Патент на изобретение №2333247
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) КОРИНЕФОРМНАЯ БАКТЕРИЯ, ПРОДУЦЕНТ L-АМИНОКИСЛОТЫ И СПОСОБ ПОЛУЧЕНИЯ L-АМИНОКИСЛОТЫ
(57) Реферат:
Изобретение относится к биотехнологии и представляет собой способ получения L-аминокислоты с использованием коринеформной бактерии, который включает культивирование коринеформной бактерии, обладающей способностью продуцировать L-аминокислоту в среде, приводящее к накоплению L-аминокислоты в среде или клетках бактерии, и отбор L- аминокислоты из среды или клеток бактерии. При этом коринеформная бактерия модифицирована таким образом, что в нее введен ген метанолдегидрогеназы, гены гексулозофосфатсинтазы и фосфогексулоизомеразы и также ген, кодирующий фактор, усиливающий активность метанолдегидрогеназы. Это придает бактерии способность утилизировать метанол. Изобретение позволяет получать L-аминокислоты с высокой степенью эффективности. 2 н. и 5 з.п. ф-лы, 4 табл.
Область техники, к которой относится изобретение Настоящее изобретение относится к промышленной микробиологической ферментации. Более конкретно, настоящее изобретение относится к методике придания способности утилизировать метанол микроорганизму, не имеющему наследственно такой способности, или усилению такой способности у микроорганизма, обладающего такой способностью на низком уровне, и к способу получения целевого вещества путем утилизации метанола при использовании микроорганизма, полученного посредством такой методики, указанной выше. Вещества, получаемые по настоящему изобретению, включают L-аминокислоты, нуклеиновые кислоты, антибиотики, витамины, ростовые факторы, физиологически активные вещества и т.д., которые традиционно получают с помощью микроорганизмов. Уровень техники К настоящему времени большинство сырьевых материалов, используемых для получения полезных веществ путем микробной ферментации, представляют собой сахара, полученные из сельскохозяйственных продуктов. Поскольку стоимость сахаров, получаемых из сельскохозяйственных продуктов, имеет тенденцию повышаться, желательно иметь недорогой материал хорошего качества, такой как альтернативный сырьевой материал для ферментации. Метанол легко растворяется в воде, является недорогим продуктом и может быть получен с достаточно высокой степенью чистоты. Кроме того, он относительно легко может быть получен из метана, который является основным компонентом природного газа. В этой связи он предпочтителен в качестве сырья для получения нужных веществ. Если метанол использовать в качестве сырья для микробной ферментации, может быть снижена не только стоимость основного сырьевого материала, но также стоимость очистки продуктов, выделяемых из растворов, образуемых в ходе ферментации, а также может быть упрощена процедура утилизации отходов в растворе. Таким образом, стоимость всего процесса получения может быть снижена. Способы получения веществ, в особенности аминокислот, с использованием метанола в качестве сырья, утилизируемого микроорганизмами, известны и включают способ с использованием микроорганизма из рода Achromobacter или Pseudomonas (Публикация патента Японии (Kokoku) № 45-25273), способ с использованием микроорганизма из рода Protaminobacter или Methanomonas (публикация выложенной заявки на патент Японии (Kokai) № 50-25790), способ с использованием микроорганизма из рода Methylobacillus (публикация выложенной заявки на патент Японии № 4-91793), способ с использованием метилотрофной бактерии, принадлежащей к роду Bacillus (публикация выложенной заявки на патент Японии № 3-505284, патент США № 6083728) и т.д. Однако известным бактериальным штаммам не свойственна высокая приобретенная продуктивность в отношении аминокислот, необходимая для практического применения бактерий. Тем временем, способ использования микроорганизмов из рода Brevibacterium, Corynebacterium, Bacillus или Escherichia представляет собой основное направление производства аминокислот из глюкозы (см. “Amino Acid Fermentation”, Ed. By H. Aida et al., the Japan Scientific Societies Press [Gakkai Shuppan Center], 1-е издание, опубликованное 30 мая 1986). Указанные бактерии, продуцирующие аминокислоты, представляют собой ценные бактериальные штаммы, создаваемые путем введения различных мутаций, в результате которых достигается максимальная продуктивность в отношении аминокислоты, проведением дополнительной работы по селекции с практической целью. Однако для этого требуется длительное время. И, кроме того, такие используемые в промышленности штаммы не могут утилизировать метанол. Краткое описание сущности изобретения Целью настоящего изобретения является получение новой коринеформной бактерии, которая обладает способностью продуцировать в ходе ферментации продукт, такой как аминокислота, при использовании метанола в качестве сырьевого материала для ферментации, путем придания способности утилизировать метанол коринеформной бактерии, которая имеет наследственную способность утилизировать сахар, но не может утилизировать метанол, или путем повышения такой способности у бактерии, имеющей уже такую способность, но на низком уровне. Другой целью настоящего изобретения является разработка способа получения целевого вещества из метанола и использование такой бактерии. Целью настоящего изобретения является разработка способа получения целевого вещества с использованием коринеформной бактерии, включающего: (А) культивирование коринеформной бактерии, имеющей способность продуцировать целевое вещество в среде, приводящее к накоплению целевого вещества в среде или клетках бактерии, и (В) отбор целевого вещества из среды или клеток бактерии, отличающегося тем, что в коринеформную бактерию вводят ген метанолдегидрогеназы, ген гексулозофосфатсинтазы и ген фосфогексулоизомеразы и указанную бактерию модифицируют таким образом, что она приобретает способность утилизировать метанол, когда среда содержит метанол в качестве источника углерода. Еще одной целью настоящего изобретения является разработка указанного выше способа, при котором в бактерию также вводят ген, кодирующий фактор, усиливающий активность метанолдегидрогеназы. Еще одной целью настоящего изобретения является разработка указанного выше способа, в рамках которого целевым веществом является L-аминокислота. Еще одной целью настоящего изобретения является разработка указанного выше способа, в рамках которого указанная L-аминокислота представляет собой L-лизин. Еще одной целью настоящего изобретения является разработка указанного выше способа, в рамках которого указанная бактерия принадлежит к роду Corynebacterium. Еще одной целью настоящего изобретения является разработка указанного выше способа, в рамках которого указанная коринеформная бактерия представляет собой Corynebacterium glutamicum. Еще одной целью настоящего изобретения является получение коринеформной бактерии, в которую введен ген метанолдегидрогеназы, ген гексулозофосфатсинтазы и ген фосфогексулоизомеразы и которая модифицирована таким образом, что приобретает способность утилизировать метанол. Еще одной целью настоящего изобретения является получение указанной выше коринеформной бактерии, в которую введен ген, кодирующий фактор, усиливающий активность метанолдегидрогеназы. Еще одной целью настоящего изобретения является получение указанной выше коринеформной бактерии, которая принадлежит к роду Corynebacterium. И еще одной целью настоящего изобретения является получение указанной выше коринеформной бактерии, которая представляет собой Corynebacterium glutamicum. В соответствии с настоящим изобретением способность утилизировать метанол может быть придана коринеформной бактерии, которая в природе не может утилизировать метанол, и таким образом может быть получен микроорганизм, способный утилизировать недорогой метанол в качестве источника углерода или источника энергии для коринеформной бактерии. Кроме того, при использовании полученного микроорганизма могут быть получены различные продукты ферментации из метанола, добавляемого в среду. ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ Авторы настоящего изобретения провели скрупулезное исследование, прежде чем достигли указанных выше целей. В результате ими было обнаружено, что при введении в коринеформную бактерию генов, кодирующих гексулозофосфатсинтазу и фосфогексулоизомеразу, а также метанолдегидрогеназу, для экспрессии указанных генов в бактерии, такой бактерии может быть придана способность утилизировать метанол или уже имеющаяся у бактерии такая способность может быть усилена, что составило сущность настоящего изобретения. Ниже приведено более подробное описание настоящего изобретения. Коринеформная бактерия по настоящему изобретению представляет собой бактерию, которая содержит ген, кодирующий метанолдегидрогеназу, введенный в нее вместе с другими генами, кодирующими гексулозофосфатсинтазу и фосфогексулоизомеразу, и которая модифицирована таким образом, что приобретает способность утилизировать метанол, или такая способность у нее повышается. Микроорганизм, который утилизирует метанол, содержит метанолоксидазу (например, метанолдегидрогеназу), и она способна к диссимиляции или ассимиляции формальдегида, получаемого при окислении метанола в условиях строгой метаболической регуляции. Это связано с тем, что формальдегид очень токсичен для организмов, и в этой связи клетки должны очень быстро его утилизировать в качестве источника углерода или источника энергии или выделять его путем детоксикации. С другой стороны, если желательно придать способность утилизировать метанол микроорганизму, который не может использовать метанол, то безусловно необходимо вводить метанолоксидазу. Однако способы соответствующего удаления формальдегида, продуцируемого за счет экспрессии активности метанолоксидазы, чрезвычайно специфичны, и в этой связи считается, что обычным микроорганизмам невозможно придать способность утилизировать метанол. Однако авторы настоящего изобретения обнаружили, что способность утилизировать метанол может быть придана даже микроорганизму, который генетически не способен его утилизировать, в частности коринеформной бактерии, если в клетках микроорганизма будет содержаться фермент, обладающий способностью окислять метанол, а также если в микроорганизм будут одновременно введены гены, кодирующие гексулозофосфатсинтазу и фосфогексулоизомеразу, для экспрессии указанных генов. Коринеформная бактерия по настоящему изобретению конкретно не ограничивается, поскольку указанные выше свойства могут быть приданы любой такой бактерии. Коринеформные бактерии включают те бактерии, которые по ранней классификации были отнесены к роду Brevibacterium, но в настоящее время объединены в род Corynebacterium (Int. J. Syst. Bacteriol., 41, 255 (1981)), и включают бактерии, принадлежащие к роду Brevibacterium, которые являются близкородственными к роду Corynebacterium. Ниже приведены примеры таких коринеформных бактерий. Corynebacterium acetoacidophilum Corynebacterium acetoglutamicum Corynebacterium alkanolyticum Corynebacterium callunae Corynebacterium glutamicum Corynebacterium lilium (Corynebacterium glutamicum) Corynebacterium melassecolaCorynebacterium thermoaminogenes Corynebacterium herculis Brevibacterium divaricatum (Corynebacterium glutamicum) Brevibacterium flavum (Corynebacterium glutamicum) Brevibacterium immariophilum Brevibacterium lactofermentum (Corynebacterium glutamicum) Brevibacterium roseum Brevibacterium saccharolyticum Brevibacterium thiogenitalis Brevibacterium album Brevibacterium cerinum Microbacterium ammoniaphilum Конкретно, примеры такой бактерии включают Corynebacterium acetoacidophilum AJ12318 (FERM BP-1172, см. патент США № 5188949) и т.п. как продуцент L-треонина; Brevibacterium lactofermentum AJ12435 (FERM BP-2294, см. патент США № 5304476), Brevibacterium lactofermentum AJ3990 (ATCC 31269 см. патент США № 4066501) и AJ110135, описанный далее, и т.п. как продуцент L-лизина; Brevibacterium lactofermentum AJ12821 (FERM BP-4172, публикация выложенной заявки на патент Японии № 5-26811, публикация выложенной заявки на патент Франции № 2701489), Brevibacterium lactofermentum AJ12475 (FERM BP-2922, см. патент США № 5272067), Brevibacterium lactofermentum AJ13029 (FERM BP-5189, см. международную патентную публикацию JP95/01586) и т.п. как продуцент L-глютаминовой кислоты; Brevibacterium lactofermentum AJ3718 (FERM P-2516, см. патент США № 3970519) и т.п. как продуцент L-лейцина; Brevibacterium flavum AJ12149 (FERM BP-759, см. патент США № 4656135) и т.п. как продуцент L-изолейцина; Brevibacterium lactofermentum AJ12341 (FERM BP-1763, см. патент США № 5188948) и т.п. как продуцент L-валина; Brevibacterium lactofermentum AJ12637 (FERM BP-4160, см. публикацию выложенной заявки на патент Франции № 2686898) и т.п. как продуцент L-фенилаланина. В результате тщательных исследований авторы настоящего изобретения смогли получить достаточную активность метанолдегидрогеназы и в то же время усилить функцию ассимиляции формальдегида, продуцируемого в ходе ферментативной реакции, как принципиального условия придания способности утилизировать метанол. Далее авторы настоящего изобретения пришли к пониманию, что усиление ферментативной активности гексулозофосфатсинтазы (ГФС) и фосфогексулоизомеразы (ФГИ), которые являются ключевыми ферментами рибулозомонофосфатного пути, будет эффективным для эффективной ассимиляции формальдегида. Таким образом, авторами было показано, что способность утилизировать метанол может иметь коринеформная бактерия, которая генетически не способна утилизировать метанол, путем введения в коринеформную бактерию генов, кодирующих ГФС и ФГИ вместе с геном метанолдегидрогеназы. Метанолдегидрогеназа (МДГ), используемая по настоящему изобретению, представляет собой фермент, обладающий ферментативной активностью, способной окислять метанол, превращая его в формальдегид. Примером МДГ, которая может использоваться по настоящему изобретению, является, без ограничения, тип МДГ, зависимый от пиррохинолинхинона (ПХХ), который, в основном, встречается в грамотрицательных бактериях. Конкретно он может включать МДГ из Methylobacterium extorquens, штамм AM1 (Biochim. Biophys. Acta, 1119:97-106 (1992)) и т.п. Кроме того, настоящее изобретение включает встречающийся в грамположительных бактериях зависимый от НАД (никотинамидадениндинуклеотида) тип МДГ, конкретно МДГ из Bacillus methanoliocus (J. Bacteriol., 174:5346-5353 (1992)), алкогольдегидрогеназу (АДГ), полученную из Bacillus stearothermophilus, штамм DSM 2334 (Biochem. J., 252:661-666). Кроме того, рассматривается также АДГ из печени быка (Biochem. J., 100:34-46 (1996)) и печени человека (Arch. Toxicol., 72:604-607 (1998)). Кроме того, может быть также создан и далее использован мутантный тип алкогольдегидрогеназы, которая действует на метанол, также путем введения мутации в ген алкогольдегидрогеназы, которая генетически не действует на метанол, для модификации субстратной специфичности. Однако в качестве МДГ, которая может соответствующим образом использоваться в настоящем изобретении, рассматривается МДГ, полученная, например, из Bacillus brevis NCIMB No. 12524, которая представляет собой метанолассимилирующую бактерию, принадлежащую к роду Bacillus. Ген, кодирующий МДГ (mdh), может быть получен из микроорганизма, который продуцирует МДГ, путем обычных методов клонирования. Так, например, ген МДГ может быть получен посредством ПЦР (полимеразно-цепьевой реакции) с использованием хромосомной ДНК из Bacillus brevis, штамм S1 (NCIMB 12524) в качестве матрицы и олигонуклеотидов, имеющих нуклеотидную последовательность, показанную в SEQ ID NO:1 и 2, в качестве праймеров. Способы получения библиотеки геномной ДНК, используемой для клонирования генов, гибридизации, ПЦР, получения плазмидной ДНК, расщепления и лигирования ДНК, трансформации и т.п., описаны в руководстве Sambrook, J., Fritsch, E.F., Maniatis, T., Molecular Cloning, Cold Spring Harbor Laboratory Press, 1.21 (1989). При этом функционирует ли ген МДГ в коринеформной бактерии, в которую вводят данный ген, можно подтвердить путем измерения активности МДГ в бактериальном лизате. Активность МДГ может быть вычислена, например, по методу определения уровня восстановления НАД+ (никотинамидадениндинуклеотида), сопровождающего окисление метанола в формальдегид, путем измерения поглощения при длине волны 340 нм. Конкретные примеры mdh гена, используемого по настоящему изобретению, включают, не ограничиваясь, ген mdh из Bacillus brevis штамм S1. Ген mdh из Bacillus methanolicus, штамм C1 (NCIMB 13114, Eur. J. Biochem., 244:426-433 (1997)) был зарегистрирован в GenBank под номером М65004 (код для входа BACMDH). Кроме того, имеется информация о существовании факторов, повышающих активность метанолдегидрогеназы (Amd: активатор метанолдегидрогеназы), таких как активатор метанолдегидрогеназы из Bacillus methanolicus, штамм C1 (Eur. J. Biochem., 244:426-433 (1997)) и продукт YqkG гена из Bacillus subtilis, штамм 168 (публикация выложенной заявки на патент Японии № 2000-69976). Указанные факторы являются эффективным средством повышения активности МДГ. Активность МДГ в бактериальных клетках может быть повышена за счет введения ДНК, кодирующей указанные факторы МДГ (ген amd), в коринеформную бактерию, имеющую ген МДГ. Ген, кодирующий активатор метанолдегидрогеназы (amd), такой как ген YqkG, может быть получен из хромосомной ДНК Bacillus subtilis, такой как Bacillus subtilis, штамм 168, путем ПЦР с использованием хромосомной ДНК в качестве матрицы и праймеров, имеющих нуклеотидную последовательность, показанную в SEQ ID NO: 11 и 12 в перечне последовательностей. В качестве конкретного примера гена yqkG, используемого по настоящему изобретению, рассматривается ген YqkG из Bacillus subtilis, штамм 168. Нуклеотидная последовательность и аминокислотная последовательность, кодируемая указанным геном, приведены в SEQ ID NO: 15 и 16. В настоящем описании приводятся способы экспрессии активности ГФС и ФГИ в бактерии. Для экспрессии активностей ГФС и ФГИ в целевой коринеформной бактерии ген, кодирующий ГФС (hps) или ФГИ (phi), может быть лигирован с вектором, который функционирует в целевой бактерии, предпочтительно с вектором мультикопийного типа, с получением рекомбинантной ДНК и далее использован для трансформации целевой бактерии. Число копий гена hps или гена phi в клетке трансформанта при этом повышается, и в результате повышается каждая из указанных ферментативных активностей. Ген hps или ген phi может быть получен из микроорганизма, который образует ГФС и ФГИ, с использованием обычных методов клонирования, аналогичных таковым, применяемым при работе с геном МДГ. В качестве микроорганизма, который образует ГФС, известны Methylomonas capsulatus (J.R.Quayle, Methods in Enzymology, 188, p. 314, 1990), Methylomonas, штамм М15 (Methods in Enzymology, 188, p. 319, 1990), Methylomonas aminofaciens, штамм 77а (Biochim. Biophys. Acta., 523, p. 236 1978), Mycobacterium gastri, штамм МВ19 (Methods in Enzymology,188, p. 393, 1990), Acetobacter methanolicus, штамм MB58 (Methods in Enzymology, 188, p. 401, 1990) и т.п. Далее, в качестве микроорганизма, который образует ФГИ, известны Methylomonas aminofaciens, штамм 77а (Agric. Biol. Chem., 41(7), p. 1133, 1977), Mycobacterium gastri (публикация выложенной заявки на патент Японии № 11-127869), которая представляет собой грамположительную факультативную метанол-ассимилирующую бактерию, и т.п. Далее, имеется сообщение о наличии гена hps и гена phi в Bacillus subtilis (J. Bacteriol., 181:7154-7160 (1999)). Кроме того, имеется сообщение о том, что в Bacillus brevis, штамм S1, который представляет собой метанол-ассимилирующую бактерию, принадлежащую к роду Bacillus, ген hps и ген phi существуют в виде тандема на хромосомной ДНК (Annual Meeting of the Society for Fermentation and Bioengineering Japan, Lecture Abstracts, p. 113 (2000); FEMS Microbiology Letters, 214, 189-193, 2002). Фрагмент ДНК, содержащий гены hps и phi, может быть получен путем ПЦР с использованной хромосомной ДНК в качестве матрицы и олигонуклеотидов, имеющих нуклеотидную последовательность, показанную в SEQ ID NO: 13 и 14, в качестве праймеров. Конкретный пример гена hps и гена phi, используемых по настоящему изобретению, включают ген hps и ген phi из Bacillus subtilis, штамм 168, и ген hps и ген phi из Bacillus brevis, штамм S1. Нуклеотидная последовательность фрагмента ДНК, включающего гены hps и phi из Bacillus brevis, штамм S1, показана в SEQ ID NO: 17. Аминокислотная последовательность, кодируемая данными генами, показана в SEQ ID NO: 18 и 19 соответственно. Bacillus methanolicus, штамм РВ1 (NCIMB 13113), и Bacillus brevis, штамм S1 (NCIMB 12524), могут быть получены из Национальной Коллекции промышленных и морских бактерий (National Collection of Industrial and Marine Bacteria) (Адрес: NCIMB Lts. Torry Research Station135, Abbey Road, Aberdeen AB9 8DG, Великобритания). Активность ГФС может быть измерена по способу, описанному в руководстве по энзимологии (Methods in Enzymology, 188, p. 397-401, 1990). Активность ФГИ может быть также измерена по другому способу, также описанному в литературе (Journal of Bacteriology, 181, p. 7154-7160 (1999)). Амплификация активности ГФС или ФГИ может быть достигнута путем введения множественных копий гена hps или гена phi в хромосомную ДНК целевой коринеформной бактерии. Для введения множественных копий гена hps или гена phi в хромосомную ДНК целевой коринеформной бактерии проводят гомологичную рекомбинацию с использованием в качестве мишени последовательности, множественные копии которой имеются на хромосомной ДНК. В качестве последовательностей, множественные копии которых имеются на хромосомной ДНК, могут использоваться повторные последовательности ДНК или инвертированные повторы, имеющиеся на конце перемещаемого элемента. Кроме того, как описано в публикации выложенной заявки на патент Японии № 2-109985, можно также включить ген hps или ген phi в транспозон, который затем подвергают трасфекции, с целью введения множественных копий генов в хромосомную ДНК. С использованием любого из указанных способов может быть достигнуто повышение активности ГФС или ФГИ как результат повышения числа копий гена hps или гена phi в трансформированном штамме. Помимо указанного выше способа амплификации гена, повышение активности ГФС или ФГИ может быть также достигнуто путем замены регуляторной последовательности экспрессии, такой как промотор гена hps или гена phi, более сильным промотором (см. публикацию выложенной заявки на патент Японии № 1-215280). Примеры сильных промоторов включают lac промотор, trp промотор, trc промотор, tac промотор, РR промотор и PL промотор фага ламбда, tet промотор, amyE промотор, veg промотор и т.д. Замена указанных промоторов повышает экспрессию гена hps или гена phi, и таким образом повышается активность ГФС или ФГИ. Усиление регуляторно экспрессируемой последовательности экспрессии может быть объединено с повышением числа копий ГФС или ФГИ. Гены mdh, hps, phi и amd, используемые по настоящему изобретению, не ограничиваются генами дикого типа, и настоящее изобретение также относится к мутантному или искусственно модифицированному гену, кодирующему генный продукт, включая замещение, делецию, инсерцию, добавление или инверсию одной или нескольких аминокислот в одном или более сайтах, с одним условием, чтобы функция кодируемого МДГ, ГФС, ФГИ или Amd белка не снижалась. Хотя указанное здесь число “несколько” аминокислот различается в зависимости от положения или типа аминокислотных остатков в трехмерной структуре белка, их число может конкретно составлять от 2 до 20, предпочтительно от 2 до 10 и более предпочтительно от 2 до 5. Кроме того, в качестве ДНК, кодирующей белок, по существу идентичный белку МДГ, настоящее изобретение относится к ДНК, гибридизируемой с последовательностью, зарегистрированной в GenBank под номером М65004 (код входа BACMDH), или с зондом, который получают из нуклеотидной последовательности в жестких условиях и который кодирует белок, имеющий активность, аналогичную активности МДГ. В качестве ДНК, кодирующей белок, по существу идентичный указанному выше белку Amd, настоящее изобретение относится к ДНК, гибридизуемой с нуклеотидной последовательностью SEQ ID NO: 15, включающей от 1 до 555 нуклеотидов, или с зондом, который может быть получен из нуклеотидной последовательности в жестких условиях и который кодирует белок, имеющий активность, аналогичную активности Amd. В качестве ДНК, кодирующей белок, по существу идентичный белку ГФС, настоящее изобретение относится к ДНК, гибридизуемой с нуклеотидной последовательностью SEQ ID NOS: 17, включающей от 508 до 1140 нуклеотидов, или с зондом, который может быть получен из нуклеотидной последовательности в жестких условиях и который кодирует белок, имеющий активность, аналогичную активности ГФС. Далее в качестве ДНК, кодирующей белок, по существу идентичный белку ФГИ, настоящее изобретение относится к ДНК, гибридизуемой с нуклеотидной последовательностью SEQ ID NO: 17, включающей от 1149 до 1700 нуклеотидов, или с зондом, который может быть получен из нуклеотидной последовательности в жестких условиях и который кодирует белок, имеющий активность, аналогичную активности ФГИ. Термин “в жестких условиях” означает условия, при которых образуется так называемый специфический гибрид, а неспецифический гибрид не образуется. Трудно четко определить такие условия с помощью цифровых значений. Однако жесткие условия включают условия, при которых ДНК, имеющие высокую гомологию, например ДНК, имеющие гомологию 50% или более, предпочтительно 80% или более, более предпочтительно 90% или более и наиболее предпочтительно 95% и более, гибридизуются друг с другом, но ДНК, имеющие гомологию ниже, чем указанные выше, не гибридизуются друг с другом. Альтернативно, жесткие условия включают условия, при которых ДНК гибридизуются друг с другом при концентрации соли, соответствующей типичным условиям промывки в случае гибридизации по Саузерну, то есть примерно 1хSSC, 0,1% ДСН, предпочтительно 0,1хSSC, 0,1% ДСН при 60°С. Для введения различных генов в коринеформную бактерию, которые могут быть получены по указанному выше способу, может быть использован, например, способ обработки реципиентных клеток хлоридом кальция с целью повышения их проницаемости для ДНК, который описан для Escherichia coli K-12 (Mandel, M. and Higa, A., J.Mol. Biol., 53, 159 (1970)), и способ получения компетентных клеток из клеток, находящихся в фазе роста, с последующим введением ДНК, который описан для Bacillus subtilis (Duncan, C.H., Wilson, G.A. and Young, F.E., Gene, 1, 153 (1977)). Кроме того, может быть также использован способ создания ДНК-содержащих реципиентных клеток в протопластах или сферопластах, которые могут легко воспринимать рекомбинантную ДНК, с последующим введением рекомбинантной ДНК в клетки, который описан для Bacillus subtilis, актиномицетов и дрожжей (Chang, S. and Choen, S.N., Molec. Gen. Genet., 168, 111 (1979); Bibb, M.J., Ward, J.M and Hopwood, O.A., Nature, 274, 398 (1978); Hinnen, A., Hicks, J.B. and Fink, G.R., Proc. Natl. Sci., USA, 75, 1929 (1978)). Кроме того, может использоваться способ электропорации (Canadian Journal of Microbiology, 43, 197 (1997)). Любой из этих способов может быть выбран соответствующим образом в зависимости от клеток, используемых в качестве реципиента. В коринеформной бактерии по настоящему изобретению в зависимости от типа целевого вещества активность фермента, участвующего в биосинтезе целевого вещества, может быть повышена. Кроме того, активность фермента, неблагоприятного для образования целевого вещества, может быть снижена или полностью устранена. В том случае, когда требуемые гены mdh, hps, phi и amd ген вводят в коринеформную бактерию, порядок введения указанных генов конкретно не ограничивается. Кроме того, бактерия по настоящему изобретению может быть получена либо путем введения указанных генов в коринеформную бактерию, обладающую способностью продуцировать целевое вещество, либо путем придания способности продуцировать целевое вещество коринеформной бактерии, в которую введены указанные гены. Коринеформная бактерия по настоящему изобретению может представлять собой бактерию, полученную методами генной рекомбинации посредством введения ДНК, содержащей генетическую информацию, нужную для биосинтеза целевого вещества. Так, например, в случае бактерий, продуцирующих L-лизин, примеры генов, которые могут быть при этом введены, включают гены, кодирующие ферменты пути биосинтеза L-лизина, такие как фосфоэнолпируваткарбоксилаза, аспартокиназа, дигидродипиколинатсинтетаза, дигидродипиколинатредуктаза, сукцинилдиаминопимелаттрансаминаза и сукцинилдиаминопимелатдеацилаза. В случае гена, кодирующего фермент, который является мишенью для ингибирования по типу обратной связи L-аспарагиновой кислотой или L-лизином, такой как фосфоэнолпируваткарбоксилаза или аспартокиназа и дигидродипиколинатсинтетаза, желательно использовать мутантный ген, кодирующий фермент, у которого чувствительность к ингибированию снижена. Пример мутантного lysC гена (lysC*), кодирующего мутантную аспартокиназу, у которой чувствительность к ингибированию снижена, включает ген, имеющийся в продуцирующей L-лизин бактерии AJ3463 (FERM P-1987), полученный из Brevibacterium lactofermentum, штамм ATCC 13869, путем мутационной обработки (международная патентная публикация WO94/25605). Кроме того, в коринеформной бактерии по настоящему изобретению может быть снижена или устранена активность фермента, который катализирует реакцию образования соединения, отличного от целевого соединения, путем ответвления от пути биосинтеза целевого вещества, или фермента, который переносит целевое вещество в клетку из среды. В том случае, когда целевым веществом является L-лизин, пример такого фермента, который катализирует реакцию образования соединения, отличного от L-лизина, путем ответвления от пути биосинтеза L-лизина, включает гомосериндегидрогеназу (см. WО95/23864). Пример фермента, который переносит L-лизин в клетки, включает лизинпермеазу (продукт гена lysI). Примеры коринеформной бактерии, в которой активность целевого фермента снижена или устранена, включают, например, штаммы с разрушенным геном, в которых ген целевого фермента на хромосоме разрушен методами генетической рекомбинации, и мутантные штаммы, которые имеют активный целевой фермент как результат наличия мутаций в регуляторной последовательности экспрессии или в кодирующем регионе гена целевого фермента на хромосоме, больше не образуются. Мутантные штаммы могут быть получены путем обработки коринеформной бактерии ультрафиолетовым облучением или мутагенным веществом, которое традиционно используют при мутационной обработке, таким как N-метил-N’-нитро-N-нитрозогуанидин (NTG) или ЭМС (EMS). Далее описывается разрушение гена lysI как пример способа разрушения гена целевого фермента на хромосоме с помощью методов генной рекомбинации. Ген lysI на хромосоме может быть разрушен путем трансформации бактерии, принадлежащей к роду Escherichia, с использованием ДНК, включающей ген lysI, модифицированный таким образом, что он больше не продуцирует лизинпермеазу, и который характеризуется ферментативной активностью (делетированный тип гена lysI), полученной в результате делеции части гена lysI, что позволяет провести рекомбинацию между делетированным типом гена lysI и геном lysI на хромосоме. Такая генная деструкция путем гомологичной рекомбинации известна, и имеются способы ее осуществления с использованием линейной ДНК, плазмиды, включающей температурочувствительный репликационный регуляторный регион и т.д. Ген lysI на хозяйской хромосоме может быть заменен делетированным типом гена lysI указанным ниже способом. Так, например, может быть получена рекомбинантная ДНК путем вставки в соответствующий вектор мутантного lysI гена и маркерного гена, придающего устойчивость к лекарственным соединениям, таким как канамицин. Затем коринеформную бактерию трансформируют рекомбинантной ДНК и трансформированный штамм культивируют в среде, содержащей лекарственное средство, для получения трансформированного штамма, в котором рекомбинантная ДНК включена в хромосомную ДНК. В случае введения указанной выше вставки достигается рекомбинация хромосомного lysI гена и рекомбинантной ДНК с новой вставкой. В результате в хромосому на обеих сторонах другой части рекомбинантной ДНК, то есть на векторной части, на температурочувствительном участке контроля репликации и маркера лекарственной устойчивости, вставляются два гена слияния, содержащие хромосомный ген lysI и делетированный тип гена lysI. В результате этого трансформированный штамм экспрессирует нормальный продукт гена lysI, поскольку в данном состоянии доминирует нормальный ген lysI. Далее, если в рекомбинантную ДНК включается, например, ген сукразы, такой рекомбинантный штамм экспрессирует сукразу и, исходя из этого, он не может расти в среде, содержащей сахарозу в качестве единственного источника углерода. Поэтому данный ген может использоваться в качестве маркера. Впоследствии, с целью сохранения только делетированного типа гена lysI на хромосомной ДНК, одну копию гена lysI удаляют из хромосомной ДНК вместе с векторным сегментом (включая маркерный ген) путем рекомбинации двух генов lysI (вторая рекомбинация). Данная стадия представляет собой случай, при котором нативный ген lysI остается на хромосомной ДНК, а делетированный ген lysI элиминируется из хромосомной ДНК, или, наоборот, случай, при котором делетированный тип гена lysI остается на хромосомной ДНК, а нативный ген lysI элиминируется из хромосомной ДНК. Исходя из этого, путем подтверждения структур гена может быть получен штамм, в котором делетированный тип гена lysI остается на хромосоме. Указанные выше гены, которые кодируют ферменты, участвующие в биосинтезе целевого вещества, могут быть также введены в коринеформные бактерии путем замены гена на хромосомной ДНК коринеформной бактерии тем же способом, что и в случае указанного выше разрушения гена. Целевое вещество, которое может быть образовано при культивировании коринеформной бактерии по настоящему изобретению, полученное указанным выше способом в среде, содержащей метанол, приводит к накоплению целевого вещества в среде или в клетках бактерии, после чего целевое вещество может быть собрано из среды или клеток бактерии. Примеры целевых веществ, которые могут использоваться по методу настоящего изобретения, включают, не ограничиваясь приведенным списком, вещество, получаемое при метаболизме метанола, и вещество, образуемое при утилизации энергии, образуемой при метаболизме метанола. Конкретно, к ним относятся, например, аминокислоты, такие как глютаминовая кислота, лизин, треонин, фенилаланин и триптофан, витамины, такие как витамин С, макромолекулярные вещества, такие как различные виды ферментов, и т.п. Выражение “способность продуцировать целевое вещество” в контексте настоящего изобретения обозначает способность коринеформной бактерии по настоящему изобретению вызывать накопление целевого вещества в среде или в клетках бактерии в таком количестве, чтобы указанное вещество могло быть собрано из них в случае культивирования бактерий в среде при поддержании определенных условий. В настоящем изобретении среда и условия культивирования могут быть соответствующим образом выбраны в зависимости от бактериального штамма и целевого вещества. Так, могут использоваться типичные среды, содержащие источник азота, неорганические ионы и другие необходимые следовые органические компоненты. Метанол может использоваться в качестве источника углерода. Вместе с метанолом могут использоваться сахариды, такие как глюкоза, лактоза, галактоза, фруктоза и гидролизат крахмала, спирты, такие как глицерин и сорбит, или органические кислоты, такие как фумаровая кислота, лимонная кислота и янтарная кислота. Неорганические аммонийные соли, такие как сульфат аммония, хлорид аммония и фосфат аммония, органический азот, такой как гидролизат соевого белка, газообразный аммиак, водный аммиак и т.п., могут использоваться в качестве источника азота. Фосфат калия, сульфат магния, ионы железа, ионы марганца и т.п. могут использоваться в небольших количествах в качестве неорганических ионов или их источника. Предпочтительно добавлять необходимые вещества, такие как L-гомосерин, витамин В1, дрожжевой экстракт и т.п., в качестве органических следовых питательных компонентов в соответствующих количествах. Культивирование может предпочтительно проводиться в условиях, подходящих для коринеформной бактерии. Обычно культивирование предпочтительно проводят в аэробных условиях в течение 16-96 часов. Температуру культивирования предпочтительно контролируют в диапазоне от 20°С до 45°С и значение pH предпочтительно контролируют в диапазоне от 5 до 8,5 в ходе культивирования. Для корректировки значения pH могут использоваться неорганические и органические кислотные или щелочные вещества, а также газообразный аммиак и т.п. Если в качестве хозяйской клетки используется термофильная бактерия, она может культивироваться в температурном диапазоне от 42°С до 60°С. Для сбора продукта метаболизма из среды после завершения культивирования по настоящему изобретению не требуется применять специальный способ. Указанный сбор может быть проведен сочетанием хорошо известных методик, таких как способы ионного обмена на смоле, способы осаждения и другие известные способы. Кроме того, в том случае, когда в качестве источника углерода используется метанол, очистка целевого вещества и процесс удаления отходов в растворе может быть упрощен в сравнении с вариантом использования сахаров, получаемых из сельскохозяйственных продуктов. ПРИМЕРЫ Ниже настоящее изобретение описывается более конкретно со ссылкой на приведенные ниже не ограничивающие примеры. Пример 1: Клонирование гена метанолдегидрогеназы Получают хромосомную ДНК обычным способом из Bacillus brevis, штамм S1 (NCIMB 12525, полученный из NCIMB), которая является метанол-ассимилирующей устойчивой к высоким температурам бактерией, относящейся к роду Bacillus. Затем клонируют ген МДГ методом ПЦР с использованием данной ДНК в качестве матрицы (см. публикацию выложенной заявки на патент Японии № 2000-69976). В качестве праймера используют MDH-BM-1 (SEQ ID NO:1) и MDH-BM-2 (SEQ ID NO:2). Указанные праймеры получают в соответствии с ранее опубликованной нуклеотидной последовательностью гена МДГ из Bacillus methanolicus, штамм C1 (зарегистрирован в GenBank под номером М65004, код доступа BACMDH). ПЦР проводят с использованием Pyrobest (Takara Shuzo) и тепловой обработки при температуре 94°С в течение 90 секунд, с последующим проведением реакций при температуре 98°С в течение 10 секунд, при 55°С в течение 30 секунд и при 70°С в течение 4 минут с повтором процедуры в течение 30 циклов, и далее проводят инкубацию при 72°С в течение 10 минут. В результате указанных реакций получают фрагмент ДНК желательного размера. После того как фрагмент ДНК очищают и делают оба конца тупыми, фрагмент ДНК клонируют в сайте SmaI шаттл-вектора pBC4 (описан далее), полученного из pHSG399 (Takara Shuzo) и включающего ориджин репликации, а ориджин репликации получают из pHM1519 (описан далее). Компетентные клетки E.coli, штамм JM109 (Takara Shuzo), трансформируют лигирующей реакционной смесью по методике, предлагаемой производителем, и отбирают несколько устойчивых к хлорамфениколу колоний. Из указанных колоний экстрагируют плазмидные ДНК и анализируют их структуры. Плазмида, в которой направление mdh гена, включенного в плазмиду, является обратным в сравнении с направлением lac промотора в векторе, обозначают как pBC-m-2 и используют в следующих экспериментах. pBC4 получают следующим образом. Плазмиду pНK4 (см. публикацию выложенной заявки на патент Японии № 5-7491), имеющую ориджин репликации, полученный из уже имеющейся плазмиды pHM1519 (Agric. Biol. Chem., 48, 2901-2903 (1984)), и которая автономно реплицируется в коринеформных бактериях, расщепляют рестрикционными ферментами BamHI и KpnI с получением генного фрагмента, содержащего ориджин репликации, в полученном фрагменте делают тупые концы с использованием набора для создания тупых концов ДНК (DNA Blunting Kit) (Takara Shuzo) и вставляют его в pHSG399 (Takara Shuzo) в сайт BamHI путем лигирования с использованием BamHI линкера (Takara Shuzo). Компетентные клетки E.coli, штамм JM109 (Takara Shuzo), трансформируют лигирующей реакционной смесью по методике, предлагаемой производителем, и отбирают затем несколько устойчивых к хлорамфениколу колоний. Плазмиду получают из образованных колоний, как было описано выше для случая получения pBC4. Пример 2: Клонирование гена, кодирующего активатор МДГ (Amd) из Bacillus subtilis Известно, что имеются факторы, активирующие ферментативную активность НАД-зависимого типа метанолдегидрогеназ, получаемых из метанол-ассимилирующей бактерии, относящейся к роду Bacillus. В публикации выложенной заявки на патент Японии № 2000-69976 указывается, что один из таких факторов имеется в Bacillus subtilis. Указанные фактор был обозначен как Amd (активатор метанолдегидрогеназы). Ген, кодирующий Amd (amd), клонируют из Bacillus subtilis традиционным способом. Конкретно клонирование проводят следующим образом. Bacillus subtilis, штамм 168, культивируют в LB среде и из полученных клеток экстрагируют хромосомную ДНК традиционным способом (Biochem. Biophys. Acta., 72, 619-629 (1963)). Проводят ПЦР с использованием хромосомной ДНК в качестве матрицы и олигонуклеотидов, сконструированных таким образом, что целевой фрагмент ДНК имеет на обоих концах сайты для рестрикционного фермента EcoRI (SEQ ID NO: 11 и 12), для амплификации фрагмента генной ДНК, содержащего amd, который представляет собой целевой ген. Для амплификации проводят цикл стадий денатурации при температуре 98°С в течение 10 секунд, стадию отжига при 55°С в течение 30 секунд и стадию наращивания при 72°С в течение 2 минут с повтором процедуры в течение 30 циклов. Использованный фермент представляет собой ДНК-полимеразу Pyrobest (Takara Shuzo), который используют в соответствии с инструкцией производителя. Амплифицированный фрагмент ДНК очищают обработкой фенолом/хлороформом и осаждают этанолом, после чего расщепляют рестрикционным ферментом EcoRI с получением фрагмента amd, имеющего сайты EcoRI на обоих концах. Отдельно рестрикционным ферментом EcoRI обрабатывают аналогичным образом pVK7, который представляет собой шаттл-вектор для Escherichia coli и Corynebacterium glutamicum. После удаления на концах фосфатных групп с помощью щелочной фосфатазы проводят лигирование с указанным выше фрагментом amd. Компетентные клетки E. Coli, штамм M109 (Takara Shuzo), трансформируют лигирующей реакционной смесью по методике, предлагаемой производителем, и отбирают несколько колоний, устойчивых к канамицину. Указанный выше вектор pVK7 конструируют (см. публикацию выложенной заявки на патент Японии № 10-266881, WO99/07853) путем лигирования pAM330, которая представляет собой криптическую плазмиду Brevabacterium lactofermentum, с pHSG299, который представляет собой вектор для Escherichia coli (KMr, см. Takeshita, S. et al., Gene, 61, 63-74, (1987)), следующим образом. Получают pAM330 из Brevabacterium lactofermentum, штамм ATCC 13869. Расщепляют pHSG299 с помощью AvaII (Takara Shuzo), затупляют концы с помощью ДНК-полимеразы T4 и затем лигируют с pAM330, расщепленной с помощью HindIII (Takara Shuzo), и затупляют концы с помощью ДНК-полимеразы T4. Таким образом, получают pVK7. pVK7 автономно реплицируется в клетках E.coli и Brevabacterium lactofermentum и содержит множественные сайты клонирования из pHSG299, lacZ’ и ген устойчивости к канамицину в качестве маркера. Из указанных колоний экстрагируют плазмидную ДНК и анализируют ее структуру. Затем плазмиду, содержащую ген amd в том же направлении, что и lac промотор в векторе, и в обратном направлении, обозначают как pVK-a соответственно и используют для дальнейших экспериментов. Пример 3: Клонирование гена hps и гена phi из метанол-ассимилирующей бактерии, относящейся к роду Bacillus Получают по описанному выше способу хромосомную ДНК из Bacillus brevis, штамм S1, которая представляет собой метанол-ассимилирующую бактерию, относящуюся к роду Bacillus. Указанную хромосомную ДНК используют в качестве матрицы в ПЦР для амплификации целевого участка ДНК. Последовательности олигонуклеотидных праймеров для ПЦР (SEQ ID NO: 13 и 14) конструируют таким образом, чтобы сайты фермента рестрикции KpnI вводились на обоих концах амплифицированного фрагмента ДНК. ПЦР проводят с использованием Pyrobest (Takara Shuzo) и тепловой обработки при температуре 94°С в течение 90 секунд, затем путем проведения реакций при 98°С в течение 10 секунд, при 55°С в течение 30 секунд и при 72°С в течение 2 минут, повторяя указанную процедуру в течение 25 циклов, и затем инкубируют при 72°С в течение 10 минут. Далее полученный фрагмент ДНК очищают традиционным способом, обрабатывают ферментом рестрикции KpnI с получением целевой ДНК, имеющей на обоих концах концы, расщепленные KpnI. Отдельно pVK7, который представляет собой шаттл-вектор для Escherichia coli и Corynebacterium glutamicum, обрабатывают ферментом рестрикции KpnI, затем обрабатывают щелочной фосфатазой и лигируют с указанным выше фрагментом ДНК с использованием лигазы Т4 (Takara Shuzo). E. Coli, штамм JM109, трансформируют лигирующей смесью описанным выше способом с получением множества колоний, устойчивых к канамицину. Отбирают несколько колоний и содержащиеся в них плазмиды исследуют для выбора такой плазмиды, в которой целевые гены hps и phi ориентированы в том же направлении, что и в lac промоторе на векторе. Указанную плазмиду обозначают как pVK-h. Пример 4: Конструирование плазмиды, содержащей hps, phi и amd Плазмиды pVK-a и pVK-h, полученные в примерах 2 и 3, обрабатывают, каждую, ферментами рестрикции ClaI и SacI. На основе pVK-a получают меньший фрагмент ДНК, содержащий ген amd. Одновременно на основе pVK-h традиционным способом получают более крупный фрагмент ДНК, содержащий hps и phi гены, и лигируют с фрагментом гена amd с использованием лигазы Т4. Компетентные клетки Escherichia coli, штамм JM109, трансформируют указанной выше реакционной смесью. Отбирают трансформанты, устойчивые к канамицину. Из нескольких десятков колоний, которые появляются на чашке с агаром, произвольно выбирают 6 колоний и анализируют структуры содержащихся в них плазмид. В результаты был сделан вывод, что все плазмиды имеют целевую структуру, то есть структуру, в которой три вида генов: amd, hps, phi перенесены на вектор pVK7. Указанная плазмида обозначается как pVK-ha. Пример 5: Получение Corynebacterium glutamicum, которой придана способность утилизировать метанол, и тестирование этой способности Два вида плазмид, сконструированных способами, описанными в примерах 1 и 4, то есть pBC-m-2 и pVK-ha, вводят в Corynebacterium glutamicum (ATTC 13869) путем электропорации (Gene Pulser производства фирмы BIO-RAD, расстояние между электродами в кювете составляет 0,1 см и параметры электрической пульсации составляют 25 мкФ, 200 U и 1,8 кВ). Полученные трансформанты могут быть отобраны на чашках с агаром СМ-2S (см. ниже состав среды), содержащим 5 мкг/л хлорамфеникола и 25 мкг/л канамицина. Трансформанты культивируют в течение ночи при 31,5°С при встряхивании в жидкой среде CM-2S, содержащей 5 мкг/л хлорамфеникола и 25 мкг/л канамицина. Культуру содержат в 3 мл культурального бульона в опытной пробирке. Среду CM-2S получают следующим образом. Смешивают все компоненты, показанные в таблице 1, доводят pH до значения 7,2 с помощью KOH и затем стерилизуют автоклавированием при 120°С в течение 20 минут. В случае агаровой среды добавляют 20 г/л агара.
Далее указанный выше культуральный бульон после культивирования в течение ночи инокулируют в количестве 1 объемн.% в среду MM-MES-RC (ниже см. состав среды) и добавляют немеченый метанол до конечной концентрации 0,2 объемн.% и культуру выдерживают при 31,5°С в течение 40 часов при встряхивании. В процессе культивирования измеряют концентрацию метанола в среде в течение соответствующего периода времени с помощью газовой хроматографии. Культуру в среде, содержащей метанол, обрабатывают в 10 мл культурального бульона в L-образной опытной пробирке. Далее в качестве контроля, в котором отсутствует фермент метанолдегидрозеназа и который в этой связи не обладает способностью утилизировать метанол, используют Corynebacterium glutamicum (АТСС 13869), в которую введен только pVK-ha путем электропорации и которую культивируют в тех же условиях и отслеживают изменение концентрации метанола в среде в процессе культивирования. Среду MM-MES-RC готовят следующим образом. Компоненты, за исключением D-рибозы и казаминокислоты, смешивают с получением раствора, имеющего 5-кратную концентрацию, раствор доводят до значения pH 7,0 с использованием NaOH и проводят стерилизацию фильтрованием. Далее готовят водные растворы, содержащие каждый 50% D-рибозы и 10% казаминокислоты, и подвергают стерилизации фильтрованием. Затем перед непосредственным использованием добавляют 50% раствор D-рибозы и 10% раствор казаминокислоты, так чтобы конечная концентрация обоих веществ составляла 5 г/л, добавляют далее 200 мл раствора, имеющего 5-кратную концентрацию, и доводят до окончательного объема 1 л стерилизованной водой.
МЭС: 2-(морфолино)этансульфоновая кислота В результате был сделан вывод, что снижение уровня метанола в среде со штаммом, несущим и pBC-m-2, и pVK-ha, было значительно выше, чем снижение уровня метанола в среде со штаммом, в который был введен только pVK-ha. Считается, что в данном эксперименте снижение уровня метанола в среде, наблюдаемое в случае штамма, в который был введен лишь pVK-ha и который не может потреблять метанол, было вызвано естественным испарением, поскольку он не содержит метанолдегидратазы. В этой связи тот факт, что в штамме, несущем и pBC-m-2, и pVK-ha, быстрее снижается содержание метанола в среде, позволяет предположить, что данный штамм приобрел способность потреблять метанол. Пример 6: Конструирование штамма Corynebacterium glutamicum, продуцирующего L-лизин Corynebacterium glutamicum, модифицированную таким образом, чтобы она могла продуцировать L-лизин, конструируют по описанному ниже способу. Corynebacterium glutamicum (ATCC 13869) используют в качестве родительского штамма. Ген аспартокиназы (lysC) в хромосоме данного штамма заменяют мутантным геном lysC (lysC*), кодирующим аспартокиназу, которая слабо чувствительна к ингибированию. Мутантный ген lysC идентифицирует в бактерии, продуцирующие лизин (AJ3463). Кроме того, ген лизинпермеазы (lysI) модифицируют в неактивный тип гена lysI за счет делеции его части. Конкретно, проводят следующие экспериментальные операции. Во-первых, элиминируют традиционным способом криптическую плазмиду pAM330, содержащуюся в родительском штамме Corynebacterium glutamicum (ATCC 13869). Затем по описанному ниже способу конструируют плазмиду pBS3C* для замены гена lysC на ген lysC*. Плазмиду pHSG299 (Takara Shuzo) расщепляют ферментом рестрикции AvaII, оба конца делают тупыми с помощью набора для создания тупых концов ДНК (DNA Blunting Kit) (Takara Shuzo), проводят дефосфорилирование щелочной фосфатазой и далее полученный фрагмент лигируют с фрагментом ДНК, содержащим ген sacB (ген левансукразы из Bacillus subtilis) с использованием ДНК-лигазы T4. Указанный фрагмент ДНК, содержащий ген sacB, получают методом ПЦР с использованием хромосомы Bacillus subtilis, штамм 168, экстрагированной традиционным способом, в качестве матрицы и праймера 3 (SEQ ID NO: 3) и праймера 4 (SEQ ID NO: 4) (Pyrobest (Takara Shuzo)). Проводят реакцию тепловой обработки при температуре 94°С в течение 90 секунд с последующим проведением реакций при температуре 98°С в течение 10 секунд, при 55°С в течение 30 секунд и при 72°С в течение 1,5 минут, повторяя процедуру в течение 25 циклов, после чего проводят инкубацию при 72°С в течение 10 минут. Амплифицированный продукт расщепляют ферментами рестрикции BglII и BamHI и оба конца делают тупыми с помощью набора DNA Blunting Kit (Takara Shuzo). Компетентные клетки Escherichia coli, штамм JM109, трансформируют лигирующей реакционной смесью. Затем отбирают трансформанты, устойчивые к канамицину. Плазмиду экстрагируют из трансформанта, для которого по виду возникающих колоний подтверждено введение гена sacB в pHSG299, и полученную плазмиду обозначают как pBS3. Затем и pBS3, и p399AK9 (плазмида, описанная в WO94/25605, которая состоит из pHSG399 (Takara Shuzo), несущей ген lysC* из L-лизин-продуцирующей бактерии, штамм AJ3463) расщепляют ферментами рестрикции EcoRI и SphI. Участок, содержащий ген sacB, и участок, содержащий ген lysC*, лигируют с использованием ДНК-лигазы Т4. Компетентные клетки Escherichia coli, штамм JM109, трансформируют лигирующей смесью. Затем отбирают трансформанты, устойчивые к канамицину. Получают плазмиды, содержащиеся в таких трансформантах, и подтверждают их структуры. Отбирают плазмиду, в которой ген lysC* из p399AK9 вставлен в pBS3, как и планировалось. Указанную плазмиду обозначают как pBC3C*. Ген lysC в Corynebacterium glutamicum (штамм ATCC 13869), из которой был элиминирован pAM330, заменяют геном lysC* с помощью процедур с использованием pBC3C*. Вначале pBC3C* вводят в штамм ATCC 13869 традиционным способом с получением штамма, который растет на среде CMDex (см. ниже состав), содержащей 10 мкг/мл канамицина. Поскольку pBC3C* не содержит никакого ориджина репликации, реплицирующегося в штамме ATCC 13869, полученный штамм, демонстрирующий устойчивость к канамицину, представляет собой штамм ATCC 13869, в котором ген lysC* в pBC3C* включен в участок гена lysC на хромосоме штамма ATCC 13869 путем гомологичной рекомбинации. Затем указанный штамм, в котором прошла одна рекомбинация, культивируют в течение ночи при 31,5°С в среде CMDex и затем наносят на среду с агаром DX-S10 (см. ниже состав). В ходе культивирования на участке lysC происходит вторая рекомбинация, и штамм, из которого векторный сегмент, содержащий ген sacB на участке pBC3*, был элиминирован, отбирают как штамм, который растет на агаровой среде и демонстрирует чувствительность к канамицину. Если ген sacB остается на хромосоме, то штамм не может расти на среде DX-S10, содержащей сахарозу, из-за активности сукразы, которая является продуктом данного гена. Определяют традиционным способом нуклеотидную последовательность участков гена lysC в рассматриваемых штаммах, полученных по описанному выше способу, и штамм, в котором было подтверждено замещение геном lysC*, обозначают как штамм 2256С*. Получают среду CMDex, как указано ниже. Все компоненты, приведенные в таблице 3, смешивают, доводят pH до значения 7,5 с помощью KOH и затем стерилизуют автоклавированием при 120°С в течение 20 минут. В случае агаровой среды добавляют 20 г/л агара. Далее готовят агаровую среду DX-S10 указанным ниже способом. Все компоненты, показанные в таблице 4, смешивают, доводят pH до значения 7,5 с помощью KOH и затем стерилизуют автоклавированием при 120°С в течение 20 минут. Далее добавляют 200 мл 50% сахарозы, прошедшей стерилизацию фильтрованием.
Далее, с целью разрушения гена lysI конструируют по указанному ниже способу плазмиду pBS3I Затем полученный по описанному выше способу фрагмент ДНК (lysI Затем ген lysI из Corynebacterium glutamicum, штамм 2256С*, инактивируют с помощью указанных ниже процедур с использованием pBC3I С помощью указанных выше процедур могут быть получены штаммы, в которых ген lysC заменен геном lysC*, и в этой связи достигается делеция гена lysI, и один такой штамм был обозначен как штамм 2256СI (AJ110135). Указанный штамм может расти, утилизируя сахариды в качестве источника углерода, и может продуцировать L-лизин в среде, как описано в примере 8. Пример 7: Введение mdh, amd, hps и phi в L-лизин-продуцирующий штамм Corynebacterium glutamicum pBC-m-2 и pVK-ha, сконструированные в примере 1 и в примере 2, вводят в Corynebacterium glutamicum, штамм AJ110135, модифицированный таким образом, что он продуцирует традиционным способом L-лизин. Штамм AJ110135, несущий две таких плазмиды, содержит все рассматриваемые гены: mdh, amd, hps и phi. Указанный штамм, содержащий плазмиды, был обозначен как MCL101. В случае обычного культивирования данного штамма, указанное культивирование проводят при температуре 31,5°С при встряхивании в среде CM-2S, содержащей антибиотики канамицин и хлорамфеникол в концентрации 25 мкг/л и 10 мкг/л соответственно. Пример 8: Определение способности утилизировать метанол у лизин-продуцирующей бактерии Corynebacterium glutamicum, штамм MCL101, в которую были введены mdh, amd, hps и phi Исследовали наличие у штамма MCL101, сконструированного в примере 7, способности утилизировать метанол, имеющийся в среде, в качестве источника углерода. Штамм MCL101 культивируют в течение ночи при 31,5°С при встряхивании в CM-2S среде, содержащей 25 мкг/л канамицина и 10 мкг/л хлорамфеникола. Указанный культуральный бульон инокулируют в количестве 1 объемн.% в среду MM-MES-RC, содержащую 25 мкг/л канамицина и 10 мкг/л хлорамфеникола, добавляют 13С-меченый метанол в конечной концентрации 0,2 объемн.%, далее добавляют среду MM-MES-RC с немеченым метанолом в конечной концентрации 0,2 объемн.% и проводят культивирование в течение 50 часов при 31,5°С со встряхиванием. После культивирования измеряют при 660 нм поглощение обоих культуральных бульонов, что отражает степень роста. Поглощение достигает значения примерно 1,7 в обоих культуральных бульонах, и между двумя штаммами не наблюдается каких-либо существенных различий в росте бактерий. Кроме того, когда концентрацию метанола после культивирования обоих культуральных бульонов измеряют методом газовой хроматографии, подтверждается, что потребляется по существу равное количество метанола в обоих культуральных бульонах. Затем культуральные бульоны центрифугируют (8000 об/мин в течение 15 мин) с получением супернатантов культуральной жидкости и далее их лиофилизируют. 60 мг каждого лиофилизированного порошка, полученного из супернатантов от обоих культуральных бульонов, растворяют в 500 мкл тяжелой воды. Измеряют содержание L-лизина в каждом растворе, составляющее, как было показано, примерно 1,3 мг в каждом растворе, и количество L-лизина в каждом растворе было по существу одинаковым. Затем каждый раствор подвергают исследованию методом 13С-ЯМР для определения уровня 13C в атомах углерода, составляющих молекулы образованного L-лизина. В результате было показано, что сигнал от каждого атома углерода в L-лизине, образованном культурой, содержащей 13С-меченый метанол, примерно в 3,3-9,9 раз выше, чем в L-лизине, образованном культурой с немеченым метанолом. Данный результат показывает, что созданный штамм MCL101 приобретает новую способность потреблять добавленный к среде 13С-меченый метанол и утилизировать его даже для образования L-лизина, и этот факт далее указывает на то, что может быть сконструирована коринеформная бактерия, которой придана способность утилизировать метанол. Несмотря на то, что настоящее изобретение описано со ссылкой на предпочтительный вариант его осуществления, вполне очевидно любому специалисту в данной области, что в него могут быть введены различные изменения и использоваться различные эквиваленты его без отхода от тематики настоящего изобретения. Каждый из приведенных ниже документов, включая документ JP2003-57171 с датой иностранного приоритета, включен полностью в настоящее описание в качестве ссылки. Пояснения к списку последовательностей SEQ ID NO: 1 и 2: Праймерные последовательности для клонирования гена mdh SEQ ID NO: 3 и 4: Праймеры для клонирования гена sacB SEQ ID NO: 5-10: Праймеры для конструирования фрагмента ДНК, содержащего ген lysI, в котором делетирована центральная часть SEQ ID NO: 11 и 12: Праймеры для клонирования yqkG (amd) SEQ ID NO: 13 и 14: Праймеры для клонирования hps-phi SEQ ID NO: 15: Нуклеотидная последовательность yqkG (amd) из Bacillus subtilis, штамм 168 SEQ ID NO: 16: Аминокислотная последовательность yqkG (amd) из Bacillus subtilis, штамм 168 SEQ ID NO: 17: Нуклеотидная последовательность hps-phi (S1) из Bacillus brevis, штамм S1 SEQ ID NO: 18: Аминокислотная последовательность ГФС из Bacillus brevis, штамм S1 SEQ ID NO: 19: Аминокислотная последовательность ФГИ из Bacillus brevis, штамм S1.
Формула изобретения
1. Способ получения L-аминокислоты с использованием коринеформной бактерии, включающей: (A) культивирование коринеформной бактерии, обладающей способностью продуцировать указанную L-аминокислоту в среде, которое приводит к накоплению L-аминокислоты в среде или клетках бактерии, и (B) отбор L-аминокислоты из среды или клеток бактерии, где в указанную коринеформную бактерию вводят ген метанолдегидрогеназы, ген гексулозофосфатсинтазы, ген фосфогексулоизомеразы и ген, кодирующий фактор, усиливающий активность метанолдегидрогеназы, и указанную бактерию модифицируют таким образом, что она приобретает способность утилизировать метанол, и указанная среда содержит метанол в качестве источника углерода. 2. Способ по п.1, отличающийся тем, что указанная L-аминокислота представляет собой L-лизин. 3. Способ по п.2, отличающийся тем, что указанная бактерия относится к роду Corynebacterium. 4. Способ по п.3, отличающийся тем, что указанная коринеформная бактерия представляет собой Corynebacterium glutamicum. 5. Коринеформная бактерия, продуцент L-аминокислоты, в которую введен ген метанолдегидрогеназы, ген гексулозофосфатсинтазы, ген фосфогексулоизомеразы и ген, кодирующий фактор, усиливающий активность метанолдегидрогеназы, и где указанная бактерия модифицируется таким образом, что приобретает способность утилизировать метанол. 6. Коринеформная бактерия по п.5, которая относится к роду Corynebacterium. 7. Коринеформная бактерия по п.6, которая представляет собой Corynebacterium glutamicum.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
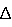 . Вначале фрагмент ДНК амплифицируют путем ПЦР с использованием хромосомной ДНК, полученной из Corynebacterium glutamicum традиционным способом, в качестве матрицы и праймера 5 (SEQ ID NO:5) и праймера 6 (SEQ ID NO:6). Отдельно амплифицируют второй фрагмент ДНК путем ПЦР с использованием хромосомной ДНК, полученной из Corynebacterium glutamicum традиционным способом, в качестве матрицы и праймера 7 (SEQ ID NO: 7) и праймера 8 (SEQ ID NO:8). ПЦР проводят с использованием LA-taq (Takara Shuzo) и тепловой обработки при 94°С в течение 5 секунд, с последующим проведением реакций при температуре 94°С в течение 30 секунд, при 52°С в течение 30 секунд и при 72°С в течение 1 минуты с повтором процедуры в течение 25 циклов, и далее проводят инкубацию при 72°С в течение 10 минут. Затем полученные по описанной выше методике первый и второй фрагменты ДНК используют в качестве матриц с праймером 9 (SEQ ID NO: 9) и праймером 10 (SEQ ID NO: 10) для достижения перекрестного ПЦР и получения при этом фрагмента ДНК гена lysI, содержащего последовательность примерно из центральной части делетированного кодирующего участка (lysI
. Вначале фрагмент ДНК амплифицируют путем ПЦР с использованием хромосомной ДНК, полученной из Corynebacterium glutamicum традиционным способом, в качестве матрицы и праймера 5 (SEQ ID NO:5) и праймера 6 (SEQ ID NO:6). Отдельно амплифицируют второй фрагмент ДНК путем ПЦР с использованием хромосомной ДНК, полученной из Corynebacterium glutamicum традиционным способом, в качестве матрицы и праймера 7 (SEQ ID NO: 7) и праймера 8 (SEQ ID NO:8). ПЦР проводят с использованием LA-taq (Takara Shuzo) и тепловой обработки при 94°С в течение 5 секунд, с последующим проведением реакций при температуре 94°С в течение 30 секунд, при 52°С в течение 30 секунд и при 72°С в течение 1 минуты с повтором процедуры в течение 25 циклов, и далее проводят инкубацию при 72°С в течение 10 минут. Затем полученные по описанной выше методике первый и второй фрагменты ДНК используют в качестве матриц с праймером 9 (SEQ ID NO: 9) и праймером 10 (SEQ ID NO: 10) для достижения перекрестного ПЦР и получения при этом фрагмента ДНК гена lysI, содержащего последовательность примерно из центральной части делетированного кодирующего участка (lysI