Патент на изобретение №2162080
|
||||||||||||||||||||||||||
(54) УЛУЧШЕННЫЙ СПОСОБ ПОЛУЧЕНИЯ 2,4-ДИАМИНО-6-АЛКИЛТИО- S -ТРИАЗИНОВ
(57) Реферат: Описывается улучшенный способ получения 2,4-диамино-6-алкилтио-s-триазинов общей формулы I, где R1 и R2 независимо друг от друга означают водород, С1-С12-алкил, С3-С12-циклоалкил, указанный циклоалкил, замещенный С1-С4алкилами, С4-С20-циклоалкил алкил, указанный С4-С20-циклоалкил алкил, замещенный С1-С4-алкилами, и R3 означает С1-С12-алкил, путем взаимодействия цианурхлорида последовательно с R1NH2, R2NH2 и реагентом, содержащим R3S-группу, в одном не смешивающемся с водой органическом растворителе в присутствии катализатора переноса фаз на стадии взаимодействия с реагентом, содержащим R3S-группу. Способ отличается тем, что в качестве реагента, содержащего R3S-группу, используют тиол в присутствии кислотосвязывающего средства, в качестве катализатора переноса фаз – четвертичные аммониевые соли и диазацилоалканы, которые используют в количестве от 1 до 10 мол.% в расчете на исходный цианурхлорид, а в качестве не смешивающегося с водой органического растворителя используют ароматический С6-С10углеводород или С4-С11алканон. Полученные соединения находят применение в качестве средств, препятствующих обрастанию микроорганизмами объектов, которые подвергаются воздействию морской воды. Технический результат – получение целевого продукта с высоким выходом и высокой степенью чистоты. 9 з.п. ф-лы, 4 табл. 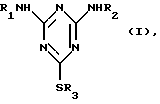
Изобретение относится к улучшенному способу получения 2,4-диамино-6-алкилтио-s-триазинов путем последовательного трехстадийного взаимодействия цианурхлорида с двумя подходящими алкиламинами и алкилмеркаптаном. Полученные соединения проявляют гербицидную и альгицидную активности и особенно полезны в качестве средств, препятствующих обрастанию микроорганизмами объектов, которые подвергаются воздействию морской воды. Известно в данной области получение замещенных 2,4-диамино-6-тио-s-триазинов путем последовательного трехстадийного добавления реагентов к цианурхлориду. Так, аналогичные трехстадийные способы описаны в патентах США N 3830810; 3766182 и 3753986, при осуществлении которых на двух первых последовательных стадиях цианурхлорид взаимодействует с одним первичным амином и затем со вторым первичным амином в растворителе или разбавителе, например алифатических углеводородах, кетонах, простых эфирах, ароматических углеводородах и тому подобных, и в присутствии кислотосвязывающего агента, причем первую стадию (то есть добавление первого амина) проводят при температуре ниже 30oC, предпочтительно в интервале от 15oC до 0oC, а второе аминирование – при температурах в интервале от 0oC до 45oC. Замещение последнего атома хлора алкилтиогруппой осуществляется путем добавления водного раствора основания к суспензии промежуточного продукта в разбавителе (то есть в смеси ацетон/вода) и затем перемешивают смесь, пока не образуется прозрачный раствор. Затем вводят двойное молярное количество алкилмеркаптана, перемешивают смесь при комнатной температуре и выделяют целевой продукт. В альтернативном варианте замещение последнего хлора на алкилтиогруппу проводят также при добавлении соответствующего производного 6-хлор-s-триазина к спиртовому или водно-спиртовому раствору меркаптида щелочного металла и нагревании полученной смеси с обратным холодильником до тех пор, пока смесь не станет нейтральной. Основной недостаток этих известных способов синтеза 2,4-ди(алкиламино)-6-алкилтио-s-триазинов заключается в том, что промежуточный продукт, полученный после проведения второй стадии, должен быть выделен и/или очищен, например, фильтрацией или перекристаллизацией до осуществления следующей стадии, если конечный продукт должен быть получен с приемлемым выходом. Однако выделение промежуточного 2,4-диамино-6-хлор-s-триазина приводит к уменьшению выхода, экономически не желательно, требует времени и вызывает загрязнение окружающей среды. Другим недостатком известных способов является необходимость изменения растворителей, например толуола, на первых двух стадиях с последующим его удалением и заменой на смесь ацетон/вода для замещения последнего атома хлора. Такая замена растворителя приводит к увеличению времени цикла, удорожанию продукта и увеличению загрязнения среды. Более того, смеси ацетона с водой затрудняют удаление хлористого натрия из конечного продукта. Наиболее близкий к заявляемому способ получения 2,4-ди(алкиламино)-6-алкилтио-s-триазинов (EP-A-0357556, 1990 г.) включает введение трех соответствующих заместителей в триазиновое кольцо в 2-фазной системе (вода и не смешивающийся с водой органический растворитель) в присутствии третичного амина на стадии введения алкилмеркаптогруппы. В примере указанного способа проиллюстрировано использование тиолата металла (тиометилата натрия) в присутствии третичного амина с введением меркаптогруппы в триазиновое кольцо на соответствующей стадии. Однако указанный способ не позволяет получить продукт высокой степени чистоты (чистота полученного продукта не выше 88,9%). В основу изобретения положена задача путем изменения технологических операций разработать улучшенный способ получения 2,4-диамино-6-алкилтио-s-триазинов, который устраняет необходимость удаления и/или очистки промежуточных продуктов, необходимость замены растворителей, а также обеспечивает получение продукта с высоким выходом и высокой степенью чистоты. Задача решена тем, что в улучшенном способе получения 2,4-диамино-6-алкилтио-s-триазинов общей формулы (I): 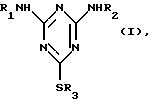 где R1 и R2 независимо друг от друга означают водород, C1-C12-алкил, C3-C12-циклоалкил, указанный циклоалкил, замещенный C1-C4-алкилами, C4-C20-циклоалкил алкил, указанный C4-C20-циклоалкил алкил, замещенный C1-C4-алкилами; и R3 означает C1-C12-алкил, путем взаимодействия цианурхлорида последовательно с R1NH2, R2NH2 и реагентом, содержащим R3S-группу, в одном не смешивающемся с водой органическом растворителе в присутствии катализатора переноса фаз на стадии взаимодействия с реагентом, содержащим R3S-группу, согласно изобретению улучшение заключается в том, что в качестве реагента, содержащего R3S-группу, используют тиол в присутствии кислотосвязывающего средства, в качестве катализатора переноса фаз – четвертичные аммониевые соли и диазациклоалканы, которые используют в количестве от 1 до 10 мол.% в расчете на исходный цианурхлорид, а в качестве не смешивающегося с водой органического растворителя используют ароматический C6-C10 углеводород или C4-C11 алканон. Предпочтительно использовать в качестве не смешивающихся с водой органических растворителей толуол, ксилол или метилизобутилкетон и, в особенности, толуол и ксилол, причем наиболее предпочтительным является ксилол. Целесообразно в качестве катализатора переноса фазы использовать трибутилметиламмоний хлорид, 1,4-диазабицикло[2,2,2]октан, бензилтриметиламмоний гидроокись, тетрабутиламмоний хлорид, тетрабутиламмоний бромид или тетрабутиламмоний кислый сернокислый, в особенности тетрабутиламмоний бромид. Предпочтительным является осуществление способа, когда R1 и R2 независимо друг от друга представляют собой водород, C1-C12-алкил, C3-C12-циклоалкил или указанный циклоалкил, замещенный C1-C4-алкильными группами. Более предпочтительно, когда R1 означает C3-C12-циклоалкил, R2 – C1-C12-алкил и R3 – C1-C12-алкил. Наиболее предпочтительно, когда R1 означает циклопропил, R2 означает трет-бутил и R3 означает метил. Согласно изобретению наиболее предпочтительными соединениями формулы (I), полученными по заявляемому способу, являются 2-(трет-бутиламино)-4-(циклопропиламино)-6-(метилтио)-s-триазин, 2,4-бис(трет-бутиламино)-6-(метилтио)-s-триазин, 2,4-бис-(циклопропиламино)-6-(метилтио)-s-триазин и 2-амино-4-трет-бутиламино-6-метилтио-s-триазин. Заявляемый способ приводит к уменьшению времени цикла, снижению загрязнений окружающей среды за счет уменьшения числа растворителей и облегчает удаление хлористого натрия из конечного продукта. Кроме того, заявляемый способ позволяет получить целевой продукт высокой степени чистоты. Результаты воспроизведения Примера 1 точно в соответствии с методикой способа Coers (EP-A-0357556) показали, что чистота конечного продукта по этому способу составляет только 88,9%, что значительно ниже по сравнению с заявляемым способом, в котором чистота полученных продуктов после проведения всех трех стадий процесса составляет от 96,0 до 99,0% Для получения соединений формулы (I) на первой стадий осуществляют взаимодействие цианурхлорида с соответствующим алкиламином, взятых в молярном отношении 1: 1 в расчете на цианурхлорид, при 0-75oC, предпочтительно при 20-50oC, в среде органического растворителя и в присутствии кислотосвязывающего агента для нейтрализации получающейся соляной кислоты. Может быть использован широкий интервал pH (то есть 1-9), но предпочтительно pH 6,5-8,0, для того, чтобы избежать образования продуктов гидролиза. Эта реакция завершается, когда поддерживается pH около 7,0 без добавления дополнительного количества кислотосвязывающего агента. На следующей стадии процесса к раствору промежуточного продукта первой стадии в органическом растворителе добавляют второй амин в молярном отношении 1:1 в расчете на исходный цианурхлорид при 20-95oC и предпочтительно при 50-75oC. Одновременно вводят кислотосвязывающий агент для нейтрализации образующейся соляной кислоты. Значение pH может находиться в широком интервале (4-12), но предпочтительно значение pH 8-10, для того, чтобы избежать образования продуктов гидролиза. Эта реакция завершается, когда сохраняется значение pH около 9,0 без добавления дополнительного количества кислотосвязывающего агента. На третьей стадии, заключающейся в замещении оставшегося хлора, к реакционному раствору, образовавшемуся на второй стадии, добавляют соответствующий катализатор переноса фазы в количестве 1-10 мол.% в расчете на исходный цианурхлорид, кислотосвязывающий агент и меркаптан в молярном отношении к исходному цианурхлориду, равном 1,0-1,3:1. Указанная реакция осуществляется при температуре 60-150oC и давлении, находящемся в интервале между атмосферным и 100 ф/дюйм2 (689,4 кПа) (атмосферное – 7 кг/см2). Однако предпочтительны температура между 90-130oC и давление в интервале между атмосферным и 50 ф/дюйм2 (344,7 кПа) (атмосферное – 3,5 кг/см2). При проведении указанных реакций в качестве кислотосвязывающих агентов применяют гидроокиси щелочных металлов или карбонаты этих металлов и гидроокиси или карбонаты щелочноземельных металлов, алкиламин, вводимый в реакцию, в избыточном количестве и третичные амины, например триалкиламины, пиридин и пиридиновые основания. Предпочтительны неорганические основания, особенно гидроокиси щелочных металлов, например гидроокись натрия. Если используют органическое основание, органический растворитель следует промывать водой с последующей нейтрализацией сточных вод и выделением органического основания. Полученные соединения проявляют гербицидную и альгицидную активности и особенно полезны в качестве средств, препятствующих обрастанию микроорганизмами объектов, которые подвергаются воздействию морской воды. В объем рассматриваемых объектов могут входить, в частности, корпуса судов, конструкции, находящиеся в воде, бакены, рыболовные сети, а также системы охлаждения и трубопроводы, через которые проходит морская вода. Триазины вообще предназначены для защиты всех материалов, находящихся в контакте с морской водой, от обрастания морскими водорослями (микроорганизмами), например, таких материалов, как древесина, целлюлоза, текстиль и кожа, краски, лаки, например краски для необрастающих покрытий и материалов для нанесения аналогичных покрытий, оптического стекла и других типов стекла, пластических масс, каучука и адгезивов, а также других материалов. Улучшенный способ получения s-триазинов формулы (I) в соответствии с настоящим изобретением иллюстрируется следующими примерами. Если иначе не оговорено, в этих примерах все части и проценты приведены по весу. Продукты, полученные в примерах, сравнивают с идентичными чистыми известными образцами соответствующих соединений методом газовой хроматографии. Пример 1 2-(трет-бутиламино)-4-(циклопропиламино)-6-(метилтио)-s-триазин Стадия 1: В герметичном реакционном сосуде, продутом азотом, осуществляют взаимодействие цианурхлорида (92,3 г, 0,5 моля) с третбутиламином (36,9 г, 0,5 моля), используя о-ксилол (370 г) в качестве растворителя. Амин добавляют в течение 30 мин. Затем для поддержания pH 6,5-8,0 при температуре 50oC добавляют в течение 90 мин 20%-ный раствор едкого натра (98,9 г, 0,49 моля). Реакция завершена, когда достигается стабильный pH около 7. Стадия 2: К о-ксилольному раствору, образовавшемуся на 1-й стадии, в течение 10-15 мин при 50oC добавляют циклопропиламин (39,6 г, 0,5 моля). В конце добавления амина реакционная температура равна 70-75oC. Для поддержания pH 8-10 при температуре 75oC в течение 30 мин добавляют 20% раствор едкого натра (98,8 г, 0,49 моля). Реакция завершена, когда достигается стабильное значение pH  9,0. По завершении второй стадии реакционная смесь разделяется на водный и органический слои и добавляется соляной раствор. Органический слой, полученный на 2-й стадии, концентрируют (до 50 вес.%) азеотропной отгонкой или вакуумной дистилляцией о-ксилола.
Стадия 3: 9,0. По завершении второй стадии реакционная смесь разделяется на водный и органический слои и добавляется соляной раствор. Органический слой, полученный на 2-й стадии, концентрируют (до 50 вес.%) азеотропной отгонкой или вакуумной дистилляцией о-ксилола.
Стадия 3:Реакционную массу, полученную на 2-й стадии (236 г), тетрабутиламмоний бромид (3,18 г, 0,01 моля) и 20%-ный раствор едкого натра (108,4 г, 0,54 моля) добавляют в реактор Parr из нержавеющей стали. Полученную смесь охлаждают до 0oC, реактор промывают азотом и герметизируют. При 0oC добавляют метилмеркаптан (27,2 г, 0,57 моля) и реакционную смесь затем нагревают до 100oC в течение 30 мин. При 100oC в реакторе создается давление 15-20 ф/дюйм2 (1,1-1,4 кг/см2) и реактор выдерживают при этих условиях и при перемешивании в течение 4-х часов. После завершения третьей стадии реакции давление сбрасывают и разделяют реакционную массу на водный и органический слои. Органический слой дважды промывают водой (по 100 г на промывку). Затем к органическому слою добавляют воду (600 г) и азеотропной отгонкой удаляют растворитель. Для осаждения конечного продукта в горячей воде добавляют диспергатор – натриевую соль формальдегид-нафталинсульфокислоты (0,7 г). Суспензию охлаждают до 50oC и фильтруют. После промывки осадка на фильтре водой (500 г) его сушат под вакуумом при 75oC, получают 124,1 г белого твердого вещества (выход 96% в расчете на цианурхлорид). Вместо формальдегид-нафталинсульфокислоты можно применять другие диспергаторы – сульфированный пропилолеат, сульфированный бутилолеат, N-метил-N-олеилтаурат. Анализ: чистота продукта – 98,2%, сравнение по стандартным образцам конечного продукта выполнено методом газовой хроматографии на колонке Hewlett Packard 5890, снабженной ДВ-5 (длина 30 м, мегабор, пленка толщиной 1 микрон). Т.пл.: 127-130oC Данные 1H ЯМР приведены в табл. 1 Данные 13C ЯМР: Химический сдвиг – Число атомов углерода 163,87-165,26 ч/млн. – 3C 51,01 ч/млн. – 1C 28,84 ч/млн. – 3C 23,25 ч/млн. – 1C 13,27-12,84 ч/млн. – 1C 7,05 ч/млн. – 2C Данные масс-спектрального анализа: отношение массы к заряду (m/z) = 253 Полученные данные подтверждают структуру. Пример 2 2-(трет-бутиламино)-4-(циклопропиламино)-6-(метилтио)-s-триазин Повторяют пример 1, но вместо о-ксилола используют метилизобутилкетон. Выход продукта (белое твердое вещество) равен 96% от теоретического. Газовая хроматография свидетельствует, что чистота продукта составляет 97,2%. Анализ данных аналогичен примеру 1, что свидетельствует о том, что структура полученного соединения такая же, как в примере 1. Пример 3 2-(трет-бутиламино)-4-(циклопропиламино)-6-(метилтио)-s-триазин Стадия 1: В герметичном сосуде, промытом азотом, осуществляют реакцию цианурхлорида с трет-бутиламином (36,9 г, 0,5 моля), используя толуол (370 г) в качестве растворителя. Амин добавляют в течение 30 мин. Затем для поддержания значения pH 6,5-8,0 при температуре 50oC в течение 90 мин добавляют 20%-ный раствор едкого натра (98,8 г, 0,49 моля). Реакция завершена, когда достигается стабильное значение pH около 7,0. Стадия 2: К раствору, полученному на 1-й стадии, при 50oC в течение 10-15 мин добавляют циклопропиламин (39,6 г, 0,5 моля). К концу добавления амина температура реакции равна 70-75oC. Для поддержания pH 8-10 при температуре 75oC в течение 30 мин добавляют 20%-ный раствор едкого натра (98,8 г, 0,49 моля). Реакция завершается, когда достигается стабильное значение pH около 9,0. После завершения 2-й стадии смесь выдерживают для разделения водного и органического слоев. Органический слой концентрируют (до 50 вес.%) азеотропной или вакуумной отгонкой толуола. Стадия 3: В реактор Parr из нержавеющей стали добавляют реакционную массу, полученную на 2-й стадии (236 г), трибутилметиламмоний хлорид (12,36 г, 0,039 моля) и 20%-ный раствор едкого натра (108,4 г, 0,54 моля). Полученную смесь охлаждают до 0oС, промывают реактор азотом и герметизируют. При 0oC добавляют метилмеркаптан (27,2 г, 0,54 моля) и затем реакционную смесь нагревают до 100oC в течение 30 мин. При 100oC в реакторе давление составляет 15-20 ф/дюйм2 (1,1-1,4 кг/см2), реактор выдерживают при этих условиях в течение 4 час при перемешивании. После завершения 3-й стадии давление сбрасывают и отделяют соляной раствор. Органический слой промывают водой (100 г) и отделяют. Затем к органическому слою добавляют воду (1200 г) и азеотропной отгонкой удаляют растворитель. Суспензию охлаждают до 50oC и отфильтровывают. После промывки осадка на фильтре водой (400 г) его сушат при 80oC под вакуумом, получая 119,2 г продукта, 96,6% от теоретического количества. Анализ: чистота 97,4% (газовая хроматография). Анализ данных аналогичен примеру 1, что свидетельствует о том, что структура полученного соединения такая же, как в примере 1. Пример 4 2-(трет-бутиламино)-4-(циклопропиламино)-6-(метилтио)-s-триазин Повторяют пример 1, но вместо тетрабутиламмоний бромида на 3-й стадии добавляют 1,4-диазабицикло[2,2,2]октан (4,94 г, 0,043 моля). Получают белое твердое вещество с выходом 97,2% от теоретического. Газовая хроматография показывает, что чистота продукта равна 97,3%. Анализ данных аналогичен примеру 1, что свидетельствует о том, что структура полученного соединения такая же, как в примере 1. Пример 5 2-(трет-бутиламино)-4-(циклопропиламино)-6-(метилтио)-s-триазин Повторяют пример 1, но на 3-й стадии вместо тетрабутиламмоний бромида применяют тетрабутиламмоний кислый сернокислый. Получают белый осадок с выходом 97,9% от теоретического. Метод газовой хроматографии показывает, что чистота продукта составляет 97,5%. Анализ данных аналогичен примеру 1, что свидетельствует о том, что структура полученного соединения такая же, как в примере 1. Пример 6 2-(трет-бутиламино)-4-(циклопропиламино)-6-(метилтио)-s-триазин Повторяют пример 1, но вместо тетрабутиламмоний бромида на 3-й стадии добавляют бензилтриметиламмоний гидроокись (8,4 г, 0,02 моля). Получают белый продукт с выходом 95,9% (от теоретического). Как свидетельствует метод газовой хроматографии, чистота продукта равна 97,5%. Анализ данных аналогичен примеру 1, что свидетельствует о том, что структура полученного соединения такая же, как в примере 1. Пример 7 2,4-бис(трет-бутиламино)-6-(метилтио)-s-триазин. Стадии 1 и 2: В герметичном сосуде, промытом азотом, осуществляют взаимодействие цианурхлорида (50 г, 0,27 моля) с трет-бутиламином (40,3 г, 0,54 моля), используя метилизобутилкетон в качестве растворителя. Амин добавляют в течение 30 мин. Затем для поддержания значения pH 8,0-10,0 при температуре 55-75oC в течение 150 мин добавляют 20%-ный раствор гидроокиси натрия (108 г, 0,54 моля). Реакция завершена, когда достигается стабильное значение pH около 9. После завершения 2-й стадии водный и органические слои расслаиваются. Органический раствор концентрируют (до 40 вес.%) азеотропной отгонкой метилизобутилкетона. Стадия 3: Реакционная масса со стадии 2 (173 г), тетрабутиламмоний бромид (7,35 г, 0,023 моля) и 20%-ный раствор едкого натра (60,0 г, 0,3 моля) добавляются в реактор Parr из нержавеющей стали. Полученную реакционную смесь охлаждают до 0oC, промывают реактор азотом и герметизируют. При 0oC добавляют метилмеркаптан (16 г, 0,33 моля) и затем нагревают реакционную смесь до 100oC в течение 30 мин. При 100oC давление в реакторе составляет 45-50 ф/дюйм2 (3,2-3,5 кг/см2), реактор выдерживают при этих условиях и при перемешивании в течение 6 часов. После завершения 3-ей стадии реакции давление сбрасывают и отделяют соляной слой. Органический слой промывают водой (350 г) и отделяют. Затем к органическому слою добавляют воду (1000 г) и удаляют растворитель азеотропной отгонкой. Остаток охлаждают до 50oC и подвергают фильтрации. После промывки осадка на фильтре с водой (300 г) его сушат под вакуумом при 80oC, получая белое вещество с температурой плавления 169-170oC. Газовая хроматография показывает, что чистота продукта составляет 96%. Т.пл.: 169-170oC Данные 1H ЯМР даны в табл. 2. Данные 13C ЯМР: Химический сдвиг – Число атомов углерода 163,6 ч/млн. – 3C 50,83 ч/млн. – 2C 29,04 ч/млн. – 6C 12,99 ч/млн. – 1C Данные масс-спектрального анализа: отношение массы к заряду (m/z) = 269 Полученные данные подтверждают структуру. Пример 8 2,4-бис(циклопропиламин)-6-(метилтио)-s-триазин Стадии 1 и 2: В герметичном сосуде, промытом азотом, осуществляют реакцию цианурхлорида с циклопропиламином (78,0 г, 1,0 моля), используя метилизобутилкетон в качестве растворителя. Амин добавляют в течение 30 мин. Затем для поддержания значения pH 8,0-10,0 при температуре 75oC в течение 165 мин добавляют 20%-ный раствор едкого натра (203,3 г, 1,0 моля). Реакция завершена, когда достигается стабильное значение pH около 9,0. По завершении 2-й стадии дают полученному продукту отстояться, водный и органические слои расслаиваются. Органический слой концентрируют (до 44 вес. %) азеотропной отгонкой метилизобутилкетона. Стадия 3: Реакционную массу, полученную на 2-й стадии (233 г), тетрабутиламмоний бромид (12,79 г, 0,04 моля) и 20%-ный раствор едкого натра (101,4 г, 0,51 моля) добавляют в реактор Parr из нержавеющей стали. Полученную смесь охлаждают до 0oC, промывают реактор азотом и герметизируют. При 0oC добавляют метилмеркаптан (30 г, 0,63 моля) и затем нагревают реакционную смесь до 100oC в течение 25 мин. При 100oC в реакторе создается давление 30-35 ф/дюйм2 (2,1-2,5 кг/см2), при этих условиях и при перемешивании реактор выдерживают в течение 6 час. После завершения 3-й стадии давление сбрасывают и отделяют соляной слой. Органический слой промывают водой (300 г) и отделяют. Затем к органическому слою добавляют воду (800 г) и удаляют растворитель азеотропной отгонкой. Остаток охлаждают до 50oC и подвергают фильтрации. После промывки осадка водой (300 г) его сушат при 80oC под вакуумом, получая белое вещество с температурой плавления 114-116oC. Данные газовой хроматографии показывают, что чистота продукта равна 96,6%. Т.пл.: 114-116oC Данные 1H ЯМР приведены в табл. 3. Данные 13C ЯМР: Химический сдвиг – Число атомов углерода 180,2-165,5 ч/млн. – 3C 23,25 ч/млн. – 2C 12,85 ч/млн. – 1C 7,01 ч/млн. – 4C Данные масс-спектрального анализа: отношение массы к заряду (m/z) = 237 Полученные данные подтверждают структуру. Пример 9 2-амино-4-трет-бутиламино-6-метилтио-s-триазин Стадия 1: В герметичном сосуде, промытом азотом, осуществляют реакцию цианурхлорида (65,5 г, 0,36 моля) с трет-бутиламином (26,5 г, 0,36 моля) с использованием о-ксилола (260 г) в качестве растворителя. Амин добавляют в течение 30 мин. Затем для поддержания значения pH 6,5-8,0 при температуре 50oC в течение 150 мин добавляют 20%-ный раствор гидроокиси натрия (71,0 г, 0,36 моля). Реакция завершена, когда достигается стабильное значение pH около 7,0. Стадия 2: К раствору в о-ксилоле, полученному на 1-й стадии при 50oC, добавляют аммиак (29%-ый, 41,6 г, 0,71 моля). В конце добавления температура поднимается до 75oC. После завершения реакции водный слой отделяют. Раствор в о-ксилоле промывают водой (230 г) и разделяют слои. Органический слой концентрируют (до 45 вес.%) азеотропной отгонкой о-ксилола. Стадия 3: Реакционную массу со 2-й стадии, тетрабутиламмоний хлорид (9,0 г, 0,028 моля) и 20%-ный раствор гидроокиси натрия (70 г, 0,35 моля) добавляют в реактор Parr из нержавеющей стали. Полученную реакционную смесь охлаждают до 0oC, реактор промывают азотом и герметизируют. При 0oC добавляют метилмеркаптан (20 г, 0,42 моля) и нагревают реакционную смесь до 100oC в течение 20 мин. При 100oC давление в реакторе равно 37 ф/дюйм2 (2,59 кг/см2), реактор выдерживают при этих условиях в течение 4 часов при перемешивании. После завершения 3-й стадии реакции давление сбрасывают, соляной раствор промывают водой (100 г) и отделяют. К органическому слою добавляют воду (600 г) и удаляют растворитель азеотропной отгонкой. Остаток охлаждают до 50oC и отфильтровывают. После промывки остатка на фильтре водой (300 г) его сушат под вакуумом при 80oC, получая 53,3 г белого вещества (70,6% от теории) с температурой плавления 143-145oC. Данные газовой хроматографии показывают, что степень чистоты конечного продукта равна 99%. Т.пл.: 143-145oC Данные 1H ЯМР приведены в табл. 4. Данные 13C ЯМР: Химический сдвиг – Число атомов углерода 164,93 ч/млн. – 3C 51,15 ч/млн. – 1C 28,98 ч/млн. – 3C 12,98 ч/млн. – 1C Данные масс-спектрального анализа: отношение массы к заряду (m/z) = 213 Полученные данные подтверждают структуру. Примеры 10-15. Используя методику примера 1 получают следующие соединения: 2-(этиламино)-4-(изопропиламино)-6-(метилтио)-s-триазин; 2-(трет-бутиламино)-4-(циклопропиламино)-6-(метилтио)-s-триазин; 2-(метиламино)-4-(циклопропилметиламино)- 6-(бутилтио)-s-триазин; 2-(октиламино)-4-(циклобутиламино)-6-(пентилтио)-s-триазин; 2-(амиламино)-4-(циклогексиламино)-6-(гексилтио)-s-триазин; 2-(гексиламино)-4-(метиламино)-6-(этилтио)-s-триазин. Примеры 16-21. Используя методику примера 7 получают следующие соединения: 2,4-бис-(изопропиламино)-6-(метилтио)-s-триазин; 2,4-бис-(додециламино)-6-(метилтио)-s-триазин; 2,4-бис-(трет-бутиламино)-6-(этилтио)-s-триазин; 2,4-бис-(амиламино)-6-(гексилтио)-s-триазин; 2,4-бис-(этиламино)-6-(бутилтио)-s-триазин; 2,4-бис-(октиламино)-6-(пропилтио)-s-триазин. Примеры 22-26 Используя методику примера 9 получают следующие соединения: 2-(амино)-4-(трет-бутиламино)-6-(метилтио)-s-триазин; 2-(амино)-4-(додециламино)-6-(метилтио)-s-триазин; 2-(амино)-4-(циклопропиламино)-6-(гексилтио)-s-триазин; 2-(амино)-4-(октиламино)-6-(децилтио)-s-триазин; 2-(амино)-4-(метиламино)-6-(нонилтио)-s-триазин. Формула изобретения
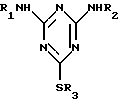 где R1 и R2 – независимо друг от друга означают водород, C1-C12-алкил, C3-C12-циклоалкил, указанный циклоалкил, замещенный C1-C4 алкилами, C4-C20-циклоалкил алкил, указанный C4-C20-циклоалкил алкил, замещенный C1-C4-алкилами; R3 означает C1-C12-алкил, путем взаимодействия цианурхлорида последовательно с R1NH2, R2NH2 и реагентом, содержащим R3S-группу, в одном не смешивающемся с водой органическом растворителе, в присутствии катализатора переноса фаз на стадии взаимодействия с реагентом, содержащим R3S-группу, отличающийся тем, что в качестве реагента, содержащего R3S-группу, используют тиол в присутствии кислотосвязывающего средства, в качестве катализатора переноса фаз четвертичные аммониевые соли и диазациклоалканы, которые используют в количестве от 1 до 10 мол.% в расчете на исходный цианурхлорид, а в качестве не смешивающегося с водой органического растворителя используют ароматический C6-C10углеводород или C4-C11алканон. 2. Способ по п.1, отличающийся тем, что в качестве не смешивающегося с водой органического растворителя используют толуол, ксилол или метилизобутилкетон. 3. Способ по п.2, отличающийся тем, что в качестве не смешивающегося с водой органического растворителя используют ксилол или толуол. 4. Способ по п.3, отличающийся тем, что в качестве не смешивающегося с водой органического растворителя используют ксилол. 5. Способ по п.1, отличающийся тем, что в качестве катализатора переноса фазы используют трибутилметиламмония хлорид, 1,4-диазабицикло[2,2,2]октан, бензилтриметиламмоний гидроокись, тетрабутиламмоний хлорид, тетрабутиламмоний бромид или тетрабутиламмоний кислый сернокислый. 6. Способ по п.5, отличающийся тем, что в качестве катализатора переноса фазы используют тетрабутиламмоний бромид. 7. Способ по п.1, отличающийся тем, что R1 и R2 независимо друг от друга означают водород, C1-C12-алкил, C3-C12-циклоалкил или указанный циклоалкил, замещенный C1-C4-алкильными группами. 8. Способ по п. 1, отличающийся тем, что в формуле I R1 означает C3-C12-циклоалкил, R2 означает C1-C12-алкил и R3 означает C1-C12-алкил. 9. Способ по п. 8, отличающийся тем, что в формуле I R1 означает циклопропил, R2 означает трет-бутил и R3 означает метил. 10. Способ по п.1, отличающийся тем, что соединениями формулы (I) являются 2-(трет-бутиламино)-4-(циклопропиламино)-6-(метилтио)-S-триазин, 2,4-бис-(трет-бутиламино)-6-(метилтио)-S-триазин, 2,4-бис-(циклопропиламино)-6-(метилтио)-S-триазин и 2-амино-4-трет-бутиламино-6-метилтио-S-триазин. РИСУНКИ
MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 15.10.2006
Извещение опубликовано: 10.01.2008 БИ: 01/2008
|
||||||||||||||||||||||||||