Патент на изобретение №2333208
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) ПРОИЗВОДНЫЕ 2-УРЕИДО-6-ГЕТЕРОАРИЛ-3Н-БЕНЗИМИДАЗОЛ-6-КАРБОНОВОЙ КИСЛОТЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ГИРАЗЫ И/ИЛИ ТОПОИЗОМЕРАЗЫ IV ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ
(57) Реферат:
Описывается соединение формулы I,
или его фармацевтически приемлемая соль, где: Q представляет собой -NH- или -О-; W представляет собой C-R4; X представляет собой СН; R1 представляет собой 5-6-членное арильное кольцо, содержащее 1-3 атома азота, где: R1 замещен 0-3 группами, независимо выбранными из R, оксо, или OR’; каждый R’ независимо выбран из атома водорода или С1-4алифатической группы; R2 выбран из атома водорода или C1-3алифатической группы; R3 выбран из C(O)NHR, C(O)N(R)2, C(O)R, CO2R, C(R’)-NOR или C(R’)=NOH; каждый R независимо выбран из Т-Ar или С1-6алифатической группы, где: указанная С1-6алифатическая группа замещена 0-3 группами, независимо выбранными из R’ или OR’; Т представляет собой (СН2)у, где у равно 0, 1 или 2; Ar выбран из: (а) 3-8-членного насыщенного, ненасыщенного или арильного кольца; (b) 3-7-членного гетероциклического кольца, содержащего 1-3 атома азота; или (с) 5-6-членного гетероарильного кольца, содержащего 1-3 атома азота где: Ar замещен 0-3 R’ группами; и R4 выбран из атомов водорода или фтора. Также описываются композиция для ингибирования активности бактериальной гиразы и/или топоизомеразы IV, способ ингибирования активности гиразы и/или топоизомеразы IV, способ снижения числа бактерий у пациента, зараженного нозокомикальными и ненозокомикальными бактериальными инфекциями. Настоящее изобретение также относится к способу получения данных соединений. Технический результат – получены новые соединения, обладающие полезными биологическими свойствами. 5 н. и 18 з.п. ф-лы, 4 табл.
Перекрестная ссылка на родственные заявки По данной заявке испрашивается приоритет в соответствии с предварительной заявкой на выдачу патента США 60/388665, поданной 13 июня 2002, и предварительной заявкой на выдачу патента США 60/429077, поданной 26 ноября 2002, содержание которых включено здесь в качестве ссылки. Область техники, к которой относится изобретение Настоящее изобретение относится к области медицинской химии и касается соединений и их фармацевтических композиций, которые ингибируют активность бактериальной гиразы и/или топоизомеразы IV. Соединения могут использоваться в качестве ингибиторов активности бактериальной гиразы и/или топоизомеразы IV. Настоящее изобретение относится также к способам лечения бактериальных инфекций у млекопитающих и способам снижения количества бактерий в биологическом образце. Предпосылки изобретения Уже давно признается наличие резистентности у бактерий к антибиотикам, и в настоящее время она рассматривается в качестве серьезной проблемы здравоохранения во всем мире. В результате наличия резистентности некоторые бактериальные инфекции либо с трудом поддаются лечению антибиотиками, либо вовсе не лечатся. Данная проблема стала особенно серьезной при недавнем развитии множественной лекарственной устойчивости у некоторых штаммов бактерий, таких как Streptococcus pneumoniae (SP), Mycobacterium tuberculosis и Enterococcus. Появление энтерококков, резистентных к ванкомицину, вызывает особую тревогу, поскольку ранее ванкомицин был единственным эффективным антибиотиком для лечения данной инфекции, и для многих инфекций считался препаратом «последнего средства». Несмотря на то, что многие другие резистентные к лекарственным препаратам бактерии не являются возбудителями заболеваний, угрожающих жизни, например, такие как энтерококки, имеется опасение того, что гены, вызывающие устойчивость, могут распространиться на более опасные для жизни микроорганизмы, такие как Staphylococcus aureus, на которые уже распространяется устойчивость к метициллину (De Clerg et al., Current Opinion in Anti-infective Investigational Drugs, 1999, 1, 1; Levy “The Challenge of Antibiotic Resistance”, Scientific American, March, 1998). Другой проблемой является то, как быстро развивается резистентность к антибиотикам. Например, до 1960 года все SP были чувствительны к пенициллину, и в 1987 году – только 0,02% штаммов SP в США были резистентными. Однако в 1995 году сообщалось, что резистентность SP к пенициллину составляла уже примерно 7% и в некоторых районах США – 30% (Lewis, FDA Consumer magazine (September, 1995); Gershman in The Medical Reporter, 1997). Больницы, в частности, являются центрами появления и распространения устойчивых к лекарственным препаратам организмов. Инфекции, имеющие место в больницах, известные как нозокомиальные инфекции, становятся все возрастающей серьезной проблемой. Из двух миллионов американцев, ежедневно инфицированных в больницах, у более чем половины имеются инфекции, возбудители которых являются резистентными, по меньшей мере, к одному антибиотику. По сообщениям Центра по контролю заболеваний, в 1992 году более чем 13000 пациентов больниц умерло в результате бактериальных инфекций, возбудители которых были резистентны к лечению антибиотиками (Lewis, “The Rise of Antibiotic-Resistant Infections”. FDA Consumer magazine, Sept., 1995). Ввиду необходимости борьбы с резистентными к лекарственным препаратам бактериями и в связи со все нарастающей нехваткой эффективных доступных лекарственных препаратов существует возрождающийся интерес к открытию новых антибиотиков. Одной привлекательной стратегией для разработки новых антибиотиков является ингибирование активности ДНК-гиразы, фермента бактерий, необходимого для репликации ДНК, и, следовательно, необходимого для роста и деления бактериальных клеток. Активность гиразы также связана с событиями, происходящими во время транскрипции, репарации и рекомбинации ДНК. Гираза представляет собой одну из топоизомераз, группы ферментов, которые катализируют взаимопревращение топологических изомеров ДНК (в основном смотри Kornberg and Baker, DNA Replication, 2d Ed., Chapter 12, 1992, W.H.Freeman and Co.; Drlica, Molecular Microbiology, 1992, 6, 425; Drlica and Zhao, Microbiology and Molecular Biology Reviews, 1997, 61, 377). Сама гираза контролирует суперспирализацию ДНК и снимает топологический стресс, который имеет место, когда цепи ДНК исходного дуплекса раскручиваются в процессе репликации. Гираза также катализирует преобразование релаксированного, замкнутого кольцевого дуплекса ДНК в отрицательно заряженную сверхспиральную форму, которая является более благоприятной для рекомбинации. Механизм сверхспирализации включает оборачивание гиразы вокруг области ДНК, двухцепочечный разрыв в данной области, прохождение второй области ДНК через место разрыва и соединение разорванных цепей. Подобный механизм расщепления характерен для топоизомеразы типа II. Реакция сверхспирализации запускается при присоединении АТФ к гиразе. Затем АТФ отщепляется в результате гидролиза в процессе реакции. Данное связывание АТФ и последующий гидролиз приводят к конформационным изменениям в связанной с ДНК гиразе, которые необходимы для проявления ее активности. Также было установлено, что степень сверхспирализации ДНК (или релаксации) зависит от соотношения АТФ/АДФ. При отсутствии АТФ гираза способна только к релаксации сверхспирализованной ДНК. Бактериальная ДНК-гираза представляет собой тетрамер массой 400 kDa, состоящий из двух субъединиц А (GyrA) и двух субъединиц В (GyrB). Связывание и расщепление ДНК связано с активностью GyrA, в то время как АТФ связывается и гидролизуется под действием белка GyrB. GyrB состоит из домена с аминоконцом, который обладает АТФазной активностью, и домена с карбоксиконцом, который взаимодействует с GyrA и ДНК. В противоположность у эукариот топоизомеразы типа II представляют собой гомодимеры, которые могут релаксировать отрицательные и положительные суперспирали, но не могут приводить к появлению отрицательных суперспиралей. В идеале антибиотик, действие которого основано на ингибировании активности ДНК-гиразы бактерий, будет избирательным для данного фермента и будет относительно неактивен для топоизомераз типа II эукариот. Широко используемые антибиотики на основе хинолонов ингибируют активность ДНК-гиразы и бактерий. Примеры хинолонов включают ранее полученные соединения, такие как налидиксовая кислота и оксолиниковая кислота, а также более поздние, более эффективные фторхинолоны, такие как норфлоксацин, ципрофлоксацин и тровафлоксацин. Данные соединения связываются с GyrA и стабилизируют расщепленный комплекс, таким образом ингибируя активность гиразы в целом, приводя к гибели клеток. Однако признается в качестве проблемы наличие резистентности к данной группе соединений (WHO Report, “Use of Quinolones in Food Animals and Potential Impact on Human Health”, 1998). Для хинолонов, а также антибиотиков других групп характерно, что у бактерий, подвергшихся воздействию более старых препаратов, быстро развивается перекрестная резистентность к более эффективным соединениям из этой же группы. Значительно меньше сведений об ингибиторах, которые связываются с GyrB. Примеры включают кумарины, новобиоцин и кумермицин А1, циклотиалидин, цинодин и клероцидин. Было показано, что кумарины очень прочно связываются с GyrB. Например, новобиоцин образует сеть из водородных связей с белком и несколько гидрофобных связей. Оказалось, что несмотря на то, что новобиоцин и АТФ связываются на связывающем сайте АТФ, имеется минимальное перекрытие в ориентации связывания двух соединений. Перекрывающиеся участки представляют собой остатки сахара молекулы новобиоцина и аденин в АТФ (Maxwell, Trends in Microbiology, 1997, 5, 102). У резистентных к кумаринам бактерий наиболее распространенная точечная мутация находится на поверхности остатка аргинина, который связывается с карбонилом кольца кумарина (Arg136 в GyrB E. coli). Несмотря на то, что ферменты с подобной мутацией обладают более низкой активностью в отношении суперспирализации и АТФазной активности, они также менее чувствительны к ингибированию под действием кумаринов (Maxwell, Mol. Microbiol., 1993, 9, 681). Несмотря на то, что кумарины являются сильными ингибиторами суперспирализации, имеющей место под действием гиразы, они не применяются широко в качестве антибиотиков. Причиной отказа от их использования в подобном качестве является их низкое проникновение в бактерии, токсичность для эукариот и низкая растворимость в воде ((Maxwell, Trends in Microbiology, 1997, 5, 102). Было бы желательно разработать новый, эффективный ингибитор GyrB, который не обладал бы вышеуказанными недостатками. Подобный ингибитор будет привлекательным кандидатом в качестве антибиотика без наличия в прошлом проблем с резистентностью, характерной для других групп антибиотиков. Движение репликативной вилки вдоль кольцевой ДНК может привести к появлению топологических изменений как впереди репликационного комплекса, так и сзади уже реплицированных областей (Champoux J. J., Annu. Rev. Biochem., 2001, 70, 369-413). Несмотря на то, что ДНК-гираза может приводить к появлению отрицательных суперспиралей для компенсации топологических стрессов впереди репликативной вилки, некоторое перекручивание может иметь место в уже реплицированной области ДНК, приводя к образованию прецепочек. Если они не удаляются, то наличие прецепочек может привести к наличию взаимосвязанных (цепочечных) дочерних молекул в конце репликации. Топоизомераза IV ответственна за отделение цепочечных дочерних плазмид, а также за удаление прецепочек, образовавшихся во время репликации, в конечном итоге приводя к расхождению дочерних молекул в дочерние клетки. Топоизомераза IV состоит из двух ParC и двух ParE субъединиц в виде С2Е2 тетрамера (где С и Е представляют собой мономеры, гомологичные соответственно мономерам А и В гиразы), для которого необходим гидролиз АТФ (на N-конце субъединицы Е) для повторного вхождения фермента в каталитический цикл. Топоизомераза IV является высококонсервативной среди бактерий и необходима для репликации бактерий (Drlica and Zhao, Microbiol. Mol. Biol. Rev., 1997, 61, 377). Несмотря на то, что меньшее внимание уделялось ингибиторам, действие которых направлено на ParE топоизомеразы IV, активно исследовалось действие более новых хинолонов на область ParC (Hooper D.C., Clin. Infect. Dis., 2000, 31 (Suppl 2): S24-28). Было показано, что моксифлоксацин и гатифлоксацин обладают более сбалансированной активностью в отношении гиразы и топоизомеразы IX, приводя к более широкому спектру действия против грамположительных бактерий, а также снижению степени резистентности, вызванной направленной мутацией. В данных случаях предрасположенность ограничивается чувствительностью второй мишени к антибиотику. Таким образом, средства, эффективно ингибирующие множественные основные мишени, могут иметь широкий спектр действия, обладать повышенной антибактериальной активностью, повышенной активностью против моносайтовых мутантов, и/или приводить к снижению уровня спонтанной резистентности. Поскольку резистентность бактерий к антибиотикам стала важной проблемой здравоохранения, имеется насущная потребность в разработке более новых и более эффективных антибиотиков. Конкретнее, имеется потребность в антибиотиках, которые представляют собой новую группу соединений, ранее не используемых для лечения бактериальной инфекции. Подобные соединения будут особенно пригодными для лечения нозокомиальных инфекций в больницах, где становится особенно распространенным появление и распространение резистентных бактерий. Сущность изобретения В настоящее время было установлено, что соединения по данному изобретению и их фармацевтически приемлемые композиции являются эффективными в качестве ингибиторов активности гиразы и/или топоизомеразы IV. Данные соединения имеют общую формулу I:
или ее фармацевтически приемлемую соль, где X, Q, W, R1, R2 и R3 имеют значения, определенные ниже. Данные соединения и их фармацевтически приемлемые композиции могут быть использованы для лечения или ослабления тяжести бактериальных инфекций. В частности, соединения по настоящему изобретению могут использоваться для лечения или ослабления тяжести инфекций мочевых путей, пневмонии, простатита, инфекций кожи и мягких тканей, внутрибрюшных инфекций или инфекций у пациентов с лихорадочной нейтропенией. Описание изобретения Настоящее изобретение относится к соединению формулы I:
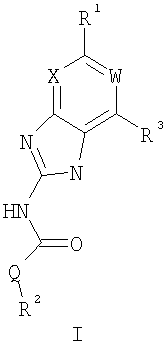
или его фармацевтически приемлемой соли, где: Q представляет собой -СН2-, -NH- или -О-; W выбран из атома азота или С-R4; X выбран из СН или CF; R1 представляет собой 5-6-членное арильное кольцо, содержащее 1-3 гетероатомов, независимо выбранных из атомов кислорода, азота или серы, где: R1 замещен 0-3 группами, независимо выбранными из R, оксо, CO2R’, OR’, N(R’)2, SR’, NO2, атома галогена, CN, C(O)N(R’)2, NR’C(О)R’, SO2R’, SO2N(R’)2 или NR’SO2R’; каждый R’ независимо выбран из атома водорода, С1-4алифатической группы или 5-6-членного насыщенного, ненасыщенного или арильного кольца, содержащего 0-3 гетероатомов, независимо выбранных из атомов азота, кислорода или серы, где: R’ замещен 0-3 группами, независимо выбранными из атома галогена, оксо, R0, N(R0)2, OR0, CO2R0, NR0C(O)R0, C(O)N(R0)2, SO2R0, SO2N(R0)2 или NR0SO2R0; каждый R0 независимо выбран из атома водорода или С1-4алифатической группы; R2 выбран из атома водорода или С1-3алифатической группы; R3 выбран из C(O)NHR, C(O)N(R)2, CH(O), C(O)R, CO2R, C(O)C(O)N(R2)R, SO2R, SO2N(R)2, SO2NHR, C(R’)=NOR, C(R’)=NOH, C(R’)=NR, C(R’)=N-N(R2)R, NO или NO2; каждый R независимо выбран из Т-Ar или С1-6алифатической группы, где: указанная C1-6алифатическая группа замещена 0-3 группами, независимо выбранными из R’, оксо, CO2R’, OR’, N(R’)2, SR’, NO2, атома галогена, CN, C(O)N(R’)2, NR’C(O)R’, SO2R’, SO2N(R’)2 или NR’SO2R’; Т представляет собой (СН2)y, где y равно 0, 1 или 2; Ar выбран из: (а) 3-8-членного насыщенного, ненасыщенного или арильного кольца; (b) 3-7-членного гетероциклического кольца, содержащего 1-3 гетероатома, независимо выбранные из атомов азота, кислорода или серы; или (с) 5-6-членного гетероарильного кольца, содержащего 1-3 гетероатома, независимо выбранные из атомов азота, кислорода или серы, где: Ar замещен 0-3 группами, независимо выбранными из R’, оксо, CO2R’, OR’, N(R’)2, SR’, NO2, атома галогена, CN, C(O)N(R’)2, NR’C(О)R’, SO2R’, SO2N(R’)2 или NR’SO2R’; и R4 выбран из атомов водорода, фтора или ОСН3. В том смысле, в котором они здесь используются, следует применять следующие определения, если не указывается иначе. Выражение «необязательно замещенный» используется взаимозаменяемо с выражением «замещенный или незамещенный». Если не указано иначе, необязательно замещенная группа может содержать заместитель в каждом замещаемом положении группы, и каждое замещение не зависит от другого. Термин «алифатический» или «алифатическая группа», в том смысле, в котором он здесь используется, означает прямую или разветвленную С1-С8 углеводородную цепь, которая является полностью насыщенной или которая содержит одну или несколько ненасыщенных связей, или моноциклический С3-С8 углеводород, или бициклический С8-С12 углеводород, который полностью насыщен, или который содержит одну или несколько ненасыщенных связей, но который не является ароматическим (также относится здесь к «карбоциклу» или «циклоалкилу»), который имеет одно место присоединения к остальной части молекулы, где любое отдельное кольцо в указанной бициклической кольцевой системе содержит 3-7 членов. Например, подходящие алифатические группы включают, но не ограничиваются прямым или разветвленным алкилом, алкенилом, алкинилом и их сочетаниями такими, как (циклоалкил)алкил, (циклоалкенил)алкил или (циклоалкил)алкенил. Термины «алкил», «алкокси», «гидроксиалкил», «алкоксиалкил» и «алкоксикарбонил», используемые самостоятельно или как часть большей группы, включают прямые или разветвленные цепи, содержащие от 1 до 12 атомов углерода. Термины «алкенил» и «алкинил», используемые самостоятельно или как часть большей группы, включают прямые или разветвленные цепи, содержащие от 2 до 12 атомов углерода. Термин «гетероатом» означает атом азота, кислорода или серы и включает любую окисленную форму атома азота и серы и кватернизованную форму любого основного азота. Также термин «атом азота» включает замещаемый азот в гетероциклическом кольце. В качестве примера в насыщенном или частично ненасыщенном кольце, содержащем 0-3 гетероатомов, выбранных из атомов кислорода, серы или азота, азот может представлять собой N (как в 3,4-дигидро-2Н-пирролиле), NH (как в пирролидиниле) или NR+ (как в N-замещенном пирролидиниле). Термин «ненасыщенный», в том смысле, в котором он здесь используется, означает группу, содержащую одну или несколько ненасыщенных связей, и включает арильные кольца. Термин «арил», используемый самостоятельно или как часть большей группы, например, в «аралкиле», «аралкокси» или «арилоксиалкиле», относится к моноциклическим, бициклическим и трициклическим кольцевым системам, содержащим в целом от 5 до 14 членов в кольце, где, по меньшей мере, одно кольцо в системе является ароматическим и где каждое кольцо в системе содержит от 3 до 7 членов в кольце. Термин «арил» можно использовать взаимозаменяемо с термином «арильное кольцо». Термин «арил» также относится к гетероарильным кольцевым системам, имеющим значения, определенные ниже. Термин «гетероцикл», «гетероциклил» или «гетероциклический», в том смысле, в котором он здесь используется, означает неароматические, моноциклические, бициклические или трициклические кольцевые системы, содержащие от 5 до 14 членов в кольце, в котором один или более членов в кольце представляет собой гетероатом, где каждое кольцо в системе содержит от 3 до 7 членов в кольце. Термин «гетероарил», используемый самостоятельно или как часть большей группы, например в «гетероаралкиле» или «гетероарилалкокси», относится к моноциклическим, бициклическим и трициклическим кольцевым системам, содержащим в целом от 5 до 14 членов в кольце, где, по меньшей мере, одно кольцо в системе является ароматическим, по меньшей мере, одно кольцо в системе содержит один или более гетероатомов, и где каждое кольцо в системе содержит от 3 до 7 членов в кольце. Термин «гетероарил» может использоваться взаимозаменяемо с термином «гетероарильное кольцо» или термином «гетероароматический». Комбинация заместителей или переменных допускается, если подобная комбинация приводит к стабильному или химически возможному соединению. Стабильное соединение или химически возможное соединение представляет таковое, которое существенно не изменяется при хранении при температуре 40°С или ниже, при отсутствии влаги или других химических реакционноспособных условий, по меньшей мере, в течение недели. Специалистам в данной области, очевидно, понятно, что некоторые соединения по данному изобретению могут быть в таутомерных формах, где все подобные таутомерные формы соединений входят в объем изобретения. Если не указано иначе, подразумевается, что структуры, представленные здесь, также включают все стереохимические формы структуры, т.е. R- и S-конфигурации для каждого асимметричного центра. Следовательно, отдельные стереохимические изомеры, а также энантиомерные и диастереоизомерные смеси настоящих соединений находятся в объеме изобретения. Если не указано иначе, подразумевается, что структуры, представленные здесь, также включают соединения, которые отличаются только по присутствию одного или более обогащенных изотопами атомов. Например, соединения, имеющие настоящие структуры, за исключением замещения водорода на дейтерий или тритий, или замещения углерода на 13С- или 14С-обогащенный углерод, входят в объем настоящего изобретения. Подобные соединения могут использоваться, например, в качестве аналитических инструментов или зондов в биологических анализах. По одному воплощению настоящее изобретение относится к соединению формулы I, где Q представляет собой -NH-. По другому воплощению настоящее изобретение относится к соединению формулы I, где Q представляет собой -О-. По другому воплощению настоящее изобретение относится к соединению формулы I, где Q представляет собой -СН2-. Предпочтительные группы R1 в формуле I выбраны из необязательно замещенного фенильного или 5-6-членного гетероарильного кольца, содержащего 1-2 атомов азота. Более предпочтительные группы R1 в формуле I выбраны из необязательно замещенного пирид-2-ила, пирид-3-ила, пирид-4-ила, пиримидин-2-ила, пиримидин-4-ила, пиримидин-5-ила, имидазол-1-ила, имидазол-2-ила, имидазол-4-ила или имидазол-5-ила. Наиболее предпочтительные группы R1 в формуле I представляют необязательно замещенные кольца, выбранные из пирид-3-ила, пирид-4-ила, пиримидин-5-ила или имидазол-1-ила. По другому воплощению R1 представляет собой пиридоновое кольцо. Более предпочтительно, R1 является 4-пиридоном. Предпочтительные заместители в группе R1 в формуле I, если они имеются, выбраны из атома галогена, оксо, R, CO2R’, OR’, N(R’)2, SR’, C(O)N(R’)2, NR’C(O)R’, SO2R’, SO2N(R’)2 или NR’SO2R’. Когда R1 замещен Т-Ar, то предпочтительные заместители включают таковые, в которых Ar представляет собой необязательно замещенное кольцо, выбранное из 5-6-членного гетероциклического кольца, содержащего 1-3 гетероатомов, независимо выбранных из атомов азота, кислорода или серы или 5-6-членного арильного кольца, содержащего 0-3 гетероатомов, независимо выбранных из атомов азота, кислорода или серы. Более предпочтительные заместители в группе R1 в формуле I, если они имеются, выбраны из оксо, атома фтора, атома хлора, N(CH3)2, NHCH2CH3, NH-циклопропила, NH2, NHC(O)CH3, C(O)NHциклопропила, метила, этила, трет-бутила, изобутила, циклопропила, изопропила, СН2фенила, СН2пиридин-2-ила, СН2пиридин-3-ила, СН2пиридин-4-ила, ОН, ОСН3, ОСН2СН3, ОСН2фенила, ОСН2пиридин-3-ила, СН2пиперидинила, СН2циклопропила или СН2СН2ОСН3. Когда два заместителя в смежных положениях R1 в формуле I взяты вместе, то они образуют необязательно замещенное кольцо, конденсированное с R1. Предпочтительными кольцами, образованными таким образом, являются 5-6-членные насыщенные, частично ненасыщенные или арильные кольца, содержащие 0-2 гетероатомов, независимо выбранных из атомов азота, кислорода или серы. Более предпочтительные кольца, конденсированные с R1, выбраны из 5-членного насыщенного кольца, содержащего два атома кислорода, или 6-членного насыщенного кольца, содержащего два атома кислорода. Предпочтительные заместители в указанном кольце, конденсированном с R1, представляют собой атом галогена и более предпочтительно атом фтора. Предпочтительные группы R2 в формуле I выбраны из метила, этила, изопропила или циклопропила. Более предпочтительными группами R2 в формуле I являются метил, циклопропил или этил. Наиболее предпочтительным R2 в формуле I является этил. Предпочтительные группы R3 в формуле I выбраны из C(O)NHR, C(O)R, C(R)=NOR, C(R)=NOH или CO2R, где: каждый R независимо выбран из необязательно замещенной С1-4алифатической группы или Т-Ar, где: Т представляет собой (СН2)y, где y равно 0, 1 или 2; и Ar представляет собой необязательно замещенное кольцо, выбранное из 5-6-членного насыщенного, ненасыщенного или арильного кольца, 5-6-членного гетероциклического кольца, содержащего 1-2 гетероатомов, независимо выбранных из атомов азота, кислорода или серы, или 5-6-членного гетероарильного кольца, содержащего 1-2 гетероатомов, независимо выбранных из атомов азота, кислорода или серы. Более предпочтительные группы R3 в формуле I выбраны из C(O)NHR, C(O)R, C(R)=NOR, C(R)=NOH или CO2R, где: каждый R независимо выбран из С1-4алифатической группы или Т-Ar, где: указанная С1-4алифатическая группа замещена 0-2 группами, независимо выбранными из атома галогена, OR’ или N(R’)2; Т представляет собой (СН2)y, где y равно 0, 1 или 2; и Ar выбран из пирролидинила, фуранила, тиазолила, тетрагидрофуранила, пиримидинила, пиразинила, пиридила, пиперидинила, имидазолила, пиридазинила, изоксазолила, пиразолила, тетрагидропиранила или циклопентена, где: Ar замещен 0-2 группами, независимо выбранными из R’, оксо, OR’ или N(R’)2. Наиболее предпочтительные группы R3 в формуле I выбраны из СО2СН3, C(R)=NOR, C(R)=NOH или C(O)NHR, где каждый R независимо выбран из следующих групп: циклопропил, СН2СН2(1-метилпирролидин-2-ил), СН2(1-этилпирролидин-2-ил), СН2СН2пирролидин-1-ил, СН2фуран-2-ил, тиазол-2-ил, СН2тетрагидрофуран-2-ил, пиримидин-2-ил, пиразин-2-ил, СН2пиридин-2-ил, пиридин-3-ил, пиридин-4-ил, СН(СН3)СН2OCH3, CH2CF3, CH2CH3, CH2CH2N(CH2CH3)2, CH2CH2N(CH3)2, CH2CH2OCH3, CH2C По другому воплощению R3, предпочтительно, представляет собой C(R)=NOR или C(R)=NOH. По другому воплощению R3, предпочтительно, представляет собой С(О)R. Предпочтительно, R4 в формуле I представляет собой атом водорода или фтора. Более предпочтительно, R4 в формуле I является атомом водорода. Соединения по настоящему изобретению подпадают под группу соединений, описанных в РСТ/US01/48855. Однако заявители установили, что наличие группы R3, имеющей значения, определенные выше, придает удивительную и неожиданно высокую ферментативную или противомикробную активность. По предпочтительному воплощению настоящее изобретение относится к соединению формулы II или II’:
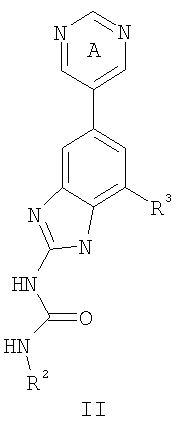
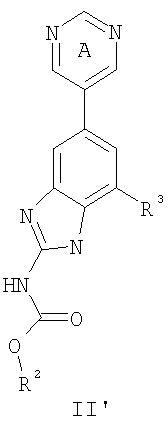
или его фармацевтически приемлемой соли, где R2 и R3 имеют значения, определенные выше, и кольцо А замещено 0-2 группами, независимо выбранными из N(R’)2, OR’, R или SR’. Предпочтительные группы R2 и R3 в формулах II и II’ являются теми же, что описаны для формулы I выше. По другому предпочтительному воплощению настоящее изобретение относится к соединению формулы III или III’.
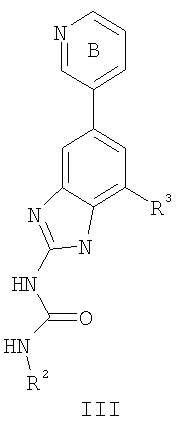
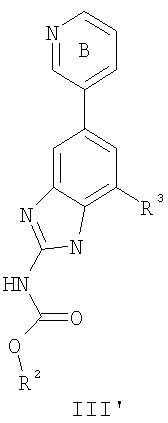
или его фармацевтически приемлемой соли, где R2 и R3 имеют значения, определенные выше, и кольцо В необязательно замещено 0-2 группами, независимо выбранными из R или оксо, где R‘, предпочтительно, представляет собой атом водорода или С1-3алифатическую группу, необязательно замещенную N(R0)2. Предпочтительные группы R2 и R3 в формулах III и III’ являются теми же, что описаны для формулы I выше. По другому предпочтительному воплощению настоящее изобретение относится к соединению формулы III-а:
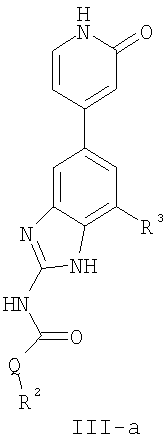
или его фармацевтически приемлемой соли, где R2 и R3 имеют значения, определенные выше, и изображенное пиридоновое кольцо замещено 0-2 группами, независимо выбранными из атома галогена, оксо, R, CO2R’, OR’, N(R’)2, SR’, C(O)N(R’)2, NR’C(O)R’, SO2R’, SO2N(R’)2 или NR’SO2R’. Предпочтительные группы R2 и R3 в формуле III-а являются теми же, что описаны для формулы I выше. Предпочтительные заместители в пиридоновом кольце в формуле III-a являются теми же, что описаны выше в качестве предпочтительных заместителей для R1 в формуле I. По одному воплощению настоящее изобретение относится к соединению формулы III-a, где Q представляет собой -NH-. По другому воплощению настоящее изобретение относится к соединению формулы III-а, где Q представляет собой -О-. По другому воплощению настоящее изобретение относится к соединению формулы I, где Q представляет собой -СН2-. По другому предпочтительному воплощению настоящее изобретение относится к соединению формулы III-b:
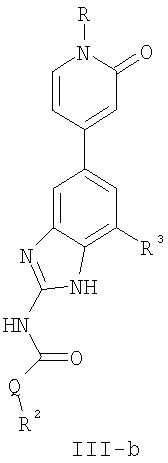
или его фармацевтически приемлемой соли, где R, R2 и R3 имеют значения, определенные выше. Предпочтительные группы R2 в формуле III-b являются теми же, что описаны для групп R2 в формуле I выше. Предпочтительные группы R3 в формуле III-b представляют те же, что описаны для групп R3 в формуле I выше. Предпочтительные заместители R в пиридоновом кольце в формуле III-b выбраны из T-Ar, где Ar представляет собой необязательно замещенное кольцо, выбранное из 5-6-членного гетероциклического кольца, содержащего 1-3 гетероатомов, независимо выбранных из атомов азота, кислорода или серы, или из 5-6-членного арильного кольца, содержащего 0-3 гетероатомов, независимо выбранных из атомов азота, кислорода или серы. Предпочтительный Ar включает фенил или пиридил. Более предпочтительные заместители R в пиридоновом кольце в формуле III-b выбраны из метила, этила, трет-бутила, изобутила, циклопропила, изопропила, СН2фенила, СН2пиридин-3-ила, СН2пиперидинила, СН2циклопропила или СН2СН2ОСН3. По одному воплощению настоящее изобретение относится к соединению формулы III-b, где Q представляет собой -NH-. По другому воплощению настоящее изобретение относится к соединению формулы III-b, где Q представляет собой -О-. По другому воплощению настоящее изобретение относится к соединению формулы I, где Q представляет собой -СН2-. По другому предпочтительному воплощению настоящее изобретение относится к соединению формулы IV:
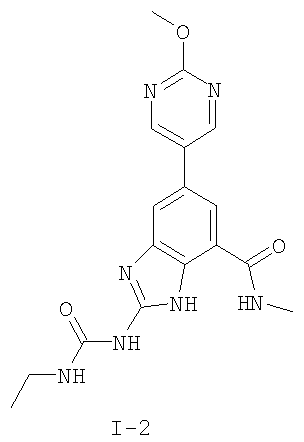
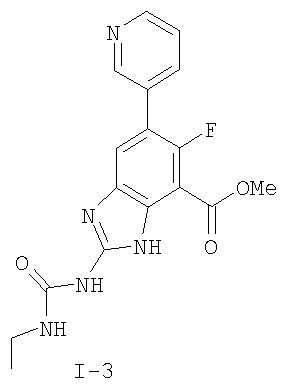
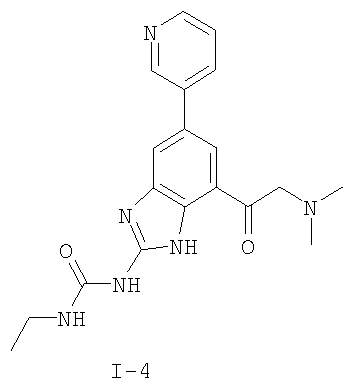
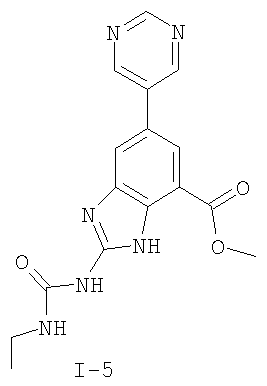
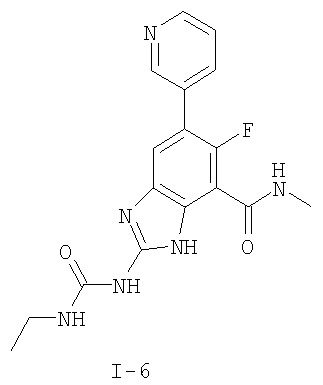
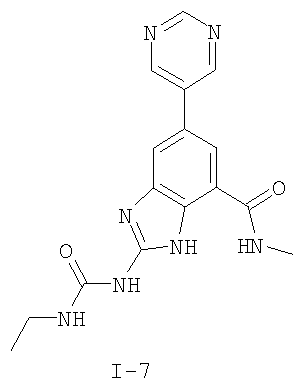
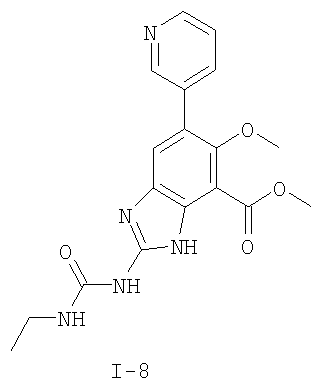
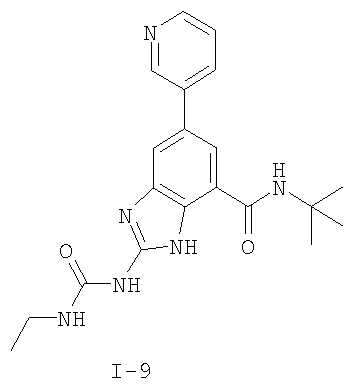
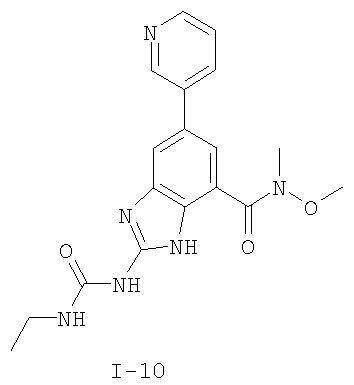
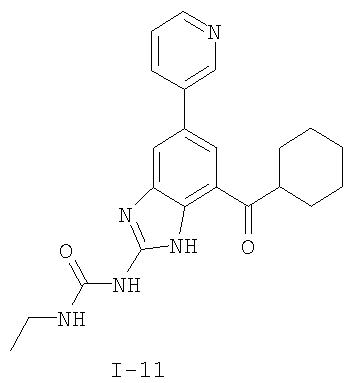
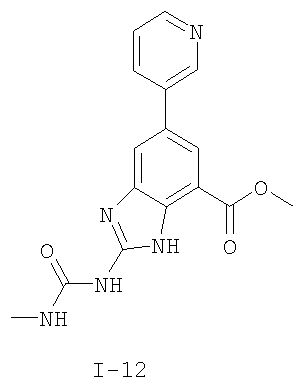
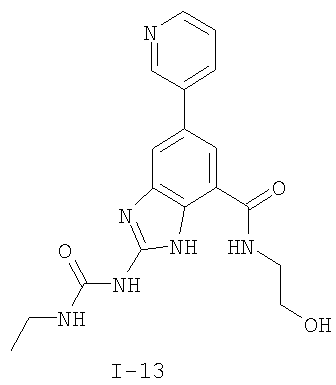
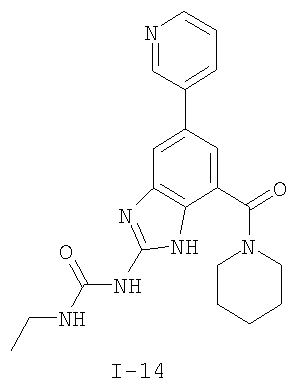
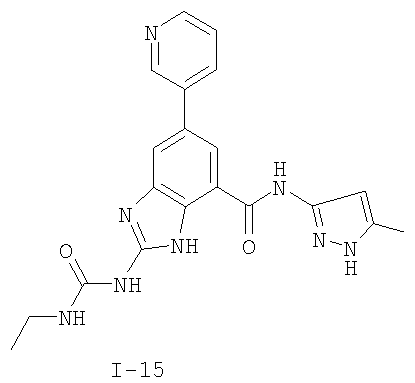
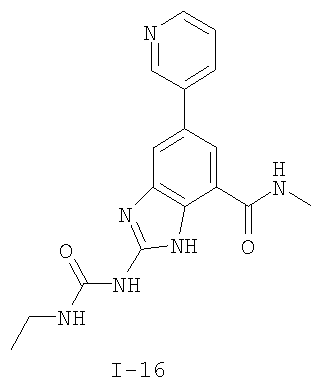
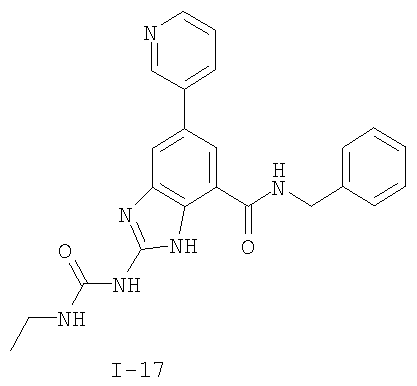
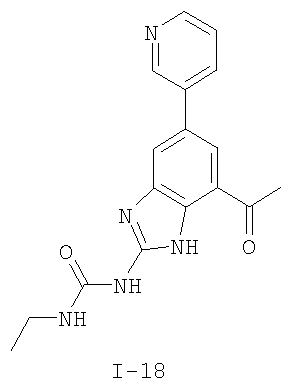
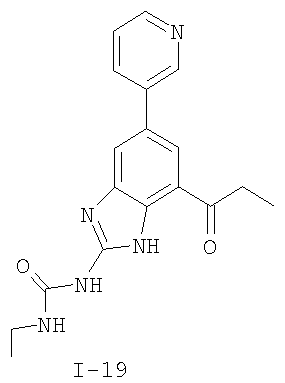
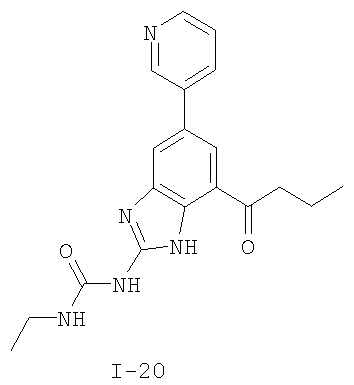
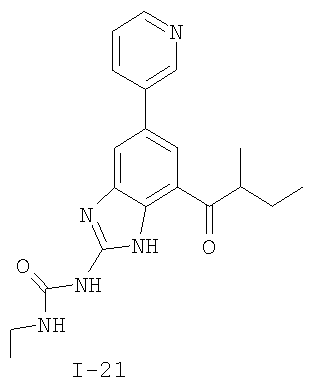
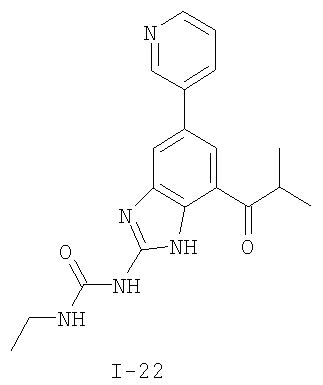
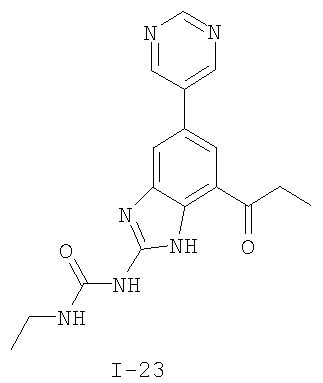
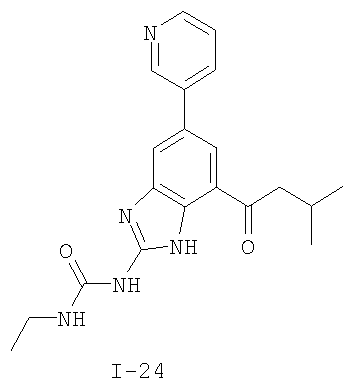
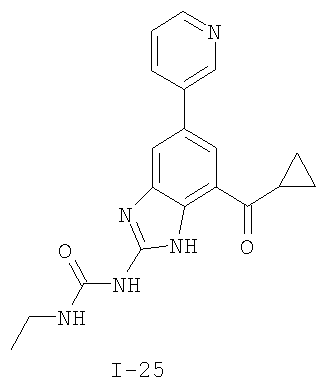
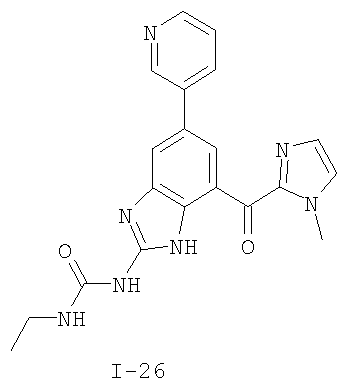
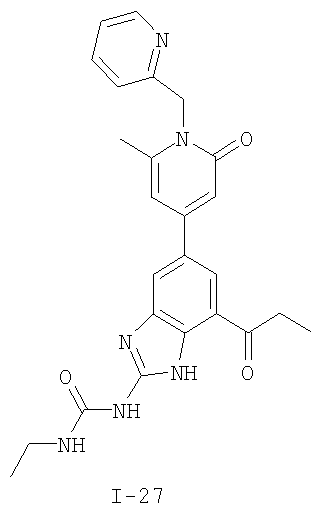
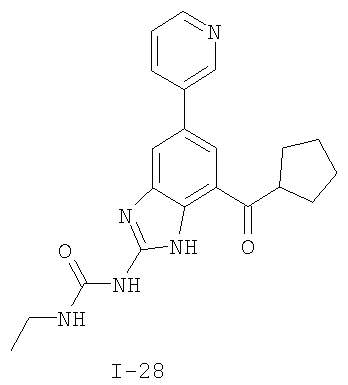
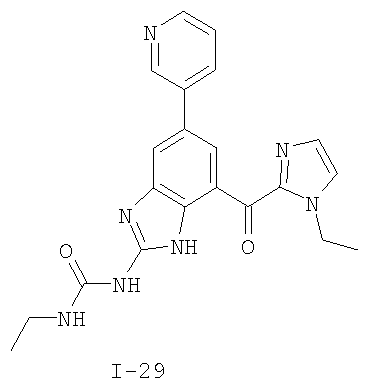
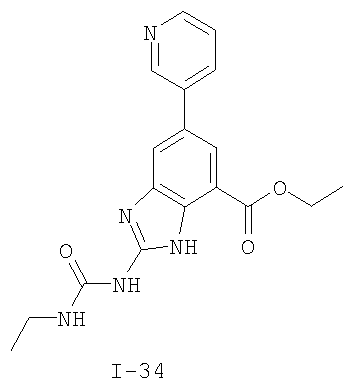
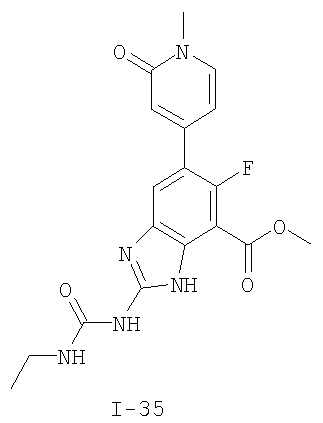
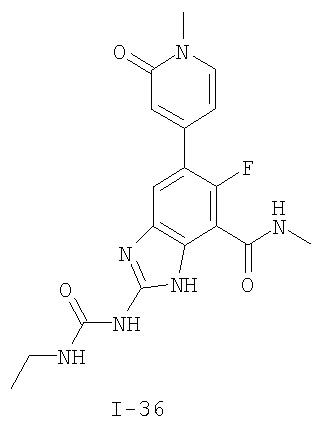
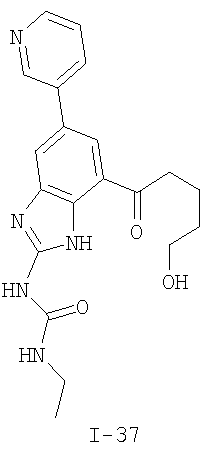
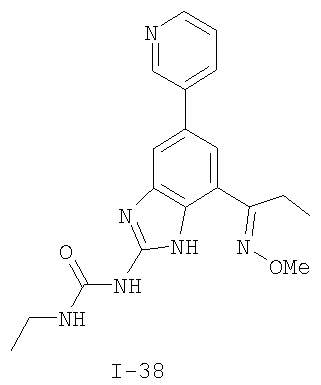
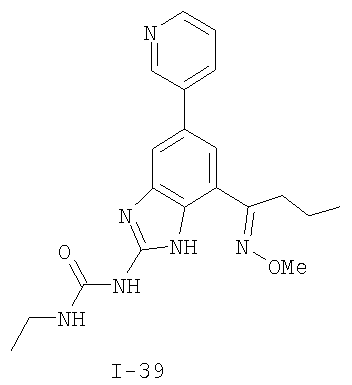
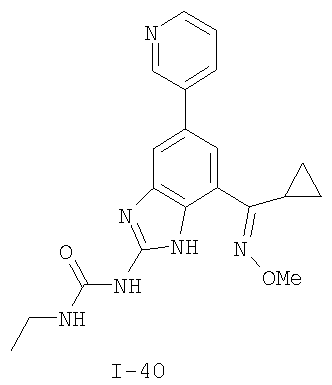
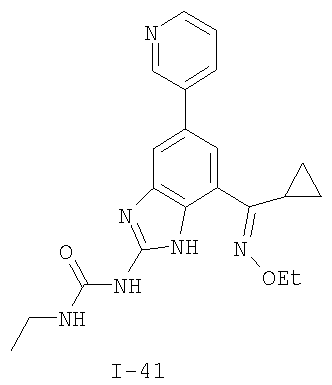
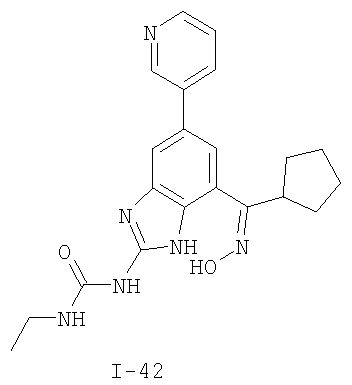
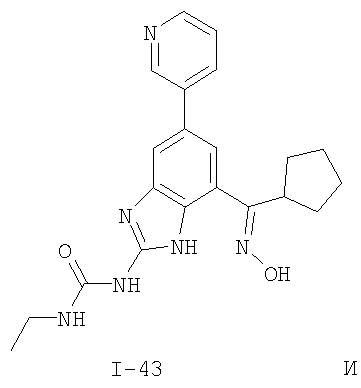
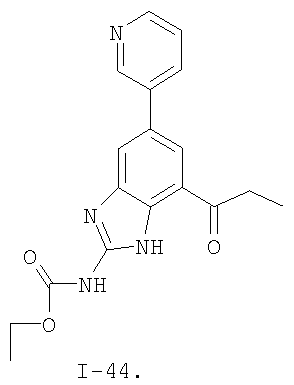
или его фармацевтически приемлемой соли, где R2 и R3 имеют значения, определенные выше, и изображенное имидазольное кольцо необязательно замещено в 4-м положении C(O)N(R’)2, оксо и/или замещено во 2-м положении R. Предпочтительные группы R2 и R3 в формуле IV являются теми же, что описаны для формулы I выше. По одному воплощению настоящее изобретение относится к соединению формулы IV, где Q представляет собой -NH-. По другому воплощению настоящее изобретение относится к соединению формулы IV, где Q представляет собой -О-. По другому воплощению настоящее изобретение относится к соединению формулы I, где Q представляет собой -СН2-. По одному воплощению настоящее изобретение относится к соединению формул I, II, III, III-a, III-b или IV, или ее любой подгруппы, где Х представляет собой СН. По другому воплощению настоящее изобретение относится к соединению формул I, II, III, III-a, III-b или IV, или ее любой подгруппы, где Х представляет собой CF. По другому воплощению настоящее изобретение относится к соединению формул I, II, III, III-a, III-b или IV, или ее любой подгруппы, где W представляет собой атом азота. По другому воплощению настоящее изобретение относится к соединению формул I, II, III, III-a, III-b или IV, или ее любой подгруппы, где W представляет собой С-R4. По другому воплощению настоящее изобретение относится к соединению формул I, II, III, III-a, III-b или IV, или ее любой подгруппы, где W представляет собой СН. Примеры структур соединений формулы I представлены в таблице 1 ниже. Таблица 1
Соединения по настоящему изобретению можно получить способами, известными специалистам в данной области для аналогичных соединений, а также – как представлено на общих схемах реакций I-IX ниже. Подробно условия, использованные для получения данных соединений, приведены в примерах. Реакционная схема I
На реакционной схеме I выше представлен общий способ получения N’-алкил-N-цианомочевин (3), пригодный для получения соединений по настоящему изобретению. На стадии (а) цианамид (2) обрабатывают этилизоцианатом в присутствии основания с получением после подкисления соединения 3. Несмотря на то, что на схеме представлена N’-этил-N-цианомочевина, специалистам в данной области, очевидно, понятно, что самые разнообразные алкилизоцианаты могут быть использованы в реакционных условий схемы I с образованием различных N’-алкил-N-цианомочевин. На стадии (а) цианамид (2) можно обработать алкилизоцианатом в присутствии различных оснований с образованием цианомочевины (3). Подходящие основания, пригодные для получения соединения 3, включают гидриды металлов, такие как NaH и KH, алкоголяты металлов, такие как трет-бутилат натрия и трет-бутилат калия, и гидроксиды металлов, такие как гидроксид натрия, гидроксид калия, гидроксид цезия и гидроксид лития. На стадии (а) основания, гидрид металла, алкоголят металла и гидроксид металла, используют для получения соли металла и цианомочевины (3), имеющей формулу 3а:
где М представляет собой натрий, Li, K, Rb или Cs. Предпочтительно, М является натрием. Стадию (а) проводят в различных растворителях, включая ТГФ, спирты, метиленхлорид, DME, EtOAc, iPrOAc, хлорбензол, метил трет-бутиловый эфир, толуол, гептан и циклогексан. Предпочтительно, растворителем, используемым на стадии (а), является безводный растворитель. Более предпочтительно, когда растворитель, используемый на стадии (а), является безводный ТГФ. Реакционная схема II
Реагенты и условия: (а) i трифторуксусный ангидрид, ii нитрат калия; b) HCl, MeOH; c) NaHCO3, тетракисфенилфосфин-Pd(O); d) H2, Pd/C; e) H2SO4. На реакционной схеме II представлен общий способ получения производных бензимидазола по настоящему изобретению. Броманилин (4) обрабатывают трифторуксусным ангидридом, затем нитратом калия с образованием нитропроизводного (5), у которого затем удаляют защиту обработкой кислотой с образованием амина (6). Затем 3-нитро-5-броманилин (6) подвергают взаимодействию с арилбороновой кислотой (7) в присутствии палладия с образованием биарильного производного (8). Нитрогруппу соединения 8 восстанавливают с образованием диамина 9, который обрабатывают N’-алкил-N-цианомочевиной с образованием бензимидазолов по данному изобретению (10). Реакции, представленные на схеме II выше, могут быть использованы на различных группах R1 и R3 по настоящему изобретению. Реакционная схема III
Реагенты и условия: (а)Pd(dppf)Cl2/KOAc, ДМСО, 80°С; b) Cu(Oac)2/пиридин, ДМФА; c) i H2, Pd/C, ii 3, H2SO4. На реакционной схеме III представлен общий способ получения соединений формулы IV, замещенных в 4-м положении С(О)N(R’)2 с использованием способов, в основном аналогичных описанным Kiyomori A. и Marcoux J.-F.; Buchwald S.L., Tetrahedron Letters, vol. 40, 1999, 2657-2660. Соединение 6 обрабатывают дибороновым эфиром в присутствии Pd(dppf)/ацетата калия в ДМСО при 80°С с получением промежуточного соединения 11. Соединение 11 обрабатывают 4-С(О)N(R’)2-имидазолом в присутствии ацетата меди с получением производного 4-С(О)N(R’)2-имидазол-1-ила, соединение 12. Нитрогруппу соединения 12 восстанавливают с образованием диамина, который, в свою очередь, обрабатывают N’-этил-N-цианомочевиной (3) с получением производного бензимидазола, соединение 13, как представлено на схеме II, стадии (е). Реакционная схема IV
На реакционной схеме IV выше представлен альтернативный общий способ получения соединений формулы I. Соединение 6 обрабатывают биспинаколадибором в присутствии Pd(dppf)/ацетата калия с получением промежуточного соединения 11, как показано выше на схеме III. Затем соединение 11 обрабатывают R1-трифлатом в присутствии (тетракистрифенилфосфин)палладия, хлорида лития и карбоната натрия с образованием соединения 14. Затем соединение 14 можно использовать для получения соединений по настоящему изобретению с использованием способов, в основном аналогичных представленным выше на схемах I-III. Реакционная схема V
Реагенты и условия: (а) галогенид R-магния, ТГФ, 0°С до комнатной температуры; b) Ac2, 80°C; c) HNO3; d) 6 н. водный раствор HCl; е) трифторуксусный ангидрид, затем KNO3; f) Na2CO3, MeOH/H2O (9:1), 65°С; g) R1 – боронат, Pd(PPh3)4, 1 н. водный раствор NaHCO3, DME, 90°C; h) SnCl22H2O, EtOH, кипячение с обратным холодильником и i) Na2SO4, EtOH/H2O (3:1), 90°С. На реакционной схеме V выше представлен общий способ получения соединений формулы I, где R3 представляет собой С(О)R. Цианопроизводное соединение 15 обрабатывают галогенидом R-магния с получением кетона, соединение 16. Нитросоединение 17 получают из соединения 16 обработкой уксусным ангидридом, затем азотной кислотой. Альтернативно, соединение 17 получают обработкой соединения 16 трифторуксусным ангидридом и нитратом калия. Затем нитропроизводное соединение 17 обрабатывают боронатом, как описано выше, с получением соединения 18. Нитрогруппу соединения 18 восстанавливают с образованием диамина, соединение 19 либо с помощью SnCl2 (стадия h), либо с помощью Na2S2O4 (стадия i). Затем диамин, соединение 19 можно использовать для получения соединений формулы I, где R3 представляет собой С(О)R, с использованием способов, в основном аналогичных представленным на схемах реакций I-IV выше. Реакционная схема VI
На реакционной схеме VI выше представлен общий способ получения соединений формулы I, где R3 представляет собой С(R)=NOR. Кетон, соединение 19 обрабатывают ацетатом калия и HCl·NH-OR с получением оксима, соединение 20. Затем соединение 20 можно использовать для получения соединений формулы I, где R3 представляет собой С(R)=NOR с использованием способов, в основном аналогичных представленным на схемах I-IV выше. Реакционная схема VII
На реакционной схеме VII представлен общий способ получения соединений формулы I, где W представляет собой CF, и R3 представляет собой СО2R. Соединение 24 получают из доступных исходных веществ с использованием способов, в основном аналогичных описанным Kim K.S. et al., J. Med. Chem. 1993, 36, 2335. Соединение 25 получают обработкой соединения 24 бромом в уксусной кислоте. Соединения по настоящему изобретению, где R3 представляет собой СО2R, можно получить из соединения 25 с использованием способов, в основном аналогичных описанным на схемах I-IV выше. Реакционная схема VIII
На реакционной схеме VIII выше представлен общий способ получения соединений по настоящему изобретению, где Q представляет собой -О-. Соединение 9, полученное по схеме II выше, обрабатывают 2-метил-2-тиопсевдомочевиной и R2-хлорформиатом с получением соединения 26. Данный способ в основном описан L.I.Kruse et al., J. Med. Chem. 1989, 32, 409-417. Реакционная схема IX
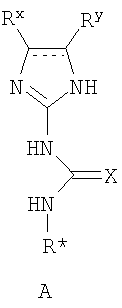
Реагенты и условия: (а) HNO3/H2SO4; b) NH4OH/диоксан/нагревание; с) диоксан/нагревание; d) Na2CO3/ДМФА/нагревание; е) H2/Pd-C/EtOH; f)способами, представленными выше. На реакционной схеме IX представлен альтернативный общий способ получения соединений по настоящему изобретению, где R1 представляет собой имидазол-1-ил. На стадии (а) дифторкетон обрабатывают азотной кислотой с получением нитросоединения 28. Затем соединение 28 обрабатывают гидроксидом аммония с получением аминонитропроизводного 29. Затем монофторпроизводное 29 можно подвергнуть взаимодействию с имидазолом 30 с образованием соединения 31. Затем соединение 31 используют для получения различных соединений по настоящему изобретению с использованием способов, описанных выше для получения соединений, в которых Q представляет собой -CH2-, -NH- или -O-. Специалистам в данной области, очевидно, понятно, что различные соединения по настоящему изобретению можно получить с использованием общего способа, представленного на схемах I-IX и примеров по синтезу, представленных ниже. По другому воплощению настоящее изобретении относится к способу получения соединения формулы А:
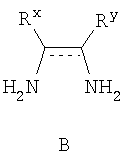
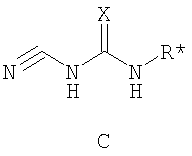
или его соли, включающему стадию взаимодействия соединения формулы В:
или его соли, с соединением формулы С:
или его соли, где указанное взаимодействие осуществляют в не основной среде, содержащей, по меньшей мере, один протонный растворитель, где: Х представляет собой атом кислорода или серы; RX и RY независимо выбраны из R5, OR5, N(R5)2, C(O)N(R5)2, CO2R5 или RX и RY, взятые вместе, образуют 5-8-членное насыщенное, частично ненасыщенное или арильное кольцо, содержащее 0-3 гетероатома, независимо выбранные из атомов азота, кислорода или серы, и где: указанное кольцо, образованное RX и RY, необязательно замещено 1-3 группами, независимо выбранными из оксо, атома галогена, R1, R3, R5, OR5, N(R5)2, OC(O)R5, NR5C(O)R5 или R6; R1 представляет собой 5-6-членное арильное кольцо, содержащее 1-3 гетероатома, независимо выбранные из атомов кислорода, азота или серы, где: R1 замещен 0-3 группами, независимо выбранными из R, оксо, CO2R’, OR’, N(R’)2, SR’, NO2, атома галогена, CN, C(O)N(R’)2, NR’C(O)R’, SO2R’, SO2N(R’)2 или NR’SO2R’; каждый R’ независимо выбран из атома водорода, С1-4алифатической группы или 5-6-членного насыщенного, ненасыщенного или арильного кольца, содержащего 0-3 гетероатомов, независимо выбранных из атомов азота, кислорода или серы, где: R’ замещен 0-3 группами, независимо выбранными из атома галогена, оксо, R0, N(R0)2, OR0, CO2R0, NR0C(O)R0, C(O)N(R0)2, SO2R0, SO2N(R0)2 или NR0SO2R0; R3 выбран из C(O)NHR, C(O)N(R)2, COR, CO2R, COCOR, SO2R, SO2N(R)2, SO2NHR, C(R)=NOH, C(R)=NOR, C(R)=NR или C(R)=N-NHR; каждый R независимо выбран из T-Ar или С1-6алифатической группы, где: указанная С1-6алифатическая группа замещена 0-3 группами, независимо выбранными из R’, оксо, CO2R’, OR’, N(R’)2, SR’, NO2, атома галогена, CN, C(O)N(R’)2, NR’C(О)R’, SO2R’, SO2N(R’)2 или NR’SO2R’; Т представляет собой (СН2)y, где y равно 0, 1 или 2; Ar выбран из: (а) 3-8-членного насыщенного, ненасыщенного или арильного кольца; (b) 3-7-членного гетероциклического кольца, содержащего 1-3 гетероатома, независимо выбранные из атомов азота, кислорода или серы; или (с) 5-6-членного гетероарильного кольца, содержащего 1-3 гетероатома, независимо выбранные из атомов азота, кислорода или серы, где: Ar замещен 0-3 группами, независимо выбранными из R’, оксо, CO2R’, OR’, N(R’)2, SR’, NO2, атома галогена, CN, C(O)N(R’)2, NR’C(О)R’, SO2R’, SO2N(R’)2 или NR’SO2R’; каждый R5 независимо выбран из атома водорода, С1-6алифатической группы или 5-6-членного насыщенного, частично ненасыщенного или арильного кольца, содержащего 0-3 гетероатомов, независимо выбранных из атомов азота, кислорода или серы, где: R5 необязательно замещен 1-3 группами, независимо выбранными из атома галогена, оксо, R0, N(R0)2, OR0, CO2R0, NR0C(O)R0, C(O)N(R0)2, SO2R0, SO2N(R0)2 или NR0SO2R0; каждый R0 независимо выбран из атома водорода, С1-6алифатической группы, фенила или 5-6-членного гетероарильного или гетероциклического кольца, содержащего 1-2 гетероатома, независимо выбранные из атомов азота, кислорода или серы; и R* выбран из R2, С1-6алифатической группы или 5-6-членного насыщенного, частично ненасыщенного или арильного кольца, содержащего 0-3 гетероатома, независимо выбранные из атомов азота, кислорода или серы, где: R2 выбран из атома водорода или С1-3алифатической группы; и R* необязательно замещен 1-3 группами, независимо выбранными из атома галогена, оксо, R0, N(R0)2, OR0, CO2R0, NR0C(O)R0, C(O)N(R0)2, SO2R0, SO2N(R0)2 или NR0SO2R0. По предпочтительному воплощению RX и RY, взятые вместе, образуют 6-7-членное насыщенное, частично ненасыщенное или арильное кольцо, содержащее 0-2 гетероатома, независимо выбранные из атомов азота, кислорода или серы, и где указанное кольцо необязательно замещено. Более предпочтительно, когда RX и RY, взятые вместе, образуют необязательно замещенное 6-членное арильное кольцо, содержащее 0-2 гетероатома, независимо выбранные из атомов азота, кислорода или серы. Наиболее предпочтительно, когда RX и RY, взятые вместе, образуют бензольное кольцо, замещенное одной группой R1 и одной группой R3. Когда RX и RY, взятые вместе, образуют необязательно замещенное 6-7-членное насыщенное кольцо, содержащее 0-2 гетероатома, независимо выбранные из атомов азота, кислорода или серы, то две аминогруппы, представленные в формуле В, предпочтительно находятся в цис-конфигурации. По другому предпочтительному воплощению R* представляет собой R2. Более предпочтительно R* является этилом. В том смысле, в котором он здесь используется, термин «нещелочная среда» означает любой растворитель или смесь, по меньшей мере, двух растворителей, сорастворителя и кислоты для доведения значения рН до ниже или равного примерно 7. Растворители, которые подходят для способа, включают, но этим не ограничиваются, воду, бензол, толуол, дихлорметан, дихлорэтан, диметилформамид, диоксан, диметилсульфоксид, диглим, момоглим, ацетонитрил, тетрагидрофуран, метанол и этанол. В том смысле, в котором он здесь используется, термин «протонный растворитель» означает содержащий протоны растворитель, такой как описан в “Advanced Organic Chemistry”, Jerry March, 3rd edition, John Wiley and Sons, N.Y. Предпочтительно, протонный растворитель выбран из воды, этанола или метанола. В альтернативном воплощении органическая кислота, такая как уксусная кислота, может служить в качестве как протонного растворителя, так и кислотного компонента в реакции. По другому предпочтительному воплощению способ проводят при рН в пределах примерно от 2 до примерно 7. Более предпочтительно, когда значение рН составляет примерно от 3 до примерно 4. Подходящие кислоты, которые можно вносить в реакционную смесь для получения нещелочной среды, включают минеральные кислоты. Примеры неорганических кислот, которые можно использовать, включают, но этим не ограничиваются, минеральные кислоты такие, как серная, соляная и азотная. Предпочтительной кислотой является серная или соляная. Более предпочтительной кислотой является серная. Способ можно проводить при температуре 20-155°С. Предпочтительно, способ проводят при температуре в пределах 40-100°С и, более предпочтительно, при 80-100°С. По другому предпочтительному воплощению настоящее изобретение относится к способу получения соединения формулы I’:
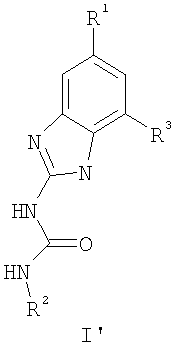
или его фармацевтически приемлемой соли, включающему стадию взаимодействия соединения формулы В’:
или его соли, с соединением формулы С’:
или его соли, в не-оснóвной среде, содержащей, по меньшей мере, один протонный растворитель, где R1, R2 и R3 имеют значения, определенные выше. По другому воплощению настоящее изобретение относится к соединению формулы С”:
где: R2 выбран из атома водорода, этила, изопропила, циклопропила или пропила; и М выбран из атомов натрия, калия, лития, цезия и рубидия. Предпочтительно, М представляет собой атом натрия или калия. Более предпочтительно, М представляет собой атом натрия. Предпочтительно, R2 является этилом. Соединения по данному изобретению являются эффективными ингибиторами активности гиразы и/или топоизомеразы IV, что определяется в тесте оценки активности ферментов. Также было показано, что данные соединения обладают противомикробной активностью по данным теста определения чувствительности к противомикробным соединениям. Активность соединения, используемого по данному изобретению в качестве ингибитора активности гиразы и/или топоизомеразы IV, можно определить в условиях in vitro, in vivo или на клеточной линии с использованием методов, известных в данной области. Подробно условия, используемые для ферментативных тестов и анализа чувствительности к противомикробным соединениям, приведены в примерах ниже. По другому воплощению изобретение относится к композиции, содержащей соединение по данному изобретению или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель, адъювант или наполнитель. Количество соединения в композициях по настоящему изобретению является таковым, что оно эффективно для определяемого ингибирования активности гиразы, топоизомеразы IV или для определяемого снижения количества бактерий в биологической пробе или у пациента. Предпочтительно, композицию по данному изобретению готовят для введения пациенту, при необходимости такого введения. Наиболее предпочтительно, когда композиция по данному изобретению изготовлена для перорального введения пациенту. Термин «биологическая проба», в том смысле, в котором он здесь используется, включает, без ограничения, клеточные культуры или их экстракты, биопсийный материал, полученный от млекопитающего, или его экстракты; кровь, слюну, мочу, фекалии, сперму, слезы и другие жидкости организма или их экстракты. Ингибирование активности гиразы и/или топоизомеразы IV в биологической пробе пригодно для различных целей, известных специалистам в данной области. Примеры подобных целей включают, но не ограничиваются переливанием крови, трансплантацией органов, хранением биологических образцов и биологическими анализами. Термин «пациент», в том смысле, в котором он здесь используется, означает животное, предпочтительно, млекопитающее и, наиболее предпочтительно, человека. Термин «фармацевтически приемлемый носитель, адъювант или наполнитель» относится к нетоксичному носителю, адъюванту или наполнителю, которые не снижают фармакологической активности соединения, с которым они составляются. Фармацевтически приемлемые носители, адъюванты или наполнители, которые можно применять в композициях по настоящему изобретению, включают, но этим не ограничиваются, ионообменники, оксид алюминия, стеарат алюминия, лецитин, сывороточные белки такие, как человеческий сывороточный альбумин, буферные вещества, такие как фосфаты, глицин, сорбиновую кислоту, сорбат калия, смеси неполных глицеридов насыщенных растительных жирных кислот, воду, соли или электролиты, такие как протамин сульфат, динатрийгидрофосфат, гидрофосфат калия, хлорид натрия, соли цинка, коллоидный диоксид кремния, трисиликат магния, поливинилпирролидон, вещества на основе целлюлозы, полиэтиленгликоль, натрий карбоксиметилцеллюлозу, полиакрилаты, воски, блок-полимеры полиэтилена-полиоксипропилена, полиэтиленгликоль и ланолин. Термин «определяемое ингибирование», в том смысле, в котором он здесь используется, означает определяемое изменение активности гиразы и/или топоизомеразы IV в пробе, содержащей указанную композицию и гиразу и/или топоизомеразу IV, по сравнению с аналогичной пробой, содержащей гиразу и/или топоизомеразу IV при отсутствии указанной композиции. В том смысле, в котором он здесь используется, термин «определяемое снижение количества бактерий» означает определяемое изменение числа бактерий в пробе, содержащей указанную композицию, по сравнению с пробой, содержащей только бактерии. «Фармацевтически приемлемая соль» означает нетоксичную соль соединения по данному изобретению, которая при введении реципиенту способна обеспечивать, прямо или опосредованно, соединение по данному изобретению или его метаболит или остаток, активные в качестве ингибитора. В том смысле, в котором он здесь используется, термин «его метаболит или остаток, активный в качестве ингибитора» означает, что его метаболит или остаток также является ингибитором активности гиразы и/или топоизомеразы IV. Фармацевтически приемлемые соли соединений по настоящему изобретению включают производные фармацевтически приемлемых неорганических и органических кислот и оснований. Примеры подходящих солей кислот включают ацетат, адипат, альгинат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, цитрат, камфорат, камфорсульфонат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептаноат, глицерофосфат, гликолат, гемисульфат, гептаноат, гексаноат, гидрохлорид, гидробромид, гидройодид, 2-гидроксиэтансульфонат, лактат, малеат, малонат, метансульфонат, 2-нафталинсульфонат, никотинат, нитрат, оксалат, пальмоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, салицилат, сукцинат, сульфат, тартрат, тиоцианат, тозилат и ундеканоат. Другие кислоты, такие как щавелевая кислота, хотя сами и не являются фармацевтически приемлемыми, могут быть использованы для получения солей, пригодных в качестве промежуточных продуктов при получении соединений по изобретению и их фармацевтически приемлемых аддитивных солей кислоты. Соли, получаемые из соответствующих оснований, включают соли щелочного металла (например, натрия и калия), щелочно-земельного металла (например, магния), аммония и N+(С1-4алкила)4. Данное изобретение также включает кватернизацию любых основных азотсодержащих групп соединений, описанных здесь. С помощью подобной кватернизации можно получить любые растворимые или диспергируемые в воде или масле продукты. Композиции по настоящему изобретению можно вводить перорально, парентерально, ингаляционно, местно, ректально, интраназально, буккально, интравагинально или с помощью имплантируемого резервуара. Термин «парентеральный», в том смысле, в котором он здесь используется, включает подкожный, внутривенный, внутримышечный, внутрисуставный, внутрисиновиальный, интрастернальный, интратекальный, внутрипеченочный, в очаг поражения и внутричерепной путь введения или инфузию. Предпочтительно, композиции вводят перорально, внутрибрюшинно или внутривенно. Стерильные инъекционные формы композиций по данному изобретению могут представлять собой водную и масляную суспензию. Данные суспензии можно получать согласно методам, известным в данной области, с использованием подходящих диспергирующих или смачивающих веществ и суспендирующих веществ. Стерильный инъекционный препарат может также представлять собой стерильный инъекционный раствор или суспензию в нетоксичном, приемлемом для парентерального введения разбавителе или растворителе, например, это может быть раствор в 1,3-бутандиоле. Среди приемлемых наполнителей и растворителей, которые можно использовать, можно указать воду, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, применяются стерильные нелетучие масла в качестве растворителя или суспендирующей среды. Для данной цели можно использовать любое мягкое, нелетучее масло, включая синтетические моно- или диглицериды. Жирные кислоты, такие как олеиновая кислота и ее глицеридные производные, являются подходящими при получении инъекционных препаратов, а также природные фармацевтически приемлемые масла, такие как оливковое или касторовое масло, особенно их полиоксиэтилированные варианты. Данные масляные растворы или суспензии могут также содержать разбавитель или диспергатор на основе спиртов с длинной цепью, такие как карбоксиметилцеллюлоза или аналогичные диспергирующие средства, которые широко используются при изготовлении фармацевтически приемлемых лекарственных форм, включая эмульсии и суспензии. Также для получения фармпрепаратов можно использовать обычно используемые поверхностно-активные вещества такие, как твины, спаны и другие эмульгаторы, или усилители биодоступности, которые широко применяются в производстве фармацевтически приемлемых твердых, жидких и других лекарственных форм. Фармацевтически приемлемые композиции по данному изобретению можно вводить перорально в виде любой лекарственной формы, приемлемой для перорального введения, включая, но этим не ограничиваясь, капсулы, таблетки, водные суспензи или растворы. В случае таблеток для перорального введения обычно используемые носители включают лактозу и кукурузный крахмал. Также, как правило, добавляют «скользящие вещества», такие как стеарат магния. Для перорального введения в виде капсул подходящие разбавители включают лактозу и сухой кукурузный крахмал. Когда для перорального применения требуются водные суспензии, то активный ингредиент смешивают с эмульгирующими и суспендирующими веществами. Если желательно, то можно также добавить подсластители, ароматизаторы или красители. Альтернативно, фармацевтически приемлемые композиции по настоящему изобретению можно вводить в виде суппозиториев для ректального применения. Их можно готовить смешением средства с подходящим нераздражающим наполнителем, который находится в твердом состоянии при комнатной температуре, но становится жидким при ректальной температуре и, следовательно, будет расплавляться в прямой кишке с высвобождением лекарственного препарата. Подобные материалы включают какао-масло, пчелиный воск и полиэтиленгликоли. Фармацевтически приемлемые композиции по данному изобретению можно также вводить местно, особенно когда мишень для лечения включает области или органы, легко доступные для местного применения, включая заболевания глаз, кожи или нижних отделов кишечного тракта. Подходящие для местного применения композиции легко приготовить для каждой из этих областей или органов. Местное применение для нижних отделов кишечного тракта можно проводить при использовании суппозитория для ректального введения (смотри выше) или в виде подходящей клизмы. Можно также использовать трансдермальные пластыри для местного применения. Для местных применений фармацевтически приемлемые композиции можно составлять в виде подходящей мази, содержащей активный компонент, суспендированный или растворенный в одном или нескольких носителей. Носители для местного применения соединений по настоящему изобретению включают, но этим не ограничиваются, минеральное масло, вазелиновое масло, белый вазелин, пропиленгликоль, полиоксиэтилен, полиоксипропилен, эмульгирующий воск и воду. Альтернативно, фармацевтически приемлемые композиции можно включать в состав подходящего лосьона или крема, содержащего активные компоненты, суспендированные или растворенные в одном или нескольких фармацевтически приемлемых носителях. Подходящие носители включают, но этим не ограничиваются, минеральное масло, сорбитан моностеарат, полисорбат 60, цетиловые эфиры воска, цетеариловый спирт, 2-октилдодеканол, бензиловый спирт и воду. Для применения в офтальмологии фармацевтически приемлемые композиции могут находиться в форме микронизированных суспензий в изотоническом, с доведенным рН стерильном физиологическом растворе или предпочтительно в виде растворов в изотоническом, с доведенным рН стерильном физиологическом растворе с консервантом или без него, таким как бензилалконий хлорид. Альтернативно, для применения в офтальмологии фармацевтически приемлемые композиции можно изготавливать в виде мази, такой как вазелин. Фармацевтически приемлемые композиции по данному изобретению также можно вводить в виде назального аэрозоля или ингаляционно. Подобные композиции получают методами, хорошо известными в области приготовления фармацевтических композиций, и их можно приготовить в виде растворов в физиологическом растворе с использованием бензилового спирта или других подходящих консервантов, усилителей всасывания для повышения биодоступности, фторуглеродов и/или других широко используемых солюбилизирующих или диспергирующих веществ. Наиболее предпочтительно, когда фармацевтически приемлемые композиции по данному изобретению изготавливают для перорального введения. Дозы в пределах примерно от 0,01 до примерно 100 мг/кг массы тела в сутки, предпочтительно от 0,5 до примерно 75 мг/кг массы тела в сутки и наиболее предпочтительно примерно от 1 до 50 мг/кг массы тела в сутки активного ингредиента являются пригодными для монотерапии в целях профилактики и лечения бактериальных инфекций, вызванных такими бактериями, как Streptococcus pneumoniae, Streptococcus pyogenes, Enterococcus faecalis, Enterococcus faecium, Klebsiella pneumoniae, Enterobacter sps., Proteus sps., Pseudomonas aeruginosa, E. coli, Serratia marcesens, Staphylococcus aureus, отрицательные на коагулазу стафилококки, Haemophilus infuenzae, Bacillus anthracis, Mycoplasma pneumoniae, Moraxella catarralis, Chlamydia pneumoniae, Legionella pneumophila, Mycobacterium tuberculosis, Helicobacter pylori, Staphylococcus epidermidis или Mycobacterium tuberculosis. Как правило, фармацевтические композиции по настоящему изобретению следует вводить примерно от 1 до 5 раз в день или, альтернативно, в виде постоянной инфузии. Или, альтернативно, композиции по настоящему изобретению можно вводить в виде пульсирующей композиции. Подобное введение можно использовать в виде длительной или кратковременной терапии. Количество активного ингредиента, которое можно смешать с носителями для получения разовой лекарственной формы, будет варьировать в зависимости от пациента, подвергаемого лечению, и конкретного пути введения. Типичный препарат будет содержать примерно от 5% до примерно 95% активного соединения (мас./мас.). Предпочтительно, подобные препараты содержат примерно от 20% до примерно 80% активного соединения. Когда композиции по данному изобретению содержат комбинацию соединения формулы I и одного или нескольких дополнительных терапевтических или профилактических средств, то соединение и дополнительное средство должны находиться в дозах, составляющих примерно от 10% до 80% от дозы, обычно применяемой при монотерапии. При улучшении состояния пациента можно вводить поддерживающую дозу соединения, композиции или комбинации по данному изобретению, если необходимо. Затем дозу или кратность введения, или оба фактора, можно снизить, как функцию симптомов, до уровня, при котором сохраняется улучшенное состояние, когда симптомы ослабляются до желаемого уровня, то лечение следует остановить. Пациенты, однако, могут получать лечение с перерывами в течение длительного периода времени при любом рецидиве симптомов заболевания. Специалистам в данной области, очевидно, понятно, что могут быть необходимы более низкие или более высокие дозы, чем указанные выше. Конкретные дозы и схемы лечения любого конкретного пациента будут зависеть от самых различных факторов, включая активность конкретного используемого соединения, возраста, массы тела, общего состояния здоровья, пола, рациона, времени введения, скорости выделения, комбинации препаратов, тяжести и течения заболевания и предрасположенности пациента к заболеванию и решения лечащего врача. В зависимости от конкретного состояния или заболевания, которое подвергается лечению или профилактике, в композициях по данному изобретению могут находиться дополнительные терапевтические средства, которые обычно применяют для лечения или профилактики заболевания. В том смысле, в котором этот термин здесь используется, дополнительные терапевтические средства, которые обычно применяют для лечения или профилактики конкретного заболевания или состояния, известны в качестве «подходящих для заболевания или состояния, которые подвергаются лечению». Подобные средства включают, но не ограничиваются антибиотиками, противовоспалительными средствами, ингибиторами матричной металлопротеазы, ингибитором липоксигеназы, антагонистом цитокинов, иммуносупрессором, противоопухолевым средством, противовирусным средством, цитокином, фактором роста, иммуномодулятором, простагландином, средством против гиперпролиферации сосудов или средством, повышающим чувствительность бактерий к антибиотикам. Примеры антибиотиков, подходящих для введения с соединениями по настоящему изобретению, и их композициями, включают хинолоны, Известны средства, которые повышают чувствительность бактерий к антибиотикам. Например, в патенте США № 5523288, патенте США № 5783561 и патенте США № 6140306 описаны способы применения комбинации бактерицидного средства/повышающего проницаемость белка (BPI) для усиления чувствительности грамположительных и грамотрицательных бактерий к антибиотику. Средства, которые повышают проницаемость наружной мембраны бактерий, описаны Vaara M. in Microbiological Reviews (1992) pp. 395-411, и сенсибилизация грамотрицательных бактерий описана Tsubery H. et al., in J. Med. Chem. (2000) pp. 3085-3092. По другому воплощению изобретение относится к способу лечения или ослабления тяжести бактериальной инфекции у пациента, включающий стадию введения указанному пациенту композиции по настоящему изобретению. По другому воплощению изобретение относится к способу ингибирования активности гиразы у пациента, включающему стадию введения указанному пациенту композиции по настоящему изобретению. По другому воплощению изобретение относится к способу ингигибрования активности топоизомеразы IV, включающему стадию введения указанному пациенту композиции по настоящему изобретению. По другому воплощению изобретение относится к способу снижения числа бактерий у пациента, включающему стадию введения указанному пациенту композиции по настоящему изобретению. По другому воплощению изобретение относится к способу ингибирования активности гиразы в биологическом образце. По другому воплощению изобретение относится к способу ингибирования активности топоизомеразы IV в биологическом образце. По другому воплощению изобретение относится к способу снижения числа бактерий в биологическом образце. По другому воплощению изобретение обеспечивает способ снижения числа бактерий в биологической пробе, но дополнительно включающий стадию контактирования указанного биологического образца со средством, которое повышает чувствительность бактерий к антибиотикам. Фармацевтические композиции и способы по данному изобретению в основном будут пригодны для борьбы с бактериальными инфекциями in vivo. Примеры бактерий, с которыми можно бороться с помощью композиций и способов по данному изобретению, включают, но этим не ограничиваются, следующие микроорганизмы: Streptococcus pneumoniae, Streptococcus pyogenes, Enterococcus faecalis, Enterococcus faecium, Klebsiella pneumoniae, Enterobacter sps. Proteus sps. Pseudomonas aeruginosa, E. coli, Serratia marcesens, Staphylococcus aureus, отрицательными на коагулазу стафилококками, Haemophilus infuenzae, Bacillus anthracis, Mycoplasma pneumoniae, Moraxella catarralis, Chlamydia pneumoniae, Legionella pneumophila, Mycobacterium tuberculosis, Helicobacter pylori, Staphylococcus epidermidis или Mycobacterium tuberculosis. По другому воплощению бактерии, с которыми можно бороться с помощью композиций и способов по данному изобретению, включают следующие микроорганизмы: Streptococcus pneumoniae, Streptococcus pyogenes, Enterococcus faecalis, Enterococcus faecium, Staphylococcus aureus, отрицательные на коагулазу стафилококки, Haemophilus infuenzae, Bacillus anthracis, Mycoplasma pneumoniae, Moraxella catarralis, Chlamydia pneumoniae, Legionella pneumophila, Mycobacterium tuberculosis, Helicobacter pylori, Staphylococcus epidermidis или Mycobacterium tuberculosis. Следовательно, композиции и способы будут пригодными для борьбы, лечения или снижения выраженности, тяжести или эффектов нозокомиальных или ненозокомиальных инфекций. Примеры нозокомиальных применений включают, но этим не ограничиваются, инфекции мочевых путей, респираторные инфекции, такие как пневмония, инфекции хирургических ран, инфекции центральной линии и бактериемию. Примеры ненозокомиальных применений включают, но этим не ограничиваются, инфекции мочевых путей, бронхиты, синуситы, пневмонию, простатит, инфекции кожи и мягких тканей, внутрибрюшинные инфекции и лечение инфекций у пациентов с лихорадочной нейтропенией. Термин «фармацевтически эффективное количество» относится к количеству, эффективному для лечения или ослабления бактериальной инфекции у пациентов. Термин «профилактически эффективное количество» относится к количеству, эффективному для профилактики или в основном ослабления бактериальной инфекции у пациента. Соединения по данному изобретению можно использовать обычным способом для борьбы с бактериальными инфекциями в условиях in vivo и для лечения заболеваний или снижения выраженности или тяжести эффектов, которые опосредуются бактериями. Подобные способы лечения, дозировки при этом и требования могут быть выбраны специалистами в данной области из доступных способов и методов. Например, соединение по данному изобретению можно комбинировать с фармацевтически приемлемым адъювантом для введения пациенту, страдающему бактериальной инфекцией или заболеванием, фармацевтически приемлемым образом и в количестве, эффективном для снижения тяжести инфекции или заболевания. Альтернативно, соединения по данному изобретению можно использовать в композициях и способах для лечения и защиты индивидуумов от бактериальных инфекций и заболеваний в течение длительного периода времени. Соединения можно использовать в подобных композициях самостоятельно или вместе с другими соединениями по изобретению способом, согласующимся с обычным применением ингибиторов ферментов в фармацевтических композициях. Например, соединение по данному изобретению можно смешать с фармацевтически приемлемыми адъювантами, обычно используемыми в вакцинах, и вводить в профилактически эффективных количествах для защиты индивидуумов от бактериальных инфекций или заболеваний в течение продолжительного периода времени. Соединения формулы I также можно вводить совместно с другими антибиотиками для повышения эффективности лечения или профилактики различных бактериальных инфекций. В случае, когда соединения по данному изобретению вводят при комбинированном лечении с другими средствами, их можно вводить пациенту последовательно или совместно. Альтернативно, фармацевтические или профилактические композиции по данному изобретению включают комбинацию соединения формулы I и другого терапевтического или профилактического средства. Дополнительные терапевтические средства, описанные выше, можно вводить отдельно, в виде части режима множественной дозировки, из содержащей ингибитор композиции. Альтернативно данные средства могут быть частью разовой лекарственной формы, смешанной вместе с ингибитором в одной композиции. Для лучшего понимания данного изобретения приводятся последующие примеры. Данные примеры предназначены только для иллюстрации, и никоим образом не ограничивают объем изобретения. ПРИМЕРЫ В том смысле, в котором он здесь используется, термин «Rt» относится ко времени удерживания в минутах, полученному для конкретного соединения, при осуществлении ВЭЖХ в следующих условиях (если не указано иначе): Колонка: Zorbax SB Phenyl, 5 мкМ, 4,6 мм × 250 мм Градиент: вода:ацетонитрил от (9:1) до (1:9) в течение 10 мин Скорость потока: 1,0 мл/мин Длина волны: 214 нм. Пример 1
Метил 5-бром-3-нитро-2N-трифторацетиламинобензоат: метил-2-амино-5-бромбензоат (5,0 г, 21,73 ммоль) добавляли в течение 5 мин к трифторуксусному ангидриду (60 мл), охлажденному до 0-5°С. После перемешивания в течение еще 15 мин добавляли нитрат калия (2,637 г, 26,08 ммоль) и полученную бежевую суспензию перемешивали в течение ночи. Реакционную смесь концентрировали при пониженном давлении с получением желтовато-коричневого твердого вещества, которое распределяли в бикарбонате натрия (водном, насыщенном растворе) и экстрагировали этилацетатом (3х). Объединенные органические слои промывали насыщенным раствором соли, высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 6,39 г указанного в заголовке соединения в виде желтовато-коричневого твердого вещества. Rt при ВЭЖХ = 3,62 мин МС (М-1 370,9). Пример 2
Метил 2-амино-5-бром-3-нитробензоат: к суспензии метил 5-бром-3-нитро-2N-трифторацетиламинобензоата (2,1 г, 5,66 ммоль) в метаноле (40 мл) добавляли соляную кислоту (20 мл, 6Н раствор). Полученную смесь нагревали при 75-80°С в течение 12 ч, затем охлаждали с получением желтой суспензии, которую фильтровали и промывали водой. Собранные твердые частицы высушивали при 50°С при пониженном давлении с получением 1,1 г указанного в заголовке соединения в виде ярко-желтого твердого вещества. 1Н ЯМР (CDCl3) д 3,93 (с, 3Н), 8,32 (д, 1Н), 8,51 (д, 1Н). Пример 3
Метил-2-амино-3-нитро-5-(3′-пиридил)бензоат: к продутой азотом смеси метил 2-амино-5-бром-3-нитробензоата (0,3 г, 1,09 ммоль) в диметиловом эфире этиленгликоля (8 мл) добавляли раствор бикарбоната натрия (1М раствор, 2,18 мл, 2,18 ммоль), 3-пиридилбороновую кислоту (0,201 г, 1,636 ммоль) и (тетракистрифенилфосфин)палладий (О) (0,125 г, 0,11 ммоль). Полученную смесь кипятили с обратным холодильником в течение 12 ч, охлаждали, разбавляли раствором бикарбоната натрия (водным, насыщенным) и экстрагировали этилацетатом (3×). Объединенные органические слои промывали насыщенным раствором соли, высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением темно-желтого твердого вещества. Сырой продукт очищали флэш-хроматографией (силикагель, гексан/этилацетат от 30 до 100%) с получением 0,069 г указанного в заголовке соединения в виде желтого твердого вещества. Rt при ВЭЖХ = 1,76 мин, МС (М+Н=274). Пример 4
Метил-2,3-диамино-5-(3′-пиридил)бензоат: к суспензии 10% Pd на угле (0,035 г) в этаноле (10 мл) добавляли суспензию метил-2-амино-3-нитро-5-(3′-пиридил)бензоата (0,167 г, 0,61 ммоль) в этаноле (15 мл). Частичную суспензию гидрировали под давлением 40 фунтов/кв. дюйм в течение 6 ч. Затем катализатор удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением 0,11 г указанного в заголовке соединения в виде светло-желтого твердого вещества. 1H ЯМР (CDCl3) Пример 5
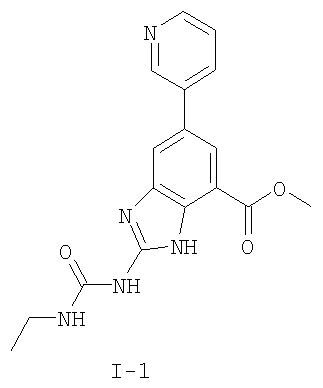
Метиловый эфир 2-(3-этилуреидо)-6-пиридин-3-ил-3Н-бензимидазол-4-карбоновой кислоты (I-1): к суспензии метил-2,3-диамино-5,3′-пиридилбензоата (0,109 г, 0,448 ммоль) в воде (1 мл) добавляли раствор серной кислоты (1Н раствор, 1,2 мл) и раствор N’-этил-N-цианомочевины (1М раствор, 0,9 мл, 0,94 ммоль). Значение pH доводили 1Н раствором серной кислоты до 3-4 и полученную реакционную смесь кипятили с обратным холодильником в течение 12 ч. Реакционную смесь охлаждали и полученную суспензию фильтровали и промывали водой. Собранные твердые частицы очищали флэш-хроматографией (силикагель от 100% метиленхлорида до 100% смеси (NH4OH/MeOH/CH2Cl2 5/10/85 об./об./об.) с получением желтовато-коричневого твердого вещества, которое перекристаллизовывали из смеси метанол-диэтиловой эфир с получением 0,009 г указанного в заголовке соединения в виде не совсем белого твердого вещества. Rt при ВЭЖХ = 1,5 мин, МС (М+Н=340). 1H ЯМР (ДМСО) д 1,13 (т, 3H), 3,24 (м, 2H), 3,97 (с, 3H), 7,48 (м, 2H), 7,91 (с, 1H), 8,03 (с, 1H), 8,14 (м, 1H), 8,56 (д, 1H), 8,92 (с, 1H) 9,92 (шир, 1H), 11,51 (шир, 1H). Пример 6
N’-Этил-N-цианомочевины натриевая соль: Способ А: при 20°С к раствору гидроксида натрия (1,5М водный раствор, 50 мл, 75,02 ммоль) добавляли цианамид (8,5 г, 202,25 ммоль), затем по каплям добавляли этилизоцианат (4 мл, 50,56 ммоль) в течение 10 мин. После перемешивания в течение 30 мин добавляли еще раствор гидроксида натрия (3М раствор, 25 мл, 75,02 ммоль) и этилизоцианат (4 мл, 50,56 ммоль). Затем полученный раствор выдерживали минимум в течение 30 мин перед непосредственным использованием без очистки. Способ В: готовили раствор трет-бутилата натрия (124,1 г) в ТГФ (500 мл, безводный) при комнатной температуре, затем охлаждали на ледяной бане. В отдельном реакционном сосуде раствор цианамида (51,76 г) в ТГФ (300 мл, безводном) объединяли с этилизоцианатом (97,5 мл) и охлаждали на ледяной бане. К полученному раствору цианамид/изоцианат добавляли раствор трет-бутилат натрия/ТГФ со скоростью, достаточной для поддержания внутренней температуры ниже 30°С. Полученное белое твердое вещество собирали фильтрованием. Затем собранное твердое вещество объединяли с ТГФ (500 мл) и полученную суспензию перемешивали на ледяной бане в течение 15 мин. Белое твердое вещество собирали фильтрованием и высушивали в вакууме с получением 151,5 г указанного в заголовке соединения (выход 91%). Rt (мин)=3,0 мин. Пример 7
Метиловый эфир 2-амино-6-фтор-5-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)-3-нитробензойной кислоты: к раствору метилового эфира 2-амино-5-бром-6-фтор-3-нитробензойной кислоты (1,0 г, 3,4 ммоль) в диоксане (25 мл) в атмосфере азота добавляли биспинаколадабор (1,3 г, 5,0 ммоль), аддукт дихлор [1,1′-бис(дифенилфосфин)ферроцен]палладия (II) и дихлорметана (0,125 г, 0,17 ммоль) и ацетат калия (1,0 г, 10,2 ммоль). Полученную смесь кипятили с обратным холодильником в течение 18 ч, охлаждали до комнатной температуры, разбавляли этилацетатом (50 мл) и фильтровали через целит®. Полученное твердое вещество растирали в гексане (40 мл) три раза с получением 0,515 г метилового эфира 2-амино-6-фтор-3-нитро-5-(4,4,5,5-тетраметил-[1,3,2]-диоксаборолан-2-ил) бензойной кислоты. К смеси метилового эфира 2-амино-6-фтор-3-нитро-5-(4,4,5,5-тетраметил-[1,3,2]диксаборолан-2-ил)бензойной кислоты в диметиловом эфире этиленгликоля (10 мл) добавляли 1-метил-2-оксо-1,2-дигидропиридин-4-иловый эфир трифторметансульфоновой кислоты (0,39 г, 1,5 ммоль), хлорид лития (0,25 г, 6,0 ммоль), карбонат натрия (1,1 мл, 2,2 ммоль 2М раствора) и (тетракистрифенилфосфин)палладий (0,18 г, 0,15 ммоль). Полученную смесь нагревали до 90°С и перемешивали в течение 18 ч. После охлаждения и концентрирования в вакууме остаток очищали хроматографией (силикагель от 0,5% метанол/дихлорметан до 2% метанол/дихлорметан) с получением указанного в заголовке соединения (0,275 г, 25%). 1H ЯМР (500 МГц, CDCl3) д 3,58 (с, 3H), 4,03 (с, 3H), 6,30 (д, 1H), 7,32 (д, 1H), 8,45 (шир.с, 2H), 8,53 (д, 1H). Пример 8
(2-Амино-5-бромфенил)циклогексилметанон: к суспензии 2-амино-5-бромбензонитрила (2,13 г, 10,80 ммоль) в сухом ТГФ (20 мл), охлажденном до 0°С, добавляли по каплям бромид циклогексилмагния (1Н раствор в ТГФ, 37,8 мл, 37,8 ммоль, 3,5 экв). Полученную желтую реакционную смесь подогревали до комнатной температуры и перемешивали в течение 20 ч. Затем реакционную смесь охлаждали до 0°С, медленно гасили насыщенным водным раствором хлорида аммония (30 мл) и разбавляли водой (30 мл) и EtOAc (50 мл). Двухфазную смесь энергично перемешивали до растворения всех твердых частиц. Фазы разделяли и водный слой экстрагировали EtOAc (25 мл). Объединенные органические экстракты промывали насыщенным раствором соли, высушивали (MgSO4), фильтровали и фильтрат концентрировали в вакууме. Сырой продукт очищали флэш-хроматографией (SiO2, от гексана до смеси гексан:EtOAc 19:1) с получением 2,43 г (80%) указанного в заголовке соединения в виде желтого твердого вещества: 1H ЯМР (CDC13, 500 МГц) д 7,84 (д, 1H), 7,35 (дд, 1H), 6,72 (д, 1H), 3,18 (м, 1H), 1,86 (м, 4H), 1,74 (м, 1H), 1,39-1,53 (м, 4H), 1,25 (м, 1H); МС (ES+) m/z (М++1) 282,07. Пример 9
(2-Амино-5-бром-3-нитрофенил)циклогексилметанон: суспензию (2-амино-5-бром-фенил)циклогексилметанона (2,40 г, 8,50 ммоль) в уксусном ангидриде (40 мл) нагревали при 80°С в течение 1 ч. Реакционную смесь концентрировали досуха, затем растворяли в дымящей азотной кислоте (18 мл). Полученный желтый раствор перемешивали при комнатной температуре в течение 2 ч. Полученный светло-оранжевый раствор выливали на лед и образовывался желтый осадок. Реакционную смесь перемешивали до тех пор, пока весь лед таял, и фильтровали с получением бледно-желтого твердого вещества. Данное твердое вещество растворяли в EtOH (10 мл) и 6 н. водном растворе соляной кислоты (20 мл). Раствор перемешивали при 80°С в течение 3 ч, охлаждали до комнатной температуры, разбавляли водой (20 мл) и подщелачивали карбонатом натрия (1 ч). Полученную смесь разбавляли гексаном (50 мл) и двухфазную смесь перемешивали до растворения всех твердых частиц. Фазы разделяли и водный слой экстрагировали гексаном (2×25 мл). Объединенные органические экстракты промывали водой, высушивали (MgSO4), фильтровали и фильтрат концентрировали в вакууме с получением указанного в заголовке соединения (1,17 г, выход 43%) в виде желтого твердого вещества: 1H ЯМР (CDC13, 500 МГц) д 8,50 (c, 1H), 8,12 (c, 1H), 3,20 (м, 1H), 1,86 (м, 4H), 1,74 (м, 1H), 1,51 (м, 2H), 1,39 (м, 2H), 1,27 (м, 1H). Пример 10
(2-Амино-3-нитро-5-пиридин-3-ил-фенил)циклогексилметанон: к раствору (2-амино-5-бром-3-нитрофенил)циклогексилметанона (600 мг, 1,83 ммоль) в DME (25 мл) последовательно добавляли 1,3-пропандиоловый циклический эфир пиридин-3-бороновой кислоты (388 мг, 2,38 ммоль), (тетраксифенилфосфин)палладий (0) (212 мг, 0,18 ммоль) и 1 н. раствор NaHCO3 (3,7 мл, 3,7 ммоль). Полученную смесь перемешивали при 90°С в течение 90 мин, затем охлаждали до комнатной температуры. Реакционную смесь разбавляли EtOAc (100 мл), промывали водой (50 мл), насыщенным раствором соли (50 мл), высушивали (MgSO4), фильтровали и фильтрат концентрировали в вакууме. Полученный остаток очищали флэш-хроматографией (SiO2, от гексана до смеси гексан:EtOAc 3:1) с получением 527 мг (89%) указанного в заголовке соединения в виде бледно-оранжевого твердого вещества: 1H ЯМР (CDCl3, 500 МГц) д 8,92 (д, 1H), 8,69 (д, 1H), 8,66 (д, 1Н), 8,31 (д, 1H), 8,20 (д, 1H), 7,71 (дд, 1H), 3,36 (м, 1H), 1,88 (м, 3H), 1,77 (м, 1H), 1,40-1,61 (м, 3H), 1,24-1,32 (м, 2H), 0,89 (м, 1H). Пример 11
1-(7-Циклогексанкарбонил-5-пиридин-3-ил-1Н-бензимидазол-2-ил)-3-этилмочевина (I-14): суспензию соединения 4 (42 мг, 0,13 ммоль) и хлорида олова (II) дигидрата (87 мг, 0,39 ммоль) в EtOH (4 мл) кипятили с обратным холодильником в течение 4 ч. Смесь охлаждали до комнатной температуры, подщелачивали насыщенным водным раствором NHCO3 (10 мл) и разбавляли EtOAc (15 мл). Добавляли целит (10 г) и полученную суспензию перемешивали (30 мин), фильтровали через слой целита и фильтрат концентрировали в вакууме. Полученный остаток разбавляли водой (5 мл) и добавляли 1 н. водный раствор N’-этил-N-цианомочевины. Для доведения значения рН до 3 добавляли по каплям достаточное количество 1 н. водного раствора серной кислоты. Полученную смесь нагревали при 100°С в течение 16 ч. Затем реакционную смесь охлаждали до комнатной температуры, подщелачивали водным раствором NaHCO3 и разбавляли EtOAc. Фазы разделяли и органический слой высушивали (MgSO4), фильтровали и фильтрат концентрировали в вакууме. Остаток очищали препаративной ВЭЖХ с получением 8 мг указанного в заголовке соединения в виде соли бис-TFA, которую превращали в соль бис-HCl с получением соединения 7 в виде бледно-желтого твердого вещества: Rt при ВЭЖХ=4,52 мин; 1H ЯМР (CD3OD, 500 МГц) д 9,40 (c, 1H), 9,08 (д, 1H), 8,95 (д, 1H), 8,48 (c, 1H), 8,27 (м, 2H), 3,72 (м, 1H), 3,36 (кв, 2H), 1,99 (м, 2H), 1,86 (м, 2H), 1,83 (м, 1H), 1,57 (м, 4H), 1,32 (м, 1H), 1,23 (т, 3H); МС (ES+) m/z (М++1) 392,2. Пример 12
Метиловый эфир 2-амино-5-бром-3-нитробензойной кислоты: к раствору 2,23 г (10,4 ммоль) метилового эфира 2-амино-3-нитробензойной кислоты в 12 мл уксусной кислоты добавляли по каплям в течение 5 мин раствор 0,53 мл (10,4 ммоль, 1 экв) брома в 2 мл уксусной кислоты. Смесь перемешивали при комнатной температуре в течение 30 мин и выливали в 100 г льда. Выпавшее в осадок желтое твердое вещество собирали вакуум-фильтрованием и высушивали с получением 2,50 г (82%) указанного в заголовке соединения в виде желтого твердого вещества. 1H ЯМР (CDCl3) д 3,95 (c, 3H), 8,35 (шир.c, 2H), 8,6 (д, 1H). Пример 13
Метиловый эфир 2-амино-3-нитро-5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)бензойной кислоты: к раствору метилового эфира 2-амино-5-бром-3-нитробензойной кислоты (0,5 г, 1,82 ммоль) в диоксане (5 мл) добавляли биспинаколадабор (0,554 г, 2,18 ммоль), аддукт дихлор[1,1′-бис(дифенилфосфин)ферроцен]палладия (II) и дихлорметана (0,133 г, 0,18 ммоль) и ацетат калия (0,535 г, 5,45 ммоль). Полученную смесь кипятили с обратным холодильником в течение 3 ч. После охлаждения и концентрирования в вакууме темное твердое вещество очищали (SiO2, от CH2Cl2 до 50% этилацетата в СН2Cl2) с получением указанного в заголовке соединения в виде оранжевого твердого вещества) (0,347 г, 57%). 1H ЯМР (500 МГц, CDCl3) д 1,32 (c, 6H), 3,91 (c, 3H), 8,3 (шир. 1H), 8,59 (c, 1H), 8,8 (c, 1H), 8,99 (шир. 1H). Пример 14
Метиловый эфир 2-амино-5-(5-метил-3-оксо-4-пиридин-2-илметилциклогекса-1,5-диенил)-3-нитробензойной кислоты: к раствору метилового эфира 2-амино-3-нитро-5-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)бензойной кислоты (0,163 г, 0,51 ммоль) в диметиловом эфире этиленгликоля (5 мл) добавляли N-(метил-2-пиридинил)-6-метил-4-трифторметилсульфонилокси-2-пиридон (0,136 г, 0,41 ммоль), бис(трифенилфосфин)палладий (II) дихлорид (0,029 г, 0,04 ммоль) и карбонат натрия (0,62 мл, 1,24 ммоль 2М раствора). Полученную смесь кипятили с обратным холодильником в течение 3 ч. После охлаждения и концентрирования в вакууме полученное твердое вещество очищали (SiO2, от 50% этилацетат/метиленхлорид до 3% метанола в 50% смеси этилацетат/метиленхлорид) с получением указанного в заголовке соединения в виде оранжевого твердого вещества (0,176 г, 89%). 1H ЯМР (500 МГц, CDCl3) д 2,41 (c, 3H), 3,39 (c, 3H), 5,5 (c, 2H), 6,28 (c, 1Н), 6,68 (c, 1H), 7,3-7,1 (м, 2H), 7,62 (т, 1H), 8,49 (c, 1Н), 8,61 (c, 1H). Пример 15
Метиловый эфир 2,3-диамино-5-(5-метил-3-оксо-4-пиридин-2-илметилциклогекса-1,5-диенил)бензойной кислоты: к суспензии 10% палладия на угле (0,045 г) в этилацетате (20 мл) добавляли метиловый эфир 2-амино-5-(5-метил-3-оксо-4-пиридин-2-илметил-циклогекса-1,5-диенил)-3-нитробензойной кислоты (0,176 г, 0,44 ммоль). Полученную смесь гидрировали под давлением 30 фунтов/кв. дюйм в течение 24 ч. Реакционную смесь фильтровали, концентрировали в вакууме и сырой осадок очищали (SiO2, от 2 до 10% метанола в метиленхлориде) с получением указанного в заголовке соединения. 1H ЯМР (500 МГц, CDCl3) д 2,5 (c, 3H), 3,99 (c, 3H), 5,3 (c, 2H), 5,38 (шир. 2H), 6,39 (c, 1H), 6,25 (c, 1H), 7,93-7,21 (м, 4H), 8,61 (c, 1H), 8,93 (c, 1H). Пример 16
Метиловый эфир 2-(3-этилуреидо)-6-(5-метил-3-оксо-4-пиридин-2-илметилциклогекса-1,5-диенил)-1Н-бензоимидазол-4-карбоновой кислоты (I-32): к смеси метилового эфира 2,3-диамино-5-(5-метил-3-оксо-4-пиридин-2-илметилциклогекса-1,5-диенил)бензойной кислоты (0,084 г, 0,23 ммоль) в 20% водном растворе диметилсульфоксида (5 мл) добавляли серную кислоту (0,5 мл) и N’-этил-N-цианомочевину (0,58 мл в 1М в NaOH, 0,58 ммоль). После доведения значения рН Пример 17
4-Гидрокси-6-метил-1-пиридин-2-илметил-1Н-пиридин-2-он: к суспензии 12,6 г (0,1 моль) 4-гидрокси-6-метил-2-пиридона в 50 мл воды добавляли 2-(аминометил)пиридин (10,3 мл, 0,1 мл) и смесь кипятили с обратным холодильником в течение 2,5 ч. Полученное светло-желтое вещество отфильтровывали из охлажденной смеси. Концентрирование фильтрата и растирание полученной смолы в дихлорметане давало второй выход указанного в заголовке соединения (19,85 г, 92%). 1H ЯМР (ДМСО-d6,ч/млн): 10,5 (c, 1H), 8,5 (д, 1H), 7,7-7,8 (т, 1H), 7,2 (т, 1H), 7,1 (д, 1H), 5,8 (c, 1Н), 5,5 (c, 1Н), 5,2 (c, 2H), 2,2 (c, 3Н). FIA: m/z-215,1 ES-/217,1 ES+. Пример 18
6-метил-2-оксо-1-пиридин-2-илметил-1,2-дигидропиридин-4-иловый эфир трифторметансульфоновой кислоты: к суспензии N-(метил-2-пиридинил)-4-гидрокси-6-метил-2-пиридона (10,0 г, 0,046 ммоль) в диметилформамиде (70 мл) добавляли N-фенил-трифторметилсульфонимид (18,2 г, 0,05 ммоль) с последующим добавлением триэтиламина (7,7 мл, 0,055 ммоль) и смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь выливали в 400 мл воды и экстрагировали этилацетатом (4×100 мл). Объединенные экстракты промывали 2 н. раствором NaOH (2×200 мл), водой (4×200 мл) и насыщенным раствором соли (1×150 мл) перед высушиванием и концентрированием. Фильтрование через слой целита размером 2 дюйма с использованием гексана в качестве элюента (отбрасывали) и затем 50% смесью этилацетат/гексан и концентрирование фильтрата давало указанное в заголовке соединение в виде белого твердого вещества (12,8 г, 80%). 1H ЯМР: (CDCl3, ч/млн): 8,5 (д, 1H), 7,7 (т, 1H), 7,3 (д, 1H), 7,2 (м, 1H), 6,4 (c, 1Н), 6,1 (c, 1H), 5,4 (c, 2H), 2,5 (c, 3H). FIA: m/z – 349,0 ES+. Пример 19
1-[4-(Циклопропилметоксииминометил)-6-пиридин-3-ил-1Н-бензимидазол-2-ил]-3-этилмочевина (I-40): к раствору 1-(4-циклопропанкарбонил-6-пиридин-3-ил-1Н-бензимидазол-2-ил)-3-этилмочевины (37,2 г, 0,106 ммоль) в сухом EtOH (3 мл) последовательно добавляли ацетат калия (84 мг, 0,848 ммоль, 8 экв), метоксиламин гидрохлорид (70,8 мг, 0,848 ммоль, 8 экв), молекулярные сита (4Å, порошкообразные). Полученную суспензию перемешивали при 50°С в течение 36 ч. Затем реакционную смесь охлаждали до комнатной температуры, разбавляли EtOAc и водой. Фазы разделяли, водный слой экстрагировали EtOAc (1×) и объединенные органические экстракты высушивали (MgSO4), фильтровали и фильтрат концентрировали в вакууме. Остаток очищали препаративной ВЭЖХ, превращали в соль бис-HCl с получением указанного в заголовке соединения в виде желтовато-коричневого твердого вещества (11,0 мг): ВЭЖХ (от 10 до 90% CH3CN, 8 мин): Rt = 4,05 мин. 1H ЯМР (CD3OD, 500 МГц) д 9,30 (c, 1H), 8,99 (д, 1Н), 8,90 (д, 1H), 8,22 (дд, 1H), 8,18 (c, 1H), 8,04 (c, 1H), 4,14 (c, 3H), 3,36 (кв, 2H), 2,04 (м, 1H), 1,23 (т, 3H), 1,15 (м, 2H), 0,92 (м, 2H). MC(ES+): m/z(M++1) 379,2. Пример 20
Этиловый эфир (7-пропионил-5-пиридин-3-ил-1Н-бензимидазол-2-ил) карбаминовой кислоты (I-44): к смеси 2-метил-2-тиопсевдомочевины (109 мг, 0,39 ммоль) и этилхлорформиата (75 мкл, 0,78 ммоль) в воде (2 мл) при 5°С добавляли в течение 40 мин 6Н водный раствор NaOH до стабилизации значения pH на 8. Затем значение pH доводили до 5 ледяной AcOH. Затем к реакционной смеси добавляли суспензию 1-(2,3-диамино-5-пиридин-3-ил-фенил)пропан-1-она (0,30 ммоль) в воде (5 мл). Реакционную смесь нагревали при 90°С в течение 18 ч, охлаждали до комнатной температуры и разбавляли водой (10 мл) и EtOAc (20 мл). Фазы разделяли и водный слой экстрагировали EtOAc (3×). Объединенные органические экстракты промывали насыщенным раствором соли, высушивали (MgSO4), фильтровали и фильтрат концентрировали в вакууме. Сырой остаток переводили в ДМСО, очищали препаративной ВЭЖХ и колоночной флэш-хроматографией (SiO2, CH2Cl2:MeOH, от 1:0 до 19:1). Затем остаток превращали в соль бис-HCl с получением указанного в заголовке соединение в виде не совсем белого твердого вещества (5,0 мг): ВЭЖХ (от 10 до 90% CH3CN, 8 мин): Rt = 3,90 мин; 1H ЯМР (CD3OD, 500 МГц) д 9,39 (д, 1H), 9,07 (д, 1H), 8,93 (д, 1H), 8,48 (д, 1H), 8,28 (д, 1Н), 8,25 (дд, 1H), 4,46 (кв, 2H), 3,35 (кв, 2H), 1,43 (т, 3H), 1,29 (т, 3H). MC (ES+): m/z(M++1) 339,1. Пример 21 Последующие соединения, представленные в таблице 2 ниже, получали способами, известными в данной области, по общим схемам I-IX, и способами, в основном аналогичными представленным в примерах 1-20 выше. Характеристики данных соединений приведены в таблице 2 ниже и включают данные 1Н-ЯМР (при 500 МГц) и данные масс-спектрометрии (МС). Номера соединений соответствуют номерам соединений, представленным в таблице 1.
Пример 22 Определение АТФазной активности гиразы Активность ДНК-гиразы гидролизовать АТФ определяли при сочетании продукции АДФ при участии пируваткиназы/лактатдегидрогеназы с окислением NADH. Данный метод был описан ранее (Tamura and Gellert, 1990, J. Biol. Chem., 265, 21342). Определение АТФазы проводили при 30°С в буферных растворах, содержащих 100 мкМ Трис, pH 7,6, 1,5 мкМ MgCl2, 150 мкМ KCl. Система сочетания содержит (конечные концентрации) 2,5 мкМ фосфоенолпирувата, 200 мкМ никотинамидадениндинуклеотида (NADH), 1 мкМ DTT, 30 мкг/мл пируваткиназы и 10 мкг/мл лактатдегидрогеназы. Добавляют 40 нМ фермента (субъединица Gyr A2B2 массой 374 kDa из Staphylococus aureus) и раствор ингибитора в ДМСО до конечной концентрации 4%, и реакционную смесь инкубируют в течение 10 мин при 30°С. Затем реакцию начинают добавлением АТФ до конечной концентрации 0,9 мкМ и определяют скорость исчезновения NADH при 340 нм в течение 10 мин. Значения Кi определяют по графику зависимости скорости от концентрации и выражают в виде среднего значения из двух параллельных значений. Было установлено, что соединения, представленные в таблице 1 выше, являются ингибиторами активности гиразы. Пример 23 Определение АТФазной активности топоизомеразы IV Превращение АТФ в АДФ под воздействием топоизомеразы IV сочетают с превращением NADH в NAD+ и определяют по изменению поглощения при 340 нм. Топоизомеразу IV инкубируют с ингибитором (конечная концентрация 4% в ДМСО) в буфере в течение 10 мин при 30°С. Реакцию начинают с добавления АТФ и регистрируют скорость превращения в течение 20 мин при 30°С на ридере для планшетов Molecular Devices SpectraMAX. Константу ингибирования Кi определяют по графику зависимости скорости от концентрации [ингибитора], построенному с использованием уравнения Моррисона для прочного связывания ингибиторов. Буфер для топоизомеразы IV из S. aureus: 100 мМ Трис, 7,5, 2 мМ MgCl2, 200 мМ К·глутамата, 2,5 мМ фосфоенолпирувата, 0,2 мМ NADH, 1 мМ DTT, 4,25 мкг/мл линеаризованной ДНК, 50 мкг/мл BSA, 30 мкг/мл пируваткиназы и 10 мкг/мл лактатдегидрогеназы (LDH). Буфер для топоизомеразы IV из E.coli: 100 мМ Трис, 7,5, 6 мМ MgCl2, 20 мМ KCl, 2,5 мМ фосфоенолпирувата, 0,2 мМ NADH, 10 мМ DTT, 5,25 мкг/мл линеаризованной ДНК, 50 мкг/мл BSA, 30 мкг/мл пируваткиназы и 10 мкг/мл лактатдегидрогеназы (LDH). Было установлено, что соединения, представленные в таблице 1, являются ингибиторами активности топоизомеразы IV. Пример 24 Тестирование чувствительности в жидких средах Соединения по данному изобретению тестировали на противомикробную активность при тестировании чувствительности в жидких средах. Данные тесты проводили, руководствуясь указаниями последнего документа NCCLS для данной практики: “M7-A5 Methods for dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically; Approved Standard – Fifth Edition (2000)”. Другие публикации, такие как “Antibiotics in Laboratory Medicine (Edited by V.Lorian, Publishers Williams and Wilkins, 1996), обеспечивали основные практические методы для тестирования антибиотиков в лабораторных условиях. Несколько отдельных колоний бактерий (3-7) с чашки после посева штрихом переносили на соответствующую обогащенную бульонную среду, такую как МНВ, с добавками, подходящими для более «требовательных» микроорганизмов. Их культивировали в течение ночи до высокой плотности с последующим 1- или 2-тысячным разведением с получением титра в пределах от 5×105 до 5×106 КОЕ/мл. Альтернативно свежесобранные колонии можно инкубировать при 37°С примерно в течение 4-8 ч до тех пор, пока мутность культуры становится равной или превышает 0,5 по стандарту МкФарланда (примерно 1,5×108 клеток на мл) и разбавляли с получением тех же значений КОЕ/мл, указанных выше. При использовании более удобного метода посевной материал готовили с использованием промышленно доступного механического устройства (BBL PROMPT System), когда тонкой палочкой с перекрестными бороздками на дне непосредственно касаются пяти колоний, с последующим приготовлением суспензии бактерий в соответствующем объеме физиологического раствора. Разведение до соответствующей плотности клеток готовили из данной клеточной суспензии. Бульон, используемый для тестирования, состоял из МНВ с добавлением 50 мг/л Са2+ и 25 мг/л Mg2+. Готовили серии стандартных разведений антибиотиков и хранили, как указано в NCCLS, стандарте М7-А5, пределы разведений, как правило, составляли от 128 мкг/мл до 0,015 мкг/мл (при 2-кратных серийных разведениях). Тестируемые соединения растворяли и разводили в день проведения опытов; использовали те же или аналогичные пределы концентраций, указанные выше. Тестируемые соединения и контроли разливали в мультилуночный планшет и тестируемые бактерии вносили так, чтобы конечный титр составлял примерно 5×104 КОЕ/лунку, и конечный объем равнялся 100 мкл. Планшеты инкубировали при 35°С в течение ночи (16-20 ч) и на глаз определяли мутность или проводили количественное определение на ридере для мультилуночных планшетов. Конечной минимальной ингибирующей концентрацией (MIC) является самая низкая концентрация препарата, при которой отсутствует рост микроорганизмов. Подобные определения также сравнивали с соответствующими табличными данными, представленными в двух вышеуказанных публикациях для гарантии того, что пределы значений антибактериальной активности находятся в приемлемых пределах для данного стандартизированного анализа. В таблице 3 представлены результаты теста определения MIC для выбранных соединений по данному изобретению, тестированных против S. aureus. Номера соединений соответствуют номерам соединений в таблице 1. Соединения, обладающие активностью, обозначенной «А», имеют значение MIC ниже или равное 0,5 мкг/мл; соединения, активность которых обозначена «В», имеют значения MIC в пределах от 0,5 до 1,0 мкг/мл; соединения с активностью, обозначенной «С», имеют значения MIC выше 1,0 мкг/мл.
В таблице 4 представлены результаты определения MIC для выбранных соединений по данному изобретению против S. pneumoniae. Номера соединений соответствуют номерам соединений в таблице 1. Соединения, обладающие активностью, обозначенной «А», имеют значение MIC ниже или равное 0,5 мкг/мл; соединения, активность которых обозначена «В», имеют значения MIC в пределах от 0,5 до 1,0 мкг/мл; соединения с активностью, обозначенной «С», имеют значения MIC выше 1,0 мкг/мл.
Несмотря на то, что заявители описали ряд воплощений настоящего изобретения, очевидно, понятно, что их основные положения можно изменять с обеспечением других воплощений, в которых применяются продукты и способы по данному изобретению.
Формула изобретения
1. Соединение формулы I
или его фармацевтически приемлемая соль, где Q представляет собой -NH- или -О-; W представляет собой C-R4; X представляет собой СН; R1 представляет собой 5-6-членное арильное кольцо, содержащее 1-3 атома азота, где R1 замещен 0-3 группами, независимо выбранными из R, оксо, или OR’; каждый R’ независимо выбран из атома водорода или С1-4алифатической группы; R2 выбран из атома водорода или С1-3алифатической группы; R3 выбран из C(O)NHR, C(O)N(R)2, C(O)R, CO2R, C(R’)=NOR или C(R’)=NOH; каждый R независимо выбран из Т-Ar или С1-6алифатической группы, где указанная С1-6алифатическая группа замещена 0-3 группами, независимо выбранными из R’ или OR’; Т представляет собой (CH2)у, где у равно 0, 1 или 2; Ar выбран из: (a) 3-8-членного насыщенного, ненасыщенного или арильного кольца; (b) 3-7-членного гетероциклического кольца, содержащего 1-3 атома азота; или (c) 5-6-членного гетероарильного кольца, содержащего 1-3 атома азота где Ar замещен 0-3 R’ группами; и R4 выбран из атомов водорода или фтора. 2. Соединение по п.1, где R1 представляет собой необязательно замещенное 5-6-членное гетероарильное кольцо, содержащее 1-2 атомов азота. 3. Соединение по п.2, где R’ замещен 0-2 группами, выбранными из атома галогена, оксо, R, или OR’. 4. Соединение по п.1, где R3 выбран из C(O)NHR, C(O)R, C(R’)=NOR, C(R’)=NOH или CO2R, где каждый R независимо выбран из необязательно замещенной С1-4алифатической группы или Т-Ar, где Т представляет собой (СН2)у, где у равно 0, 1 или 2; и Ar представляет собой необязательно замещенное кольцо, выбранное из 5-6-членного насыщенного, ненасыщенного или арильного кольца, 5-6-членного гетероциклического кольца, содержащего 1-2 атома азота, или 5-6-членного гетероарильного кольца, содержащего 1-2 атома азота. 5. Соединение по п.4, где каждый R независимо выбран из С1-4алифатической группы или Т-Ar, где указанная С1-4алифатическая группа замещена 0-2 группами, независимо выбранными из OR’; Т представляет собой (СН2)у, где у равно 0, 1 или 2; и Ar выбран из пирролидинила, пиримидинила, пиразинила, пиридила, пиперидинила, имидазолила, пиридазинила, пиразолила, или циклопентена, где Ar замещен 0-2 R’группами. 6. Соединение по п.4, где указанное соединение имеет формулу II или II’:
или его фармацевтически приемлемая соль, где кольцо А замещено 0-2 группами, независимо выбранными из OR’ или R. 7. Соединение по п.4, где указанное соединение имеет формулу III или III’:
или его фармацевтически приемлемая соль, где каждое кольцо В замещено 0-2 группами, независимо выбранными из оксо или R. 8. Соединение по п.1, где указанное соединение имеет формулу III-а:
или его фармацевтически приемлемая соль, где изображенное пиридоновое кольцо замещено 0-2 группами, независимо выбранными из атома оксо, R или OR’. 9. Соединение по п.8, где указанное соединение имеет формулу III-b:
или его фармацевтически приемлемая соль. 10. Соединение по п.9, где Q представляет собой -NH-. 11. Соединение по п.6, где R2 представляет собой этил. 12. Соединение по п.7, где R2 представляет собой этил. 13. Соединение по п.10, где R2 представляет собой этил. 14. Соединение по п.1, выбранное из группы, включающей:
15. Композиция для ингибирования активности бактериальной гиразы и/или топоизомеразы IV, содержащая эффективное количество соединения по п.1 и фармацевтически приемлемый носитель, адъювант или наполнитель. 16. Композиция по п.15, содержащая, кроме того, дополнительно антибиотик, выбранный из хинолинов, бета-лактамов, макролидов, гликопептидов или липопептидов или средство, повышающее чувствительность бактерий к антибиотикам. 17. Способ ингибирования активности гиразы и/или топоизомеразы IV в биологическом образце или у пациента, включающий стадию контактирования указанного биологического образца с: a) композицией по п.15 или b) соединением по п.1. 18. Способ снижения числа бактерий у пациента, зараженного нозокомикальными и ненозокомикальными бактериальными инфекциями, включающий стадию введения указанному пациенту: a) композиции по п.15 или b) соединения по п.1. 19. Способ по п.18, где бактериальная инфекция, которая подвергается лечению, характеризуется наличием одного или более из следующих микроорганизмов: Streptococcus pneumoniae, или Staphylococcus aureus. 20. Способ по п.19, где бактериальная инфекция, которая подвергается лечению, выбрана из одного или нескольких следующих состояний: инфекции мочевых путей, респираторной инфекции, инфекции хирургических ран, инфекции центральной линии, бактериемии, бронхита, синусита, пневмонии, инфекции кожи и мягких тканей, внутрибрюшинной инфекции или бактериальной инфекции у пациентов с лихорадочной нейтропенией. 21. Способ по п.20, включающий, кроме того, стадию введения указанному пациенту дополнительного терапевтического средства, выбранного из антибиотика, противовоспалительного средства, ингибитора матричной металлопротеазы, ингибитора липоксигеназы, антагониста цитокинов, иммуносупрессора, противоопухолевого средства, противовирусного средства, цитокина, фактора роста, иммуномодулятора, простагландина, средства против гиперпролиферации сосудов, либо в виде части сложной лекарственной формы вместе с указанным соединением, либо в виде отдельной лекарственной формы. 22. Способ получения соединения формулы А:
или его соли, включающий стадию взаимодействия соединения формулы В:
или его соли, с соединением формулы С:
где указанное взаимодействие осуществляют при pH от 2 до 7 протонном растворителе, выбранном из воды метанола и этанола; в присутстввие минеральной кислоты, выбранной из серной кислоты или соляной кислоты при температуре 40-100°С, и где X представляет собой атом кислорода; RX и RY, взятые вместе, образуют 6-членное арильное кольцо, где указанное кольцо, образованное RX и RY необязательно замещено 1-3 группами, независимо выбранными из атома галогена или R1; R1 представляет собой 5-6-членное арильное кольцо, содержащее 1-3 атома азота, где R1 замещен 0-3 группами, независимо выбранными из R’, оксо, или OR’; каждый R’ независимо выбран из атома водорода или С1-4алифатической группы; R3 выбран из C(O)NHR, C(O)N(R)2, COR, CO2R, C(R’)=NOH или C(R’)=NOR; каждый R независимо выбран из Т-Ar или С1-6алифатической группы, где указанная С1-6алифатическая группа замещена 0-3 группами, независимо выбранными из R’ или OR’; Т представляет собой (CH2)у, где у равно 0, 1 или 2; Ar выбран из: (a) 3-8-членного насыщенного, ненасыщенного или арильного кольца; (b) 3-7-членного гетероциклического кольца, содержащего 1-3 атома азота; или (c) 5-6-членного гетероарильного кольца, содержащего 1-3 атома азота, где Ar замещен 0-3 группами, независимо выбранными из R; R2 выбран из атома водорода или С1-3алифатической группы. 23. Способ получения по п.24 соединения формулы I’:
или его фармацевтически приемлемой соли, где R1 представляет собой 5-6-членное арильное кольцо, содержащее 1-3 атома азота где R1 замещен 0-3 группами, независимо выбранными из R, оксо, или OR’; каждый R’ независимо выбран из атома водорода или С1-4алифатической группы; R2 выбран из атома водорода или С1-3алифатической группы; R3 выбран из C(O)NHR, C(O)N(R)2, C(O)R, CO2R, C(R’)=NOH или C(R’)=NOR; каждый R независимо выбран из Т-Ar или С1-6алифатической группы, где указанная С1-6алифатическая группа замещена 0-3 группами, независимо выбранными из R’ или OR’; Т представляет собой (СН2)у, где у равно 0, 1 или 2; Ar выбран из: (a) 3-8-членного насыщенного, ненасыщенного или арильного кольца; (b) 3-7-членного гетероциклического кольца, содержащего 1-3 атома азота; или (c) 5-6-членного гетероарильного кольца, содержащего 1-3 атома азота, где Ar замещен 0-3 группами, независимо выбранными из R’.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
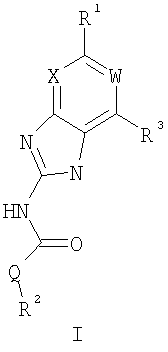
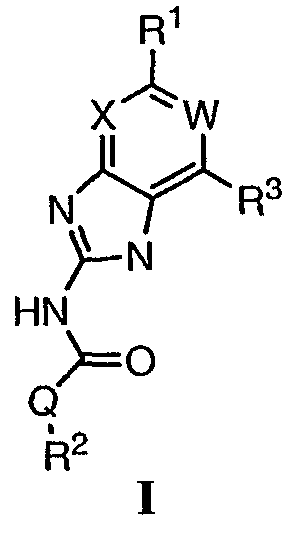
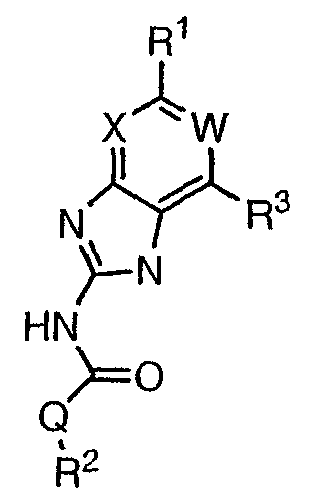
 CH, CH2циклопропил, 1-этилпиперидин-3-ил, CH(CH2CH3)CH2OCH3, CH(CH3)CH2OCH3, дигидрофуран-2-он-3-ил, 1-метил-1,5-дигидро-имидазол-4-он-2-ил, пиридазин-4-ил, имидазол-2-ил, 3Н-пиридин-4-он-2-ил, пиримидин-5-ил, циклопентен-4-ил, 1-метилимидазол-2-ил, тетрагидропиранил, СН2(3-метил-изоксазол-5-ил) или СН2(1,3-диметилпиразол-5-ил).
CH, CH2циклопропил, 1-этилпиперидин-3-ил, CH(CH2CH3)CH2OCH3, CH(CH3)CH2OCH3, дигидрофуран-2-он-3-ил, 1-метил-1,5-дигидро-имидазол-4-он-2-ил, пиридазин-4-ил, имидазол-2-ил, 3Н-пиридин-4-он-2-ил, пиримидин-5-ил, циклопентен-4-ил, 1-метилимидазол-2-ил, тетрагидропиранил, СН2(3-метил-изоксазол-5-ил) или СН2(1,3-диметилпиразол-5-ил).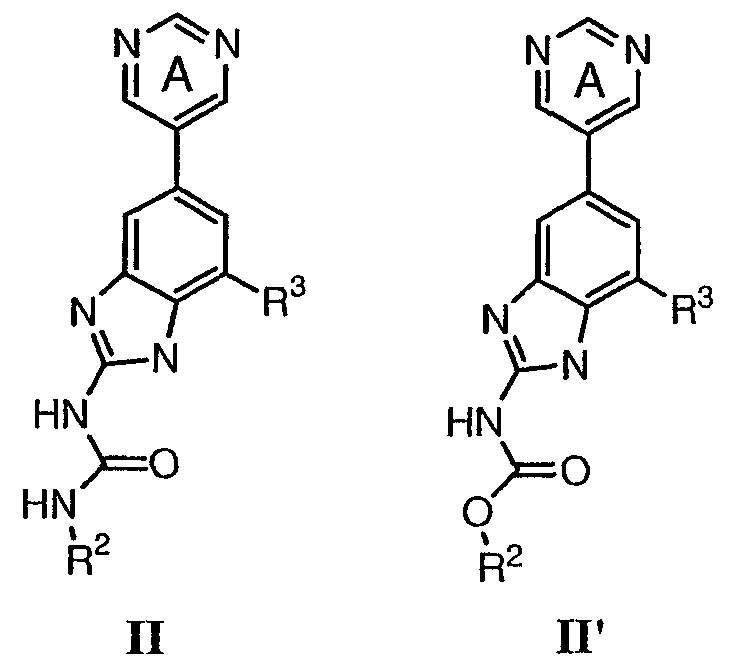
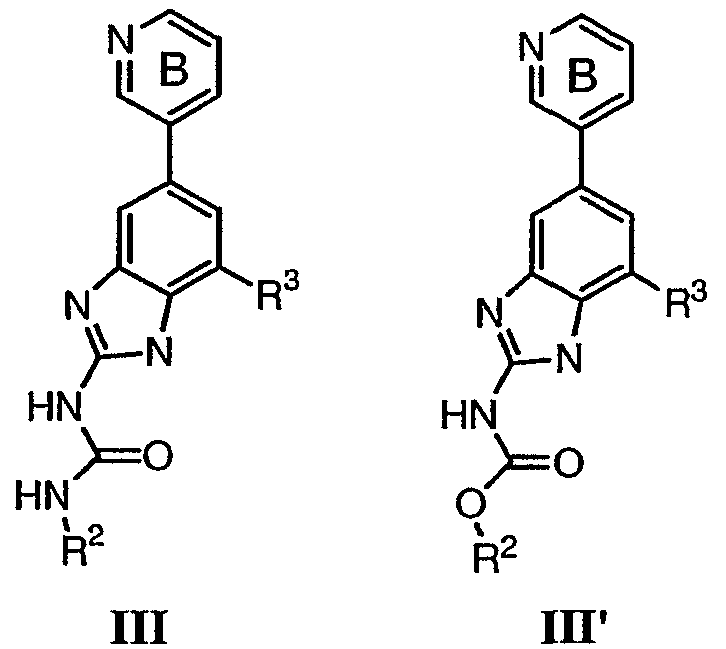
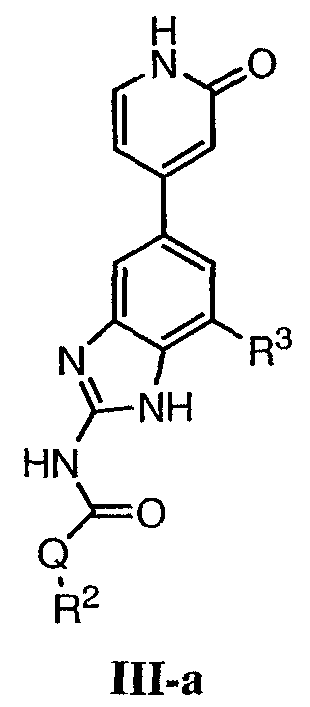
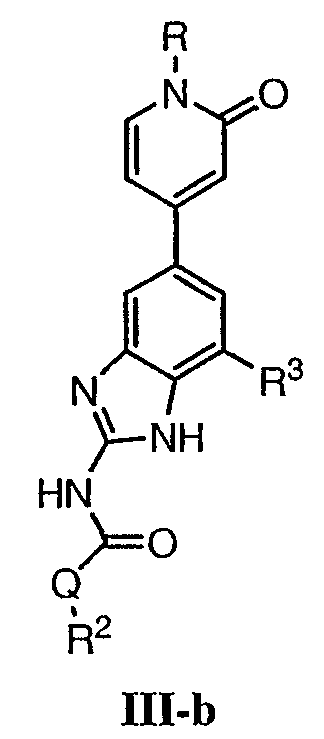
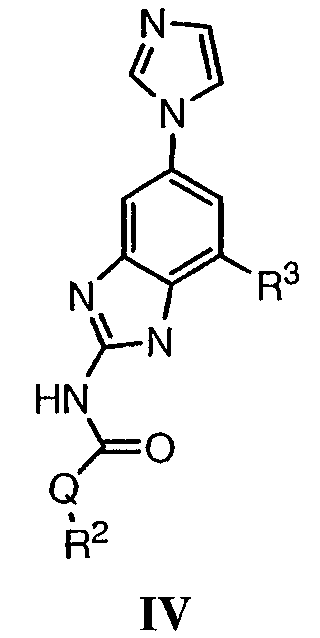
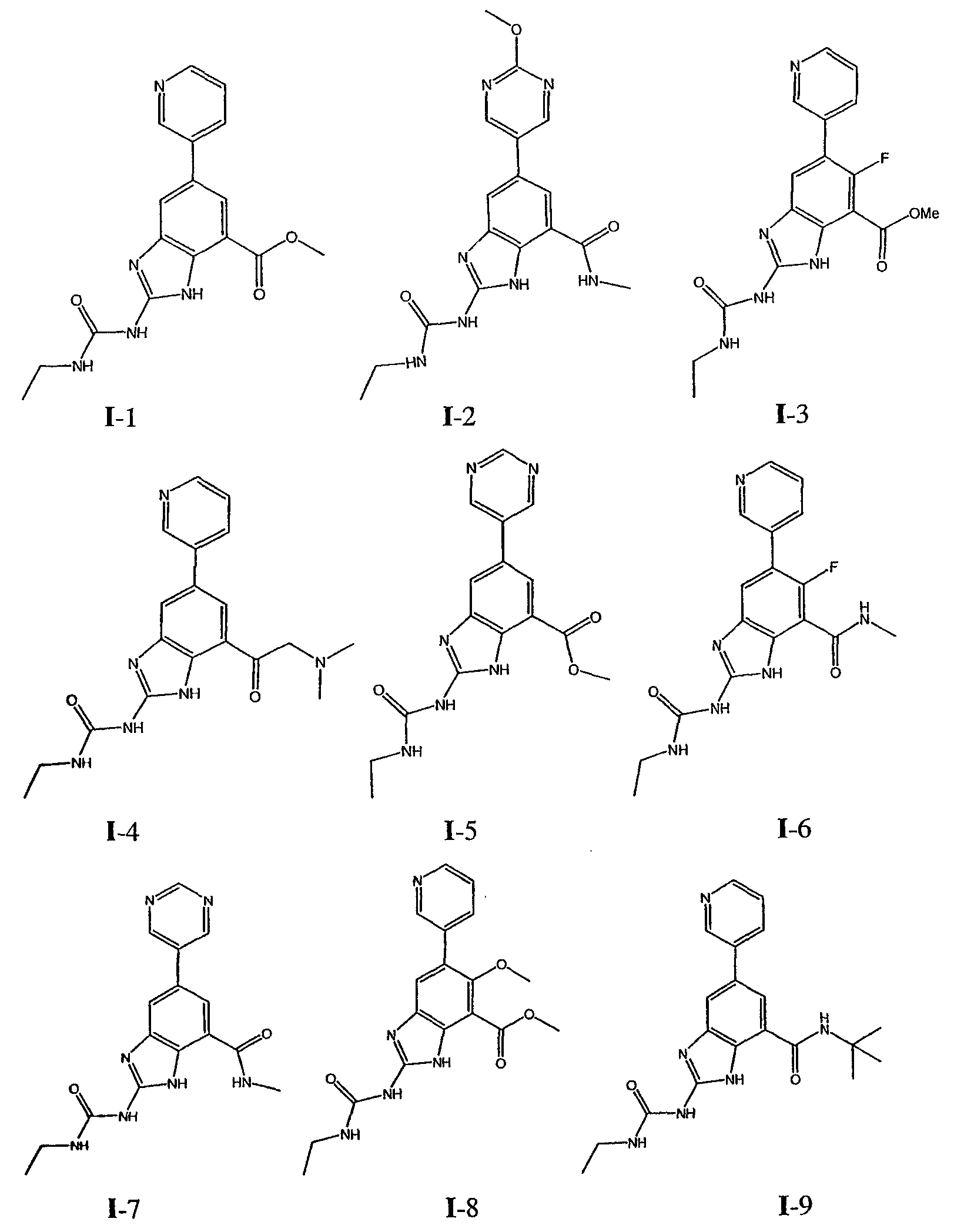
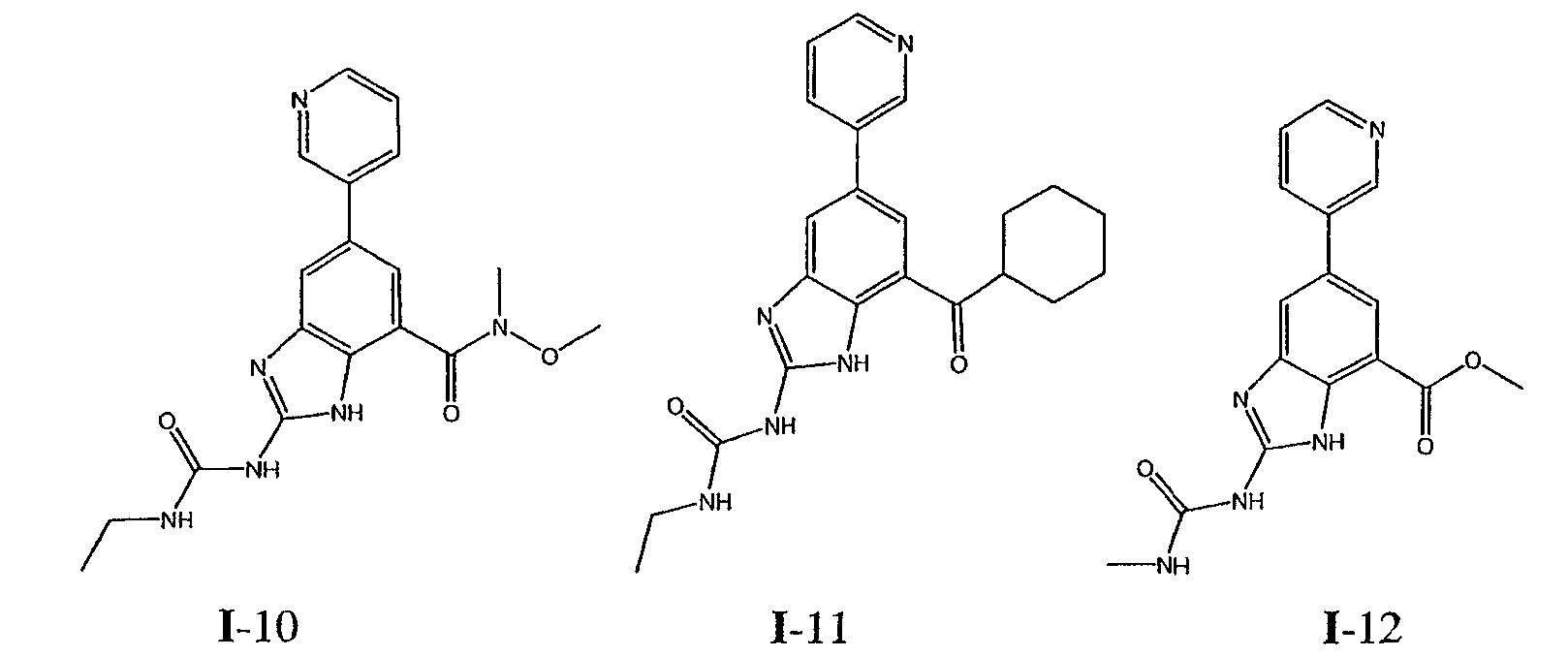
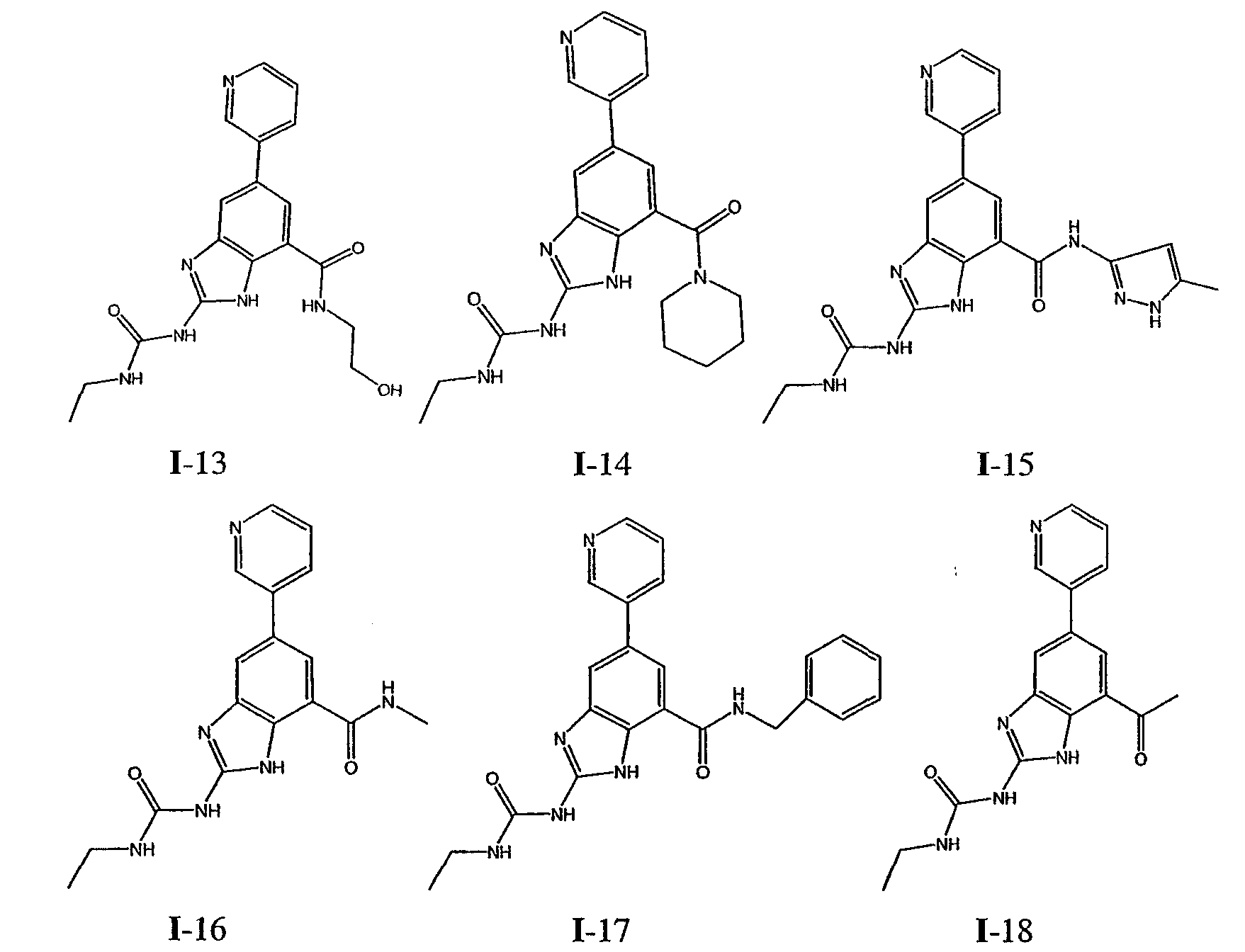
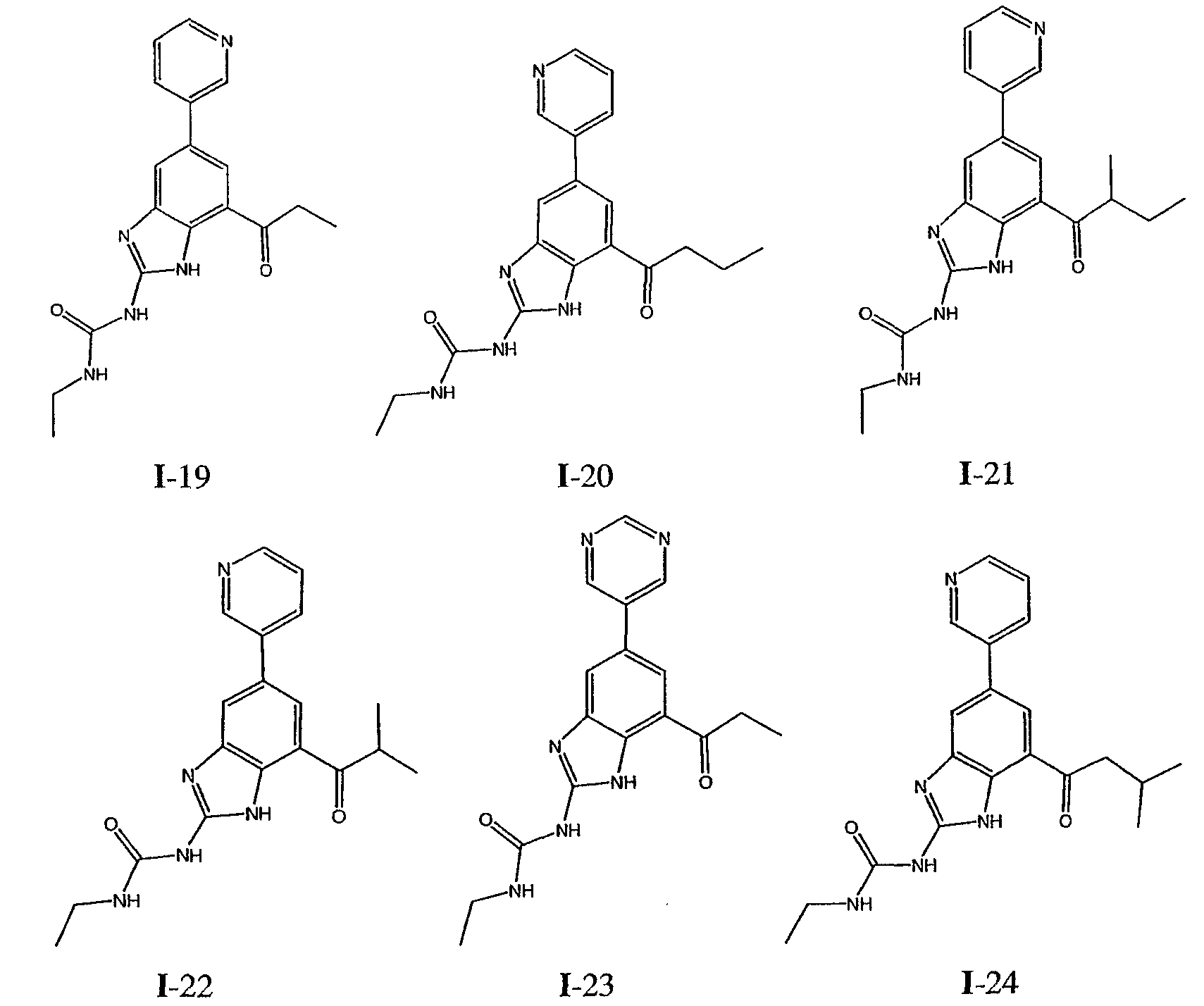
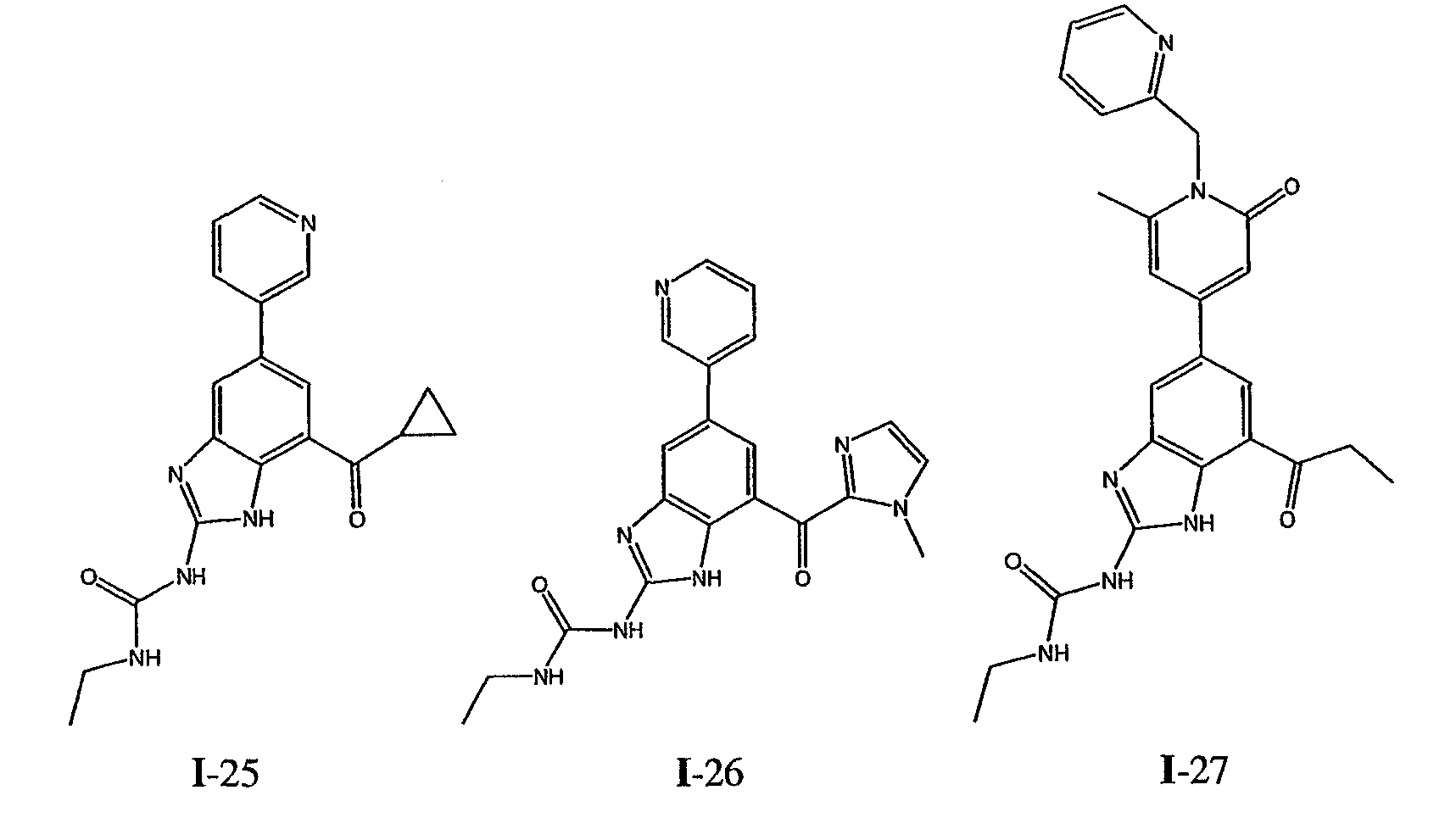
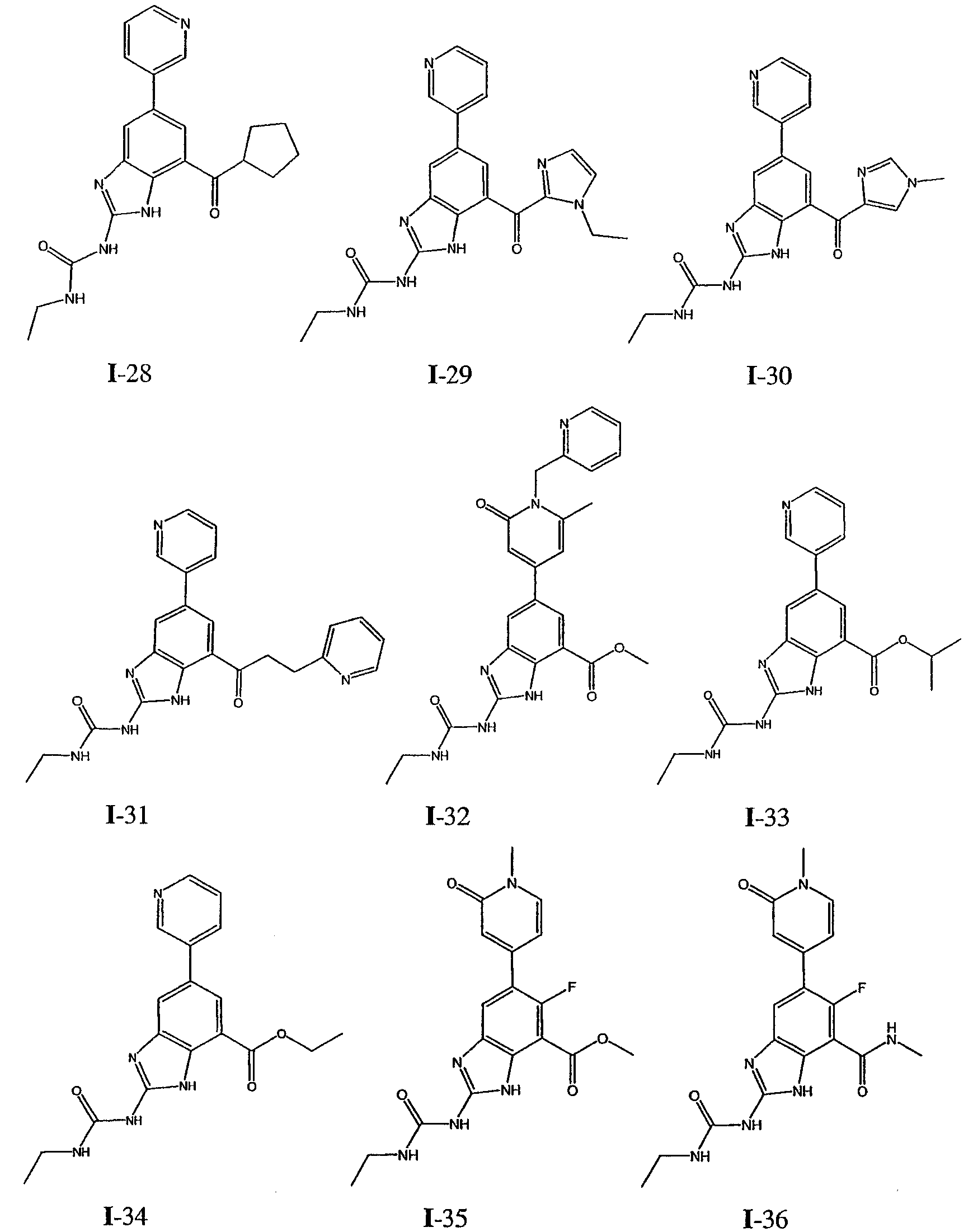
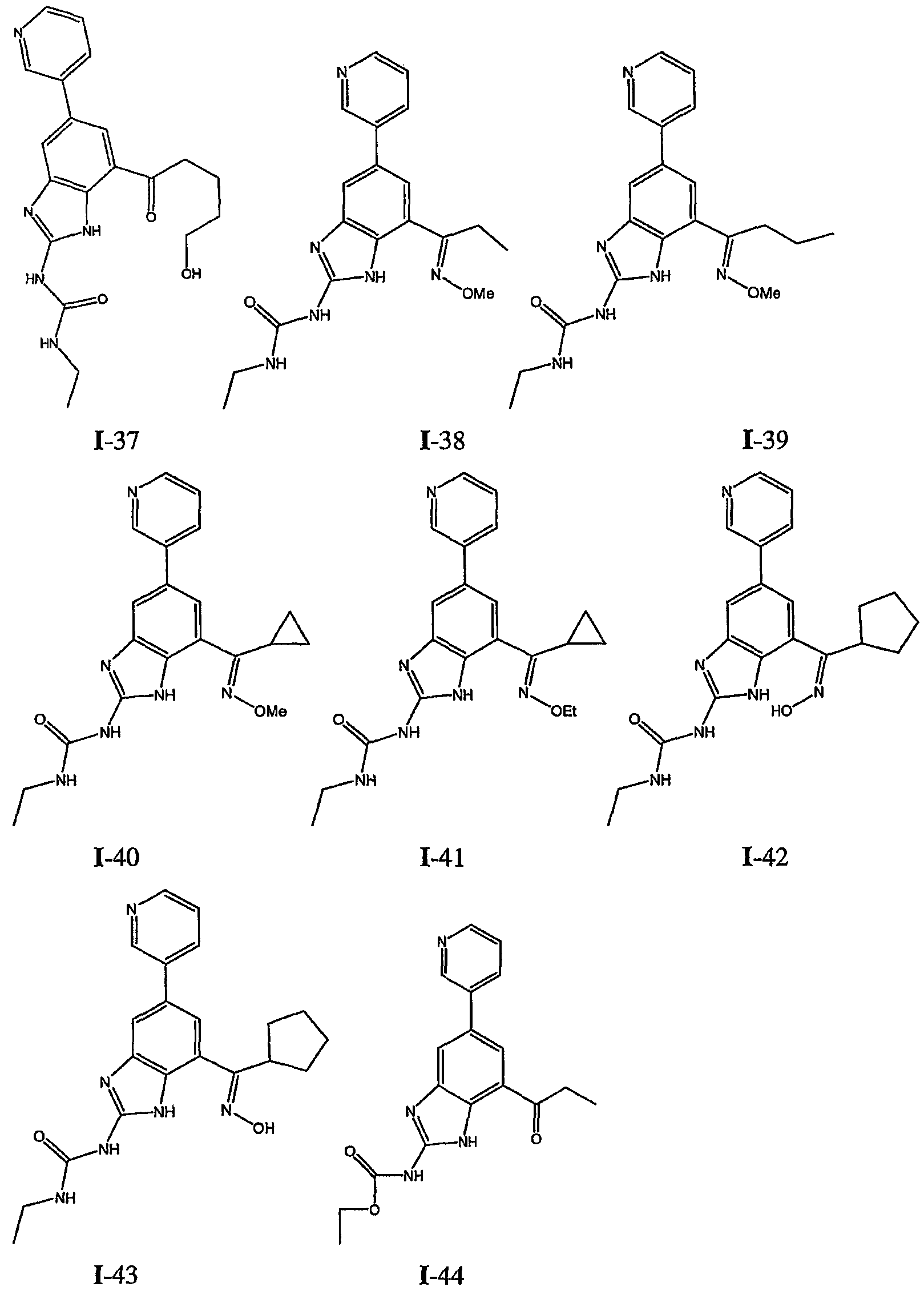
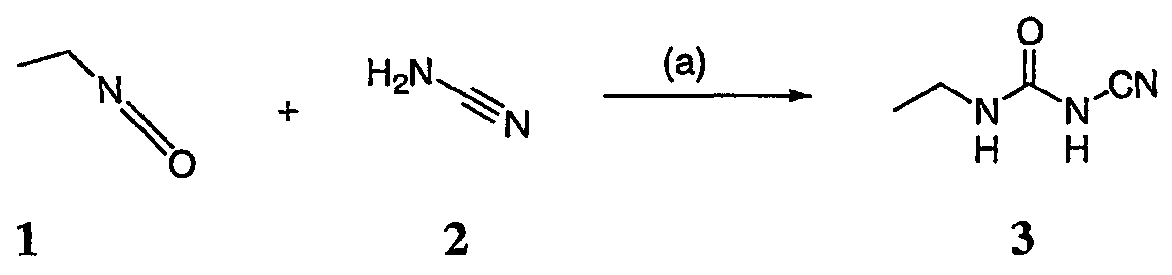
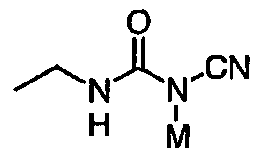
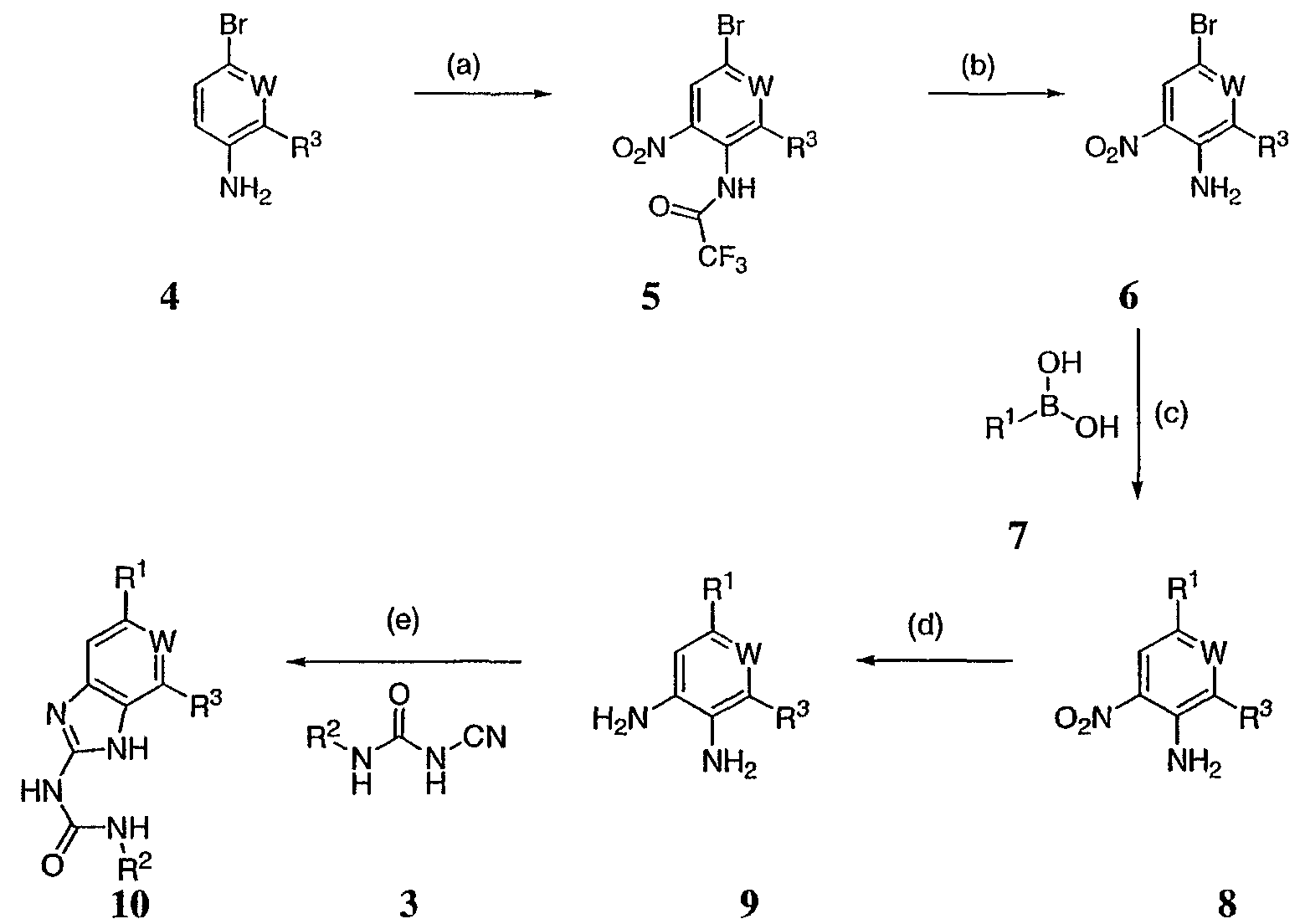
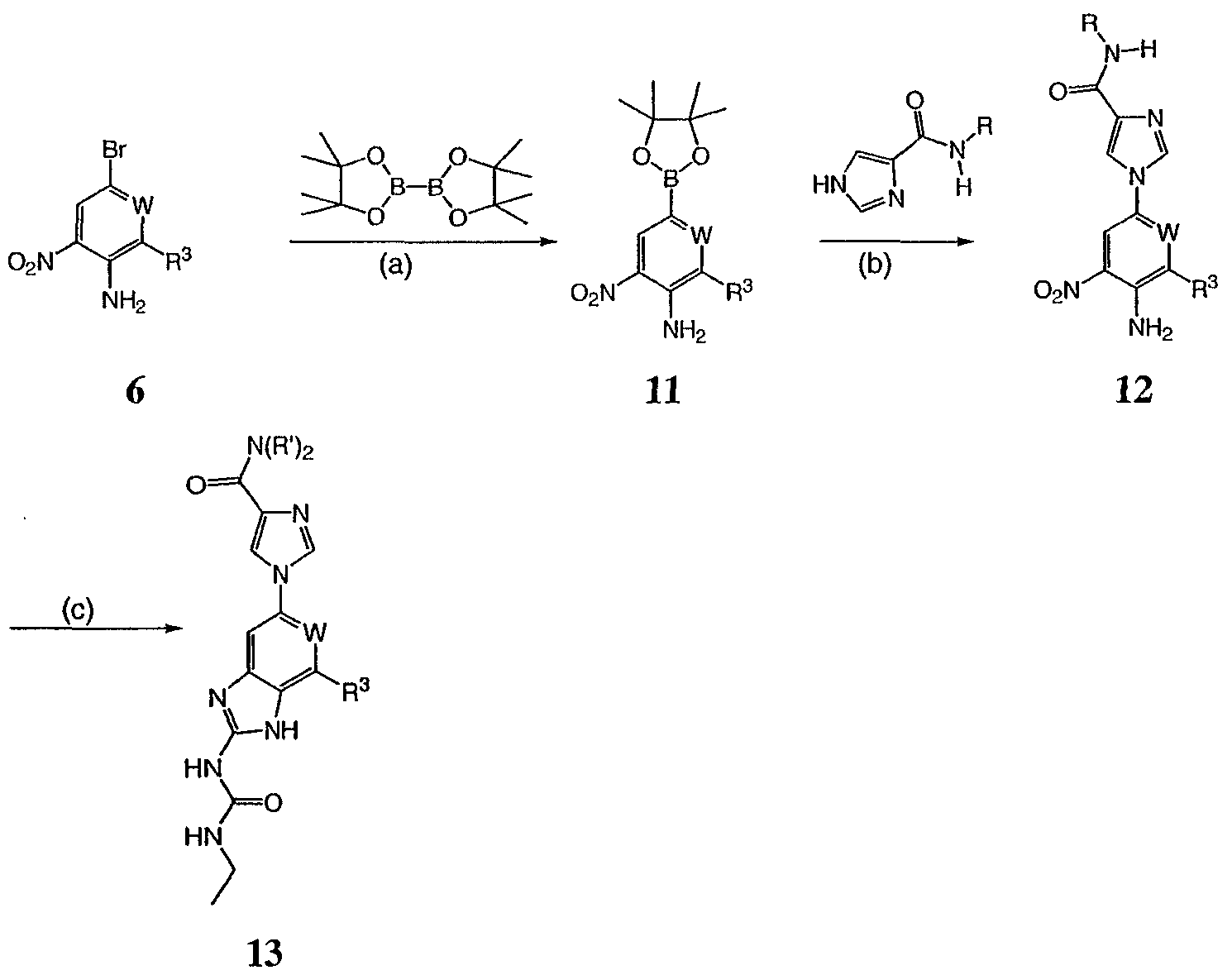
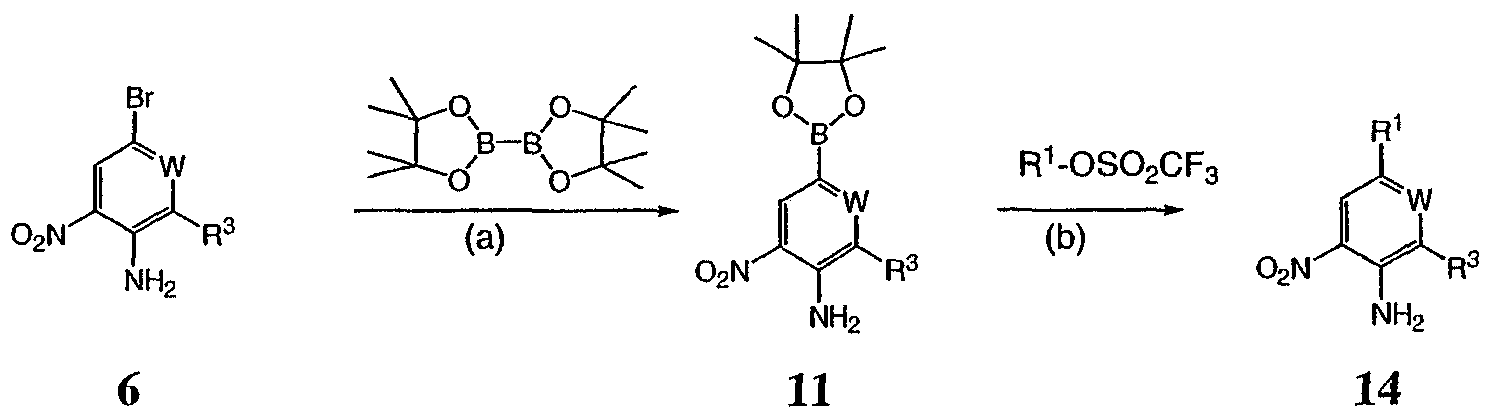
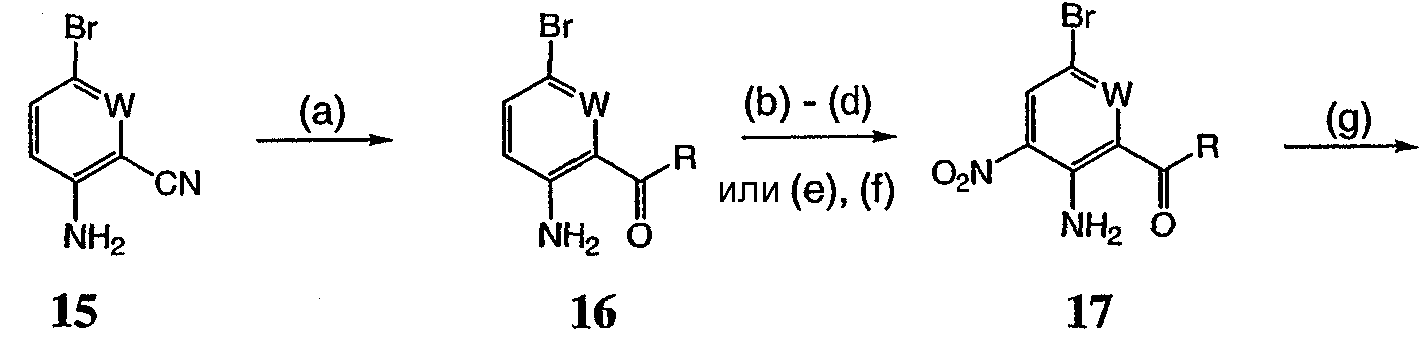
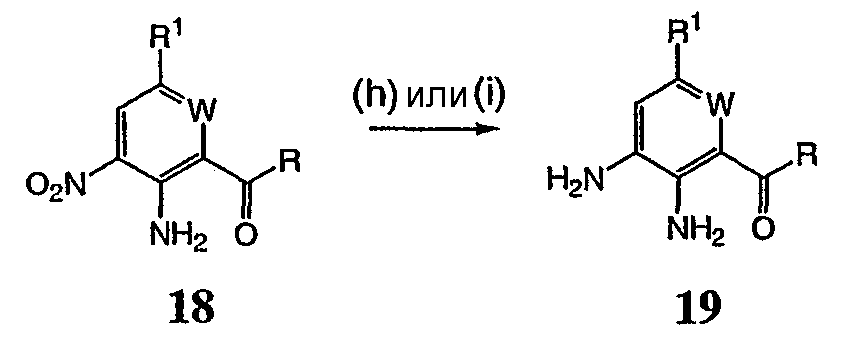
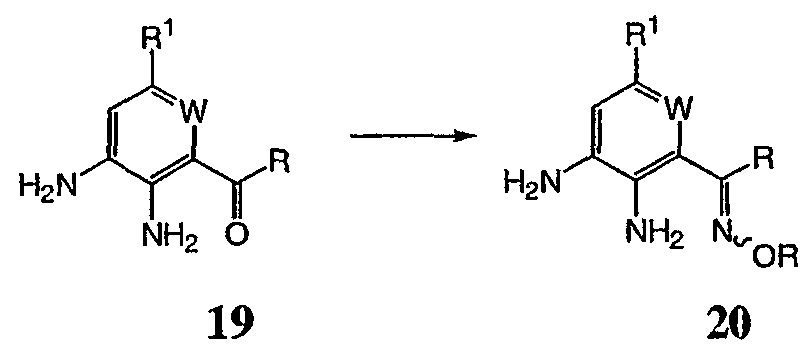
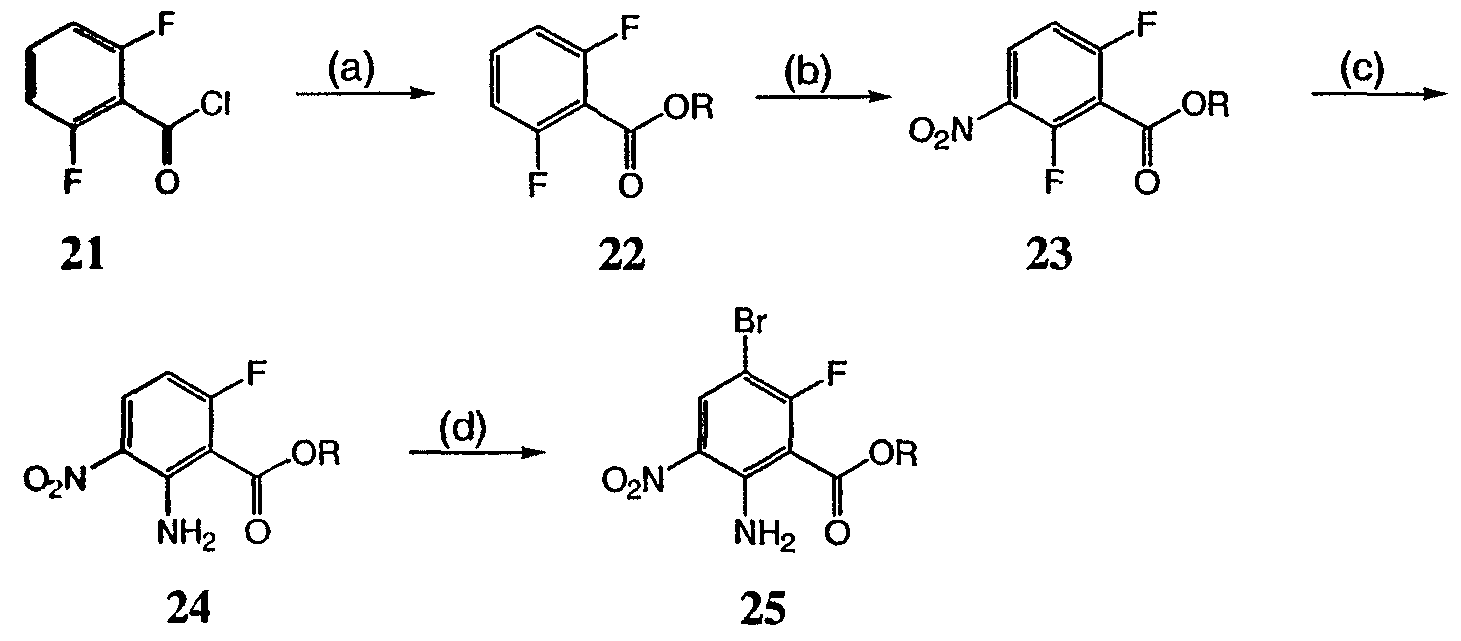
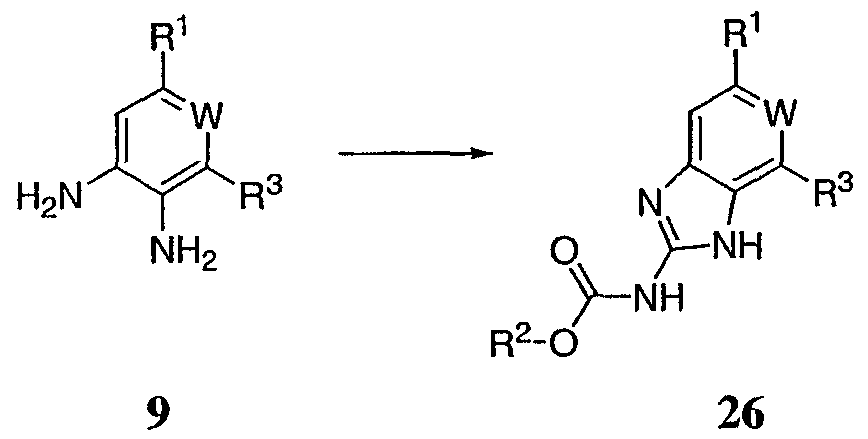
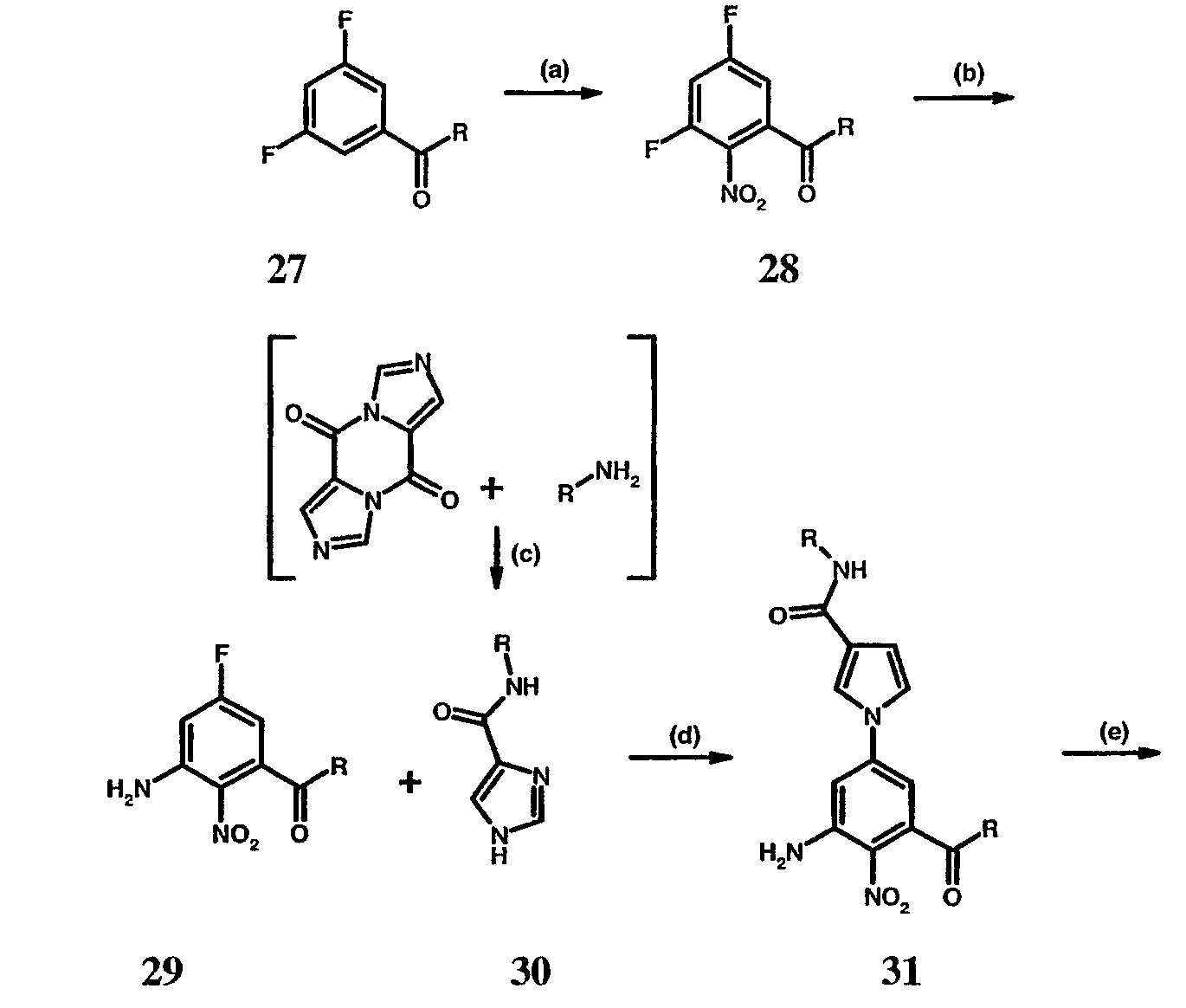
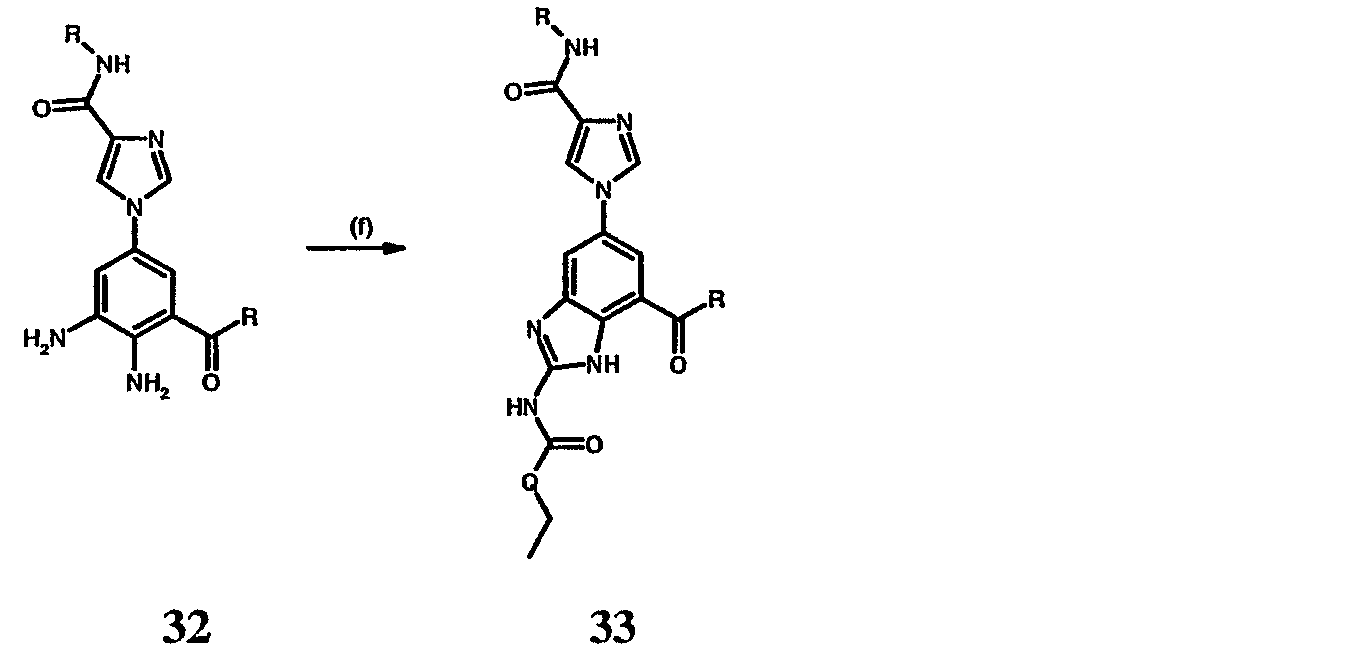
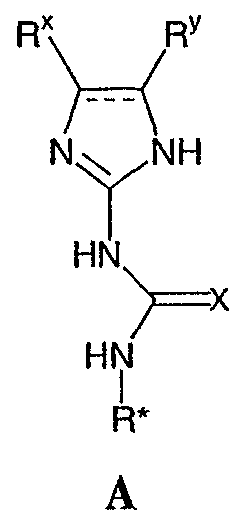
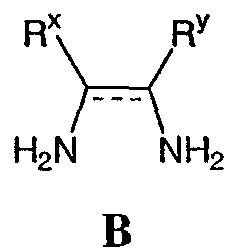
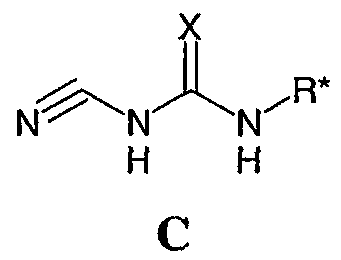
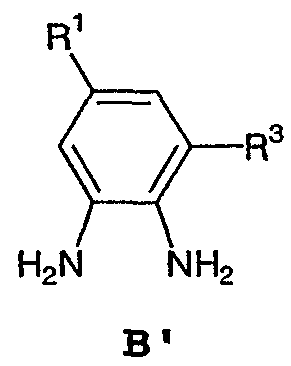
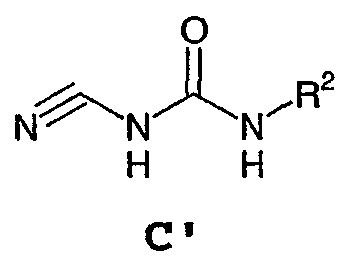
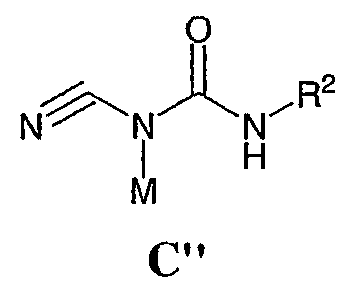
 -лактамы, макролиды, гликопептиды и липопептиды.
-лактамы, макролиды, гликопептиды и липопептиды.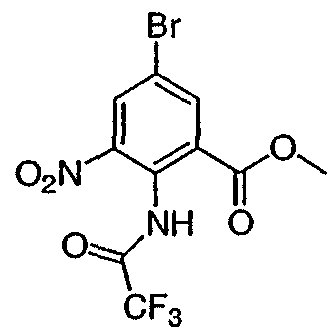
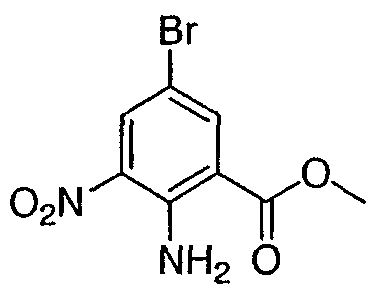
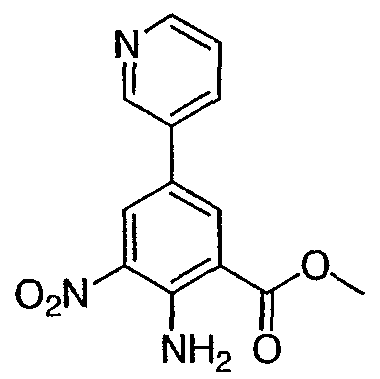
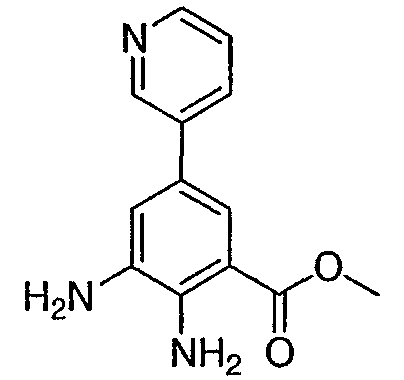
 ,54 (шир, 1H), 3,91 (с, 3H), 5,76 (шир, 1H), 7,08-7,81(м, 7H), 8,54 (м, 1H), 8,79 (м, 1H).
,54 (шир, 1H), 3,91 (с, 3H), 5,76 (шир, 1H), 7,08-7,81(м, 7H), 8,54 (м, 1H), 8,79 (м, 1H).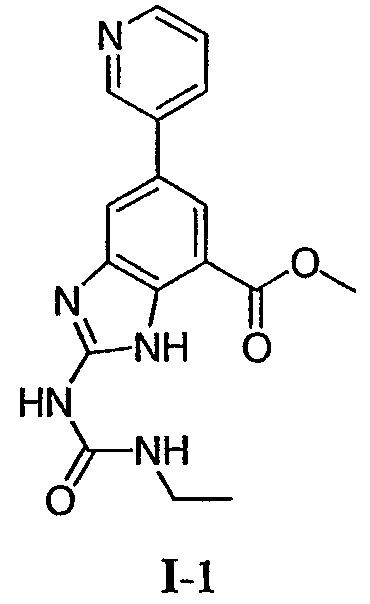
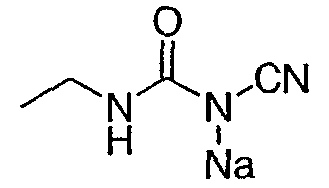
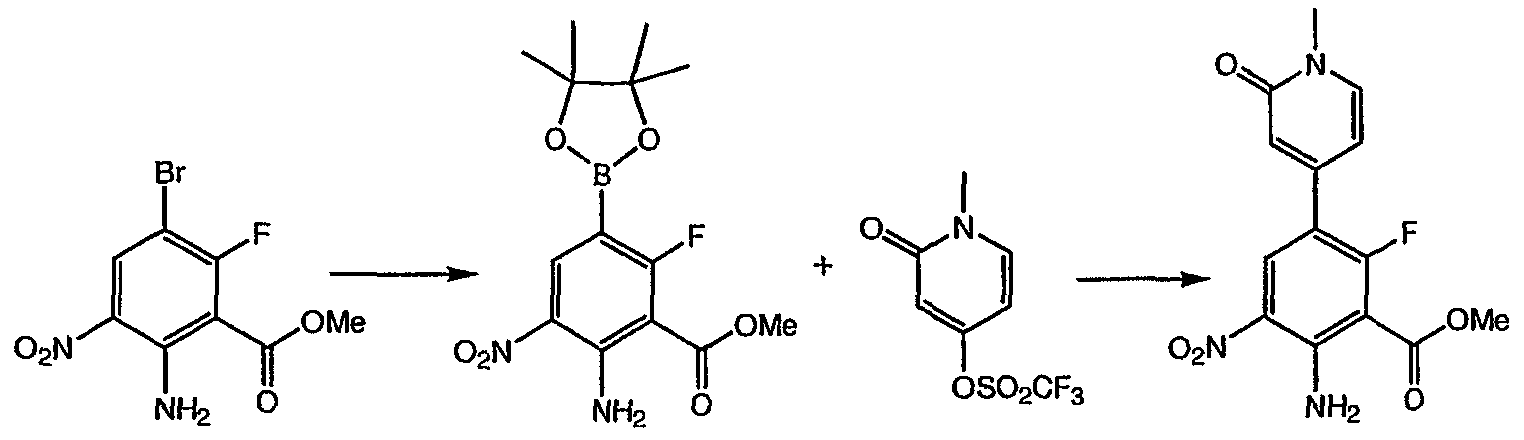
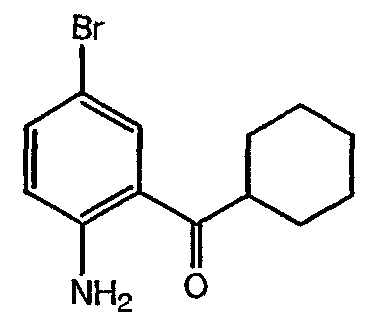
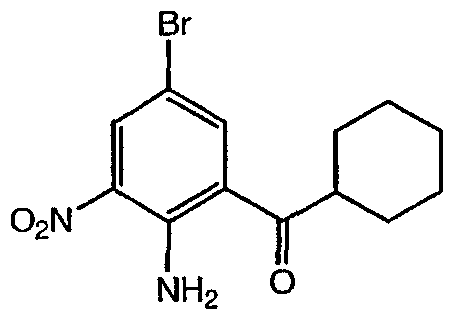
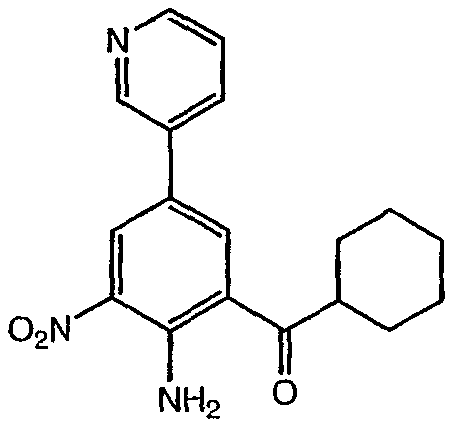
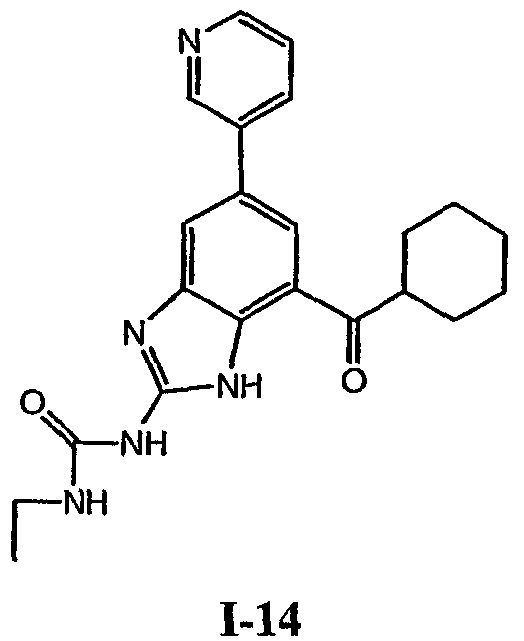
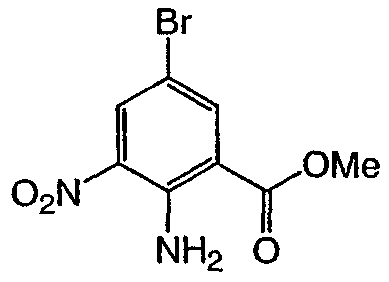
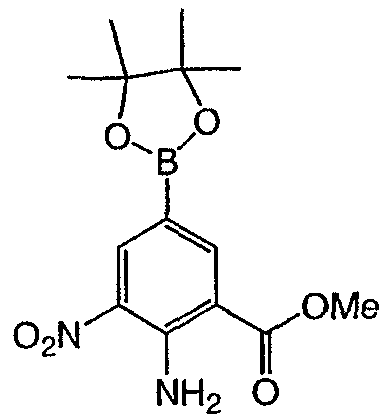
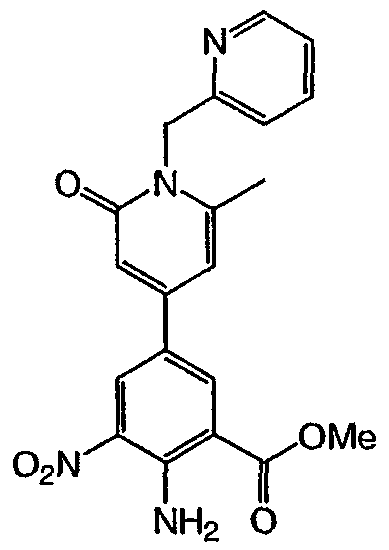
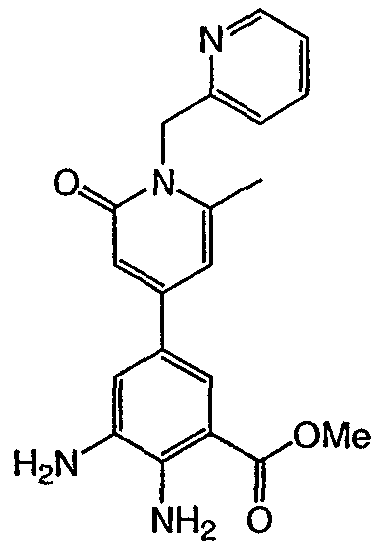
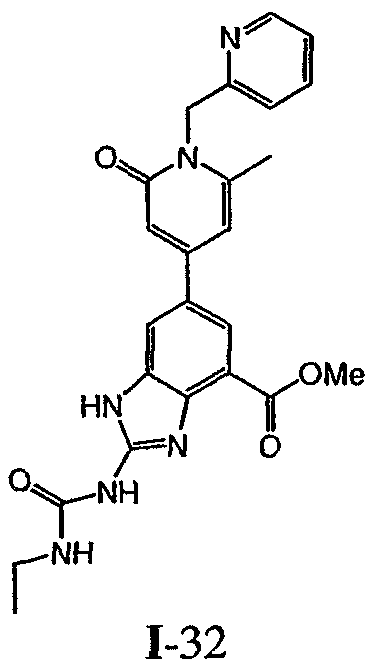
 до 3 добавлением дополнительного количества серной кислоты полученную смесь кипятили с обратным холодильником в течение 3 ч. После охлаждения до комнатной температуры смесь подщелачивали насыщенным водным раствором карбоната натрия и разбавляли водой. Полученную суспензию фильтровали и дополнительно промывали водой. Твердые частицы растворяли в тетрагидрофуране, высушивали над сульфатом натрия, фильтровали и концентрировали досуха. Водный маточный раствор экстрагировали этилацетатом, промывали насыщенным раствором соли, высушивали над сульфатом натрия, фильтровали и высушивали в вакууме. Свободное основание выделяли из маточного раствора, переводили в этилацетат и метанол, подкисляли избытком безводной HCl и концентрировали досуха с получением указанного в заголовке соединения в виде дигидрохлорида (желтовато-коричневое твердое вещество, 0,036 г). 1H ЯМР (500 МГц, ДМСО) д 1,12 (т, 3H), 2,41 (c, 3H), 3,22 (м, 2H), 3,97 (c, 3H), 5,38 (c, 2H), 6,6 (c, 1H), 6,68 (c, 1H), 7,8-7,25 (м, 2H), 7,48 (шир. 1H), 7,79 (т, 1H), 7,94 (c, 1H), 8,03 (c, 1H), 8,49 (c, H), 9,93 (шир. 1H), 11,53 (шир. 1H). Rt при ВЭЖХ: 3,7 мин [градиент от 10 до 90% в течение 12 мин, 1 мл/мин 0,1% TFA (YMC 3×150)]. МС: М+Н=461, М-Н=459. Rt МС/ВЭЖХ=2,24 мин.
до 3 добавлением дополнительного количества серной кислоты полученную смесь кипятили с обратным холодильником в течение 3 ч. После охлаждения до комнатной температуры смесь подщелачивали насыщенным водным раствором карбоната натрия и разбавляли водой. Полученную суспензию фильтровали и дополнительно промывали водой. Твердые частицы растворяли в тетрагидрофуране, высушивали над сульфатом натрия, фильтровали и концентрировали досуха. Водный маточный раствор экстрагировали этилацетатом, промывали насыщенным раствором соли, высушивали над сульфатом натрия, фильтровали и высушивали в вакууме. Свободное основание выделяли из маточного раствора, переводили в этилацетат и метанол, подкисляли избытком безводной HCl и концентрировали досуха с получением указанного в заголовке соединения в виде дигидрохлорида (желтовато-коричневое твердое вещество, 0,036 г). 1H ЯМР (500 МГц, ДМСО) д 1,12 (т, 3H), 2,41 (c, 3H), 3,22 (м, 2H), 3,97 (c, 3H), 5,38 (c, 2H), 6,6 (c, 1H), 6,68 (c, 1H), 7,8-7,25 (м, 2H), 7,48 (шир. 1H), 7,79 (т, 1H), 7,94 (c, 1H), 8,03 (c, 1H), 8,49 (c, H), 9,93 (шир. 1H), 11,53 (шир. 1H). Rt при ВЭЖХ: 3,7 мин [градиент от 10 до 90% в течение 12 мин, 1 мл/мин 0,1% TFA (YMC 3×150)]. МС: М+Н=461, М-Н=459. Rt МС/ВЭЖХ=2,24 мин.