Патент на изобретение №2333211
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) N,N
(57) Реферат:
Изобретение относится к новым N,N
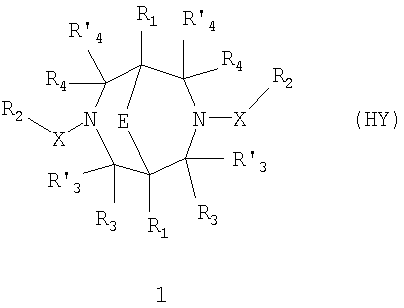
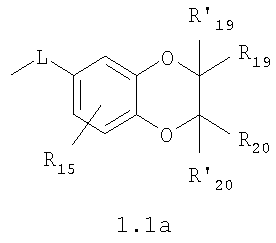
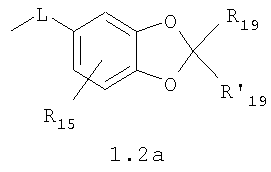
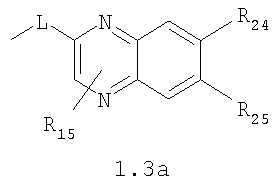
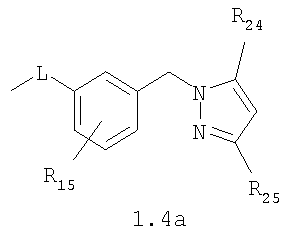
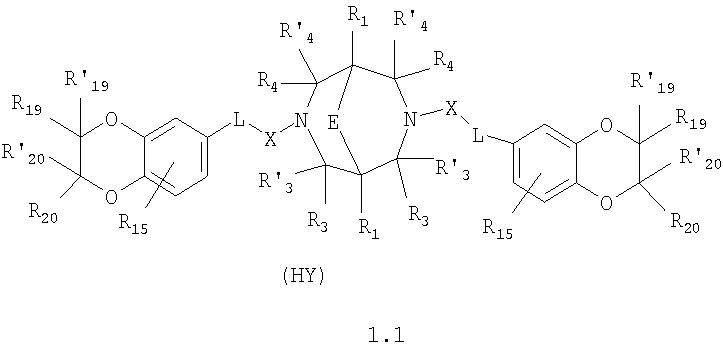
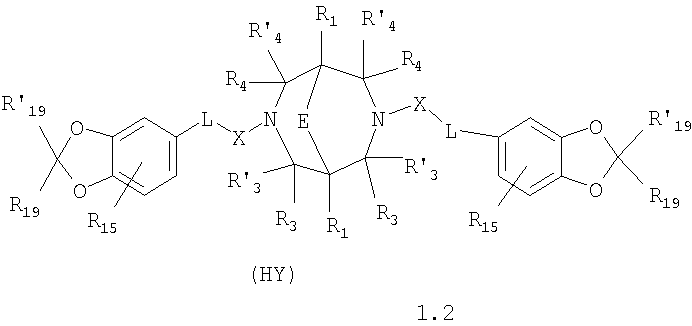
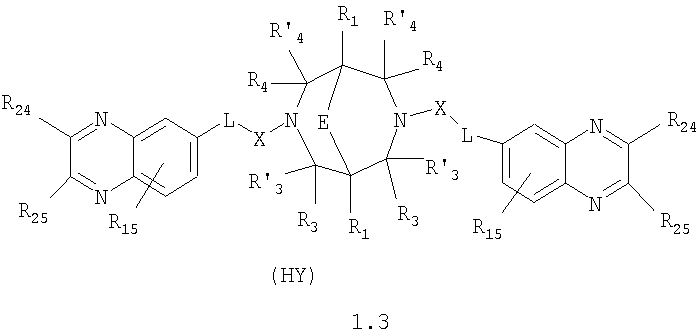
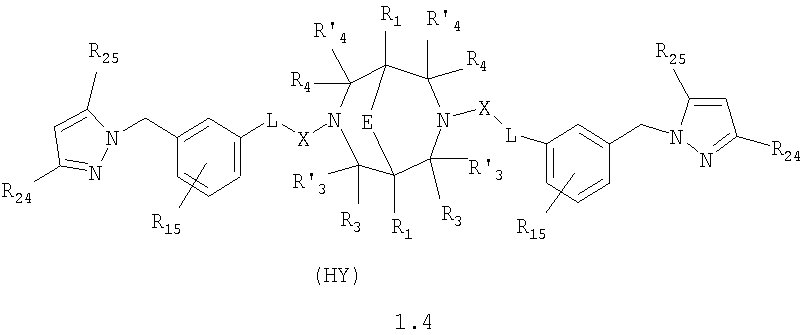
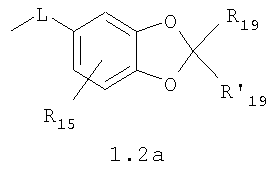
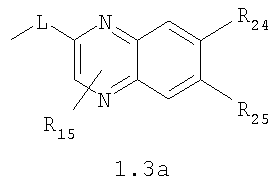
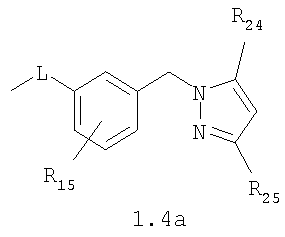
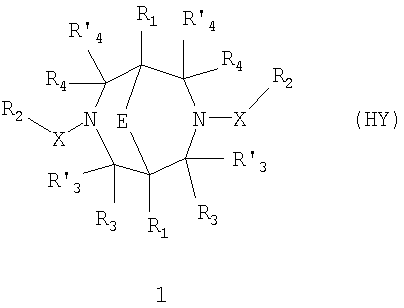
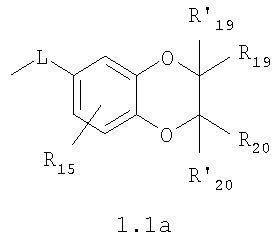
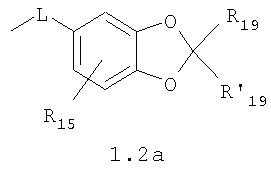
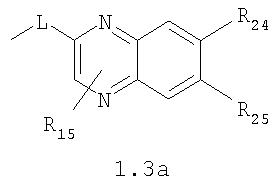
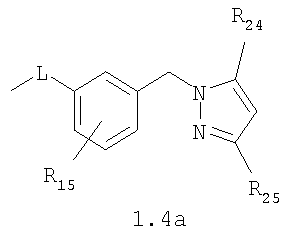
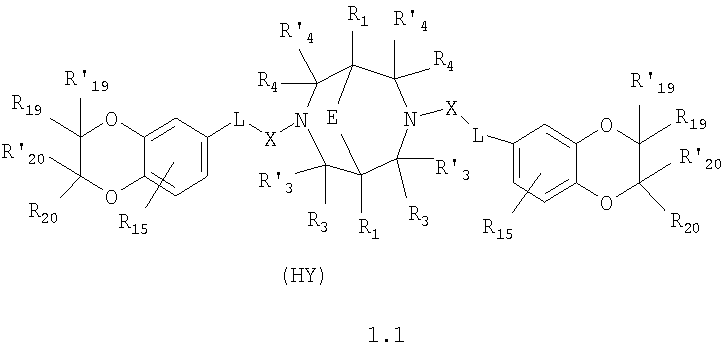
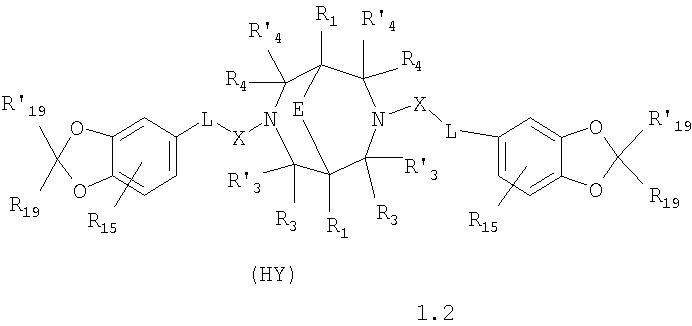
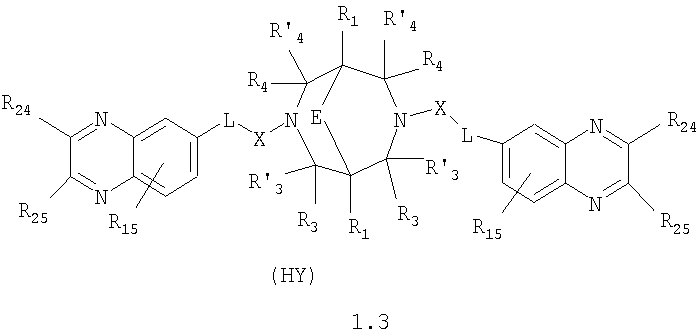
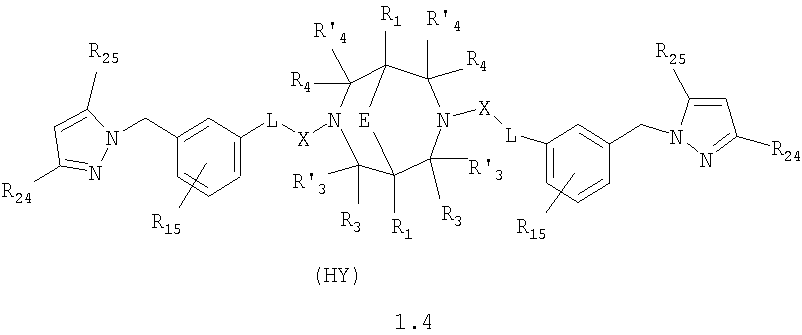
где HY здесь и далее представляет фармакологически приемлемую кислоту; Е означает R1 означает Н, низший алкил, C1-С10алкокси; R2 в совокупности представлено общими формулами (1.1а), (1.2а), (1.3а), (1.4а):
В которых L означает CHR11, R11 означает H, NH2; R15 означает Н, низший алкил, С1-С10алкокси; R19, R R24 и R25 могут быть одинаковыми или различными и каждый независимо означает Н, низший алкил, С1-С10алкокси; R3 и R R4 и R X означает группу общей формулы: (CH2)m-Z, в которой m=0, a Z означает ацетил, либо X представляет валентную связь. Соединения I обладают свойством модулировать активности АМРА-рецепторов, что позволяет использовать их в фармацевтических композициях. 3 н. и 9 з.п. ф-лы, 2 табл.
Данное изобретение относится в целом к новым производным N,N Глутаматергическая система, к которой относятся и АМРА-рецепторы, является основной возбуждающей нейромедиаторной системой в мозге млекопитающих, и в том числе и человека, и участвует в реализации целой серии физиологических и патологических процессов. Известно, что широкий круг психоневрологических заболеваний, таких как БП, БА и подобные им нейродегенеративные расстройства, связан с нарушением регуляции этих процессов (Doble A. Pharmacology and Therapeutics. 1999, V.81, N3, pp.163-221). АМРА-рецепторы неравномерно распределены в головном мозге. Высокая концентрация этих рецепторов была обнаружена в поверхностных слоях новой коры (неокортексе) и в гиппокампе [Monaghan, Brain Res., 1984, V.324, pp.160-164]. Исследования на животных и человеке показали, что эти структуры в основном отвечают за сенсомоторные процессы и представляют собой матрицу для высокоповеденческих реакций. Таким образом, за счет АМРА-рецепторов осуществляется передача сигналов в нейросетях мозга, ответственных за совокупность когнитивных процессов. По причинам, изложенным выше, лекарства, усиливающие функционирование АМРА-рецепторов, участвуют в регуляции процессов, формирующих память, а также процессов, отвечающих за восстановление нервных клеток. В экспериментах было показано [Arai, Brain Res., 1992, V.598, pp.173-184], что усиление АМРА-опосредованного синаптического ответа увеличивает индукцию долговременного потенцирования (LTP). LTP – это увеличение прочности синаптических контактов, которое сопровождает постоянную физиологическую активность в мозге, типичную во время процессов обучения. Вещества, которые усиливают функционирование АМРА-рецепторов, содействуют индукции LTP [Granger, Synapse, 1993, V.15, pp.326-329; Arai, Brain Res., V.638, pp.343-346]. Существует много доказательств того, что LTP является физиологической основой памяти. Например, вещества, которые блокируют LTP, препятствуют механизмам запоминания у животных и людей [Cerro, Neuroscience, 1992, V.46, pp.1-6]. На данный момент известно много соединений, активирующих АМРА-рецепторы. Например, анирацетам [Ito, J. Physiol, 1990, V.424, рр.533-543]. Этими авторами было показано, что анирацетам усиливает синаптический сигнал на нескольких сайтах гиппокампа, никак не действуя на NMDA-опосредованные сигналы [Staubli, 1990, Psychobiology, V.18, рр.377-381; Xiao, Hippocampus, 1991, V.1, pp.373-380]. Анирацетам имеет свойства «быстрой атаки», но он непригоден для длительного применения из-за отсутствия продолжительного эффекта, что является характерной особенностью для «поведенчески-значимых (релевантных)» лекарств. Это лекарство работает только в больших концентрациях (0.1 мМ) и, как было показано [Guenzi, J.Chromatogr., 1990, V.530, рр.397-406], при периферийном применении он превращается в анизоил-GABA (около 80% лекарства), который уже не имеет анирацетам-подобных эффектов. К сожалению, в большинстве случаев соединения, обладающие нейропротекторной активностью, либо действуют в больших дозах, либо обладают повышенной токсичностью. Сравнительно недавно был открыт довольно широкий класс веществ, которые по своему физиологическому действию являются аллостерическими модуляторами АМРА-рецепторов. Эти соединения более стабильны и более эффективны, чем известные ранее, как было показано в экспериментах [Staubli, PNAS, 1994, V.91: pp.11158-11162]. В связи с бурным развитием исследований, связанных с изучением фармакологического действия подобных соединений, недавно был установлен экспериментальный факт, что интенсивный ионный ток, который вызван действием таких аллостерических модуляторов на АМРА-рецепторы с последующей деполяризацией постсинаптической мембраны, запускает механизм экспрессии генов, отвечающих за синтез нейротропинов NGF (nerve growth factor) и BDNF (brain-derived neurotrophic factor) – факторов роста нервной ткани [Legutko В., Neuropharmacology, 2001, V.40, pp.1019-1027; Ebadi, Neurochemistry International, 2000, V.30, pp.347-374]. Процесс экспрессии генов, отвечающих за синтез нейротропина, имеет огромное значение при лечении нейродегенеративных расстройств и других психоневрологических заболеваниях. Так, было показано [Siuciak, Brain Research, 1994, V.633, pp.326-330], что BDNF имеет антидепрессантный эффект в поведенческих моделях отчаяния и уменьшает концентрацию глюкозы в крови у мышей, страдающих сахарным диабетом [Ono, J. Biochem. and Bioph. Res. Commun., 1997, Vol.238, pp.633-637]. Задачей, на решение которой направлено предлагаемое изобретение, является расширение арсенала средств, которые могут быть использованы в качестве новых эффективных аллостерических модуляторов АМРА-рецепторов. В результате проведенных исследований, направленных на поиск таких соединений, в том числе запускающих механизм экспрессии генов, отвечающих за синтез нейротропинов – факторов роста нервной ткани, в частности, среди соединений, обладающих подобной активностью, изобретатели обнаружили широкую группу новых производных N,N Другой аспект изобретения представляет физиологическая активность этих соединений, проявляющаяся в способности при различных концентрациях вызывать положительное аллостерическое модулирование АМРА-рецептора или антагонистическое действие. Это дает основание рассматривать настоящие соединения, с одной стороны, как положительные аллостерические модуляторы, могущие иметь когнитивно-усиливающие свойства, с другой – как потенциальные блокаторы АМРА-рецепторов при более высоких концентрациях. Дополнительный аспект изобретения заключается в представлении фармацевтических композиций, включающих эффективное количество производных N,N Техническим результатом настоящего изобретения является создание новых производных N,N
в которой HY здесь и далее представляет фармакологически приемлемую кислоту; Е представляет карбонильную группу; R1 представляет Н, низший алкил, C1-С10алкокси; R2 в совокупности представлено общими формулами (1.1а), (1.2а), (1.3а), (1.4а):
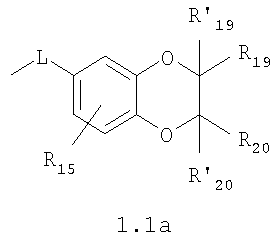
В которых L представляет собой CHR11, карбонильную группу; R11 представляет Н, амино; R15 представляет Н, низший алкил, C1-С10алкокси; R19, R R24 и R25 могут быть одинаковыми или различными и каждый независимо представляет Н, низший алкил, C1-С10алкокси; R3 и R R4 и R X представляет группу общей формулы: (CH2)m-Z, в которой m=0, a Z означает ацетил, либо X представляет валентную связь. Используемый в приведенных выше определениях и последующем описании термин “низший алкил” означает алкильную группу с прямой или разветвленной цепью, содержащую от 1 до 10 атомов углерода, примерами которой являются метил, этил, изопропил, трет-бутил, изопентил и аналогичные. Термин “алкокси” означает группу AlkO-, в которой алкильный фрагмент является таким, как определенная выше алкильная группа. Примеры алкокси-групп включают метокси, бутокси, изопропилокси и аналогичные группы. Термин “фармакологически приемлемые кислоты” охватывает все фармакологически приемлемые кислоты, как неорганические (например, соляную, серную, фосфорную и т.д.); так и органические (например, муравьиную, уксусную, щавелевую, лимонную, винную, малеиновую, янтарную, n-толуол-сульфокислоту, метилсерную и т.д.). Предпочтительные варианты воплощения изобретения. Среди соединений формулы (1), составляющих один из объектов настоящего изобретения, предпочтительными являются следующие четыре группы соединений, который могут быть представлены формулами (1.1), (1.2), (1.3) и (1.4), приведенными ниже. В частности, предпочтительными соединениями являются: 1.1. N,N
1.2. N,N
1.3. N,N
1.4. N,N
в которых HY здесь и далее представляет фармакологически приемлемую кислоту; Е, R1, R3, R Наиболее предпочтительным соединением формулы 1.1 (в виде фармакологически приемлемых солей и/или свободных оснований) является: 3,7-бис(2,3-дигидро-1,4-бензодиоксин-6-илкарбонил)-1,5-диметил-3,7-диазабицикло[3.3.1]нонан-9-он. Наиболее предпочтительным соединением формулы 1.2 (в виде фармакологически приемлемых солей и/или свободных оснований) является: 3,7-бис(1,3-бензодиоксол-5-илкарбонил)-1,5-диметил-3,7-диазабицикло[3.3.1]нонан-9-он. Наиболее предпочтительными соединениями формулы 1.3 (в виде фармакологически приемлемых солей и/или свободных оснований) являются: 1,5-диметил-3,7-бис(хиноксалин-6-илкарбонил)-3,7-диазабицикло[3.3.1]нонан-9-он; 3,7-бис[(2,3-диметилхиноксалин-6-ил)карбонил]-1,5-диметил-3,7-диазабицикло[3.3.1]нонан-9-он. Наиболее предпочтительным соединением формулы 1.4 (в виде фармакологически приемлемых солей и/или свободных оснований) является: N,N Ниже изобретение описывается более подробно с помощью примеров получения конкретных соединений. Исходные реагенты, как и конечные продукты, получаются известными в литературе способами или являются промышленно доступными. Схемы синтеза конечных соединений представлены ниже: Схема 1:
Схема 2:
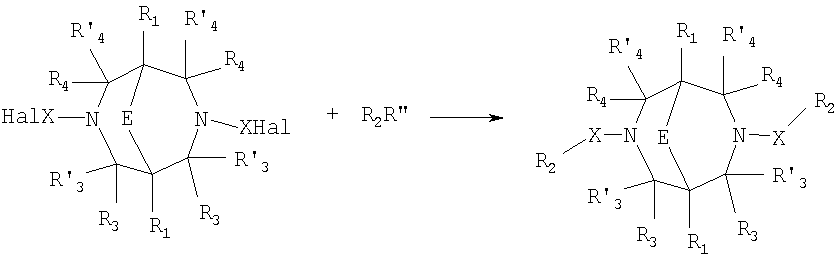
Где в случае схемы 1 R Структуры полученных соединений подтверждались данными химического, спектрального анализов и других физико-химических характеристик. Приведенные ниже примеры иллюстрируют, но не ограничивают данное изобретение. Пример 1. 3,7-бис(2,3-дигидро-1,4-бензодиоксин-6-илкарбонил)-1,5-диметил-3,7-диазабицикло[3.3.1]нонан-9-он (соединение 1, соответствующее общей формуле 1.1). К раствору 39,8 г (0,2 моль) хлорангидрида бензодиоксановой кислоты в абсолютном бензоле прибавляют при перемешивании 55,2 г (0,4 моль) мелкоизмельченного, сухого карбоната калия и 24,1 г (0,1 моль) гидрохлорида 1,5-диметил-3,7-диазабицикло[3.3.1]нонан-9-она. Кипятят в течение шести часов. После этого отфильтровывают осадок. Фильтрат упаривают досуха. Остаток наносят на хроматографическую колонку с силикагелем. В качестве элюэнта используют хлороформ. Отбирают фракцию с Rf=0.43 на силуфоле в системе CHCl3-EtOH (50:1). Отгоняют растворитель и получают прозрачное масло, которое около 30 мин кипятят в этиловом эфире. После чего оно кристаллизуется. Выход: 72%. ПМР-спектр (CDCl3 Пример 2. 3,7-бис(1,3-бензодиоксол-5-илкарбонил)-1,5-диметил-3,7-диазабицикло[3.3.1]нонан-9-он (соединение 2, соответствующие общей формуле 1.2). К раствору 37,г (0,2 моль) хлорангидрида пиперониловой кислоты в абсолютном бензоле прибавляют при перемешивании 55,2 г (0.4 моль) мелкоизмельченного, сухого карбоната калия и 24,1 г (0,4 моль) гидрохлорида 1,5-диметил-3,7-диазабицикло[3.3.1]нонан-9-она. Кипятят в течение семи часов. После этого отфильтровывают осадок. Фильтрат упаривают досуха. Остаток наносят на хроматографическую колонку с силикагелем. В качестве элюэнта используют хлороформ. Отбирают фракцию с Rf=0.4 на силуфоле в системе CHCl3-EtOH (50:1). Отгоняют растворитель и получают прозрачное масло, которое около 30 мин кипятят в этиловом эфире. После чего оно кристаллизуется. Выход: 69%. ПМР-спектр (CDCl3 Пример 3. 3,7-бис[(2,3-диметилхиноксалин-6-ил)карбонил]-1,5-диметил-3,7-диазабицикло[3.3.1]нонан-9-он (соединение 3, соответствующее общей формуле 1.3). К раствору 40,4 г (0,2 моль) 2,3-диметилхиноксалин-6-карбоновой кислоты в абсолютном диметилформамиде прибавляют при перемешивании и охлаждении на ледяной бане 35,64 г (0.22 моль) CDI (N,N Выход: 58%. ПМР-спектр (CDCl3 Пример 4. N,N К раствору 52 г (0,2 моль) 4-метокси-3-(1Н-пиразол-1-илметил)бензойной кислоты в абсолютном диметилформамиде прибавляют при перемешивании и охлаждении на ледяной бане 35,64 г (0,22 моль) CDI. Перемешивание осуществляют шесть часов. После чего к реакционной смеси добавляют раствор 24,1 г (0,1 моль) гидрохлорида 1,5-диметил-3,7-диазабицикло[3.3.1]нонан-9-она в DBU. Нагревают до 50°С и греют в течение семи часов. После этого отфильтровывают осадок. Фильтрат упаривают досуха. Остаток наносят на хроматографическую колонку с силикагелем. В качестве элюэнта используют хлороформ. Отбирают фракцию с Rf=0.68 на силуфоле в системе CHCl3-EtOH (50:1). Отгоняют растворитель и получают игольчатые белые кристаллы. Выход: 79%. ПМР-спектр (CDCl3 Пример 5. 3,7-бис[(хиноксалин-6-ил)карбонил]-1,5-диметил-3,7-диазабицикло[3.3.1]нонан-9-он(соединение 5, соответствущее общей формуле 1.3). К раствору 34,8 г (0,2 моль) хиноксалин-6-карбоновой кислоты в абсолютном диметилформамиде прибавляют при перемешивании и охлаждении на ледяной бане 35,64 г (0.22 моль) CDI. Перемешивание осуществляют пять часов. После чего к реакционной смеси добавляют раствор 24,1 г (0,1 моль) гидрохлорида 1,5-диметил-3,7-диазабицикло[3.3.1]нонан-9-она в DBU. Нагревают до 50°С и греют в течение восьми часов. После этого отфильтровывают осадок. Фильтрат упаривают досуха. Остаток наносят на хроматографическую колонку с силикагелем. В качестве элюэнта используют хлороформ. Отбирают фракцию с Rf=0.52 на силуфоле в системе CHCl3-EtOH (20:1). Отгоняют растворитель и получают прозрачное масло, которое со временем кристаллизуется. Выход: 64%. ПМР-спектр (CDCl3): 0.97 с. (6Н); 3.00 д. (2Н, J 12 Гц); 3.34 д. (2Н, J 12 Гц); 4.22 д. (2Н, J 12 Гц) 4.84 д. (2Н, J 12 Гц); ароматические протоны [7.80 д. (1H, J=5.8 Гц), 8.18 с. (1H), 8.20 д. (1H, J=5.8 Гц)]. Пример 6. Основание 3,7-бис{N-[4-метокси-3-(1H-пиразол-1-илметил)бензил]глицил}-1,5-диметил-3,7-диазабицикло[3.3.1]нонан-9-он. К раствору 48 г (0,15 моль) 3,7-бис(хлорацетил)-1,5-диметил-3,7-диазабицикло[3.3.1]нонан-9-она в абсолютном диметилформамиде прибавляют при перемешивании раствор 64 г (0.3 моль) [4-метокси-3-(1Н-пиразол-1-илметил)бензил]амина в DBU. Перемешивание осуществляют в течение 30 мин. Затем упаривают растворитель примерно на 2/3 от общего объема. Остаток разбавляют пятикратным избытком воды по отношению к растворителю и экстрагируют 4 раза хлористым метиленом. Экстракты объединяют, упаривают досуха и наносят на хроматографическую колонку с силикагелем. В качестве элюэнта используют хлороформ. Отбирают фракцию с Rf=0.73 на силуфоле в системе CHCl3-EtOH (50:1). Отгоняют растворитель и получают прозрачное масло, которое перекристаллизовывают из диэтилового эфира. Выход: 85%. ПМР-спектр (CDCl3 Следующий аспект изобретения составляют соединения общей формулы I, обладающие свойством модулирования активности АМРА-рецепторов. Авторы использовали определение потенцирующего эффекта новых соединений на каинат-вызванные трансмембранные токи в нейронах Пуркинье мозжечка крыс для направленного поиска соединений, способных потенцировать ответы АМРА-рецепторов (и тем самым вызывать улучшение памяти и когнитивных функций). Метод оценки потенцирующих АМРА-рецепторы свойств соединений, позволяющих им влиять на глутаматергическую медиаторную систему ЦНС. Эксперименты по оценке действия веществ на АМРА-рецепторы были проведены методом patch-clamp на свежеизолированных нейронах Пуркинье, выделенных из мозжечков крыс (12-15-дневных). Для выделения использовали модифицированный метод. Срезы мозжечка толщиной 400-600 мкм помещались в термостатируемую камеру объемом 10 мл. Раствор для выделения имел следующий состав (в мМ): NaCl 150.0, KCl 5.0, CaCl2 2.0, MgSO4×7H2O 2.0, HEPES 10.0, глюкоза 15.0, рН 7.42. Срезы инкубировались в этом растворе в течение 60 минут, после чего этот раствор заменяли аналогичным раствором, содержащим проназу (2 мг/мл) и коллагеназу (1 мг/мл), и инкубировали в течение 70 минут. После отмывки первоначальным раствором в течение 20 минут срезы помещались в чашку Петри и разъединялись механическим способом при помощи пастеровской пипетки. Растворы непрерывно продувались 100% О2 при t 34°C. Нейроны Пуркинье помещались в рабочую камеру объемом 0.6 мл. Рабочий раствор имел состав (в мМ): NaCl 150.0, KCl 5.0, CaCl2 2.6, MgSO4×7H2O 2.0, HEPES 10.0, глюкоза 15.0, рН 7.36. Трансмембранные токи вызывались активацией АМРА-рецепторов аппликацией растворов агониста этих рецепторов – каиновой кислоты (КК) методом быстрой суперфузии. Каиновая кислота является агонистом АМРА-рецепторов и используется для изучения свойств АМРА-рецепторов, поскольку сама АМРА вызывает слишком сильную десенситизацию рецепторов и в таких экспериментах не используется. Регистрация токов была осуществлена при помощи боросиликатных микроэлектродов (сопротивление 1.5-2.5 мОм), заполненных следующим составом (в мМ): KCl 100.0, EGTA 11.0, CaCl2 1.0, MgCl2 1.0, HEPES 10.0, ATP 5.0, рН 7.2. Для регистрации использовали прибор ЕРС-9 (НЕКА, Germany). Запись токов осуществлялась на жесткий диск ПК Pentium-4 при помощи программы Pulse, также закупленной в фирме НЕКА. Обработка результатов осуществлялась при помощи программы Pulsefit (НЕКА). Аппликация КК вызывает в нейронах Пуркинье трансмембранные входящие токи. Добавление в перфузируемый раствор соединений формулы I вызывает увеличение амплитуды токов. Это увеличение зависит от соединения, от его концентрации, от времени, прошедшего после начала аппликации вещества. Пример 1. Соединение 1 в дозе 0.001 мкМ вызывает увеличение каинат-вызванных токов на 40-50%, в дозе 0.01 мкМ на 80%, в дозе 0.1 мкМ – на 0%, а в дозе 1 мкМ – блок на -20-40%. Отмывка в течение 3-5 минут возвращает амплитуду ответов к контрольному значению. Пример 2. Соединение 2 в дозе 0.00001 мкМ вызывает увеличение каинат-вызванных токов на 10%, в дозе 0.0001 мкМ – на 50-90%, в дозе 0.001 мкМ – на 60-140%, в дозе 0.01 мкМ – на 0-20%, а в дозе 0.1 мкМ – блок на -20-40%). Отмывка в течение 4-6 минут возвращает амплитуду ответов к контрольному значению. Полученные результаты представлены в таблице 1.
Как видно из представленной таблицы, соединения общей формулы I обладают свойствами потенцировать токи, вызываемые активацией АМРА-рецепторов. Соединения превосходят по активности стандартное вещество Мемантин в 30000-3000000 раз по тесту потенцирования АМРА-ответов. При этом они не оказывают заметного нейротоксического действия (LD50 составляет от 90 до 900 мг/кг в интервале активных исследуемых доз), что делает их ценными для применения в медицине, особенно при лечении нейродегенеративных заболеваний, таких как болезнь Альцгеймера. Проведение эксперимента по определению острой токсичности. Острую токсичность веществ определяли на белых беспородных мышах-самцах массой 22-26 г. Вещества вводили внутрибрюшинно в виде раствора или суспензии в 1% растворе крахмала. Срок наблюдения составлял 14 суток. Токсичность в виде LD50 рассчитывали по методу Литчфилда и Вилкоксона [Litchfild J.T., Wilcoxon F.J., J. Pharmacol. Exp. Ther, 1949, v.96, pp.99-114]. Примеры токсичности заявляемых соединений приведены в таблице 2.
В результате проведенных испытаний установлено, что LD50 исследованных соединений составляет от 90 до 900 мг/кг. В соответствии с классификацией токсичности химических веществ эти соединения относятся к умеренно и малотоксичным веществам. Хотя острая токсичность данных соединений несколько превосходит значение острой токсичности Мемантина, сравнение параметров терапевтического индекса, определяемого как отношение LD50/ED50 (Д.Н.Плутицкий и др. – Хим. Фарм. Ж., 1986, N 10, 1209-1231), показывает, что большинство из исследованных соединений имеет существенное преимущество перед Мемантином. Как это обычно принято в медицине, соединения формулы I согласно настоящему изобретению рекомендуется применять в виде композиций, составляющих соответственно следующий аспект изобретения. Фармацевтическая композиция согласно изобретению приготавливается с помощью общепринятых в данной области техники приемов и включает фармакологически эффективное количество активного агента, представляющего соединение формулы I или его фармацевтически приемлемую соль (называемые далее “активное соединение”), составляющее обычно от 5 до 30 вес.%, в сочетании с одной или более фармацевтически приемлемыми вспомогательными добавками, такими как разбавители, связующие, разрыхляющие агенты, адсорбенты, ароматизирующие вещества, вкусовые агенты. В соответствии с известными методами фармацевтические композиции могут быть представлены различными жидкими или твердыми формами. Примеры твердых лекарственных форм включают, например, таблетки, пилюли, желатиновые капсулы и др. Примеры жидких лекарственных форм для инъекций и парентерального введения включают растворы, эмульсии, суспензии и др. Композиции, как правило, получают с помощью стандартных процедур, предусматривающих смешение активного соединения с жидким или тонкоизмельченным твердым носителем. Композиции согласно изобретению в форме таблеток содержат от 5 до 30% активного соединения и наполнитель(и) или носитель(и). В качестве таковых для таблеток применяются: а) разбавители: свекловичный сахар, лактоза, глюкоза, натрия хлорид, сорбит, маннит, гликоль, фосфат кальция двузамещенный; б) связующие вещества: магниевый силикат алюминия, крахмальная паста, желатин, трагакант, метилцеллюлоза, карбоксиметилцеллюлоза и поливинилпирролидон; в) разрыхлители: декстроза, агар, альгиновая кислота или ее соли, крахмал, твин. ПРИМЕР 1. 100 мг таблетки, содержащие по 0.1 мг соединения 2
Таблетка может быть сформирована посредством прессовки или формовки активного ингредиента с одним или более дополнительными ингредиентами. Получение прессованных таблеток осуществляется на специальной установке. Активный ингредиент в свободной форме, такой как порошок или гранулы, в количестве 10 г (количество вещества, необходимое для получения 10000 таблеток) перемешивается со связующим веществом – трагакантом (200 г), смешивается с разбавителем – лактозой (590 г), в смесь добавляется разрыхляющее вещество – альгиновая кислота (200 г) и отдушка – лимонная кислота (50 г). Для желатиновых капсул используются дополнительно красители и стабилизаторы. В качестве красителей используются: тетразин, индиго; в качестве стабилизаторов могут быть представлены: натрия метабисульфит, натрия бензоат. Предлагаемые желатиновые капсулы содержат от 1 до 20% активного ингредиента. ПРИМЕР 2. 500 мг капсулы, содержащие по 0.5 мг соединения 2
5 г активного вещества (соединения 2) (количество, необходимое для приготовления 10000 капсул) тонко измельчают и смешивают в смесителе с глицерином (1000 г) и сахарным сиропом (3490 мг). После перемешивания в смесь добавляют мятное масло (400 г), бензоат натрия (100 г), аскорбиновую кислоту (50 г) и тетразин (50 г). Желатиновые капсулы приготовляют капельным методом. Этот метод позволяет осуществлять одновременное капельное дозирование раствора лекарственного вещества и нагретой желатиновой массы (900 г желатина) в охлажденное вазелиновое масло. В результате образуются бесшовные шарообразные желатиновые капсулы, заполненные лекарственной смесью, полностью готовые к употреблению, содержащие 50 мг активного вещества. Инъекционные формы композиции предпочтительно представляют собой изотонические растворы или суспензии. Вышеуказанные формы могут стерилизоваться и содержать добавки, такие как консерванты: натрия метабисульфит, бензойная кислота, натрия бензоат, смесь метилпарабена и пропилпарабена; стабилизаторы: абрикосовая и аравийская камедь, декстрин, крахмальный клейстер, метилцеллюлоза, твин; соли, регулирующие осмотическое давление (хлорид натрия), или буферы. Кроме того, они могут содержать другие терапевтически полезные вещества. ПРИМЕР 3. 2 мл ампулы, содержащие по 1 мг соединения 1
Для приготовления инъекционных форм активное соединение 1 (1 г; количество, необходимое для изготовления 1000 ампул) тонко измельчают и смешивают в смесителе с мятным маслом (400 мл), затем добавляют метилцеллюлозу (10 г), смешивают с 0,9% раствором хлорида натрия (1600 мл) и добавляют бензойную кислоту (10 г). Полученный раствор фасуют в ампулы по 2 мл и стерилизуют паром в течение 30 мин. Следующий аспект изобретения составляет способ воздействия на АМРА-рецепторы введением эффективного количества соединения общей формулы I. Назначаемая для приема доза активного компонента (соединения формулы I или его фармацевтически приемлемых солей) варьируется в зависимости от многих факторов, таких как возраст, пол, вес пациента, симптомы и тяжесть заболевания, конкретно назначаемое соединение, способ приема, форма препарата, в виде которой назначается активное соединение. Обычно общая назначаемая доза составляет от 0.1 до 20 мг в день. Общая доза может быть разделена на несколько доз, например, для приема от 1 до 4 раз в день. При оральном назначении интервал общих доз активного вещества составляет от 0.1 до 20 мг в день, предпочтительно от 0.1 до 10 мг. При парентеральном приеме интервал назначаемых доз составляет от 0.5 до 20 мг в день, предпочтительно от 0.5 до 10 мг, а при внутривенных инъекциях – от 0.05 до 5.0 мг в день, предпочтительно от 0.05 до 2.5 мг. Точная доза может быть выбрана лечащим врачом.
Формула изобретения
1. N,N
в которой HY здесь и далее представляет фармакологически приемлемую кислоту; Е представляет карбонильную группу; R1 представляет Н, низший алкил, С1-С10алкокси; R2 в совокупности представлено общими формулами (1.1а), (1.2а), (1.3а), (1.4а):
в которых L представляет собой CHR11, карбонильную группу; R11 представляет Н, амино; R15 представляет Н, низший алкил, С1-С10алкокси; R19, R R24 и R25 могут быть одинаковыми или различными и каждый независимо представляет Н, низший алкил, С1-С10алкокси; R3 и R R4 и R X представляет группу общей формулы: (CH2)m-Z, в которой m=0, a Z означает ацетил, либо X представляет валентную связь; 2. Соединения по п.1, представляющие производные N,N
в которых HY здесь и далее представляет фармакологически приемлемую кислоту; Е, R1, R3, R 3. Соединения по п.1, представляющие производные N,N
в которых HY здесь и далее представляет фармакологически приемлемую кислоту; Е, R1, R3, R 4. Соединения по п.1, представляющие производные N,N-замещенные 3,7-диазабицикло[3.3.1]нонанов общей формулы (1.3):
в которых HY здесь и далее представляет фармакологически приемлемую Е, R1, R3, R 5. Соединения по п.1, представляющие производные N,N
в которых HY здесь и далее представляет фармакологически приемлемую кислоту; Е, R1, R3, R 6. Соединение по п.2, представляющее 3,7-бис(2,3-дигидро-1,4-бензодиоксин-6-илкарбонил)-1,5-диметил-3,7-диазабицикло[3.3.1]нонан-9-он. 7. Соединение по п.3, представляющее 3,7-бис(1,3-бензодиоксол-5-илкарбонил)-1,5-диметил-3,7-диазабицикло[3.3.1]нонан-9-он. 8. Соединения по п.4, представляющие 1,5-диметил-3,7-бис(хиноксалин-6-илкарбонил)-3,7-диазабицикло[3.3.1]нонан-9-он; 3,7-бис[(2,3-диметилхиноксалин-6-ил)карбонил]-1,5-диметил-3,7-диазабицикло[3.3.1]нонан-9-он. 9. Соединение по п.5, представляющее N,N 10. Соединения общей формулы 1 по любому из пп.1-9, обладающие свойством модулирования активности АМРА-рецепторов. 11. Фармацевтическая композиция, обладающая свойством модулировать активности АМРА-рецепторов, содержащая соединение общей формулы I по любому из пп.1-9 в эффективном количестве и фармацевтически приемлемый носитель. 12. Способ воздействия на АМРА-рецепторы введением эффективного количества соединения общей формулы 1 по п.1 в дозе от 0,1 до 20 мг в день.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 -ЗАМЕЩЕННЫЕ 3, 7-ДИАЗАБИЦИКЛО[3.3.1]НОНАНЫ, ОБЛАДАЮЩИЕ ФАРМАКОЛОГИЧЕСКОЙ АКТИВНОСТЬЮ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ И СПОСОБ ИХ ПРИМЕНЕНИЯ
-ЗАМЕЩЕННЫЕ 3, 7-ДИАЗАБИЦИКЛО[3.3.1]НОНАНЫ, ОБЛАДАЮЩИЕ ФАРМАКОЛОГИЧЕСКОЙ АКТИВНОСТЬЮ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ И СПОСОБ ИХ ПРИМЕНЕНИЯ -замещенным 3,7-диазабицикло[3.3.1]нонанам общей формулы 1:
-замещенным 3,7-диазабицикло[3.3.1]нонанам общей формулы 1: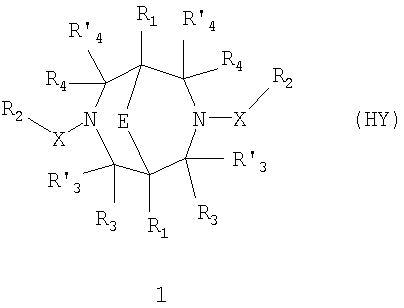
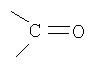 ;
;
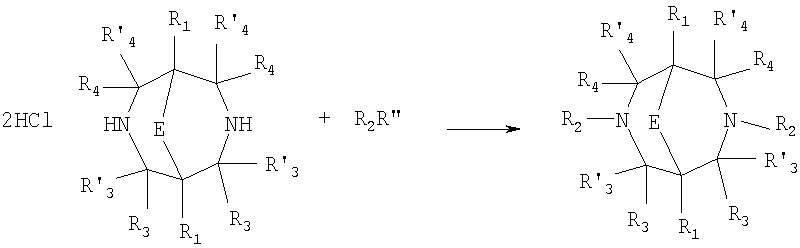
 , м.д.): 1.00 с. (6Н); 3.00 д. (2Н, J 12 Гц); 3.34 д.(2Н, J 12 Гц); 4.22 д. (2Н, J 12 Гц), 4.84 д. (2Н, J 12 Гц); 4.3 с. (8Н); ароматические протоны [7.06 д.д. (2Н, J2,1 8.4 Гц, J2,3 2.4 Гц); 7.09 д. (2Н, J3,2 2.4 Гц)].
, м.д.): 1.00 с. (6Н); 3.00 д. (2Н, J 12 Гц); 3.34 д.(2Н, J 12 Гц); 4.22 д. (2Н, J 12 Гц), 4.84 д. (2Н, J 12 Гц); 4.3 с. (8Н); ароматические протоны [7.06 д.д. (2Н, J2,1 8.4 Гц, J2,3 2.4 Гц); 7.09 д. (2Н, J3,2 2.4 Гц)].