 60 (75%) с Тпл=129-133°С (Тпл(литер.)=125-128°С). Масс-спектр, m/z: 428, [М]+. Спектр ЯМР 1Н (CDCl3, По методике, описанной в примере 1, измеряли аналогичные спектральные (фиг.24) и кинетические (фиг.25) характеристики, свидетельствующие о фотохромных свойствах фотохромного мономера. Пример 18 Фотохромный олигомер – олиго(Шиффово) основание IV получали по схеме, представленной на фиг.26. В двугорлую коническую колбу, снабженную системой для ввода и вывода аргона, ловушкой Дина-Старка, капельной воронкой, магнитной мешалкой и обратным холодильником, помещали 103,7 мг (0,2 ммоль) 2,2-бис[4-(3-аминофенокси)фенил]гексафторпропана, 83,3 мг (0,2 ммоль) мономера 5а, 2 мл ДМФА и 2 мл толуола. При перемешивании и сильном токе аргона колба была нагрета до 140°С. Реакционную смесь выдерживали при этой температуре в течение 4 часов, периодически прикапывая в общей сложности 2 мл толуола, при этом в ловушке Дина-Старка конденсируется азеоторп толуола с водой. Затем олигомер осаждали этанолом, отфильтровывали на фильтре Шотта, промывали этанолом, сушили. Выход составил 0,14 г (70%). Образец фотохромного полимера готовили путем совместного растворения полученного олигомера и полиметилметакрилата (10 мас.% от массы сухого полимера) в хлороформе. Затем измеряли спектрально-кинетические характеристики полученного образца по методике, описанной в примере 1. Спектральные (фиг.27) и кинетические (фиг.28) характеристики свидетельствуют о фотохромных свойствах синтезированного фотохромного олигомера. На фиг.29 показана цикличность окрашивания УФ светом через светофильтр УФС-8 и обесцвечивание через комбинацию двух светофильтров ПС-7 и ЖС-18 (периодическая смена фильтров). Из фиг.29 следует, что фотохромный полимер обладает высокой цикличностью без потери фотохромных свойств. Пример 19 Аналогично примеру 18 из мономера 21 получили полимер IVa (Фиг.30). Пример 20 Аналогично примеру 18 из мономера 29 получили полимер IVb (Фиг.31). Пример 21 Аналогично примеру 18 из мономера 35 получили полимер IVc (Фиг.32). Пример 22 Фотохромный сополимер V синтезирован из 3-аллил-4,5-бис-(2,5-диметил-3-тиенил)-1,3-оксазол-2-она (соединение 38) из класса II и метилметакрилата по схеме, представленной на фиг.33. Раствор 0,173 г (0,5 ммоль) 3-аллил-4,5-бис-(2,5-диметил-3-тиенил)-1,3-оксазол-2-она (соединения 38) в 1 г (10 ммоль) метилметакрилата помещали в ампулу с 0,0017 г (0,1 мас.% от суммы мономеров) инициатора – азоизобутиронитрила. Ампулу заполняли аргоном и нагревали при 60°С в течение 20 часов. Получали стеклообразный сополиметилметакрилат V Mw=80000. Сополимер растворим в хлороформе. Образец фотохромного полимера для измерения спектрально-кинетических характеристик готовили путем совместного растворения фотохромного полимера и поликарбоната, который использовался в качестве полимерного связующего, в хлороформе. Затем раствор наносился на кварцевую подложку методом центрифугирования. В результате была получена фотохромная пленка, для которой были измерены по методике примера 1 спектры поглощения формы А (фиг.34, кривая 1) и формы В (фиг.34, кривая 2), а также кинетические кривые фотоокрашивания (фиг.35, кривая 1) и фотообесцвечивания (фиг.35, кривая 2). Полученные спектрально-кинетические данные свидетельствуют о фотохромизме полученного образца фотохромной пленки. Пример 23 Фотохромный сополимер Va (фиг.36) получен из соединения 38 и бутилметилметакрилата аналогично примеру 22. Mw=87000. Полимер растворим в хлороформе. Спектрально-кинетические характеристики образца фотохромного полимера, изготовленного по методике, представленной в примере 22, свидетельствуют о практически приемлемом фотохромизме. Пример 24 Фотохромный сополимер Vb (фиг.37) получен из соединения 41 и бутилметилметакрилата аналогично примеру 22. Mw=72000. Полимер растворим в хлороформе. Спектрально-кинетические характеристики образца фотохромного полимера, изготовленного по методике, представленной в примере 22, свидетельствуют о практически приемлемом фотохромизме. Пример 25 Фотохромный сополимер Vc (фиг.38) получен из соединения 47 и бутилметилметакрилата аналогично примеру 22. Mw=85000. Полимер растворим в хлороформе. Спектрально-кинетические характеристики образца фотохромного полимера, изготовленного по методике, представленной в примере 22, свидетельствуют о практически приемлемом фотохромизме. Пример 26 Фотохромный сополимер Vd (фиг.39) получен из соединения 53 и бутилметилметакрилата аналогично примеру 15. Mw=76000. Полимер растворим в хлороформе. Спектрально-кинетические характеристики образца фотохромного полимера, изготовленного по методике, представленной в примере 22, свидетельствуют о практически приемлемом фотохромизме. Пример 27 Сложный полиэфир VII синтезировали из 1,2-бис(5-гидроксиметил-2-метилтиофен-3-ил)гексафторпентена (соединение 60) и дихлорангидрида терефталевой кислоты по схеме, представленной на фиг.40. В трехгорлую колбу емкостью 25 мл, снабженную мешалкой, обратным холодильником и вводом для аргона, помещали 0,27 г (0,63 ммоль) 1,2-бис(5-гидроксиметил-2-метилтиофен-3-ил)гексафторпентена (соединение 61), 0,128 г (0,63 ммоль) дихлорангидрида терефталевой кислоты (соединение VI), 3,2 мл дихлорэтана. Температуру реакционной смеси доводили до 40°С и медленно, в течение нескольких минут в реакционную смесь прикапывали 0,154 мл пиридина. Продолжительность реакции составляла 2 часа. Затем реакционную смесь выливали в метанол, осадок сушили под вакуумом в течение 24 часов при 40°С. Выход полимера VIII составил 98%. ИК (KBr, см-1): С=O 1710. Mn=35000. Полимер растворим в тетрагидрофуране, диметилформамиде, этаноле, ацетоне, при нагревании – в хлороформе, не растворим в толуоле. Образец фотохромного полимера для измерения спектрально-кинетических характеристик готовили путем совместного растворения фотохромного полимера и поликарбоната, который использовался в качестве полимерного связующего, в хлороформе. Затем раствор наносили на кварцевую подложку методом центрифугирования. В результате была получена фотохромная пленка, для которой были измерены по методике примера 1 спектры поглощения формы А (фиг.41, кривая 1) и формы В (фиг.41, кривая 2), а также кинетические кривые фотоокрашивания (фиг.42, кривая 1) и фотообесцвечивания (фиг.42, кривая 2). Полученные спектрально-кинетические данные свидетельствуют о фотохромизме полученного образца фотохромной пленки. Пример 28 Сложный полиэфир VIIa (фиг.43) получали из соединения V и дихлорангидрида изофталевой кислоты аналогично примеру 22. Выход составил 93%. Mn=28000. Полимер растворим в хлороформе, тетрагидрофуране, в диметилформамиде, толуоле, этаноле и ацетоне. Спектрально-кинетические характеристики образца фотохромного полимера, изготовленного по методике, представленной в примере 22, свидетельствуют о практически приемлемом фотохромизме. Пример 29 Сложный полиэфир VIIb (фиг.44) получали из соединения 61 и дихлорангидрида 4,4′-дифенил дикабоновой кислоты аналогично примеру 22. Выход составил 89%. Mn=22000. Полимер растворим в хлороформе, тетрагидрофуране, в диметилформамиде, не растворим в толуоле, этаноле и ацетоне. Спектрально-кинетические характеристики образца фотохромного полимера, изготовленного по методике, представленной в примере 5, свидетельствуют о практически приемлемом фотохромизме. Пример 30 Сложный полиэфир VIIc (фиг.21) получали из соединения V и дихлорангидрида 4,4′-дифенилоксид дикабоновой кислоты аналогично примеру 22. Выход составил 91%. Mn=24000. Полимер растворим в хлороформе, тетрагидрофуране, диметилформамиде, толуоле, этаноле и ацетоне. Спектрально-кинетические характеристики образца фотохромного полимера, изготовленного по методике, представленной в примере 22, свидетельствуют о практически приемлемом фотохромизме. Пример 31 Сложный полиэфир VIId (фиг.46) получали из соединения 61 и дихлорангидрида 4,4′-дифенилметан дикабоновой кислоты аналогично примеру 22. Выход составил 96%. Mn=30000. Полимер растворим в хлороформе, тетрагидрофуране, диметилформамиде, толуоле, этаноле и ацетоне. Спектрально-кинетические характеристики образца фотохромного полимера, изготовленного по методике, представленной в примере 22, свидетельствуют о практически приемлемом фотохромизме.
Формула изобретения
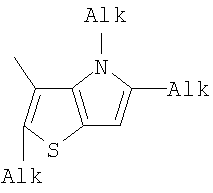
1. Фотохромные мономеры общей формулы
Q= Alk=CH3-С10H21; X=Cl, Br, I, F, NH2, CH2OH, CH2Cl, CH2Br, CHO, CO2H. 2. Способ получения фотохромных мономеров по п.1, включающий ацилирование производных бензотиофена, а также пироллотиофена, тиенотиофена или фуранотиофена дихлорангидридом глутаровой кислоты в хлористом метилене в присутствии хлористого алюминия и последующую восстановительную циклизацию полученных дикетонов под действием TiCl4, Zn в ТГФ в присутствии пиридина с дальнейшим формилированием продуктов реакции дихлорметиловым эфиром в нитробензоле в присутствии хлористого алюминия. 3. Фотохромные мономеры общей формулы
X=CH2, O, S, NAlk; Y=O, S, NAlk; n=0-6; Q= Alk=CH3-C10H21. 4. Способ получения фотохромных мономеров по п.3, включающий обработку производных 1,3-оксазолан-2-она и тиофена, а также бензотиофена, пироллотиофена, тиенотиофена или фуранотиофена аллиламином и дальнейшую обработку продуктов реакции трифторуксусной кислотой. 5. Фотохромные мономеры общей формулы
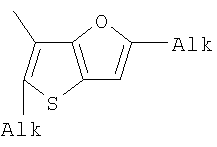
Alk=CH3-C10H21. 6. Способ получения фотохромных мономеров по п.5, включающий обработку тетрагидропиранильного производного бромтиофена булитлитием и последующее взаимодействие с перфторциклопентеном с дальнейшим гидролизам продукта реакции при действии соляной кислоты. 7. Фотохромный сополимер-олиго(Шиффово) основание на основе фотохромных мономеров по п.1
8. Способ получения фотохромного олигомера по п.7, включающий взаимодействие 2,2-бис[4-(3-аминофенокси)фенил]гексафторпропана с диальдегидным производным 1,2-циклопентена при повышенной температуре с одновременной отгонкой воды в виде азеотропа. 9. Конденсационные и полимеризационные сложные фотохромные полиэфиры на основе фотохромных мономеров по п.3. 10. Способ получения фотохромных полимеров по п.9, включающий взаимодействие аллильного производного 1,3-оксазалон-2-она с производным акриловой кислоты при повышенной температуре в присутствии инициатора. 11. Конденсационные и полимеризационные сложные фотохромные полиэфиры на основе фотохромных мономеров по п.5. 12. Способ получения фотохромных полимеров по п.11, включающий взаимодействие 1,2-бис(5-гидроксиметил-2-метилтиофен-3-ил)гексафторциклопентана с дихлорангидридом кислоты при повышенной температуре в присутствии пиридина. 13. Применение фотохромного олигомера по п.7 в полимерном связующем в качестве двухфотонной регистрирующей среды для трехмерной оптической памяти и фотопереключателей оптических сигналов. 14. Применение фотохромных полимеров по п.9 в полимерном связующем в качестве двухфотонной регистрирующей среды для трехмерной оптической памяти и фотопереключателей оптических сигналов. 15. Применение фотохромных полимеров по п.11 в полимерном связующем в качестве двухфотонной регистрирующей среды для трехмерной оптической памяти и фотопереключателей оптических сигналов.
РИСУНКИ
|
||||||||||||||||||||||||||
 ;
;  ;
;  ;
; 
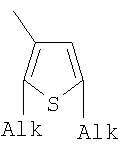 ;
;  ;
;  ;
;  ;
;  ;
; 
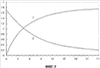
 =0,5 (М.Irie. In: Organic Photochromic and Thermochromic Compounds. Eds. J.С.Crano and R.J.Guglielmetti, N.Y. and L., Plenum Press. 1999. V.1. p.207), и следовательно, светочувствительность фотохромной среды не достигает максимальных значений.
=0,5 (М.Irie. In: Organic Photochromic and Thermochromic Compounds. Eds. J.С.Crano and R.J.Guglielmetti, N.Y. and L., Plenum Press. 1999. V.1. p.207), и следовательно, светочувствительность фотохромной среды не достигает максимальных значений. n=0,0008) на длине волны излучения полупроводникового лазера (830 нм).
n=0,0008) на длине волны излучения полупроводникового лазера (830 нм). , м.д., J/Гц): 2,0-2,2 (м, 2Н, СН2), 2,62 (с, 6Н, 2×СН3), 3,1-3,2 (уш.м., 4Н, 2×СН2), 7,3-7,5 (уш.м., 4Н, 4×СНаром.), 7,85-8,2 (уш.м., 4Н, 4×СНаром.). Найдено (%): С – 70,42; Н – 5,16; S – 16,29. C23H20O2S2. Вычислено (%): С – 70,37; Н – 5,14; S – 16,34.
, м.д., J/Гц): 2,0-2,2 (м, 2Н, СН2), 2,62 (с, 6Н, 2×СН3), 3,1-3,2 (уш.м., 4Н, 2×СН2), 7,3-7,5 (уш.м., 4Н, 4×СНаром.), 7,85-8,2 (уш.м., 4Н, 4×СНаром.). Найдено (%): С – 70,42; Н – 5,16; S – 16,29. C23H20O2S2. Вычислено (%): С – 70,37; Н – 5,14; S – 16,34. /см-1: 2120 оч.с.(N3). Найдено (%): С – 48,52; Н – 4,20; N – 19,02; S – 14,81. C9H9N3O2S. Вычислено (%): С – 48,42; Н – 4,06; N – 18,82; S – 14,36.
/см-1: 2120 оч.с.(N3). Найдено (%): С – 48,52; Н – 4,20; N – 19,02; S – 14,81. C9H9N3O2S. Вычислено (%): С – 48,42; Н – 4,06; N – 18,82; S – 14,36.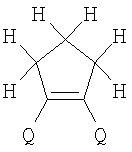
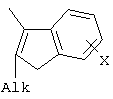 ;
;  ;
;  ;
; 
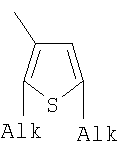 ;
; 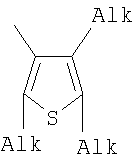 ;
; 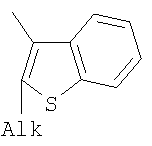 ;
; 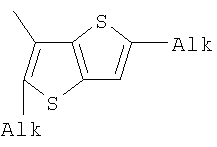 ;
;  ;
;