Патент на изобретение №2341263
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) ЛЕКАРСТВЕННЫЕ ФОРМЫ, СОДЕРЖАЩИЕ AG013736
(57) Реферат:
Изобретение относится к лекарственным средствам и касается способа лечения рака молочной железы у человека, включающего введение соединения формулы 1 или его фармацевтически приемлемой соли, где соединение вводят в количестве от 5 до 20 мг на дозу с частотой дозирования два раза в сутки и где способ дополнительно включает совместное введение доцетаксела. Также раскрыт способ лечения рака поджелудочной железы, включающий введение соединения формулы 1 или его фармацевтически приемлемой соли в количестве от 5 до 20 мг на дозу с частотой дозирования два раза в сутки, при этом способ дополнительно включает совместное введение гемцитабина. Предложенный способ лечения рака обеспечивает значительную регрессию опухоли. 2 н. и 6 з.п. ф-лы, 2 ил., 4 табл.
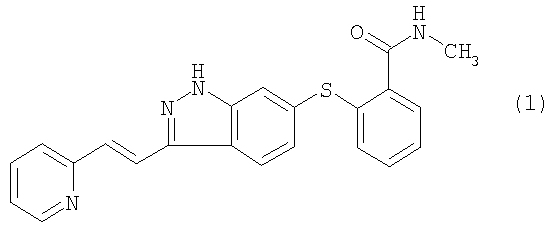
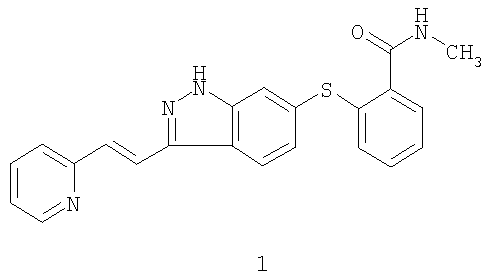
Испрашивается приоритет данной заявки согласно предварительной заявке на патент США № 60/460695, поданной 3 апреля 2003 года, и предварительной заявке на патент США № 60/491771, поданной 31 июля 2003 года, содержание которых во всей их полноте включено в данное описание изобретения ссылкой. Предшествующий уровень техники Данное изобретение относится к ингибиторам VEGFR (рецептор васкулярного эндотелиального фактора роста), которые полезны для лечения аномального клеточного роста, такого как рак, у млекопитающих. Изобретение также относится к способу использования таких соединений в лечении аномального клеточного роста у млекопитающих, особенно людей, и к фармацевтическим композициям, содержащим такие соединения. Соединение 6-[2-(метилкарбамоил)фенилсульфанил]-3-Е-[2-(пиридин-2-ил)этенил]индазол, имеющее формулу 1
является эффективным и селективным ингибитором тирозинкиназ VEGFR/PDGFR (рецептор васкулярного эндотелиального фактора роста/рецептор тромбоцитарного фактора роста) с широкой предклинической активностью в ксенотрансплантатных моделях рака ободочной кишки, меланомы, рака молочной железы и рака легкого (Hu-Lowe D, Heller, D, Brekken J, Feeley R, Amundson K, Haines M, Troche Г, Kim Y, Gonzales D, Herrman M, Batugo M, Vekich S, Kania R, McTigue M, Gregory S, Bender S, Shalinsky D., Pharmacological Activities of AG 013736, a Small Molecule Inhibitor of VEGF/PDGF Receptor Tyrosine Kinases; Proc. Am. Assoc. Cancer Res.2002: abstract #5357). Было показано, что предклинический опухолевый сосудистый ответ, который оценивали методом магнитно-резонансной визуализации с динамическим усилением контраста, соответствует индексу роста опухоли (Wilmes LJ, Hylton НМ, Wang D, Fleming LM Gibbs J, Kirn Y, Dillon R, Brasch RC, Park JW, Li K-L, Henry RG, Partridge SC, Shalinsky DR, Hu-Lowe D, McShane TM, and Pallavicini MG., AG 013736, a Novel VEGFR TK Inhibitor Suppresses Tumor Growth and Vascular Permeability in Human БТ474 Breast Cancer Xenografts in Nude Mice”; Proc. Am. Assoc. Cancer Res.2003: Abstract #3772). Краткое изложение сущности изобретения Согласно данному изобретению предложены лекарственные формы и способы лечения с использованием соединения формулы 1:
которое согласно систематической номенклатуре может называться 6-[2-(метилкарбамоил)фенилсульфанил]-3-Е-[2-(пиридин-2-ил)этенил]индазолом. В одном воплощении данного изобретения предложена лекарственная форма для введения млекопитающему, содержащая соединение формулы 1, его фармацевтически приемлемую соль, сольват или пролекарство или их смесь в количестве, эффективном для обеспечения величины 24-часовой AUC (площадь под кривой) s плазме крови не более 4500 нг·ч/мл соединения формулы 1 или его активных метаболитов после введения млекопитающему. Величины 24-часовой AUC в плазме крови могут быть определены как описано в разделе “Подробное описание изобретения”. В конкретных аспектах этого воплощения верхний предел величины 24-часовой AUC в плазме крови составляет не более 4000 нг·ч/мл, или не более 3000 нг·ч/мл, или не более 2500 нг·ч/мл, или не более 2000 нг·ч/мл, или не более 1500 нг·ч/мл, или не более 1000 нг·ч/мл, или не более 800 нг·ч/мл, или не более 700 нг·ч/мл. Предпочтительно, и в комбинации с любым из указанных верхних пределов величина 24-часовой AUC в плазме крови составляет по меньшей мере 10 нг·ч/мл, или по меньшей мере 25 нг·ч/мл, или по меньшей мере 50 нг·ч/мл, или по меньшей мере 75 нг·ч/мл, или по меньшей мере 100 нг·ч/мл, или по меньшей мере 125 нг·ч/мл. Рассматриваемые диапазоны величин 24-часовой AUC в плазме крови включают диапазоны от любого из указанных нижних пределов до любого из указанных верхних пределов. Конкретные неограничивающие примеры предпочтительных диапазонов включают диапазоны от 25 до 4500 нг·ч/мл, от 50 до 2500 нг·ч/мл, от 75 до 1000 нг·ч/мл, от 100 до 800 нг·ч/мл и от 125 до 700 нг·ч/мл. В другом воплощении данного изобретения предложена лекарственная форма, содержащая соединение формулы 1, как оно определено выше, его фармацевтически приемлемую соль, сольват или пролекарство или их смесь в количестве не более 30 мг. Следует иметь в виду, что когда все соединение или часть соединения присутствует в лекарственной форме в виде соли, сольвата или пролекарства, его количество представляет собой эквивалентное количество соединения формулы 1, которое специалист в данной области без труда вычислит исходя из молекулярных масс. В конкретных аспектах этого воплощения верхний предел количества составляет не более 20 мг, или не более 15 мг, или не более 12 мг, или не более 10 мг, или не более 8 мг, или не более 7 мг. Предпочтительно, и в комбинации с любым из указанных верхних пределов это количество составляет по меньшей мере 0,5 мг, или по меньшей мере 1 мг, или по меньшей мере 1,5 мг, или по меньшей мере 2 мг, или по меньшей мере 2,5 мг, или по меньшей мере 3 мг. Рассматриваемые диапазоны включают диапазоны от любого из указанных нижних пределов до любого из указанных верхних пределов. Конкретные неограничивающие примеры предпочтительных диапазонов включают диапазоны от 0,5 до 30 мг, от 1 до 20 мг, от 1,5 до 15 мг, от 2 до 10 мг, от 2,5 до 8 мг и от 3 до 7 мг. Согласно данному изобретению предложен также способ лечения аномального клеточного роста у млекопитающего, включая человека, путем введения этому млекопитающему соединения формулы 1, как оно определено выше, его фармацевтически приемлемой соли, сольвата или пролекарства или их смеси в количестве, эффективном для обеспечения величины 24-часовой AUC в плазме крови не более 4500 нг·мг/мл соединения формулы 1 или его активных метаболитов после введения млекопитающему. Величины 24-часовой AUC в плазме крови могут быть определены как описано в разделе “Подробное описание изобретения”. В конкретных аспектах этого воплощения верхний предел величины 24-часовой AUC в плазме крови составляет не более 4000 нг·ч/мл, или не более 3000 нг·ч/мл, или не более 2500 нг·ч/мл, или не более 2000 нг·ч/мл, или не более 1500 нг·ч/мл, или не более 1000 нг·ч/мл, или не более 800 нг·ч/мл, или не более 700 нг·ч/мл. Предпочтительно, и в комбинации с любым из указанных верхних пределов величина 24-часовой AUC в плазме крови составляет по меньшей мере 10 нг·ч/мл, или по меньшей мере 25 нг·ч/мл, или по меньшей мере 50 нг·ч/мл, или по меньшей мере 75 нг·ч/мл, или по меньшей мере 100 нг·ч/мл, или по меньшей мере 125 нг·ч/мл. Рассматриваемые диапазоны величины 24-часовой AUC в плазме крови охватывают диапазоны от любого из указанных нижних пределов до любого из указанных верхних пределов. Конкретные неограничивающие примеры предпочтительных диапазонов включают диапазоны от 25 до 4500 нг·ч/мл, от 50 до 2500 нг·ч/мл, от 75 до 1000 нг·ч/мл, от 100 до 800 нг·ч/мл и от 125 до 700 нг·ч/мл. Согласно данному изобретению предложен также способ лечения аномального клеточного роста у млекопитающего, включая человека, путем введения этому млекопитающему соединения формулы 1, как оно определено выше, его фармацевтически приемлемой соли, сольвата или пролекарства или их смеси в количестве не более 30 мг на дозу. Следует иметь в виду, что когда все соединение или часть соединения присутствует в лекарственной форме в виде соли, сольвата или пролекарства, его количество представляет собой эквивалентное количество соединения формулы 1, которое специалист в данной области без труда вычислит исходя из молекулярных масс. В конкретных аспектах этого воплощения верхний предел этого количества составляет не более 20 мг, или не более 15 мг, или не более 12 мг, или не более 10 мг, или не более 8 мг, или не более 7 мг. Предпочтительно, и в комбинации с любым из указанных верхних пределов это количество составляет по меньшей мере 0,5 мг, или по меньшей мере 1 мг, или по меньшей мере 1,5 мг, или по меньшей мере 2 мг, или по меньшей мере 2,5 мг, или по меньшей мере 3 мг. Рассматриваемые диапазоны охватывают диапазоны от любого из указанных нижних пределов до любого из указанных верхних пределов. Конкретные не ограничивающие примеры предпочтительных диапазонов включают диапазоны от 0,5 до 30 мг, от 1 до 20 мг, от 1,5 до 15 мг, от 2 до 10 мг, от 2,5 до 8 мг и от 3 до 7 мг. В конкретном воплощении любого из описанных здесь способов по изобретению аномальный клеточный рост представляет собой рак, включая, но не ограничиваясь ими, рак легкого, рак кости, рак поджелудочной железы, рак кожи, рак в области головы или шеи, кожную или внутриглазную меланому, рак матки, рак яичника, рак прямой кишки, рак анальной области, рак желудка, рак ободочной кишки, рак молочной железы, карциному фаллопиевых труб, карциному эндометрия, карциному шейки матки, карциному влагалища, карциному вульвы, болезнь Ходжкина, рак пищевода, рак тонкого кишечника, рак эндокринной системы, рак щитовидной железы, рак паращитовидной железы, рак надпочечника, саркому мягкой ткани, рак уретры, рак пениса, рак простаты, хронический или острый лейкоз, лимфоцитарную лимфому, рак мочевого пузыря, рак почки или мочеточника, почечно-клеточный рак, карциному почечной лоханки, новообразования центральной нервной системы (ЦНС), первичную ЦНС лимфому, опухоли позвоночного столба, глиому ствола мозга, аденому гипофиза и их комбинации. В еще одном воплощении указанного способа указанный аномальный клеточный рост представляет собой доброкачественное пролиферативное заболевание, включая псориаз, доброкачественную гипертрофию простаты или рестеноз, но не ограничиваясь ими. В еще одном воплощении данного изобретения предложен способ ингибирования миграции раковых клеток, опосредованной рецепторами тромбоцитарного фактора роста ВВ (PDGFR ВВ), у млекопитающего путем введения этому млекопитающему терапевтически приемлемого количества соединения формулы 1. В еще одном воплощении данного изобретения предложен способ ингибирования c-KIT активности у млекопитающего путем введения этому млекопитающему терапевтически приемлемого количества соединения формулы 1. В других конкретных воплощениях любого из описанных здесь способов по изобретению способ дополнительно включает введение млекопитающему некоторого количества одного или более веществ, выбранных из противоопухолевых агентов, антиангиогенных агентов, ингибиторов сигнальной трансдукции и антипролиферативных агентов, причем эти количества вместе эффективны в лечении указанного аномального клеточного роста. Такие вещества включают вещества, раскрытые в международных публикациях WO 00/38715, WO 00/38716, WO 00/38717, WO 00/38718, WO 00/38719, WO 00/33730, WO 00/38665, WO 00/37107 и WO 00/38786, содержание которых во всей их полноте включено в данное описание изобретения ссылкой. Примерами противоопухолевых агентов являются митотические ингибиторы, например производные алкалоидов барвинка, такие как винбластин, винорелбин, виндесин и винкристин; колхины аллокохин, галихондрин, колхициновая кислота N-бензоилтриметил-метиловый эфир, доластатин 10, мэйстансин, ризоксин, таксаны, такие как паклитаксел (Taxol Антиангиогенные агенты включают ингибиторы ММР-2 (матриксной металлопротеиназы 2), ингибиторы ММР-9 (матриксной металлопротеиназы 9) и ингибиторы СОХ-11 (циклооксигеназы II). Примеры полезных ингибиторов СОХ-11 включают CELEBREX Примеры ингибиторов ММР включают AG-3340, RO 32-3555, RS 13-0830 и соединения, указанные в следующем списке: 3-([4-(4-фтор-фенокси)бензолсульфонил]-(1-гидроксикарбамоил-циклопентил)амино]пропионовая кислота, 3-экзо-3-[4-(4-фтор-фенокси)бензолсульфониламино]-8-окса-бицикло[3.2.1]октан-3-карбоновой кислоты гидроксиамид, (2R,3R)-1-[4-(2-хлор-4-фтор-бензилокси)бензолсульфонил]-3-гидрокси-3-метил-пиперидин-2-карбоновой кислоты гидроксиамид, 4-[4-(4-фтор-фенокси)бензолсульфониламино]тетрагидропиран-4-карбоновой кислоты гидроксиамид, 3-[[4-(4-фтор-фенокси)бензолсульфонил]-(1-гидроксикарбамоил-циклобутил)-амино]-пропионовая кислота, 4-[4-(4-хлор-фенокси)бензосульфониламино]тетрагидропиран-4-карбоновой кислоты гидроксиамид, 3-[4-(4-хлор-фенокси)бензолсульфониламино]тетрагидропиран-3-карбоновой кислоты гидроксиамид, (2R,3R)-1-[4-(4-фтор-2-метил-бензилокси)бензолсульфонил]-3-гидрокси-3-метил-пиперидин-2-карбоновой кислоты гидроксиамид, 3-[[4-(4-фтор-фенокси)бензосульфонил]-(1-гидроксикарбамоил-1-метилэтил)амино]пропионовая кислота, 3-[[4-(4-фтор-фенокси)бензолсульфонил]-(4-гидроксикарбамоил-тетрагидропиран-4-ил)амино]пропионовая кислота, 3-экзо-3-[4-(4-хлор-фенокси)бензолсульфониламино]-8-окса-бицикло[3.2.1]октан-3-карбоновой кислоты гидроксиамид, 3-эндо-3-[4-(4-фтор-фенокси)бензолсульфониламино]-8-окса-бицикло[3.2.1]октан-3-карбоновой кислоты гидроксиамид и 3-[4-(4-фтор-фенокси)бензолсульфониламино]тетрагидрофуран-3-карбоновой кислоты гидроксиамид, и фармацевтически приемлемые соли, сольваты и пролекарства указанных соединений. Примеры ингибиторов сигнальной трансдукции включают агенты, которые могут ингибировать EGFR (рецептор эпидермального фактора роста) ответы, такие как антитела к EGFR, антитела к EGF и молекулы, которые являются ингибиторами EGFR; ингибиторы VEGF (васкулярный эндотелиальный фактор роста); и ингибиторы рецептора erbВ2, такие как органические молекулы или антитела, которые связываются с рецептором erbВ2, например HERCEPTIN Ингибиторы EGFR раскрыты, например, в WO 95/19970 (дата публикации 27 июля 1995), WO 98/14451 (дата публикации 9 апреля 1998), WO 98/02434 (дата публикации 22 января 1998) и патенте США 5747498 (дата выдачи 5 мая 1998). EGFR-ингибирующие агенты включают, без ограничений, моноклональные антитела С225 и anti-EGFR 22Mab (ImClone Systems Incorporated of New York, New York, USA), соединения ZD-1839 (AstraZeneca), BIBX-1382 (Boehringer Ingelheim), MDX-447 (Medarex Inc. of Annandale, New Jersey, USA) и OLX-103 (Merck & Co. of Whitehouse Station, New Jersey, USA), VRCTC-310 (Ventech Research) и слитый с EGF токсин (Seragen Inc. of Hopkinton, Massachusetts). Комбинировать или вводить вместе с соединениями формулы 1 можно также ингибиторы VEGF, например SU-5416 и SU-6668 (Sugen Inc. of South San Francisco, California, USA). Ингибиторы VEGF раскрыты, например, в WO 99/24440 (дата публикации 20 мая 1999), Международной заявке PCT/IB 99/00797 (дата подачи 3 мая 1999), WO 95/21613 (дата публикации 17 августа 1995), WO 99/61422 (дата публикации 2 декабря 1999), патенте США 5834504 (дата выдачи 10 ноября 1998), WO 98/50356 (дата публикации 12 ноября 1998), патенте США 5883113 (дата выдачи 16 марта 1999), патенте США 5886020 (дата выдачи 23 марта 1999), патенте США 5792783 (дата выдачи 11 августа 1998), WO 99/10349 (дата публикации 4 марта 1999), WO 97/32856 (дата публикации 12 сентября 1997), WO 97/22596 (дата публикации 26 июня 1997), WO 98/54093 (дата публикации 3 декабря 1998), WO 98/02438 (дата публикации 22 января 1998), WO 99/16755 (дата публикации 8 апреля 1999) и WO 98/02437 (дата публикации 22 января 1998), которые все включены в данное описание изобретения во всей их полноте ссылкой. Другими примерами некоторых конкретных ингибиторов VEGF являются IM862 (Cytran Inc. of Kirkland, Washington, USA), анти-VEGF моноклональные антитела бевацизумаб (Genentech, inc. of South San Francisco, California) и Angiozyme В комбинации с соединением формулы 1 можно вводить ингибиторы ErbВ2-рецептора, такие как GW-2S2974 (Glaxo Wellcome ple), и моноклональные антитела AR-209 (Aronex Pharmaceuticals Inc. of The Woodlands, Texas, USA) и 2В-1 (Chiron). Такие ингибиторы erbB2 включают ингибиторы, описанные в WO 98/02434 (дата публикации 22 января 1998), WO 99/35146 (дата публикации 15 июля 1999), WO 99/35132 (дата публикации 15 июля 1999), WO 98/02437 (дата публикации 22 января 1998), WO 97/13760 (дата публикации 17 апреля 1997), WO 95/19970 (дата публикации 27 июля 1995), патенте США 5537453 (дата выдачи 24 декабря 1996) и патенте США 5877305 (дата выдачи 2 марта 1999), каждый из которых во всей полноте включен в данное описание изобретения ссылкой. Ингибиторы ErbВ2-рецептора, полезные в настоящем изобретению, также описаны в предварительной заявке на патент США № 60/117341 (дата подачи 27 января 1999) и в предварительной заявке на патент США № 60/117, 346, дата подачи 27 января 1999, которые обе во всей их полноте включены в данное описание изобретения ссылкой. Другие антипролиферативные агенты, которые можно использовать, включают ингибиторы фермента фарнезил-протеинтрансфераза и ингибиторы рецепторной тирозинкиназы PDGFR, в том числе соединения, раскрытые и заявленные в следующих заявках на патент США: 09/221946 (дата подачи 28 декабря 1998), 09/454058 (дата подачи 2 декабря 1999), 09/501163 (дата подачи 9 февряля 2000), 09/539930 (дата подачи 31 марта 2000), 09/202796 (дата подачи 22 мая 1997), 09/384339 (дата подачи 26 августа 1999) и 09/383755 (дата подачи 26 августа 1999), и соединения, раскрытые и заявленные в следующих предварительных заявках на патент США: 60/168207 (дата подачи 30 ноября 1999), 60/170119 (дата подачи 10 декабря 1999), 60/177718 (дата подачи 21 января 2000), 60/168217 (дата подачи 30 ноября 1999) и 60/200834 (дата подачи 1 мая 2000). Каждая из вышеуказанных заявок и предварительных заявок на патент США во всей ее полноте включена в данное описание изобретения ссылкой. Соединение формулы 1 можно также использовать с другими агентами, полезными в лечении аномального клеточного роста или рака, включая, без ограничений, агенты, способные усиливать противоопухолевые иммунные ответы, такие как антитела к CTLA4 (антиген 4 цитотоксических лимфоцитов) и другие агенты, способные блокировать CTLA4, и антипролиферативные агенты, например другие ингибиторы фарнезил-протеинтрансферазы. Конкретные антитела к CTLA4, которые можно использовать в настоящем изобретении, включают антитела, описанные в предварительной заявке на патент США 60/113647 (дата подачи 23 декабря 1998), который во всей его полноте включен в данное описание изобретения ссылкой. В еще одном воплощении данного изобретения предложена фармацевтическая композиция, содержащая соединение формулы 1 или его фармацевтически приемлемую соль, сольват или пролекарство и терапевтически эффективное количество доцетаксела. В еще одном воплощении данного изобретения предложен способ лечения аномального клеточного роста у млекопитающего, включая человека, путем введения млекопитающему соединения формулы 1 или его фармацевтически приемлемой соли, сольвата или пролекарства и терапевтически эффективного количества доцетаксела. Соединение формулы 1 и доцетаксел можно вводить по отдельности или в одной и той же композиции и можно вводить по одной и той же схеме дозировки или при разных схемах дозировки, как желательно. Определения Используемое здесь понятие “аномальный клеточный рост” относится, если не указано иное, к клеточному росту, который не зависит от нормальных регуляторных механизмов (например, потеря контактного ингибирования). Это понятие охватывает аномальный рост (1) опухолевых клеток (опухолей), которые пролиферируют из-за экспрессии мутантной тирозинкиназы или повышенной экспрессии рецепторной тирозинкиназы; (2) доброкачественных и злокачественных клеток других пролиферативных заболеваний, при которых происходит аберрантная активация тирозинкиназы; и (4) любых опухолей, которые пролиферируют под действием рецепторныхтирозинкиназ. Используемый здесь термин “лечение” означает, если не указано иное, реверсирование, облегчение, сдерживание прогрессирования или предупреждение расстройства или состояния, к которому этот термин применяется, или одного или более симптомов такого расстройства или состояния. Используемый здесь термин “проведение лечения” относится, если не указано иное, к акту лечения как “лечения”, которое определено непосредственно выше. Используемая здесь фраза “фармацевтически приемлемая(ые) соль(и)”, если не указано иное, включает в себя соли, образованные кислотными или основными группами, которые могут присутствовать в соединении. Соединения, которые являются основными по своей природе, способны образовывать множество солей с различными неорганическими и органическими кислотами. Кислотами, которые можно использовать для получения фармацевтически приемлемых солей присоединения кислоты таких основных соединений, являются кислоты, которые образуют нетоксичные соли присоединения кислоты, то есть соли, содержащие фармакологически приемлемые анионы, такие как соли ацетат, бензолсульфонат, бензоат, бикарбонат, бисульфат, битартрат, борат, бромид, кальциевый эдетат, камсилат, карбонат, хлорид, клавуланат, цитрат, дигидрохлорид, эдетат, эдисилат, эстолат, эзилат, этилсукцинат, фумарат, глуцептат, глюконат, глутамат, гликолиларсанилат, гексилрезорцинат, гидрабамин, гидробромид, гидрохлорид, йодид, изотионат, лактат, лактобионат, лаурат, малат, малеат, манделат, мезилат, метилсульфат, мукат, напсилат, нитрат, олеат, оксалат, памоат (эмбонат), пальмитат, пантотенат, фосфат/дифосфат, полигалактуронат, салицилат, стеарат, субацетат, сукцинат, таннат, тартрат, теоклат, тозилат, триэтиодод и валерат. Используемый здесь термин “пролекарство”, если не указано иное, означает соединения, являющиеся предшественниками лекарственного средства, которые после введения высвобождают лекарственное средство in vivo в результате химического или физиологического процесса (например, пролекарство в условиях физиологического рН превращается в целевую форму лекарственного средства). Изобретение также охватывает меченные изотопами соединения, которые идентичны соединениям формулы 1, за исключением того, что один или более атомов заменены атомом, имеющим атомную массу или массовое число, отличающееся от атомной массы или массового числа, обычно обнаруживаемых в природе. Примеры изотопов, которые могут быть введены в соединения по изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы фтора и хлора, например 2H, 3H, 13С, 14С, 15N, 18O, 17O, 31P, 32Р, 35S, 18F и 36Cl соответственно. Соединения по настоящему изобретению, их пролекарства и фармацевтически приемлемые соли и сольваты указанных соединений или указанных пролекарств, которые содержат вышеуказанные изотопы и/или другие изотопы других атомов, входят в объем данного изобретения. Некоторые меченные изотопами соединения по настоящему изобретению, например соединения, в которые введены радиоактивные изотопы, такие как 3H и 14C, полезны в анализах на распределение лекарственного средства и/или субстрата в тканях. Изотопы тритий, то есть 3H, и углерод-14, то есть 14С, особенно предпочтительны благодаря простоте их получения и детектирования. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий, то есть 2H, вследствие большей метаболической стабильности может давать определенные терапевтические преимущества, например увеличение периода полужизни in vivo или снижение требований к дозировке и, следовательно, может быть предпочтительным в некоторых обстоятельствах. Меченные изотопами соединения формулы 1 по данному изобретению и их пролекарства в общем могут быть получены путем проведения процедур, описанных для немеченого соединения, заменяя немеченый реагент легко доступным меченым реагентом. Краткое описание графических материалов На Фиг.1 представлены метаболиты соединения формулы 1, идентифицированные у собак после введения однократной пероральной дозы 14С-меченого соединения. На Фиг.2 представлены метаболиты соединения формулы 1, идентифицированные у мышей после введения однократной пероральной дозы 14С-меченного соединения. Подробное описание изобретения Соединение формулы 1 может быть получено, как описано в патентах США №№ 6531491 и 6534524 (дата выдачи 11 марта 2003 и 18 марта 2003 соответственно), которые во всей их полноте включены в данное описание ссылкой. Некоторые исходные вещества могут быть получены способами, известными специалистам в данной области, и некоторые синтетические модификации могут быть сделаны способами, известными специалистам в данной области. Соединение формулы 1 способно образовывать множество разных солей с различными неорганическими и органическими кислотами. Хотя такие соли должны быть фармацевтически приемлемыми для введения млекопитающим, на практике часто желательно сначала выделить соединение формулы 1 из реакционной смеси в виде фармацевтически неприемлемой соли, а затем просто превратить последнюю обратно в соединение – свободное основание путем обработки щелочным реагентом и последующего превращения этого свободного основания в фармацевтически приемлемую соль присоединения кислоты. Соли присоединения кислоты соединений-оснований по данному изобретению легко можно получить путем обработки соединения-основания по существу эквивалентным количеством выбранной минеральной или органической кислоты в среде водного растворителя или в подходящем органическом растворителе, таком как метанол или этанол. При осторожном выпаривании растворителя легко можно получить желаемую соль в виде твердого вещества. Желаемую соль кислоты можно также осадить из раствора свободного основания в органическом растворителе добавлением к этому раствору подходящей минеральной или органической кислоты. Введение соединения формулы 1 можно осуществлять любым способом, который обеспечивает доставку соединения к месту действия. Эти способы включают пероральные пути, интрадуоденальные пути, парентеральную инъекцию (в том числе внутривенную, подкожную, внутримышечную, внутрисосудистую инъекцию или инфузию), местное и ректальное введение. Соединение может быть предоставлено, например, в форме, подходящей для перорального введения в виде таблетки, капсулы, пилюли, порошка, препарата пролонгированного высвобождения, раствора, суспензии, для парентеральной инъекции в виде стерильного раствора, суспензии или эмульсии, для местного введения в виде мази или крема или для ректального введения в виде суппозитория. Соединение может быть в стандартных лекарственных формах, подходящих для однократного введения точных доз. Предпочтительно лекарственные формы содержат традиционный фармацевтический носитель или эксципиент и соединение формулы 1 в качестве активного ингредиента. Кроме того, лекарственные формы могут содержать другие медицинские или фармацевтические агенты, носители, адъюванты и т.д. Примерами форм для парентерального введения являются растворы или суспензии в стерильных водных растворах, например водных растворах пропиленгликоля или декстрозы. Такие лекарственные формы могут быть подходящим образом забуферены, если это желательно. Подходящие фармацевтические носители включают инертные разбавители или наполнители, воду и различные органические растворители. Фармацевтическая композиция может, если желательно, содержать дополнительные ингредиенты, такие как корригенты, связующие агенты, эксципиенты и тому подобные. Так, для перорального введения можно использовать таблетки, содержащие различные эксципиенты, такие как лимонная кислота, вместе с различными разрыхлителями, такими как крахмал, алгиновая кислота и некоторые сложные силикаты, и связующими агентами, такими как сахароза, желатин и аравийская камедь. Дополнительно, для таблетирования часто используют смазывающие агенты, такие как стеарат магния, лаурилсульфат натрия и тальк. Твердые композиции подобного типа можно также использовать в мягких и твердых желатиновых капсулах. Предпочтительные вещества для них включают лактозу или молочный сахар и высокомолекулярные полиэтиленгликоли. Когда для перорального введения желательны водные суспензии или эликсиры, активное соединение в них можно комбинировать с различными подсластителями или корригентами, красящими веществами или красителями и, если желательно, эмульгирующими агентами или суспендирующими агентами вместе с разбавителями, такими как вода, этанол, пропиленгликоль, глицерин или их комбинации. В предпочтительных воплощениях лекарственных форм по данному изобретению лекарственная форма представляет собой пероральную лекарственную форму, более предпочтительно таблетку или капсулу. В предпочтительных воплощениях способов по данному изобретению соединение формулы 1 вводят перорально, например с использованием описанной здесь пероральной лекарственной формы. Эти способы включают введение соединения формулы 1 в любом желаемом режиме дозировки. В одном конкретном воплощении соединение вводят один раз в день (quaque die, или QD), предпочтительно два раза в день (bis in die, или BID), хотя более или менее частое введение входит в объем данного изобретения. Соединение можно вводить млекопитающему, включая человека, предпочтительно натощак (без пищи или питья в пределах 2 часов до и после введения). Особенно предпочтительна дозировка BID натощак. Способы приготовления различных лекарственных форм с конкретным количеством соединения формулы 1 известны или очевидны специалистам в данной области. Например, смотри Remington’s Pharmaceutical Sciences. Mack Publishing Company, Easter, Pa., 15th Edition (1975). Величины 24-часовой AUC в плазме крови могут быть определены непосредственно измерением концентраций соединения формулы 1 или его активных метаболитов в плазме крови, например методом жидкостной хроматографии – тандемной масс-спектрометрии (ЖХ-МС/МС) при различных интервалах времени, и вычислением площади под кривой зависимости концентрации в плазме от времени. В данной области хорошо известны подходящие методы вычисления AUC, такие как, например, метод трапецеидальной аппроксимации,
где n означает число точек на графике, и ti и Ci означают время и концентрацию (значения х и у) для i-той точки на графике. Величины 24-часовой AUC могут быть определены путем нормализации измеренных концентраций в плазме крови согласно схеме дозирования. Для получения стандартов концентраций в раствор данного разведения добавляют бисульфит натрия в качестве стабилизатора. Соединение формулы 1 имеет полезные свойства, относящиеся к модуляции и/или ингибированию киназной активности, ассоциированной с VEGF-R, FGF-R (рецептор фактора роста фибробластов), комплексами CDK (циклинзависимая киназа), СНК1 (чекпойнт киназа 1), CSF-R (рецептор фактора стволовых клеток) и/или LCK (лимфоцит-специфичная протеинтирозинкиназа). Как показано ниже в примерах, соединение формулы 1 способно индуцировать апопотоз эндотелиальных клеток пуповины человека (HUVEC) in vitro, ингибировать опосредованное VEGF фосфорилирование Akt и eNOS (эндотелиальная синтаза оксида азота) в HUVEC, демонстрировать продолжительное ингибирующее действие на фосфорилирование VEGFR-2 в HUVEC после удаления соединения и ингибировать индуцированную PDGF ВВ миграцию раковых клеток на матриксном белке фибронектине. Соединение формулы 1 может обладать активностью против управляемого PDGFR развития опухоли посредством ингибирования миграции и инвазии. Соединение формулы 1 также проявляет более сильную активность в ингибировании роста опухоли в комбинации с Taxol Настоящее изобретение также относится к способам модулирования или ингибирования протеинкиназной активности, например в ткани млекопитающего, путем введения соединения формулы 1. Активность соединения по изобретению как модулятора протеинкиназной активности, такой как активность киназ, может быть измерена любым из методов, доступных специалистам в данной области, в том числе анализами in vivo и/или in vitro. Примеры подходящих анализов для измерения активности включают анализы, описанные в Parast С. et al., Biochemistry, 37, 16788-16801 (1998); Jeffrey et al., Nature, 376, 313-320 (1995); WIPO Международной публикации WO 97/34876; и WIPO Международной публикации WO 96/14843. Эти свойства можно оценить, например, используя одну или более методик биологического тестирования, изложенных ниже в примерах. Приведенные ниже примеры и получения дополнительно иллюстрируют и демонстрируют лекарственные формы и способы по настоящему изобретению. Следует иметь в виду, что объем настоящего изобретения никоим образом не ограничен объемом нижеследующих примеров. Пример 1 Соединение формулы 1 тестировали на: (1) эффективность in vivo по нескольким схемам: один раз в сутки, еженедельная доза по выходным и дозирование с перерывами, (2) эффективность в комбинации с доцетакселем на ксенотрансплантатных моделях, (3) фосфорилирование eNOS и Akt in vitro в эндотелиальных клетках, (4) концентрация оксида азота и родственных продуктов в клеточной культуре и in vivo и (5) использование сигнала от c-Kit в качестве потенциального биомаркера соединения в клетках цельной крови. БИОЛОГИЧЕСКОЕ ТЕСТИРОВАНИЕ; ФЕРМЕНТНЫЙ АНАЛИЗ Стимуляция клеточной пролиферации ростовыми факторами, такими как VEFG, FGF (фактор роста фибробластов) и другие, зависит от индукции аутофосфорилирования каждой из соответствующих им рецепторных тирозинкиназ. Поэтому способность ингибитора протеинкиназы блокировать клеточную пролиферацию, индуцированную этими ростовыми факторами, коррелирует непосредственно с его способностью блокировать аутофосфорилирование рецептора. Для измерения активности соединения в отношении ингибировании протеинкиназ были разработаны нижеследующие конструкции. Конструкция VEGF-R2 для анализа: Эта конструкция определяет способность тестируемого соединение ингибировать тирозинкиназную активность. Конструкция (VEGF-R2 Конструкция FGF-R1 (рецептор типа 1 фактора роста фибробластов) для анализа: Внутриклеточный киназный домен рецептора FGF-R1 человека экспрессировали в системе экспрессии с использованием бакуловирусного вектора, начиная с эндогенного остатка метионина 456 до глутамата 766 в соответствии с системой нумерации остатков Mohammadi et al., Mol. Cell. Biol., 16, 977-989 (1996). Кроме того, эта конструкция также имеет следующие 3 аминокислотные замены: L457V, С488А и C584S. Конструкция LCK для анализа: Тирозинкиназу LCK экспрессировали в клетках насекомого в виде N-концевой делеции, начиная с аминокислотного остатка 223 и заканчивая остатком 509 на конце белка, со следующими двумя аминокислотными заменами на N-конце: Р233М и C224D. Конструкция СНК-1 для анализа: Полноразмерную СНК-1 человека с С-концевой His-меткой (FL-CHK-1) экспрессировали с использованием системы бакуловирусы/клетки насекомого. Она содержит 6 гистидиновых остатков (6 × Mis-метка) на С-конце 476-аминокислотной последовательности СНК-1 человека. Этот белок очищали общепринятыми хроматографическими методами. Конструкция CDK2/Cyclin А для анализа: CDK2 очищали, используя опубликованную методологию (Rosenblatt et al., J. Mol. Biol., 230, 1317-1319 (1993)), из клеток насекомого, которые были инфицированы экспрессионным бакуловирусным вектором. Циклин A (Cyclin А) очищали из клеток Е. coli, экспрессирующих полноразмерный рекомбинантный циклин А, и конструкцию, представляющую собой усеченный циклин А, генерировали ограниченным протеолизом и очищали, как описано ранее (Jeffrey et al., Nature, 376, 313-320 (1995)). Конструкция CDK4/Cyclin D для анализа: Комплекс CDK4 и циклина D3 человека или комплекс циклина D1 и белка слияния CDK4 и глутатион-S-трансферазы (GST-CDK4) человека очищали, используя традиционные биохимические хроматографические методы, из клеток насекомого, которые были коинфицированы соответствующими экспрессионными бакуловирусными векторами. Анализ VEGF-R2: Двойной спектрофотометрический (FLVK-P) анализ Продуцирование ADP (аденозиндифосфата) из АТР (аденозинтрифосфата), которая сопровождает перенос фосфорила, было сопряжено с окислением NADH (никотинамид-аденин-динуклеотид, восстановленная форма) с использованием фосфоенолпирувата (PEP) и системы, содержащей пируваткиназу (РК) и лактатдегидрогеназу (LDH). Мониторинг окисления NADH проводили путем отслеживания снижения поглощения при 340 нм (е340=6,22 см-1·мМ-1) с использованием спектрофотометра Beckman DU 650. Условия анализа на фосфорилированный VEGF-R2 ELISA анализ (твердофазный иммуноферментный анализ): Мониторинг образования фосфогастрина осуществляли, используя биотинилированный пептид гастрина (1-17) в качестве субстрата. Биотинилированный фосфогастрик иммобилизовали, используя покрытые стрептавидином 96-луночные титрационные микропланшеты, с последующим детектированием с использованием анти-фосфотирозин-антител, конъюгированных с пероксидазой хрена. Активность пероксидазы хрена контролировали с использованием диаммониевой соли 2,2′-азино-ди-[3-этилбензатиазолинсульфоната(6)] (ABTS). Типичные растворы для анализа содержали: 2 мМ биотинилированного пептида гастрина; 5 мМ DTT; 20 мМ АТР; 26 мм MgCl2 и 2 мМ MnCl2 в 200 мМ HEPES, pH 7,5. Анализ инициировали 0,8 нМ фосфорилированным VEGF-R2 Анализ FGF-R: Спектрофотометрический анализ проводили, как описано выше для VEGF-R2, за исключением следующих изменений в концентрации: FGF-R=50 нМ, АТР=2 мМ и поли(Е4Y1)=15 мМ. Анализ LCK: Спектрофотометрический анализ проводили, как описано выше для VEGF-R2, за исключением следующих изменений в концентрации: LCK=60 нМ, MgCl2=0 мМ, поли(Е4Y1)=20 мМ. Анализ СНК-1: Продуцирование ADP из АТР, которое сопровождает перенос фосфорила на синтетический субстрат пептид Syntide-2 (PLARTLSVAGLPGKK), было сопряжено с окислением NADH с использованием фосфоенолпирувата (PEP) под действием пируваткиназы (РК) и лактатдегидрогеназы (LDH). Окисление NADH контролировали, отслеживая снижение поглощения при 340 нм ( Анализ пролиферации HUVEC: Этот анализ определяет способность тестируемого соединения ингибировать стимулированную фактором роста пролиферацию эндотелиальных клеток пуповинной вены человека (HUVEC). Клетки HUVEC (пассажи 3-4, Clonetics, Corp.) размораживали в культуральную среду EGM2 (Clonetics Corp) в колбах Т75. Свежую среду EGM2 добавляли в колбы 24 часа спустя. Четыре или пять дней спустя клетки подвергали воздействию другой культуральной среды (среда F12K, дополненная 10% сыворотки плода коровы (FBS), добавкой для роста эндотелиальных клеток (ECGS, 60 мкг/мл) и гепарином (10 мкг/м). Экспоненциально растущие клетки HUVEC использовали в последующих экспериментах. От десяти до двенадцати тысяч клеток HUVEC высевали в 96-луночные чашки в 100 мкл обогащенной культуральной среды (описанной выше). Клеткам давали возможность прикрепиться в течение 24 часов в этой среде. Среду затем удаляли отсасыванием и в каждую лунку добавляли 115 мкл минимальной среды (F12K+1% FBS). Через 18 часов 15 мкл тестируемого агента, растворенного в 1% DMSO (диметилсульфоксид) в минимальной среде, или только этот носитель добавляли в каждую лунку для обработки; конечная концентрация DMSO составляла 0,1%. Один час спустя 20 мкл 150 нг/мл hrVEGF165 в минимальной среде добавляли во все лунки за исключением лунок, содержащих необработанные контроли; конечная концентрация VEGF составляла 20 нг/мл. Клеточную пролиферацию количественно оценивали через 72 часа по восстановлению красителя МТТ, при этом клетки подвергали воздействию МТТ (Promega Corp.) в течение 4-5 часов. Восстановление красителя останавливали добавлением стоп-раствора (Promega Corp.), и поглощение при 570 и 630 нм определяли на спектрофотометрическом считывателе 96-луночных планшетов. Анализ пролиферации раковых клеток (MV522): Для определения того, может ли ингибитор протеинкиназ иметь терапевтическую полезность в блокировании ангиогенеза для лечения рака, важно продемонстрировать, что этот ингибитор не блокирует неспецифически клеточную пролиферацию клеток, которые не экспрессируют киназный рецептор. Это делают путем проведения анализов на пролиферацию с использованием раковых клеток. Протокол для оценки клеточной пролиферации раковых клеток подобен протоколу, который использовали для оценки клеток HUVEC. Две тысячи легочных раковых клеток (линия MV522, получены из UCSD (Раковый центр Калифорнийского университета в Сан-Диего)) высевали в среду для выращивания клеток (среда RPMI1640, дополненная 2 мМ глутамина и 10% FBS). Клеткам давали возможность прикрепляться в течение 1 дня, после чего добавляли тестируемые агенты и/или носители. Клетки обрабатывали одновременно теми же тестируемыми агентами, которые использовали в анализе HUVEC. Клеточную пролиферацию оценивали количественно анализом восстановления красителя МТТ через 72 часа после воздействия тестируемых агентов. Определение эффективности в отношении c-Kit: Для определения действенности ингибитора в отношении c-Kit использовали клетки NCl-H526 (АТСС (Американская коллекция типовых культур)). Клетки выращивали до суб-конфлюэнтности и инкубировали в минимальной среде в течение 18 часов. Добавляли ингибитор и инкубировали клетки в течение 45 мин при 37°С в присутствии 2,3% альбумина и 1 мМ Na3VO4 (Sigma). SCF, фактор роста c-Kit, добавляли в культуру в конечной концентрации 50 нг/мл. Пять минут спустя клетки промывали два раза холодным PBS и лизировали лизирующим буфером (50 мМ Tris (трис-буфер), 150 мМ NaCl, 1 мм PMSF (фенилметилсульфонилфторид), 1% NP40, 1 мМ Na2VO4 и коктейль ингибиторов протеаз). Иммунопреципитацию осуществляли с использованием 1 мг общего белка из каждого лизата инкубированием в течение ночи при 4°С с 4 мкг/мл антител CD117 ab-3 (K45, Neomarkers). Комплекс с антителом конъюгировали с гранулами белка А на следующее утро. SDS-PAGE (электрофорез в полиакриламидном геле с додецилсульфатом натрия) и вестерн-блоттинг проводили с использованием антител 4G10 против фосфотирозина (Upstate Biotechnology) для фосфорилированных рецепторов, или антител sc-1493 против c-Kit-рецептора (С-14, Santa Cruz) при 1:1000. Блоты визуализировали хемилюминесцентными реагентами ECL Plus. Фосфовизуализатор (Storm 846, Molecular Dynamics) использовали для количественного определения сигналов в блотах. Снижение популяции c-kit-положительных клеток в общей численности клеток периферической крови животного и млекопитающего можно использовать в качестве биомаркера активности соединения формулы 1. Измерение фосфорилирования eNOS и Akt: HUVEC (Clonetics) использовали для определения эффективности действия ингибитора против eNOS и Akt. Клетки выращивали до суб-конфлюэнтности и инкубировали в минимальной среде в течение 18 часов. Добавляли ингибитор и инкубировали клетки в течение 45 мин при 37°С в присутствии 2,3% альбумина и 1 мМ Na2VO4 (Sigma). В культуральную среду добавляли VEGF при 50 нг/мл. Пять минут спустя клетки промывали два раза холодным PBS и лизировали лизирующим буфером (50 мМ Tris, 150 мМ NaCl, 1 мМ PMSF, 1% NP40, 1 мМ Na2VO4 и коктейль ингибиторов протеаз). Общий белок в диапазоне 30-40 нг анализировали Вестерн-блоттингом. Фосфорилирование eNOS и Akt оценивали с использованием антител PhosphoeNOS (Ser 1177) #9571 или Phospho-Akt (Ser 473) #9271 (оба от Cell Signaling). Детектирование белка осуществляли с использованием антител NOS3 (С-20) sc-654 (Santa Cruz) или Akt #9272 (Cell Signaling). Всем требуются вторичные анти-кроличьи антитела, связанные с пероксидазой хрена, используемые при 1:3000. Блоты визуализировали хемилюминесцентным субстратом Super Signal West Dura (Pierce). Для количественного определения сигналов в блотах использовали Alpha Imager 8800 от Alpha Innotech. Анализ РК (фармакокинетики) у мышей: Фармакокинетику (например, всасывание и элиминацию) лекарственных средств у мышей анализировали в приведенном ниже эксперименте. Тестируемые соединения готовили в виде суспензии в 0,5% носителе CMC (карбоксиметилцеллюлоза) или в виде раствора в носителе, представляющем собой смесь PEG400 и подкисленной H2O 30:70. Эту суспензию или этот раствор вводили перорально (п.о.) или внутрибрюшинно (в.б.) самкам мышей С3Н (n=4). Образцы крови брали из глазниц в точках времени: час 0 (перед введением дозы), 0,5 ч, 1,0 ч, 2,0 ч и 4,0 ч после введения дозы. Плазму получали из каждого образца центрифугированием при 2500 об/мин в течение 5 мин. Тестируемое соединение экстрагировали из плазмы методом органической преципитации белка. Для каждого времени отбора крови 50 мкл плазмы объединяли с 1,0 мл ацетонитрила, перемешивали на вортексе в течение 2 мин и затем центрифугировали при 4000 об/мин в течение 15 мин для осаждения белка и экстракции тестируемого соединения. Затем ацетонитрильный супернатант (экстракт, содержащий тестируемое соединение) вливали в новые пробирки и упаривали на нагревательной плитке (25°С) в струе газа N2. В каждую пробирку, содержащую высушенный экстракт тестируемого соединения, добавляли 125 мкл подвижной фазы (60:40, 0,025 М NH4H2PO4+2,5 мл/л ТЕА:ацетонитрил). Тестируемое соединение ресуспендировали в подвижной фазе перемешиванием на вортексе и большую часть белка удаляли центрифугированием при 4000 об/мин в течение 5 мин. Каждый образец вливали во флакон для ВЭЖХ (высокоэффективная жидкостная хроматография) для анализа тестируемого соединения. Анализ проводили на системе ВЭЖХ Hewlett Packard 1100 с УФ-детекцией. 95 мкл из каждого образца впрыскивали на колонку Phenomenex-Prodigy с обращенной фазой С-18, 150×3,2 мм и элюировали градиентом 45%-50% ацетонитрила за 10 мин. Концентрацию тестируемого соединения в плазме (мкг/мл) определяли сравнением со стандартной кривой (площадь пика против концентрации в мкг/мл) с использованием известных концентраций тестируемого соединения, экстрагированных из образцов плазмы способом, описанным выше. Наряду со стандартами и не идентифицированными образцами выполняли анализ трех групп (n=4) контролей качества (0,25 мкг/мл, 1,5 мкг/мл и 7,5 мкг/мл), чтобы убедиться в надежности анализа. Стандартная кривая имела R2>0,99, и все контроли качества были в пределах 10% от их ожидаемых значений. Количественные показатели тестируемых образцов наносили на график для визуального отображения с использованием программного обеспечения Kaleidagraph и их фармакокинетические параметры определяли с использованием программного обеспечения WIN NONLIN. Анализ микросом печени человека (HLM): Метаболизм соединения в микросомах печени человека измеряли аналитическим анализом жидкостная хроматография/масс-спектрометрия, как описано ниже. Сначала микросомы печени человека (HLM) размораживали и разбавляли до 5 мг/мл холодного 100 мМ калий-фосфатного (KPO4) буфера. Соответствующие количества KPO4 буфера, NADH-регенерирующего раствора (содержащего B-NADH, глюкозо-6-фосфат, глюкозо-6-фосфатдегидрогеназу и MgCl2 и HLM предварительно инкубировали в стеклянных пробирках размером 13×100 мм при 37°С в течение 10 мин (3 пробирки на тестируемое соединение – три повтора). Тестируемое соединение (конечная концентрация 5 мМ) добавляли в каждую пробирку для инициирования реакции и смешивали осторожным перемешиванием на вортексе, а затем инкубировали при 37°С. При t=0, 2 ч, образец 250 мкл отбирали из каждой инкубационной пробирки в отдельные стеклянные пробирки 12×75 мм, содержащие 1 мл охлажденного льдом ацетонитрила с 0,05 мкМ резерпина. Образцы центрифугировали при 4000 об/мин в течение 20 мин для осаждения белка и соли (Beckmnan Allegra 6KR, S/M ALK98D06, #634). Супернатант переносили в новые стеклянные пробирки 12×75 мм и упаривали на центрифужном вакуумном испарителе Speed-Vac. Образцы разводили в 200 мкл смеси 0,1% муравьиной кислоты/ацетонитрил (90/10) и интенсивно перемешивали на вортексе до растворения. Затем образцы переносили в отдельные полипропиленовые микроцентрифужные пробирки и центрифугировали при 14000× g в течение 10 мин (Fisher Micro 14, S/N MO 017580). Для каждого повтора (#1-3) в каждой временной точке (0 и 2 ч) аликвоту образца каждого тестируемого соединения объединяли в одном ВЭЖХ флаконе (в сумме 6 образцов) для ЖХ-МС (жидкостная хроматография-масс-спектрометрия) анализа, который описан ниже. Объединенные образцы соединения впрыскивали в ЖХ-МС систему, состоящую из ВЭЖХ с диодной матрицей Hewlett-Packard HP1100 и тройного квадрупольного масс-спектрометра Micromass Quattro II, работающего в SIR-режиме электрораспыльной ионизации с регистрацией положительный ионов (запрограммирован для специфического сканирования в отношении ионов молекулы каждого тестируемого соединения). Пик каждого тестируемого соединения интегрировали в каждой временной точке. Для каждого соединения площадь пика в каждой временной точке (n=3) усредняли и эту среднюю площадь пика в точке 2 ч делили на среднюю площадь пика в точке 0 ч с получением процента тестируемого соединения, оставшегося в точке 2 ч. Анализы апоптоза HUVEC in vitro Количественная оценка апоптоза твердофазным иммуноферментным анализом (ELISA): Апоптоз клеток HUVEC измеряли с использованием Cell Death Detection Elisa PLUS (каталожный номер №1775425, Roche Biochemicals, Mannheim, Германия), который определяет количество цитоплазматических гистон-ассоциированных фрагментов ДНК в клеточных лизатах. Эту процедуру выполняли с незначительными модификациями инструкций изготовителя. Кратко, клетки HUVEC, выращенные в минимальной среде, обрабатывали Соединением А в различных концентрациях в присутствии VEGF (20 нг/мл). Цитозольную фракцию клеток в разные точки времени собирали и использовали в качестве источника антигена в сэндвич-анализе ELISA с первичными анти-гистон mAb (моноклональное антитело), нанесенными на титрационный микропланшет, и вторичными анти-ДНК mAb, связанными с пероксидазой. Число апоптических клеток определяли добавлением хромогенного пероксидазного субстрата и измерением абсорбции на спектрофотометре при 405 нм (стандартная длина волны 490 нм). Визуализация апоптоза методом TUNEL: Детектирование апоптических клеток in situ проводили с использованием метода опосредованного терминальной дезоксинуклетидтрансферазой мечения дезоксиуридинрифосфатом 3′-концов ДНК, образующихся в местах разрывов ДНК (TdT-mediated dUTP nick end labeling (TUNEL)). Кратко, клетки HUVEC, выращенные в 8-луночных слайд-камерах Lab-Tek, выдерживали в минимальной среде в течение ночи и затем обрабатывали в течение 6 часов Соединением А в различных концентрациях. Затем клетки фиксировали в 4% параформальдегиде, проницаемость их мембран нарушали Triton X-100 и инкубировали в течение 1 часа в смеси терминальной трансферазы и нуклеотидов, включая Fluorescin-dUTP (флуоресцин-2′-дезоксиуридин-5′-трифосфат) (система Deadend Fluorometric TUNEL, Promega, каталожный номер G3250) в соответствии с инструкциями производителя. Клетки подвергали контрастному окрашиванию раствором йодида пропидия (PI). Позитивно окрашенные меченные флуоресцеином и PI клетки визуализировали и фотографировали методом флуоресцентной микроскопии. Анализ PDGF-опосредованной миграции клеток: В этом анализе использовали клетки U87MG. 6-Луночные планшеты предварительно инкубировали в течение ночи с 0,5 нг/мл фибронектина. На следующий день клетки U87MG высевали в каждую лунку и оставляли их расти до конфлюэнтности. Клетки инкубировали в течение ночи с минимальной средой, содержащей 0,1% FBS. Кончиком пипетки делали примерно 1 см царапину, и промывали клетки минимальной средой. Затем планшеты инкубировали с 0,5 нг/мл фибронектина в течение 1 часа и снова промывали. Вводили экспериментальную среду, содержащую 100 нг/LI rhPDGF BB и Соединение А в минимальной среде. Клетки фотографировали между часами 0 и 15 и миграцию визуализировали. Анализ фосфорилирования клеточного VEGFR-2 и молекул, следующих за ним по цепи передачи сигнала: HUVEC (Clonetics) культивировали до суб-конфлюэнтности и инкубировали в минимальной среде (F12K плюс 0,1% FBS) в течение 18 часов. Соединение А добавляли к клеткам в присутствии 2,3% альбумина и 1 мМ Na2VO4 (Sigma). Сорок пять минут спустя к культуре добавляли VEGF с конечной концентрацией 50 нг/мл. Пять минут спустя клетки промывали холодным PBS и лизировали лизирующим буфером (50 мм Tris, 150 мм NaCl, 1 мМ PMSF, 1% NP40, 1 мм Na2VO4 и коктейль из ингибиторов протеаз). Один миллиграмм общего белка из лизата иммунопреципитировали с использованием anti-Flk-1 C-1158 (Santa Cruz). Комплекс с антителом конъюгировали с гранулами белка А и проводили анализ SDS PAGE/Western (электрофорез в полиакриламидном геле с додецилсульфатом натрия и вестерн-блоттинг) и фосфорилированный VEGFR-2 и белок детектировали антителами против фосфотирозина 4G10 (Upstate Biotechnology) и против Flk-1 С-20 (Santa Cruz) соответственно. Для eNOS и Akt клетки обрабатывали так же, как описано выше. Вестерн-блоттинг осуществляли с использованием общего количества белка 30-40 мкг. Фосфорилирование eNOS и Akt зондировали при использовании антител Phospho-eNOS (Ser 1177, #9571) или Phospho-Akt (Ser 473, #9271) (Cell signaling). Белки определяли с использованием NOS3 С-20 (sc-654, Santa Cruz) или Akt антитела #9272 (Cell signaling). Связанные с пероксидазой хрена антитела к кроличьим IgG использовали в качестве вторичных антител. Все блоты визуализировали с помощью хемилюминесцентного субстрата Super Signal West Dura (Pierce). Сигнал количественно определяли, используя Alpha Imager 8800 от Alpha Innotech. Эксперименты по отмывке: клетки HUVEC обрабатывали, как описано выше. После инкубации с Соединением А (10 нМ) в течение 45 мин и стимуляции VEGF (50 нг/мл) в течение 5 мин супернатант удаляли, промывали и заменяли минимальной средой, содержащей VEGF и Na3VO4. Клетки дополнительно инкубировали в течение желательного отрезка времени, после чего лизировали и процессировали с использованием иммунопреципитации и вестерн-блоттинга на фосфорилированный и общий VEGFR-2 (смотри выше). В другом эксперименте клетки обрабатывали VEGF в течение всего срока обработки, как указано выше, и фосфорилирование VEGFR-2 и общий VEGFR-2 в желательных точках времени оценивали аналогичным образом. Сигналы в процессе отмывки количественно определяли денситометрией. Интенсивности максимальной стимуляции (5 мин) в каждом эксперименте нормализовали от каждого к каждому, и интенсивность cpoccpo-VEGFR-2 на каждой точке времени сравнивали между двумя экспериментами, что давало возможность определить выход по фосфорилированию VEGFR-2 относительно клеток, которые не обрабатывали, но VEGF-стимулировали. Модели опухолей: Для моделей MV522 (карцинома ободочной кишки) и MDA-MB-231 (карцинома молочной железы) человека бестимусным мышам (n=812) имплантировали (подкожно) 5×106 клеток/сайт. Для мышиной модели легочной карциномы Льюиса фрагменты опухоли (1-2 мм2) имплантировали в правый бок мышей B6D2F1. Введение лекарственного средства обычно начинали в день 7 (MV522), или когда средний размер опухоли достигал 150-200 мм3 (MDA-MB-231). Готовили препарат соединения формулы 1 в 0,5% СМС/Н2О и вводили его перорально дважды в день. Готовили препарат доцетаксела в 7% EtOH/3% полисорбата/90% Н2О и вводили его еженедельно внутривенно. Лечение обычно продолжали в течение 3-4 недель. Геометрическую длину и ширину опухоли измеряли три раза в неделю с использованием электронного циркуля. Объем опухоли вычисляли как произведение 0,4× [Длина × (Ширина)2]. Данные записывали как среднее ±SEM (среднеквадратическая ошибка). В конце исследований опухоли и ткани вырезали, взвешивали их и собирали для анализа. Для анализа концентрации лекарственного средства собирали плазму. Результаты показаны в Таблицах 1-3.
Другими ферментами, которые прошли скрининг, но были выше предела для вычисления Кi, являются: cMet, LCK, c-Src, FAK, Pyk2, IRL, ВТК, CDK1, CDK2, CDK4, PKA, PKC, PLK и Chk1.
Комбинированные группы демонстрируют увеличение частоты случаев полной и частичной регрессии опухоли. Скорость роста опухоли снижалась в большей степени при комбинировании агентов. Комбинированное лечение было в равной степени переносимым по сравнению с лечением отдельно одним агентом. Пример 2 Соединение формулы 1, 6-[2-(метилкарбамоил)фенилсульфанил]-3-Е-[2-(пиридин-2-ил)этенил]индазол, вводили в варьирующих дозах пациентам с солидными опухолями. Тридцать пациентов (13 мужчин, 17 женщин) лечили, используя соединение формулы 1 в пероральной лекарственной форме в форме таблеток по схеме BID (дважды в день) или QD (ежедневно). Каждый цикл составлял 28 дней. Конкретными диагнозами были опухоли молочной железы (11), щитовидной железы (5), почечно-клеточные (5), легкого (4) и другие (5). Фармакокинетические данные получали жидкостной хроматографией-тандемной масс-спектрометрией (ЖХ/МС-МС). Пробы крови брали в день 15 цикла через 1/2 часа, 1 час, 2 часа, 4 часа, 8 часов и 12 часов после введения. Фармакокинетические результаты (средние значения в день 15) показаны в Таблице 4. Если не указано иное, пациенты были не голодными. Числа в скобках представляют собой коэффициенты варьирования, выраженные в процентах. В этой Таблице Сmax представляет собой максимальную наблюдаемую концентрацию соединения формулы 1 в плазме крови, AUC (0-24) (площадь под кривой в интервале 0-24 часа) означает 24-часовую AUC концентрации в плазме крови, и T1/2 представляет собой период полужизни, определенный из графика зависимости концентрации от времени. Обозначение “# пациентов с ФК” указывает число пациентов, в отношении которых были получены фармакокинетические данные.
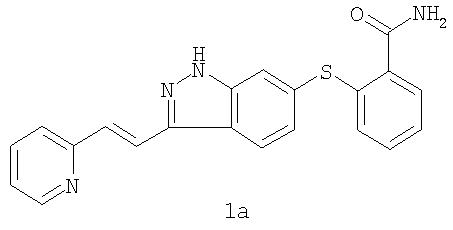
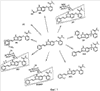
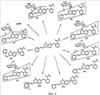
Кроме того, пациенты в первой группе (n=6) получали индивидуализированные дозы, изменяющиеся от 10 мг BID до 30 мг BID (ФК не показана). Экспозиции плазмы были выше (примерно 49%) и вариабельность у каждого пациента была снижена в состоянии натощак при сравнении с сытым состоянием. Было определено, что максимальная толерантная доза (MTD) в настоящее время составляет 5 мг дважды в день натощак. Ограничивающими дозу проявлениями токсичности (DLT) при дозах больше, чем MTD были гипертензия (HTN), судороги, повышенные результаты тестов на функцию печени, панкреатит, апноэ и стоматит. Кроме того, 2 пациента с немелкоклеточным раком легкого (NSCLC), давших реакцию на лечение, имели фатальное кровохарканье, одно через 3 недели после прекращения лечения соединением. Наблюдалась также не ограничивающая дозу протеинурия. При дозах, меньших или равных MTD, DLT были ограничены стоматитом второй степени у 1 пациента. Не ограничивающую дозу гипертензию наблюдали у 7 из 14 пациентов, и с ней справились обычными лекарствами против гипертензии. Наблюдалось два продолжительных частичных ответа по критериям REClST (критерии оценки ответа при солидных опухолях) (при почечно-клеточной опухоли и кистозной аденоидной опухоли гайморовой полости) и стабильное заболевание, длящееся 4 месяца или дольше (диапазон от 4 до 13 и более месяцев) у 5 пациентов этой совокупности, получившей тяжелое предшествующее лечение. Используя метод dceMRI (магнитно-резонансная визуализация с динамическим увеличением контрастности), проводили предварительный анализ 21 пациентов с целью измерения сосудистых эффектов, индуцированных соединением формулы 1, в исходной точке и в день 2, день 28 и день 56. Процент изменения среднего Кtrans (P.S.Tofts, G.Brix, D.L.Buckley, J.L.Evelhoch, E.Henderson, M.V.Knopp, H.B.W.Larsson, T.Lee, N.A.Mayr, G.J.M.Parker, R.E.Port, J.Taylorand R.M.Weisskoff, Estimating Kinetic Parameters from Dynamic Contrast-Enhanced T1-Weighted MRI of a Diffusable Tracer: Standardized Quantities and Symbols, Journal of Magnetic Resonance Imaging, 10: 223-232 (1999)) и исходной площадей под кривой интенсивность контраста Х время (IAUC) вычисляли для опухоли каждого индекса (n=1-4 на пациента). Сосудистый ответ опухоли определяли как больше или равно 50% снижению величин исходных параметров ко дню 2. Острые (день 2) снижения сосудистого ответа опухоли (более или равно 50% снижения Кtrans и IAUC) наблюдались у 6 из 18 обследованных пациентов, а 11 из 18 пациентов продемонстрировали большее или равное 40% снижение как Ktrans, так и IAUC. Из-за технических проблем со сканами 3 из 21 набора изображений не были оценены. Это пример показывает, что соединение формулы 1 является высокоактивным агентом, судя по клиническому ответу и острым сосудистым изменениям опухоли. Пример 3 После перорального введения [14С]-меченного соединения формулы 1 в дозе 30 мг свободного основания/кг интактным или канюлированным в желчевыводящий проток гончим собакам наблюдался экстенсивный метаболизм. Пути биотрансформации включали оксигенацию (моно- или ди-), глюкуронирование, глюкозилирование и оксигенацию с последующим либо сульфатированием, либо глюкозилированием. На Фиг.1 показаны идентифицированные метаболиты. В плазме М12 (N-оксид) является единственным детектируемым метаболитом. В моче основным метаболитом является М5 (депиридинилкарбоновая кислота). Основные желчные метаболиты включают М8 (сульфат) и М12. Химическая структура основного фекального метаболита М1 остается неизвестной. Паттерны экскреции для радиоактивности от 14С у гончих собак после введения одной пероральной дозы соединения были сходными у самцов и самок, причем радиоактивность выводилась главным образом с фекалиями. Средние величины выхода для интактных самцов составляли 85,5% в фекалиях и 5,3% в моче по сравнению с величинами выхода, составляющими 80,9% в фекалиях и 7,0% в моче у интактных самок. У канюлированных в желчевыводящий проток собак-самцов выводились относительно небольшие доли радиоактивности с желчью (выход 8,3%), причем дополнительная радиоактивность выводилась с фекалиями (52,7%) и мочой (11,3%). Из объединенной суммарной радиоактивности мочи и желчи от собак с канюлей в желчевыводящем протоке следует, что приблизительно 20% введенной радиоактивности всасывалось из желудочно-кишечного тракта. Суммарные средние величины выхода во всех образцах составляли 92,4% и 92,6% для интактных самцов и самок соответственно и 89,6% для самцов с канюлей в желчевыводящем протоке. Профилирование и выяснение структуры метаболитов осуществляли с использованием ВЭЖХ, сопряженной линейно с радио-ВЭЖХ-детектором ( Пример 4 Соединение формулы 1 подвергается экстенсивному метаболизму у мышей CD-1 после единственного перорального введения [14С]-меченного соединения. Низкий процент неизмененного лекарственного средства выводился с мочой и фекалиями, и наблюдалось множество разных метаболитов фазы I и фазы II. Пути биотрансформации включали оксигенацию (моно- или ди-), глюкуронирование, глюкозилирование и оксигенацию с последующим либо глюкуронированием, либо глюкозилированием. Идентифицированные метаболиты показаны на Фиг.2. В плазме неизмененное лекарственное средство и М12 (N-оксид) представляли собой два основных компонента. М7 (глюкуронид) является наиболее значительным метаболитом как в моче, так и в фекалиях. После одного перорального введения свободного основания в дозе 50 мг/кг [14C]AG-013736 самцам мышей CD-1 большая часть радиоактивности, полученной от [14С], выводилась с фекалиями. Средние (n=2) выведения радиоактивности (% дозы) через 48 часов после введения дозы составляли 65,8% в фекалиях и 12,7% в моче. Скорость выведения радиоактивности с экскрементами была быстрой, причем примерно 72% дозы выводилось в пределах 24 часов после введения дозы. Профилирование радиоактивности и характеризацию структуры метаболитов осуществляли с использованием методов LC-RAM-MS. Кроме метаболитов, показанных на Фиг.1 и 2, другие известные метаболиты включают активный деметилированный метаболит формулы 1а.
Пример 5 Ангиогенез оценивали путем измерения плотности микрососудов опухоли (MVD) с использованием иммуногистохимии. Замороженные срезы опухоли окрашивали на маркер сосудистой поверхности CD-31, и количество сосудов в нескольких полях среза ткани количественно определяли вручную. Исследования показали, что введение соединения формулы 1 перорально дважды в сутки в течение 2-3 недель уменьшает число кровеносных сосудов в подвергнутых лечению опухолях на 70% по сравнению с контрольными опухолями. Это снижение плотности микрососудов после лечения наблюдалось во всех использованных моделях опухолей, включая LLC (легочная карцинома Льюиса), MV522 и M24met. При непрерывной доставке осмотическим насосом Alzet в модель опухоли LLC соединение формулы 1 вызывает значительное ингибирование роста. Данные трех исследований показали, что максимальное ингибирование роста опухоли (MGI), которое может быть достигнуто под действием этого класса агента в модели LLC, составляло 78%. При низких концентрациях в плазме, таких как 55±17 нг/мл (N=3), достигалось 90% максимального ингибирования роста. Эту концентрацию обозначили как биологически активную концентрация (ВАС). 50% максимального ингибирования роста были связаны с концентрацией в плазме, составляющей 28±11 нг/мл (N=3). Эта концентрация обозначена как минимальная эффективная концентрация (МЕС). В 1 группе исследований 70% MGI, вызванного непрерывной инфузией соединения, ассоциировалось с AUC (0-24) 574 нг·ч/мл, тогда как в том же исследовании AUC (0-24) 720 нг·ч/мл после перорального введения дважды в сутки соответствовало 40% MGI. Из этих результатов следует, что противоопухолевая эффективность, наблюдаемая в этой модели, зависит от минимума концентрации и что у мышей постоянная низкая концентрация соединения может быть достаточной для получения максимальной противоопухолевой эффективности. Соединение формулы 1 было эффективным в качестве единственного агента в ксенотрансплантатной модели карциномы молочной железы человека MDA-MB-231. При подготовке к исследованию эффективности комбинации соединения формулы 1 и доцетаксела в этой модели проводили предварительное исследование на нативных голых мышах, чтобы определить воздействие потенциальных взаимодействий одного лекарственного средства с другим на фармакокинетику и толерантность. После внутривенного введения 15 или 30 мг/кг доцетаксела один раз в неделю в течение 3 недель у животных, которым вводили доцетаксел, было выявлено уменьшение массы тела (7% и 11% соответственно) по сравнению с контролем. Отсутствие разницы в массе тела было отмечено между животными, которым вводили доцетаксел один, и животными, которым вводили комбинацию доцетаксела и соединения формулы 1 (30 мг/кг/день в течение 16 дней перорально). Введение доцетаксела не влияло на AUC соединения формулы 1, тогда как значения Сmax AG-013736 значительно снижались в группах комбинированной терапии по сравнению с соединением формулы 1 одним. Гистологическое исследование выбранных тканей (печень, почки, сердце, селезенка, желудок, тонкий и толстый кишечник, яичники, грудина, сустав) выявили отсутствие эффектов в органе-мишени у мышей, которых в этом исследовании лечили соединением формулы 1 в качестве единственного агента. Изменения, отмеченные у мышей, которых лечили доцетакселем, включали фолликулярный некроз яичников и минимальную-легкую гипоцеллюлярность костного мозга. Комбинированное лечение соединением формулы 1 и доцетакселем не усугубляло воздействие доцетаксела на яичник, но была отмечена повышенная интенсивность гипоцеллюлярности костного мозга (от минимальной до умеренной) у животных, которым давали комбинацию соединения формулы 1 и доцетаксела. Кроме того, у животных, которых лечили комбинацией, наблюдалось кровоизлияние в костный мозг, по-видимому, вторичный эффект повышения интенсивности гипоцеллюлярности. Соединение формулы 1 и доцетаксел комбинировали для оценки эффективности в модели опухоли MDA-MB-231. Соединение формулы 1 одно (25, 5, и 1 мг/кг, перорально, два раза в сутки, давали в течение 3 недель) приводило к дозозависимому ингибированию роста опухоли. Доцетаксел один (внтривенно, еженедельно) в дозе 20 и 10 мг/кг, но не 2 мг/кг, также был эффективным. Оказалось, что могло иметь место полезное терапевтическое взаимодействие между соединением формулы 1 и доцетакселем. Эта польза была более заметной при комбинировании этих агентов как при высоких, так и при средних дозах. Число случаев частичной регрессии (от 16% до 97% снижения размера опухоли) и полного ответа при комбинациях с высокой и средней дозой были гораздо больше, чем в группах, которых лечили индивидуальным агентом одним при одинаковых дозах. Из-за ограниченности групп и относительно коротких временных рамок исследования этот результат не является окончательным. Соединение формулы 1 хорошо переносилось при всех дозах. Имело место 3%-7% снижение средней массы тела в группе с комбинацией высоких доз (25 мг/кг соединения 1 и 20 мг/кг доцетакселя) после третьей дозы этого химиотерапевтического агента по сравнению со всеми остальными группами. Фармакокинетический анализ продемонстрировал, что присутствие доцетаксела не влияет на величины AUC соединения формулы 1, но значения Сmax значительно снижались в группах, получающих комбинацию, по сравнению с группой, получающей соединение формулы 1 одно. Противоопухолевую эффективность соединения формулы 1 в комбинации с доцетакселем исследовали на модели LLC. Модель LLC высокоустойчива к доцетакселу. При указанной величине MTD (30 мг/кг, один раз в неделю, внутривенно) с этим цитотоксическим агентом наблюдалась небольшая задержка роста опухоли (TGD) (TGD=3,2 суток). Всех мышей подвергли эвтаназии в пределах 28 суток эксперимента из-за больших первичных опухолей. Напротив, единственный агент, представляющий собой соединение формулы 1, вызывал дозозависимую и статистически значимую задержку роста опухоли (13,4 суток при 10 мг/кг и 15,4 суток при 30 мг/кг, перорально, два раза в сутки). Однако этот агент только замедлял, но не останавливал метастазирование в легкое. TGD (20,4 суток) в группе с комбинацией высоких доз, но не в группе с комбинацией низких доз (TGD=15,2 суток), статистически отличалась от наблюдаемой с любым единственным агентом одним (Р=0,0079 и Р=0,254 соответственно). Больше животных (3 из 10) достигали объективной конечной точки в группе с комбинацией высоких доз, но не в группе с комбинацией низких доз. В заключение, терапия комбинацией высоких доз соединения формулы 1 и доцетаксела может вызывать большую задержку роста первичной опухоли и метастазирования, чем любая монотерапия, но не приводит к полному излечению. Одно исследование с использованием соединения формулы 1 на модели опухоли MV522 продемонстрировало, что единственная ежесуточная (QD) пероральная доза 60 мг/кг приводит к такому же эффекту ингибирования роста опухоли, к какому приводит 30 мг/кг перорально два раза в день (р=0,154). Кроме того, оказалось, что противоопухолевая эффективность не компрометируется при введении пероральных доз 30 мг/кг два раза в сутки в течение 5 дней подряд с последующими 2 днями перерыва по сравнению с ежесуточным пероральным введением два раза в сутки с использованием той же концентрации дозы (р=0,223). Из этих результатов следует, что в этой неклинической модели опухоли соединение формулы 1 можно давать по схеме либо QD, либо с определенными перерывами и ожидать достижения значительной противоопухолевой эффективности. Исследовали длительность времени ингибирования рецептора и концентрации соединения формулы 1, требуемые для достижения противоопухолевой эффективности в ксенотрансплантатной модели MV522. Результаты показали, что при пероральном введении (QD или BID), приблизительно 24-часовое ежесуточное воздействие выше EC50 (5 нг/мл) было необходимо для обеспечения не менее чем 50% противоопухолевой эффективности. Минимум 4-часовое ежесуточное воздействие при концентрации в плазме не менее 40-60 нг/мл было необходимо для достижения 90% ингибирования роста опухоли. Воздействие за пределами вышеуказанного порога не гарантировало дополнительную эффективность. Как в группе BID, так и в группе QD имела место аналогичная потеря массы тела, и в обоих случаях менее 5%. Таким образом, при условии соответствующей дозы и соответствующего времени воздействия режим QD может быть столь же эффективным, как и режим BID. Было также показано, что непрерывное воздействие через насосы Alzet давало более высокую противоопухолевую эффективность соединения формулы 1 по сравнению с введением с перерывами. Доставка насосами при 10 мг/мл приводит к постоянному среднему системному воздействию 30 нг/мл, которое приводит к стазу опухоли. Напротив, насыщающие дозы (перорально, BID), которые дают концентрации соединения формулы 1 в плазме выше предполагаемых ЕС50, могут вызывать только задержку роста опухоли. Таким образом, в лечение опухоли непрерывное системное воздействие соединением формулы 1 оказывается более эффективным, чем режим перорального введения два раза в сутки. Исследовали также противоопухолевую эффективность соединения формулы 1 с использованием режима введения с перерывами. Группами лечения были следующие: введение носителя один раз в сутки, введение носителя с перерывами, суточная доза 30 мг/кг (BID) и доза с перерывами 30 мг/кг. Схема введения с перерывами была следующая: Цикл-1 (введение в дни 1218 и невведение в дни 1928) и Цикл 2 (введение в дни 2936 и невведение в дни 3744). Введение начинали, когда средний размер опухоли был 250 мм3. Всем давали AG-013736 (перорально два раза в день). В целом, имела место значительная разница между введением с перерывами и ежедневным введением два раза в сутки, причем режим постоянного ежедневного введения был более эффективным в обеспечении задержки роста. В группе введения лекарственного средства с перерывами опухоли возобновляли нормальную скорость роста в пределах 3-4 дней после прекращения введения. Однако ингибирование роста опухоли возобновлялось в пределах 2 дней Цикла-2 введения. Как и ожидалось, ни в одной группе рецидива опухоли не наблюдалось. Исследовали также противоопухолевую эффективность соединения формулы 1 (AG-013736) в комбинации с гемцитабином (GEM) у пациентов с запущенным раком поджелудочной железы. Все пациенты в фазе I исследования получали 1000 мг/м2 гемцитабина в виде 30-минутных инфузий на сутки 1, 8 и 15 с последующей неделей перерыва в лечении. AG-013736 давали в дозе 5 мг перорально BID, начиная Цикл 1, сутки 3 (Ц1СЗ). Пациенты, удовлетворяющие критериям исследования, не получали предварительной химиотерапии какими-либо ингибиторами VEGF/VEGFR или лечения антиангиогенными препаратами. Полные фармакокинетические (ФК) профили собирали на Ц1С1 (гемцитабин один), Ц1С14 (установившийся режим, AG-013736 один) и Ц1С15 (GEM+AG-013736). В фазе II клинического исследования пациентов рандомизировали в группы AG-013736 или AG-013736+GEM, начиная с Ц1С1. Результаты и эффективность: 8 пациентов лечили в данном исследовании. Радиологическая оценка выявила 2 пациентов, давших частичный ответ на лечение, и 4 пациентов со стабильным ответом. Среднее количество циклов равно 3. Лечение 4 пациентов продолжили: Цикл 6 (2 пациента) и Цикл 2 (2 пациента). Выводы: данная комбинация является безопасной и эффективной, причем у 2 пациентов наблюдали значительную регрессию опухоли. Терапия была хорошо переносимой при токсичности, которая легко поддавалась контролю. Несмотря на то что данное изобретение было проиллюстрировано ссылкой на конкретные и предпочтительные воплощения, специалисты в данной области поймут, что результатом рутинного экспериментирования и практикования данного изобретения могут быть вариации и модификации. Поэтому приведенное выше описание не должно рассматриваться как ограничивающее данное изобретение, которое определяется пунктами прилагаемой формулы изобретения и их эквивалентами.
Формула изобретения
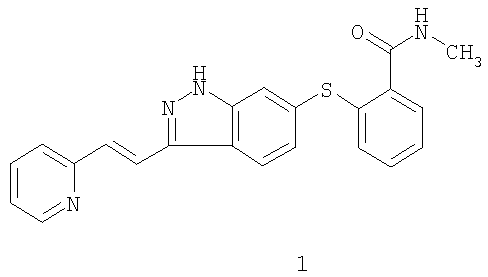
1. Способ лечения рака у человека, включающий введение человеку соединения формулы 1
или его фармацевтически приемлемой соли, где рак представляет собой рак молочной железы, и где соединение вводят в количестве от 5 до 20 мг на дозу с частотой дозирования два раза в сутки, и где способ дополнительно включает совместное введение доцетаксела. 2. Способ по п.1, где количество составляет от 5 до 7 мг. 3. Способ по п.1, где количество составляет 5 мг, 7 мг или 10 мг. 4. Способ по п.1, при котором соединение вводят перорально. 5. Способ лечения рака у человека, включающий введение человеку соединения формулы 1
или его фармацевтически приемлемой соли, где рак представляет собой рак поджелудочной железы, и где соединение вводят в количестве от 5 до 20 мг на дозу с частотой дозирования два раза в сутки, и где способ дополнительно включает совместное введение гемцитабина. 6. Способ по п.5, где количество составляет от 5 до 7 мг. 7. Способ по п.5, где количество составляет 5 мг, 7 мг или 10 мг. 8. Способ по п.5, при котором соединение вводят перорально.
РИСУНКИ
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 ), доцетаксел (Taxotere
), доцетаксел (Taxotere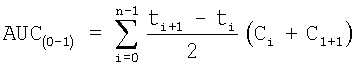
 50) цитозольного домена рецептора 2 васкулярного эндотелиального фактора роста человека (VEGF-R2), у которого отсутствуют 50 центральных остатков из 68 остатков киназного встроенного домена, экспрессировали в системе бакуловирусы/клетки насекомого. Из 1356 остатков полноразмерного VEGF-R2, VEGF-R2
50) цитозольного домена рецептора 2 васкулярного эндотелиального фактора роста человека (VEGF-R2), у которого отсутствуют 50 центральных остатков из 68 остатков киназного встроенного домена, экспрессировали в системе бакуловирусы/клетки насекомого. Из 1356 остатков полноразмерного VEGF-R2, VEGF-R2 340=6,22 см–1·мм-1), используя спектрофотометр НР8452. Типичные реакционные растворы содержали: 4 мМ PEP; 0,15 мМ NADH; 28 единиц LDH/мл; 16 единиц РК/мл: 3 мМ DTT; 0,125 мМ Syntide-2; 0,15 мм АТР; 25 мм MgCl2 в 50 мм TRIS, pH 7,5; и 400 мм NaCl. Анализы инициировали 10 нМ FL-CHK-1. Значения Ki определяли измерением исходной активности фермента в присутствии варьированных концентраций тестируемого соединения. Данные анализировали с использованием программного обеспечения Enzyme Kinetic и Kaleidagraph.
340=6,22 см–1·мм-1), используя спектрофотометр НР8452. Типичные реакционные растворы содержали: 4 мМ PEP; 0,15 мМ NADH; 28 единиц LDH/мл; 16 единиц РК/мл: 3 мМ DTT; 0,125 мМ Syntide-2; 0,15 мм АТР; 25 мм MgCl2 в 50 мм TRIS, pH 7,5; и 400 мм NaCl. Анализы инициировали 10 нМ FL-CHK-1. Значения Ki определяли измерением исходной активности фермента в присутствии варьированных концентраций тестируемого соединения. Данные анализировали с использованием программного обеспечения Enzyme Kinetic и Kaleidagraph. -RAM) и MS-детектированием с электрораспылительной ионизацией (ESI) и химической ионизацией при атмосферном давлении (APCl) с регистрацией положительных или отрицательных ионов.
-RAM) и MS-детектированием с электрораспылительной ионизацией (ESI) и химической ионизацией при атмосферном давлении (APCl) с регистрацией положительных или отрицательных ионов.