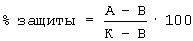Патент на изобретение №2238950
|
||||||||||||||||||||||||||
(54) ПРОИЗВОДНЫЕ ГЕМИНА И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ПОЛУЧЕНИЯ, ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ
(57) Реферат:
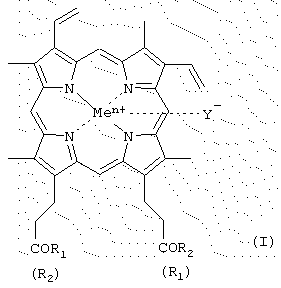
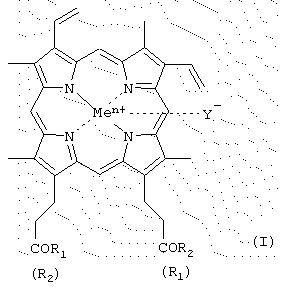
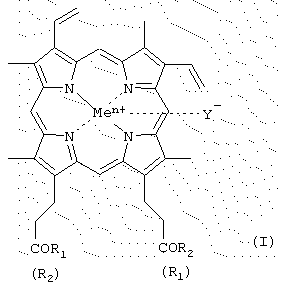
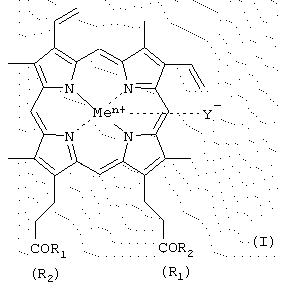
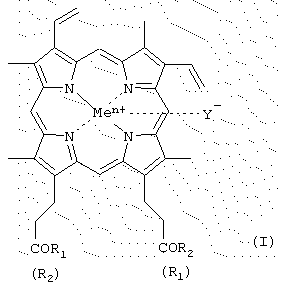
Изобретение относится к производным гемина или их фармацевтически приемлемым солям – ингибиторам протеолитических ферментов и представляющим собой соединения общей формулы (I) где R1 и R2 – заместители, которые могут представлять собой аминокислоты, производные аминокислот, пептиды, состоящие из 1-15 аминокислотных остатков, производные пептидов, состоящих из 1-15 аминокислотных остатков, а Ме=Fe3+, Y–=Cl–, R1=-LeuLeuValPheOMe, R2=-OH; R1=-ValPheOMe, R2=-OH; R1=-LeuHisOMe, R2=-OH; R1=-LeuHisAlaOMe, R2=-OH; R1=-LeuHisNHC10H20COOMe, R2=-ОН; R1=-LeuHisNHC10H20COOH, R2=-OH; R1=-LeuHisNHC10H20COOMe, R2=-LeuHisNHC10H20COOMe; R1=-Lys(Tfa)AlaAlaOMe, R2=-OH; R1=-ValPheOMe, R2=-LeuHisOMe; R1=-LeuLeuValPheOMe, R2=-LeuHisOMe; R1=-LeuLys(Tfa)LeuOMe, R2=-OH; R1=-LeuLys(Tfa)LeuOMe, R2=-LeuHisOMe; R1=-Lys(Tfa)AlaAlaOMe, R2=-AlaHisLys(Cbz)LeuOMe; R1=-GlyOBzl, R2=-GlyOBzl; R1=-HisOMe, R2=-HisOMe; R1=-LeuHisOMe, R2=-LeuHisOMe; R1=-LeuHisLeuGlyCys(Bzl)OBzl, R2=-LeuHisLeuGlyCys(Bzl)OBzl; R1=-LeuHisOMe, R2=-OEt; R1=-LeuHisLeuGlyCys(Bzl)OBzl, R2=-OEt; R1=-OBzl, R2=-OBzl; R1=-OBzl, R2=-OH; R1=-AlaOMe, R2=-OBzl; R1=-HisOMe, R2=-OBzl; R1=-LeuHisOMe, R2=-OBzl; R1=-LeuHisLeuGlyCys(Bzl)OBzl, R2=-OBzl; R1=-LeuHisAlaLys(Cbz)GlyCys(Bzl)OBzl, R2=-OBzl; R1=-LeuHisLys(Cbz)OMe, R2=-OH; R1=-LeuHis(Bzl)Lys(Cbz)OMe, R2=-OH; R1=-LeuHisOMe, R2=-OMe; R1=-LeuHis(Bzl)Lys(Cbz)OMe, R2=-OMe; R1=-AlaLeuAlaPheAlaCys(Bzl)OMe, R2=-LeuHis(Bzl)Lys(Cbz)OMe; R1=-AlaLeuAlaPheAlaCys(Bzl)OBzl, R2=-LeuHis(Bzl)Lys(Cbz)OMe; R1=-LeuHisAlaLys(Cbz)Cys(Bzl)OBzl, R2=-LeuHis(Bzl)Lys(Cbz)OMe; R1=-LeuHisOMe, R2=-OMe; R1=-GlyProArgGlyGlyOMe, R2=-OH; R1=-ArgProProGlyPheSer(Bzl)PheArgGlyGlyOMe, R2=-OH, двум способам получения производных гемина общей формулы I, фармацевтической композиции, обладающей способностью ингибировать протеолитические ферменты и применению производных гемина формулы I, ранее известных, обозначенных выше, в качестве ингибиторов протеолитических ферментов: протеиназы ВИЧ, пепсина, трипсина, химотрипсина. 5 н. и 9 з.п. ф-лы, 2 табл., 2 ил. Изобретение относится к химии и медицине, а именно к новым ингибиторам протеолитических ферментов, в частности ВИЧ-протеиназы, пепсина и химотрипсина, а также способу их получения. Известно применение в медицине ингибиторов протеолитических ферментов, например, для лечения острых панкреатитов, а также ВИЧ-инфекции. В последнем случае лечебный эффект достигается путем использования ингибиторов протеиназы ВИЧ псевдопептидной природы (ифавиренц, криксиван). Кроме того, известно применение ингибиторов протеолитических ферментов в фармацевтике в составе лекарственных форм препаратов пептидно-белковой природы для предотвращения их деградации в желудочно-кишечном тракте. Так как известные синтетические неприродные ингибиторы протеолитических ферментов обладают рядом отрицательных побочных эффектов при их терапевтическом применении, то задачей настоящего изобретения явилось создание новых ингибиторов ферментов, представляющих собой конъюгаты природных соединений. Было найдено, что поставленная задача решается хотя бы одним из производных гемина общей формулы (I) где R1 и R2 – заместители, которые могут представлять собой аминокислоты, производные аминокислот, пептиды, состоящие из 1-15 аминокислотных остатков, производные пептидов, состоящих из 1-15 аминокислотных остатков, а R1=-ValPheOMe, R2=-OH (V); R1=-ValPheOMe, R2=-ValPheOMe (VI); R1=-LeuValPheOMe, R2=-OH (VII); R1=-LeuLeuValPheOMe, R2=-OH (VIII); R1=-LeuLeuValPheOMe, R2=-LeuLeuValPheOMe (IX); R1=-GlyProArgOMe, R2=-OH (X); R1=-ArgOMe, R2=-OH (XI); R1=-ArgOMe, R2=-ArgOMe (XII); R1=-LeuValPheOH, R2=-OH (XIII); R1=-ValSerGlnAsnLeuOH, R2=-OH (XIV); R1=-ArgArgTrpHisArgLeuLysGlu(OMe)OH, R2=-OH (XV); R1-лизиновый кор, R2=-OH (XVI); R1=-GluAspLeuOH, R2=-OH (XVII); карбоксильная группа порфирина может быть модифицирована метиловым или другим С2-С8-эфиром или физиологически приемлемой солью; Y– представляет собой Сl–, СН3СОО–; Me представляет собой Fe, или их фармацевтически приемлемых солей, обладающих способностью ингибировать протеолитические ферменты (протеиназы ВИЧ, пепсина, трипсина, химотрипсина), а также интерферониндуцирующей и противовирусной активностью. Еще одним объектом данного изобретения является способ получения новых и известных псевдопептидов общей формулы (I).
Получение пептидных производных гемина, особенно содержащих длинные пептиды и/или с карбоксильными и другими функциональными группами сопровождается рядом сложностей при их синтезе в растворе. Это определяется неполным протеканием реакции образования амидных связей между гемином и пептидом, плохой растворимостью незащищенных и длинных пептидов в органических растворителях, отсутствием селективности при получении моно- и бис-производных гемина, что приводит к трудностям при выделении целевого продукта – геминпептида. Твердофазный метод синтеза пептидных производных гемина имеет ряд преимуществ перед классическим в растворе, а именно позволяет повысить выход и облегчает выделение целевого продукта (имеются в виду известные преимущества твердофазного синтеза вообще перед синтезом в растворе). Кроме того, синтез на твердой фазе позволяет селективно получать 6(7)-монопроизводные гемина. Данный подход в общем заключается в синтезе пептида на полимере с последующим ацилированием аминогруппы пептида на полимере активированным производным гемина с последующим отщеплением продукта от полимера. Синтез на полимере Trilar имеет ограничения при выборе стратегии и предусматривает применение только кислотолабильных N Новые производные гемина, в частности соединение (XVII), получают твердофазным методом с применением полимера Меррифилда и трет-бутилоксикарбонильной промежуточной защиты
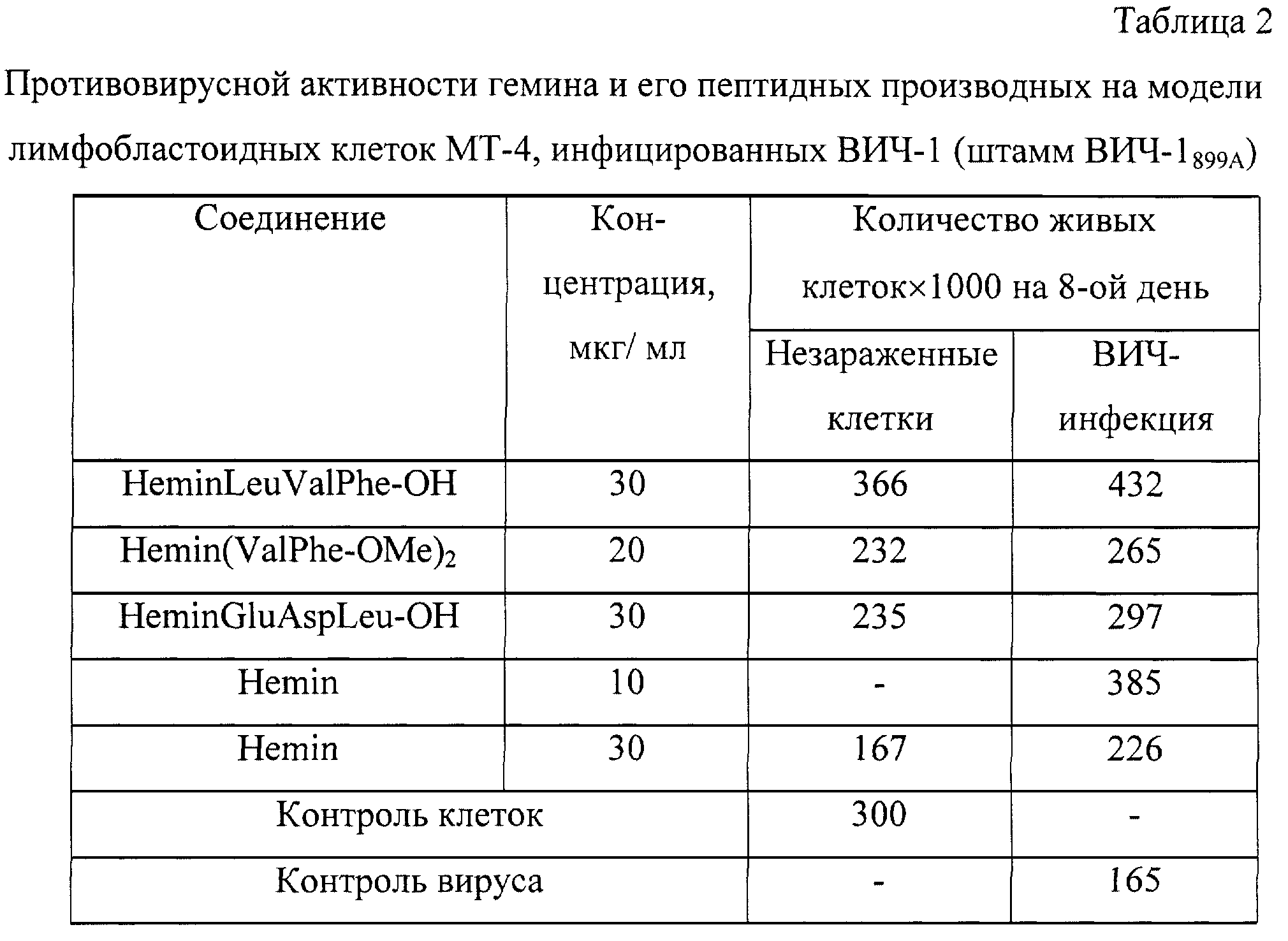
Отщепление производных гемина от полимера и деблокирование боковых защитных групп проводят смесью уксусной кислоты, трифторэтанола и дихлорметана в соотношении 1:1:8. Предложенный способ твердофазного синтеза производных порфиринов может быть использован не только для синтеза геминпептидов, но и для синтеза производных любых карбоксилсодержащих порфиринов и металлопорфиринов, в том числе с использованием альтернативных способов активации карбоксильной группы порфирина (металлопорфирина). Синтез производных гемина отражен в примерах 1-16. Предлагаемые производные гемина получены с высокими выходами и высокой степенью чистоты и охарактеризованы УФ-, ИК-спектроскопией, масс-спектрометрией, ТСХ. Индивидуальность полученных соединений проверяют методом ТСХ на пластинах Silufol UV-254 (Чехия) в системе растворителей хлороформ-метанол 9:1 (1), на пластинах Alufol (нейтр., “Merck”, Германия) в системе хлороформ-метанол 9:1 (2), на пластинах Kieselgel (“Merck”, Германия) в системах: хлороформ-метанол 9:1 (3), пиридин-вода-хлороформ-гексан 10:10:10:1 (4), н-бутанол-вода-уксусная кислота 4:5:1 (5), хлороформ-метанол-уксусная кислота 9:1:0.1 (6), хлороформ-метанол-уксусная кислота 7:3:1 (7), на бумаге в системе н-бутанол-вода-уксусная кислота 4:5:1 (8). Хроматограммы проявляют нингидрином, хлор-толидиновым реактивом и по свечению в УФ-свете. Выходы продуктов твердофазного синтеза рассчитывают по отношению к содержанию исходной аминокислоты в полимере. Пептидилполимеры для количественного аминокислотного анализа гидролизуют смесью 12 н. соляная кислота-пропионовая кислота (1:1) в течение двух часов при 140 Количественный аминокислотный анализ гидролизатов проводят на автоматическом аминокислотном анализаторе Biotronic IC5001 (Германия). Масс-спектры высокого разрешения получают на времяпролетном масс-спектрометре с лазерной десорбционной ионизацией (TOF MALDI) Vision 2000 (Thermo Bioanalysis, Англия) и магнитно-секторном приборе с прямой конфигурацией и электроспрей-ионизацией Finnigan MAT 900S (США). Электронные спектры снимают на приборе Jasco model UV/VS 7800 Spectrophotometer (Япония). ПМР-спектры регистрируют на приборе Bruker MSL 200 (200 МГц) (Германия) в CDCl3, CD3OD D2O. ИК-спектры снимают на приборах Perkin ELMER FT-IR Spectrometer 1725X (Щвейцария) и Hitachi 250-70 IR Spectrometer (Япония). Температуру плавления определяют на термоплавильном столике фирмы Boetius (Германия). В качестве полимерных носителей в синтезе производных гемина используют сополимер стирола с 1% дивинилбензола с 2-хлортритилхлоридной якорной группой с содержанием Сl– 1.58 ммоль/г (фирма “Bachem”, Германия), а также хлорметилированный сополимер стирола и 1% дивинилбензола, содержащий 1.05 ммоль/г Сl– Пример 1. N К раствору 0.79 мл (5.46 ммоль) Et3N и 4.0 мл DMFA прибавляют 1.19 г (5.46 ммоль) HCl Пример 2. N Раствор 1.10 г (2.91 ммоль) Boc-Val-Phe-OMe в TFA оставляют на 25 мин при 20 Пример 3. N Синтез, выделение и очистку пептида (IV) осуществляют в соответствии с методикой, описанной для пептида (III), исходя из 0.25 г (0.51 ммоль) Вос-Leu-Val-Phe-OMe. Выход 0.19 г (61.6%), Rf 0.60 (1). ИК-спектр, Пример 4. N Раствор 0.10 г (0.26 ммоль) Boc-Val-Phe-OMe в TFA оставляют на 25 мин при 20 Пример 5. N Соединение (VI) получают по методике, описанной для соединения (V) из 0.10 г (0.26 ммоль) Boc-Val-Phe-OMe и 6,7-бис-N-оксисукцинимидного эфира протогемина IX в 2 мл DMF, синтезированного из 0.09 г (0.14 ммоль) гемина, 0.03 г (0.28 ммоль) N-гидроксисукцинимида и 0.06 г (0.28 ммоль) DCC. Продукт очищают на колонке (170 Пример 6. N Соединение (VII) получают аналогично соединению (V) из 0.10 г (0.20 ммоль) Boc-Leu-Val-Phe-OMe и 6(7)-моно-N-оксисукцинимидного эфира протогемина IX в 2 мл DMF, синтезированного из 0.18 г (0.29 ммоль) гемина. Сырой продукт очищают препаративной хроматографией аналогично соединению (V). Для введения противоиона геминпептид (VII) обрабатывают в соответствии с методикой, описанной для соединения (V). Выход 0.02 г (12.4%), Rf 0.41 (3). Масс-спектр, m/z: [М+3Н]+ 992.26. Пример 7. N Соединение (VIII) получают аналогично соединению (V) из 0.12 г (0.20 ммоль) Boc-Leu-Leu-Val-Phe-OMe и 6(7)-моно-N-оксисукцинимидного эфира протогемина IX в 2.5 мл DMF, синтезированного из 0.18 г (0.29 ммоль) гемина. Сырой продукт очищают препаративной хроматографией аналогично соединению (V). Для введения противоиона геминпептид (VIII) обрабатывают в соответствии с методикой, описанной для соединения (V). Выход 0.09 г (41.0%), Rf 0.5 (3), Rf 0.05 (2). Электронный спектр, Пример 8. N Соединение (IX) получают аналогично соединению (VI) из 0.10 г (0.20 ммоль) Boc-Leu-Leu-Val-Phe-OMe и 6,7-бис-N-оксисукцинимидного эфира протогемина IX в 2 мл DMF, синтезированного из 0.07 г (0.11 ммоль) гемина. Сырой продукт очищают препаративной хроматографией аналогично соединению (V). Для введения противоиона геминпептид (IX) обрабатывают в соответствии с методикой, описанной для соединения (V). Выход 0.09 г (32.0%), Rf 0.7 (3), Rf 0.9 (2). Электронный спектр, Пример 9. N Деблокирование аминокомпонента и синтез геминпептида (X) осуществляют в соответствии с условиями, описанными для соединения (V), исходя из 0.04 г (0.09 ммоль) Boc-Gly-Pro-Arg-OMe и 6(7)-моно-N-оксисукцинимидного эфира протогемина IX в 2 мл DMF, синтезированного из 0.09 г (0.13 ммоль) гемина. Целевое вещество выделяют на колонке (300 Пример 10. N К раствору 0.12 мл (0.80 ммоль) Еt3N и 1 мл DMF прибавляют 0.10 г (0.40 ммоль) HCl Пример 11. N Соединение (XII) получают аналогично соединению (XI) из 0.10 г (0.40 ммоль) HCl Пример 12. N –) полимера, предварительно набухшего в 3 мл DCE, прибавляют свежеприготовленный раствор 0.12 г (0.30 ммоль) Fmoc-Phe-OH и 0.11 мл (0.75 ммоль) Et3N в 4.5 мл DCE. Смесь перемешивают 25 мин при 20 Пример 13. N Присоединение стартовой аминокислоты Leu к 0.33 г (0.52 ммоль Сl–) полимера с 2-хлортритилхлоридной якорной группой осуществляют по методике, описанной для соединения (XIII), с использованием 0.12 г (0.33 ммоль) Fmoc-Leu-OH и 0.12 мл (0.83 ммоль) Et3N. Содержание стартовой аминокислоты в полимере 0.28 ммоль/г. К раствору 0.10 г (0.27 ммоль) Fmoc-Asn-OH в 3 мл DMF при охлаждении и перемешивании добавляют 0.04 г (0.27 ммоль) 1-НОВТ и 0.06 г (0.27 ммоль) DCC. Смесь перемешивают в течение 20 мин, прибавляют к деблокированному пептидилполимеру и перемешивают в течение 30 мин. Оставляют на 24 ч при 20 Пример 14. N Присоединение стартовой аминокислоты Glu к 0.6 г (0.95 ммоль Сl–) полимера с 2-хлортритилхлоридной якорной группой осуществляют по методике, описанной для соединения (XIII), с использованием 0.36 г (0.95 ммоль) Fmoc-Glu(OMe)-OH и 0.33 мл (2.38 ммоль) Et3N. Содержание глутаминовой кислоты в полимере 0.22 ммоль/г. Твердофазный синтез октапептидилполимера осуществляют аналогично описанному выше синтезу трипептидилполимера при получении геминпептида (XIII). Реакции образования пептидной связи на полимере проводят с использованием трехкратных избытков (по отношению к содержанию стартовой глутаминовой кислоты в полимере) Fmoc-производных аминокислот, в том числе Fmoc-Lys(methoxy-Trt)-OH и Fmoc-His(Trt)-OH, в присутствии эквивалентных количеств DCC 1-HOBT (по 0.40 ммоль) в среде DMF в течение 24 ч. Присоединение остатка гемина осуществляют в соответствии с методикой, описанной в примере синтеза соединения (XIII). Отщепление целевого геминпептида с одновременным деблокированием боковых функциональных групп проводят действием на пептидилполимер смеси AcOH-TFE-DCM (2:1:7) в течение 3 ч при перемешивании. Далее полимер промывают смесью AcOH-TFE-DCM (2:1:7) (4 Пример 15. Конъюгат лизинового кора с протогемином IX (XVI). Присоединение стартовой аминокислоты Lys к 0.30 г (0.47 ммоль Сl–) полимера с 2-хлортритилхлоридной якорной группой осуществляют по методике, описанной для соединения (XIII), с использованием 0.18 г (0.30 ммоль) Fmoc-Lys(Fmoc)-OH и 0.11 мл (0.75 ммоль) Et3N. Содержание стартовой аминокислоты в полимере 0.11 ммоль/г. К раствору 0.12 г (0.20 ммоль) Fmoc-Lys(Fmoc)-OH в 2 мл DMF при охлаждении и перемешивании добавляют 0.03 г (0.20 ммоль) 1-HOBT и 0.04 г (0.20 ммоль) DCC. Смесь перемешивают в течение 20 мин, прибавляют к деблокированному пептидилполимеру и перемешивают в течение 30 мин. Оставляют на 24 ч при 20 Пример 16. N К 1.0 г хлорметилированного сополимера стирола 1% DVB (1.05 ммоль/г Сl–) прибавляют 0.38 г (1.05 ммоль) Boc-Leu-OCs в 7 мл DMF, перемешивают при 40 Пример 17. Натриевая соль N В 0.2 мл раствора 0.01М NaOH добавляют 2 мг (0.002 ммоль) соединения (V). Суспензию перемешивают до полного растворения вещества. Получают водорастворимую натриевую соль N Далее, настоящее изобретение относится к применению известных соединений формулы (I) где R1=R2 или R1 Y– представляет собой Cl–; Me представляет собой Fe3+; а также R1=-LeuLeuValPheOMe, R2=-OH; R1=-ValPheOMe, R2=-OH; R1=-LeuHisOMe, R2=-OH; R1=-LeuHisAlaOMe, R2=-OH; R1=-LeuHisNHC10H20COOMe, R2=-OH; R1=-LeuHisNHC10H20COOH, R2=-OH; R1=-LeuHisNHC10H20COOMe, R2=-LeuHisNHC10H20COOMe; R1=-Lys(Tfa)AlaAlaOMe, R2=-OH; R1=-ValPheOMe, R2=-LeuHisOMe; R1=-LeuLeuValPheOMe, R2=-LeuHisOMe; R1=-LeuLys(Tfa)LeuOMe, R2=-OH; R1=-LeuLys(Tfa)LeuOMe, R2=-LeuHisOMe; R1=-Lys(Tfa)AlaAlaOMe, R2=-AlaHisLys(Cbz)LeuOMe; R1=-GlyOBzl, R2=-GlyOBzl; R1=-HisOMe, R2=-HisOMe; R1=-LeuHisOMe, R2=-LeuHisOMe; R1=-LeuHisLeuGlyCys(Bzl)OBzl, R2=-LeuHisLeuGlyCys(Bzl)OBzl; R1=-LeuHisOMe, R2=-OEt; R1=-LeuHisLeuGlyCys(Bzl)OBzl, R2=-OEt; R1=-OBzl, R2=-OBzl; R1=-OBzl, R2=-OH; R1=-AlaOMe, R2=-OBzl; R1=-HisOMe, R2=-OBzl; R1=-LeuHisOMe, R2=-OBzl; R1=-LeuHisLeuGlyCys(Bzl)OBzl, R2=-OBzl; R1=-LeuHisAlaLys(Cbz)GlyCys(Bzl)OBzl, R2=-OBzl; R1=-LeuHisLys(Cbz)OMe, R2=-OH; R1=-LeuHis(Bzl)Lys(Cbz)OMe, R2=-OH; R1=-LeuHisOMe, R2=-OMe; R1=-LeuHis(Bzl)Lys(Cbz)OMe, R2=-OMe; R1=-AlaLeuAlaPheAlaCys(Bzl)OMe, R2=-LeuHis(Bzl)Lys(Cbz)OMe; R1=-AlaLeuAlaPheAlaCys(Bzl)OBzl, R2=-LeuHis(Bzl)Lys(Cbz)OMe; R1=-LeuHisAlaLys(Cbz)Cys(Bzl)OBzl, R2=-LeuHis(Bzl)Lys(Cbz)OMe; R1=-LeuHisOMe, R2=-OMe; R1=-GlyProArgGlyGlyOMe, R2=-OH; R1=-ArgProProGlyPheSer(Bzl)PheArgGlyGlyOMe, R2=-OH, или их фармацевтически приемлемых солей в качестве ингибиторов протеолитических ферментов: протеиназы ВИЧ, пепсина, трипсина, химотрипсина, а также в качестве индукторов интерферона и противовирусных средств. Ингибирующая активность заявляемых соединений оценивалась в тестах по их влиянию на скорость гидролиза соответствующих субстратов протеиназой ВИЧ-1 пепсином, химотрипсином и трипсином. Пример 19. Исследование влияния гемина и его производных гемина на функционирование протеиназы ВИЧ-1. cat, Km и Кi2)-Glu-Ala-Nle-OH, H-Lys-Ala-Arg-Val-Nle-Phe(p-NO2)-Glu-Ala-Nle-OH, H-Lys-Ala-Arg-Val-Nle-Phe(p-NO2)-Glu-Ala-Met-OH, H-Ala-Thr-His-Arg-Val-Tyr-Phe(p-NO2)-Val-Arg-Lys-Ala-OH, H-Ala-Thr-His-Arg-Val-Tyr-Phe(p-NO2)-Val-Arg-OH, содержащие в качестве хромофорной метки п-нитрофенилаланин. При гидролизе субстрата в условиях рН 6.2, 37 Пример 20. Исследование влияния гемина и его производных на функционирование пепсина. 2)-Phe-APM в условиях рН 4.0, 37 В экспериментах используют субстрат в концентрации: [S]0 0.15-6.00 мМ в 0.1 М ацетатном буфере при концентрации ингибитора и фермента соответственно [I]0 0.33-8.00 мкМ и [Е]0 8.44 Пример 21. Исследование влияния гемина и его производных на функционирование химотрипсина. Активность химотрипсина определяют, регистрируя уменьшение величины оптической плотности при В экспериментах используют субстрат в концентрации: [S]0 0.133-0.267 мМ при концентрации фермента [Е]0 1.33 Пример 22. Исследование влияния гемина и его производных на функционирование трипсина. Активность трипсина определяют с помощью субстрата p-нитроанилида N-бензоил-DL-аргинина (BAPNA). Спектрофотометрически регистрируют возрастание величины оптической плотности при гидролизе субстрата с образованием p-нитроанилина при В экспериментах используют субстрат в концентрации: [S]0 0.568-1.08 мМ при концентрации фермента [Е]0 8.5 В результате проведенной работы в ряду исследованных соединений обнаружены вещества, обладающие ингибирующей способностью в отношении протеиназы ВИЧ-1 в выбранных условиях (табл. 1), причем значения констант ингибирования составляют 10-6-10-8 М. Показано, что протопорфирин IX и пептид (II) в данных условиях не проявляют ингибирующего действия. Показано также, что заявляемые соединения являются ингибиторами эндогенных протеиназ. Гемин является слабым ингибитором химотрипсина (Кi 0.101М), ингибирующая активность производных гемина на 1-2 выше. Для гемина величина константы ингибирования трипсина i составляет 6.43 Таким образом, заявляемые соединения, соответствующие общей формуле, ингибируют протеиназы, например, пепсин, химотрипсин, трипсин и протеиназу ВИЧ-1 и, следовательно, предлагаемые соединения перспективны в качестве потенциальных лекарственных, в том числе противовирусных средств, что подтверждают результаты дальнейших биологических и биохимических исследований. Пример 23. Влияние гемина и его производных на репродукцию вируса иммунодефицита человека при острой инфекции лимфобластоидных клеток. (фиг. 1). Исследование проводили на культуре лимфобластоидных клеток человека МТ-4. В эксперименте использовали вирус иммунодефицита человека, тип 1, штаммы ВИЧ-1899А и ИВ76 из коллекции Института вирусологии им. Д.И.Ивановского РАМН. Изучали действие предлагаемых соединений на репродукцию вируса иммунодефицита человека в концентрациях: 0.03-30.0 мкМ. Препарат сравнения – азидотимидин (АЗТ) (фирма “Wellcome” Великобритания), в дозах 0.03-30.0 мкМ. Соединения вносили в культуру клеток МТ-4 за 1 час до внесения вируса в дозе 1000 ТЦИД50. Оценку жизнеспособности клеток МТ-4 проводили на 3, 5, 7, 8 дни эксперимента. Основным параметром эффективности действия исследуемых соединений являлась жизнеспособность клеток. где А – число жизнеспособных клеток в опытной культуре; В – число жизнеспособных клеток в инфицированной культуре (контроль вируса); К – число жизнеспособных клеток в контрольной неинфицированной культуре. В предварительных экспериментах было показано, что исследуемые соединения являются малотоксичными и обладают выраженными свойствами подавлять репликацию ВИЧ-1. В результате проведенных экспериментов на примере гемина и некоторых его производных показано, что предлагаемые соединения обладают способностью ингибировать репликацию ВИЧ-1 в культуре клеток МТ-4 при острой инфекции, причем АЗТ в концентрации 1.0 мкМ обеспечивал 75% защиту клеток от ВИЧ-1 штамм 899А, гемин был неэффективен, а защитный эффект пептидных производных гемина достигал 52-86% (табл. 2). При исследовании действия гемина и его производных, в концентрациях 0.5 и 1.0 мкМ при одновременном с заражением внесении их в культуры лимфобластоидных клеток HPBALL ВИЧ-1 (изолят ИВ26) на репликацию ВИЧ в зависимости от времени их применения показано, что предлагаемые соединения одинаково эффективны как на ранних (до 6 часов), так и на поздних стадиях репликации вируса ВИЧ-1. При этом можно отметить, что максимальный антивирусный эффект порфиринопептидов отмечался после введения в культуру клеток через 4 часа, а гемина – через 20 часов. При этом уже через 2 часа после внесения вируса в культуру клеток в цитоплазме чувствительных клеток обнаруживается линейная форма ДНК провируса ВИЧ, а к 6 часам в ядре уже обнаруживаются как линейные, так и циркулярные формы вирусной ДНК. Пример 24. Изучение дозово-зависимого эффекта гемина и его производных на репликацию ВИЧ-1. В серии экспериментов для установления минимально эффективной концентрации были исследованы концентрации 10.0, 1.0, 0.5, 0.1 мкМ. Препаратом сравнения служил гемин, взятый в тех же концентрациях. Наряду с противовирусной активностью исследована токсичность предлагаемых соединений в использованных концентрациях, определяемая по 50% ингибирующей концентрации (ИК 50). Токсичность гемина проявляется уже в концентрации 1 мкМ (деструкция 25% клеток): ИК 50 – 50-20 мкМ. Производные гемина оказались значительно менее токсичными для клеток МТ-4. Так, при концентрации до 10 мкМ токсичность для них практически отсутствует: ИК 50 – 50-30 мкМ. Пример 25. Определение противовирусного эффекта по подавлению синцитиеобразования и по степени защиты от цитодеструктивного действия ВИЧ-1 (штамм BRU). Гемин в концентрации 0.1-1.0 мкМ подавлял образование синцития на 50%, при 10 мкМ – на 75%. В то же время производные гемина в концентрации 0.5 мкМ подавляли синцитиеобразование уже на 75%, а при концентрации 1.0 мкМ фокусов синцитиев в инфицированных клетках не обнаруживалось. Максимальная защита, достигнутая при использовании гемина составила 63.4% при концентрации 10.0 мкМ, а для его производных – 94.3% при той же концентрации. Индекс эффективности (ИЭ 50) для гемина составил 1.0 мкМ (0.95), а для производных гемина – 0.4 мкМ. Химиотерапевтический индекс (ХТИ), или индекс селективности, демонстрирует более высокую активность производных гемина: ХТИ гемина составляет 21, ХТИ порфиринопептидов – 75. Аналогичные исследования были проведены со свежевыделенным изолятом ВИЧ-1 ИВ64, обладающим большей агрессивностью. В случае применения гемина подавление образования синцития наблюдалось лишь при 10 мкМ (75%), в то же время производные гемина подавляли синцитиеобразование уже при концентрации 0.5 мкМ (75%), а при 1.0 мкМ синцитий не обнаруживался. В этом случае индекс эффективности производных гемина составил 0.66 мкМ. Таким образом, полученные результаты, показывают, что заявляемые соединения обладали защитным эффектом, выраженным в разной степени, от цитодеструктивного действия ВИЧ-1 в концентрациях 1-30 мкМ. Причем влияние соединений на сохранение жизнеспособности клеток было сопоставимо с действием препарата сравнения – азидотимидина. Пример 26. Исследование острой токсичности гемина и его производных на модели экспериментальной инфекции, вызванной вирусом энцефаломиокардита мышей (ЕМС). Мышам весом 10 г внутрибрюшинно однократно вводили растворы исследуемых веществ в дозе 1.0, 10.0, 20.0 и 50.0 мг/кг. При дозе 20.0 мг/кг и выше у мышей через 24 часа отмечались признаки токсического действия: вялость, озноб, взъерошенная шерсть. Однако в последующие дни симптомы исчезли, и животные выжили. Доза вещества 10 мг/кг гибели мышей не вызывала. Пример 27. Изучение интерферониндуцирующей активности производных гемина. Исследована возможность индукции интерферона гемином и его производными у мышей. Уровень интерферона определяли в сыворотке крови мышей через 5 и 24 часа после введения испытуемых веществ в дозах 0.1-5.0 мг/кг (фиг. 2). Для определения интерферона сыворотки мышей в двукратном разведении наносили на монослой клеток L-929-перевиваемой линии фибробластов мышей. Через 24 часа инкубации при 37 Установлено, что предлагаемые соединения, введенные внутрибрюшинно, обладают способностью индуцировать синтез “раннего” интерферона у мышей. Через 5 часов после введения уровень интерферона в крови колебался от 80 до 160 ед/мл-1. Полученные данные позволяют предположить, что исследуемые вещества обладают иммуномодулирующими свойствами и в развитии противовирусного эффекта может играть роль стимуляция интерферонообразования.
При исследовании связывания гемина и его производных в присутствии поверхностно-активного вещества – проксанола (сополимер полиэтоксиэтилена с полиоксипропиленом с молекулярной массой 8000 г/ммоль) с основными типами лейкоцитов показано, что гемин за время инкубирования в течение 30 мин вызывал разрушение клеток. При этом наблюдался выход ДНК, в результате чего раствор приобретал вязкую консистенцию, вследствие чего оценить связывание гемина с клетками не представлялось возможным. В отличие от гемина его производные в проксаноле не разрушали клетки, и независимо от разведения (1:50 и 1:100), количество связавшегося порфиринопептида (в пересчете на одну клетку) было одинаковым. Спектральными методами оценивалось влияние гемина и его производных в проксаноле и самого проксанола на целостность клеточной мембраны. Показано, что ни проксанол в концентрации 1%, ни гемин или геминпептид в более низких концентрациях (8 мкмоль) не приводят к заметным нарушениям в проницаемости клеточной мембраны. Настоящее изобретение относится также к фармацевтической композиции, содержащей в качестве активного ингредиента как новые, так и известные соединения общей формулы (I) где R1 и R2 – заместители, которые могут представлять собой аминокислоты, производные аминокислот, пептиды, состоящие из 1-15 аминокислотных остатков, производные пептидов, состоящих из 1-15 аминокислотных остатков, а Y– представляет собой Сl–, СН3СОО–; или их фармацевтически приемлемые соли, обладающей способностью ингибировать протеолитические ферменты, конкретнее протеиназу ВИЧ и пепсин, противовирусным действием, в частности против ВИЧ-инфекции, интерферониндуцирующей активностью. Фармацевтические композиции настоящего изобретения могут быть использованы в виде фармацевтических препаратов (например, в твердой, полутвердой или жидкой формах), содержащих предлагаемые в настоящем изобретении соединения в качестве активных ингредиентов в смеси с органическим или неорганическим носителем или наполнителем, приемлемым для внутримышечного, внутривенного, интраназального, перорального, сублингвального, ингаляционного и интраректального введения. Активный ингредиент может быть включен в фармацевтическую композицию вместе с обычно используемыми нетоксичными, фармацевтически приемлемыми носителями, пригодными для изготовления растворов, таблеток, пилюль, капсул, суппозиториев, эмульсий, суспензий, мазей и любых других лекарственных форм. В качестве носителей могут быть использованы вода, глюкоза, лактоза, аравийская камедь, желатин, крахмал, триксилит магния, тальк, кукурузный крахмал, мочевина и другие носители, пригодные для изготовления твердых, мягких или жидких препаратов. При этом в качестве добавок могут быть использованы стабилизаторы, загустители, красители и отдушки. Активное целевое соединение вводят в фармацевтическую композицию в количестве, достаточном для получения нужного эффекта в зависимости от нозологии, течения и тяжести заболевания. При изготовлении разовой лекарственной формы, количество активного ингредиента, используемого в комбинации с носителем, может варьировать в зависимости от реципиента, подвергающегося лечению, от конкретного способа введения лекарственного средства. Пример 28. Примеры лекарственных форм. А. Желатиновые капсулы. Состав вводимого в капсулу порошка, мг: Соединение, соответствующее общей формуле (I), 1-35 Оксид магния 50 Крахмал 100-200 Указанные выше ингредиенты смешивают и смесь вводят в твердые желатиновые капсулы в количестве 151-285 мг. Б. Таблетированная форма. Таблетированную форму получают, используя приведенные ниже ингредиенты, мг: Соединение, соответствующее общей формуле (I) 1-35 Крахмал картофельный 100 Поливинилпирролидон 10 Магния стеарат 2 Лактоза 48-82 Аэросил 5 Компоненты смешивают и прессуют для образования таблеток весом 200 мг каждая. В. Аэрозольная форма. Состав аэрозольной смеси, рассчитанной на 10 приемов, мг: Соединение, соответствующее общей формуле (I) 10-40 Магния сульфата 150 Лактоза 110-140 Соединение смешивают с наполнителями и помещают в специальное устройство для распыления. Г. Суппозитории. В качестве суппозиторной основы могут быть использованы: основы, не растворимые в воде – масло какао; основы, растворимые в воде или смешиваемые с водой – желатино-глицериновые или полиэтиленоксидные; комбинированные основы – мыльно-глицериновые. Пример состава суппозитория: Соединение, соответствующее общей формуле (I) 1-35 Масло какао – количество, необходимое для получения суппозитория. При необходимости возможно изготовление ректальных, вагинальных и уретральных суппозиториев с соответствующими наполнителями. Д. Мази. В качестве мазевой основы могут быть использованы: углеводородные мазевые основы – вазелин белый и желтый (Vaselinum album, Vaselinum flavum), вазелиновое масло (Oleum Vaselini), мазь белая и жидкая (Unguentum album, Unguentum flavum), а в качестве добавок для придания более плотной консистенции твердый парафин и воск; абсорбтивные мазевые основы – гидрофильный вазелин (Vaselinum hydrophylicum), ланолин (Lanolinum), кольдкрем (Unguentum leniens); мазевые основы, смываемые водой – гидрофильная мазь (Unguentum hydrophylum); водорастворимые мазевые основы – полиэтиленгликолевая мазь (Unguentum Glycolis Polyaethyleni), бентонитовые основы и другие. Пример состава мази, г: Соединение, соответствующее общей формуле (I) 0,1-0,5 Вазелин 10 Мази изготавливают по соответствующей технологии. Е. Раствор для инъекций. В качестве растворителя при приготовлении раствора для инъекций могут быть использованы 0,9% раствор натрия хлорида, дистиллированная вода, раствор новокаина. Производные гемина могут быть использованы в липосомальной форме (с применением, например, летицина) или солюбилизированными в разрешенных кровезаменителях, например проксаноле. Форма выпуска – ампулы, флаконы, шприц-тюбики, “insert”. Состав раствора для инъекций: Соединение, соответствующее общей формуле (I), 1-5 Вода дистиллированная 1-2 Возможно изготовление различных лекарственных форм для инъекций – стерильных растворов, стерильных порошков и таблеток. Производные гемина в свободной форме или в виде фармацевтически приемлемой соли в эффективном количестве в фармацевтически приемлемом носителе, в том числе в липосомальной форме или в виде комплекса с белком плазмы, обладают полезными фармакологическими свойствами, поэтому они могут использоваться в качестве основы фармацевтических препаратов, например, в качестве противовирусных и иммуномодулирующих средств и/или добавки к лекарственным формам пептидных препаратов. Предложен удобный способ получения всех предлагаемых препаратов. Список сокращений Tfa – трифторацетил Cbz – бензилоксикарбонил OBut – трет-бутиловый эфир Вос – трет-бутилоксикарбонил ТЦИД – тканевая цитопатическая доза Формула изобретения
1. Производные гемина или их фармацевтически приемлемые соли-ингибиторы протеолитических ферментов, представляющие собой соединения общей формулы (I) где R1 и R2 – заместители, которые могут представлять собой аминокислоты, производные аминокислот, пептиды, состоящие из 1-15 аминокислотных остатков, производные пептидов, состоящих из 1-15 аминокислотных остатков, а Y– представляет собой Сl–, СН3СОО–; Me представляет собой Fe; за исключением соединений, где Me=Fe3+, Y–=Сl–, R1=-LeuLeuValPheOMe, R2=-OH; R1=-ValPheOMe, R2=-OH; R1=-LeuHisOMe; R2=-OH; R1=-LeuHisAlaOMe, R2=OH; R1=-LeuHisNHC10H20COOMe, R2=-OH; R1=-LeuHisNHC10H20COOH, R2=-OH; R1=-LeuHisNHC10H20COOMe; R2=-LeuHisNHC10H20COOMe; R1=-Lys(Tfa)AlaAlaOMe, R2=-OH; R1=-ValPheOMe, R2=-LeuHisOMe; R1=-LeuLeuValPheOMe, R2=-LeuHisOMe; R1=-LeuLys(Tfa)LeuOMe, R2=-OH; R1=-LeuLys(Tfa)LeuOMe, R2=-LeuHisOMe; R1=-Lys(Tfa)AlaAlaOMe, R2=-AlaHisLys(Cbz)LeuOMe; R1=-GlyOBzl; R2=-GlyOBzl; R1=-HisOMe, R2=-HisOMe; R1=-LeuHisOMe, R2=-LeuHisOMe; R1=-LeuHisLeuGlyCys(Bzl)OBzl, R2=-LeuHisLeuGlyCys(Bzl)OBzl; R1=-LeuHisOMe, R2=-OEt; R1=-LeuHisLeuGlyCys(Bzl)OBzl, R2=-OEt; R1=-Obzl; R2=-OBzl; R1=-OBzl, R2=-OH; R1=-AlaOMe, R2=-OBzl; R1=-HisOMe, R2=-Obzl; R1=-LeuHisOMe, R2=-OBzl; R1=-LeuHisLeuGlyCys(Bzl)OBzl, R2=-OBzl; R1=-LeuHisAlaLys(Cbz)GlyCys(Bzl)OBzl, R2=-OBzl; R1=-LeuHisLys(Cbz)Ome; R2=-OH; R1=-LeuHis(Bzl)Lys(Cbz)OMe, R2=-OH; R1=-LeuHisOMe, R2=-Ome; R1=-LeuHis(Bzl)Lys(Cbz)OMe, R2=-OMe; R1=-AlaLeuAlaPheAlaCys(Bzl)Ome; R2=-LeuHis(Bzl)Lys(Cbz)OMe; R1=-AlaLeuAlaPheAlaCys(Bzl)Obzl; R2=-LeuHis(Bzl)Lys(Cbz)OMe; R1=-LeuHisAlaLys(Cbz)Cys(Bzl)Obzl; R2=-LeuHis(Bzl)Lys(Cbz)OMe; R1=-GlyProArgGlyGlyOMe, R2=-OH; R1=-ArgProProGlyPheSer(Bzl)PheArgGlyGlyOMe, R2=-OH. 2. Производные гемина или их фармацевтически приемлемые соли общей формулы (I) по п.1, где R1=-ValPheOMe, R2=-ValPheOMe (VI); R1=-LeuValPheOMe, R2=-OH (VII); R1=-LeuLeuValPheOMe, R2=-LeuLeuValPheOMe (IX); R1=-GlyProArgOMe, R2=-OH (X); R1=-ArgOMe, R2=-OH (XI); R1=-ArgOMe, R2=-ArgОМе (XII); R1=-LeuValPheOH, R2=-OH (XIII); R1=-ValSerGlnAsnLeuOH, R2=-OH (XIV); R1=-ArgArgTrpHisArgLeuLysGlu(OMe)OH, R2=-OH (XV); R1=-лизиновый кор, R2=-OH (XVI); R1=-GluAspLeuOH, R2=-OH (XVII); Y–=Cl–; Me=Fe3+. 3. Производные гемина или их фармацевтически приемлемые соли по пп.1 и 2, являющиеся ингибиторами протеиназы ВИЧ. 4. Производные гемина или их фармацевтически приемлемые соли по пп.1 и 2, являющиеся ингибиторами пепсина. 5. Производные гемина или их фармацевтически приемлемые соли по пп.1 и 2, являющиеся ингибиторами химотрипсина. 6. Производные гемина или их фармацевтически приемлемые соли по пп.1 и 2, обладающие противовирусной активностью, в том числе против ВИЧ-инфекции. 7. Производные гемина или их фармацевтически приемлемые соли по пп.1 и 2, обладающие интерферониндуцирующими свойствами. 8. Способ получения производных гемина или их фармацевтически приемлемых солей по п.1, представляющих собой соединения общей формулы (I) где R1 и R2 – заместители, которые могут представлять собой аминокислоты, производные аминокислот, пептиды, состоящие из 1-15 аминокислотных остатков, производные пептидов, состоящих из 1-15 аминокислотных остатков, а Y– представляет собой Сl–, СН3СОО–; Me представляет собой Fe, твердофазным методом, заключающимся в том, что N-защищенную стартовую аминокислоту присоединяют к полимеру с хлортритилхлоридной якорной группой для твердофазного синтеза пептидов, деблокирование и присоединение по N-концу аминокислот, защищенных по боковым функциональным группам защитами тритильного типа, повторяют до достижения пептидилполимером необходимой длины, затем полученный продукт деблокируют и вводят во взаимодействие с активированным производным протогемина IX в ДМФА, геминпептид одновременно деблокируют и отщепляют от полимера в мягких кислых условиях и вводят противоион Сl– или СН3СОО–. 9. Способ получения производных гемина или их фармацевтически приемлемых солей по п.8, где R1=-LeuValPheOH, R2=-OH (XIII); R1=-ValSerGlnAsnLeuOH, R2=-OH (XIV); R1=-ArgArgTrpHisArgLeuLysGlu(OMe)OH, R2=-OH (XV); R1=-лизиновый кор, R2=-OH (XVI); Y–=Cl–; Me=Fe3+, а активированное производное протогемина IX представляет собой N-оксисукцинимидный эфир. 10. Способ получения производных гемина или их фармацевтически приемлемых солей общей формулы (I) по п.1, где R1=R2 или R1 в том числе R1=-ValPheOMe, R2=-OH (V); R1=-ValPheOMe, R2=-ValPheOMe (VI); R1=-LeuValPheOMe, R2=-OH (VII); R1=-LeuLeuValPheOMe, R2=-OH (VIII); R1=-LeuLeuValPheOMe, R2=-LeuLeuValPheOMe (IX); Y–=Сl–, СН3СОО–; Me=Fe3+, отличающийся тем, что конденсацию производного гемина проводят с пептидом требуемой длины, получаемым методом смешанных ангидридов с использованием алкилхлорформиата в присутствии N-метилморфолина. 11. Фармацевтическая композиция, обладающая способностью ингибировать протеолитические ферменты, содержащая в качестве активного ингредиента соединение общей формулы (I) где R1 и R2 – заместители, которые могут представлять собой аминокислоты, производные аминокислот, пептиды, состоящие из 1-15 аминокислотных остатков, производные пептидов, состоящих из 1-15 аминокислотных остатков, а Y– представляет собой Сl–, СН3СОО–; Me представляет собой Fe, в эффективном количестве и в фармацевтически приемлемом носителе, в том числе в липосомальной форме, а также в виде фармацевтически приемлемой соли. 12. Фармацевтическая композиция по п.11, обладающая способностью ингибировать протеолитические ферменты: ВИЧ-протеиназу, пепсин, трипсин, химотрипсин, обладающая противовирусными свойствами и интерферониндуцирующей активностью. 13. Применение производных гемина общей формулы (I) где R1=R2, или R1 Y– представляет собой Сl–; Me представляет собой Fe3+; а также R1=-LeuLeuValPheOMe, R2=-OH; R1=-ValPheOMe, R2=-OH; R1=-LeuHisOMe, R2=-OH; R1=-LeuHisAlaOMe, R2=-OH; R1=-LeuHisNHC10H20COOMe, R2=-OH; R1=-LeuHisNHC10H20COOH, R2=-OH; R1=-LeuHisNHC10H20COOMe, R2=-LeuHisNHC10H20COOMe; R1=-Lys(Tfa)AlaAlaOMe, R2=-OH; R1=-ValPheOMe, R2=-LeuHisOMe; R1=-LeuLeuValPheOMe, R2=-LeuHisOMe, R1=-LeuLys(Tfa)LeuOMe, R2=-OH; R1=-LeuLys(Tfa)LeuOMe, R2=-LeuHisOMe, R1=-Lys(Tfa)AlaAlaOMe, R2=-AlaHisLys(Cbz)LeuOMe; R1=-GlyOBzl, R2=-GlyOBzl; R1=-HisOMe, R2=-HisOMe; R1=-LeuHisOMe, R2=-LeuHisOMe; R1=-LeuHisLeuGlyCys(Bzl)OBzl, R2=-LeuHisLeuGlyCys(Bzl)OBzl; R1=-LeuHisOMe, R2=-OEt; R1=-LeuHisLeuGlyCys(Bzl)OBzl, R2=-OEt; R1=-OBzl, R2=-OBzl; R1=-OBzl, R2=-OH; R1=-AlaOMe, R2=-OBzl; R1=-HisOMe, R2=-OBzl, R1=-LeuHisOMe, R2=-OBzl; R1=-LeuHisLeuGlyCys(Bzl)OBzl, R2=-OBzl; R1=-LeuHisAlaLys(Cbz)GlyCys(Bzl)OBzl, R2=-OBzl; R1=-LeuHisLys(Cbz)OMe, R2=-OH; R1=-LeuHis(Bzl)Lys(Cbz)OMe, R2=-OH; R1=-LeuHisOMe, R2=-OMe, R1=-LeuHis(Bzl)Lys(Cbz)OMe, R2=-OMe; R1=-AlaLeuAlaPheAlaCys(Bzl)OMe, R2=-LeuHis(Bzl)Lys(Cbz)OMe; R1=-AlaLeuAlaPheAlaCys(Bzl)OBzl, R2=-LeuHis(Bzl)Lys(Cbz)OMe; R1=-LeuHisAlaLys(Cbz)Cys(Bzl)OBzl, R2=-LeuHis(Bzl)Lys(Cbz)OMe; R1=-GlyProArgGlyGlyOMe, R2=-OH; R1=-ArgProProGlyPheSer(Bzl)PheArgGlyGlyOMe, R2=-OH, или их фармацевтически приемлемых солей в качестве ингибиторов протеолитических ферментов: протеиназы ВИЧ, пепсина, трипсина химотрипсина. 14. Применение производных гемина общей формулы (I) или их фармацевтически приемлемых солей по п.12 в качестве индукторов интерферона и противовирусных средств. РИСУНКИ
|
||||||||||||||||||||||||||
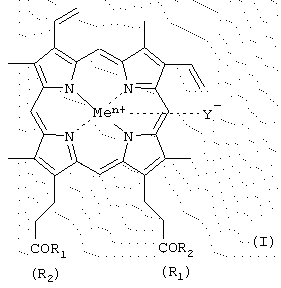
 -карбоксильная группа аминокислот или пептидов и боковые группы аминокислот или пептидов могут быть модифицированы, причем возможно, что R1=R2 или R1
-карбоксильная группа аминокислот или пептидов и боковые группы аминокислот или пептидов могут быть модифицированы, причем возможно, что R1=R2 или R1 R2=OH; карбоксильная группа порфирина может быть модифицирована метиловым или другим C2-C8-эфиром или физиологически приемлемой солью; Y– представляет собой Cl–, СН3СОО–; Me представляет собой Fe, за исключением соединений, где
R2=OH; карбоксильная группа порфирина может быть модифицирована метиловым или другим C2-C8-эфиром или физиологически приемлемой солью; Y– представляет собой Cl–, СН3СОО–; Me представляет собой Fe, за исключением соединений, где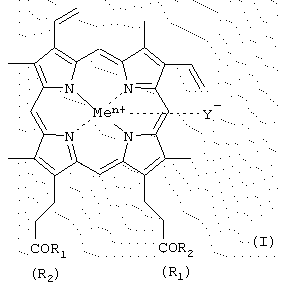
 С.
С. 2; 2) обработка 20% раствором пиперидина в DMF 3 мин
2; 2) обработка 20% раствором пиперидина в DMF 3 мин  H-Phe-OMe, оставляют на 15 мин при 0
H-Phe-OMe, оставляют на 15 мин при 0 , см-1, вазелиновое масло: 3260 (NH-), 1745 (СО сл. эф.), 1632 (амид I), 1515 (амид II). Спектр 1Н-ЯМР (CDCl3),
, см-1, вазелиновое масло: 3260 (NH-), 1745 (СО сл. эф.), 1632 (амид I), 1515 (амид II). Спектр 1Н-ЯМР (CDCl3),  , м. д.: 1.4 (с, 9Н, трет. Вu), 1.6 (с, 6Н, изо-Pr.), 2.05 (к, 1Н, -СН-СОО), 3.1 (д, 2Н, -CH2-), 3.7 (с, 3Н, -O-СН3), 4.0 (т, 4Н, N-CH-CON), 4.9 (д, 4Н, 4NH), 6.6 (д, 6Н, (СН3)2-СН-), 7.1-7.3 (м, 5Н, Ph). Масс-спектр, m/z: [M]+ 605.6.
, м. д.: 1.4 (с, 9Н, трет. Вu), 1.6 (с, 6Н, изо-Pr.), 2.05 (к, 1Н, -СН-СОО), 3.1 (д, 2Н, -CH2-), 3.7 (с, 3Н, -O-СН3), 4.0 (т, 4Н, N-CH-CON), 4.9 (д, 4Н, 4NH), 6.6 (д, 6Н, (СН3)2-СН-), 7.1-7.3 (м, 5Н, Ph). Масс-спектр, m/z: [M]+ 605.6. max, нм, СНСl3, (
max, нм, СНСl3, (
 (Швеция), элюируя целевое вещество смесью хлороформ-метанол (1:1). Выход 0.08 г (72.0%), Rf 0.63 (4). Электронный спектр,
(Швеция), элюируя целевое вещество смесью хлороформ-метанол (1:1). Выход 0.08 г (72.0%), Rf 0.63 (4). Электронный спектр,