Патент на изобретение №2343157
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) АНАЛОГИ КОЛХИКОЗИДА
(57) Реферат:
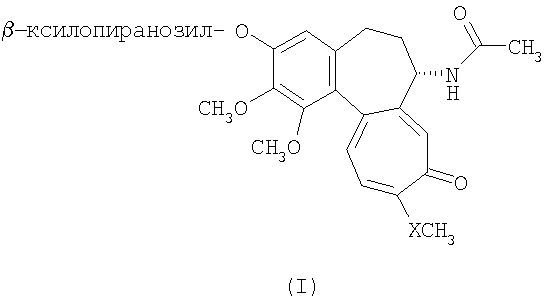
Настоящее изобретение относится к производным колхицина общей формулы (I), где X представляет собой кислород или серу, в частности – к 3-О-
Данное изобретение относится к производным колхицина, в частности к 3-деметил- и 3-деметилтио-производным колхицина, обладающим миорелаксантной, противовоспалительной и противоподагрической активностью. Уровень техники Расслабляющие лекарственные средства снижают мышечный тонус и используются в терапии для лечения контрактур и мышечного спазма. Мышечный спазм является одним из главных факторов, ответственных за хроническую боль; он характеризуется некоторыми патологиями опорно-двигательного аппарата, а также воспалительно-ревматическими и дегенеративными ортопедическими патологиями; при воздействии на суставы мышечный спазм помимо боли вызывает ригидность, которая снижает подвижность сустава и гибкость части, подверженной его действию. Поэтому исследование молекул, проявляющих миорелаксантные и спазмолитические свойства, по-прежнему представляет значительный интерес для клинического применения. Как известно, колхицин представляет собой псевдоалкалоид, который в течение определенного периода времени широко использовался для лечения подагры. Применение 3-деметилтиоколхицингликозида, тиоколхикозида также широко распространено в терапии для лечения контактур и воспалительных состояний, которые воздействуют на мышечную систему (Ortopedia e traumatologia Oggi XII, n. 4, 1992). Ранее было показано, что активность тиоколхикозида обусловлена его способностью взаимодействовать со стрихнин-чувствительными глициновыми рецепторами; следовательно, соединения, обладающие глицин-имитирующей активностью, могут применяться в ревматологической ортопедической области благодаря их миорелаксантным свойствам. Описание изобретения Данное изобретение относится к производным колхицина общей формулы (I):
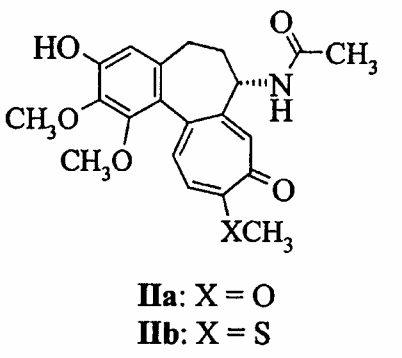
где Х представляет собой кислород или серу. В описании данного изобретения соединение, в котором Х представляет собой кислород, определено как (Ia), в то время как соединение формулы (I), в котором Х представляет собой серу, определено как (Ib). D- и L-изомеры включены в соединения формулы (I). D и L изомеры соединения (Ib), 3-О- Соединения данного изобретения получены взаимодействием D- или L-ксилопиранозилфторида с 3-О-деметилколхицином (IIa) и 3-О-деметилтиоколхицином (IIb)
в соответствии с общим способом, описанным в ЕР 0789028. Точнее, 3-О-деметилколхицин (IIa) или 3-О-деметилтиоколхицин (IIb) подвергается взаимодействию с D- или L-ксилопиранозилфторидом (III) или его защищенной формы, предпочтительно перацетатом. Реакция проводится в полярных апротонных растворителях, предпочтительно выбранных из ацетонитрила и хлорированных растворителей, при температурах в интервале от 0°С до температуры кипения растворителя, предпочтительно при комнатной температуре, в присутствии основания, предпочтительно 1,1,3,3-тетраметилгуанидина. Реакция обычно завершается в интервале времени от 10 минут до 2 часов. Гидролиз защитных групп может проводиться без выделения промежуточных продуктов. В частности, было показано, что Миорелаксантная активность оценивалась в “rota-rod” опыте. Самцы швейцарских мышей (Swiss male mice) массой 20-25 г обрабатывались интраперитонеально
Кроме того, соединение данного изобретения значительно менее токсично. Фактически DL50 данного соединения равно 80 (63-94) мг/кг и.п., в то время как DL50 тиоколхикозида равно 20 мг/кг. Данные результаты показывают, что соединение данного изобретения помимо более высокой активности обладает соотношением токсичность/активная доза, значительно более благоприятным по сравнению с тиоколхинозидом.
Соединения данного изобретения могут вводиться в фармацевтические препараты, предназначенные для перорального, внутривенного, внутримышечного, чрескожного и местного применения, в сочетании со стандартными наполнителями способами, описанными, например, в Remington’s Pharmaceutical Sciences Handbook, XVII Ed., Mack Pub., N.Y., U.S.A. Среди наполнителей, применяемых для приготовления липосомальных форм для парентерального или местного введения, особенно предпочтительными являются природные и синтетические фосфолипиды. Дозировки могут находиться в интервале от 5 до 50 мг в сутки в зависимости от заболевания и способа введения. Далее изобретение будет описано более подробно с помощью нескольких примеров. Экспериментальная часть Температуры плавления были измерены на аппарате Buchi 510. ЯМР спектры были записаны с помощью Bruker АС 200. Пример 1 – 3-О-(2′,3′,4′-О-триацетил- 3-О-Деметилтиоколхицин (IIb) (0,5 ммоль) и 2,3,4-О-триацетил- 1Н-ЯМР (CDCl3) – (ч./млн) 7,06 (NH, д, 7,4 Гц), 7,06 (H12, д, 10,3 Гц), 7,27 (Н11, д, 10,3 Гц), 7,33 (Н8, с), 6,71 (Н4, с), 4,71-4,55(Н7, м), 2,60-1,90 (Н5-Н6, м), 3,90 (2-OMe, с), 3,66 (1-ОМе, с), 2,44 (SMe, с), 2,00 (ацетамид), 5,28-5,18, 5,08-4,98 (Н1′, H2′, H3′, H4′, м), 4,30(Н5’a, ддд, 4,3, 7,0, 12,1, Гц), 3,58 (H5’b, ддд 4,3, 7,0, 12,1 Гц), 2,12 (ОАс), 2,11 (ОАс), 2,10 (ОАс). Пример 2 – 3-О-(2′,3′,4′-О-триацетил- 3-О-Деметилтиоколхицин (IIb) (0,5 ммоль) и 2,3,4-О-триацетил- 1Н-ЯМР (CDCl3) (ч./млн) 7,34 (NH, д, 7,9 Гц), 7,07 (H12, д, 10,7 Гц), 7,30 (Н11, д, 10,7 Гц), 7,37 (Н8, с), 6,71 (Н4, с), 4,71-4,55 (Н7, м), 2,60-1,80 (Н5-Н6, м), 3,88 (2-OMe, с), 3,64 (1-ОМе, с), 2,44 (SMe, с), 2,00 (ацетамид), 5,28-5,18 е 5,10-4,90 (Н1′, H2′, H3′, H4′, м), 4,25 (Н5’a, ддд, 4,3, 4,4, 12,1, Гц), 3,58 (H5’b, ддд 4,3, 4,4, 12,1 Гц), 2,14 (ОАс), 2,11 (ОАс), 2,10 (ОАс). Пример 3 – Общий способ удаления защиты в этаноле Сырой продукт (0,5 теоретического ммоля), полученный в примере 1 или 2, растворяют в этаноле (4 мл) и 1 н. NaOH (2 мл) при комнатной температуре. Ход реакции контролируют ТСХ. После исчезновения исходных веществ растворитель выпаривают и остаток очищают хроматографией на силикагеле. Продукт может быть далее кристаллизован из смеси метанол/изопропанол. Пример 4 – Общий способ удаления защиты в ацетоне Сырой продукт, полученный в примере 1 или 2 (1 теоретический моль), суспендируют с карбонатом калия в ацетоне (30 мл) и воде (10 мл). Смесь кипятят с обратным холодильником до исчезновения исходного вещества. Растворитель выпаривают и продукт очищают хроматографией. Продукт может быть далее кристаллизован из метанола и диизопропилового эфира. Пример 5 – 3-О- Продукт получают в соответствии со способом удаления защиты, описанным в примере 3 или 4, с 45% выходом и очищают хроматографией на силикагеле (элюирование с градиентом CH2Cl2/MeOH). Т. пл. 193°С; [ 1Н-ЯМР (CDCl3) ч./млн 8,64 (NH, д, 7,6 Гц), 7,15 (H12, д, 10,2 Гц), 7,28 (Н11, д, 10,6 Гц), 7,03 (Н8, с), 6,85 (Н4, с), 4,37-4,25 (Н7, м), 2,60-1,80 (Н5-Н6, м), 3,84 (2-OMe, с), 3,55 (1-ОМе, с), 2,42 (SMe, с), 1,86 (ацетамид), 4,97 (H1′, 6,6 Гц), 3,20-3,90 (Н2′, H3′, H4′, H5′, м) 4,40-5,60 (ОН). Пример 6 – 3-О- Продукт получают в соответствии со способом удаления защиты, описанным в примере 3 или 4, с 45% и очищают хроматографией на силикагеле (элюирование с градиентом CH2Cl2/MeOH). Т. пл. 220°С; [ 1Н-ЯМР (CDCl3) ч./млн 8,64 (NH, д, 7,3 Гц), 7,17 (H12, д, 10,2 Гц), 7,29 (Н11, д, 10,2 Гц), 7,03 (Н8, с), 6,87 (Н4, с), 4,23-4,41 (Н7, м), 2,70-1,90 (Н5-Н6, м), 3,84 (2-OMe, с), 3,55 (1-ОМе, с), 2,42 (SMe, с), 1,86 (ацетамид), 5,02 (H1′, 6,9 Гц), 3,20-3,90 (Н2′, H3′, H4′, H5′, м) 4,90-5,60 (ОН).
Формула изобретения
1. Соединения общей формулы (I)
где X представляет собой кислород или серу. 2. Соединение по п.1, где X представляет собой кислород. 3. Соединение по п.1, где X представляет собой серу. 4. Соединение, выбранное из 3-O- 3-О- 5. Применение соединения по любому из пп.1-4 для приготовления миорелаксантных лекарственных средств. 6. Применение соединения по любому из пп.1-4 для приготовления лекарственных средств для лечения воспалительных состояний, которые воздействуют на мышечную систему. 7. Применение соединения по любому из пп.1-4 для приготовления противоподагрических лекарственных средств. 8. Фармацевтические композиции для лечения воспалительных состояний, которые воздействуют на мышечную систему, содержащие соединение по любому из пп.1-4 в смеси с подходящими наполнителями и/или носителями. 9. Фармацевтические композиции по п.8 для местного применения. 10. Фармацевтические композиции по п.8 для парентерального применения. 11. Фармацевтические композиции по п.9 или 10, в которых наполнители выбраны из натуральных и синтетических фосфолипидов.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 -D-ксилопиранозил-3-О-деметилтиоколхицину и 3-О-
-D-ксилопиранозил-3-О-деметилтиоколхицину и 3-О-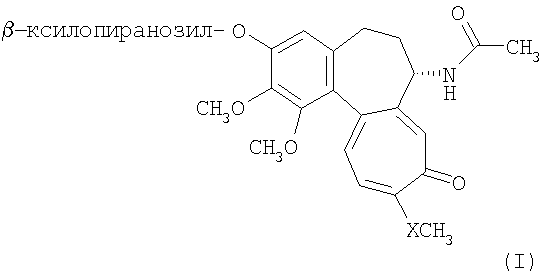
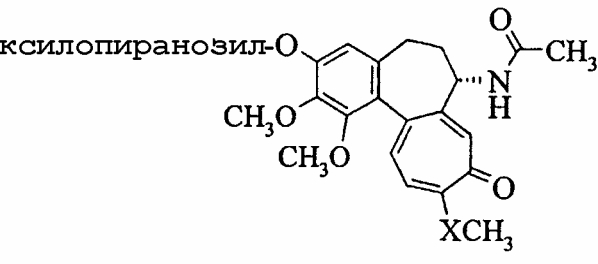
 -D-ксилопиранозилфторид (0,75 ммоль), полученный в соответствии с методикой публикации Hayashi et al., Chemistry Lett. 1984, 1747, при комнатной температуре в атмосфере азота при перемешивании суспендируют в сухом ацетонитриле (10 мл). К смеси добавляют тетраметилгуанидин (1,5 ммоль), полученная суспензия становится прозрачной и приобретает красную окраску. Добавляют этерат трифторида бора (4 ммоль), после чего раствор становится бесцветным. Ход реакции контролируют ТСХ (СН2Cl2:MeOH 9:1). После исчезновения исходных веществ (30 мин.) реакцию гасят добавлением насыщенного раствора гидрокарбоната натрия (10 мл). Фазы разделяют и водную фазу экстрагируют этилацетатом (3х10 мл). Объединенные органические фазы промывают насыщенным раствором гидросульфата калия (15 мл), раствором соли (15 мл) и сушат над сульфатом магния. Растворитель выпаривают и продукт очищают хроматографией на силикагеле. Альтернативно, из сырого продукта удаляют защитные группы.
-D-ксилопиранозилфторид (0,75 ммоль), полученный в соответствии с методикой публикации Hayashi et al., Chemistry Lett. 1984, 1747, при комнатной температуре в атмосфере азота при перемешивании суспендируют в сухом ацетонитриле (10 мл). К смеси добавляют тетраметилгуанидин (1,5 ммоль), полученная суспензия становится прозрачной и приобретает красную окраску. Добавляют этерат трифторида бора (4 ммоль), после чего раствор становится бесцветным. Ход реакции контролируют ТСХ (СН2Cl2:MeOH 9:1). После исчезновения исходных веществ (30 мин.) реакцию гасят добавлением насыщенного раствора гидрокарбоната натрия (10 мл). Фазы разделяют и водную фазу экстрагируют этилацетатом (3х10 мл). Объединенные органические фазы промывают насыщенным раствором гидросульфата калия (15 мл), раствором соли (15 мл) и сушат над сульфатом магния. Растворитель выпаривают и продукт очищают хроматографией на силикагеле. Альтернативно, из сырого продукта удаляют защитные группы.