Патент на изобретение №2162085
|
||||||||||||||||||||||||||
(54) СПОСОБ ПОЛУЧЕНИЯ ЭНАНТИОМЕРНО ЧИСТЫХ СОЕДИНЕНИЙ ИМИДАЗОЛИЛА, ЭНАНТИОМЕРНО ЧИСТАЯ КИСЛАЯ АДДИТИВНАЯ СОЛЬ ИМИДАЗОЛИЛА И D- ИЛИ L-ПИРОГЛУТАМИНОВОЙ КИСЛОТЫ, МОНОГИДРАТ ГИДРОХЛОРИДА ЭНАНТИОМЕРНО ЧИСТЫХ СОЕДИНЕНИЙ ИМИДАЗОЛИЛА
(57) Реферат: Настоящее изобретение относится к способу получения энантиомерно чистого соединения имидазолила общей формулы I, энантиомерно чистой аддитивной соли имидазолила и моногидрату гидрохлорида энантиомерно чистого соединения имидазолила общей формулы I, где R1 имеет указанные в формуле значения. Полученные соединения обладают биологической активностью в отношении 5-НГ антагонистов. 3 с. и 14 з.п.ф-лы, 1 табл. 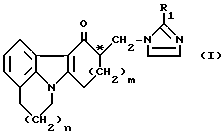
Настоящее изобретение относится к способу получения энантиомерно чистого соединения имидазолила, а также кислой аддитивной соли этого соединения. 4,5,6,8,9,10-гексагидро-10-[(2-метил-1Н-имидазол-1-ил) метил-11Н – пиридо[3,2,1-jk] карбазол-11-он известен из Европейского патента N 0297651 и заявки N 0601345. В первой публикации описаны общий класс соединений, включая вышеуказанное соединение имидазолила и гомологичные ему соединения, их получение и применение в качестве 5-НТ антагонистов. Во второй заявке речь идет о применении соединения, выбранного из этого типа, для лечения некоторых заболеваний. Различные биологически активные вещества, применяемые в фармакологических составах для лечения людей или животных, содержат в своей молекулярной структуре хиральный центр, что порождает оптический изомеризм. Специалистам хорошо известно, что часто лишь один из энантиомеров обладает необходимой биологической активностью. Наличие в составе или композиции другого оптического антипода может вызвать или усилить некоторые побочные явления и причинить вред реципиенту, т. е. организму человека или животного. Поэтому считается все в большей степени желательным вводить биологически активное вещество в виде по существу чистого энантиомера, который наиболее ярко проявляет необходимую биологическую активность. Вот почему разделение рацемата на составляющие его энантиомеры часто является важной стадией в технологии изготовления фармакологически активных веществ. Установлено, что R-(-)-энантиомер вышеуказанного соединения имидазолила, известный также под тривиальным названием цилансетрон, особенно полезен в составах, приведенных в Европейской заявке N 0601345. Поэтому желательно разработать способ отделения R-энантиомера от рацемата. По существу известны три способа разделения рацематов на соответствующие энантиомеры. Первый из них – разделение на основе различий в физических свойствах, например в кристаллической структуре, – применяется лишь изредка. Более поздний способ разделения предусматривает введение энзимов с целью селективной химической модификации энантиомера в рацемате с последующим отделением модифицированного энантиомера от немодифицированного. Третий, наиболее распространенный способ разделения заключается в реакции с промышленно выпускаемым оптически активным реактивом с получением диастереомеров, обладающих различными физическими свойствами. Полученные таким путем диастереомеры можно разделить, например, кристаллизацией, после чего выделить нужный энантиомер химической обработкой. Специалистам хорошо известно, что разделение энантиомеров путем приготовления диастереомеров – весьма сложная задача. Даже опытные исследователи находят, что некоторые соединения не поддаются химическому разделению как отдельными разделяющими агентами, так и их сочетаниями при различных условиях реакции. Как правило, исследователи в области разделения энантиомеров начинают опыты с использования тех реактивов и условий реакции, которые ранее уже показали свою эффективность при разделении подобных соединений. Наиболее распространенный способ разделения рацематов вышеописанных соединений имидазолила заключается в реакции с какой-либо оптически активной кислотой с последующим разделением полученных диастереомеров, предпочтительно кристаллизацией. В Европейской заявке N 0297651 описано применение (+)ди-0,0′-n-толуил-D-винной кислоты. По всей видимости, эта оптически активная карбоновая кислота оптимальна для разделения подобных рацематов, поскольку та же кислота использовалась и для разделения химически близкого соединения имидазолила – 1,2,3,9-тетрагидро-9-метил-3-[(2-метил-1Н-имидазол-1-ил)метил] -4Н- карбазол-4-она, или ондансетрона (см., например, патент Нидерландов 190373, пример XX). Это особенно примечательно с учетом того обстоятельства, что разделение с помощью (+)-ди-0,0′-n-толуил-D-винной кислоты связано с рядом недостатков, например, с использованием малоэффективного растворителя – смеси диметилформамида с водой. Раствор, разбавленный такой смесью, экономически невыгоден или даже неприемлем. Более того, как растворитель диметилформамид обладает такими общеизвестными недостатками, как высокая точка кипения и заметная токсичность (подозревается канцерогенность). Наряду с вышеуказанной оптически активной ди-0,0′-n-толуил-D-винной кислотой промышленно выпускается ряд хиральных дикарбоновых, хиральных сульфоновых и хиральных монокарбоновых кислот, например, дибензоил-L-винная, L-винная, L-яблочная, D-камфор-10-сульфоновая, D-хинная, 2,3:4,6-ди-O-изопропилиден-2-кето-L-гулоновая, L-миндальная, R-2-(4-гидроксифенокси) пропионовая кислоты и (-)-1,3,2-диоксофосфоринан-5,5-диметил-2-гидрокси-4-фенил-2-оксид. Однако, как будет показано в примерах, эти либо никак не способствуют осаждению аддитивной соли одним из энантиомеров, либо не обогащают один из энантиомеров в осадке. Задачей настоящего изобретения является создание экономичного способа получения энантиомерно чистых соединений имидазолила, отвечающего следующим требованиям: а) использование эффективного растворителя и ведение реакции без разбавления; б) легкость регенерации дорогостоящей хиральной кислоты. Поставленная задача достигается тем, что способ получения энантиомерно чистого соединения имидазолила общей формулы 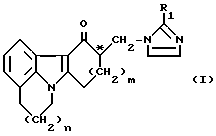 где: n=0 или 1 m=1 или 2 R1 – водород, метил или этил; и C* – хиральный центр, а также его фармакологически приемлемой кислой аддитивной соли, в котором а) добавляют карбоновую кислоту в оптически активной форме к раствору рацемической смеси вышеуказанного соединения I с последующим отделением кристаллизованной кислой аддитивной соли указанной смеси энантиомеров соединения I, обогащенной одним из энантиомеров, от маточной жидкости, обогащенной другим энантиомером; б) после обогащения кристаллизованной кислой аддитивной соли нежелательным энантиомером отделяют смесь энантиомеров в маточной жидкости от указанной оптически активной карбоновой кислоты с последующим добавлением рацемической смеси указанной карбоновой кислоты к раствору полученной смеси изомеров I и отделением кристаллизованной кислой аддитивной соли указанной смеси, обогащенной по целевому энантиомеру, от маточной жидкости; в) произвольно перекристаллизовывают продукт до достижения нужной степени энантиомерной чистоты; г) затем преобразуют эту кислую аддитивную соль целевого энантиомера в целевое энантиомерно чистое соединение имидазолила общей формулы I или его фармацевтически приемлемую кислую аддитивную соль, отличающийся тем, что в качестве указанной карбоновой кислоты используют пироглутаминовую кислоту. Когда образующаяся кислая аддитивная соль обогащается по целевому энантиомеру, ее можно выделить и после доведения до нужной степени энантиомерной чистоты путем последующей обработки преобразовать в целевое энантиомерно чистое соединение имидазола или его фармацевтически приемлемую кислую аддитивную соль. Для удобства такая прямая кристаллизация целевого энантиомера предпочтительна. Когда кислая аддитивная соль, образованная при добавлении оптически активной пироглутаминовой кислоты, обогащается нежелательным энантиомером, используется прием взаимного разделения (Eliel, E.L., Wilen, S.H. and Mander, L. N. в сб. Stereochemistry or Organic Compaunds, John Wiley & Sons, Inc., New York (1994), 325). Он заключается в том, что после первой стадии разделения, на которой кислая аддитивная соль обогащается нежелательным энантиомером, оптически активную пироглутаминовую кислоту удаляют из сухого осадка, полученного из маточной жидкости, например, путем экстракции растворителем – системой дихлорметан – вода. Затем осуществляют вторую стадию, добавляя рацемическую пироглутаминовую кислоту в раствор полученной смеси изомеров I, что приводит к кристаллизации кислой аддитивной соли целевого энантиомера. С учетом того обнаруженного нами (см. примеры) обстоятельства, что химически близкое соединение имидазолила – ондастерон – нельзя выделить в его оптических антиподах с помощью активной пироглутаминовой кислоты, удивительно то, что вышеуказанный целевой энантиомер общей формулы I легко выделить с помощью пироглутаминовой кислоты в оптически активной форме, после чего при желании добавить рацемическую пироглутаминовую кислоту, при соблюдении вышеуказанных требований. Неожиданным оказался и тот факт, что пироглутаминовая кислота столь благотворно воздействует на разделение рацемата соединения имидазолила формулы I, с учетом весьма посредственных результатов применения множества других разделяющих агентов. Энантиомерно чистое соединение имидазолила в соответствии с настоящим изобретением относится к оптически активным соединениям, имеющим избыток энантиомера (ИЭ) свыше 90%. Кристаллическую кислую аддитивную соль целевого энантиомерно чистого соединения имидазолила, получаемую в соответствии с изобретением, можно преобразовать в чистый собственно энантиомер приемами, хорошо известными в расщеплении солей. Как правило, можно проводить расщепление под действием какого-либо основания, получая свободное энантиомерно чистое имидазолильное основание. При желании указанное имидазолильное основание можно перевести в фармацевтически приемлемую кислую аддитивную соль путем обработки кислотой – соляной, малеиновой или иной, как описано в Европейской заявке N 601345. Настоящее изобретение относится, в частности, и к способу получения цилансетрона, т. е. энантиомерно чистого соединения имидазолила общей формулы I, где m и n оба равны 1, R1 – метил, и атом C* имеет R – конфигурацию. Процесс кристаллизации, т.е. отдаления кристаллизующейся кислой аддитивной соли целевого энантиомера или, по меньшей мере, рацемата, обогащенного целевым энантиомером, предпочтительно осуществляют в спиртовом растворителе. В качестве спиртовых растворителей для процесса кристаллизации можно использовать метанол и этанол. В способе в соответствии с изобретением используемая оптически активная кислота D-пироглутаминовая [R-2-пирролидон-5-карбоновая кислота] при прямой кристаллизации и L-пироглутаминовая кислота [S-2-пирролидон-5-карбоновая кислота] при взаимном разделении цилансетрона предпочтительно вводится в количестве от 0,2 до 1,5 эквивалента в расчете на исходную рацемическую смесь. Отношение объема растворителя к количеству энантиомеров в разделяемой смеси может варьироваться в достаточно широких пределах. При прямой кристаллизации отношение количества растворителя к количеству энантиомеров может составлять от около 3: 1 до 15:1 (отношение между объемом растворителя и массой энантиомеров в нем). Предпочтительно это отношение составляет от около 5:1 до около 10:1. В предпочтительном варианте осуществления изобретения объем растворителя и масс энантиомеров соотносится как 7:1. При взаимном разделении отношение количества растворителя к количеству энантиомеров составляет около 3:1 до 15:1 на первой стадии и от 5:1 до 15:1 на второй стадии. Предпочтительно оно равно от около 5:1 до около 10:1 на первой стадии и от 7: 1 до 12: 1 на второй стадии. В предпочтительном варианте осуществления изобретения объем растворителя и масс энантиомеров соотносится как около 7:1 на первой стадии и 10:1 на второй стадии. Раствор, содержащий энантиомеры, можно получить путем растворения смеси энантиомеров в растворителе. Обычно растворение осуществляют при температуре от около 25oC до 80oC, предпочтительно от около 50oC до около 60oC. Кристаллизацию можно проводить при температуре от около -20oC до +20oC, но обычно ее осуществляют при температуре от около -10oC до около 0oC. Впрочем, выход целевого энантиомера остается неудовлетворительным – теоретически менее 50% от исходного рацемата. В качестве дополнительного признака настоящего изобретения нами установлено, что маточную жидкость или смесь маточных жидкостей от процессов кристаллизации можно подвергать дополнительной обработке, включая стадию рацемизации, что позволяет повысить общий выход целевого энантиомера до более 50% в результате последующей кристаллизации, как описано выше. Следовательно, настоящее изобретение относится также к описанному выше способу, отличающемуся тем, что маточную жидкость или смесь маточных жидкостей после отделения кристаллической кислой аддитивной соли подвергают последующей обработке путем (I) отщепления растворенной кислой аддитивной соли с образованием раствора смеси энантиомеров соединения имидазолила общей формулы I, как представлено выше, содержащей пониженное количество целевого энантиомера, и (II) последующего преобразования указанного раствора в рацемическую смесь под действием основания. В случае взаимного разделения кислую аддитивную соль, обогащенную нежелательным энантиомером, можно при желании добавить к маточной жидкости или их смеси. В качестве основания при рацемизации предпочтительно служит неорганическое основание, например гидрооксид щелочного металла. После вышеописанной рацемизации восстановленный рацемат можно вновь подвергнуть указанному процессу кристаллизации с применением оптически активной пироглутаминовой кислоты, при желании с последующим вводом рацемической пироглутаминовой кислоты с целью обеспечить дополнительный выход энантиомерно чистого соединения имидазолила. При желании маточную жидкость или смесь маточных жидкостей от этой кристаллизации можно вновь рацемировать и т.д. и т. п., тем самым можно заметно увеличить общий выход целевого энантиомера. Технологически и экономически целесообразно добавлять восстановленный рацемат к исходному рацемату при следующей обработке с тем, чтобы в конечном счете практически исключить потери сырья. Кислая аддитивная соль энантиомерно чистого соединения имидазолила общей формулы I, в частности цилансетрона и D-пиролутаминовой кислоты, обладает существенной новизной. Таким образом, настоящее изобретение относится также к этой кислой аддитивной соли, которую можно получить вышеописанным способом кристаллизации. Далее изобретение будет описано более подробно со ссылками на нижеследующие конкретные примеры. Пример 1 Получение (R)-(-)-4,5,6,8,9,10-гексагидро-10 – [2-метил-1Н-имидазол-1-ил)метил] -11Н-пиридо-[3,2,1-jk] -карбазол- 11-он гидрохлорида моногидрата (цилансетрона) прямым разделением 25,00 г (RS)-4,5,6,8,9,10-гексагидро-10-[(2-метил-1Н-имидазол-1- ил)метил] -11Н-пиридо-[3,2,1-jk] -карбазол-11-она и 10,11 г R-2-пирролидон-5-карбоновой кислоты (D-пироглутаминовой кислоты) в 175 мл метанола нагревают до 50oC. Полученную суспензию диастереомерных солей перемешивают 1 час при этой температуре. Смесь охлаждают до 0oC и перемешивают 1 час при этой температуре. Твердую фазу отсасывают, промывают холодным метанолом и сушат. Выход: 25,91 г. Процесс кристаллизации повторяют еще дважды с расходом метанола 5 мл на 1 г получаемой соли в первый раз и 10 мл во второй раз. Выход: 11,91 г. Маточные жидкости от трех этапов кристаллизации сливают вместе и используют на новой стадии. 10,00 г полученной соли перемешивают 15 мин с 200 мл воды, 50 мл дихлорметана и 6,00 г бикарбоната натрия. После разделения фаз водную фазу дважды экстрагируют 25 мл дихлорметана. Дихлорметановые фазы сливают и упаривают досуха. Полученное сухое вещество растворяют в 60 мл изопропанола и добавляют к раствору 2,5 мл концентрированной соляной кислоты при комнатной температуре. Перемешивают 1 час и отсасывают образовавшееся твердое вещество, промывают холодным изопропанолом и петролейным эфиром в отношении 40:65 и сушат. Выход целевого соединения: 7,93 г (ИЭ 94%). Точка плавления: 219oC. [  ]D25= – 6,9 (C=1,8; метанол).
Пример 2 ]D25= – 6,9 (C=1,8; метанол).
Пример 2Получение (R)-(-)-4,5,6,8,9,10-гексагидро-10 – [2-метил-1Н-имидазол-1-ил)метил] -11Н-пиридо-[3,2,1-jk] -карбазол-11-она гидрохлорида моногидрата (цилансетрона) взаимным разделением 25,00 г (RS)-4,5,6,8,9,10-гексагидро-10[(2-метил-1Н- имидазол-1-ил)метил] -11Н-пиридо-[3,2,1-jk] -карбазол-11-она и 10,11 г S-2-пирролидон-5-карбоновой кислоты (L-пироглутаминовой кислоты) в 175 мл метанола нагревают до 50oC. Полученную суспензию диастереометрических солей перемешивают 1 час при этой температуре. Смесь охлаждают до 0oC и перемешивают 1 час при этой температуре. Твердое вещество отсасывают, промывают холодным метанолом и сушат. Выход: 18,5 г. Метанол выпаривают из маточной жидкости. Остаток перемешивают 15 мин с 200 мл воды, 50 мл дихлорметана и 6,00 г бикарбоната натрия. После разделения фаз водную фазу дважды экстрагируют 25 мл дихлорметана. Дихлорметановые фазы сливают и упаривают досуха. Полученное сухое вещество (11,50 г) и 4,75 г RS-пирролидон-5-карбоновой кислоты (D,L-пироглутаминовой кислоты) растворяют в 115 мл метанола и нагревают с обратным холодильником. Раствор охлаждают до комнатной температуры и перемешивают при ней 1 час. Отсасывают образовавшееся твердое вещество, промывают холодным метанолом и сушат. Выход: 6,00 г (ИЭ 97%). 5,00 г полученной таким образом соли перемешивают 15 мин со 100 мл воды, 25 мл дихлорметана и 3,00 г бикарбоната натрия. После разделения фаз водную фазу дважды экстрагируют 12,5 мл дихлорметана. Дихлорметановые фазы сливают и упаривают досуха. Полученное сухое вещество растворяют в 30 мл изопропанола. К раствору добавляют 1,25 мл концентрированной соляной кислоты при комнатной температуре. После 1 ч перемешивания полученное твердое вещество отсасывают, промывают холодным изопропанолом и петролейным эфиром 1 в отношении 40:65 и сушат. Выход целевого соединения: 3,95 г (ИЭ 98%). Точка плавления: 219oC. Пример 3 Рацемизация слитых маточных жидкостей до (R,S)-(-)- 4,5,6,8,9,10-гексагидро-10-[(2-метил-1Н-имидазол-1-ил)метил] -11Н- пиридо-[3,2,1-jk]-карбазол-11-она и извлечение второго сбора R-энантиомера прямым разделением. Метанол выпаривают из слитых маточных жидкостей по примеру 1. Остаток перемешивают с 250 мл воды, 100 мл дихлорметана и 10,00 г бикарбоната натрия. После разделения фаз водную фазу экстрагируют 50 мл дихлорметана. Дихлорметановые фазы сливают и упаривают досуха. Полученное твердое вещество растворяют в 90 мл метанола и 20 мл воды. С целью рацемизации добавляют раствор 20 г гидроксида калия в 5 мл воды. После 30 мин перемешивания реакционную смесь нейтрализуют 2 N соляной кислоты. К этому раствору добавляют 500 мл воды. Водно-метанольную фазу экстрагируют 100 мл дихлорметана, затем дважды по 50 мл. Слитые дихлорметановые фазы упаривают досуха. К полученному сухому веществу добавляют 6,1 г R-2 пирролидон-5-карбоновой кислоты и 75 г метанола. Температуру поднимают до 50oC. Полученную суспензию диастереомерных солей перемешивают 1 час при этой температуре. Смесь охлаждают до 0o С и перемешивают 1 час при этой температуре. Твердое вещество отсасывают, промывают холодным метанолом и сушат. Выход аддитивной соли – 7,49 г. Этот процесс кристаллизации повторяют еще дважды с расходом метанола на 1 г получаемой соли 5 мл в первом случае и 10 мл во втором случае. Выход – 4,97 г. Полученную соль перемешивают 15 мин со 100 мл воды, 25 мл дихлорметана и 3,00 г бикарбоната натрия. После разделения фаз водную фазу дважды экстрагируют 15 мл дихлорметана. Слитые дихлорметановые фазы упаривают досуха. Полученное сухое вещество растворяют в 30 мл изопропанола. К этому раствору добавляют 1,3 мл концентрированной соляной кислоты при комнатной температуре. После 1 ч перемешивания полученное твердое вещество отсасывают, промывают холодным изопропанолом и петролейным эфиром в отношении 40:65 и сушат. Дополнительный выход целевого соединения – 3,12 г (ИЭ 95%). Точка плавления – 219oC. Таким же образом можно рацемизировать и кристаллизировать (прямым или взаимным разделением) маточные жидкости по примеру 2 в смеси с кислой аддитивной солью, обогащенной по нежелательному энантиомеру. Пример 4 Попытки разделения R,S-1,2,3,9-тетрагидро-9-метил-3-[(2- метил-1Н-имидазол-1-ил)метил]-4Н-карбазол-4-она (ондасетрона) 0,50 г (R, S)-1,2,3,9-тетрагидро-9-метил-3-[(2-метил-1Н-имидазол-1-ил)метил] -4Н-карбазол-4-она и 0,22 г R-2-пирролидон-5-карбоновой кислоты нагревают в 5,0 мл метанола до 50oC. Полученный светлый раствор охлаждают до 0oC за 30 мин. После 1 ч перемешивания при 0oC полученные кристаллы отсасывают, промывают холодным метанолом и сушат. Выход – 0,02 г. Высокоэффективная жидкостная хроматография показывает отношение R/S 1:1. Это означает, что обогащение не имеет места. Опыт повторяют в тех же условиях, но вместо 5,0 мл метанола берут 1,5 мл. Выход – 0,12 г. Отношение R/S также составляет 1:1. Пример 5 Сравнительные опыты По процедуре, описанной в примере 1, исследуют выделение цилансетрона из рацемата с применением ряда промышленно выпускаемых оптически активных кислот. Полученные результаты приведены в прилагаемой таблице. Из этих результатов можно заключить следующее: Вывод: только применение D-пироглутаминовой кислоты (R-2-пирролидон-5-карбоновой кислоты) позволяет добиться требуемого обогащения по R-энантиомеру. Формула изобретения
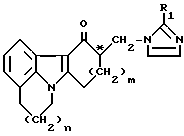 где n = 0 или 1; m = 1 или 2; R1 – водород, метил или этил; С* – хиральный центр, а также его фармацевтически приемлемой кислой аддитивной соли, а) путем добавления карбоновой кислоты в оптически активной форме к раствору рацемической смеси вышеуказанного соединения I с последующим отделением кристаллизованной кислой аддитивной соли указанной смеси энантиомеров соединения I, обогащенной в отношении одного энантиомера, от маточной жидкости, обогащенной в отношении другого энантиомера; b) в случае получения кристаллизованной кислой аддитивной соли, обогащенной нежелательным изомером, с последующим удалением оптически активной кислоты от смеси энантиомеров в полученной маточной жидкости, с последующим добавлением рацемической смеси указанной карбоновой кислоты к раствору полученной смеси изомеров I и отделением кристаллизованной кислой аддитивной соли указанной смеси, обогащенной целевым энантиомером, от маточной жидкости; с) необязательной перекристаллизацией продукта до достижения нужной степени энантиомерной чистоты; d) преобразованием этой кислой аддитивной соли целевого энантиомера в целевое энантиомерно чистое соединение имидазолила общей формулы I или его фармацевтически приемлемую кислую аддитивную соль, отличающийся тем, что в качестве указанной карбоновой кислоты используют пирроглутаминовую кислоту. 2. Способ получения энантиомерно чистого соединения имидазолила общей формулы I по п.1, в котором n, m, R1 и C* имеют те же значения, что в п.1, а также его фармацевтически приемлемой кислой аддитивной соли, а) путем добавления оптически активной карбоновой кислоты к раствору рацемической смеси вышеуказанного соединения I с последующим отделением кристаллизованной кислой аддитивной соли из указанной смеси, обогащенной целевым энантиомером, от маточной жидкости; b) необязательной перекристаллизацией продукта до достижения нужной степени энантиомерной чистоты; с) превращением полученной кислой аддитивной соли в целевое энантиомерно чистое соединение имидазолила общей формулы I или его фармацевтически приемлемую кислую аддитивную соль, отличающийся тем, что в качестве оптически активной карбоновой кислоты используют D-пироглутаминовую кислоту. 3. Способ получения энантиомерно чистого соединения имидазолила общей формулы I по п.1, в котором n, m, R1 и C* имеют те же значения, что в п.1, а также его фармацевтически приемлемой кислой аддитивной соли, а) путем добавления оптически активной карбоновой кислоты к раствору рацемической смеси вышеуказанного соединения I с последующим отделением кристаллизованной кислой аддитивной соли из указанной смеси, обогащенной целевым энантиомером, от маточной жидкости; b) отделением оптически активной кислоты от смеси энантиомеров в маточной жидкости, добавлением рацемической смеси указанной карбоновой кислоты, отделением кристаллизованной кислой аддитивной соли указанной смеси энантиомеров соединения формулы I, обогащенной целевым энантиомером, от маточной жидкости; с) необязательной перекристаллизацией продукта до достижения нужной степени энантиомерной чистоты; d) преобразованием полученной кислой аддитивной соли желаемого энантиомера в целевое энантиомерно чистое соединение имидазолила общей формулы I или его фармацевтически приемлемую кислую аддитивную соль, отличающийся тем, что в качестве карбоновой кислоты используют пирроглутаминовую кислоту, а в качестве ее оптически активной формы – L-форму указанной карбоновой кислоты. 4. Способ по пп.1 – 3, отличающийся тем, что получают соединение формулы I, где n = 1, m = 1, R1 – метил; атом C* имеет R-конфигурацию. 5. Способ по пп.1 – 4, отличающийся тем, что пирроглутаминовую кислоту в оптически активной форме вводят в количестве 0,2 – 1,5 эквивалента в расчете на исходную рацемическую смесь. 6. Способ по любому из предшествующих пунктов, отличающийся тем, что кристаллизацию проводят в спиртовом растворителе. 7. Способ по п.6, отличающийся тем, что кристаллизацию проводят в метаноле или этаноле. 8. Способ по пп.1 – 7, отличающийся тем, что маточную жидкость или смесь маточных жидкостей после отделения кристаллизующейся кислой аддитивной соли подвергают последующей обработке путем (I) расщепления растворенной кислой аддитивной соли с получением раствора смеси энантиомеров соединения имидазолила формулы I по п.1 с пониженным содержанием целевого энантиомера и (II) последующего преобразования указанного раствора в рацемическую смесь под действием основания. 9. Способ по п.8, отличающийся тем, что для рацемизации применяют неорганическое основание, предпочтительно гидроксид щелочного металла. 10. Способ по пп.1 – 9, отличающийся тем, что соединение общей формулы I выделяют в виде его гидрохлорида моногидрата. 11. Кислая аддитивная соль энантиомерно чистого соединения имидазолила общей формулы I по п.1, где n, m, R1 и C* имеют те же значения, что в п.1, и D- или L-пироглутаминовой кислоты. 12. Кислая аддитивная соль энантиомерно чистого соединения имидазолила общей формулы I и D-пироглутаминовой кислоты по п.11. 13. Кислая аддитивная соль энантиомерно чистого соединения имидазолила общей формулы I и L-пироглутаминовой кислоты по п.11. 14. Кислая аддитивная соль по п.12, где n, m, R1 и C* имеют значения, указанные в п.4. 15. Кислая аддитивная соль по п.13, где n, m, R1 и C* имеют значения, указанные в п.4. 16. Соединение общей формулы I, где n, m, R1 и C* имеют значения, указанные в п.1, в виде его гидрохлорида моногидрата. 17. Соединение по п.16, где n, m, R1 и C* имеют значения, указанные в п. 4. РИСУНКИ
|
||||||||||||||||||||||||||