Патент на изобретение №2162090
|
||||||||||||||||||||||||||
(54) ПРОТИВОПАРАЗИТАРНЫЕ МАРКФОРТИНЫ И ПАРАГЕРКВАМИДЫ
(57) Реферат: Описываются новые соединения – 15-алкил-14-гидроксисоединения формулы III, где n1 = 1 – 3. Они могут быть использованы в качестве противопаразитарных средств. 11 с. и 9 з.п. ф-лы, 15 ил. 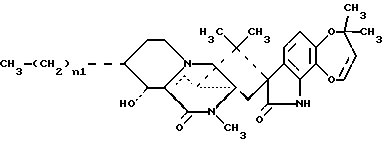
Изобретение относится к замещенным маркфортинам и парагерквамидам, которые могут быть использованы в качестве противопаразитарных средств. 2. Описание предшествующего уровня техники Маркфортины являются известными соединениями (см. Journal of the Chemical Society Chemical Communications, 601-602 (1980) для маркфортина A; и Tetrahedron Letters, 22, 1977-1980 (1981) для маркфортинов B и C). Эти соединения представляют собой продукты метаболизма грибков Penicillium roqueforti. Маркфортины имеют структуру, родственную структуре парагерквамидов, которые также являются известными соединениями. Парагерквамиды раскрываются в Tetrahedron Letters, 22, 135-136 (1981) и Journal of Antibiotics, 44, 492-497 (1991). В патентах США 4866060 и 4923867 раскрывается использование маркфортинов A, B и C и некоторых их производных для лечения и предупреждения паразитарных болезней у животных. В WO 92/22555 (опубликованном 23 декабря 1992) описано производное маркфортина или парагерквамида (т. е. частичная формула (III)), замещенное в положении 14 метилом или метилом и гидрокси, однако в этой работе не описан способ получения таких соединений 14-метил-14- гидроксимаркфортина. В Journal of Antibiotics, 43, 1380-1386 (1990) раскрывается парагерквамид A, который имеет следующую структуру: 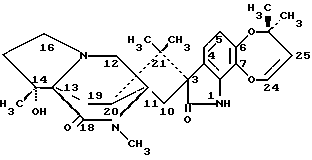 Маркфортин A имеет следующую структуру: 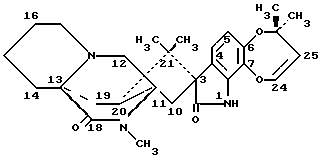 Маркфортин B имеет следующую структуру: 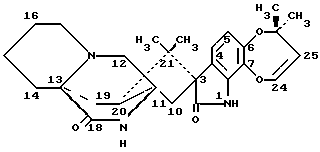 Маркфортин C имеет следующую структуру: 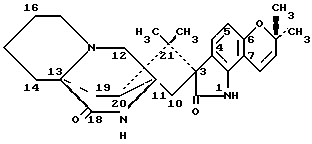 Маркфортин D имеет следующую структуру: 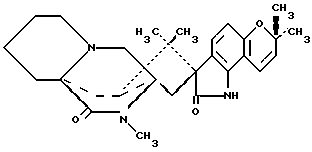 В WO 91/09961 (опубликованном 11 июля 1991) раскрываются производные маркфортина и парагерквамида и их 12a-N-оксиды, а также получение VM 29919 (парагерквамида) и VM 55596 (12a-N-оксида парагерквамида) inter alia из Penicillium Sp. IMI 332995. В патенте США 4873247 раскрываются производные парагерквамида и штамм MF 5123 Penicillium charlessi (АТСС 20841), используемый для продуцирования парагерквамида. В патенте 4978656 (а также в EP 390532-A, EP-301742-A) раскрываются различные синтетические производные парагерквамида, а также получение парагерквамида из MF 5123 Penicillium charlessi (АТСС 20841). В Международной публикации WO 92/22555 (опубликованной 23 декабря 1992) в общих чертах раскрываются соединения 14  -гидроксимаркфортина и способ использования 14-гидрокси-14-метилмаркфортинов для продуцирования противопаразитарных лекарственных средств. Однако в этой публикации не приводится описания каких-либо способов получения соединений 14-гидроксимаркфортина или 14-гидрокси-14 -гидроксимаркфортина и способ использования 14-гидрокси-14-метилмаркфортинов для продуцирования противопаразитарных лекарственных средств. Однако в этой публикации не приводится описания каких-либо способов получения соединений 14-гидроксимаркфортина или 14-гидрокси-14 -метилмаркфортина.
В Международной публикации WO 94/29319 раскрываются 14-замещенных маркфортина и их производные.
15-Алкил-14-гидроксисоединения (III), где n1 = 0, являются известными соединениями, см. Международную публикацию WO 94/29319.
Краткое описание изобретения -метилмаркфортина.
В Международной публикации WO 94/29319 раскрываются 14-замещенных маркфортина и их производные.
15-Алкил-14-гидроксисоединения (III), где n1 = 0, являются известными соединениями, см. Международную публикацию WO 94/29319.
Краткое описание изобретенияРаскрываются 15-алкил-14-гидроксисоединения формулы (III), где n1 = 1-3, их N-оксиды и фармацевтически приемлемые соли. Также раскрываются фторсоединения формулы (VIII), где n2 = 0-3, их N-оксиды и фармацевтически приемлемые соли. Кроме того, раскрываются 15-алкил-16-гидроксисоединения формулы (X), где n1 = 0-3, их N-оксиды и фармацевтически приемлемые соли. Раскрываются также соединения парагерквамида B формулы (XIII), где n1 = 0-3, их N-оксиды и фармацевтически приемлемые соли. Раскрывается 14,15-дегидро-16-оксопарагерквамид B. Раскрываются также 2-дезоксо-15-алкильные соединения формулы (XXI), где R14 обозначает -H или C1-C4 алкил, a R15 обозначает -H или C1-C4 алкил; их N-оксиды и фармацевтически приемлемые соли. Кроме того, раскрывается 2-дезоксосоединение формулы (XXIII), которое представляет собой 2-дезоксомаркфортин A, и его фармацевтически приемлемые соли. Помимо этого раскрываются 14-гидрокси-2-дезоксопарагерквамиды формулы (XXV), их N-оксиды и фармацевтически приемлемые соли. Раскрываются соединения, выбранные из группы, включающей 15 -этил-14-гидрокси-17-оксомаркфортин A, 14-гидрокси-15-винил-17-оксомаркфортин A, 14-гидрокси-15 -(1′,2′-дигидpoкcиэтил)-17-oкcoмapкфopтин A, 14-гидрокси-15-гидроксиметил-17-оксомаркфортин A, 15-фторметил-14-гидрокси-17-оксомаркфортин A, 14,15-дегидро-15-метилмаркфортин A, 14-гидрокси-16,17-диоксо-15-метилмаркфортин A, 14-гидpoкcи-16-oкco-15-мeтилпapaгepквaмид B, 16,17- диоксомаркфортин A, 16-оксопарагерквамид B (XVI), 14-гидрокси-15-метил-17-оксомаркфортин.
Раскрываются 1,2-дегидросоединения (XXIX).
Раскрываются также 2-алкил-2-дезоксосоединения (XXXI).
Подробное описание изобретения.
Заявленные соединения получают известными способами из известных исходных соединений или из соединений, которые могут быть легко получены из известных соединений способами, хорошо известными специалистам. Новые соединения настоящего изобретения получают из известных исходных соединений с использованием известных химических реакций в новой последовательности.
В СХЕМЕ A представлен предпочтительный способ получения 15-алкил-14-гидроксисоединений (III). Исходное 14-гидрокси-,-ненасыщенное соединение (I) является известным соединением, см. Международную публикацию WO 94/2931.9. 14-гидрокси-,-ненасыщенные соединения (I) могут быть превращены в соответствующие 15-алкил-17-оксосоединения (II) посредством реакции с алкилирующими агентами, такими как реактив Гриньяра или алкилкупраты; при этом предпочтительным является реактив Гриньяра формулы CH3-(CH2)n1-Mg-X0, где n1 = 0-3, а X0 представляет галоген. Предпочтительно, если n1 = 1, а X0 представляет – Br. В предпочтительном способе 14-гидрокси-,-ненасыщенное соединение (I) подвергают реакции с бромидом этилмагния и иодидом меди (I) в условиях стандартной реакции 1,4-присоединения, в результате чего получают 15-алкил-17- оксосоединения (II). Затем 15-алкил-17-оксосоединения (II) подвергают реакции восстановления, обычно используемой специалистами для восстановления карбонильной группы, с образованием алкиленового радикала, например, реакции восстановления с использованием боран-диметилсульфидного комплекса или других восстанавливающих агентов, таких как боран-ТГФ комплекс или алюмогидрид лития. При этом в реакции восстановления предпочтительно использовать комплекс боран-диметилсульфида. В случае использования 15-алкил-14-гидроксисоединений (III), предпочтительно, если в этих соединениях n1 = 1. 15-Алкил-14-гидроксисоединения (III), где n1 = 0, являются известными, см. Международную публикацию WO 94/29319.
В СХЕМЕ B представлен способ получения фторсоединений формулы (VIII). 14-гидрокси- ,-ненасыщенное исходное соединение (I) превращают в соответствующее ненасыщенное соединение (IV) посредством реакции присоединения Гриньяра, аналогичной реакции, используемой для алкилирования 14- гидрокси-,-ненасыщенного соединения (I) в СХЕМЕ A, за исключением того, что вместо CH3-(CH2)n1-Mg-X0 (СХЕМА A) используют CH2= CH-(CH2)n2-Mg-X0/иодид меди, где n2 = 0-3. Затем ненасыщенное соединение (IV) превращают в соответствующее дигидроксисоединение (V) путем окисления двойной связи ненасыщенной части C15 боковой цепи посредством реакции с окисляющим агентом, таким как тетроксид осмия (каталитическое количество) и N-оксид 4-метилморфолина; при этом предпочтительным окисляющим агентом является тетроксид осмия и N-оксид 4-метилморфолина. После этого дигидроксисоединения (V) превращают в соответствующие гидроксиалкильные соединения (VI) путем окисления с последующим восстановлением. При этом предпочтительным окисляющим агентом является периодат натрия, а восстанавливающим агентом боргидрид натрия. Гидроксиалкильные соединения (VI) превращают в соответствующие фтор-оксосоединения (VII) посредством реакции с фторирующим агентом, таким как фторид тетрабутиламмония и п-толуолсульфонилфторид. Эндоциклическую двойную связь фтор-оксосоединений (VII) восстанавливают известными способами, предпочтительно, с использованием комплекса “борана-тетрагидрофурана”, в результате чего получают нужное фторсоединение (VIII). При этом предпочтительными фторсоединениями (VIII) являются соединения, в которых n2 = 1.
В СХЕМЕ C представлен способ получения 15-алкил-16- гидроксисоединений (X). Сначала хорошо известными методами с использованием трифторида диэтиламиносеры (DAST) удаляют 14- гидроксильную группу с образованием функциональной 14,15- дегидрогруппы, в результате чего получают  14-15-алкильные соединения (IX). 14-15-алкильные соединения (IX) подвергают реакции гидроксилирования с гидроксилирующим агентом, пред почтительно, диоксидом селена, в условиях нагревания с обратным холодильником в инертном растворителе, таком как п-диоксан, в результате чего получают нужные 15-алкил-16-гидроксисоединения (X). При этом предпочтительными являются такие 15-алкил-16- гидроксисоединения (X), в которых n1 = 0.
В СХЕМЕ D представлен способ получения соединений 15-алкилпарагерквамида B (XIII). Исходные 15-алкил-14-гидрокси соединения (III) окисляют посредством реакции с кислородом в присутствии катализатора, такого как платина-на-угле, в результате чего получают соответствующие соединения 15-алкил-16,16-диоксо-маркфортина A (XI). Затем соединения 15-алкил-16,17-диоксо-маркфортина B (XI), имеющие 6-членное диоксо-кольцо, восстанавливают до соединений, имеющих 5-членное кольцо, и получают соединения 15-алкил-16-оксо-парагерквамида B (XII) путем обработки перкислотой, предпочтительно, м-хлорпербензойной кислотой. Затем 16-оксогруппу, присутствующую в соединениях 15-алкил-16-оксо-парагерквамида B (XII), удаляют с использованием восстанавливающего агента, предпочтительно, алюмогидрида лития/алюмохлорида лития, в результате чего получают нужные соединения 15-алкил-парагерквамида B (XIII). При этом предпочтительными являются такие 15-алкил-парагерквамиды B (XIII), в которых n1 = 0.
В СХЕМЕ E представлен способ получения различных оксосоединений, таких как 16,17-диоксомаркфортин A (XV), 16- оксопарагерквамид B (XVI) и 14,15-дегидро-16-оксо-парагерквамид B (XVII), способами, описанными в Примерах 13 и 14.
В СХЕМЕ F представлены способы получения 2-дезоксо-14- гидроксисоединений (XXI) с использованием в качестве исходных соединений 14-гидрокси-,-ненасыщенных кетонов (XVIII), где R14 обозначает H или C1-C4 алкил, a R15 обозначает H или C1-C4 алкил. 15 -Двойную связь, присутствующую в 14-гидрокси-,-ненасыщенных амидах (XVIII), восстанавливают посредством реакции с соответствующим литиевым реагентом R15-Li в присутствии бромида лития, в результате чего получают 14-гидрокси-17-оксосоединения (XIX). Если необходимо, то в процессе этой реакции C15-положение может быть алкилировано. Затем 17-оксогруппу в 14-гидрокси-17-оксосоединениях (XIX) восстанавливают с использованием комплекса боран-диметилсульфида (как описано выше в СХЕМЕ A), см. Пример 15. В результате этой реакции восстановления получают 14-гидроксисоединение (XX), а также соединение, в котором 2- и 17-карбонильные группы являются восстановленными, т. е. нужное 2-дезоксо-14-гидроксисоединение (XXI).
В СХЕМЕ G представлен способ продуцирования 2-дезоксосоединений (XXIII), см. Пример 16.
В СХЕМЕ H представлен способ получения соответствующих 14-гидрокси-2-дезоксопарагерквамидов (XXV).
Альтернативно и предпочтительно, производные 2-дезоксомаркфортина (XXIII), 14-гидрокси-2-дезоксопарагерквамида B и 14-гидроксимаркфортина A (XXV) могут быть получены с выходом 40-70% способом, описанным в СХЕМЕ O. В СХЕМЕ O амид (XXVI) подвергают реакции с соответствующим алкилхлорформиатом или ангидридным производным путем обработки либо гидридом калия, либо гидридом натрия, в результате чего получают соответствующий имид (XXVII), который затем восстанавливают с использованием боргидрида натрия и получают соответствующее 2-гидроксисоединение (XXVIII). В имиде (XXVII) азот в N-1 должен быть защищен (см. R17 формулы XXVII), как известно специалистам, после восстановления C-2-карбонила. Предпочтительными защитными группами являются фенил, 4-нитрофенил и трет-бутилфлуоренилметил. Затем 2-гидроксисоединение (XXVIII) подвергают снятию защиты различными методами, известными специалистам, и получают соответствующее 1,2-дегидросоединение (XXIX), которое может быть восстановлено с использованием боргидрида натрия, в результате чего получают соответствующие 2-дезоксомаркфортин (XXIII), 14-гидрокси-2-дезоксопарагерквамид B и 14-гидроксимаркфортин A (XXV) с полным выходом 40-70%. В случае, когда R17 представляет трет-бутил, то 2-дезоксомаркфортин (XXIII), 14-гидрокси-2-дезоксопарагерквамид B и 14-гидроксимаркфортин A (XXV) могут быть получены более коротким способом, т. е. посредством реакции имида (XXVII) с боргидридом натрия при нагревании с обратным холодильником в глиме и диглиме.
2-Алкил-2-дезоксопараквамид A (XXXI) получают из соответствующего 1,2-дегидромаркфортина A (XXIX) посредством реакции с подходящим алкиллитиевым реагентом способом, известным специалистам. Противопаразитарными соединениями являются 15-алкил-14-гидроксисоединения (III), фторсоединения (VIII), 15-алкил-16- гидроксисоединения (X), 15-алкилпарагерквамид B (XIII), 2-дезоксо- 14-гидроксисоединения (XXI), 2-дезоксомаркфортин (XXIII), 14- гидрокси-2-дезоксопарагерквамид B и 14-гидроксимаркфортин A (XXV), 14,15-дегидро-16-оксопарагерквамид B (XVII), 1,2-дегидросоединение (XXIX) и 2-алкил-2-дезоксосоединение (XXXI), их N-оксиды и фармацевтически приемлемые соли, если таковые существуют.
Противопаразитарными соединениями являются амины, которые при взаимодействии с кислотами достаточной силы образуют кислотно-аддитивные соли. Фармацевтически приемлемыми солями являются соли неорганических и органических кислот. При этом фармацевтически приемлемые соли являются более предпочтительными, чем соответствующие свободные амины, поскольку они образуют более растворимые в воде соединения и более кристаллические соединения. Предпочтительными фармацевтически приемлемыми солями являются соли метансульфоновой, соляной, бромистоводородной, серной, фосфорной, азотной, бензойной, лимонной, винной, фумаровой, малеиновой кислот, CH3-(CH2)n-COOH, где n = 0-4, HOOC-(CH2)n-COOH, где n определен выше.
Противопаразитарными соединениями являются амины, из которых путем взаимодействия с перкислотами, такими как м-хлорпербензойная кислота, могут быть получены соответствующие 12a-N-оксиды, известные специалистам.
Противопаразитарные соединения настоящего изобретения являются неожиданно сильнодействующими противопаразитарными средствами против эндопаразитов и эктопаразитов, особенно против гельминтов и членистоногих, вызывающих множество паразитарных болезней у человека, животных и растений.
Паразитарные болезни могут быть вызваны как эндопаразитами, так и эктопаразитами. Эндопаразитами являются такие паразиты, которые обитают внутри организма-хозяина или внутри какого-либо органа (такого как желудок, легкие, сердце, кишечник и т. п.) или просто под кожей. Эктопаразитами являются такие паразиты, которые обитают на внешней поверхности тела хозяина, но которые при этом извлекают из организма хозяина питательные вещества.
Эндопаразитарные болезни, обычно называемые гельминтозом, возникают вследствие инфекции организма хозяина паразитическими червями, именуемыми гельминтами. Гельминтоз является широко распространенным заболеванием, создающим серьезные экономические проблемы из-за заражения домашних животных, таких как свиньи, овцы, лошади, крупный рогатый скот, козы, собаки, кошки и домашняя птица. Многие из этих болезней являются следствием инфекции группой гельминтов, которые называются нематодами и которые способны заражать различные виды животных во всем мире. Эти болезни являются достаточно серьезными и часто приводят к гибели зараженных животных. Наиболее распространенные нематоды, заражающие животных, относятся к родам Haemonchus, Trichostrongylus, Ostertagia, Nematodirus, Cooperia, Ascaris, Bunostomum, Oesophagostomum, Chabertia, Trichuris, Strongylus, Trichonema, Dictyocaulus, Capillaria, Heterakis, Toxocara, Ascaridia, Oxyuris, Ancylostoma, Uncinaria, Toxoscaris и Parascaris. Многие паразиты являются специфическими видами (заражают только одного хозяина), а большинство из них также имеет преимущественный участок заражения животного. Так, например, нематоды рода Haemonchus и Ostertagia поражают, главным образом, желудок, а нематоды рода Nematodirus и Cooperia поражают кишечник. Некоторые паразиты предпочитают заселяться в таких органах, как сердце, глаза, легкие, кровеносные сосуды и т. п., а некоторые являются подкожными паразитами. Гельминтоз может приводить к слабости, потере веса, анемии, кишечным расстройствам, нарушению питания и к поражению других органов. Без лечения эти болезни могут привести к гибели животного.
Заражение членистоногими эктопаразитами, такими как клещи, вши, жигалки, жигалки коровьи, мухи мясные синие, блохи и т. п., также создает серьезные проблемы. Заражение этими паразитами приводит к потере крови, кожным поражениям и может препятствовать нормальному приему пищи, что может служить причиной потери веса. Заражение этими паразитами может также приводить к передаче серьезных болезней, таких как энцефалит, анаплазмоз, оспа свиней и т. п., которые являются смертельными.
Животные могут быть одновременно заражены несколькими видами паразитов, поскольку заражение одним видом паразита может ослаблять животное и делать его восприимчивым к заражению другим видом паразита. Поэтому для лечения таких болезней особенно предпочтительно использовать соединение с широким спектром активности. Противопаразитарные соединения настоящего изобретения обнаружили неожиданно высокую активность против этих паразитов, и кроме того, они также являются активными против дирофиляриоза у собак (возбудители Dirofilaria), Nematospiroides и Syphacia у грызунов, жалящих насекомых и мигрирующих личинок двукрылых насекомых, таких как Hypoderma sp. у крупного рогатого скота и Gastrophilus у лошадей.
Противопаразитарные соединения могут быть также использованы против эндо- и эктопаразитов, являющихся возбудителями паразитарных болезней человека. Примерами таких эндопаразитов, которые поражают человека, являются желудочно-кишечные паразиты рода Ancylostoma, Necator, Ascaris, Strongyloides, Trichinella, Capillaria, Trichuris, Enterobius и т. п. Другими эндопаразитами, которые поражают человека, являются паразиты, обитающие в крови и других органах. Примерами таких паразитов могут служить филярии Wucheria, Brugia, Onchocerca и т. п., а также кишечные гельминты, находящиеся во внекишечной стадии, такие как Strongylides и Trichineila. Эктопаразитами человека являются членистоногие, такие как иксодовые клещи, блохи, вши и т. п., и так же, как в случае домашних животных, заражение этими паразитами может приводить к переносу серьезных и даже смертельных болезней. Противопаразитарные соединения настоящего изобретения обладают активностью против этих эндо- и эктопаразитов, и кроме того, они обладают также активностью против жалящих насекомых и других двукрылых насекомых, досаждающих человеку. Для перорального или парентерального введения противопаразитарные соединения могут быть использованы в дозе от 0,05 до 20 мг/кг массы тела животного.
Противопаразитарные соединения настоящего изобретения могут быть также использованы против насекомых-вредителей, обитающих в помещениях, например, таких как Blatella sp. (таракан), Tineola sp. (платяная моль), Attagenus sp. (антренус), Musca domestica (муха комнатная) и против Solenopsis Invicta (муравей Рихтера).
Кроме того, противопаразитарные соединения настоящего изобретения могут быть использованы против насекомых-вредителей культурных растений, таких как тля (Acyrthiosiphon sp. ), саранча и долгоносик хлопковый, а также против насекомых-вредителей, поражающих зерно, находящееся на хранении, таких как Tribolium sp., и против неполовозрелых стадий насекомых (например, личинок), обитающих на растительной ткани. Противопаразитарные соединения настоящего изобретения могут быть также использованы в качестве нематоцида для борьбы с почвенными нематодами, которые могут приносить вред сельскому хозяйству.
Для использования в качестве противопаразитарных средств в целях борьбы с паразитами животных соединения настоящего изобретения могут быть введены системно, либо перорально, либо путем инъекции, или местно в виде жидкого состава для пропитки, или в виде шампуня.
Для перорального введения противопаразитарные соединения настоящего изобретения могут быть использованы в виде капсул, таблеток или болюсов, пропитанных составом, либо альтернативно, они могут быть подмешаны в корм для животных. Эти капсулы, таблетки и пропитанные болюсы содержат активный ингредиент в сочетании с подходящим носителем, таким как крахмал, тальк, стеарат магния или дикальцийфосфат. Такие стандартные лекарственные формы получают путем тщательного смешивания активного ингредиента с подходящими тонко измельченными инертными ингредиентами, включая разбавители, наполнители, дезинтегрирующие агенты, суспендирующие агенты и/или связующие вещества, так что в результате такого смешивания получают однородный раствор или суспензию. Инертным ингредиентом является такой ингредиент, который не реагирует с противопаразитарными соединениями и который является нетоксичным для животного, подвергающегося лечению. Подходящими инертными ингредиентами являются крахмал, лактоза, тальк, стеарат магния, растительные камеди и масла или т. п. Количество активных и неактивных ингредиентов, содержащихся в указанных лекарственных формах, может широко варьироваться в зависимости от ряда факторов, таких как размер и вид животного, подвергающегося лечению, а также природа и тяжесть заражения. Активный ингредиент может быть также введен в виде добавки в пищу путем простого смешивания противопаразитарных соединений с кормом или путем их нанесения на поверхность пищи. Альтернативно, активный ингредиент может быть смешан с инертным носителем, и полученная композиция может быть затем либо смешана с пищей, либо непосредственно введена животному. Подходящими инертными носителями являются кукурузная мука, мука из цитрусовой пульпы, отходы ферментации, соевая крупа, сухое зерно и т. п. Активные ингредиенты тщательно смешивают с этими инертными носителями путем измельчения, размешивания, размалывания или взбалтывания так, чтобы конечная композиция содержала от 0,001 до 5,0 мас.% активного ингредиента.
Альтернативно, противопаразитарные соединения настоящего изобретения могут быть введены парентерально путем инъекции лекарственной формы, содержащей активный ингредиент, растворенный в инертном жидком носителе. Инъекция может быть внутримышечной, внутрирубцовой, внутритрахеальной или подкожной. Подходящая для инъекции лекарственная форма содержит активный ингредиент, смешанный с соответствующим инертным жидким носителем. Подходящими жидкими носителями являются растительные масла, такие как арахисовое масло, масло из семян хлопчатника, кунжутное масло и т. п., а также органические растворители, такие как кеталь в виде золя, глицеринформаль и т. п. В качестве альтернативы могут быть также использованы водные парентеральные композиции. Предпочтительными жидкими носителями являются растительные масла. Эти лекарственные формы получают путем растворения или суспендирования активного ингредиента в жидком носителе так, чтобы полученный в результате препарат содержал от 0,005 до 20% масс. активного ингредиента.
Для местного применения могут быть использованы жидкие составы или шампуни, содержащие противопаразитарные соединения настоящего изобретения в виде водного раствора или суспензии. Эти лекарственные формы обычно содержат суспендирующий агент, такой как бентонит, а в большинстве случаев, они также содержат противовспенивающее средство. Подходящие препараты содержат от 0,005 до 20 мас.% активного ингредиента. Предпочтительные препараты содержат от 0,5 до 5 мас.% противопаразитарных соединений.
Противопаразитарные соединения настоящего изобретения могут быть использованы, главным образом, в качестве противопаразитарных средств для лечения и/или предупреждения гельминтоза у домашних животных, таких как крупный рогатый скот, овцы, лошади, собаки, кошки, козы, свиньи и домашняя птица. Они могут быть также использованы для предупреждения и лечения паразитарного заражения этих животных эктопаразитами, такими как иксодовые клещи, клещи, вши, блохи и т. п. Эти соединения являются также эффективными при лечении паразитарных заражений человека. Для лечения таких заражений, противопаразитарные соединения настоящего изобретения могут быть использованы отдельно, либо в комбинации друг с другом или с другими неродственными противопаразитарными средствами. Доза противопаразитарных соединений настоящего изобретения, необходимая для получения наилучшего эффекта, зависит от нескольких факторов, таких как вид и размер животного, тип и тяжесть заражения, способ введения и конкретно используемые противопаразитарные соединения. Обычно, хорошие результаты дает пероральная доза противопаразитарного соединения, составляющая от 0,005 до 50 мг на кг массы тела животного и вводимая в виде разовой дозы или в виде дробной дозы с интервалами в несколько дней. Разовая доза одного из противопаразитарных соединений обычно дает прекрасный результат, однако, для борьбы с повторным заражением или в случае заражения чрезвычайно устойчивым видом паразита, могут быть введены повторные дозы. Способы введения противопаразитарных соединений животным известны специалистам в области ветеринарии.
Противопаразитарные соединения настоящего изобретения могут быть также использованы для борьбы с насекомыми-вредителями, поражающими культурные растения на корню или в условиях хранения. В этих целях противопаразитарные соединения настоящего изобретения используют в виде растворов для опрыскивания, дустов, эмульсий и т. п. путем нанесения на растущие растения или на собранный урожай. Способы использования противопаразитарных соединений в этих целях хорошо известны специалистам по агротехнике.
Точная доза и частота введения зависят от конкретно используемых противопаразитарных соединений, конкретных условий обработки, тяжести поражения, возраста, веса и общего физического состояния пациента и других показаний для данного индивидуума, обычно принимаемых во внимание и хорошо известных специалистам; при этом доза и схема введения более точно могут быть определены путем измерения уровня или концентрации противопаразитарных соединений в крови пациента и/или реакции пациента в ответ на конкретные условия лечения.
ОПРЕДЕЛЕНИЯ И УСЛОВНЫЕ ОБОЗНАЧЕНИЯ 14-15-алкильные соединения (IX). 14-15-алкильные соединения (IX) подвергают реакции гидроксилирования с гидроксилирующим агентом, пред почтительно, диоксидом селена, в условиях нагревания с обратным холодильником в инертном растворителе, таком как п-диоксан, в результате чего получают нужные 15-алкил-16-гидроксисоединения (X). При этом предпочтительными являются такие 15-алкил-16- гидроксисоединения (X), в которых n1 = 0.
В СХЕМЕ D представлен способ получения соединений 15-алкилпарагерквамида B (XIII). Исходные 15-алкил-14-гидрокси соединения (III) окисляют посредством реакции с кислородом в присутствии катализатора, такого как платина-на-угле, в результате чего получают соответствующие соединения 15-алкил-16,16-диоксо-маркфортина A (XI). Затем соединения 15-алкил-16,17-диоксо-маркфортина B (XI), имеющие 6-членное диоксо-кольцо, восстанавливают до соединений, имеющих 5-членное кольцо, и получают соединения 15-алкил-16-оксо-парагерквамида B (XII) путем обработки перкислотой, предпочтительно, м-хлорпербензойной кислотой. Затем 16-оксогруппу, присутствующую в соединениях 15-алкил-16-оксо-парагерквамида B (XII), удаляют с использованием восстанавливающего агента, предпочтительно, алюмогидрида лития/алюмохлорида лития, в результате чего получают нужные соединения 15-алкил-парагерквамида B (XIII). При этом предпочтительными являются такие 15-алкил-парагерквамиды B (XIII), в которых n1 = 0.
В СХЕМЕ E представлен способ получения различных оксосоединений, таких как 16,17-диоксомаркфортин A (XV), 16- оксопарагерквамид B (XVI) и 14,15-дегидро-16-оксо-парагерквамид B (XVII), способами, описанными в Примерах 13 и 14.
В СХЕМЕ F представлены способы получения 2-дезоксо-14- гидроксисоединений (XXI) с использованием в качестве исходных соединений 14-гидрокси-,-ненасыщенных кетонов (XVIII), где R14 обозначает H или C1-C4 алкил, a R15 обозначает H или C1-C4 алкил. 15 -Двойную связь, присутствующую в 14-гидрокси-,-ненасыщенных амидах (XVIII), восстанавливают посредством реакции с соответствующим литиевым реагентом R15-Li в присутствии бромида лития, в результате чего получают 14-гидрокси-17-оксосоединения (XIX). Если необходимо, то в процессе этой реакции C15-положение может быть алкилировано. Затем 17-оксогруппу в 14-гидрокси-17-оксосоединениях (XIX) восстанавливают с использованием комплекса боран-диметилсульфида (как описано выше в СХЕМЕ A), см. Пример 15. В результате этой реакции восстановления получают 14-гидроксисоединение (XX), а также соединение, в котором 2- и 17-карбонильные группы являются восстановленными, т. е. нужное 2-дезоксо-14-гидроксисоединение (XXI).
В СХЕМЕ G представлен способ продуцирования 2-дезоксосоединений (XXIII), см. Пример 16.
В СХЕМЕ H представлен способ получения соответствующих 14-гидрокси-2-дезоксопарагерквамидов (XXV).
Альтернативно и предпочтительно, производные 2-дезоксомаркфортина (XXIII), 14-гидрокси-2-дезоксопарагерквамида B и 14-гидроксимаркфортина A (XXV) могут быть получены с выходом 40-70% способом, описанным в СХЕМЕ O. В СХЕМЕ O амид (XXVI) подвергают реакции с соответствующим алкилхлорформиатом или ангидридным производным путем обработки либо гидридом калия, либо гидридом натрия, в результате чего получают соответствующий имид (XXVII), который затем восстанавливают с использованием боргидрида натрия и получают соответствующее 2-гидроксисоединение (XXVIII). В имиде (XXVII) азот в N-1 должен быть защищен (см. R17 формулы XXVII), как известно специалистам, после восстановления C-2-карбонила. Предпочтительными защитными группами являются фенил, 4-нитрофенил и трет-бутилфлуоренилметил. Затем 2-гидроксисоединение (XXVIII) подвергают снятию защиты различными методами, известными специалистам, и получают соответствующее 1,2-дегидросоединение (XXIX), которое может быть восстановлено с использованием боргидрида натрия, в результате чего получают соответствующие 2-дезоксомаркфортин (XXIII), 14-гидрокси-2-дезоксопарагерквамид B и 14-гидроксимаркфортин A (XXV) с полным выходом 40-70%. В случае, когда R17 представляет трет-бутил, то 2-дезоксомаркфортин (XXIII), 14-гидрокси-2-дезоксопарагерквамид B и 14-гидроксимаркфортин A (XXV) могут быть получены более коротким способом, т. е. посредством реакции имида (XXVII) с боргидридом натрия при нагревании с обратным холодильником в глиме и диглиме.
2-Алкил-2-дезоксопараквамид A (XXXI) получают из соответствующего 1,2-дегидромаркфортина A (XXIX) посредством реакции с подходящим алкиллитиевым реагентом способом, известным специалистам. Противопаразитарными соединениями являются 15-алкил-14-гидроксисоединения (III), фторсоединения (VIII), 15-алкил-16- гидроксисоединения (X), 15-алкилпарагерквамид B (XIII), 2-дезоксо- 14-гидроксисоединения (XXI), 2-дезоксомаркфортин (XXIII), 14- гидрокси-2-дезоксопарагерквамид B и 14-гидроксимаркфортин A (XXV), 14,15-дегидро-16-оксопарагерквамид B (XVII), 1,2-дегидросоединение (XXIX) и 2-алкил-2-дезоксосоединение (XXXI), их N-оксиды и фармацевтически приемлемые соли, если таковые существуют.
Противопаразитарными соединениями являются амины, которые при взаимодействии с кислотами достаточной силы образуют кислотно-аддитивные соли. Фармацевтически приемлемыми солями являются соли неорганических и органических кислот. При этом фармацевтически приемлемые соли являются более предпочтительными, чем соответствующие свободные амины, поскольку они образуют более растворимые в воде соединения и более кристаллические соединения. Предпочтительными фармацевтически приемлемыми солями являются соли метансульфоновой, соляной, бромистоводородной, серной, фосфорной, азотной, бензойной, лимонной, винной, фумаровой, малеиновой кислот, CH3-(CH2)n-COOH, где n = 0-4, HOOC-(CH2)n-COOH, где n определен выше.
Противопаразитарными соединениями являются амины, из которых путем взаимодействия с перкислотами, такими как м-хлорпербензойная кислота, могут быть получены соответствующие 12a-N-оксиды, известные специалистам.
Противопаразитарные соединения настоящего изобретения являются неожиданно сильнодействующими противопаразитарными средствами против эндопаразитов и эктопаразитов, особенно против гельминтов и членистоногих, вызывающих множество паразитарных болезней у человека, животных и растений.
Паразитарные болезни могут быть вызваны как эндопаразитами, так и эктопаразитами. Эндопаразитами являются такие паразиты, которые обитают внутри организма-хозяина или внутри какого-либо органа (такого как желудок, легкие, сердце, кишечник и т. п.) или просто под кожей. Эктопаразитами являются такие паразиты, которые обитают на внешней поверхности тела хозяина, но которые при этом извлекают из организма хозяина питательные вещества.
Эндопаразитарные болезни, обычно называемые гельминтозом, возникают вследствие инфекции организма хозяина паразитическими червями, именуемыми гельминтами. Гельминтоз является широко распространенным заболеванием, создающим серьезные экономические проблемы из-за заражения домашних животных, таких как свиньи, овцы, лошади, крупный рогатый скот, козы, собаки, кошки и домашняя птица. Многие из этих болезней являются следствием инфекции группой гельминтов, которые называются нематодами и которые способны заражать различные виды животных во всем мире. Эти болезни являются достаточно серьезными и часто приводят к гибели зараженных животных. Наиболее распространенные нематоды, заражающие животных, относятся к родам Haemonchus, Trichostrongylus, Ostertagia, Nematodirus, Cooperia, Ascaris, Bunostomum, Oesophagostomum, Chabertia, Trichuris, Strongylus, Trichonema, Dictyocaulus, Capillaria, Heterakis, Toxocara, Ascaridia, Oxyuris, Ancylostoma, Uncinaria, Toxoscaris и Parascaris. Многие паразиты являются специфическими видами (заражают только одного хозяина), а большинство из них также имеет преимущественный участок заражения животного. Так, например, нематоды рода Haemonchus и Ostertagia поражают, главным образом, желудок, а нематоды рода Nematodirus и Cooperia поражают кишечник. Некоторые паразиты предпочитают заселяться в таких органах, как сердце, глаза, легкие, кровеносные сосуды и т. п., а некоторые являются подкожными паразитами. Гельминтоз может приводить к слабости, потере веса, анемии, кишечным расстройствам, нарушению питания и к поражению других органов. Без лечения эти болезни могут привести к гибели животного.
Заражение членистоногими эктопаразитами, такими как клещи, вши, жигалки, жигалки коровьи, мухи мясные синие, блохи и т. п., также создает серьезные проблемы. Заражение этими паразитами приводит к потере крови, кожным поражениям и может препятствовать нормальному приему пищи, что может служить причиной потери веса. Заражение этими паразитами может также приводить к передаче серьезных болезней, таких как энцефалит, анаплазмоз, оспа свиней и т. п., которые являются смертельными.
Животные могут быть одновременно заражены несколькими видами паразитов, поскольку заражение одним видом паразита может ослаблять животное и делать его восприимчивым к заражению другим видом паразита. Поэтому для лечения таких болезней особенно предпочтительно использовать соединение с широким спектром активности. Противопаразитарные соединения настоящего изобретения обнаружили неожиданно высокую активность против этих паразитов, и кроме того, они также являются активными против дирофиляриоза у собак (возбудители Dirofilaria), Nematospiroides и Syphacia у грызунов, жалящих насекомых и мигрирующих личинок двукрылых насекомых, таких как Hypoderma sp. у крупного рогатого скота и Gastrophilus у лошадей.
Противопаразитарные соединения могут быть также использованы против эндо- и эктопаразитов, являющихся возбудителями паразитарных болезней человека. Примерами таких эндопаразитов, которые поражают человека, являются желудочно-кишечные паразиты рода Ancylostoma, Necator, Ascaris, Strongyloides, Trichinella, Capillaria, Trichuris, Enterobius и т. п. Другими эндопаразитами, которые поражают человека, являются паразиты, обитающие в крови и других органах. Примерами таких паразитов могут служить филярии Wucheria, Brugia, Onchocerca и т. п., а также кишечные гельминты, находящиеся во внекишечной стадии, такие как Strongylides и Trichineila. Эктопаразитами человека являются членистоногие, такие как иксодовые клещи, блохи, вши и т. п., и так же, как в случае домашних животных, заражение этими паразитами может приводить к переносу серьезных и даже смертельных болезней. Противопаразитарные соединения настоящего изобретения обладают активностью против этих эндо- и эктопаразитов, и кроме того, они обладают также активностью против жалящих насекомых и других двукрылых насекомых, досаждающих человеку. Для перорального или парентерального введения противопаразитарные соединения могут быть использованы в дозе от 0,05 до 20 мг/кг массы тела животного.
Противопаразитарные соединения настоящего изобретения могут быть также использованы против насекомых-вредителей, обитающих в помещениях, например, таких как Blatella sp. (таракан), Tineola sp. (платяная моль), Attagenus sp. (антренус), Musca domestica (муха комнатная) и против Solenopsis Invicta (муравей Рихтера).
Кроме того, противопаразитарные соединения настоящего изобретения могут быть использованы против насекомых-вредителей культурных растений, таких как тля (Acyrthiosiphon sp. ), саранча и долгоносик хлопковый, а также против насекомых-вредителей, поражающих зерно, находящееся на хранении, таких как Tribolium sp., и против неполовозрелых стадий насекомых (например, личинок), обитающих на растительной ткани. Противопаразитарные соединения настоящего изобретения могут быть также использованы в качестве нематоцида для борьбы с почвенными нематодами, которые могут приносить вред сельскому хозяйству.
Для использования в качестве противопаразитарных средств в целях борьбы с паразитами животных соединения настоящего изобретения могут быть введены системно, либо перорально, либо путем инъекции, или местно в виде жидкого состава для пропитки, или в виде шампуня.
Для перорального введения противопаразитарные соединения настоящего изобретения могут быть использованы в виде капсул, таблеток или болюсов, пропитанных составом, либо альтернативно, они могут быть подмешаны в корм для животных. Эти капсулы, таблетки и пропитанные болюсы содержат активный ингредиент в сочетании с подходящим носителем, таким как крахмал, тальк, стеарат магния или дикальцийфосфат. Такие стандартные лекарственные формы получают путем тщательного смешивания активного ингредиента с подходящими тонко измельченными инертными ингредиентами, включая разбавители, наполнители, дезинтегрирующие агенты, суспендирующие агенты и/или связующие вещества, так что в результате такого смешивания получают однородный раствор или суспензию. Инертным ингредиентом является такой ингредиент, который не реагирует с противопаразитарными соединениями и который является нетоксичным для животного, подвергающегося лечению. Подходящими инертными ингредиентами являются крахмал, лактоза, тальк, стеарат магния, растительные камеди и масла или т. п. Количество активных и неактивных ингредиентов, содержащихся в указанных лекарственных формах, может широко варьироваться в зависимости от ряда факторов, таких как размер и вид животного, подвергающегося лечению, а также природа и тяжесть заражения. Активный ингредиент может быть также введен в виде добавки в пищу путем простого смешивания противопаразитарных соединений с кормом или путем их нанесения на поверхность пищи. Альтернативно, активный ингредиент может быть смешан с инертным носителем, и полученная композиция может быть затем либо смешана с пищей, либо непосредственно введена животному. Подходящими инертными носителями являются кукурузная мука, мука из цитрусовой пульпы, отходы ферментации, соевая крупа, сухое зерно и т. п. Активные ингредиенты тщательно смешивают с этими инертными носителями путем измельчения, размешивания, размалывания или взбалтывания так, чтобы конечная композиция содержала от 0,001 до 5,0 мас.% активного ингредиента.
Альтернативно, противопаразитарные соединения настоящего изобретения могут быть введены парентерально путем инъекции лекарственной формы, содержащей активный ингредиент, растворенный в инертном жидком носителе. Инъекция может быть внутримышечной, внутрирубцовой, внутритрахеальной или подкожной. Подходящая для инъекции лекарственная форма содержит активный ингредиент, смешанный с соответствующим инертным жидким носителем. Подходящими жидкими носителями являются растительные масла, такие как арахисовое масло, масло из семян хлопчатника, кунжутное масло и т. п., а также органические растворители, такие как кеталь в виде золя, глицеринформаль и т. п. В качестве альтернативы могут быть также использованы водные парентеральные композиции. Предпочтительными жидкими носителями являются растительные масла. Эти лекарственные формы получают путем растворения или суспендирования активного ингредиента в жидком носителе так, чтобы полученный в результате препарат содержал от 0,005 до 20% масс. активного ингредиента.
Для местного применения могут быть использованы жидкие составы или шампуни, содержащие противопаразитарные соединения настоящего изобретения в виде водного раствора или суспензии. Эти лекарственные формы обычно содержат суспендирующий агент, такой как бентонит, а в большинстве случаев, они также содержат противовспенивающее средство. Подходящие препараты содержат от 0,005 до 20 мас.% активного ингредиента. Предпочтительные препараты содержат от 0,5 до 5 мас.% противопаразитарных соединений.
Противопаразитарные соединения настоящего изобретения могут быть использованы, главным образом, в качестве противопаразитарных средств для лечения и/или предупреждения гельминтоза у домашних животных, таких как крупный рогатый скот, овцы, лошади, собаки, кошки, козы, свиньи и домашняя птица. Они могут быть также использованы для предупреждения и лечения паразитарного заражения этих животных эктопаразитами, такими как иксодовые клещи, клещи, вши, блохи и т. п. Эти соединения являются также эффективными при лечении паразитарных заражений человека. Для лечения таких заражений, противопаразитарные соединения настоящего изобретения могут быть использованы отдельно, либо в комбинации друг с другом или с другими неродственными противопаразитарными средствами. Доза противопаразитарных соединений настоящего изобретения, необходимая для получения наилучшего эффекта, зависит от нескольких факторов, таких как вид и размер животного, тип и тяжесть заражения, способ введения и конкретно используемые противопаразитарные соединения. Обычно, хорошие результаты дает пероральная доза противопаразитарного соединения, составляющая от 0,005 до 50 мг на кг массы тела животного и вводимая в виде разовой дозы или в виде дробной дозы с интервалами в несколько дней. Разовая доза одного из противопаразитарных соединений обычно дает прекрасный результат, однако, для борьбы с повторным заражением или в случае заражения чрезвычайно устойчивым видом паразита, могут быть введены повторные дозы. Способы введения противопаразитарных соединений животным известны специалистам в области ветеринарии.
Противопаразитарные соединения настоящего изобретения могут быть также использованы для борьбы с насекомыми-вредителями, поражающими культурные растения на корню или в условиях хранения. В этих целях противопаразитарные соединения настоящего изобретения используют в виде растворов для опрыскивания, дустов, эмульсий и т. п. путем нанесения на растущие растения или на собранный урожай. Способы использования противопаразитарных соединений в этих целях хорошо известны специалистам по агротехнике.
Точная доза и частота введения зависят от конкретно используемых противопаразитарных соединений, конкретных условий обработки, тяжести поражения, возраста, веса и общего физического состояния пациента и других показаний для данного индивидуума, обычно принимаемых во внимание и хорошо известных специалистам; при этом доза и схема введения более точно могут быть определены путем измерения уровня или концентрации противопаразитарных соединений в крови пациента и/или реакции пациента в ответ на конкретные условия лечения.
ОПРЕДЕЛЕНИЯ И УСЛОВНЫЕ ОБОЗНАЧЕНИЯНиже приводятся определения и объяснения терминов, используемых в настоящей заявке, включая описание и формулу изобретения. I. Обозначения, используемые в формулах, и определение символов B данном описании и формуле изобретения химические формулы, представляющие различные соединения или фрагменты их молекул, могут содержать, помимо точно определенных структурных особенностей, переменные заместители. Эти переменные заместители обозначаются буквой или буквой с подстрочным индексом, например, “Z1” или “Ri“, где “i” означает целое число. Эти переменные заместители являются одновалентными или двухвалентными, то есть они представляют группу, присоединенную в формуле посредством одной или двух химических связей. Так, например, группа Zi представляет двухвалентный радикал в формуле CH3-C(= Z1)H. Группы Ri и Rj представляют одновалентные переменные заместители в формуле CH3-CH2-C(Ri)(Rj)-H. Если химические соединения описаны линейной формулой, как, например, вышеприведенные формулы, то переменные заместители, заключенные в скобки, связаны с атомом, находящимся непосредственно слева от заместителя в скобках. Если два или несколько следующих друг за другом заместителей заключены в скобки, то каждый из этих последовательных заместителей связаны с непосредственно предшествующим и расположенным слева от них атомом, который не заключен в скобки. Так, например, в вышеуказанной формуле оба Ri и Rj связаны с предшествующим атомом углерода. Кроме того, для любой молекулы с установленной системой нумерации атомов углерода, как, например, в стероидах, эти атомы углерода обозначаются Ci, где “i” означает целое число, соответствующее номеру атома углерода. Так, например, C6 представляет положение 6 или номер атома углерода в стероидном ядре стероида, как это традиционно обозначается специалистами по стероидной химии. Аналогичным образом, “R6” представляет переменный заместитель (одновалентный или двухвалентный) в положении C6. Линейные химические формулы или их части представляют атомы в линейной цепи. Символ “-” обычно означает связь между двумя атомами в цепи. Так, например, формула CH3-O-CH2-CH(Ri)-CH3 представляет 2-замещенное-1-метоксипропановое соединение. Аналогично, символ “=” означает двойную связь, например, CH2= C(Ri)-O-CH3, а символ “  ” означает тройную связь, например, HCC-CH(Ri)-CH2-CH3 . Карбонильные группы могут быть представлены двумя путями: -CO или -C(= O)-, при этом первое обозначение предпочтительней из-за своей простоты.
Химические формулы циклических (кольцевых) соединений или их молекулярных фрагментов могут быть представлены линейно. Так, например, соединение 4-хлор-2-метилпиридин может быть выражено следующей линейной формулой: N*= C(CH3)-CH=CCl-CH=C*Н, где, как это обычно принято, атомы, обозначенные звездочкой, связаны друг с другом, образуя в результате кольцо. Аналогичным образом, циклический молекулярный фрагмент 4-(этил)-1-пиперазинил может быть представлен -N*-(CH2)2-N(C2H5)-CH2– C*H2.
Жесткая циклическая (кольцевая) структура для любых соединений, рассматриваемых в настоящем описании, определяет ориентацию по отношению к плоскости кольца для заместителей, связанных с каждым атомом углерода жесткого циклического соединения. Для насыщенных соединений, которые имеют два заместителя, связанные с атомом углерода, являющимся частью циклической системы, -C(X1)(X2)-, два заместителя могут, быть либо в аксиальном, либо в экваториальном положении по отношению к кольцу, и эти положения могут изменяться между аксиальным и экваториальным положением. Однако положения двух заместителей по отношению к кольцу и по отношению друг к другу остается фиксированным. Один из заместителей может иногда находиться в плоскости кольца (экваториальное положение), а не выше или ниже этой плоскости (аксиальное положение), тогда как другой заместитель всегда находится “выше” первого заместителя. В структурных химических формулах, описывающих такие соединения, заместитель (X1), который находится “ниже” другого заместителя (X2), идентифицируется как имеющий альфа ()-конфигурацию, и его присоединение к атому углерода обозначается прерывистой линией, черточкой или пунктирной линией, т. е. символом “–“, “-” или “…”. Соответствующий заместитель (X2), присоединенный “выше” другого заместителя (X1), идентифицируется как находящийся в бета ()-конфигурации и показан сплошной линией связи с атомом углерода.
Если переменный заместитель является двухвалентным, то при определении радикала валентности могут быть взяты вместе или отдельно, либо и то, и другое. Так, например, переменный радикал Ri, связанный с атомом углерода как -C(=Ri)-, может быть двухвалентным и определен как оксогруппа (например, образующая карбонильную группу (-CO)), или как два отдельно связанных одновалентных переменных заместителя -Ri-j и -Ri-k. Если переменный двухвалентный радикал Ri определяют как состоящий из двух переменных одновалентных заместителей, то для обозначения такого двухвалентного радикала используют форму ” -Ri-j: -Ri-k” или некий ее вариант. В этом случае оба -Ri-j и -Ri-k связаны с атомом углерода, образуя -C( -Ri-j)( -Ri-k)-. Так, например, если двухвалентный радикал представлен R6, то -C(=R6)- определяется как состоящий из двух одновалентных переменных заместителей, где эти два одновалентных заместителя являются -R6-1: -R6-2, … -R6-9: -R6-10 и т. д. , образуя -C( -R6-1) -R6-2)-, … -C( -R6-9)( -R6-10)-, и т. п. Аналогично, для двухвалентного радикала R11, -C(=R11)-, двумя переменными одновалентными заместителями являются -R11-1: -R11-2. Для кольцевых заместителей, для которых отдельных – и -ориентаций не существует (например, благодаря присутствию в кольце двойной углерод-углеродной связи), и для заместителя, связанного с атомом углерода, не являющегося частью кольца, также используется вышеуказанное обозначение, но при этом символы и опускаются.
Точно так же двухвалентный радикал может быть определен как два отдельных одновалентных переменных заместителя, при этом два отдельных одновалентных переменных заместителя могут быть определены как взятые вместе и образующие двухвалентный радикал. Так, например, в формуле: -C1(Ri)H-C2(Rj)H- (C1 и C2 произвольно обозначают первый и второй атом углерода, соответственно) Ri и Rj могут быть определены как взятые вместе и образующие (1) вторую связь между C1 и C2, либо (2) двухвалентную группу, такую как окса (-O-), и, таким образом, указанная формула описывает эпоксид. Если Ri и Rj, взятые вместе, образуют более сложную молекулярную структуру, такую как группа -X-Y-, то ее ориентация может быть такой, что C1 в вышеуказанной формуле будет связан с X, а C2 будет связан с Y. Таким образом, понятие “… .Ri и Rj, взятые вместе, с образованием -CH2-CH2-O-CO-…”, означает лактон, в котором карбонил связан с C2. Однако, если говорят, что “…Ri и Rj, взятые вместе, образуют -CO-O-CH2-CH2-“, то обозначенная структура означает лактон, в котором карбонил связан с C1.
Количество атомов углерода в переменных заместителях может быть указано двумя способами. В первом способе используют префикс перед названием радикала, например, “C1-C4“, где “1” и “4” являются целыми числами, указывающими на минимальное и максимальное число атомов углерода в радикале. Этот префикс отделен от радикала пробелом. Например, “C1-C4 алкил” означает алкил, имеющий от 1 до 4 атомов углерода (включая его изомерные формы, если это не оговорено особо). Если имеется лишь один префикс, то этот префикс указывает на полное число атомов углерода в радикале. Так, например, 2-C4 алкоксикарбонил представляет собой группу CH3-(CH2)n-O-CO-, где n = 0, 1 или 2. Во втором способе указывается число атомов углерода для каждой части радикала отдельно, для чего обозначение “Ci-Cj” заключают в скобки и помещают непосредственно (без пробела) перед той частью радикала, к которой относится данное обозначение. Такое необязательное обозначение (C1-C3)алкоксикарбонил имеет то же самое значение, что и обозначение C2-C4 алкоксикарбонил, поскольку “C1-C3” относится только к количеству атомов углерода алкоксигруппы. Аналогично, хотя оба C2-C6 алкоксиалкил и (C1-C3)алкокси(C1-C3)алкил обозначают алкоксиалкильные группы, содержащие от 2 до 6 атомов углерода, однако, эти два определения отличаются друг от друга, поскольку в соответствии с первым определением, одна алкокси- или алкильная часть содержит 4 или 5 атомов углерода, тогда как в соответствии с последним определением, любая из этих групп ограничена лишь 3 атомами углерода.
Если заявленные соединения имеют достаточно сложный (циклический) заместитель, то в конце названия/обозначения этого конкретного заместителя дается примечание (в скобках), которое относится к тому же самому названию/обозначению в одной из СХЕМ, где также представлена структурная формула этого заместителя.
II. Определения ” означает тройную связь, например, HCC-CH(Ri)-CH2-CH3 . Карбонильные группы могут быть представлены двумя путями: -CO или -C(= O)-, при этом первое обозначение предпочтительней из-за своей простоты.
Химические формулы циклических (кольцевых) соединений или их молекулярных фрагментов могут быть представлены линейно. Так, например, соединение 4-хлор-2-метилпиридин может быть выражено следующей линейной формулой: N*= C(CH3)-CH=CCl-CH=C*Н, где, как это обычно принято, атомы, обозначенные звездочкой, связаны друг с другом, образуя в результате кольцо. Аналогичным образом, циклический молекулярный фрагмент 4-(этил)-1-пиперазинил может быть представлен -N*-(CH2)2-N(C2H5)-CH2– C*H2.
Жесткая циклическая (кольцевая) структура для любых соединений, рассматриваемых в настоящем описании, определяет ориентацию по отношению к плоскости кольца для заместителей, связанных с каждым атомом углерода жесткого циклического соединения. Для насыщенных соединений, которые имеют два заместителя, связанные с атомом углерода, являющимся частью циклической системы, -C(X1)(X2)-, два заместителя могут, быть либо в аксиальном, либо в экваториальном положении по отношению к кольцу, и эти положения могут изменяться между аксиальным и экваториальным положением. Однако положения двух заместителей по отношению к кольцу и по отношению друг к другу остается фиксированным. Один из заместителей может иногда находиться в плоскости кольца (экваториальное положение), а не выше или ниже этой плоскости (аксиальное положение), тогда как другой заместитель всегда находится “выше” первого заместителя. В структурных химических формулах, описывающих такие соединения, заместитель (X1), который находится “ниже” другого заместителя (X2), идентифицируется как имеющий альфа ()-конфигурацию, и его присоединение к атому углерода обозначается прерывистой линией, черточкой или пунктирной линией, т. е. символом “–“, “-” или “…”. Соответствующий заместитель (X2), присоединенный “выше” другого заместителя (X1), идентифицируется как находящийся в бета ()-конфигурации и показан сплошной линией связи с атомом углерода.
Если переменный заместитель является двухвалентным, то при определении радикала валентности могут быть взяты вместе или отдельно, либо и то, и другое. Так, например, переменный радикал Ri, связанный с атомом углерода как -C(=Ri)-, может быть двухвалентным и определен как оксогруппа (например, образующая карбонильную группу (-CO)), или как два отдельно связанных одновалентных переменных заместителя -Ri-j и -Ri-k. Если переменный двухвалентный радикал Ri определяют как состоящий из двух переменных одновалентных заместителей, то для обозначения такого двухвалентного радикала используют форму ” -Ri-j: -Ri-k” или некий ее вариант. В этом случае оба -Ri-j и -Ri-k связаны с атомом углерода, образуя -C( -Ri-j)( -Ri-k)-. Так, например, если двухвалентный радикал представлен R6, то -C(=R6)- определяется как состоящий из двух одновалентных переменных заместителей, где эти два одновалентных заместителя являются -R6-1: -R6-2, … -R6-9: -R6-10 и т. д. , образуя -C( -R6-1) -R6-2)-, … -C( -R6-9)( -R6-10)-, и т. п. Аналогично, для двухвалентного радикала R11, -C(=R11)-, двумя переменными одновалентными заместителями являются -R11-1: -R11-2. Для кольцевых заместителей, для которых отдельных – и -ориентаций не существует (например, благодаря присутствию в кольце двойной углерод-углеродной связи), и для заместителя, связанного с атомом углерода, не являющегося частью кольца, также используется вышеуказанное обозначение, но при этом символы и опускаются.
Точно так же двухвалентный радикал может быть определен как два отдельных одновалентных переменных заместителя, при этом два отдельных одновалентных переменных заместителя могут быть определены как взятые вместе и образующие двухвалентный радикал. Так, например, в формуле: -C1(Ri)H-C2(Rj)H- (C1 и C2 произвольно обозначают первый и второй атом углерода, соответственно) Ri и Rj могут быть определены как взятые вместе и образующие (1) вторую связь между C1 и C2, либо (2) двухвалентную группу, такую как окса (-O-), и, таким образом, указанная формула описывает эпоксид. Если Ri и Rj, взятые вместе, образуют более сложную молекулярную структуру, такую как группа -X-Y-, то ее ориентация может быть такой, что C1 в вышеуказанной формуле будет связан с X, а C2 будет связан с Y. Таким образом, понятие “… .Ri и Rj, взятые вместе, с образованием -CH2-CH2-O-CO-…”, означает лактон, в котором карбонил связан с C2. Однако, если говорят, что “…Ri и Rj, взятые вместе, образуют -CO-O-CH2-CH2-“, то обозначенная структура означает лактон, в котором карбонил связан с C1.
Количество атомов углерода в переменных заместителях может быть указано двумя способами. В первом способе используют префикс перед названием радикала, например, “C1-C4“, где “1” и “4” являются целыми числами, указывающими на минимальное и максимальное число атомов углерода в радикале. Этот префикс отделен от радикала пробелом. Например, “C1-C4 алкил” означает алкил, имеющий от 1 до 4 атомов углерода (включая его изомерные формы, если это не оговорено особо). Если имеется лишь один префикс, то этот префикс указывает на полное число атомов углерода в радикале. Так, например, 2-C4 алкоксикарбонил представляет собой группу CH3-(CH2)n-O-CO-, где n = 0, 1 или 2. Во втором способе указывается число атомов углерода для каждой части радикала отдельно, для чего обозначение “Ci-Cj” заключают в скобки и помещают непосредственно (без пробела) перед той частью радикала, к которой относится данное обозначение. Такое необязательное обозначение (C1-C3)алкоксикарбонил имеет то же самое значение, что и обозначение C2-C4 алкоксикарбонил, поскольку “C1-C3” относится только к количеству атомов углерода алкоксигруппы. Аналогично, хотя оба C2-C6 алкоксиалкил и (C1-C3)алкокси(C1-C3)алкил обозначают алкоксиалкильные группы, содержащие от 2 до 6 атомов углерода, однако, эти два определения отличаются друг от друга, поскольку в соответствии с первым определением, одна алкокси- или алкильная часть содержит 4 или 5 атомов углерода, тогда как в соответствии с последним определением, любая из этих групп ограничена лишь 3 атомами углерода.
Если заявленные соединения имеют достаточно сложный (циклический) заместитель, то в конце названия/обозначения этого конкретного заместителя дается примечание (в скобках), которое относится к тому же самому названию/обозначению в одной из СХЕМ, где также представлена структурная формула этого заместителя.
II. ОпределенияПротивопаразитарными соединениями настоящего изобретения являются: 15-алкил-14-гидроксисоединения (III), фторсоединения (VIII), 15-алкил-16-гидроксисоединения (X), 15-алкил-парагерквамид B (XIII), 2-дезоксо-14-гидроксисоединения (XXI), 2-дезоксосоединения (XXIII), 14-гидрокси-2-дезоксопарагерквамидные соединения (XXV), 14,15-дегидро-16-оксопарагерквамид B (XVII), 1,2-дегидросоединение (XXIX) и 2-алкил-2-дезоксосоединение (XXXI), их N-оксиды и фармацевтически приемлемые соли, если таковые существуют. Все температуры приведены в градусах Цельсия. ТГФ означает тетрагидрофуран. Солевой раствор относится к водному насыщенному раствору хлорида натрия. Хроматография (колоночная и флэш-хроматография), используемая для очистки/разделения соединений, описана указанием (в скобках) носителя, элюента. Следует отметить, что соответствующие фракции объединяют и концентрируют, в результате чего получают нужное соединение. ЯМР означает спектроскопию ядерного (протонного) магнитного резонанса, при этом химические сдвиги измеряют по отношению к сигналу эталона-тетраметилсилана и выражают в миллионных долях магнитного поля (м. д.) (  ).
МС означает масс-спектрометрию, которую выражают в единицах m/e, mz или масса/заряд. [M+H] + означает положительный ион-радикал + атом водорода. E1 означает электронный удар. CI (ХИ) означает химическую ионизацию. FAB означает бомбардировку быстрыми атомами.
HRMS (ВРМС) означает высокоразрешающую масс-спектрометрию.
Термин “фармацевтически приемлемый” относится к свойствам и/или веществам, которые являются приемлемыми для пациента с фармакологической/токсикологической точки зрения, а также для химиков-фармацевтов с физической/химической точки зрения, т. е. с точки зрения состава используемых веществ, технологии изготовления лекарственных средств, стабильности, переносимости пациентом и биологической доступности.
Фармацевтически приемлемыми анионными солями являются соли метансульфоновой, соляной, бромистоводородной, серной, фосфорной, азотной, бензойной, лимонной, винной, фумаровой, малеиновой кислот, CH3-(CH2)n-COOH, где n = 0-4, HOOC-(CH2)n-COOH, где n определено выше.
Если используются два растворителя, то их соотношения выражены в объемных отношениях (объем/объем).
Если твердое вещество растворено в растворителе, то растворимость выражают в виде отношения (масса/объем) твердого вещества к растворителю.
ПРИМЕРЫ ).
МС означает масс-спектрометрию, которую выражают в единицах m/e, mz или масса/заряд. [M+H] + означает положительный ион-радикал + атом водорода. E1 означает электронный удар. CI (ХИ) означает химическую ионизацию. FAB означает бомбардировку быстрыми атомами.
HRMS (ВРМС) означает высокоразрешающую масс-спектрометрию.
Термин “фармацевтически приемлемый” относится к свойствам и/или веществам, которые являются приемлемыми для пациента с фармакологической/токсикологической точки зрения, а также для химиков-фармацевтов с физической/химической точки зрения, т. е. с точки зрения состава используемых веществ, технологии изготовления лекарственных средств, стабильности, переносимости пациентом и биологической доступности.
Фармацевтически приемлемыми анионными солями являются соли метансульфоновой, соляной, бромистоводородной, серной, фосфорной, азотной, бензойной, лимонной, винной, фумаровой, малеиновой кислот, CH3-(CH2)n-COOH, где n = 0-4, HOOC-(CH2)n-COOH, где n определено выше.
Если используются два растворителя, то их соотношения выражены в объемных отношениях (объем/объем).
Если твердое вещество растворено в растворителе, то растворимость выражают в виде отношения (масса/объем) твердого вещества к растворителю.
ПРИМЕРЫИспользуя нижеследующее описание, каждый специалист может осуществить настоящее изобретение во всей его полноте без излишнего экспериментирования. В нижеследующих подробных примерах описано получение различных соединений и/или осуществление различных способов настоящего изобретения, однако при этом следует отметить, что эти примеры приводятся лишь в иллюстративных целях и не должны рассматриваться как некое ограничение объема изобретения. Для любого специалиста очевидно, что могут быть внесены определенные изменения, касающиеся используемых методов, реагентов и реакционных условий. Способ N 1. Получение и выделение маркфортина A Процесс получения посевного материала для ферментации Посевной материал для ферментации инокулировали с использованием агаровых пластин изолята Penicillium sp. UC 7780 (NRRL 18887), которые хранили в атмосфере жидкого азота. Три пластины размораживали и использовали в качестве инокулята. GS-7 состояла из глюкозы и муки из семян хлопчатника (которая продается фирмой Traders Protein, Procter & Gamble Oilseed Products Co. , Memphis, TN, U.S.A. под товарным знаком “Pharmamedia””. Для гидрирования компонентов среды использовали водопроводную воду, не содержащую примесей, и pH среды доводили до 7,2 путем добавления гидроксида аммония. Среду разливали в плотно закрывающиеся колбы без перегородок в количестве 300 мл на 1000 мл-колбу и стерилизовали в автоклаве в течение 30 минут при 121oC. Каждую плотно закрывающуюся колбу, содержащую 300 мл среды GS-7, инокулировали тремя агаровыми пластинками Penicilium sp. UC 7780 (NRRL 18887) и встряхивали на роторном шейкере при 250 об/мин, в течение 36 часов при 22oC. Процесс получения вторичного посевного материала Зрелые посевные культуры использовали в качестве инокулята для вторичной среды при норме засева 0,3%, Вторичная среда содержала 25 г моногидрата глюкозы (который продается фирмой C. P.C. International под товарным знаком “Cerelose”), 25 г муки из семян хлопчатника (которая продается под товарным знаком “Pharmamedia”), 329,8 мг MgCl2  6H2O; 11,4 мг MnSO4H2O; 3,2 мг FeSO47H2O; 1,8 мг Na2MoO42H2O; 367,6 мг CaCl22H2O; 84,2 мг NaCl; 5,8 мг KCl; 0,1 мг ZnSO47H2O; 0,1 мг CoCl26H2O; 3,1 мг CuSO45H2O и 0,5 мл силиконового пеногасителя (который продается под товарным знаком SAG-471 Antifoam) на один литр воды, очищенной методом обратного осмоса.
Компоненты среды в количестве, достаточном для 200 литров вторичной посевной среды, подвергали гидратации в воде, очищенной методом обратного осмоса, до дост. объема 190 л в 250-литровом ферментере. После получения состава, pH среды доводили до 7,2 путем добавления NH4OH, а затем среду стерилизовали в течение 30 минут при 121oC. Две плотно закрывающиеся колбы со зрелой первичной посевной культурой использовали в качестве инокулята при норме засева 0,3%. Вторичную посевную культуру инкубировали при 22oC с аэрацией 125 slm (л пос. культуры в мин.) при противодавлении 5 фунт/кв. дюйм (34,5 кПа) и при 250 об./мин. в течение 36 часов.
Процесс производственной ферментации 6H2O; 11,4 мг MnSO4H2O; 3,2 мг FeSO47H2O; 1,8 мг Na2MoO42H2O; 367,6 мг CaCl22H2O; 84,2 мг NaCl; 5,8 мг KCl; 0,1 мг ZnSO47H2O; 0,1 мг CoCl26H2O; 3,1 мг CuSO45H2O и 0,5 мл силиконового пеногасителя (который продается под товарным знаком SAG-471 Antifoam) на один литр воды, очищенной методом обратного осмоса.
Компоненты среды в количестве, достаточном для 200 литров вторичной посевной среды, подвергали гидратации в воде, очищенной методом обратного осмоса, до дост. объема 190 л в 250-литровом ферментере. После получения состава, pH среды доводили до 7,2 путем добавления NH4OH, а затем среду стерилизовали в течение 30 минут при 121oC. Две плотно закрывающиеся колбы со зрелой первичной посевной культурой использовали в качестве инокулята при норме засева 0,3%. Вторичную посевную культуру инкубировали при 22oC с аэрацией 125 slm (л пос. культуры в мин.) при противодавлении 5 фунт/кв. дюйм (34,5 кПа) и при 250 об./мин. в течение 36 часов.
Процесс производственной ферментацииПроизводственная среда состояла из 50 г свеклосахарной мелассы, 16 г рыбной муки (которая продается под товарным знаком Menhaden Select Fish Meal), 10 г дрожжевого экстракта (который продается под товарным знаком Fidco), 329,8 мг MgCl2 6H2O; 11,4 мг MnSO4H2O; 3,29 мг FeSO47H2O; 1,8 мг Na2MoO42H2O; 367,6 мг CaCl22H2O; 84,2 мг NaCl; 5,8 мг KCl; 0,1 мг ZnSO47H2O; 0,1 мг CoCl26H2O; 3,1 мг CuSO45H2O и 0,5 мл силиконового пеногасителя (который продается под товарным знаком SAG-4 71 Antifoam) на один литр воды, очищенной методом обратного осмоса.
Компоненты среды в количестве, достаточном для 5000 литров среды, подвергали гидратации в воде, очищенной методом обратного осмоса, до дост. объема 4700 литров в 5000-литровом ферментере. После получения состава pH среды доводили до 7,0 путем добавления KOH, а затем среду стерилизовали в течение 30 минут при 123oC. Зрелую вторичную посевную культуру использовали в качестве инокулята при норме засева 1,0%. Культуру инкубировали при 22oC с аэрацией 2500 slm (литр культуры в мин.) при противодавлении 5 фунт/кв. дюйм (34,5 кПа) и при 250 об./мин. в течение 96 часов.
Выделение маркфортина A.
Сбраживаемую среду в объеме 4900 л собирали в сборник путем пропускания через смеситель с большими сдвиговыми усилиями. После переноса добавляли 4% масс. /объем диатомовой земли и 1/2 объема метиленхлорида. Затем собранный объем фильтровали через фильтр-пресс. Осадок на фильтре 2 раза промывали 10% объемом метиленхлорида.
Полученный фильтрат декантировали для удаления водной фазы. Оставшуюся метиленхлоридную фазу, обогащенную продуктом, концентрировали до объема 44 л. Затем концентрат подвергали конечной очистке через фильтр с использованием 20%-ного объема концентрата (9 л) метиленхлорида и диатомовой земли.
Затем 53 л очищенного концентрата дополнительно очищали для отделения маркфортина A от других компонентов с помощью хроматографии на силикагеле и кристаллизации.
Перед хроматографией очищенный концентрат разделяли на четыре приблизительно равные аликвоты. Каждую аликвоту хроматографировали на вновь упакованной колонке диаметром 9 дюймов (22,5 см), приготовленной с использованием 25 кг сухого силикагеля (объем слоя 59 л), Загруженные колонки элюировали 120литрами 10% ацетона в метиленхлориде, 120 литрами 20% ацетона в метиленхлориде, 120 литрами 30% ацетона в метиленхлориде, 160 литрами 40% ацетона в метиленхлориде и 130 литрами ацетона, в результате чего собирали 30 и 40% элюаты в виде 20-литровых фракций. Элюаты контролировали с помощью ТСХ с использованием, например, системы растворителей, содержащей 6% изопропанола и 0,3% гидроксида аммония в метиленхлориде, для проявления пластинок с силикагелем (Whatman LK6DF). Фракции маркфортина A (содержащие небольшое количество маркфортина D, который хроматографируется вместе с D), кристаллизовали из ацетона. Соответствующие фракции (40-100 л) концентрировали при пониженном давлении до объема приблизительно 5 л. Раствор (или жидкую суспензию) переносили в роторный испаритель и концентрирование продолжали при пониженном давлении. Во время концентрации добавляли несколько 1-литровых порций ацетона до тех пор, пока метиленхлорид полностью не исчезнет. Полученную ацетоновую суспензию (объемом приблизительно 1 л) помещали на ночь в холодильник, после чего кристаллы маркфортина A собирали, промывали несколькими небольшими порциями холодного ацетона и сушили в вакууме. Эти кристаллы могут в качестве примеси содержать несколько процентов маркфортина D. После повторной перекристаллизации из метиленхлорида/ацетона (с вытеснением метиленхлорида, как описано выше) получали чистый маркфортин A. Выделение маркфортина D Сбраживаемую среду в объеме 4900 л собирали в сборник путем пропускания через смеситель с большими сдвиговыми усилиями. После переноса добавляли 4% масс. /объем диатомовой земли и 1/2 объема метиленхлорида. Затем собранный объем фильтровали через фильтр-пресс. Осадок на фильтре 2 раза промывали 10% объемом метиленхлорида. Полученный фильтрат декантировали для удаления водной фазы. Оставшуюся метиленхлоридную фазу, обогащенную продуктом, концентрировали до объема 44 л. Затем концентрат подвергали конечной очистке через фильтр с использованием 20%-ного объема концентрата (9 л) метиленхлорида и диатомовой земли. Затем 53 л очищенного концентрата дополнительно очищали с помощью хроматографии на силикагеле и кристаллизации для отделения маркфортина A от других компонентов. Перед проведением хроматографии очищенный концентрат разделяли на четыре приблизительно равные аликвоты. Каждую аликвоту хроматографировали на вновь упакованной колонке диаметром 9 дюймов (22,5 см), приготовленной с использованием 25 кг сухого силикагеля (объем слоя 59 л). Загруженные колонки элюировали 120 литрами 10% ацетона в метиленхлориде, 120 литрами 20% ацетона в метиленхлориде, 120 литрами 30% ацетона в метиленхлориде, 160 литрами 40% ацетона в метиленхлориде и 130 литрами ацетона, собирая 30 и 40%-элюаты в виде 20-литровых фракций. Элюаты контролировали с помощью ТСХ с использованием, например, системы растворителей, содержащей 6% изопропанола и 0,3% гидроксида аммония в метиленхлориде, для проявления пластинок с силикагелем (Whatman LK6DF). Фракции маркфортина A, содержащие маркфортин D, концентрировали. Один грамм этого материала растворяли в муравьиной кислоте (20 мл, 93%) и выдерживали в течение 16 часов при 20-25oC. После удаления летучих компонентов при пониженном давлении остаток подвергали хроматографии на силикагеле (1:20 MeOH:CH2Cl2), в результате чего получали маркфортин D (100 мг) в виде белого твердого продукта. Структура этого продукта может быть подтверждена с помощью ЯМР-спектроскопии и масс-спектрометрии. BPMC (FAB) M/Z [M+H] для C28H35N3O3+H вычислено: 462,2756, измерено: 462,2739. Способ 1A. Получение и выделение маркфортинов A и C Процесс получения первичного посевного материала для ферментации Посевной материал для ферментации инокулировали с использованием агаровых пластин изолята Penicillium sp. UC 7780 (NRRL 18887), которые хранили в атмосфере жидкого азота. Три пластины размораживали и использовали в качестве инокулята для 100 мл посевной среды GS-7. Среда GS-7 состояла из глюкозы и муки, приготовленной из семян хлопчатника (которая продается фирмой Traders Protein, Procter & Gamble Oilseed Products Co., Memphis, TN, U.S.A. под товарным знаком “Pharmamedia”), причем каждый из этих компонентов добавляли в концентрации 25 г/л водопроводной воды. После получения нужного состава среды, pH GS-7 доводили до 7,2 путем добавления NH4OH. Среду автоклавировали в 100 мл-овых объемах в 500-миллилитровых колбах для ферментации, не имеющих перегородок, в течение 30 минут. Стерильную среду GS-7 инокулировали, как описано выше, и встряхивали при 250 об./мин. в течение 35-58 часов при 23oC. Процесс производственной ферментации (колба-шейкер). Зрелые посевные культуры использовали в качестве инокулята для производственной среды при норме засева 1%. Производственная среда состояла из 45 г глюкозы, 25 г ферментативно гидролизованного казеина (который продается фирмой Sheffield Products, Norwich, N.Y., U.S.A. под товарным знаком Peptonized Milk Nutrient), 2,5 г дрожжевого экстракта (который продается Difco Laboratories, Detroit, MI под товарным знаком BACTO Yeast Extract Code: 0127) на литр водопроводной воды. После получения нужного состава pH производственной среды доводили до 7,0 путем добавления гидроксида калия. Эту среду затем автоклавировали в течение 30 минут в 100 мл-овых объемах, содержащихся в 500-миллилитровых колбах с перегородками. Стерильную производственную среду инокулировали, как описано выше, и встряхивали при 250 об./мин. в течение 7-14 дней при 21oC. Процесс производственной ферментации (чаны Labraferm). Зрелые посевные культуры использовали в качестве инокулята для стерильной производственной среды при норме засева 0,5%. Производственная среда описана выше. После доведения pH до 7,0 путем добавления KOH 10 л этой среды автоклавировали в течение 90 минут в 12-литровых чанах Labraferm (New Brunswick Scientific Co. , Inc.). Эти чаны инокулировали при норме засева 0,5% и их содержимое размешивали при 500 об./мин. в течение 5-9 дней при 20oC. Скорость воздушного потока поддерживали на уровне 10-15 л/мин. Выделение Маркфортинов A и C Весь сбраживаемый бульон (35 л) мацерировали (измельчали) на низкой скорости в большом промышленном смесителе Waring Blender, а затем смешивали с равным объемом метиленхлорида. Полученную смесь оставляли на ночь на хранении в холодильнике, а затем подвергали центрифугированию для расслоения эмульсии. Прозрачный метиленхлоридный слой сливали и выпаривали при пониженном давлении. Концентрированный раствор остатка (37,4 г) в метиленхлориде наносили на колонку, упакованную суспензией силикагеля (1 кг) в метиленхлориде. Колонку элюировали возрастающими концентрациями ацетона в метиленхлориде (10%, 20%, 30%, 40% и 50% ацетона). Фракции контролировали с помощью ТСХ и подходящие фракции выпаривали и кристаллизовали из ацетона, в результате чего получали маркфортин A и маркфортин C. Способ 1B. Получение и выделение маркфортинов A и C Процесс получения посевного материала для ферментации. Посевной материал для ферментации инокулировали с использованием агаровых пластин изолята Penicillium sp. UC 7780 (NRRL 18887), которые хранили в атмосфере жидкого азота. Три пластины размораживали и использовали в качестве инокулята для 100 мл посевной среды GS-7. Среда GS-7 состояла из глюкозы и муки, приготовленной из семян хлопчатника (которая продается фирмой Traders Protein, Procter & Gamble Oilseed Products, Co., Memphis, TN, U.S.A. под товарным знаком “Pharmamedia”), причем каждый из этих компонентов добавляли в концентрации 25 г/л водопроводной воды. После получения нужного состава среды pH GS-7 доводили до 7,2 с использованием NH4OH. Среду автоклавировали в течение 30 минут в 100 мл-овых объемах в 500-миллилитровых, не имеющих перегородок колбах для ферментации. Стерильную среду GS-7 инокулировали, как описано выше, и встряхивали при 250 об./мин. в течение 35-58 часов при 23oC. Процесс производственной ферментации (колба-шейкер). Зрелые посевные культуры использовали в качестве инокулята для производственной среды при норме засева 1%. Производственная среда состояла из 20 г глюкозы, 15 мл глицерина, 20 г муки из семян хлопчатника (которая продается фирмой Traders Protein, Procter & Gamble Oilseed Products Co., Memphis, TN, U. S.A. под товарным знаком “Pharmamedia”), 10 г соевой муки и 3 г K2HPO4 на литр водопроводной воды. После получения нужного состава pH производственной среды доводили до 6,8 путем добавления гидроксида калия. Затем эту среду автоклавировали в течение 30 мин. в 100 мл-овых объемах, содержащихся в 500-миллилитровых колбах с перегородками для ферментации. Стерильную производственную среду инокулировали, как описано выше, и встряхивали на шейкере при 250 об./мин. в течение 7-14 дней при 21oC. Процесс производственной ферментации (чаны Labraferm): Зрелые посевные культуры использовали в качестве инокулята для стерильной производственной среды при норме засева 0,5%. Производственная среда описана выше. После доведения pH до 7,0 путем добавления KOH, 10 л этой среды автоклавировали в течение 90 минут в 12-литровых чанах Labraferm. (New Brunswick Scientific Co. , Inc.). Эти чаны инокулировали при норме засева 0,5% и размешивали при 500 об./мин. в течение 5-9 дней при 20oC. Скорость потока воздуха поддерживали в пределах 10-15 л/мин. Выделение маркфортинов A и C Весь сбраживаемый бульон (35 л) мацерировали на низкой скорости в большом промышленном смесителе Waring Biender, а затем смешивали с равным объемом метиленхлорида. Полученную смесь оставляли на хранении на ночь а холодильнике, а затем подвергали центрифугированию для расслоения эмульсии. Прозрачный метиленхлоридный слой сливали и выпаривали при пониженном давлении. Концентрированный раствор остатка (37,4 г в метиленхлориде наносили на колонку, упакованную суспензией силикагеля (1 кг) в метиленхлориде. Колонку элюировали возрастающими концентрациями ацетона в метиленхлориде (10%, 20%, 30%, 40% и 50% ацетона). Фракции контролировали с помощью ТСХ и подходящие фракции выпаривали и кристаллизовали из ацетона, в результате чего получали маркфортин A и C. Синтез 14-замещенных маркфортинов B результате обработки маркфортина A (формула 1A, Схема I) йодистым цианом получали смесь (формула 5) 16 -иод-17– цианомаркфортина A и 16-иод-17-цианомаркфортина A, которая может быть разделена с помощью хроматографии на силикагеле. Дегидроиодирование этой смеси с использованием гидроксида калия в метаноле приводило к образованию 16,17-дегидро-17-цианомаркфортина A (Формула 6), который окисляли с использованием диоксида селена с получением 17-кетомаркфортина A (Формула 7). Введение двойной связи между C15 и C16 осуществляли путем селенирования 16-положения (фенил-селенилхлорид и LDA) с последующим окислением селенового промежуточного соединения перекисью водорода. В результате последующего элиминирования фенилселеновой кислоты получали 15,16-дегидро-17-кетомаркфортин A (Формула 8). Это соединение является ключевым промежуточным соединением в синтезе 14-гидроксимаркфортина A (Формула 10), в который это промежуточное соединение может быть превращено двумя различными способами.
В первом способе аллильное окисление 14-положения указанного соединения с использованием бис(триметилсилил)амида калия и 2-фенилсульфонил-3-фенилоксазиридина осуществляют путем окисления 16-положения с получением смеси нужного 14-гидрокси-15,16-дегидро-17-кетомаркфортина A (Формула 9a) и 14,15-дегидро-16-гидрокси-17-кетомаркфортина A (Формула 9b). Эти два продукта разделяют с помощью хроматографии на силикагеле. Соединение формулы 9a восстанавливают с использованием алюмогидрида лития в ТГФ с получением 14-гидроксимаркфортина A (Формула 10), т. е. соединения настоящего изобретения. Альтернативно, соединение Формулы 8 (Схема J) окисляют с помощью диоксида селена в диоксане, в результате чего получают смесь 14-гидрокси-15,16-дегидро-17-кетомаркфортина A (Формула 9A) и 15,16-дегидро-14,17-дикетомаркфортина A (Формула 11) 2:1. Эту смесь разделяют с помощью хроматографии на силикагеле. Каждое из этих соединений независимо превращают в 14-гидрокси-17-кетомаркфортин A (Формула 12a): соединение Формулы 9a – путем восстановления 15,16-двойной связи с использованием триэтилборгидрида лития; соединение Формулы 11 – путем восстановления карбонила в положении 14 с использованием боргидрида лития. В последнем случае также продуцируется равное количество 14-гидрокси-17-кетомаркфортина A (Формула 12b), которое удаляют с помощью хроматографии. Соединение Формулы 12a восстанавливают, используя комплекс борана-тетрагидрофурана (ТГФ), в результате чего получают 14-гидроксимаркфортин A (Формула 10).
14-Гидрокси-15,11-дегидро-17-кетомаркфортин A (Формула 9a. Схема K) восстанавливают триэтилборгидридом лития, в результате чего получают 14-гидрокси-17-кетомаркфортин A (Формула 12a). Это соединение превращают в 14,17-дикетомаркфортин A (Формула 13) посредством реакции окисления Сверна с использованием оксалилхлорида и ДМСО. После обработки бромидом метилмагния в реакции Гриньяра получают смесь 14-гидрокси-14-метил-17-кетомаркфортина A (Формула 14a) и 14-гидрокси-14-метил-17-кетомаркфортина A (Формула 14b), которую разделяют с помощью хроматографии на силикагеле. Соотношение продуктов зависит от используемого растворителя, так, например, метиленхлорид дает отношение 6: 1, тогда как ТГФ дает отношение > 50:1, соответственно. После восстановления соединения Формулы 13a с использованием алюмогидрида лития получают 14-гидрокси-14-метилмаркфортин A (Формула 15).
В результате окисления по Сверну 14-гидроксимаркфортина A (Формула 10, Схема L) получают 14-кетомаркфортин A (Формула 16), который восстанавливают с использованием боргидрида натрия, в результате чего получают 14-гидроксимаркфортин A (Формула 17). После обработки 14-кетомаркфортина A (Формула 16) бромидом этилмагния в реакции Гриньяра получают 14-гидрокси-14-этилмаркфортин A (Формула 19). После обработки 14-гидроксимаркфортина A (Формула 10) м-хлорпероксибензойной кислотой получают N-оксид 14-гидроксимаркфортина A (Формула 18). 14-метилмаркфортин A может быть получен из 14-гидрокси-14-метилмаркфортина A путем дегидроксилирования. Так, например, 14-гидрокси- 14-метилмаркфортин A обрабатывают фенилхлортионоформиатом в присутствии основания. Это тионоформиатное производное 14-гидрокси-14-метилмаркфортина A восстанавливают гидридом три-н-бутилолова, в результате чего получают 14-метилмаркфортин A.
Альтернативно, 14-гидроксимаркфортин A может быть синтезирован из маркфортина A (Схема M). После обработки маркфортина A бикарбонатом натрия и иодом в водном тетрагидрофуране получают 17-кетомаркфортин A (Формула 7), который может быть дисульфенилирован с использованием LDA и фенилдисульфида с получением 16-дитиофенил-17-кетомаркфортина A (Формула 20, Схема M) с выходом 60%. После окисления м-хлорпероксибензойной кислотой получают 16-тиофенил-16-сульфоксифенил-17-кетомаркфортин A (Формула 21), который подвергают реакции элиминирования в кипящем толуоле с получением 15,16-дегидро-16-тиофенил-17-кетомаркфортин A (Формула 22). После обработки м-хлорпероксибензойной кислотой получают 15,16-дегидро-16-сульфоксифенил-17-кетомаркфортин A (Формула 23), который подвергают перегруппировке с использованием диэтиламина в метаноле и в результате получают 15,16-дегидро-14-гидрокси- 17-кетомаркфортин A (Формула 9a).
14-Гидрокси-15-метилмаркфортин A (Формула 35, Схема N) может быть синтезирован из 15,16-дегидро-14-гидрокси-17- кетомаркфортина A (Формула 9a. Схема N). Так, например, 15,16-дегидро-14-гидрокси-17-кетомаркфортин A (Формула 9a) обрабатывают либо бромидом метилмагния, либо диметилкупратом лития с получением 15 -метил-14-гидрокси-17- кетомаркфортина A (Формула 34), который восстанавливают с использованием комплекса борана-диметилсульфида и получают 15 -метил-14-гидроксимаркфортин A (Формула 35). Посредством реакции окисления Сверна с использованием оксалилхлорида и ДМСО 15 -метил-14-гидрокси-17- кетомаркфортин A (Формула 34) превращают в 15 -метил-14,17-дикетомаркфортин A (Формула 36). После обработки бромидом метилмагния в реакции Гриньяра получают 15-метил-14-гидрокси-14-метил-17-кетомаркфортин A (Формула 37), который восстанавливают с использованием комплекса борана-диметилсульфида и получают 15-метил-14-гидрокси-14-метилмаркфортин A (Формула 38).
Нижеописанные способы могут быть использованы для получения 14-замещенных производных маркфортина B, C и D.
Получение 1: 16-Иод-17-цианомаркфортин A в виде смеси диастереомеров (Формула 5)Твердый йодистый циан (11,7 г, 76,5 ммоль) добавляли к раствору маркфортина A (10,5 г, 22 ммоль) в CHCl3 (150 мл) и полученную реакционную смесь нагревали с обратным холодильником до тех пор, пока весь маркфортин A не был израсходован (примерно 5 часов). Полученный черный раствор охлаждали до температуры 20-25oC, разбавляли CH2Cl2 (100 мл), промывали насыщенным NaHCO3, а затем промывали раствором Na2SO3. Органическую фазу отделяли, сушили MgSO4 и концентрировали досуха. Полученное неочищенное твердое вещество подвергали хроматографии на силикагеле (EtOAc:гексан, 3:2), в результате чего получали 16-иод-17-цианомаркфортин A (12,5 г, 90%) в виде белого порошкообразного твердого вещества. Структура полученного вещества может быть подтверждена с помощью спектроскопии ядерного магнитного резонанса и масс-спектрометрии. Получение 2: 16,17-Дегидро-17-цианомаркфортин A (Формула 6) 16-иод-17-цианомаркфортин A (9,5 г, 15 ммоль) растворяли в MeOH (150 мл), а затем добавляли водный раствор KOH (45%, 3 мл). Реакционную смесь перемешивали в течение 2 часов при температуре 20-25oC. После добавления воды полученный белый осадок собирали путем фильтрации, промывали водой и сушили в течение ночи в вакууме, в результате чего получали 16,17-дегидро-17-цианомаркфортин A (6,6 г, 75%) в виде белого порошка. Структура этого продукта может, быть подтверждена спектроскопией ядерного магнитного резонанса и масс-спектрометрией. МС (FAB) (бомбардировка быстрыми атомами): M/Z [M+H]: 501 Получение 3: 17-Кетомаркфортин A (Формула 7) Диоксид селена (2,9 г, 26 ммоль) добавляли к раствору 16,17-дегидро-17-цианомаркфортина A (6,0 г, 10 ммоль) в 95% EtOH (100 мл) и реакционную смесь перемешивали в течение 2 часов при температуре 20-25oC. Затем реакцию гасили путем добавления насыщенного NaHCO3 (100 мл). Полученную смесь экстрагировали CH2Cl2 (2 х 200 мл). Экстракты объединяли, сушили (MgSO4) и концентрировали с получением 7 г неочищенного продукта. Этот продукт очищали с помощью хроматографии на силикагеле (EtOAc) и получали 17-кетомаркфортин A (3,6 г, 75%) в виде белого твердого вещества. Структура этого вещества может быть подтверждена посредством спектроскопии ядерного магнитного резонанса и масс-спектрометрии. BPMC (FAB) M/Z: (M+H) для C28H33N3O5+H вычислено: 492,2498, измерено: 492,2478. Целевое соединение может быть синтезировано альтернативным и более предпочтительным методом с использованием п-толуолсульфоновой кислоты. Так, например, моногидрат п-толуолсульфоновой кислоты. (1 г) добавляли к раствору 16,17-дегидро-17-цианомаркфортина A (10 г) в 95% MeOH (50 мл) и реакционную смесь перемешивали в течение 1 часа при 20-25oC. К полученной смеси добавляли триэтиламин (2 мл), а затем растворитель выпаривали. Остаток растирали с 10%-ным водным раствором карбоната натрия (100 мл), после чего образовавшееся твердое вещество фильтровали и сушили, в результате чего получали целевое соединение в виде твердого продукта (выход 90%). Структура этого продукта может быть подтверждена посредством спектроскопии ядерного магнитного резонанса и масс-спектрометрии. Получение 4: 15,1б-Дегидро-17-кетомаркфортин A (Формула 8) Раствор диизопропиламида лития получали из раствора н-бутиллития (1,6 М, 9,9 мл, 15,4 ммоль) в гексане и диизопропиламине (2,2 мл, 15,7 ммоль). Этот раствор разбавляли безводным тетрагидрофураном (ТГФ, 20 мл) и охлаждали до температуры -78oC. После добавления по каплям раствора 17-кето-маркфортина A (2,0 г, 4,1 ммоль) в безводном ТГФ (20 мл) реакционную смесь оставляли на 1 час для нагревания до температуры -40oC. Затем смесь снова охлаждали до -78oC и по каплям обрабатывали фенилхлоридом селена (19 мг, 5,2 ммоль) в ТГФ (10 мл). Через 5 минут реакцию гасили насыщенным NaHCO3, экстрагировали CH2Cl2, сушили (MgSO4) и концентрировали, в результате чего получали желтое твердое вещество, которое может быть использовано без дополнительной очистки. Полученное вещество растворяли в ТГФ (150 мл) и обрабатывали H2O2 (30%, 1,5 мл) при температуре 0oC. После удаления охлаждающей бани реакционную смесь перемешивали в течение 30 минут при температуре 20-25oC. Затем реакцию гасили путем добавления NaOH (1 н, 100 мл). Полученную смесь экстрагировали CH2Cl2 (2 х 200 мл). После этого экстракты объединяли, сушили (MgSO4) и концентрировали с получением неочищенного продукта. Этот продукт очищали посредством хроматографии на силикагеле (EtOAc), в результате чего получали 15,16-дегидро-17-кетомаркфортин A (1,3 г, 65%) в виде белого твердого продукта. Структура этого продукта может быть подтверждена с помощью спектроскопии ядерного магнитного резонанса и масс-спектрометрии. BPMC (FAB) M/Z [M+H] для C28H31N3O5 + H вычислено: 490,2342, измерено: 490, 2345. Получение 5: Получение 14 -гидрокси-15,16-дегидро-17-кетомаркфортина A (Формула 9a) с использованием оксазиридинаРаствор бис(триметилсилил)амида калия в толуоле (0,5 М, 1 мл, 0,5 ммоль) по каплям добавляли к раствору 15,16-дегидро-17-кетомаркфортина A (66 мг, 0,14 ммоль) в ТГФ (2 мл) при температуре -78oC. Полученный мутный, слегка желтоватый раствор нагревали до температуры -40oC в течение 1 часа. Реакционную смесь охлаждали до -78oC, перемешивали в течение 15 минут, а затем по каплям обрабатывали раствором 2-фенил-сульфонил-3-фенилоксазиридина (42 мг, 0,16 ммоль) в ТГФ (2 мл).После перемешивания смеси в течение 5 минут реакцию гасили путем добавления NaHCO3. Затем смесь экстрагировали CH2Cl2 (2 х 25 мл). Экстракты объединяли, сушили (MgSO4) и концентрировали с получением неочищенного продукта. Этот продукт очищали посредством препаративной тонкослойной хроматографии на силикагеле (EtOAc) и получали 14 -гидрокси-15,16-дегидро-17-кетомаркфортин A (8 мг, 12%) в виде белого твердого вещества. Структура этого вещества может быть подтверждена с помощью спектроскопии ядерного магнитного резонанса и масс-спектрометрии. BPMC (FAB) M/Z [M+H] для C28H31N3O6 + H вычислено: 506,2291, измерено: 506,2280. Аналогичным образом был получен 14,15-дегидро-16-гидрокси-17-кетомаркфортин A (14 мг, 20%). Структура этого соединения может быть подтверждена с помощью спектроскопии ядерного магнитного резонанса.
Получение 6: Получение 14-гидрокси-15,16-дегидро-17-кетомаркфортина A (Формула 9a), 15,16-дегидро-14,17-дикетомаркфортина A (Формула 11) и 14,15-дегидро-16,17-дикетомаркфортина A (Формула 24) с использованием диоксида селена.
15,16-Дегидро-17-кетомаркфортин A (1,29 г, 2,6 ммоль) растворяли в п-диоксане (30 мл) и обрабатывали диоксидом селена (390 мг). Полученную смесь нагревали с обратным холодильником в течение 1 часа и растворитель выпаривали в вакууме. Образовавшийся остаток растирали с метиленхлоридом (30 мл) и фильтровали. Затем фильтрат концентрировали и остаток подвергали хроматографии на силикагеле (MeOH:EtOAc, 1:20), в результате чего получали 14-гидрокси-15,16-дегидро-17-кетомаркфортин A (430 мг, 32%) в виде твердого продукта. 15,16-Дегидро-14,17-дикетомаркфортин A (Формула 11, 212 мг, 16%) был также получен в результате хроматографии. 14,15-Дегидро-16,17-дикетомаркфортин A (Формула 24, 106 мг, 8%) также был получен после хроматографии. Структура этих продуктов может быть подтверждена посредством ядерного магнитного резонанса и масс-спектрометрии.
Получение 7: 15, 16-Дегидро-14,17-дикетомаркфортин A (Формула 11)14 -Гидрокси-15,16-дегидро-17-кетомаркфортин A (60 мг, Формула 9a) растворяли в метиленхлориде (10 мл) и обрабатывали диоксидом марганца (60 мг). Полученную смесь перемешивали 1 час при температуре 20-25oC и концентрировали. Остаток подвергали препаративной тонкослойной хроматографии на силикагеле (50%-ный метиленхлорид в EtOAc), в результате чего получали 15,16- дегидро-14,17-дикетомаркфортин A (Формула 11, 35 мг, 60%). Структура этого продукта может быть подтверждена с помощью спектроскопии ядерного магнитного резонанса и масс-спектрометрии.
Получение 8: 14-Гидроксимаркфортин A (Формула 10)14 -Гидрокси-15,16-легидро-17-кетомаркфортин A (20 мг, 0,040 ммоль) растворяли в ТГФ (5 мл) и обрабатывали раствором алюмогидрида лития (1 М, 0,11 мл, 0,11 ммоль) в ТГФ при температуре 0oC. Полученную смесь перемешивали в течение 0,5 часа при температуре 0oC, а затем добавляли раствор NaHCO3 (10%). Полученную смесь экстрагировали CH2Cl2 (2 х 10 мл). Затем экстракты объединяли, сушили (MgSO4) и растворитель удаляли при пониженном давлении. Остаток подвергали препаративной тонкослойной хроматографии на силикагеле (10% MeOH в EtOAc) и получали целевое соединение. BPMC (FAB, M/Z) [M+H] для C28H35N3O5 + H вычислено: 494,2655, измерено: 494,2653.
Получение 9: 14-гидрокси-17-кетомаркфортин A (Формула 12a)14 -гидрокси-15,16-дегидро-17-кетомаркфортин A (Формула 9a, 50 мг, 0,1 ммоль) растворяли в ТГФ (5 мл) и обрабатывали раствором триэтилборгидрида лития в ТГФ (1 М, 0,7 мл) при -78oC. Полученную смесь перемешивали в течение 0,5 часа при температуре -78oC. Реакцию гасили путем добавления MeOH (1 мл) и смесь концентрировали. Полученное твердое вещество хроматографировали на силикагеле (MeOH/CH2Cl2, 1:20) и получали 14-гидрокси-17-кетомаркфортин A (43 мг, 86%) в виде белого твердого вещества. Структура этого вещества может быть подтверждена с помощью ЯМР-спектроскопии и масс-спектрометрии. BPMC (FAB) M/Z [M+H] для C28H33N3O6 + H вычислено: 508,2447; измерено: 508,2437.
Получение 10: Получение 14-гидрокси-17-кетомаркфортина A(Формула 12а) из 15,16-дегидро-14,17-дикетомаркфортина A (Формула 11) 15,16-Дегидро-14,17-дикетомаркфортин A (470 мг, 0,93 ммоль) растворяли в ТГФ и обрабатывали раствором боргидрида лития в ТГФ (1 М, 2 мл) при комнатной температуре. Полученную смесь перемешивали в течение 2 часов, а затем добавляли раствор NaHCO3 (10%). Затем смесь экстрагировали CH2Cl2 (2 х 20 мл). Экстракты объединяли, сушили (MgSO4), а растворитель выпаривали. Образовавшийся остаток содержал смеси двух эпимеров, которые сразу разделяли с помощью хроматографии на силикагеле (MeOH/EtAOC, 1:20), в результате чего получали 14 -гидрокси-17-кетомаркфортин A (90 мг, 19%) и 14-гидрокси-17- кетомаркфортин A (94 мг, 20%). Структура этих продуктов может быть подтверждена посредством ЯМР-спектроскопии и масс-спектрометрии.
Получение 11: Получение 14-гидроксимаркфортина A (Формула 10) из 14-гидрокси-17-кетомаркфортина A (Формула 12a)14 -Гидрокси-17-кетомаркфортин A (413 мг, 0,81 ммоль) растворяли в ТГФ (20 мл) и обрабатывали раствором ТГФ-боранового комплекса в ТГФ (1 М, 2,43 мл) при температуре 0oC. Смесь перемешивали в течение 2,25 часа. После перемешивания в течение еще 0,5 часа к смеси добавляли MeOH (3 мл). После выпаривания растворителя остаток хроматографировали на силикагеле (MeOH:EtOAc, 1: 16), в результате чего получали 14-гидроксимаркфортин A (250 мг, выход 92% исходя из выделенного исходного продукта) и 14-гидрокси-17-кетомаркфортин A (исходный продукт, 140 мг, 34%).
Получение 12: 14,17-Дикетомаркфортин A (Формула 13)Раствор оксалилхлорида (40 мкл) в безводном CH2Cl2 (5 мл) обрабатывали диметилсульфоксидом (45 мкл) при температуре -78oC. Полученную смесь перемешивали в течение 1 часа при температуре -78oC. После этого по каплям добавляли раствор 14 -гидрокси-17-кетомаркфортина A (27 мг) в CH2Cl2 (2 мл). Реакционную смесь перемешивали в течение 20 минут при температуре -78oC. К этой реакционной смеси добавляли триэтиламин (0,3 мл) и смесь нагревали до комнатной температуры в течение 20 минут. Затем смесь распределяли между 10%. Na2CO3 (10 мл) и CH2Cl2 (10 мл). Органический слой сушили (MgSO4) и концентрировали. Остаток подвергали колоночной хроматографии на силикагеле (MeOH: CH2Cl2, 1:20) и получали 14,17-дикетомаркфортин A (22 мг, 80%) в виде белого твердого продукта. Структура этого продукта может быть подтверждена с помощью ЯМР-спектроскопии и масс-спектрометрии. BPMC (FAB) M/Z [M+H] для C28H31N3O6 + H вычислено: 506,2291; найдено: 506,2280.
Получение 13: 14-гидрокси-14-метил-17-кетомаркфортин A (Формула 14a)Раствор 14,17-дикетомаркфортина A (16 мг, 0,032 ммоль) в CH2Cl2 (5 мл) при температуре -78oC обрабатывали раствором бромида метилмагния (3 М, 0,16 мл, 0,48 ммоль) в Et2O при -78oC. Полученную смесь перемешивали в течение 0,5 часа при -78oC. Реакцию гасили путем добавления нескольких капель 10% Na2CO3. Полученную смесь разбавляли CH2Cl2 (10 мл), сушили (MgSO4) и концентрировали. Остаток хроматографировали на силикагеле (MeOH/CH2Cl2, 1:20) и получали 14 -гидрокси-14-метил-17-кетомаркфортин A (8 мг, 50%, Rf=0,25) в виде белого твердого продукта. Структура этого продукта может быть подтверждена посредством ЯМР-спектроскопии и масс-спектрометрии. BPMC (FAB) M/Z [M+H] для C29H35N3O6 + H вычислено: 522,2604; измерено: 522,2620. Из полученного слоя был также получен 14-гидрокси-14-метил-17-кетомаркфортин A (1,2. мг, 7%, Rf= 04) в виде белого твердого продукта. Структура этого продукта может быть подтверждена с помощью ЯМР-спектроскопии и масс-спектрометрии. BPMC (FAB) M/Z [M+H] для C29H35N3O6 + H вычислено: 522,2604; измерено: 522,2630. Полученное таким образом отношение 6:1 продуктов увеличивается более чем до 50:1, а выход увеличивается до 80% в том случае, если в качестве реакционного растворителя вместо CH2Cl2 используется ТГФ.
Получение 14: 14-Гидрокси-14-метилмаркфортин A (Формула 15)Раствор 14 -гидрокси-14-метил-17-кетомаркфортина A (5 мг, 0,01 ммоль) в ТГФ (5 мл) обрабатывали раствором алюмогидрида лития (1 М, 0,03 мл, 0,03 ммоль) в ТГФ при температуре 0oC. Полученную смесь перемешивали в течение 0,5 часа при 0oC, а затем добавляли раствор NaHCO3 (10%). Смесь экстрагировали CH2Cl2 (2 х 5 мл). Экстракты объединяли, сушили (MgSO4), а растворитель выпаривали. Остаток подвергали препаративной тонкослойной хроматографии на силикагеле (MeOH/CH2Cl2, 1:20) и получали 14-гидрокси-14 -метилмаркфортин A (2 мг, 40%). Структура этого продукта может быть подтверждена посредством ЯМР-спектроскопии и масс-спектрометрии. BPMC (FAB) M/Z [M+H] для C29H37N3O5 + H вычислено: 508,2811; измерено: 508,2816.
Получение 15: 14-Кетомаркфортин A (Формула 16)Раствор оксалилхлорида (150 мкл) в безводном CH2Cl2 (20 мл) обрабатывали ДМСО (170 мкл) при температуре -78oC. Полученную смесь перемешивали 1 час при -78oC. Затем по каплям добавляли раствор 14 -гидроксимаркфортина A (110 мг) в CH2Cl2 (5 мл). Реакционную смесь перемешивали в течение 20 минут при температуре -78oC. К этой реакционной смеси добавляли триэтиламин (1 мл) и смесь нагревали до комнатной температуры в течение 20 минут. Затем смесь распределяли между 10% Na2CO3 (20 мл) и CH2Cl2 (20 мл). Органический слой сушили (MgSO4) и концентрировали. Остаток хроматографировали на силикагеле (MeOH: CH2Cl2, 1: 25) и получали 14-кетомаркфортин A (82 мг, 75%) в виде белого твердого продукта. Структура этого продукта может быть подтверждена с помощью ЯМР-спектроскопии и масс-спектрометрии. BPMC (FAB) M/Z [M+H] для C28H33N3O5 + H вычислено: 492,2498; измерено: 492,2510.
Получение 16: 14-Гидроксимаркфортин A (Формула 17)Раствор 14-кетомаркфортина A (10 мг) в MeOH (2 мл) обрабатывали боргидридом натрия (5 мг) при температуре 0oC. Смесь перемешивали в течение 0,5 часа при температуре 0oC, а затем добавляли раствор NaHCO3 (10%). Полученную смесь экстрагировали CH2Cl2 (2 х 10 мл). Экстракты объединяли, сушили (MgSO4) и растворитель выпаривали. Остаток подвергали препаративной тонкослойной хроматографии на силикагеле (MeOH/EtOAc, 1:16), в результате чего получали 14 -гидроксимаркфортин A (5 мг, 50%). Структура этого продукта может быть подтверждена посредством ЯМР-спектроскопии и масс-спектрометрии. BPMC (FAB) M/Z [M+H] для C28H35N3O5 + H вычислено: 494,2655; измерено: 494,2653.
Получение 17: N-оксид 14-гидроксимаркфортина A (Формула 18)Раствор 14 -гидроксимаркфортина A (15 мг) в CH2Cl2 (3 мл) обрабатывали м-хлорпероксибензойной кислотой (15 мг) при 0oC. Полученную смесь перемешивали в течение 0,5 часа при температуре 0oC, обрабатывали триэтиламином (30 мкл) и концентрировали. Остаток подвергали препаративной тонкослойной хроматографии на силикагеле (MeOH: CH2Cl2, 1:8) и получали N-оксид 14-гидроксимаркфортина A (12 мг, 80%). Структура данного продукта может быть подтверждена посредством масс-спектрометрии. BPMC (FAB) M/Z [M+H] для C28H35N3O6 + H вычислено: 510,2604; измерено: 510,2615.
Получение 18: 14-Гидрокси-14 -этилмаркфортин A (Формула 19)Раствор 14-кетомаркфортина A (25 мг, 0,05 ммоль) в ТГФ (5 мл) при -78oC обрабатывали раствором бромида этилмагния (3 М, 0,15 мл 0,45 ммоль) в Et2O при температуре -78oC. Полученную смесь перемешивали в течение 0,5 часа при -78oC. Затем реакционную смесь нагревали до комнатной температуры в течение 20 минут. Реакцию гасили путем добавления 10% Na2CO3 (несколько капель). Полученную смесь разбавляли CH2Cl2 (10 мл), сушили (MgSO4) и концентрировали. Остаток подвергали хроматографии на силикагеле (MeOH: CH2Cl2, 1:20) и получали 14 -гидрокси-14 -этилмаркфортин A (10 мг, 45%) в виде белого твердого продукта. Структура этого продукта может быть подтверждена с помощью ЯМР-спектроскопии и масс-спектрометрии. BPMC (FAB) M/Z [M+H] для C30H39N3O5 + H вычислено: 522,2968; измерено: 522,2983.
Получение 19: Получение 14-метилмаркфортина A из 14-гидрокси-14-метилмаркфортина AРаствор бис(триметилсилил)амида калия в толуоле (0,5 М, 1 мл, 0,5 ммоль) по каплям добавляли к раствору 14 – гидрокси-14-метилмаркфортина A (66 мг, 0,14 ммоль) в ТГФ (2 мл) при -78oC. Полученный светло-желтый мутный раствор нагревали до температуры -40oC в течение 1 часа. Реакционную смесь охлаждали до -78oC, перемешивали 15 минут и по каплям обрабатывали раствором фенилхлортионоформиата (0,094 мл, 0,7 ммоль) в ТГФ (2 мл). Через 10 минут баню из сухого льда удаляли. Еще через 3 часа реакцию гасили путем добавления NaHCO3. Смесь экстрагировали CH2Cl2 (2х25 мл). Экстракты объединяли, сушили (MgSO4) и концентрировали, в результате чего получали неочищенный продукт. Этот продукт очищали с помощью препаративной тонкослойной хроматографии на силикагеле (EtOAc) и получали 14-O-фенокситиокарбонил-14-метилмаркфортин A.
К раствору 14-O-фенокситиокарбонил-14-метилмаркфортина A (64 мг, 0,1 ммоль) в толуоле (5 мл) добавляли AlBN (3,3 мг), а затем гидрид трибутилолова (54 мкл, 0,2 ммоль). Полученную смесь нагревали с обратным холодильником в течение 3 часов. После выпаривания растворителя остаток очищали с помощью препаративной тонкослойной хроматографии (силикагель, EtOAc), в результате чего получали 14-метилмаркфортин A. Структура этого соединения может быть подтверждена посредством спектроскопии ядерного магнитного резонанса и масс-спектрометрии.
Получение 20: Альтернативный синтез 17-кетомаркфортина A (Формула 7)К маркфортину A (65 г, 0,136 моль) и бикарбонату натрия (137 г, 1,63 моль) в тетрагидрофуране (ТГФ, 2 л) и воде (1,25 л) при нагревании с обратным холодильником в течение одного часа по каплям добавляли раствор иода (206 г, 0,81 моль) в ТГФ (1,25 л). (Альтернативный способ предусматривает перемешивание смеси при комнатной температуре в течение 16 часов). После медленного охлаждения до температуры окружающей среды (2,5 часа) реакцию гасили путем добавления насыщенного тиосульфата натрия (Na2S2O3, 1,5 л)) и смесь экстрагировали этилацетатом (2 х 1 л). Объединенные органические слои промывали насыщенным тиосульфатом натрия (1 л), сушили (MgSO4), фильтровали, выпаривали и сушили в течение ночи в вакуумной печи (65oC), в результате чего получали 62 г неочищенного 17-кетомаркфортина A (Формула 7) в виде желтого твердого продукта. 1H-ЯМР (300 МГц, CDCl3): 7,68 (с, 1H), 6,80 (д, 1H), 6,70 (д, 1H), 6,32 (д, 1H), 4,90 (д, 1H), 3,75 (кв, 2H), 3,23 (т, 1H), 3,09 (с, 3H), 2,80 (д, 1H), 2,65 (д, 1H), 2,49-2,21 (м, 2H), 2,08 (д, 1H), 1,98-1,45 (м, 5H), 1,46 (с, 3H), 1,44 (с, 3H), 1,09 (с, 3H), 0,90 (с, 3H).
Альтернативно, вместо иода может быть использован ICI.
Получение 21: 16-Дитиофенил-17-кетомаркфортин A (Формула 20)Неочищенный 17-кетомаркфортин A (5 г, 10,2 ммоль) в ТГФ (150 мл) при температуре -78oC через канюлю вводили в раствор LDA, который был получен путем добавления н-бутиллития (1,6 М, 24,8 мл, 0,04 моль) по каплям к диизопропиламину (5,7 мл, 0,041 моль) в ТГФ (100 мл) при температуре 0oC. Реакционную смесь медленно нагревали до -50oC в течение одного часа. Полученную красно-коричневую мутную смесь обрабатывали фенилдисульфидом (4,4 г, 0,02 моль). Затем реакцию сразу гасили насыщенным раствором бикарбоната натрия (100 мл) и экстрагировали метиленхлоридом (CH2Cl2, 300 мл). Органическую фазу сушили (MgSO4), концентрировали (до 8 г) и хроматографировали на силикагеле (120 г, 60% этилацетат/гексан), в результате чего получали целевое соединение в виде беловатого твердого вещества (4,4 г, 61% исходя из маркфортина A). FAB-MC: 708 (M+H). 1H-ЯМР (300 МГц, CDCl3) 7,74 (с, 1H), 7,71 (д, 2H), 7,64(д, 2H), 7,45-7,30 (м, 6H), 6,81 (д, 1H), 6,72 (д, 1H), 6,32 (д, 1H), 4,91 (д, 1H), 3,70 (кв, 2H), 3,16 (т, 1H,), 3,01 (с,.3H), 2,75 (д, 1H,), 2,53 (дт, 1H), 2,35 (дт, 1H), 2,15-1,50 (м, 5H), 1,47 (с, 3H), 1,45 (с, 3H), 1,06 (с, 3H,), 0,82 (с, 3H).
Получение 22: 16-Тиофенил-16-сульфоксифенил-17-кетомаркфортин A (Формула 21)К 16-дитиофенил-17-кетомаркфортину A (10 г, 14 ммоль) в CH2Cl2 (250 мл) при -78oC в атмосфере азота в течение 15 минут по каплям добавляли м-хлорпероксибензойную кислоту (64%, 4,2 г, 15,5 ммоль) в CH2Cl2 (200 мл). Реакцию сразу гасили насыщенным тиосульфатом натрия (200 мл), а затем разбавляли насыщенным NaHCO3 (200 мл) и экстрагировали CH2Cl2 (200 мл). После сушки (MgSO4) и концентрирования при пониженном давлении получали 11 г неочищенного 16-тиофенил-16-сульфоксифенил-17-кетомаркфортина A (Формула 21). 1H-ЯМР (300 МГц, CDCl3): 8,0-7,29 (м, 11H), 6,80 (д, 1H), 6,70 (д, 1H), 6,31 (д, 1H), 4,90 (д, 1H), 3,68 (д, 1H), 3,41 (д, 1H), 3,14 (т, 1H), 3,07 (с, 3H), 2,82 (дт, 1H), 2,80-2,65 (м, 2H), 2,16 (дт, 1H), 2,05-1,1 (м, 4H), 1,47 (с, 3H), 1,43 (с, 3H), 0,96 (с, 3H), 0,83 (с, 3H).
Получение 23: 16-Тиофенил-15,16-дегидро-17-кетомаркфортин A (Формула 22)Неочищенный 16-тиофенил-16-сульфоксифенил-17-кетомаркфортин A (Формула 21, 11 г) в толуоле (250 мл) нагревали с обратным холодильником в течение 45 минут, а затем охлаждали до комнатной температуры, разбавляли насыщенным бикарбонатом натрия (300 мл) и экстрагировали EtOAc (300 мл). Органический слой сушили (MgSO4) и концентрировали, в результате чего получали 10,6 г неочищенного 16-тиофенил-15,16-дегидро-17-кетомаркфортина A (Формула 22). FAB-MC: 598 (M+H); BPMC M/Z (M++H, C34H35N3O5S + H1) вычислено: 598,2376, набл, 598,2287. 1H-ЯМР (300 МГц, CDCl3): 8,18 (с, 1H), 7,55-7,45 (м, 2H), 7,29-7,45 (м, 3H), 6,83 (д, 1H), 6,70 (д, 1H), 6,34 (д, 1H), 5,92 (дт, 1H), 4,91 (д, 1H), 3,87 (кв, 2H), 3,30 (дд, 1H), 3,21 (т, 1H), 3,08 (с, 3H), 2,80 (д, 1H), 2,35 (дд, 1H), 2,10 (д, 1H), 2,03 (дд, 1H), 1,78 (дд, 1H), 1,46 (с, 3H), 1,44 (с, 3H), 1,11 (с, 3H), 0,88 (с, 3H).
Получение 24: 16-Сульфоксифенил-15, 16-дегидро-17-кетомаркфортин A (Формула 23)К неочищенному 16-тиофенил-15,16-дегидро-17-кетомаркфортину A (формула 22, 10,6 г) в метиленхлориде (300 мл) при -78oC по каплям добавляли м-хлорпероксибензойную кислоту (64%, 2,8 г) в CH2Cl2 (125 мл). Реакцию гасили насыщенным тиосульфатом натрия (300 мл) и насыщенным бикарбонатом натрия (300 мл), а затем смесь экстрагировали метиленхлоридом (300 мл). Органический слой сушили (MgSO4), фильтровали и концентрировали, в результате чего получали 13 г неочищенного 16-сульфоксифенил-15,16-дегидро-17-кетомаркфортина A (Формула 23). 1H-ЯМР (300 МГц, CDCl3): 7,75-7,3 (м, 5H), 6,81 (с, 1H), 6,75-6,6 (м, 2H), 6,31 (д, 1H), 4,90 (д, 1H), 3,78-3,58 (м, 2H), 3,22 (т, 1H), 2,98 (с, 3H), 2,88-2,45 (м, 2H), 2,12-1,55 (м, 5H), 1,46 (с, 3H), 1,44 (с, 3H), 1,12 (с, 3H), 0,88 (с, 3H).
Получение 25: 14-Гидрокси-15,16-дегидро-17-кетомаркфортин A (Формула 9a)К неочищенному 16-сульфоксифенил-15,16-дегидро-17-кетомаркфортину A (Формула 23, 13 г) в водном растворе MeOH (10:1, 300 мл) добавляли диэтиламин (15 мл). После нагревания с обратным холодильником в течение 0,5 часа реакционную смесь охлаждали до комнатной температуры, разбавляли водой (450 мл) и экстрагировали CH2Cl2 (500 мл). После сушки (MgSO4) и концентрирования остаток хроматографировали на силикагеле (130 г, элюент 30% ацетон/CH2Cl2), в результате чего получали 14 -гидрокси-15,16-дегидро-17-маркфортин A (Формула 9a, 3,6 г, 50% выход исходя из 16-дитиофенил-17-кетомаркфортина A) в виде белого твердого вещества.
Получение 26: 14-Гидрокси-14 -винилмаркфортин A (Формула 30)Раствор 14-кетомаркфортина A (200 мг, 0,4 ммоль) в ТГФ (5 мл) при -78oC обрабатывали раствором бромида винилмагния (1 М, 4,0 мл, 4 ммоль) в ТГФ при температуре -78oC. Полученную смесь перемешивали в течение 2 часов при -78oC, а затем нагревали до комнатной температуры. Затем смесь перемешивали при комнатной температуре еще 2 часа. Реакцию гасили путем добавления 10% Na2CO3 (3 мл). После этого смесь разбавляли CH2Cl2 (30 мл), промывали насыщенным раствором хлорида аммония, сушили (MgSO4) и концентрировали. Остаток хроматографировали на силикагеле (гексан/ацетон, 6: 4) и получали 14 -гидрокси-14 -винилмаркфортин A (120 мг, 60%, Rf=0,45) в виде белого твердого вещества.
1H-ЯМР (300 МГц, CDCl3): 7,86 (с, NH), 6,78 и 6,67 (д, J = 8,1Гц, C4-H и C5-H), 6,32 (д, J = 7,7Гц, C24-H), 6,58 (дд, J = 17,4, 10,9Гц, 1H, винил), 5,43 (д, J = 17,4Гц, 1H, винил), 5,18 (д, J = 10,9Гц, 1H, винил), 4,89 (д, J = 7,7Гц, C25-H), 3,7 (шир., 1H), 3,11 (с, 3H, N-Ме), 2,95 (т, 1H, C20-H), 2,8-1,5 (м, 12H), 1,44 (с, 6H, C27-H и C28-H), 1,08 (с, 3H), 0,82 (с, 3H);МС (FAB) M/Z (M+H]:520 Получение 27; N-Оксид 14 -гидрокси-14-метилмаркфортина A (Формула 32)Раствор 14 -гидроксимаркфортина A (30 мг) в CH2Cl2 (3 мл) обрабатывали м-хлорпероксибензойной кислотой (20 мг) при 0oC. Смесь перемешивали в течение 0,5 часа, а затем распределяли между 5% водным раствором бикарбоната натрия (10 мл) и метиленхлоридом (20 мл). Затем слои разделяли, а водный слой экстрагировали метиленхлоридом (10 мл). Объединенные экстракты сушили сульфатом магния, фильтровали и выпаривали в вакууме при 0oC, а затем обрабатывали триэтиламином (30 мкл) и концентрировали, в результате чего получали целевое соединение в виде твердого продукта (20 мг).
1H-ЯМР (300 МГц, CD3OD) 6,91 и 6,70 (д, J = 8,1Гц, C4-H и C5-H), 6,36 (д, J = 7,7Гц, C24-H), 4,91 (д, J = 7,7Гц, C25-H), 4,08 и 3,76 (АВ кв, J = 12,9Гц, 2H, C12-H), 3,5-3,1 (м, 4H), 3,12 (с, 3H, N-Me), 2,8-1,6 (м, 7H), 1,46 и 1,44 (2с, 6H, C27-H и C28-H), 1,50 (с, 3H, C14-Me), 1,20 (с, 3H), 0,93 (с, 3H).
Получение 28: 14-Гидрокси-15-метилмаркфортин A (Формула 35)14 -гидрокси-15-метил-17-кетомаркфортин A-(90 мг, 0,18 ммоль), растворенный в ТГФ (10 мл), обрабатывали боран- диметилсульфидным комплексом (12 М, 0,18 мл) при 0oC. Полученную смесь перемешивали в течение 2 часов при 0oC, а затем добавляли MeOH (0,4 мл) и перемешивали еще 1 час. После выпаривания растворителя остаток подвергали хроматографии на силикагеле (ацетон/метиленхлорид, 30: 70), в результате чего получали 14-гидрокси-15-метилмаркфортин A (20 мг) в виде твердого вещества.
1H-ЯМР (300 МГц, CDCl3) 8,39 (с, NH), 6,79 и 6,70 (д, J = 8,1Гц, C4-H и C5-H), 6,36 (д, J = 7,7Гц, C24-H), 4,91 (д, J = 7,7Гц, C25-H), 3,81 (шир, 1H, C14-H), 3,67 (д, J = 11,7Гц, C12-H), 3,03 (т, 1H, C20-H), 3,11 (с, 3H, N-Ме), 2,68 и 1,86 (д, 2H, J = 15,7Гц, C10-H), 2,7-1,2 (м, 8H), 1,44 (2с, 6H, C27-H и C28-H), 1,02 (д, 3H, J = 6,8Гц, C15-Me), 1,11 (с, 3H), 0,85 (с, 3H), BPMC (FAB) M/Z [M+H] для C29H37N3O5 + H вычислено: 508,2811, измерено: 508,2840.
Получение 29: 14,17-Дикето-15-метилмаркфортин A (Формула 36)Раствор оксалилхлорида (40 мкл) в безводном CH2Cl2 (5 мл) обрабатывали ДМСО (45 мкл) при -78oC. Полученную смесь перемешивали в течение 1 часа при -78oC. К смеси по каплям добавляли раствор 14 -гидрокси-15-метил-17- кетомаркфортина A (27 мг) в CH2Cl2 (2 мл). Реакционную смесь перемешивали в течение 20 минут при -78oC. После добавления триэтиламина (0,3 мл) реакционную смесь нагревали до комнатной температуры в течение 20 минут. Затем смесь распределяли между 10% Na2CO3 (10 мл) и CH2Cl2 (10 мл). Органический слой сушили (MgSO4) и концентрировали. Остаток хроматографировали на силикагеле (MeOH: CH2Cl2 1:20) и получали 14,17-дикетомаркфортин A (22 мг, 80%) в виде белого твердого продукта. Структура этого продукта может быть подтверждена посредством ЯМР-спектроскопии и масс-спектрометрии. BPMC (FAB) M/Z (M+H] для C28H31N3O6 + H вычислено: 506,2291; измерено: 506,2280.
Получение 30: 14-Гидрокси-14-метил-15-метил- 17-кетомаркфортин A (Формула 37)Раствор 14,17-дикето-15 -метилмаркфортина A (25 мг, 0,05 ммоль) в CH2Cl2 (5 мл) при -78oC обрабатывали раствором бромида метилмагния (3 М, 0,2 мл, 0,6 ммоль) в Et2O при -78oC. Полученную смесь перемешивали в течение 0,5 часа при -78oC. Реакцию гасили путем добавления 10% Na2CO3 (несколько капель). Полученную смесь разбавляли CH2Cl2 (10 мл), сушили (MgSO4) и концентрировали. Остаток подвергали хроматографии на силикагеле (MeOH:CH2Cl2, 1:25) и получали 14 – гидрокси-14-метил-15-метил-17-кетомаркфортин A (16 мг, 62%) в виде белого твердого продукта.
1H-ЯМР (300 МГц, CDCl3) 8,13 (с, 1H), 6,78 (д, 1H), 6,70 (д, 1H), 6,33 (д, 1H), 4,91 (д, 1H), 3,75 (кв, 2H), 3,16 (т, 1H), 3,05 (с, 3H), 2,78 (д, 1H), 2,68-2,57 (м, 1H), 2,42-2,0 (м, 6H), 1,64 (с, 3H), 1,45 (с, 3H), 1,44 (с, 3H), 1,11 (с, 3H), 1,04 (д, 3H), 0,92 (д, 3H).
Получение 31: 14-Гидрокси-14-метил-15-метилмаркфортин A (Формула 38)14 -гидрокси-14-метил-15-метил-17-кетомаркфортин A (15 мг, 0,028 ммоль) растворяли в ТГФ (10 мл) и обрабатывали боран-диметилсульфидным комплексом (10 М, 0,02 мл) при 0oC. Полученную смесь перемешивали в течение 2 часов при 0oC, а затем добавляли MeOH (0,4 мл) и эту смесь перемешивали еще 1 час. После выпаривания растворителя остаток подвергали хроматографии на силикагеле (ацетон: метиленхлорид = 30: 70), в результате чего получали 14-гидрокси-14-метил-15 -метилмаркфортин A (4 мг, 29%) в виде твердого вещества.
1H-ЯМР (300 МГц, CDCl3) 7,82 (с, 1H), 6,79 (д, 1H), 6,67 (д, 1H), 6,33 (д, 1H), 4,90 (д, 1H), 3,65 (д, 1H), 3,09 (с, 3H), 2,98 (т, 1H), 2,69 (д, 1H), 2,60-2,22 (м, 7H), 2,06 (дд, 1H), 1,87 (д, 1H), 1,85-1,75 (м, 1H), 1,44 (с, 6H), 1,43 (с, 3H), 1,10 (с, 3H), 0,94 (д, 3H), 0,86 (с, 3H).
Пример 1: 15-Этил-14-гидрокси-17-оксомаркфортин A (II)К иодиду меди (1) (0,18 г, 0,95 ммоль) в ТГФ (10 мл) при 0oC по каплям добавляли бромид этилмагния (1 М в ТГФ, 2 мл, 2 ммоль). После перемешивания в течение 0,25 часа при температуре 0oC реакционную смесь по каплям обрабатывали 14 -гидрокси-15,16-дегидро-17-оксомаркфортином A (1, 0,1 г, 0,2 ммоль) в ТГФ (5 мл) при 0oC. Через 1 час реакцию гасили насыщенным хлоридом аммония (25 мл), а затем экстрагировали этилацетатом (2 х 25 мл). Объединенные органические экстракты сушили сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением остатка. Этот остаток хроматографировали (силикагель; метанол/метиленхлорид = 4:96) и получали целевое соединение.
ЯМР (400 МГц, CDCl3) 7,75, 6,80, 6,70, 6,32, 4,91, 4,66, 3,75, 3,20, 3,06, 2,79, 2,09, 2,40-1,50, 1,48, 1,44, 1,11, 1,02 и 0,90 ; MC (FAB, M/Z) [M+H] = 536.
Пример 2: 15 -Этил-14-гидроксимаркфортин A (III)15 -Этил-14-гидрокси-17-оксомаркфортин A (I Пример 1, 40 мг, 0,075 ммоль) растворяли в ТГФ (5 мл) и обрабатывали комплексом “боран-диметилсульфид” (10 М, 0,08 мл, 0,8 ммоль) при 0oC. Полученную смесь перемешивали в течение 1 часа при 0oC, а затем реакцию гасили метанолом (0,2 мл) и смесь перемешивали еще 0,25 часа при температуре 20-25oC. После удаления растворителя остаток хроматографировали (силикагель; метанол/метиленхлорид, 4/96), в результате чего получали целевое соединение,ЯМР (400 МГц, CDCl3) 7,85, 6,80, 6,67, 6,33, 4,90, 3,92, 3,67, 3,10, 3,01, 2,69, 1,87, 2,65-1,20, 1,45, 1,44, 1,12, 0,97 и 0,88 BPMC (FAB, M/Z) [M+H] для C30H39N3O5+H вычислено: 522,2968, измерено: 522,2958.
Пример 3: 14-гидрокси-15-винил-17-оксомаркфортин A (IV)Целевое соединение получали в соответствии с общей методикой, описанной в Примере 1, и после внесения несущественных изменений с использованием бромида винилмагния (1 М в ТГФ, 39,5 мл, 0,04 моль) вместо бромида этилмагния, ЯМР (400 МГц, CDCl3) 7,69, 6,80, 6,71, 6,32, 4,91, 6,11-5,95, 5,32- 5,20, 4,50, 3,21, 3,08, 3,07-3,0, 2,80, 2,10, 2,66, 2,32, 2,20-1,80, 1,46, 1,44, 1,11 и 0,89 .
Пример 4: 14-Гидрокси-15-(1′,2′-дигидроксиэтил) -17-оксомаркфортин A (V)Раствор тетроксида осмия (2,5/97,5) в 2-метил-2-пропаноле, 1,9 мл), N-оксид 4-метилморфолина (1,9 г, 0,016 моль) и 14 -гидрокси-15-винил-17-оксомаркфортин A (IV, Пример 3, 1,9 г, 0,0035 моль) объединяли и перемешивали в течение 6 часов при температуре 20-25oC в ацетоне/воде (9:1, 100 мл). Реакционную смесь распределяли между водой (200 мл) и метиленхлоридом (250 мл). Органический слой сушили сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением остатка. Этот остаток хроматографировали (силикагель; метанол/метиленхлорид = 10/90) и получали целевое соединение. BPMC (FAB, M/Z) [M+H] для C30H37N3O8 + H вычислено: 568,2659, измерено: 568,2670.
Пример 5: 14-гидрокси-15-гидроксиметил-17- оксомаркфортин A (VI)К 14 -гидрокси-15-(1′,2′-дигидроксиэтил)-17-оксомаркфортину A (V, Пример 4, 1 г, 1,8 ммоль) в этаноле (100 мл) при 0oC добавляли по каплям периодат натрия (0,68 г, 40 мл воды). После перемешивания в течение 10 минут при 0oC к смеси добавляли боргидрид натрия и полученную смесь перемешивали в течение еще 10 минут при 0oC. Реакционную смесь гасили солевым раствором (150 мл) и экстрагировали метиленхлоридом (200 мл). Органический экстракт сушили сульфатом магния, фильтровали и концентрировали при пониженном давлении, в результате чего получали остаток, который затем хроматографировали (силикагель; метанол/метиленхлорид, 5/95) и получали целевое соединение,ЯМР (400 МГц, CDCl3) 7,73, 6,81, 6,71, 6,32,.4,92, 4,72, 4,06, 3,83, 3,76, 3,21, 3,06, 2,90-2,30, 2,80, 2,10, 2,22, 2,01, 1,46, 1,44, 1,12 и 0,89 ; MC (FAB, M/Z) [M+H] = 538.
Пример 6: 15-Фторметил-14-гидрокси-17-оксомаркфортин A (VII)14 -гидрокси-15-гидроксиметил-17-оксомаркфортин A (VI, Пример 5, 0,06 г, 0,1 ммоль), фторид тетрабутиламмония (1 М в ТГФ, 0,66 мл, 0,66 ммоль) и п-толуолсульфонилфторид (0,075 г, 0,43 ммоль) объединяли и нагревали с обратным холодильником в ТГФ (10 мл) в течение 0,5 часа. Затем смесь охлаждали и концентрировали. Концентрат хроматографировали (на силикагеле) и получали целевое соединение,ЯМР (400 МГц, CDCl3) 7,93, 6,80, 6,70, 6,32, 4,90, 4,80-4,50, 4,67, 3,75, 3,21, 3,06, 2,78, 2,15, 2,70-1,50, 1,46, 1,44, 1,12 и 0,89 .
Пример 7: 15-Фторметил-14-гидроксимаркфортин A (VII)15 -Фторметил-14-гидрокси-17-оксомаркфортин A (VII, Пример 6, 15 мг, 0,027 ммоль) растворяли в ТГФ (5 мл) и обрабатывали комплексом “боран-тетрагидрофуран” (1 М в ТГФ, 0,15 мл, 0,15 ммоль) при температуре 0oC. Полученную смесь перемешивали в течение 1,5 часа при температуре 0oC, а затем реакцию гасили путем добавления метанола (0,75 мл) и смесь перемешивали в течение еще 0,25 часа при 20-25oC. После удаления растворителя образовывался остаток. Этот остаток подвергали хроматографии (силикагель; метанол/метиленхлорид = 5/95) и получали целевое соединение,ЯМР (400 МГц, CDCl3) 7,57, 6,80, 6,68, 6,33, 4,90, 4,75-4,30, 4,09, 4,80, 3,50, 3,12, 3,05, 2,70, 1,88, 2,80-1,40, 1,45, 1,44, 1,12 и 0,86 ; BPMC (FAB, M/Z) [M+H] для C29H36FN3O5 + H вычислено: 526,2717, измерено: 526,2727.
Пример 8: 14,15-Дегидро-15-метилмаркфортин A (IX)Трифторид диэтиламиносеры (DAST, 0,15 мл, 1,1 ммоль) при 20-25oC по каплям добавляли к 14 -гидрокси-15-метилмаркфортину A (III, n1 = 0, 0,2 г, 0,39 ммоль), который затем растворяли в метиленхлориде (15 мл). После перемешивания в течение 5 минут реакционную смесь распределяли между водой (25 мл) и метиленхлоридом (25 мл). Органический слой сушили сульфатом магния, фильтровали, концентрировали при пониженном давлении и хроматографировали (на силикагеле), в результате чего получали целевое соединение.
ЯМР (400 МГц, CDCl3) 7,67, 6,81, 6,68, 6,33, 4,90, 5,46, 3,66, 3,14, 3,10, 2,70, 1,88, 2,75-2,54, 2,30, 1,92, 1,78, 1,46, 1,44, 1,12 и 0,86 .
Пример 9: 14,15-Дегидро-16-гидрокси-15-метилмаркфортин A (X)Диоксид селена (8 мг, 0,07 ммоль) и 14,15-дегидро-15- метилмаркфортин A (IX, Пример 8, 30 мг, 0,06 ммоль) нагревали с обратным холодильником в п-диоксане в течение 1,5 часа. Остаток концентрировали при пониженном давлении и хроматографировали (на силикагеле), в результате чего получали целевое соединение. ЯМР (400 МГц, CDCl3) 7,60, 6,81, 6,69, 6,32, 4,90, 5,55, 3,75, 2,53, 3,67, 3,14, 3,10, 2,88-2,70, 2,30, 1,90, 1,95-1,50, 1,46, 1,45, 1,11 и 0,87 ; MC (FAB, M/Z) [M+H] = 506.
Пример 10: 14-Гидрокси-16,17-диоксо-15-метилмаркфортин A (XI)14 -гидрокси-15-метилмаркфортин A (III, 100 мг) растворяли в диоксане/воде (3:1, 20 мл) и обрабатывали платиной на угле (10%, 1 г). Полученную смесь помещали в атмосферу кислорода (с использованием баллона), а затем перемешивали в течение 16 часов при 20-25oC. После этого катализатор отфильтровывали и раствор распределяли между водным раствором бикарбоната натрия (10%) и метиленхлоридом. Органический слой отделяли, сушили сульфатом магния и концентрировали. Концентрат подвергали хроматографии (силикагель; метанол/метиленхлорид; 5/95). После объединения соответствующих фракций и концентрирования было получено четыре соединения, а именно: (1) 14-гидрокси-16,17-диоксо-15-метилмаркфортин A, ЯМР (400 МГц, CDCl3) 8,35, 6,82, 6,71, 6,32, 4,90, 4,53, 3,90, 3,76, 3,4-3,3, 3,26, 3,00, 2,80, 2,14, 2,20, 1,98, 1,45, 1,43, 1,31, 1,12 и 0,86 ; BPMC (FAB, M/Z) [M+H] для C2H3N3O7 + H вычислено: 536,2397, измерено: 536,2392; (2) 14-гидрокси-16-оксо-15-метилпарагерквамид B.
ЯМР (400 МГц, CDCl3) 7,81, 6,82, 6,72, 6,33, 4,91, 4,94, 3,73, 3,53, 3,4-3,3, 3,26, 3,06, 2,82, 2,04, 2,9-2,8, 1,9-2,1, 1,46, 1,44, 1,27, 1,11 и 0,88 ; BPMC (FAB, M/Z) [M+H] для C28H33N3O6 + H вычислено: 508,2447, измерено: 508,2453; (3) 14-гидрокси-17-оксо-15-метилмаркфортин A, ЯМР (400 МГц, CDCl3) 7,89, 6,80, 6,71, 6,32, 4,91, 4,35, 3,65, 3,20, 3,06, 2,79, 2,09, 1,9-2,5, 1,46, 1,44, 1,13, 1,12 и 0,88 ; (4) 14-гидрокси-16-гидрокси-17-оксо-15-метилмаркфортин A, BPMC (FAB, M/Z) [M+H] для C29H35N3O7 + H вычислено: 538,2553, измерено: 538,2544.
Пример 11: 14-Гидрокси-16-оксо-15-метилпарагерквамид B (XII)14 -гидрокси-16,17-диоксо-15-метилмаркфортин A (XI, Пример 10) растворяли в метиленхлориде (5 мл) и обрабатывали м-хлорпероксибензойной кислотой (65% чистоты, 30 мг). Полученную смесь перемешивали в течение 1,5 часа при температуре 20-25oC. Затем смесь распределяли между метиленхлоридом (20 мл) и карбонатом калия (10% водный раствор, 20 мл). Органический слой отделяли, сушили сульфатом магния и концентрировали. Концентрат хроматографировали (силикагель; метанол/метиленхлорид = 5/95) и получали целевое соединение,ЯМР (400 МГц, CDCl3) 7,81, 6,82, 6,72, 6,33, 4,91, 4,94, 3,73, 3,53, 3,4-3,3, 3,26, 3,06, 2,82, 2,04, 2,9-2,8, 1,9-2,1, 1,46, 1,44, 1,27, 1,11 и 0,88 .
Пример 12: 14-Гидрокси-15-метилпарагерквамид B (XIII)Алюмогидрид лития (1 М раствор в ТГФ, 0,21 мл) в ТГФ (10 мл) при температуре -60oC обрабатывали хлоридом алюминия (15 мг, 3 порции). Полученную смесь перемешивали и нагревали до -25oC, а затем медленно добавляли 14 -гидрокси-16-оксо-15-метилпарагерквамид B (XII, Пример 11, 20 мг, 2 мл в ТГФ). После этого смесь перемешивали при температуре -25oC в течение 20 минут. К смеси добавляли метанол (0,8 мл), а затем цианборгидрид натрия (50 мг). Полученную смесь нагревали до 20-25oC и концентрировали. Концентрат распределяли между метиленхлоридом (20 мл) и карбонатом калия (10% водный раствор, 20 мл). Органический слой отделяли, сушили сульфатом магния и концентрировали. Концентрат подвергали хроматографии (силикагель; ацетон/гексан = 40/60), в результате чего получали целевое соединение.
ЯМР (400 МГц, CDCl3) 7,56, 6,82, 6,69, 6,32, 4,90, 4,42, 3,64, 2,62, 3,08, 3,04, 2,9-1,5, 1,46, 1,45, 1,12, 1,08 и 0,86 ; BPMC (FAB, M/Z) [M+H] для C28H35N3O5 + H вычислено: 494,2662, измерено: 494,2655.
Пример 13: 16,17-Диоксомаркфортин A (XV), 16-оксопарагерквамид B (XVI), 15-гидрокси-16-оксопарагерквамид B, 15,16-диоксопарагерквамид BМаркфортин A (XIV, 1,1 г, 2,3 ммоль) растворяли в диоксане/воде (3/1, 150 мл) и обрабатывали платиной-на-угле (10%, 10 г). Полученную смесь перемешивали в атмосфере кислорода (баллонный кислород) при температуре 20-25oC в течение 48 часов. После отфильтровывания катализатора полученную смесь распределяли между метиленхлоридом и водой. Органическую фазу отделяли, сушили сульфатом магния, фильтровали и выпаривали при пониженном давлении с получением остатка. Этот остаток хроматографировали на силикагеле (ацетон/метиленхлорид, 30/70) и получали соединения, а именно: (1) 16,17-диоксомаркфортин A, ЯМР (400 МГц, CDCl3) 7,69, 6,81, 6,74, 6,32, 4,92, 3,95, 3,80, 3,32, 3,15, 3,14-2,70, 2,19-1,86, 1,47, 1,45, 1,12 и 0,88 ;(2) 16-оксопарагерквамид B, ЯМР (400 МГц, CDCl3) 7,81, 6,80, 6,71, 6,32, 4,91, 3,74, 3,52, 3,29, 3,08, 3,0-2,85, 2,80, 2,00, 2,55-2,49, 2,08-1,75, 1,46, 1,44, 1,10 и 0,88 ; BPMC (FAB, M/Z) [M+H] для C27H31N3O5 + H вычислено: 478,2342, измерено: 478,2384;(3) 15-гидрокси-16-оксопарагерквамид B, ЯМР-комплекс представлен диастереомерами. BPMC (FAB, M/Z) [M+H] для C27H31N3O6 + H вычислено: 494,2291, измерено: 494,2292; (4) 15,16-диоксопарагерквамид B, ЯМР (400 МГц, CDCl3) 7,60, 6,83, 6,74, 6,32, 4,92, 4,12, 3,84-3,70, 3,46, 3,14, 2,89, 2,13, 2,50, 2,25, 1,95, 1,47, 1,45, 1,11 и 0,90 ; BPMC (FAB, M/Z) [M+H] для C27H29N3O6 + H вычислено: 492,2134, измерено: 492,2141.
Пример 14: 14,15-Дегидро-16-оксопарагерквамид B (XVII)Смесь диизопропиламида лития, которую получали из н-бутиллития (1,6 М в гексане, 1,2 ммоль, 0,78 мл) и диизопропиламина (1,3 ммоль, 0,17 мл) в ТГФ (4 мл), охлаждали до -60oC. Затем по каплям добавляли смесь 16-оксопарагерквамида B (XVI, Пример 13, 0,15 г, 0,3 ммоль) в ТГФ (1,5 мл) и реакционную смесь нагревали до температуры -20oC в течение 0,25 часа. Полученную смесь по каплям обрабатывали фенилселенилхлоридом (0,075 г, 0,39 ммоль) в ТГФ (1 мл) и через 5 минут гасили насыщенным бикарбонатом натрия (20 мл). Реакционную смесь экстрагировали метиленхлоридом (30 мл), сушили сульфатом магния, фильтровали и концентрировали при пониженном давлении, в результате чего получали твердое вещество, которое использовали без дополнительной очистки. Это вещество растворяли в ТГФ (8 мл) и обрабатывали перекисью водорода (30%, 0,12 мл) при 0oC. После удаления охлаждающей бани реакционную смесь перемешивали в течение 0,25 часа при температуре 20-25oC. Реакцию гасили гидроксидом натрия (1 н, 10 мл), а затем смесь экстрагировали метиленхлоридом (30 мл), сушили сульфатом магния, фильтровали, концентрировали при пониженном давлении и хроматографировали на силикагеле (метанол/метиленхлорид, 5/95), в результате чего получали целевое соединение. ЯМР (400 МГц, CDCl3) 7,78, 7,36, 6,25, 6,81, 6,70, 6,32, 4,91, 3,96, 3,60, 3,36, 3,09, 2,88, 2,09, 2,36, 1,56, 1,46, 1,45, 1,06 и 0,88 ; BPMC (FAB, M/Z) [M+H] для C27H29N3O5 + H вычислено:476, 2185, измерено: 476, 2195. Пример 15: 14 -гидрокси-15-метил-2-дезоксомаркфортин A (XXI)К 14 -гидрокси-15-метил-17-оксомаркфортину A (XIX, 21 г, 0,04 ммоль) в ТГФ (1,3 л) при 0oC по каплям добавляли боран-диметилсульфидный комплекс (12 М, 40 мл, 0,48 моль). Полученную смесь перемешивали в течение 2,5 часа при 0oC, а затем гасили путем медленного добавления по каплям метанола (50 мл). После удаления растворителя при пониженном давлении остаток подвергали хроматографии на силикагеле (метанол/метиленхлорид, 3/97), в результате чего получали 14-гидрокси-15-метилмаркфортин A и 14-гидрокси-15-метил-2- дезоксомаркфортин A,ЯМР (400 МГЦ, CDCl3) 6,66, 6,40, 6,29, 4,79, 3,92, 3,41, 3,78, 3,55, 2,92, 2,62, 2,35, 2,25, 2,26, 2,15, 2,10-1,40, 1,40, 1,04, 1,02, 0,89, 0,91; BPMC (FAB, M/Z) [M+H] для C29H39N3O4 + H вычислено: 494,3019, измерено: 494,3208.
Пример 16: 2-Дезоксомаркфортин A (XXIII)К маркфортину A (XXII, 0,16 г, 0,335 ммоль) в ТГФ (25 мл) при 0oC по каплям добавляли алан-N,N-диметилэтиламиновый комплекс (0,5 М, 2,6 мл, 13,4 ммоль). Полученную смесь перемешивали в течение 1 часа при 0oC, а затем медленно по каплям гасили метанолом (5 мл). Затем растворитель удаляли при пониженном давлении и остаток подвергали хроматографии на силикагеле (ацетон/метиленхлорид = 30/70), в результате чего получали целевое соединение. ЯМР (400 МГц, CDCl3) 6,67, 6,40, 6,29, 4,79, 3,91, 3,40, 3,57, 2,36, 2,95, 2,30-2,05, 1,95-1,25, 1,39, 0,88, 0,85; CMR (магнитный резонанс с химическим сдвигом (ХМР) (CDCl3, 100 МГц) 175,40, 146,26, 143,77, 139,74, 137,06, 126,78, 120,11, 114,88, 114,19, 79,67, 63,66, 61,13, 61,05, 60,74, 56,23, 54,68, 45,77, 41,92, 32,15, 31,94, 31, 83, 30,30, 26,28, 26,19, 23,38, 21,13, 19,75; BPMC (FAB, M/Z) [M+H] для C28H37N3O3 + H вычислено: 464,2913, измерено: 464,2929.
Пример 17: C-2-Дезоксопарагерквамид AК парагерквамиду A (0,05 г, 0,1 ммоль) в тетрагидрофуране (ТГФ, 6 мл) при температуре 20-25oC в атмосфере азота по каплям добавляли алан-N,N-диметиламиновый комплекс (0,5 М в толуоле, 2 мл, 1 ммоль). Полученную реакционную смесь перемешивали в течение 0,5 часа, а затем гасили метанолом (1 мл). Затем смесь концентрировали при пониженном давлении и получали остаток, который хроматографировали на силикагеле (ацетон/метиленхлорид, 30/70), в результате чего получали целевое соединение, ЯМР (400 МГц, CDCl3) 6,69, 6,30, 4,80, 3,94, 3,51, 3,39, 3,19, 2,92, 2,53, 2,38-2,12, 2,08, 1,95-1,74, 1,65, 1,43, 0,92 и 0,89 ; BPMC (FAB, M/Z) [M+H] для C28H37N3O4 + H вычислено: 480,2862, измерено: 480,2869.
Пример 18: N(1)-Феноксикарбонилмаркфортин A (XXVII)Маркфортин A (XXVI, 2,4 г, 5,0 ммоль) в ТГФ (120 мл) и гидриде калия (35 мас. %, 0,7 г, 6,2 ммоль) перемешивали в течение 1 часа при 20-25oC. К смеси добавляли фенилхлорформиат (1,2 мл, 9,6 ммоль). Полученную смесь перемешивали в течение 0,5 часа, а затем гасили раствором карбоната калия (10%, 50 мл) и экстрагировали этилацетатом (150 мл). Органический слой сушили сульфатом магния, фильтровали и концентрировали. Остаток растирали с эфиром/гексаном и образовавшийся осадок фильтровали, собирали и сушили, в результате чего получали целевое соединение в виде твердого продукта. ЯМР (400 МГц, CDCl3) 0,89, 1,08, 1,2-3,0, 1,45, 1,49, 3,06, 3,69, 4,83, 6,26, 6,89 и 7,2-7,5 ; BPMC (FAB, m/z) [M+H] для C35H39N3O6 + H+ вычислено: 598,2917, измерено: 598,2919.
Пример 19: N(1)-трет-Бутоксикарбонилмаркфортин A (XXVII)Маркфортин A (XXVI) в ТГФ/метиленхлориде (50 мл/50 мл) и гидриде калия (35 мас. %, 0,62 г, 5,5 ммоль) перемешивали в течение 1 часа при 20-25oC. К смеси добавляли ди-трет- бутилдикарбонат (3,4 г, 15,6 ммоль). Полученную смесь перемешивали в течение 0,5 часа, а затем гасили раствором карбоната калия (10%, 50 мл) и экстрагировали этилацетатом (150 мл). Органический слой отделяли, сушили сульфатом магния, фильтровали и концентрировали. Концентрат растирали с эфиром/гексаном, а затем осадок фильтровали, собирали и сушили, в результате чего получали целевое соединение. ЯМР (400 МГц, CDCl3) 0,83, 1,05, 1,2-3,0, 1,46, 1,53, 1,59, 3,12, 3,67, 4,85, 6,28 и 6,82 ; BPMC (FAB, m/z) [M+H] для C35H43N3O6C + H+ вычислено: 578,3230, измерено: 578,3230.
Пример 20: N(1)-4′-Нитрофеноксикарбонилмаркфортин A (XXVII)Целевое соединение было получено в соответствии с общей методикой, описанной в Примерах 18 и 19, и после внесения несущественных изменений с использованием 4-нитрофенил-хлорформиата (423 мг, 2,1 ммоль). ЯМР (400 МГц, CDCl3) 0,89, 1,08, 1,2-3,0, 1,45, 1,49, 3,06, 3,69, 4,83, 6,26, 6,92, 7,50 и 8,33 ; BPMC (FAB, m/z) [M+H] для C35H38N4O8 + H+ вычислено: 643,2767, измерено: 643,2766.
Пример 21: N(1)-9′-Флуоренилметилоксикарбонилмаркфортин A (XXVII)Целевое соединение получали в соответствии с общей методикой, описанной в Примерах 18-20, и после внесения несущественных изменений с использованием 9-флуоренилметилхлорформиата (1,6 г, 6 ммоль), ЯМР (400 МГц, CDCl3) 7,78, 7,66, 7,42, 6,89, 6,20, 4,82, 4,70-4,60, 4,39, 3,16, 2,85, 1,45 и 1,43 ; MC (FAB, M/Z) (M+H] = 700.
Пример 22: N(1)-трет-Бутоксикарбонилпарагерквамид A (XXVII)Парагерквамид A (XXV, 70 мг, 0,14 ммоль) в ТГФ (10 мл) и гидриде калия (35 мас.%, 0,062 г, 0,55 ммоль) перемешивали в течение 2 часов при 20-25oC. К смеси добавляли ди-трет-бутилдикарбонат (86 мг, 0,42 ммоль). Полученную смесь перемешивали в течение 0,5 часа, а затем гасили 10% раствором карбоната калия (50 мл) и экстрагировали этилацетатом (150 мл). Органический слой сушили сульфатом магния, фильтровали и концентрировали. Концентрат очищали посредством препаративной тонкослойной хроматографии (метанол/метиленхлорид, 5/95), в результате чего получали целевое соединение. ЯМР (400 МГц, CDCl3) 0,89. 1,02, 1,42, 1,46, 1,59, 1,63, 1,2-3,3, 3,06, 3,69, 4,83, 6,26 и 6,80 .
Пример 23: N(1)-4′-Нитрофеноксикарбонилпарагерквамид A (XXVII)Целевое соединение было получено в соответствии с общей методикой, описанной в Примере 22, и после внесения несущественных изменений с использованием 4-нитрофенилхлорформиата (814 мг, 4,2 ммоль), ЯМР (400 МГц, CDCl3) 0,85, 0,94, 1,2-3,9, 1,40, 1,47, 3,02, 4,79, 5,85, 6,18, 7,35 и 8,29 ; BPMC (FAB, m/z) (M+H] для C35H38N4O9 + H+ вычислено: 659,2717, измерено: 659,2732.
Пример 24: N(1)-4′-Нитрофеноксикарбонил-14-гидрокси-14-метилмаркфортин A (XXVII)14 -гидрокси-14-метилмаркфортин A (XXVI, n=2, R14=Me, R15=H, R16=OH, 0,188 г, 0,37 ммоль) в ТГФ (30 мл) и гидриде натрия (60 мас.%, 0,075 г, 1,875 ммоль) перемешивали в течение 2 часов при температуре 20-25oC. К смеси добавляли 4-нитрофенилхлорформиат (150 мг, 0,74 ммоль). Полученную смесь перемешивали в течение 0,5 часа, а затем гасили буферным раствором с pH 7 (15 мл) и экстрагировали этилацетатом (50 мл). Органический слой отделяли и сушили сульфатом магния, а затем фильтровали и концентрировали, в результате чего получали целевое соединение.
ЯМР (400 МГц, CDCl3) 0,92, 1,07, 1,2-3,0, 1,44, 1,47, 1,48, 3,13, 3,67, 4,87, 6,25, 6,92, 7,50 и 8,35 ; BPMC (FAB, m/z) [M+H] для C36H42N4O9 + H+ вычислено: 673,2873, измерено: 673,2866.
Пример 25: N(1)-4′-Нитрофеноксикарбонил-14 – гидрокси-15-метилмаркфортин A (XXVII)Целевое соединение было получено в соответствии с общей методикой, описанной в Примерах 18-24, и после внесения несущественных изменений с использованием 14 -гидрокси- 15-метилмаркфортина A (XXVI, n=2, R14=H, R15=Me, R16=OH, 1,98 г, 3,9 ммоль) и 4-нитрофенилхлорформиата (150 мг, 0,74 ммоль).
ЯМР (400 МГц, CDCl3) 0,92, 1,02, 1,09, 1,2-3,0, 1,44, 1,47, 3,14, 3,68, 3,95, 4,87, 6,24, 6,92, 7,50 и 8,34 ; BPMC (FAB, m/z) [M+H] для C36H40N4O9 + H+ вычислено: 673,2873, измерено: 673,2866.
Пример 26: N(1)-Фeнoкcикapбoнил-2 -гидpoкcи-2- дeзoкcoмapкфортин A (XXVIII)N(1)-Феноксикарбонилмаркфортин A (Пример 18, 2,4 г, 4,0 ммоль) растворяли в метаноле (100 мл) и обрабатывали боргидридом натрия (540 мг) при 0oC в течение 15 минут. Реакционную смесь гасили путем добавления карбоната калия (10%, 100 мл). Полученный осадок сушили и получали целевое соединение, ЯМР (400 МГц, CDCl3) 0,85, 0,92, 1,3-2,7, 1,37, 1,48, 3,06, 3,18, 3,43, 3,61, 4,75, 5,87, 6,28, 6,84 и 7,2-7,5 ; BPMC (FAB, m/z) [M+H] для C35H41N3O6 + H вычислено: 600,3073, измерено: 600,3080.
Пример 27: N(1)-4′-Нитрофеноксикарбонил-2-гидрокси-2-дезоксомаркфортин A (XXVIII)Целевое соединение было получено в соответствии с общей методикой, описанной в Примере 26, и с использованием N(1)- 4′-нитрофеноксикарбонилмаркфортина A (Пример 20, 0,5 г, 0,78 ммоль). ЯМР (400 МГц, CDCl3) 0,82, 0,89, 1,3-2,7, 1,39, 1,47, 3,06, 3,18, 3,59, 4,78, 5,85, 6,20, 6,84, 7,36 и 8,28 ; BPMC (FAB, m/z) [M+H] для C35H40N4O8 + H+ вычислено: 645,2924, измерено: 649,2925.
Пример 28: N(1)-9′-Флуоренилметилоксикарбонил-2-гидрокси-2-дезоксомаркфортин A (XXVIII)Целевое соединение получали в соответствии с общей методикой, описанной в Примере 26, и после внесения несущественных изменений с использованием N(1)-9′-флуоренилметилоксикарбонилмаркфортина A (Пример 21, 30 мг, 0,043 ммоль). ЯМР (400 МГц, CDCl3) 7,88-7,20, 6,72, 6,64, 6,38, 4,76, 4,28, 3,01, 2,85 и 2,60 .
Пример 29: N(1)-4′-Нитрофеноксикарбонил-2 -гидрокси- 2-дезоксопарагерквамид A (XXVIII)Целевое соединение получали в соответствии с общей методикой, описанной в Примере 26, и после внесения несущественных изменений с использованием N(1)-4′-нитрофеноксикарбонилпарагерквамида A (Пример 23, 1,0 г, 1,52 ммоль). ЯМР (400 МГц, CDCl3) 0,85, 0,93, 1,3-2,7, 1,40, 1,47, 1,63, 3,02, 3,2-3,6, 4,79, 5,85, 6,18, 6,88, 7,35 и 8,28 ; BPMC (FAB, m/z) [M+H] для C35H40N4O9 + H+ вычислено: 661,2873, измерено: 661,2877.
Пример 30: N(1)-4′-Нитрофеноксикарбонил-14-гидрокси-14-метил-2-гидрокси-2-дезоксомаркфортин A (XXVIII)Целевое соединение получали в соответствии с общей методикой, описанной в Примере 26, и после внесения несущественных изменений с использованием N(1)-4′-нитрофеноксикарбонил-14 -гидрокси-14-метилмаркфортина A (Пример 24, 180 мг, 0,27 ммоль).
ЯМР (400 МГц, CDCl3) 0,85, 0,91, 1,3-2,7, 1,40, 1,45, 1,48, 3,09, 3,1-3,7, 4,79, 5,88, 6,21, 6,87, 7,36 и 8,29 ; BPMC (FAB, m/z) [M+H] для C36H42N4O9 + H+ вычислено: 675,3030, измерено: 675,3031.
Пример 31: N(1)-4′-Нитрофеноксикарбонил-14-гидрокси-15-метил-2-гидрокси-2-дезоксомаркфортин A (XXVIII)Целевое соединение получали в соответствии с общей методикой, описанной в Примере 26, и после внесения несущественных изменений с использованием N(1)-4′-нитрофеноксикарбонил-14 -гидрокси-15-метилмаркфортина A (Пример 25, 2 г, 2,97 ммоль).
ЯМР (400 МГц, CDCl3) 0,85, 0,92, 1,01, 1,3-2,7, 1,39, 1,48, 3,05, 3,1-3,7, 4,79, 5,86, 6,19, 6,87, 7,36 и 8,29 ; BPMC (FAB, m/z) [M+H] для C36H42N4O9 + H+ вычислено: 675,3030, измерено: 675,3036.
Пример 32: 1,2-Дегидромаркфортин A (XXIX)Метод A: N(1)-Феноксикарбонил-2 -гидрокси-2-дезоксомаркфортин A (XXVIII, Пример 26, 1 г, 1,67 ммоль) растворяли в глиме (15 мл) и обрабатывали гидроксидом натрия (1 н, 20 мл). Полученную смесь нагревали с обратным холодильником в течение 1-2 часов. После охлаждения до 20-25oC к смеси добавляли карбонат калия (10%, 60 мл). Полученный осадок собирали и сушили, в результате чего получали целевое соединение,ЯМР (400 МГц, CDCl3) 0,67, 1,25, 1,3-2,7, 1,44, 1,47, 3,04, 3,70, 4,91, 6,48, 6,95 и 8,18 ; BPMC (FAB, m/z) [M+H] для C28H35N3O3 + H+ вычислено: 462,2756, измерено: 462,2762.
Метод B:N(1)-4′-Нитрофеноксикарбонил-2 -гидрокси-2-дезоксомаркфортин A (XXVIII, Пример 27, 50 мг, 0,08 ммоль) растворяли в глиме (1 мл) и обрабатывали гидроксидом натрия (1 н, 1 мл). Смесь перемешивали в течение 1 часа при 20-25oC. После добавления к смеси карбоната калия (10%, 5 мл) ее экстрагировали этилацетатом (20 мл). Органический слой отделяли, сушили сульфатом магния, концентрировали и получали целевое соединение.
Метод C:К N(1)-9′-флуоренилметилоксикарбонил-2 -гидрокси-2- дезоксомаркфортину A (XXVIII, Пример 28, 30 мг, 0,043 ммоль) в ДМФ (3 мл) при 20-25oC по каплям добавляли фторид тетрабутиламмония (1,0 М в ТГФ, 0,04 мл, 0,04 ммоль). После перемешивания в течение 10 минут реакционную смесь гасили карбонатом калия (10%, 30 мл) и экстрагировали этилацетатом (30 мл). Органический экстракт сушили сульфатом магния, фильтровали и концентрировали, в результате чего получали остаток, который затем хроматографировали на силикагеле (метанол/метиленхлорид, 5/95) и получали целевое соединение.
Пример 33: 1,2-Дегидропарагерквамид A (XXIX)Целевое соединение получали в соответствии с общей методикой, описанной в Примере 32, методах A и B, и после внесения несущественных изменений с использованием N(1)-4′- нитрофеноксикарбонил-2 -гидрокси-2-дезоксопарагерквамида A (XXVIII, Пример 29, 880 мг, 1,33 ммоль).
ЯМР (400 МГц, CDCl3) 0,69, 1,22, 1,3-2,7, 1,43, 1,45, 1,66, 2,97, 3,22, 3,62, 4,90, 6,29, 6,94 и 8,13 ; BPMC (FAB, M/Z) [M+H] для C28H35N3O4 + H+ вычислено: 478,2705, измерено: 478,2717.
Пример 34: 1,2-Дегидро-14-гидрокси-14-метилмаркфортин A (XXIX)Целевое соединение получали в соответствии с общей методикой, описанной в Примере 32, методах A и B, и после внесения несущественных изменений с использованием N(1)-4′- нитрофеноксикарбонил-14 -гидрокси-15-метил-2 -гидрокси-2-дезоксомаркфортина A (XXVIII, Пример 31, 150 мг, 0,22 ммоль).
ЯМР (400 МГц, CDCl3) 0,68, 1,21, 1,3-2,7, 1,44, 1,46, 1,47, 3,05, 3,65, 4,91, 6,46, 6,93 и 8,19 .
Пример 35: 1,2-Дегидро-14-гидрокси-15-метилмаркфортин A (XXIX)Целевое соединение получали в соответствии с общей методикой, описанной в Примере 32, методах A и B, и после внесения несущественных изменений с использованиемN(1)-4′-нитрофеноксикарбонил-14 -гидрокси-15-метил-2-гидрокси-2-дезоксомаркфортина A (XXVIII, Пример 31, 2 г, 2,96 ммоль).
ЯМР (400 МГц, CDCl3) 0,69, 1,04, 1,24, 1,3-2,7, 1,45, 1,47, 3,02, 3,69, 3,85, 4,92, 6,48, 6,95 и 8,19 .
Пример 36: 2-Дезоксомаркфортин A (XXIV)Метод A: 1,2-Дегидромаркфортин A (XXIV, Пример 32, 220 мг, 0,48 ммоль) растворяли в метаноле (10 мл) и обрабатывали боргидридом натрия (30 мг) при температуре 0oC в течение 15 минут. Реакционную смесь гасили путем добавления карбоната калия (10%, 20 мл). Полученный осадок сушили и получали целевое соединение. ЯМР (400 МГц) аналогичен ЯМР Примера 16. Метод B: N(1)-трет-Бутоксикарбонилмаркфортин A (XXVII, Пример 19, 100 мг, 0,17 ммоль) растворяли в диглиме. (5 мл) и обрабатывали боргидридом натрия (20 мг) при температуре 20-25oC. Затем смесь нагревали с, обратным холодильником в течение 0,5 часа. После охлаждения до температуры 20-25oC к смеси добавляли карбонат калия (10%, 10 мл). Полученный осадок сушили и получали целевое соединение. Пример 37: 2-Дезоксопарагерквамид A (XXX) Метод A: 1,2-Дегидропарагерквамид A (XXIX, Пример 33, 1,5 г, 3,14 ммоль) растворяли в метаноле (30 мл) и обрабатывали боргидридом натрия (250 мг) при 0oC в течение 15 минут. Реакционную смесь гасили карбонатом калия (10%, 60 мл) и экстрагировали этилацетатом (100 мл). Органический экстракт сушили сульфатом магния, фильтровали, концентрировали и получали остаток, который хроматографировали на силикагеле (метанол/метиленхлорид, 5/95), в результате чего получали целевое соединение. ЯМР (400 МГц) аналогичен ЯМР, указанному в Примере 17. Метод B: N(1)-трет-Бутоксикарбонилпарагерквамил A (XXVII, Пример 22, 30 мг, 0,05 ммоль) растворяли в глиме (2 мл) и обрабатывали боргидридом натрия (20 мг) при температуре 20-25oC. Затем смесь нагревали с обратным холодильником в течение 4 часов. После того как смесь охлаждалась до 20-25oC, к ней добавляли карбонат калия (10%, 5 мл) -и экстрагировали этилацетатом (10 мл). Органический слой сушили сульфатом магния, фильтровали, концентрировали и получали остаток, который затем хроматографировали на силикагеле (метанол/метиленхлорид, 5/95), в результате чего получали целевое соединение. Метод C: К раствору парагерквамида A (XXVI, 1 г, 2 ммоль) в ТГФ (полученному в результате дистилляции из бензофенона и металлического калия, 40 мл) в атмосфере азота одной порцией добавляли гидрид натрия (60% в масле, 0,24 г, 6 ммоль). Полученную реакционную смесь перемешивали в течение 0,75 часа при температуре 20-25oC, а затем охлаждали до 0oC и обрабатывали одной порцией 9-флуоренилметилхлорформиата (0,8 г, 3 ммоль). Через 5 минут реакцию гасили путем добавления фосфатно-буферного раствора (pH= 7; поставляемого от ЕМ Science, 40 мл), а затем смесь разбавляли водой (40 мл) и экстрагировали этилацетатом (2 х 50 мл). Объединенные органические экстракты сушили сульфатом магния, фильтровали и концентрировали при пониженном давлении, в результате чего получали неочищенный N(1)-9′-флуоренилметилоксикарбонилпарагерквамид A (XXVII, 1,4 г, 2 ммоль), который затем растворяли в метаноле, охлаждали до 0oC и обрабатывали одной порцией боргидрида натрия (0,3 г, 7,9 ммоль). Через 10 минут реакционную смесь гасили бикарбонатом натрия (насыщенный, 100 мл) и экстрагировали этилацетатом (2 х 50 мл). Объединенные органические экстракты сушили сульфатом магния, фильтровали, концентрировали при пониженном давлении и получали неочищенный N(1)-9′-флуоренилметилоксикарбонил-2 -гидрокси-2-дезоксопарагерквамид A (XXVIII, 1,4 г, 2 ммоль), который растворяли в ТГФ (50 мл) при температуре 20-25oC и обрабатывали фторидом тетрабутиламмония (1,0 М в ТГФ, 8 мл, 8 ммоль). После перемешивания в течение 0,5 часа реакционную смесь гасили водой (50 мл) и экстрагировали этилацетатом (2 х 50 мл). Объединенные органические экстракты сушили сульфатом магния, фильтровали, концентрировали при пониженном давлении и получали 1,2-дегидропарагерквамид A (XXIX, 0,96 г, 2 ммоль). Это соединение растворяли в метаноле при температуре 0oC и обрабатывали одной порцией боргидрида натрия (0,5 г, 13 ммоль). Через 10 минут реакцию гасили бикарбонатом натрия (насыщенный водный раствор, 100 мл) и смесь экстрагировали этилацетатом (2 х 50 мл). Объединенные органические экстракты сушили сульфатом магния, фильтровали, концентрировали при пониженном давлении и хроматографировали на силикагеле (ацетон/метиленхлорид, 30/70), в результате чего получали целевое соединение.
Пример 38: 2-Дезоксо-14-гидрокси-14-метилмаркфортин A (XXX)Целевое соединение получали в соответствии с общей методикой, описанной в Примере 38 (метод A), и после внесения несущественных изменений с использованием 1,2-дегидро-14 -гидрокси-14-метилмаркфортина A (XXIX, Пример 34, 50 мг, 1 ммоль).
ЯМР (400 МГц, CDCl3) 0,86, 0,90, 1,3-2,7, 1,42, 1,46, 2,94, 3,40, 3,52, 3,93, 4,79, 6,29, 6,39 и 6,66 ; BPMC (FAB, M/Z) [M+H] для C29H39N3O4 + H+ вычислено: 494,3018, измерено: 494,3018.
Пример 39: 2-Дезоксо-14-гидрокси-15-метил-2-гидрокси-2-дезоксомаркфортин A (XXX)Целевое соединение было получено в соответствии с общей методикой, описанной в Примере 37 (метод A), и после внесения несущественных изменений с использованием 1,2-дегидро-14 -гидрокси-15-метилмаркфортина A (XXIX, Пример 35, 1,0 г, 2,0 ммоль).
ЯМР (400 МГц) аналогичен ЯМР, описанному в Примере 15.
Пример 40: 2 -Метил-2-дезоксомаркфортин A (XXXI)1,2-Дегидромаркфортин A (XXIX, Пример 32, 42 мг, 0,09 ммоль) растворяли в ТГФ (10 мл) и обрабатывали метиллитием (литий-бромидный комплекс, 1,5 М в эфире, 0,06 мл) при температуре -78oC в течение 15 минут. Полученную смесь нагревали до 20-25oC, а затем гасили карбонатом калия (10%, 5 мл) и экстрагировали этилацетатом (20 мл). Органический экстракт сушили сульфатом магния, фильтровали и концентрировали, в результате чего получали остаток, который хроматографировали (силикагель; метанол/метиленхлорид, 5/95) и получали целевое соединение. ЯМР (400 МГц, CDCl3) 0,81, 0,92, 1,17, 1,3-2,7, 1,41, 1,43, 3,02, 3,63, 3,95, 4,79, 6,29, 6,38 и 6,63 ; BPMC (FAB, M/Z) [M+H] для C29H39N3O3 + H вычислено: 478,3069, измерено: 478,3083.
Формула изобретения
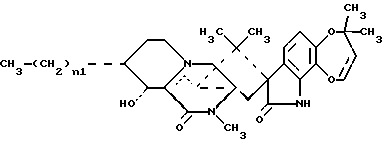 где n1 = 1 – 3. 2. 15-Алкил-14-гидроксисоединение формулы III по п.1, где n1 = 1. 3. Фторсоединения формулы VIII 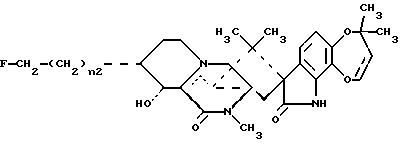 где n2 = 0 – 3. 4. Фторсоединение формулы VIII по п.3, где n2 равно 0 или 1. 5. Фторсоединение формулы VIII по п.3, где n2 = 1. 6. 15-Алкил-14-гидроксисоединения формулы X 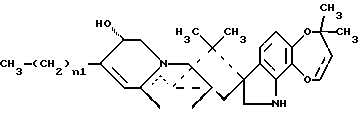 где n1 = 0 – 3. 7. 15-Алкил-16-гидроксисоединение формулы X по п.6, где n1 = 0. 8. Соединения парагерквамида B формулы XIII 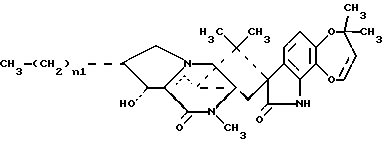 где n1 = 0, 3. 9. Соединения парагерквамида B формулы XIII по п.8, где n1 = 0. 10. 14,15-Дегидро-16-оксопарагерквамид B. 11. 2-Дезоксо-15-алкилсоединения формулы XXI 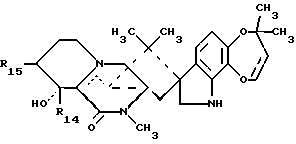 где R14 представляет H или C1 – C4-алкил; R15 представляет H или C1 – C4-алкил. 12. 2-Дезоксо-15-алкилсоединения формулы XXI по п.11, которым является 14 -гидрокси-15-метил-2-дезоксомаркфортин А.
13. 2-Дезоксосоединение формулы XXIII, которым является 2-дезоксомаркфортин А.
14. 14-Гидрокси-2-дезоксопарагерквамид B и 14-гидрокси-2-дезоксомаркфортин – соединения формулы XXV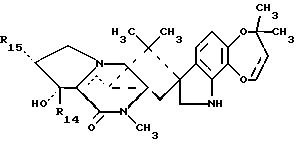 где n = 1 или 2; R14 представляет H или C1 – C4-алкил; R15 представляет H или C1 – C4-алкил. 15. 14-Гидрокси-2-дезоксопарагерквамидное соединение формулы XXV по п. 14, которым является С-2-дезоксопарагерквамид А и 2-дезоксо-14 -гидрокси-14-метилмаркфортин А.
16. Соединения, выбранные из группы, включающей: 15-этил-14-гидрокси-17-оксомаркфортин А, 14-гидрокси-15-винил-17-оксомаркфортин А, 14-гидрокси-15-(1′,2′-дигидроксиэтил)-17-оксомаркфортин А, 14-гидрокси-15-гидроксиметил-17-оксомаркфортин А, 15-фторметил-14-гидрокси-17-оксомаркфортин А, 14,15-дегидро-15-метилмаркфортин А, 14-гидрокси-16,17-диоксо-15-метилмаркфортин А, 14-гидрокси-16-оксо-15-метилпарагерквамид B, 16,17-диоксомаркфортин А, 16-оксопарагерквамид B (XVI).
17. 1,2-Дегидросоединение формулы XXIX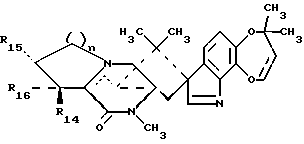 где n = 1 или 2; R14 представляет H и C1 – C4-алкил; R15 представляет H и C1 – C4-алкил; R16 представляет H и -OH. 18. 1,2-Дегидросоединение XXIX по п.17, которым являются 1,2-дегидромаркфортин А, 1,2-дегидропарагерквамид А, 1,2-дегидро-14 -гидрокси-14-метилмаркфортин А и 1,2-дегидро-14-гидрокси-15-метилмаркфортин А.
19. 2-Алкил-2-дезоксосоединение XXXI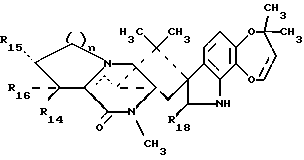 где n = 1 или 2; R14 представляет H и C1 – C4-алкил; R15 представляет H и C1 – C4-алкил; R16 представляет H и -OH; R18 представляет C1 – C4-алкил. 20. 2-Алкил-2-дезоксосоединение XXXI по п. 19, которым является 2 -метил-2-дезоксомаркфортин А.
РИСУНКИ
|
||||||||||||||||||||||||||