Патент на изобретение №2162089
|
||||||||||||||||||||||||||
(54) НОВЫЕ ПИРРОЛОКАРБАЗОЛЫ
(57) Реферат: Изобретение относится к новым пирролокарбазолам общей формулы I, где R1 означает водород, низший алкил; R2 означает тиофенил; R3 и R4 независимо друг от друга означают водород, низший алкил, или их фармацевтически приемлемым солям. Соединения I подавляют рост опухоли и могут быть использованы в медицине. 4 з.п.ф-лы, 1 табл. 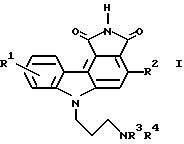
Изобретение относится к новым пирролокарбазоловым производным, их получению, фармацевтическим композициям, содержащим такие производные, а также к их применению при лечении злокачественных заболеваний. Более конкретно один из предметов настоящего изобретения относится к пирролокарбазоловым производным формулы I: 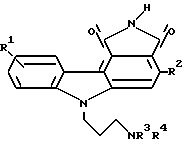 где R1 обозначает водородный атом или низший алкил; R2 обозначает гетероарил, R3 и R4 каждый независимо друг от друга обозначает водородный атом или низший алкил, и к их фармацевтически приемлемым солям. Предпочтительно настоящее изобретение относится к некоторым соединениям формулы I, в частности, включающим в себя те соединения, у которых R1 обозначает водородный атом, а R2 обозначает тиофенил. Изобретение также относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединения формулы I, или ее фармацевтически приемлемой соли в смеси по меньшей мере с одним фармацевтически приемлемым эксципиентом. Кроме того, настоящее изобретение относится к способу лечения злокачественных заболеваний, в частности мелкоклеточной карциномы легкого, карциномы толстой кишки и новообразований почек и предстательной железы у млекопитающих, в частности у человека, введением в организм млекопитающего, нуждающегося в таком лечении, терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Термин “низший алкил” означает монорадикальную разветвленную или неразветвленную насыщенную углеводородную цепь, содержащую от 1 до 6 углеродных атомов, в частности, метила, этила, н-пропила, изопропила, н-бутила, трет. -бутила, н-пентила, н-гексила. Термин “гетероарил” относится к одновалентному ненасыщенному ароматическому карбоциклическому радикалу, у которого имеется единственное кольцо из 5 или 6 атомов, одним из которых является гетероатом, выбираемый из N, O или S, например тиофенил, фуранил, пирролил или пиридил, и который может быть, но необязательно, моно-, ди- или тризамещенными независимыми друг от друга заместителями, такими как гидроксигруппа, низший алкил, низшая алкоксигруппа, атомы хлора и фтора, трифторметил и/или цианогруппа. Термин “необязательный” или “необязательно” означает, что описываемое далее обстоятельство может иметь место или его может не быть и что данное описание охватывает примеры, когда упомянутое обстоятельство имеет место, и примеры, когда этого нет. “Фармацевтически приемлемой солью” может быть любая соль, полученная из минеральной или органической кислоты. Термин “фармацевтически приемлемый анион” означает анион таких кислотноаддитивных солей. Эту соль и/или анион выбирают с таким расчетом, чтобы они не были нежелательными в биологическом или других отношениях. Такие анионы являются производными минеральных кислот, в частности соляной кислоты, бромистоводородной кислоты, серной кислоты (дающей сульфатные и бисульфатные соли), азотной кислоты, фосфорной кислоты и тому подобного, и органических кислот, в частности уксусной кислоты, пропионовой кислоты, гликолевой кислоты, пировиноградной кислоты, щавелевой кислоты, яблочной кислоты, малоновой кислоты, янтарной кислоты, малеиновой кислоты, фумаровой кислоты, винной кислоты, лимонной кислоты, бензойной кислоты, коричной кислоты, миндальной кислоты, метансульфокислоты, этансульфокислоты, салициловой кислоты, п-толуолсульфокислоты и тому подобного. Термин “лечение” означает любое лечение заболевания млекопитающих, включая: (i) профилактику заболевания, то есть действие, предотвращающее развитие клинических симптомов заболевания; (ii) подавление заболевания, то есть задержку развития клинических симптомов, и/или (iii) ослабление заболевания, то есть действие, вызывающее регрессию клинических симптомов. Термин “эффективное количество” означает дозу, достаточную для лечения болезненного состояния, подвергаемого лечению. Дозу изменяют в зависимости от конкретного пациента, заболевания и производимого лечения. Соединения формулы I имеют наименования и нумерацию в соответствии с изложенным ниже: 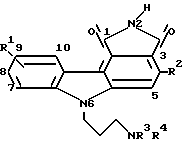 Так, например, соединение формулы I, в котором R1 обозначает водородный атом, R2 обозначает 3-тиофенил, R3 обозначает водородный атом, а R4 обозначает метил, предпочтительное соединение по изобретению называется 1,3-диоксо-6-(3-метилами-нопропил)-1,2,3,6-тетрагидро-4-(тиофен-3- ил)-пирроло[3,4-c]карбазол. Другие примеры приведены в таблице. Они имеют соответственно нижеследующие наименования: 1. 1,3-диоксо-6-(3-этиламинопропил)-1,2,3,6-тетрагидро- 4-(тиофен-3-ил)-пирроло[3,4-c]карбазол. 2. 1,3-диоксо-8-метил-6-(3-метиламинопропил)-1,2,3,6- тетрагидро-4-(тиофен-2-ил)-пирроло[3,4-c]карбазол. 3. 1,3-диоксо-6-(3-метиламинопропил)-1,2,3,6-тетрагидро- 4-(фуран-3-ил)-пирроло[3,4-c]карбазол. 4. 1,3-диоксо-6-[3-(диметиламино)-пропил] -1,2,3,6-тетрагидро- 4-(пиррол-3-ил)-пирроло[3,4-c]карбазол. Терминами “растворитель”, “инертный органический растворитель” или “инертный растворитель” обозначены растворители, инертные в условиях проведения реакции, которая описана с использованием указанных терминов (включая, например, бензол, толуол, ацетонитрил, тетрагидрофуран (ТГФ), диметилформамид (ДМФ), хлороформ, хлористый метилен (или дихлорметан), диэтиловый эфир, метанол, пиридин и тому подобное). Если не оговорено иное, растворители, используемые в реакциях по настоящему изобретению, представляют собой органические растворители. Термин “в необходимом количестве” означает добавление количества, которое достаточно для достижения указываемого эффекта, например, для доведения объема раствора до желаемого (т.е. до 100%). Если не оговорено иное, описанные в настоящей заявке реакции проводят под атмосферным давлением и в температурном интервале от 5 до 100oC (предпочтительно от 10 до 50, а наиболее предпочтительно при “комнатной” температуре или температуре “окружающей среды”, например при 20oC). Если не оговорено иное, продолжительность реакции и условия ее проведения также следует понимать как приблизительные, в частности ее проводят под приблизительно атмосферным давлением в температурном интервале от примерно 5 до примерно 100oC (предпочтительно от примерно 10 до примерно 50oC и наиболее предпочтительно при примерно 20oC) и в течение периода времени от примерно 1 до примерно 10 ч (предпочтительно в течение примерно 5 ч). Параметры, приведенные в примерах, следует принимать как конкретные, а не приблизительные. Соединения и промежуточные продукты по настоящему изобретению можно выделять и очищать, если это желательно, согласно любой подходящей процедуре разделения или очистки, как фильтрование, экстракция, кристаллизация, хроматография на колонках, тонкослойная хроматография или толстослойная хроматография, или с использованием сочетания этих процедур. Конкретные иллюстрации пригодных процедур разделения и выделения могут быть найдены по ссылке на приведенные ниже примеры. Однако, что очевидно, могут быть также использованы другие эквивалентные процедуры разделения или выделения. Соединения формулы I могут быть получены в соответствии с процедурами, описанными ниже со ссылкой на схему реакции 1. Если не оговорено иное, значения заместителей, например R1, R2, R3 и R4, аналогичны указанным в разделе “Краткое описание сущности изобретения”. Необязательно замещенные N,N-ди(низший алкил)аминометил-индолы формулы I являются коммерчески доступными соединениями или же могут быть легко получены любым специалистом в данной области техники с помощью общепринятой методологии синтеза. Так, например, N,N-диметиламинометилиндол поставляется фирмой Aldrich Chemical Company, Милуоки, штат Висконсин. Как показано на схеме реакции 1, стадия 1, конверсию необязательно замещенного N,N-ди(низший алкил)аминометилиндола формулы 1 в необязательно замещенную фосфониевую соль формулы 2 проводят по методу, описанному в Canadian J. Chem., том 51, стр. 792 (1973). Соединение формулы 1, предпочтительно N,N-диметиламинометилиндол, растворяют в протонном растворителе, предпочтительно в метаноле, и проводят реакцию с избытком йодистого метила при температуре между 10 и 50oC (предпочтительно при 25oC) в течение от 1 до 10 ч (предпочтительно 3 ч). Затем к продукту добавляют примерно 1 мол.экв. трифенилфосфина в полярном растворителе (предпочтительно в диметилформамиде) и смесь выдерживают при 100-150oC, предпочтительно при температуре кипения с обратным холодильником, в течение от 6 до 24 ч, предпочтительно 16 ч. Когда реакцию практически завершают, необязательно замещенную фосфониевую соль формулы 2 выделяют и очищают с помощью обычных средств, предпочтительно кристаллизацией. Как показано на схеме реакции 1, стадия 2, реакцию необязательно замещенной фосфониевой соли формулы 2 с гетероарилальдегидом формулы R2CHO проводят в присутствии основания с получением винилиндола формулы 3. Необязательно замещенную фосфониевую соль формулы 2 растворяют в полярном апротонном растворителе (предпочтительно в ДМСО) и ее реакцию с альдегидом формулы 2CHO проводят в присутствии затрудненного основания (предпочтительно 1,5-диазадицикло[4.3.0] нон-5-ена или 1,8-диазадицикло[5.4.0] ундец-7-ена). Эту реакцию проводят при 20-100oC (предпочтительно 80oC) в течение приблизительно 2 ч с последующим перемешиванием при примерно 20oС в течение 6-48 ч (предпочтительно около 16 ч). Когда реакцию практически завершают, винилиндол формулы 3 выделяют и очищают с помощью обычных средств, предпочтительно хроматографией на силикагеле или кристаллизацией. Как показано на схеме реакции 1, на стадии 3 проводят реакцию винилиндола формулы 3 с 1-иодо-3-(трет.-бутил)-дифенилсилилоксипропаном и удаляют защитную группу, получая N-гидроксипропилвинилиндол формулы 4. Винилиндол формулы 3 растворяют в полярном апротонном растворителе (предпочтительно в ДМФ или ДМСО) и обрабатывают гидридом щелочного металла, например гидридом калия или гидридом натрия (предпочтительно гидридом калия), при температуре 0-50oC (предпочтительно 25oC). По истечении периода реакции от 5 мин до 3 ч (предпочтительно 15 мин) добавляют 1-иодо-3-(трет. -бутил)-дифенилсилилоксипропан и реакционную смесь перемешивают при той же температуре в течение примерно 1-24 ч (предпочтительно около 16 ч). Когда реакцию практически завершают, силилзащищеннное соединение выделяют и очищают обычными средствами, предпочтительно хроматографией на силикагеле. Силильную группу удаляют обработкой тетрабутиламмонийфторидом или пиридин-фтористоводородной кислотой в тетрагидрофуране или диметоксиэтане при 20-30oC в течение от 1 до 12 ч (предпочтительно 2 ч). N-гидроксипропилвинилиндол формулы 4 предпочтительно очищать хроматографией на силикагеле. Как показано на схеме реакции 1, стадия 4, проводят конверсию гидроксильной группы соединения формулы 4 в ди(низший алкил)аминогруппу с получением соединения формулы 5, у которого R3 и R4 обозначают низшие алкилы. Спирт формулы 4 растворяют в хлористом метилене или хлороформе (предпочтительно в хлористом метилене) и обрабатывают затрудненным основанием (например, 2,6-лутидином или 2,4,6-коллидином, предпочтительно 2,6-лутидином), а затем трифторметансульфоновым ангидридом при температуре между -10 и 20oC (предпочтительно при 0oC) в течение от 15 мин до 1 ч (предпочтительно 30 мин). Далее проводят реакцию продукта с избытком амина формулы R3R4NH, где R3 и R4 обозначают низшие алкилы, при температуре 0-40oC (предпочтительно 25oC), в течение примерно 3 ч с последующей реакцией при приблизительно 0oC в течение 6-24 ч (предпочтительно 12 ч). Когда реакцию практически завершают, амин формулы 5 выделяют и либо очищают обычными средствами, предпочтительно хроматографией на силикагеле, либо используют непосредственно на стадии 5. Как показано на схеме реакции 1, стадия 4а, проводят конверсию гидроксильной группы соединения формулы 4, у которого по меньшей мере один из R3 и R4 обозначает водородный атом, в защищенную аминогруппу с получением соединения формулы 5а. Спирт формулы 4 растворяют в хлористом метилене или хлороформе (предпочтительно в хлористом метилене) и обрабатывают затрудненным основанием (например, 2,6-лутидином или 2,4,6-коллидином, предпочтительно 2,6-лутидином), а затем трифторметансульфоновым ангидридом при температуре между -10 до 20oC (предпочтительно при 0oC) в течение от 15 мин до 1 ч (предпочтительно 30 мин). Далее проводят реакцию продукта с избытком амина формулы R3R4NH, где по меньшей мере один из R3 и R4 обозначает водородный атом, при температуре 0-40oC (предпочтительно 25oC) в течение приблизительно 3 ч, с последующей реакцией при примерно 0oC в течение 6-24 ч (предпочтительно 12 ч). Когда реакцию практически завершают, виниламин выделяют и либо очищают обычными средствами, предпочтительно хроматографией на силикагеле, либо используют непосредственно при проведении следующей реакции. Полученный промежуточный амин представляет собой первичный или вторичный амин (т.е. амин, у которого по меньшей мере один из R3 и R4 обозначает водородный атом). Его защищают растворением в третичном основании (предпочтительно в пиридине) и проводят реакцию с трифторуксусным ангидридом в течение от 5 мин до 4 ч (предпочтительно 30 мин) при температуре около 25oC. Когда реакцию практически завершают, винилтрифторацетамидное соединение формулы 5а выделяют и очищают обычными средствами, предпочтительно хроматографией на силикагеле. По другому варианту промежуточный амин, у которого по меньшей мере один из R3 и R4 обозначает водородный атом, защищают растворением в инертном растворителе в присутствии третичного основания (предпочтительно триэтиламина) и проводят реакцию с ди-трет.-бутилдикарбонатом с получением трет.-бутоксикарбаматного производного. Как показано на схеме реакции 1, стадия 5, конверсию виниламина формулы 5 или защищенного виниламина, в частности винилтрифторацетамидного соединения формулы 5а, в соединение формулы 1 проводят реакцией с малеимидом. Виниламин или защищенный виниламин формулы 5 или 5а растворяют в ароматическом углеводороде (предпочтительно в толуоле) и кипятят с обратным холодильником с 2-3 мол.экв. (предпочтительно с 2 мол.экв.) малеимида в течение 6-24 ч (предпочтительно 16 ч). Когда реакция практически завершена, аддукт Дильса-Альдера выделяют и очищают предпочтительно хроматографией на силикагеле. Этот аддукт растворяют в инертном растворителе (например, в бензоле, толуоле, хлористом метилене, предпочтительно в бензоле) и обрабатывают 2-3 мол.экв. (предпочтительно 2 мол. экв.) дихлордицианобензохинона при температуре 20-50oC (предпочтительно 25oC) в течение от 15 мин до 3 ч (предпочтительно 30 мин). Когда реакция практически завершена, полученный карбазол выделяют обычным путем. Если исходный виниламин представляет собой соединение формулы 5а, защитную группу Y удаляют. Удаление защитной группы можно производить с помощью средств, которые известны. В случае, например, присутствия трифторацетатной защитной группы с целью отщепления этой трифторацетатной защитной группы карбазол обрабатывают неорганическим основанием (гидроксидом натрия, гидроксидом калия и т.п., предпочтительно гидроксидом натрия) в протонном растворителе (например, в метаноле, этаноле или их смеси), смешанном с тетрагидрофураном, в течение примерно 15 мин при приблизительно 25oC. В другом варианте, если амин защищен трет.-БОС-группой, защитную группу удаляют обработкой кислотой. Полученный пирролокарбазол формулы I выделяют и очищают предпочтительно хроматографией на силикагеле. Можно провести конверсию соединений формулы I в соответствующие кислотноаддитивные соли. Такую конверсию ведут обработкой стехиометрическим количеством соответствующей кислоты, в частности соляной кислоты (например, 3 мол. экв. с получением тригидрохлоридной соли). Обычно свободное основание растворяют в полярном органическом растворителе, например в метаноле или этаноле, и кислоту добавляют в воде, метаноле или этаноле. Температуру поддерживают на уровне от 0 до 50oC. Соответствующая соль выпадает в осадок самопроизвольно или может быть выделена из раствора добавлением меньшего количества полярного растворителя. Кислотноаддитивные соли соединения формулы I можно разлагать до соответствующих свободных оснований обработкой избытком подходящего основания, в частности аммиаком или бикарбонатом натрия, обычно в присутствии водного растворителя и при температуре между 0 и 50oC. Свободное основание выделяют обычными средствами, в частности экстракцией органическим растворителем. Предпочтительны соединения формулы I, у которых R1 обозначает водородный атом, а R2 обозначает 3-тиофенил. Предпочтительны также те соединения, у которых R3 обозначает водородный атом, а R4 обозначает низший алкил, предпочтительно метил. Кроме того, предпочтительны также соединения, которые сочетают в себе вышеуказанные отличительные особенности. Наиболее предпочтительным является соединение 1,3-диоксо-6-(3-метиламинопропил)-1,2,3,6-тетрагидро-4-(тиофен-3- ил)-пирроло[3,4-c]карбазол. Соединения по настоящему изобретению представляют собой ингибиторы протеинкиназы C, которые могут быть использованы в качестве химиотерапевтических агентов для лечения млекопитающих, в частности, людей, страдающих различными злокачественными заболеваниями, включая мелкоклеточную карциному легкого, карциному толстой кишки и новообразования молочной железы, соответствующие клеточным линиям MCS7, MDA-MB435 и MDA-N, и резко выраженные опухоли ПКС, в частности, такие, которые соответствуют обозначению СНО/ПКС-  . Различные соединения по изобретению проявляют более сильное действие, направленное против некоторых новообразований, чем другие, что подтверждается обычно применяемыми методами.
Активность в отношении ингибирования протеинкиназы C in vitro рассчитывают измерением включения 32P из . Различные соединения по изобретению проявляют более сильное действие, направленное против некоторых новообразований, чем другие, что подтверждается обычно применяемыми методами.
Активность в отношении ингибирования протеинкиназы C in vitro рассчитывают измерением включения 32P из  –32P АТФ в синтетические пептидные субстраты.
Для химиотерапевтических агентов действие in vivo, в частности, при лечении злокачественных заболеваний определяют испытаниями с подавлением новообразований, например, как это изложено Maneckjee и др. в Proc. Natl. Acad. Sci. USA, том 89, 1169-1173 (февраль 1992 г.). Можно осуществлять вариации такого испытания, например, используя клетки карциномы толстой кишки НТ-29, клетки МКЛК Н82, клетки СНО/ПКС- –32P АТФ в синтетические пептидные субстраты.
Для химиотерапевтических агентов действие in vivo, в частности, при лечении злокачественных заболеваний определяют испытаниями с подавлением новообразований, например, как это изложено Maneckjee и др. в Proc. Natl. Acad. Sci. USA, том 89, 1169-1173 (февраль 1992 г.). Можно осуществлять вариации такого испытания, например, используя клетки карциномы толстой кишки НТ-29, клетки МКЛК Н82, клетки СНО/ПКС- и СНО/ПКС-. и СНО/ПКС-.Соединения формулы I вводят в организм в терапевтически эффективной дозе, например в дозе, достаточной для лечения вышеописанных болезненных состояний. Соединения по изобретению или их фармацевтически приемлемые соли можно вводить в организм по любому из приемлемых вариантов введения в организм агентов, которые используют с аналогичными целями. Несмотря на то, что размеры доз соединений по изобретению для человека все еще необходимо оптимизировать, суточная доза обычно составляет от примерно 0,1 до 20,0 мг/кг веса тела, предпочтительно примерно от 0,5 до 10,0 мг/кг веса тела и наиболее предпочтительно от примерно 1,0 до 5,0 мг/кг веса тела. Так, например, при введении в организм пациента весом 70 кг доза должна находиться в интервале от примерно 7,0 до 1400 мг/день, предпочтительно от примерно 35,0 до 700 мг/день и наиболее предпочтительно от примерно 70 до 350 мг/день. Количество вводимого в организм активнодействующего соединения зависит, разумеется, от конкретного пациента и болезненного состояния, при котором проводят лечение, серьезности заболевания, варианта и графика введения, а также мнения врача, прописывающего лекарство. При применении соединений по настоящему изобретению для лечения вышеуказанных состояний можно прибегать к любому фармацевтически приемлемому варианту введения в организм. Соединения формулы I можно вводить в организм либо индивидуально, либо в сочетании с фармацевтически приемлемыми эксципиентами, в том числе в виде твердых, полутвердых, жидких или аэрозольных дозированных препаративных форм, в частности в виде таблеток, капсул, порошков, жидкостей, суспензий, суппозиториев, аэрозолей и т.п. Соединения формулы I можно также вводить в организм в дозированных препаративных формах с постоянным или регулируемым выделением, включая инъекции веществ замедленного всасывания, осмотические насосы, пилюли, трансдермальные (в том числе и электротранспортные) накладки и т.п., применяемые для пролонгированного введения в организм соединения с заданной скоростью, предпочтительно в единичных дозированных формах, пригодных для однократного введения в организм точных доз. Такие композиции, как правило, включают в себя обычный фармацевтический носитель или эксципиент и соединение формулы I или его фармацевтически приемлемую соль. Кроме того, эти композиции могут включать в себя другие лекарственные агенты, фармацевтические агенты, носители, адъюванты и т.п., такие как агенты, устойчивые к большинству лекарственных средств. В зависимости от намечаемого варианта введения в организм фармацевтически приемлемая композиция обычно содержит примерно от 0,1 до 90 вес.%, предпочтительно примерно от 0,5 до 50 вес.%, соединения или соли формулы I, а ее остальная часть приходится на фармацевтически приемлемые эксципиенты, носители и т.п. Одним из предпочтительных путей введения в организм при состояниях, подробно описанных выше, является пероральный с использованием обычного режима суточной дозы, которую можно регулировать в соответствии со степенью заболевания. При таком пероральном введении в организм фармацевтически приемлемую, нетоксичную композицию готовят введением любого из обычно применяемых эксципиентов, например такого, как маннит, лактоза, крахмал, стеарат магния, натрийсахарин, тальк, целлюлоза, натрийтранскармеллоза, глюкоза, желатин, карбонат магния и т.п. Такие композиции используют в виде растворов, суспензий, таблеток, диспергируемых таблеток, пилюль, капсул, порошков, препаратов постоянного выделения и т.п. В предпочтительном варианте композиции должны использоваться в виде пилюль или таблеток, вследствие чего такая композиция помимо активнодействующего вещества содержит разбавитель, в частности лактозу, сахарозу, вторичный кислый фосфат кальция или т. п.; смазывающее вещество, в частности стеарат магния или т.п., и связующий компонент, например крахмал, аравийскую камедь, поливинилпирролидон, желатину, целлюлозу, их производные и т.п. Жидкие фармацевтически приемлемые для введения в организм композиции могут быть приготовлены, например, растворением, диспергированием и т.п. активнодействующего вещества, которое определено выше, и необязательных фармацевтических адъювантов в носителе, в частности, таком, как вода, солевой раствор, водная декстроза, глицерин, гликоли, этанол и т.п., благодаря чему образуется раствор или суспензия. Если желательно, фармацевтическая композиция, предназначенная для введения в организм, может также содержать незначительные количества нетоксичных вспомогательных веществ, в частности смачивающих агентов, эмульгаторов или солюбилизирующих агентов, агентов регулирования величины pH и т.п., например ацетата натрия, цитрата натрия, производных циклодекстрина, сорбитанмонолаурата, триэтаноламинацетата, триэтаноламинолеата и т.п. Существующие методы приготовления таких дозированных препаративных форм известны или очевидны для любого специалиста в данной области техники, например, из работы “Remington’s Pharmaceutical Sciences”, Mack Publishing Company, Easton, Pennsylvania, издание 15-е, 1975. В любом случае композиция или препарат, предназначенный для введения в организм, содержит активнодействующее вещество в количестве, эффективном для ослабления симптомов у подвергаемого лечению пациента. Могут быть приготовлены дозированные препаративные формы или композиции, содержащие активнодействующее вещество в интервале от 0,005 до 95%, а остальное приходится на долю нетоксичного носителя. Для перорального введения в организм фармацевтически приемлемую нетоксичную композицию готовят введением любого из обычно применяемых эксципиентов, как, например, маннит, лактоза, крахмал, стеарат магния, тальк, производные целлюлозы, натрийтранскармеллоза, глюкоза, сахароза, карбонат магния, натрийсахарин, тальк и т.п. материалы фармацевтических сортов. Эти композиции используют в виде растворов, суспензий, таблеток, капсул, порошков, препаратов с постоянной скоростью выделения и т.п. Такие композиции могут содержать 0,01-95% активнодействующего вещества, предпочтительно 0,1-50%. Твердую дозированную препаративную форму, раствор или суспензию, например, в пропиленкарбонате, растительных маслах или триглицеридах предпочтительно инкапсулировать в желатиновую капсулу. Такие диэфирные растворы, их приготовление и инкапсулирование описаны в американских патентах 4328245, 4409239 и 4410545. Жидкую дозированную препаративную форму, раствор, например, в полиэтиленгликоле с целью упростить дозирование при введении в организм можно разбавлять достаточным количеством фармацевтически приемлемого жидкого носителя, в частности водой. В соответствии с другим вариантом жидкие или полутвердые пероральные композиции могут быть приготовлены растворением или диспергированием активнодействующего соединения или его соли в растительных маслах, гликолях, триглицеридах, пропиленгликолевых сложных эфирах (например, в пропиленкарбонате) и т.п. и инкапсулированием этих растворов или суспензий в желатиновые капсулы с твердыми или мягкими оболочками. Другие пригодные композиции описаны в американских патентах 28819 и 4358603. Парентеральные пути введения в организм обычно характеризуются инъекцией либо подкожно, внутримышечно, либо внутривенно. Впрыскиваемые препараты могут быть приготовлены в обычных формах, то есть либо как жидкие растворы или суспензии, в твердых формах, пригодных для приготовления раствора или суспензии в жидкости перед инъекцией, либо как эмульсии. Приемлемыми эксципиентами служат, например, вода, солевой раствор, декстроза, глицерин, этанол и т.п. Кроме того, если это желательно, фармацевтические композиции, предназначенные для введения в организм, могут также содержать незначительные количества нетоксичных вспомогательных веществ, в частности смачивающих агентов или эмульгаторов, буферных агентов для регулирования pH, агентов, повышающих растворимость, и т.п., например таких, как ацетат натрия, сорбитанмонолаурат, триэтаноламинолеат, циклодекстрины и так далее. Согласно более современному подходу в области парентеральных средств введения в организм применяют имплантацию системы медленного выделения или постоянного выделения, которые поддерживают постоянную концентрацию (см., например, американский патент 3710795). Процентное содержание активнодействующего вещества в таких парентеральных композициях находится в большой зависимости от их конкретной природы, а также от активности соединения и нужд пациента. Однако в растворе применяемое процентное содержание активнодействующего компонента составляет от 0,01 до 10%, и оно выше в твердом препарате, который предназначен для последующего разбавления до вышеуказанного процентного содержания. Предпочтительно растворная композиция включает в себя 0,2-2% активнодействующего вещества. Можно также использовать для введения в организм назальные растворы активнодействующего вещества индивидуально или в сочетании с фармацевтически приемлемыми эксципиентами. Композиции с активнодействующим соединением или его солью можно также вводить в организм через дыхательные пути в виде аэрозоля или раствора с помощью распылителя или микродисперсного порошка для вдувания либо индивидуально, либо в сочетании с таким инертным носителем, как лактоза. В таком случае диаметр частиц композиции составляет менее 50 мкм, предпочтительно менее 10 мкм. Сущность и возможность практического выполнения настоящего изобретения для любого специалиста в данной области техники станут более понятны из нижеприведенных препаратов и примеров. Их следует рассматривать не как ограничивающие объем настоящего изобретения, а как его иллюстрацию. Пример 1 Получение соединений формулы 2 1А. R1 обозначает водородный атом Раствор 5,4 г 2-N,N-диэтиламинометилиндола [Acta. Chim. Acad. Sci. Hung. , том 34, стр. 439 (1962)] в 40 мл метанола смешивали с 15 мл йодистого метила и оставляли на 3 ч. В результате выпаривания растворителя получали стеклоподобный материал, к которому добавляли 11,6 г трифенилфосфина и 100 мл диметилформамида (ДМФ). Смесь кипятили с обратным холодильником в течение ночи, затем большую часть ДМФ отгоняли под пониженным давлением и остаток растворяли с растиранием в 75 мл бензола. Полученные кристаллы отфильтровывали, промывали небольшим количеством бензола и сушили в вакууме, получая 10,4 г индол-2-метилтрифенилфосфонийиодида. 1Б. Варьирование значений R1 Осуществляя процедуру примера 1А и заменяя 2-диэтиламинометилиндол 2-диэтиламинометилиндоламин, замещенными желаемым R1-заместителем, получали соответствующие соединения формулы 2, у которых R1 обозначает метил, этил, н-пропил, изопропил, н-бутил, трет.-бутил, н-пентил, н-гексил и т.п., например: 4-метилиндол-2-метилтрифенилфосфонийиодид; 5-этилиндол-2-метилтрифенилфосфонийиодид; 6-н-пропилиндол-2-метилтрифенилфосфонийиодид и 7-н-бутилиндол-2-метилтрифенилфосфонийиодид. Пример 2 Получение соединений формулы 3 2А. R1 обозначает водородный атом и R2 обозначает тиофен-3-ил В раствор 1,04 г индол-2-метилтрифенилфосфонийиодида в 60 мл диметилсульфоксида добавляли 175 мкл тиофен-3-карбоксальдегида, а затем 250 мкл 1,8-диазадицикло[5.4.0]ундец-7-ена. Смесь перемешивали в токе азота при 40oC в течение 1 ч, а затем при 80oC в течение 2 ч и, наконец, перемешивали в течение ночи при 20oC. Смесь выливали в воду и экстрагировали диэтиловым эфиром. Органический слой сушили, под пониженным давлением удаляли растворитель и остаток очищали кристаллизацией из метанола. Получали 200 мг (44%) 2-[2-(тиофен-3-ил)-винил]-индола. 2Б. Варьирование значений R1 и R2 Осуществляя процедуру примера 2А с необязательной заменой индол-2-метилтрифенилфосфонийиодида другими соединениями формулы 2, полученными, в частности, по изложенному выше в примере 1, и с необязательной заменой тиофен-3-карбоксальдегида другими соединениями формулы R2CHO, получали нижеследующие соединения формулы 3: 2-[2-(тиофен-2-ил)-винил]-индол; 2-[2-(фуран-3-ил)-винил]-индол; 2-[2-(пиррол-3-ил)-винил]-индол; 2-[2-(пирид-3-ил)-винил]-индол; 4-метил-2-[2-(тиофен-3-ил)-винил]-индол; 5-этил-2-[2-(тиофен-3-ил)-винил]-индол; 6-н-пропил-2-[2-(тиофен-3-ил)-винил]-индол и 7-н-бутил-2-[2-(тиофен-3-ил)-винил]-индол. Пример 3 Получение соединений формулы 4 3А. R1 обозначает водородный атом и R2 обозначает тиофен-3-ил 200 мг 2-[2-(тиофен-3-ил)-винил] -индола растворяли в 3 мл диметилформамида и раствор обрабатывали при 25oC в течение 15 мин 40 мг гидрида калия. Затем добавляли 500 мг 1-иод-3-(трет.-бутилдифенилсилилокси)-пропана и реакционную смесь перемешивали в течение ночи при 25oC. После разделения смеси между диэтиловым эфиром и водой органический слой сушили и при пониженном давлении удаляли растворитель. Остаток очищали препаративной тонкослойной хроматографией на силикагеле, элюируя смесью гексана с этилацетатом в соотношении 5: 1. Получали 330 мг (71%) 1-[3-(трет.-бутилдифенилсилилокси)-пропил]-2-[2-(тиофен-3- ил)-винил]-индола. Этот материал растворяли в 2 мл тетрагидрофурана и при 25oC 2 ч обрабатывали 2 мл 1 М раствора тетрабутиламмонийфторида. После разделения смеси между диэтиловым эфиром и водой органический слой сушили и при пониженном давлении удаляли растворитель. Остаток очищали препаративной тонкослойной хроматографией на силикагеле, элюируя смесью гексана с этилацетатом в соотношении 2:1, получая 121 мг (68%) 1-(3-гидроксипропил)-2-[2-(тиофен-3-ил)-винил]-индола. 3Б. Варьирование значений R1 и R2 Осуществляя процедуру примера 3А с заменой 2-[2-(тиофен-3-ил)-винил]-индола другими соединениями формулы 3, в частности, полученными по примеру 2, получали соответствующие гидроксипропиловые соединения формулы 4. Пример 4 Получение соединений формулы 5а 4А. R1 обозначает водородный атом, R2 обозначает тиофен-3-ил, R3 обозначает водородный атом и R4 обозначает метил Раствор 121 мг 1-(3-гидроксипропил)-2-[2-(тиофен-3-ил)-винил]-индола в 3 мл хлористого метилена обрабатывали 120 мкл 2,6-лутидина и охлаждали до 0oC. Добавляли 100 мкл трифторметансульфонового ангидрида, после перемешивания в течение 30 мин вводили 5 мл 40%-ного водного раствора метиламина и реакционную смесь перемешивали при 25oC 3 ч. После перемешивания еще в течение 12 ч при 0oC реакционную смесь разделяли между хлористым метиленом и водой, органический слой сушили и под пониженным давлением удаляли растворитель. В результате препаративной тонкослойной хроматографии на силикагеле (элюируя 10%-ным раствором метанола в хлористом метилене) получали 83 мг (67%) 1-[3-(метиламино)-пропил] -2-[2-(тиофен-3-ил)-винил] -индола. Этот продукт растворяли в 3 мл хлористого метилена, содержавшего 150 мкл пиридина, и добавляли 40 мкл трифторуксусного ангидрида. По истечении 30 мин смесь разделяли между диэтиловым эфиром и водным раствором бикарбоната натрия, органический слой сушили и при пониженном давлении удаляли растворитель. В результате препаративной тонкослойной хроматографии на силикагеле (элюируя смесью гексана с этилацетатом в соотношении 3:1) получали 80 мг (73%) 1-[3-(N-метилтрифторметилацетамидо)-пропил]-2-[2-(тиофен-3-ил)-винил]-индола. 4Б. Варьирование значений R1, R2, R3 и R4 Осуществляя процедуру примера 4А с необязательной заменой 1-(3-гидроксипропил)-2-[2-(тиофен-3-ил)-винил]-индола соединениями формулы 4, в частности, полученными по примеру 3, получали соответствующие соединения формул 5 и 5А. Пример 5 Получение соединений формулы I 5А. R1 обозначает водородный атом, R2 обозначает тиофен-3-ил, R3 обозначает водородный атом и R4 обозначает метил Раствор 80 мг 1-[3-(N-метилтрифторметилацетамидо)-пропил] – 2-[2-(тиофен-3-ил)-винил] -индола в 2 мл толуола обрабатывали 40 мг малеимида. После кипячения с обратным холодильником в течение ночи растворитель выпаривали при пониженном давлении и остаток очищали препаративной тонкослойной хроматографией на силикагеле, получая 20 мг (20%) аддукта Дильса-Альдера. 1H-ЯМР-спектрограмма (CDCl3): 7,96 (m, 1H), 7,35-7,15 (m, 4H), 7,13 (m, 1H), 7,02 (m, 1H), 4,40 (d, 1H), 4,13 (t, 2H), 3,73 (m, 2H), 3,45 (t, 2H), 3,12 (m, 2H), 3,06 (s, 3H), 2,02 (s, 2H). Этот материал растворяли в 3 мл бензола и обрабатывали 20 мг дихлордицианобензохинона (ДДХ). По истечении 20 мин добавляли еще 10 мг ДДХ. По истечении еще 10 мин смесь наносили на пластину для препаративной тонкослойной хроматографии и элюировали смесью гексана с этилацетатом в соотношении 2:1. Таким образом в виде желтой пеноподобной массы получали 18 мг 1,3-диоксо-6-(N-метилтрифторметилацетамидо)-1,2,3,6- тетрагидро-4-(тиофен-3-ил)-пирроло[3,4-c]карбазола. Этот продукт растворяли в 4 мл смеси метанола с ТГФ в соотношении 1:1 и добавляли 1 мл 1 М раствора NaOH. После перемешивания 15 мин смесь разделяли между хлористым метиленом и водой. Органический слой отделяли и под пониженным давлением удаляли растворитель. В результате препаративной тонкослойной хроматографии остатка (элюируя 10%-ным раствором метанола в хлористом метилене) получали 15 мг 1,3-диоксо-6-(3-метиламинопропил)-1,2,3,6-тетрагидро- 4-(тиофен-3-ил)-пирроло[3,4-c]карбазола. 1H-ЯМР-спектрограмма (d6-DMSO): 8,96 (d, H), 8,00 (s, 1H), 7,95 (m, 1H), 7,74 (d, 1H), 7,62 (m, 3H), 7,35 (t, 1H), 4,59 (t, 2H), 2,55 (t, 2H), 2,29 (s, 3H), 1,97 (t, 2H). Молекулярная масса для C22H19N3O2S: вычислено – 389,1197, найдено – 389,1198. 5Б. Варьирование значений R1, R2 и R3 Осуществляя процедуру примера 5А с заменой 1-[3-(N-метилтрифторметилацетамидо)-пропил] -2-[2-(тиофен-3-ил)-винил] -индола другими соединениями формулы 5 или 5а, полученными, в частности, по примеру 4Б, получали соответствующие пирролокарбазоловые производные формулы I. Пример 6 Данный пример иллюстрирует приготовление типичной фармацевтической композиции для перорального введения в организм, содержащей активнодействующее соединение формулы I, например 1,3-диоксо-6-(3-метиламинопропил)-1,2,3,6-тетрагидро-4-(тиофен-3- ил)-пирроло[3,4-c]карбазол. Компоненты – Количество на таблетку, мг Активнодействующее соединение – 200 Лактоза, высушенная распылением – 148 Стеарат магния – 2 Вышеуказанные компоненты смешивают и вводят в желатиновую капсулу с твердой оболочкой. При приготовлении композиций данного примера для перорального введения в организм в качестве активнодействующего соединения могут быть использованы другие соединения формулы I, в частности, полученные в соответствии с примерами 1-6. Пример 6А Данный пример иллюстрирует приготовление другой типичной фармацевтической композиции для перорального введения в организм, содержащей активнодействующее соединение формулы I, например 1,3-диоксо-6-(3-метиламинопропил)-1,2,3,6-тетрагидро-4-(тиофен-3- ил)-пирроло[3,4-c]карбазол. Компоненты – Количество на таблетку, мг Активнодействующее соединение – 400 Кукурузный крахмал – 50 Лактоза – 145 Стеарат магния – 5 Вышеуказанные компоненты смешивают до однородной массы и прессуют с формованием таблеток с одной риской. При приготовлении композиций данного примера для перорального введения в организм в качестве активнодействующего соединения могут быть использованы другие соединения формулы I, в частности, полученные в соответствии с примерами 1-6. Пример 7 Данный пример иллюстрирует приготовление типичной фармацевтической композиции, содержащей активнодействующее соединение формулы I, например 1,3-диоксо-6-(3-метиламинопропил)-1,2,3,6- тетрагидро-4-(тиофен-3-ил)-пирроло[3,4-c]карбазол. Готовят суспензию для перорального введения в организм нижеследующего состава: Компоненты – Количество на таблетку Активнодействующее соединение – 1,0 г Фумаровая кислота – 0,5 г Хлористый натрий – 2,0 г Метилпарабен – 1,0 г Гранулированный сахар – 25,5 г Сорбит (70%-ный раствор) – 12,85 г Камедь Veegum K (фирмы Vanderbilt Co.) – 1,0 г Корригент – 0,035 мл Красители – 0,5 мг Дистиллированная вода в необходимом количестве – до 100 мл При приготовлении композиций данного примера для перорального введения в организм в качестве активнодействующего соединения могут быть использованы другие соединения формулы I, в частности, полученные в соответствии с примерами 1-6. Пример 8 Данный пример иллюстрирует приготовление типичной фармацевтической композиции, содержащей активнодействующее соединение формулы I, например 1,3-диоксо-6-(3-метиламинопропил)-1,2,3,6- тетрагидро-4-(тиофен-3-ил)-пирроло[3,4-c]карбазол. Готовят препарат для инъекций, величину pH которого с помощью буфера доводят до 7,4, нижеследующего состава: Компоненты – Количество Активнодействующее соединение – 0,2 г Буферный раствор (0,4 М) ацетата натрия – 2,0 мл HCl (1 н.) – в необходимом количестве до величины pH 7,4 Вода (дистиллированная, стерилизованная) в необходимом количестве – до 20,0 мл При приготовлении композиций данного примера для инъекций в качестве активнодействующего соединения могут быть использованы другие соединения формулы I, в частности, полученные в соответствии с примерами 1-6. Пример 9 Данный пример иллюстрирует приготовление типичной фармацевтической композиции, содержащей активнодействующее соединение формулы I, например 1,3-диоксо-6-(3-метиламинопропил)-1,2,3,6- тетрагидро-4-(тиофен-3-ил)-пирроло[3,4-c]карбазол. Готовят суппозиторий общим весом 2,5 г нижеследующего состава: Активнодействующее соединение – 500 мг Продукт Witepsol H-15* – остальное (* триглицериды насыщенной растительной жирной кислоты; продукция фирмы Riches-Nelson, Inc., Нью-Йорк, штат Нью-Йорк). При приготовлении композиций данного примера в форме суппозиториев в качестве активнодействующего соединения могут быть использованы другие соединения формулы I, в частности, полученные в соответствии с примерами 1-6. Пример 10 Испытание на определение ингибирующего действия in vitro с использованием протеинкиназы C Ингибирующее действие на протеинкиназу C (ПКС) рассчитывают измерением включения 32P из –32P АТФ в синтетические пептидные субстраты. Ингибирующий потенциал измеряют с использованием 1- изофермента ПКС из головного мозга крысы и синтетического пептидного субстрата ala-lys-arg-arg-arg-leu-ser-ser-leu-arg-ala.
С помощью – 32P АТФ (>5000 Ки/ммоль) определяют пики для реакционной смеси, содержащей 25 мМ трис-HCl (pH 7,5), 2,5 мМ Mg(NO3), 1,0 мМ этиленгликольтетрауксусной кислоты, 20 мкМ субстрата, 1 мкг/мл фосфатидилсерина (ФС), 5 10-6 М диацилглицерина (ди-C8) и 50 мкМ АТФ с обеспечением приблизительно 106 СРМ на реакцию и 0,08 мкг/мл ПКС в 50-микролитровом объеме на ячейку. Испытание проводят с использованием или без использования испытываемого соединения, добавляемого в различных концентрациях. По истечении пяти минут инкубации при комнатной температуре реакцию прекращают добавлением 0,2 объема 50%-ного раствора трихлоруксусной кислоты. Затем на ионообменную хроматографическую бумагу Whatman P-81 наносят пробу 30-микролитрового образца из каждой ячейки (контрольной и с испытываемым соединением), после чего 32-P-инкорпорирование подсчитывают с помощью жидкостного сцинцилляционного счетчика Beckman LS 5000 TA. Процентную степень ингибирования ПКС, активированной 510-6 М диC8 и 1 мкг/мл фосфатидилсерина, определяют по формуле 10-6 М диацилглицерина (ди-C8) и 50 мкМ АТФ с обеспечением приблизительно 106 СРМ на реакцию и 0,08 мкг/мл ПКС в 50-микролитровом объеме на ячейку. Испытание проводят с использованием или без использования испытываемого соединения, добавляемого в различных концентрациях. По истечении пяти минут инкубации при комнатной температуре реакцию прекращают добавлением 0,2 объема 50%-ного раствора трихлоруксусной кислоты. Затем на ионообменную хроматографическую бумагу Whatman P-81 наносят пробу 30-микролитрового образца из каждой ячейки (контрольной и с испытываемым соединением), после чего 32-P-инкорпорирование подсчитывают с помощью жидкостного сцинцилляционного счетчика Beckman LS 5000 TA. Процентную степень ингибирования ПКС, активированной 510-6 М диC8 и 1 мкг/мл фосфатидилсерина, определяют по формуле% ингибирования = 1,0-[(образец СРМ – базальный СРМ)/(общий СРМ – базальный СРМ)] 100и определяют концентрацию, необходимую для 50%-ного ингибирования. В процессе испытания по такому методу соединения по настоящему изобретению проявляют себя как активные ингибиторы протеинкиназы C. Так, например, ИК50 1,3-диоксо-6-(3-метиламинопропил)-1,2,3,6- тетрагидро-4-(тиофен-3-ил)-пирроло[3,4-c]карбазола составляет 30 нМ. Пример 11 Испытание на определение действия in vivo с использованием ксенотрансплантата мелкоклеточной легочной карциномы Данная процедура является модификацией процедуры, описанной Maneckjee и др. в Proc. Natl. Acad. Sci. USA, том 89, 1169-1173 (февраль 1992 г.). Клетки мелкоклеточной легочной карциномы Н82 (МКЛК) берут из замороженной биомассы, оттаивают и выращивают в RPMI. Перед инъекцией эти клетки трипсинизируют, подсчитывают и повторно суспендируют в смеси забуференного фосфатом физиологического раствора (ЗФР): препарата солюбилизированной базальной мембраны (Matrigel  ) в соотношении 1:2 с доведением концентрации до 5105 или 1,5106 клеток/мл. За день до провокационной пробы самки “голых” мышей с удаленной вилочковой железой в возрасте 4-5 недель (разновидности Harlan Sprague Dawley) получают по 200 P на каждую особь облучения до провокационной пробы и подкожной инъекцией в бок им вводят по 0,2 мл МКЛК на каждую особь (концентрация 1105 или 3105 клеток МКЛК/особь). Группы по 30 мышей обрабатывают внутрибрюшинно по одному разу в день испытываемым соединением в виде 0,2%-ного раствора (солюбилизированным в ДМСО и разбавленным до конечной концентрации носителя 20% ДМСО в ЗФР) из расчета по 10 мг/кг. Такие процедуры осуществляют, начиная с 2 ч после провокационной пробы, и продолжают в течение 45 дней. В качестве контрольных животных используют мышей, которых обрабатывали носителем и не обрабатывали вообще.
Статистический анализ: для сопоставления степени проявления опухолей у животных различных групп прибегают к точному испытанию Фишера [Kendall M., Stuart A., The Advanced Theory of Statistics, том 2 (MacMillan Pub. Co. NY, 1979)]. Для сопоставления разницы в продолжительности жизни применяют U испытание Mann Whitney [Hollander N., Wolfe D.A., Nonparametric Statistical Methods (John Wiley and Sons, Inc., NY, 1973)] и для сопоставления промежутков времени, необходимых для достижения каждой из опухолей объема 2000 мм3, используют испытание с логарифмическим рядом [Kalbfleisch J.D., Prentice R. L. , The Statistical Analysis of Failure Time Data (John Whiley and Sons, Inc., NY, 1980)].
В ходе проведения испытания по такому методу соединения по настоящему изобретению подавляют рост опухоли.
Пример 12 ) в соотношении 1:2 с доведением концентрации до 5105 или 1,5106 клеток/мл. За день до провокационной пробы самки “голых” мышей с удаленной вилочковой железой в возрасте 4-5 недель (разновидности Harlan Sprague Dawley) получают по 200 P на каждую особь облучения до провокационной пробы и подкожной инъекцией в бок им вводят по 0,2 мл МКЛК на каждую особь (концентрация 1105 или 3105 клеток МКЛК/особь). Группы по 30 мышей обрабатывают внутрибрюшинно по одному разу в день испытываемым соединением в виде 0,2%-ного раствора (солюбилизированным в ДМСО и разбавленным до конечной концентрации носителя 20% ДМСО в ЗФР) из расчета по 10 мг/кг. Такие процедуры осуществляют, начиная с 2 ч после провокационной пробы, и продолжают в течение 45 дней. В качестве контрольных животных используют мышей, которых обрабатывали носителем и не обрабатывали вообще.
Статистический анализ: для сопоставления степени проявления опухолей у животных различных групп прибегают к точному испытанию Фишера [Kendall M., Stuart A., The Advanced Theory of Statistics, том 2 (MacMillan Pub. Co. NY, 1979)]. Для сопоставления разницы в продолжительности жизни применяют U испытание Mann Whitney [Hollander N., Wolfe D.A., Nonparametric Statistical Methods (John Wiley and Sons, Inc., NY, 1973)] и для сопоставления промежутков времени, необходимых для достижения каждой из опухолей объема 2000 мм3, используют испытание с логарифмическим рядом [Kalbfleisch J.D., Prentice R. L. , The Statistical Analysis of Failure Time Data (John Whiley and Sons, Inc., NY, 1980)].
В ходе проведения испытания по такому методу соединения по настоящему изобретению подавляют рост опухоли.
Пример 12Испытание на определение действия in vivo с использованием ксенотрансплантата карциномы толстой кишки Действие на карциному толстой кишки определяют осуществлением процедуры примера 11 с заменой клеток мелкоклеточной легочной карциномы Н82 клетками рака толстой кишки НТ-29, выращенными до концентрации 5 106 клеток/мл и введенным в концентрации 1106 клеток/особь.
В ходе проведения испытания по такому методу соединения по настоящему изобретению подавляют рост опухоли.
Хотя настоящее изобретение описано со ссылкой на конкретные варианты его выполнения, любому специалисту в данной области техники совершенно очевидно, что в них можно вносить различные изменения и одни их составляющие можно заменять их эквивалентами, не выходя при этом за рамки и сущность изобретения. Кроме того, могут быть произведены многочисленные модификации для применения конкретных ситуаций, материала, рассматриваемого состава, способа, стадии или стадий способа к объекту, существу и рамкам настоящего изобретения. Все такие модификации следует рассматривать как охватываемые объемом прилагаемой формулы изобретения. Все патенты и публикации, упомянутые выше, приведены в качестве ссылок.
Формула изобретения
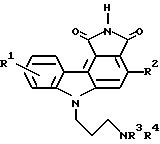 где R1 обозначает водородный атом или низший алкил; R2 обозначает тиофенил; R3 и R4 независимо друг от друга обозначают водородные атомы или низшие алкилы, или его фармацевтически приемлемая соль. 2. Соединение по п.1, где R1 обозначает водородный атом. 3. Соединение по п.2. где R2 обозначает 3-тиофенил. 4. Соединение по п.3, где R3 обозначает водородный атом и R4 обозначает низший алкил. 5. Соединение по п.4, где R3 обозначает водородный атом и R4 обозначает метил, а именно 1,3-диоксо-6-(3-метиламинопропил)-1,2,3,6-тетрагидро-4-(тиофен-3-ил)-пирроло[3,4-с]карбазол. РИСУНКИ
MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 04.08.2006
Извещение опубликовано: 20.12.2007 БИ: 35/2007
|
||||||||||||||||||||||||||