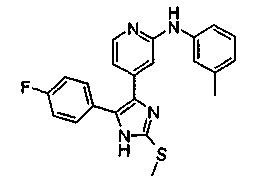
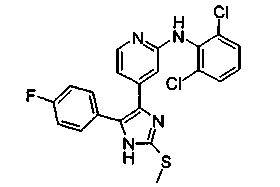
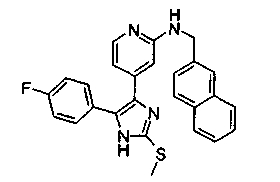
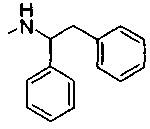
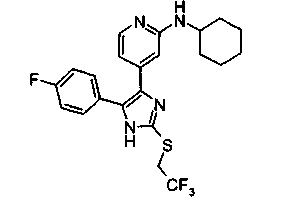
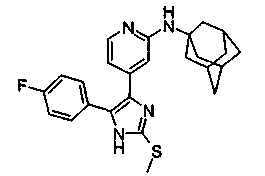
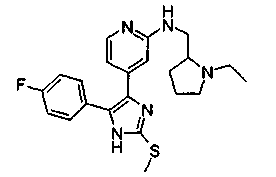
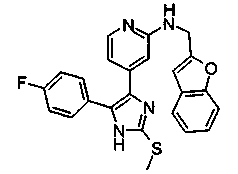
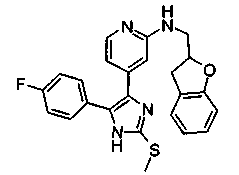
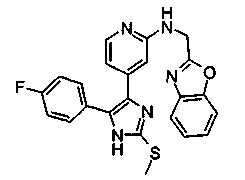
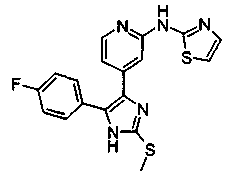
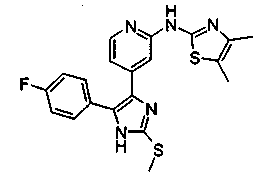
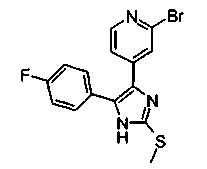
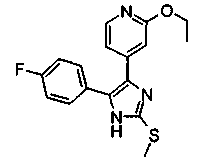
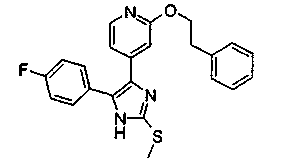
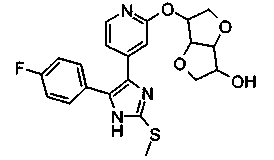
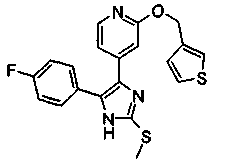
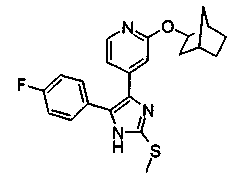
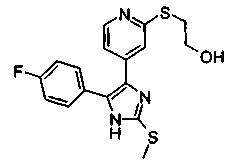
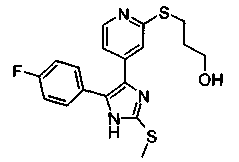
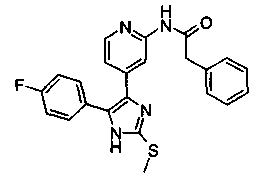
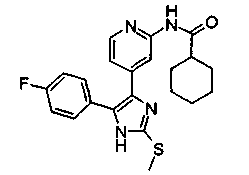
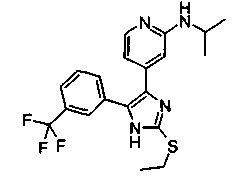
Патент на изобретение №2331638
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) 2-ТИОЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА И ИХ ПРИМЕНЕНИЕ В ФАРМАЦЕВТИКЕ
(57) Реферат:
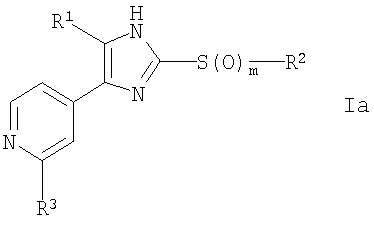
Изобретение относится к 2-тиозамещенному производному имидазола формулы I, где R1 представляет собой арил, который является замещенным атомом галогена или галоген-С1-С6-алкилом; R2 выбран из группы, включающей а) арил-С1-С4-алкил и b) C1-С6-алкил; R3 выбран из группы, включающей (a) NR4R10, (b) NR7COR10, (с) OR10, (d) NH2; R4 представляет собой H; R5 и R6, которые могут быть одинаковыми или различными, представляют собой H, галоген, ОН, С1-С6-алкокси, C1-С6-алкил или галоген-C1-С6-алкил; R7 представляет собой R4; R10 имеет одно из следующих значений: (а) А-В, (b)-(e), (f) C1-С6-алкил, который замещен 2 фенильными группами; А представляет собой линейный или разветвленный C1-С6-алкилен; В выбран из группы, включающей (а) H, (b)-(е), (f) OC1-С6-алкил, (g) ОН; Ну представляет собой 3-10-членный неароматический, моно-, би- или трициклический карбоцикл, который может быть или может не быть конденсирован с бензольным кольцом; Ar представляет собой 5- или 6-членный ароматический гетероцикл, который имеет 1 гетероатом, выбранный из группы, состоящей из О, S и N, и который может не быть конденсирован с бензольным кольцом; Het представляет собой 5- или 6-членный неароматический гетероцикл, который имеет 1 гетероатом, который представляет собой О, который может не быть конденсирован с бензольным кольцом; m равно 0, 1 или 2; или его оптические изомеры или физиологически приемлемые соли. Соединения формулы I применяют при получении фармацевтической композиции, обладающей ингибирующей активностью в отношении высвобождения цитокинов. Технический результат – 2-тиозамещенные производные имидазола, обладающие ингибирующим высвобождением цитокина действием. 3 н. и 10 з.п. ф-лы, 4 табл.
(56) (продолжение): CLASS=”b560m”mitogen-activated protein kinase. J. MED. CHEM., 1999, v.42, p.2180-2190. JEFFREY C.BOEHM et al. 1-Substituted 4-aryl-5-pyridinylimidazoles: a new class of cytokine suppressive drugs with low 5-lipoxygenase and cyclooxygenase inhibitory potency. J. MED. CHEM. 1996, v.39, p.3929-3937. WO 93/14081 A1, 22.07.1993. WO 2002/066458 A, 29.08.2002. DE 10114775 A, 10.10.2002. SU 1259962 A3, 23.09.1986.
Настоящее изобретение относится к 2-тиозамещенным производным имидазола, обладающим иммуномодулирующим и ингибирующим высвобождение цитокина действием, к фармацевтическим композициям, включающим данные соединения и их применению в фармацевтике. Фармакологически активные соединения имидазола с противовоспалительной активностью уже известны. Так, среди прочих, соединения, имеющие 4,5-ди(гетеро)арилимидазольные фрагменты, были исследованы более подробно и было описано их различное фармацевтическое действие. Также известны соединения, которые являются замещенными во 2-положении. В патенте США 4585771 описаны производные 4,5-дифенилимидазола, которые замещены во 2-положении пирролильным, индолильным, имидазолильным или тиазолильным радикалом и которые обладают противовоспалительной и противоаллергической активностью. В патентах США 4461770, 4528298 и 4584310 (ЕР 004648А) описаны производные 4-(5-арил)-5-(4-гетероарил)имидазола, которые замещены во 2-положении по тио-, сульфинильной или сульфонильной группе замещенным или незамещенным алифатическим углеводородом и которые, помимо прочего, обладают противовоспалительным действием. Соединения имидазола, обладающие иммуномодулирующим и ингибирующим высвобождение цитокина действием, описаны в WO 02/066458, WO 02/076951 и DE 10222103. Кроме того, в WO 96/03387, EP 005545 (US 4440776, 4355039, 4269847), EP 236628 (патент США 4686231), патенте Германии 3504678, патенте США 4190666, патенте США 4402960 и патенте США 4585771 описаны производные имидазола, обладающие противовоспалительным действием. В патентах ЕР 372445 (патент США 5318984; патент США 5166214) и патенте США 5364875 описаны соединения имидазола, обладающие антигиперхолестеринемической активностью. Патент WO 00/17192 (патент Германии 19842833) относится к производным 4-гетероарил-5-фенилимидазола, которые замещены во 2-положении фенилалкилтио группой. Данные соединения действуют как противовоспалительные соединения и ингибиторы высвобождения цитокина. В WO 99/03837 и WO 93/14081 описаны 2-замещенные имидазолы, которые ингибируют синтез ряда воспалительных цитокинов. Соединения, описанные в WO 93/14081, имеют во 2-положении присоединенный через атом серы фосфорсодержащий заместитель или арильный, или гетероарильный заместитель. В WO 91/10662 и WO 91/13876 описаны производные имидазола, которые ингибируют трансферазу ацил-коэнзим А:холестерин О-ацил и связывание тромбоксана TxA2. В WO 95/00501 описаны производные имидазола, которые можно использовать в качестве ингибиторов циклооксигеназы. В J.Med.Chem., 1996, 39, 3927-37, описаны соединения, обладающие 5-липоксигеназа- и циклооксгеназа-ингибирующим действием, 2-(4-метилсульфинилфенил)-4-(4-фторфенил-5-(пирид-4-ил)имидазол, также обладающий цитоксинингибирующим действием. Кроме того, 2-тиозамещенные производные имидазола описаны в патенте Японии 01-040467, патенте России 1415725, Acta Chim., 1969, 61-77, J.Pract. Chem., 1972, 314, 785-792 и патенте Германии 10114775, Indian J.Chem., Sect.B, 1983, 22B(3), 268-269, Bioorganic & Medical Chem. Lett., Vol.5, No.2, 177-180, 1995, Phosphorus Sulfur 1988, 35(1-2), 83-88, Arch.Biochem.Biophys., Vol. 297, 258-164, 1992, J.Med.Chem. 1995, 38, 1067-1083, Helv.Chim.Acta 82, 1999, 290-296, Helv.Chim.Acta 81, 1998, 1585-1595. Несмотря на то, что известны многочисленные соединения, все еще сохраняется необходимость в соединениях, обладающих противовоспалительным действием, которые ингибируют высвобождение цитокина. Задачей настоящего изобретения является разработка таких соединений. Неожиданно было установлено, что некоторые 2-замещенные производные имидазола обладают высокой иммуномодулирующей и/или ингибирующей высвобождение цитокина активностью. Соответственно, настоящее изобретение относится к 2-тиозамещенным производным имидазола формулы I
где R1 представляет собой С1-С6-алкил, С3-С7-циклоалкил или арил, который является незамещенным или замещенным атомом галогена, С1-С6-алкилом или галоген-С1-С6-алкилом; R2 выбран из группы, включающей a) арил-С1-С4-алкил, где арильный радикал может иметь один, два или три заместителя, независимо друг от друга выбранных из группы, состоящей из С1-С6-алкила, С1-С6-алкокси, галогена, С1-С6-алкилсульфанила, С1-С6-алкилсульфинила, С1-С6-алкилсульфонила и гидроксила, и b) С1-С6-алкил, который является незамещенным или замещенным CN или галогеном; с) d) R3 выбран из группы, включающей a) NR4R10; b) NR7COR10; c) NR7COOR10; d) NR7CONR7R10; e) NR7CONR7COR10; f) OR10; g) S(O)mR10; h) галоген; i) OH; j) N3; k) NH2; l) SH; где R3 не является ОН, галогеном, С1-С6-алкилтио или С1-С6-алкокси, если R2 представляет собой фенил-С1-С4-алкил, и фенильный радикал имеет С1-С6-алкилсульфанильный, С1-С6-алкилсульфинильный или С1-С6-алкилсульфонильный заместитель; R4 представляет собой Н или физиологически отщепляемую группу, R5 и R6, которые могут быть одинаковыми или различными, представляют собой Н, галоген, ОН, С1-С6-алкокси, С1-С6-алкил, галоген-С1-С6-алкил, С1-С6-алкилсульфанил, NH2, С1-С6-алкиламино или ди-С1-С6-алкиламино; R7 представляет собой R4, С1-С6-алкил или бензил; R10 имеет одно из следующих значений:
f) С1-С6-алкил, который замещен 2 или 3 фенильными группами; g) трифторметил (в частности, если R3 представляет собой один из радикалов b)-f)). А представляет собой линейный или разветвленный С1-С6-алкилен, С2-С6-алкенилен или С3-алкинилен; В выбран из группы, включающей
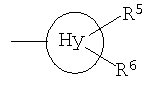
f) OC1-С6-алкил; g) NR11R12; h) OH; i) галоген; j) С1-С6-алкилсульфанил; R11 и R12, которые могут быть одинаковыми или различными, представляют собой Н, С1-С6-алкил или фенил; Hy представляет собой 3-10-членный неароматический, моно-, би- или трициклический карбоцикл, который может быть или может не быть конденсированным с бензольным кольцом; Ar представляет собой 5- или 6-членный ароматический гетероцикл, который имеет 1, 2 или 3 гетероатома, независимо друг от друга выбранных из группы, состоящей из О, S и N, и который может быть или может не быть конденсирован с бензольным кольцом; Het представляет собой 5- или 6-членный неароматический гетероцикл, который имеет 1, 2 или 3 гетероатома, независимо друг от друга выбранных из группы, состоящей из О, S и N, который может быть или может не быть конденсирован с бензольным кольцом, и который может быть или может не быть мостиковым бициклическим или трициклическим; m равно 0,1 или 2; n равно 1, 2, 3, 4 или 5 и их таутомерам, оптическим изомерам и физиологически приемлемым солям. Если соединения согласно изобретению имеют центры асимметрии, в объем изобретения входят как рацематы, так и оптические изомеры (энантиомеры, диастереомеры). Для соединений согласно изобретению возможно существование следующего таутомерного равновесия:
Данное изобретение охватывает обе таутомерные формы. Изобретение также охватывает физиологически приемлемые соли соединений формулы I. В настоящем случае они в частности представляют кислотно-аддитивные соли. Для получения кислотно-аддитивных солей используют неорганические кислоты, такие как хлористоводородная кислота, серная кислота или фосфорная кислота, или органические кислоты, такие как винная кислота, лимонная кислота, малеиновая кислота, фумаровая кислота, яблочная кислота, миндальная кислота, аскорбиновая кислота, глюконовая кислота и тому подобные. Термин «алкил» (также в сочетании с другими группами, такими как фенилалкил, алкилсульфонил, алкокси и т.д.) охватывает линейные и разветвленные алкильные группы, имеющие от 1 до 6 или от 1 до 4 атомов углерода, такие как метил, этил, н- и изопропил, н-, изо- и трет-бутил, втор-бутил, н-пентил, изоамил, неопентил и н-гексил. Соответственно, это применяется к термину «С1-С6-алкилен». Термин «карбоцикл» охватывает насыщенные или ненасыщенные неароматические моноциклические, бициклические и трициклические углеводороды. Углеводороды могут быть конденсированы с одним или двумя бензольными кольцами. Моноциклические углеводороды представляют собой С3-С6-циклоалкил, такие как циклопропил, циклопентил, циклогексил. Примерами би- и трициклических углеводородов и бензоконденсированных карбоциклов являются инданил, декалинил, тетралинил, флуоренил, дигидроантрацнил, дибензосуберенил, норборнил или адамантил. Примерами замещенных карбоциклов являются метилциклопропил или метилциклогексил. Предпочтение отдается незамещенным радикалам. Термин «арил» охватывает ароматические кольцевые системы, такие как фенил или нафтил. Термин «галоген» означает атом фтора, хлора, брома или иода, в частности, атом фтора или атом хлора. Термин «галоген-С1-С6-алкил» охватывает моно- и полигалогенированные линейные и разветвленные алкильные группы, имеющие от 1 до 6 и, в частности, от 1 до 4 атомов углерода. Предпочтительно присутствуют 1, 2, 3, 4 или 5 атомов галогена. Предпочтительными атомами галогена являются F и Cl. Примерами галоген-С1-С6-алкила являются -CH2Cl, -CH2CH2Cl, -CH2CCl3, -CF3, -CHF2, -CH2F, -CH2CF3 и CF2CF3. Предпочтительным является CF3. Физиологически отщепляемая группа представляет собой группу, которая может отщепляться от остатка молекулы при физиологических условиях, ферментативно или химически. Примерами являются -COR14, -CO2R14, -CONH2, -CONHR14, -CHR16-OR14, -CHR16-O-COR14, -COC(R16)2-OH, -COR15, SO2R15 и -SO2R14, где R14 представляет собой С1-С6-алкил или CF3, R15 представляет собой фенил или толил (в частности п-толил), и R16 представляет собой Н или С1-С6-алкил. Ароматический 5- или 6-членный гетероцикл в частности представляет собой незамещенный (R5, R6=H) или замещенный 2-пиридил, 3-пиридил, 4-пиридил, 2- или 3-тиенил, 2- или 3-фурил, тиазолил, имидазолил, оксазолил, изотиазолил, триазолил или пиримидил. Предпочтительными заместителями являются одна или две группы, независимо друг от друга выбранные из группы, включающей галоген, в частности Cl, и С1-С6-алкил. Заместитель(и) присоединены к атому углерода или атому азота ароматического радикала. Предпочтительными являются незамещенные радикалы. Примерами замещенных радикалов являются хлортиенил, в частности 5-хлорфур-2-ил, примерами конденсированных радикалов являются бензофуранил, бензотиазолил и бензотиофен. Неароматический 5- или 6-членный гетероцикл может быть насыщенным или ненасыщенным. Предпочтительно это незамещенный или замещенный тетрагидрофуранил, тетрагидропиранил, пирролидинил, N-метилпирролидинил, N-этилпирролидинил, пиперазинил или морфолинил, где гетероцикл может быть присоединен через гетероатом азота или кольцевой атом углерода или может быть замещенным. Предпочтительными заместителями являются один или два радикала, независимо друг или друга выбранные из группы, включающей галоген, в частности Cl, и С1-С6-алкил. Предпочтение отдается незамещенным радикалам.
представляет собой замещенный или незамещенный циклопропил, циклобутил, циклогептил и, в частности, циклопентил и циклогексил. R5 и R6 предпочтительно, независимо друг от друга, представляют собой Н, галоген или С1-С6-алкил. Примерами замещенных циклоалкильных групп являются метилциклопропил или метилциклогексил. Предпочтение отдается незамещенным радикалам. Фенил-С1-С4-алкил представляет собой, в частности, бензил, 1-фенилэтил или 2-фенилэтил. R1 предпочтительно представляет собой фенильный радикал и, в частности, галоген-, CF3– или С1-С6-алкил-замещенный фенильный радикал, при этом особенно предпочтительным является фторзамещенный фенильный радикал. Заместитель предпочтительно находится в 3- и, в частности, в 4-положении. Примерами замещенных фенильных радикалов являются 4-фторфенил, 2,4-дифторфенил, 3-трифторметил, 3-толил или 3-хлорфенил. R2 предпочтительно представляет собой бензильный, С3-С6-циклоалкильный, С4-С7-метилциклоалкильный или С1-С6-алкильный радикал, где фенильная группа в бензильном радикале может быть замещенной, как указано выше. Предпочтительными заместителями фенильной группы в бензильном радикале являются С1-С6-алкилсульфанил, С1-С6-алкилсульфинил и С1-С6-алкилсульфонил. Примерами R2 являются CH3, CH3CH2, (CH3)2CH, CH2CN, CH2CF3, CF3 и циклопропил. R3 предпочтительно представляет собой радикал формулы
где R4, R5 и R6, а также А являются такими, как определено выше. R5 и R6 предпочтительно представляют собой Н, метил, метокси или хлор. Если фенильное кольцо в данной группе является замещенным, радикалы R5 и R6 предпочтительно расположены в 3- и/или 4-положении. Кроме того, R3 предпочтительно представляет собой а) NR4R10, где R10 представляет собой циклопропил, циклопропилметил, циклопентил, циклогексил или циклогептил; b) NR4R10, где R10 представляет собой С1-С6-алкил, в частности, метил, этил или изопропил, или представляет собой 3,3-дифенилпропил или 1,3-дифенилпроп-2-ил; с) NR4R10, где R10 представляет собой А-В, и В представляет собой ОН, С1-С6-алкокси, NR11R12 или фенил; d) NR7COR10, где R10 представляет собой А-В, и В представляет собой фенил; е) NR7COR10, где R10 представляет собой С1-С6-алкил. А предпочтительно представляет собой С1-С6-алкилен и, в частности, этилиден. Особенно предпочтительным вариантом осуществления являются соединения формулы I, в которой R1 представляет собой 4-фторфенил, R2 представляет собой С1-С6-алкил или бензил, где фенильная группа в бензильном радикале может быть замещенной, как указано выше; R3 представляет собой радикал формулы
где R4, R5 и R6, а также А являются такими, как определено выше, и m равно 0. Следующим предпочтительным вариантом осуществления являются соединения формулы I, в которой R2 представляет собой С1-С6-алкил, в частности метил, и R1 представляет собой галогенфенил или галоген-С1-С6-алкилфенил, в частности, 4-фторфенил, 2,4-дифторфенил, 4-трифторметилфенил или 3-трифторметилфенил. Далее R3 предпочтительно представляет собой группы, определенные ниже: а) галоген, в частности F или Cl; b) OH или OC1-C6-алкил, в частности метокси и изопропокси, с) фениламино; d) фенил- или нафтил-C1-C6-амино, где фенильная группа может быть замещена 1 или 2 галогенами, в частности F или Cl, С1-С6-алкокси или С1-С6-алкилом. Аминогруппа дополнительно может быть замещена С1-С6-алкилом. Примерами таких радикалов являются бензиламино, 4-метоксибензиламино, 4-метилбензиламино, 4-хлорбензиламино, 3,4-дихлорбензиламино, 2-фенилэтиламино, 1-фенилэтиламино, 1-нафт-1-ил-амино, 1-нафт-2-иламино; 1-фенилпроп-3-иламино, 3-фенилпропиламино, -4-изобутилфенил)этиламино; е)
где А представляет собой С1-С2-алкилен, R5 и R6 представляют собой Н, и
представляет собой тиенил, фурил, 2-, 3- или 4-пиридил, тиазолил, оксазолил, бензотиофенил или бензофуранил, где гетероциклические радикалы могут быть замещены галогеном, в частности F или Cl, или С1-С6-алкилом. f)
где А, R5,R6 и
являются такими, как определено в пункте е). g) NH-C1-C6-алкил, который замещен 2 или 3 фенильными группами, например, 3,3-дифенилпропиламино, 1,3-дифенилпроп-2-иламино; h)
где А представляет собой С1-С2-алкилен, R5 и R6 представляют собой Н, и
представляет собой тетрагидрофуранил, тетрагидропиранил, пирролидин, N-метил или N-этилпирролидин; i)
где R5,R6 и
являются такими, как определено в пункте h); j) -NHCOOR10, где R10 представляет собой С3-С6-циклоалкил; k) -NH-CO-NHR10, где R10 представляет собой С3-С6-циклоалкил; l) NH-COR10, где R10 представляет собой С3-С6-циклоалкил-С1-С4-алкил, например, циклопентилметил, циклогексилметил; m) NHCONHCOфенил; n) -A-С3-С6-циклоалкил, где А представляет собой С1-С2-алкилен, например, циклопентилметиламино, циклогексилметиламино. Соединения согласно изобретению могут быть получены соответствующим образом в соответствии со способами, описанными в данной области и указанными в начале данного описания, в частности, в WO 00/17192. Установлено, что особенно подходящим является способ получения в соответствии со следующим двухстадийным способом. На первой стадии сначала получают замещенный имидазол-2-тион формулы II. На второй стадии его подвергают взаимодействию таким образом, что получают 2-тиозамещенные производные имидазола формулы I с введением требуемого заместителя R2.
1) Получение имидазол-2-тиона Имидазол-2-тионы, где R3=Н, галоген (Br, Cl, F), O-алкил или S-алкил, получают в соответствии со способами А или В. В качестве примера, способ А проиллюстрирован для соединений, в которых R1 представляет собой 4-фторфенил, и R3 представляет Н; способ В проиллюстрирован для соединений, в которых R1 представляет собой 4-фторфенил, и R3 представляет Cl, (25а), F (25b) или О-алкил (25с, 25d). Номера в скобках относятся к номерам примеров. 2-Тиозамещенные производные имидазола, где R3=NR4R10 получают не из соответствующих имидазол-2-тионов, где R3=NR4R10, а другим образом в соответствии со способом С. 2-Тиозамещенные производные имидазола, где R3=О-алкил или S-алкил, могут быть получены как в соответствии со способом С, так и в соответствии со способом В. Способ А Синтез замещенных имидазол-2-тионов, где R3 представляет собой Н, проводят в соответствии с реакционным путем по схеме 1 с использованием изоникотината и 4-фторфениоацетонитрила в качестве исходных веществ. Исходные вещества преобразуют в процессе реакции конденсации с помощью металлического натрия в спирте, например, этаноле, в 2-циано-2-(4-фторфенил)-1-(4-пиридил)этанон (IIIa). Цианогруппу затем удаляют гидролизом, например, с использованием бромистоводородной кислоты, и декарбоксилированием, получая 2-(4-фторфенил)-1-(4-пиридил)этанон (IVa). На следующей стадии IVa превращают обработкой смесью хлорида аммония/ацетата натрия в спиртовом растворителе, таком как метанол, в оксим (Va). Взаимодействием с п-толуолсульфонилхлоридом в пиридине последний преобразуют в тозилат (VIa). Из тозилата получают тионовое соединение (IIa) обработкой этоксидом натрия и взаимодействием образующегося промежуточного азирена с тиоцианатом калия. Схема 1. Синтез тионов согласно изобретению в соответствии со способом А
Способ В Получение соединений согласно изобретению, в которых пиридиновый радикал имеет галогеновый, О-алкильный или S-алкильный заместитель, проводят в соответствии со схемой 2 через соответствующие 2-галогенпиридилзамещенные имидазолтионы (способ В). Получение данных имидазолтионов проиллюстрировано с использованием в качестве примера 2-фторзамещенного пиридинового соединения (R3=2-F), где R1=п-фторфенил. Имидазолтионы, содержащие в положении 4 алкильный и циклоалкильный радикалы (R1 = С1-С4-алкил, С3-С7-циклоалкил), получают аналогичным образом, исходя из подходящим образом замещенных 2-фтор- Схема 2.
Оксимы кетонов восстанавливают в спиртовом растворе в присутствии водорода и минеральных кислот, например, HCl, с использованием палладия-на-угле, получая аммониевые соли аминокетонов (VIIIb) (23 b). Альтернативно, другие оксимы кетонов могут быть восстановлены в спиртовом растворе в присутствии минеральных кислот, например, H2SO4, с использованием цинковой пыли, с получением соответствующих аммониевых кетонов (23f). Данные аммониевые кетоны дают, после действия тиоцианатов щелочных металлов, например, тиоцианата калия, в сухом диметилформамиде (ДМФ) при кипении с обратным холодильником, имидазолтионы формулы IIb, где R3=F, Cl, Br, О-алкил или S-алкил, в виде желтых твердых веществ (24 b). Получение соединений согласно изобретению, в которых пиридиновый радикал содержит или простой эфирный (R3=OR10), тиоэфирный (R3=SR10), или амино (R3=NR4R10) заместитель, осуществляют в соответствии со схемой 4 или схемой 5 через соответствующие 2-галогенпиридил-замещенные имидазолтионы (способ С, см. ниже). 2) Получение 2-тиоимидазольных соединений Соединения 2-имидазолтиона формулы II, полученные в соответствии со способом А или В путем замещения атома серы во 2-положении, преобразуют в соединения формулы I согласно изобретению. Замещение, как иллюстративно показано для некоторых соединений на схеме 3, проводят обычным образом с использованием реакции нуклеофильного замещения. В данном случае соединение IIa или IIb подвергают взаимодействию с R2-X в инертном полярном растворителе, таком как спирт. Х представляет собой легко замещаемую группу, такую как Hal, в частности Cl, Br, I, метилсульфонил, тозил и т.д. Подходящие способы известны специалисту в данной области и описаны, например, в WO 00/17192, EP 0372445 и патенте США 4440776. Соединения R2-X являются известными или могут быть получены известными способами, как описано, например, в WO 00/17192. Схема 3 3. Замещение серы алкилгалогенидами и арилгалогенидами или спиртовыми сульфонатами. 3.1. Способ С Соединения согласно изобретению, в которых R3 представляет собой амино заместитель (R3=NR4R10), получают из 2-тиоимидазолов с использованием 4(5)-(2-галогенпиридин-4-ил) замещения. Способ (способ С) проиллюстрирован на схеме 4 с использованием в качестве примера 2-бензиламино (R3=NH-CH2-Ph), где R1=п-фторфенил. Исходные вещества (Ib) могут быть получены описанным выше способом. Схема 4 4.1. 4-(2-Аминипиридин-4-ил)-замещенные 2-тиоимидазолы
4.2. 4-(2-Арилоксипиридин-4-ил)-замещенные 2-тиоимидазолы
4.3. 4-(2-Арилтиопиридин-4-ил)-замещенные 2-тиоимидазолы
4.4. 4-(2-Арилсульфонилпиридин-4-ил)-замещенные 2-тиоимидазолы
Целесообразно проводить реакцию в рассматриваемом амине, который предпочтительно используют в количестве от 5 до 20 мольных эквивалентов на мольный эквивалент соединения (Ib). Температура реакции обычно колеблется от 100 до 200°С. При желании, также возможно использование инертного растворителя, такого как диоксан, диметилформамид, диэтилацетамид, тетраэтилмочевина, метилпирролидон и т.д., и подходящих вспомогательных добавок, таких как карбонаты щелочных металлов или галогениды одновалентной меди (для нейтрализации высвобождающегося количества кислоты или для катализа элиминирования галогена). Соединения согласно изобретению, в которых R3 представляет собой алкоксильный заместитель или алкилтио заместитель (R3=O-C1-C6-алкил, S-C1-C6-алкил), могут быть получены не только способом В (исходя из подходящим образом замещенных пиколинов), но и способом С, исходя из 4(5)-(2-галогенпиридин-4-ил)-замещенных 2-тиоимидазолов. Способ D Соединения согласно изобретению, в которых R3 представляет собой алкоксильный заместитель (R3=O-C1-C6-алкил), могут быть получены не только способом В или С, но также и способом D, исходя из 4(5)-(2-галогенпиридин-4-ил)-замещенных 2-тиоимидазолов. Способ проиллюстрирован на схеме 5 с использованием в качестве примера 2-изопропилоксипиридиновых соединений (R3=ОСН(СН3)2), где R1=п-фторфенил. Исходные вещества (Ib) могут быть получены описанным выше способом. Схема 5 4-(2-Алкоксипиридин-4-ил)-замещенные 2-тиоимидазолы
Целесообразно проводить реакцию в спирте, который предпочтительно используют в количестве от 5 до 20 мольных эквивалентов на мольный эквивалент соединения (Ib), в случае низших спиртов также вплоть до ста мольных эквивалентов, в присутствии сильной кислоты, такой как HCl или трифторуксусная кислота, метансульфоновая кислота и т.д. Температура реакции обычно находится в диапазоне температур кипения низших спиртов, в случае высших спиртов в диапазоне от 100 до 200°С. Было установлено, что выгодным является, например, насыщение спирта газообразным HCl или повторное насыщение в процессе реакции. Альтернативно, замещение фтора на алкоксильную группу во 2-положении пиридильного заместителя может быть осуществлено на более ранней стадии синтеза, например, на стадии оксимов кетонов или аминокетонов. В таких случаях реакции протекают в условиях, сопоставимых с условиями, только что описанными для промежуточного соединения Ib (22c). Способы Е, F и G Соединения согласно изобретению, в которых R3 представляет собой амидный заместитель (R3=NR7COR10), во-первых, получают из 4(5)-(2-галогенпиридин-4-ил)-замещенных 2-тиоимидазолов. Способ (способ Е) проиллюстрирован на схеме 6.1 с использованием в качестве примера 2-бензоиламидо (R3=NHCOPh), R1=п-фторфенил. Во-вторых, после гидролиза амидов до аминозамещенных (R3=NR7Н, NHR10) 2-тиоимидазолов и их повторного ацилирования или преобразования в амиды, мочевины и уретаны, могут быть получены дополнительные амидные заместители (способ F). Это проиллюстрировано на схеме 6.2. В третьих, 2-аминопиридиновые соединения-предшественники могут быть получены из 4(5)-(2-галогенпиридин-4-ильных) соединений через 4(5)-(2-азидопиридин-4-ильные) соединения (способ G). В таком варианте галоген нуклеофильно замещают азидом щелочного металла и азидную группу преобразовывают в аминогруппу способами восстановления, см. схему 6.3. Способ Н Данный интересный вариант также открывает доступ к алкилированным аминам из альдегидных или кетонных соединений-предшественников. Если преобразование азидогруппы в аминогруппу проводить в условиях гидрирования с использованием катализатора гидрирования в присутствии данных альдегидов и кетонов, получают алкилированные амины, где R3=NH-CH2-B или NHCH(алкил)-В (способ Н, схема 6.4). Такой же результат получают, когда азид расщепляют фосфином с получением фосфимида, и данные имиды, полученные реакцией аза-Виттига с альдегидом (или кетоном), восстанавливают в амины с использованием комплексных гидридов (способ Н, схема 6.5). Исходные вещества (Ib) могут быть получены описанным выше способом. Схема 6 6.1. 4(2-Амидопиридин-4-ил)-замещенные 2-тиоимидазолы
Целесообразно проводить реакцию в рассматриваемом амиде, который, предпочтительно, используют в количестве от 5 до 20 мольных эквивалентов на мольный эквивалент соединения (Ib). Температура реакции обычно колеблется от 100 до 200°С. При желании, также возможно использование инертного растворителя, такого как диоксан, диметилформамид, диэтилацетамид, тетраэтилмочевина, метилпирролидон и т.д., и подходящих вспомогательных добавок, таких как карбонаты щелочных металлов или галогениды одновалентной меди (для нейтрализации высвобождающегося количества кислоты или для катализа элиминирования галогена). 2-Аминопиридиновые соединения могут быть получены из 2-амидоацилпиридинов путем гидролиза (6.2) или, кроме того, замещением на азид в 2-фторзамещенных соединениях и последующим восстановлением 2-азидопиридинов (6.3), например, гидрированием на палладие-на-угле в спиртовых растворителях.
Возможны дальнейшие преобразования полученных аминов (Ik, Id) посредством получения производных (способ F). При этом используются реакции аминов Id и Ik как с ангидридами кислот, так и с хлорангидридами кислот с получением дополнительных амидов, а также реакции с хлорформиатными сложными эфирами с получением уретанов, с изоцианатами с получением мочевин и с ацилизоцианатами с получением ацилмочевин. Образование данных производных проиллюстрировано на схеме 7. Схема 7 Преобразование аминов (Ik, Id) в амиды, уретаны и мочевины
Соединения согласно изобретению демонстрируют иммуномодулирующую и ингибирующую высвобождение цитокина активность in vitro и in vivo. Цитокины представляют собой белки, такие как TNF- Соединения согласно изобретению можно вводить или в качестве индивидуальных терапевтически активных соединений, или в виде смеси с другими терапевтически активными соединениями. Соединения можно вводить сами по себе; однако, обычно, их вводят в состав лекарственного средства и вводят в виде фармацевтических композиций, т.е. в виде смесей активных соединений с подходящими фармацевтическими носителями или разбавителями. Соединения или композиции можно вводить перорально или парентерально; предпочтительно их вводят в виде пероральных препаративных лекарственных форм. Тип фармацевтической композиции или носителя, или разбавителя зависит от желаемой формы введения. Пероральные композиции, например, могут быть представлены в виде таблеток или капсул и могут включать в себя обычные эксципиенты, такие как связующие вещества (например, сироп, аравийская камедь, желатин, сорбит, трагакант или поливинилпирролидон), наполнители (например, лактоза, сахар, кукурузный крахмал, фосфат кальция, сорбит или глицерин), глиданты (например, стеарат магния, тальк, полиэтиленгликоль или диоксид кремния), разрыхляющие вещества (например, крахмал) или смачивающие агенты (например, лаурилсульфат натрия). Жидкие пероральные препараты могут иметь вид водных или масляных суспензий, растворов, эмульсий, сиропов, эликсиров или спреев и тому подобное. Они также могут представлять собой сухой порошок, влагосодержание которого восстанавливают с использованием воды или другого подходящего носителя. Такие жидкие препараты могут включать обычные добавки, например, суспендирующие агенты, вкусовые агенты, разбавители или эмульгаторы. Для парентерального введения возможно применение растворов или суспензий с обычными фармацевтическими носителями. Соединения или композиции согласно изобретению можно вводить млекопитающим (человек или животное) в дозе от примерно 0,5 мг до 100 мг на кг массы тела в сутки. Их можно вводить в виде одной отдельной дозы или в виде множества доз. Спектр активности соединений в качестве ингибиторов высвобождения цитокинов был исследован с использованием приведенной ниже системы испытаний, как описано C.Donat и S.Laufer в Arch.Pharm.Pharm.Med.Chem., 333, suppl.1, 1-40, 2000. Испытание in vivo с использованием цельной крови человека Исследуемое вещество добавляли к образцам калий-EDTA цельной крови человека (по 400 мкл каждый) и образцы предварительно инкубировали в инкубаторе с CO2 (5% CO2; 95% насыщенный влагой воздух) при 37°С в течение 15 минут. Образцы затем стимулировали 1 мкг/мл LPS (E.coli 026:B6) при 37°С в инкубаторе с CO2 (5% CO2; 95% насыщенный влагой воздух) в течение 4 часов. Реакцию останавливали, помещая образцы на лед, добавляя DPBS буфер и затем центрифугируя при 1000 g в течение 15 минут. Затем с использованием ELISA определяли количество IL-1 Тест in vitro с использованием PBMCs 1) Моноядерные клетки (PBMCs) из калий-ЭДТА цельной крови человека, разведенной 1:3, выделяли градиентным по плотности центрифугированием (Histopaque®-1.077). Клетки промывали дважды буфером DPBS, повторно суспендировали в макрофаговой среде SFM и доводили до числа клеток 1×106 клеток/мл. Полученную суспензию PBMCs (образец в каждом случае 390 мкл) и исследуемое вещество предварительно инкубировали при 37°С в инкубаторе с CO2 (5% CO2; 95% насыщенный влагой воздух) в течение 15 минут. Образцы затем стимулировали в каждом случае 1 мкг/мл LPS (E.coli 026:B6) при 37°С в инкубаторе с CO2 (5% CO2; 95% насыщенный влагой воздух) в течение 4 часов. Реакцию останавливали, помещая образцы на лед, добавляя DPBS буфер и затем центрифугируя при 15880 g в течение 12 минут. Затем с использованием ELISA определяли количество IL-1 2) Киназный анализ При 37°С микротитровальные планшеты покрывали в течение одного часа 50 мкл раствора АТF2 (20 мкг/мл). Планшеты промывали три раза водой и в лунки добавляли 50 мкл киназной смеси (50 мМ трис-HCl, 10 мМ MgCl2, 10 мМ Результаты испытаний in vitro показаны ниже в таблице 1.
Приведенные ниже примеры иллюстрируют изобретение, не ограничивая его. Пример 1 а) 4-(4-Фторфенил)-5-пиридин-4-ил-1,3-дигидроимидазол-2-тион 2-(4-фторфенил)-3-гидрокси-3-пиридин-4-илакрилонитрил (а1) Смесь этилизоникотината (75,8 г; 0,5 моль) и 4-фторфенилацетонитрила (67,6 г; 0,5 моль) добавляли по каплям к раствору металлического натрия (17,3 г; 0,7 моль) в абсолютном этаноле (250 мл). Реакционную смесь перемешивали при 100°С в течение 15 минут. Затем реакционную смесь охлаждали на ледяной бане и добавляли 600 мл дистиллированной воды. Когда смесь подкисляли концентрированной HCl (90 мл), при рН 1 образовывался желтый осадок гидрохлорида а1. Осадок отфильтровывали, промывали H2O и сушили при пониженном давлении над Р2О5. Т.пл. 226°С. 2-(4-Фторфенил)-1-пиридин-4-илэтанон (а2) Раствор а1 (40,6 г; 0,15 моль) в 48%-ной бромистоводородной кислоте (130 мл) перемешивали при кипении с обратным холодильником в течение 19 часов. Смесь охлаждали на ледяной бане и полученный осадок (4-фторфенилуксусной кислоты) отфильтровывали и промывали Н2О. Когда фильтрат нейтрализовали водным аммиаком (80 мл), получали а2 в виде темно-зеленого осадка, который отфильтровывали, промывали Н2О и сушили при пониженном давлении над Р2О5; светло-серый/бежевый порошок. Т.пл. 215°С. Оксим 2-(4-фторфенил)-1-пиридин-4-илэтанона (а3) В суспензию а2 (21,5 г; 0,1 моль) в 50%-ном метаноле (350 мл) вводили ацетат натрия (36,1 г; 0,44 моль) и гидрохлорид гидроксиламина (22,0 г; 0,32 моль). Реакционную смесь перемешивали при кипении с обратным холодильником в течение 1 часа. Когда прохладный раствор охлаждали на ледяной бане, а3 получали в виде бежевого осадка, который отфильтровывали, промывали Н2О и сушили при пониженном давлении над Р2О5. Т.пл. 155°С. О-[(4-Метилфенил)сульфонил]оксим 2-(4-фторфенил)-1-пиридин-4-илэтанона (а4) В атмосфере аргона а3 (10,1 г; 0,04 моль) растворяли в абсолютном пиридине (50 мл). Раствор охлаждали до 6°С и понемногу с течение времени добавляли толуолсульфохлорид (10,1 г; 0,05 моль). По окончании прибавления реакционную смесь перемешивали при комнатной температуре в течение 20 часов. Затем смесь выливали в 500 мл ледяной воды. Осадок (а4) отфильтровывали, промывали холодной Н2О и сушили в сушильной камере при 50°С. Т.пл. 201°С. а) 4-(4-Фторфенил)-5-пиридин-4-ил-1,3-дигидроимидазол-2-тион (1а) В атмосфере аргона раствор а4 (10,0 г; 0,03 моль) в абсолютном этаноле (56 мл) охлаждали до 5°С и добавляли по каплям свежеприготовленный раствор металлического натрия (0,75 г; 0,03 моль) в абсолютном этаноле (30 мл). Реакционную смесь перемешивали при 5°С в течение 5 часов. После добавления диэтилового эфира (500 мл) перемешивание продолжали в течение 30 минут. Осадок (TosOH) отфильтровывали и промывали диэтиловым эфиром (4×50 мл). Объединенную эфирную фазу экстрагировали 10% соляной кислотой (3×90 мл). Водный экстракт концентрировали примерно до объема 40 мл и добавляли тиоцианат калия (5,0 г; 0,05 моль). Реакционную смесь перемешивали при кипении с обратным холодильником в течение 1 часа. Когда смесь нейтрализовали 5%-ным раствором бикарбоната натрия (270 мл), получали а5 в виде бежевого осадка, который отфильтровывали, промывали Н2О и сушили в сушильной камере при 60°С. Выход 5,6 г (79%); т.пл. 382°С. 1Н-ЯМР (ДМСО-d6): Соответствующим образом были получены следующие соединения. 1b: 3-(4-фторфенил)-5-пиридин-4-ил-1,3-дигидроимидазол-2-тион 1с 4-(4-хлорфенил)-5-пиридин-4-ил-1,3-дигидроимидазол-2-тион 1d: 4-(4-бромфенил)-5-пиридин-4-ил-1,3-дигидроимидазол-2-тион 1е: 4-фенил-5-пиридин-4-ил-1,3-дигидроимидазол-2-тион Пример 2 1-Хлорметил-4-метилсульфанилбензол (2) 4-Метилсульфанилбензиловый спирт (30,5 г; 0,2 моль) растворяли в дихлорметане (180 мл). Раствор тионилхлорида (23,8 г; 0.2 моль) в дихлорметане (120 мл) добавляли по каплям к первоначальной смеси, которую выдерживали при кипении с обратным холодильником. Реакционную смесь перемешивали при кипении с обратным холодильником дополнительно в течение 2 часов. Раствор охлаждали до комнатной температуры, промывали Н2О (2×250 мл), сушили над N2SO4 и концентрировали. Маслянистый осадок (6) очищали колоночной хроматографией (Al2O3, CH2Cl2). 1Н-ЯМР (CDCl3): Пример 3 1-Хлорметил-4-метансульфинилбензол (3) Раствор 2 (17,3 г; 0,1 моль) в ледяной уксусной кислоте (150 мл) охлаждали до 10°С. К первоначально загруженному раствору добавляли по каплям раствор Н2О2 (35% раствор 35%-ной концентрации; 13,1 г; 0,13 моль) в ледяной уксусной кислоте. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Смесь охлаждали на ледяной бане, добавляли лед (200 г) и смесь нейтрализовали аммиачной водой (290 мл). Водную фазу экстрагировали этилацетатом (2×300 мл). Органическую фазу промывали Н2О (2×300 мл), сушили над Na2SO4 и концентрировали. Растиранием и охлаждением маслянистого осадка получали 3 в кристаллическом виде. 1Н-ЯМР (CDCl3): Пример 4 1-Хлорметил-4-метансульфонилбензол (4) м-Хлорпербензойную кислоту (70%; 8,6 г; 0,04 моль) вводили в раствор 3 (3,0 г; 0,02 моль) в хлороформе (50 мл). Реакционную смесь перемешивали при кипении с обратным холодильником в течение 4 часов. Смесь охлаждали до комнатной температуры и фильтровали. Фильтрат промывали насыщенным раствором NaHCO3 (2×) и сушили над Na2SO4. После концентрирования органической фазы в остатке получали 4 в виде кристаллического белого твердого вещества. Т.пл. 102°С. 1Н-ЯМР (CDCl3): Пример 5 Метил-5-хлорсульфонил-2-гидроксибензоат (5а) 5а получали из метилсалицилата (10,0 г; 65,7 ммоль) с использованием способа, описанного для синтеза 5с. 1Н-ЯМР (CDCl3): Метил 5-хлор-3-хлорсульфонил-2-гидроксибензоат (5b) 5b получали из метил(5-хлор)салицилата (16,0 г; 85,7 ммоль) с использованием способа, описанного для синтеза 5с. 1Н-ЯМР (CDCl3): Этил 3-хлорсульфонил-4-метоксибензоат (5с) Раствор этил(4-метокси)бензоата (15,7 г; 87,2 ммоль) в CCl4 (60 мл) охлаждали до -15°С и добавляли по каплям в течение 15 минут хлорсульфоновую кислоту (17,5 мл; 263 ммоль), что приводило к повышению температуры до -10°С. По окончании прибавления реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем нагревали при 50°С до тех пока, пока исходное вещество не переставало обнаруживаться по данным тонкослойной хроматографии. При охлаждении льдом и интенсивном перемешивании реакционную смесь добавляли к суспензии льда (50 г) в CCl4 (100 мл). Смесь интенсивно перемешивали в течение 3 минут. Органическую фазу отделяли и водную фазу экстрагировали CH2Cl2 (3×100 мл). Объединенные органические экстракты промывали насыщенным раствором NaCl (3×), сушили над Na2SO4 и концентрировали. Растирание маслянистого коричневого остатка с диэтиловым эфиром приводило к осаждению 5с в виде кристаллического белого вещества. 1Н-ЯМР (CDCl3): Пример 6 2-Гидрокси-5-меркаптобензойная кислота (6а) 6а получали из 5а (0,50 г; 2,0 ммоль) с использованием способа, описанного для синтеза 7с, без алкилирования диметилсульфатом. 1Н-ЯМР (ДМСО-d6): Пример 7 2-Гидрокси-5-метилсульфанилбензойная кислота (7а) 7а получали из 5а (10,0 г; 40,0 ммоль) с использованием способа, описанного для синтеза 7с. 1Н-ЯМР (CDCl3): 5-Хлор-2-гидрокси-3-метилсульфанилбензойная кислота (7b) 7b получали из 5b (13,0 г; 45,6 ммоль) с использованием способа, описанного для синтеза 7с. 1Н-ЯМР (ДМСО-d6): 4-Метокси-3-метилсульфанилбензойная кислота (7с) Трифенилфосфин (20,5 г; 78,2 ммоль) постепенно вводили в раствор 5с (5,1 г; 18,3 ммоль) в толуоле (50 мл). Реакционную смесь перемешивали при комнатной температуре в течение 4,5 часов. Осадок (трифенилфосфиноксид) отфильтровывали и желтый фильтрат экстрагировали водный раствором гидроксида натрия 10%-ной концентрации (4×). К объединенному водному экстракту добавляли диметилсульфат (2 мл) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Полученный осадок растворяли путем нагревания до температуры кипения с обратным холодильником. Прозрачный раствор охлаждали и доводили до рН 1 с использованием соляной кислоты 20%-ной концентрации. Осадок (7с) отфильтровывали, промывали Н2О и сушили при пониженном давлении над CaCl2. 1Н-ЯМР (CD3OD): 4-Гидрокси-3-метилсульфанилбензойная кислота (7d) Суспензию 7с (0,5 г, 2,5 ммоль) в смеси ледяная уксусная кислота/48%-ная бромистоводородная кислота (1+1,7 мл) перемешивали, нагревая при кипении с обратным холодильником, в течение 6 часов. Реакционную смесь охлаждали, добавляли к Н2О (20 мл) и доводили до рН 2 с использованием раствора Na2CO3 10%-ной концентрации. Водный раствор экстрагировали диэтиловым эфиром (4×20 мл). Объединенный органический экстракт промывали насыщенным раствором NaCl (2×), сушили над Na2SO4 и концентрировали. При хранении при комнатной температуре грязновато-коричневый маслянистый остаток (7d) кристаллизовался. Кристаллы растирали с Н2О, отфильтровали и сушили. 1Н-ЯМР (CDCl3): Пример 8 2-Гидроксиметил-4-метилсульфанилфенол (8а) 8а получают из 7а (1,5 г; 8,1 ммоль) с использованием способа, описанного для синтеза 8с. 1Н-ЯМР (CDCl3): 4-Хлор-2-гидроксиметил-6-метилсульфанилфенол (8b) 8b получают из 7b (2,2 г; 10,1 ммоль) с использованием способа, описанного для синтеза 8с. 1Н-ЯМР (ДМСО-d6): 4-Гидроксиметил-2-метилсульфанилфенол (8с) При охлаждении льдом раствор 7d (1,37 г; 7,4 ммоль) в абсолютном тетрагидрофуране (ТГФ; 15 мл) добавляли к суспензии LiAlH4 95%-ной чистоты (0,55 г; 14 ммоль) в абсолютном ТГФ в трехгорлой колбе (которая была высушена нагреванием и продувкой аргоном) таким образом, что происходит только умеренное выделение газа. По окончании прибавления охлаждение удаляли и реакционную смесь перемешивали при комнатной температуре и при 55-65°С в течение следующего 21 часа. При охлаждении льдом к реакционной смеси добавляли ледяную воду. Осадок Al(OH)3 растворяли с помощью добавления серной кислоты 10%-ной концентрации и кислый водный раствор (рН 1) экстрагировали диэтиловым эфиром (3×50 мл). Объединенные эфирные экстракты экстрагировали водным раствором гидроксида натрия 10%-ной концентрации (2×25 мл). Объединенный раствор гидроксида натрия нейтрализовали соляной кислотой 20%-ной концентрации. Осадок (8с) отфильтровывали, промывали Н2О и сушили. Дополнительную порцию 8с получали экстракцией нейтрального водного раствора диэтиловым эфиром. Эфирный экстракт промывали насыщенным раствором NaCl, сушили над Na2SO4 и концентрировали, получая кристаллическое белое твердое вещество. 1Н-ЯМР (CDCl3): Пример 9 2-Гидрокси-5-метилсульфанилбензальдегид (9а) Указанное в заголовке соединение получали в качестве побочного продукта при синтезе 8а. 1Н-ЯМР (CDCl3): Пример 10 4-(3-Хлорэтил)бензолсульфонилхлорид (10а) При охлаждении льдом (2-хлорэтил)бензол (14,0 г; 0,1 моль) добавляли по каплям в течение 40 минут к хлорсульфоновой кислоте (72 г). Раствор коричневого цвета перемешивали при комнатной температуре в течение 24 часов, охлаждали на ледяной бане и понемногу добавляли ко льду, где образовывалось вязкое вещество, которое не удается отфильтровать. Водный раствор экстрагировали этилацетатом (3×). Объединенный органический экстракт промывали раствором NaHCO3 10%-ной концентрации, сушили над Na2SO4 и концентрировали. Маслянистый остаток помещали в смесь трет-бутилметиловый эфир/петролейный эфир. Раствор потирали стеклянной палочкой и охлаждали. Белые кристаллы отфильтровывали и сушили. Дополнительное количество реакционного продукта получали из маточного раствора. Неочищенный продукт использовали без дополнительной очистки для синтеза 11а. 1Н-ЯМР (CDCl3): 4-(3-Хлорпропил)бензолсульфонилхлорид (10b) 10b получали из (3-хлорпропил)бензола (15,5 г; 0,1 моль) с использованием способа, описанного для синтеза 10а. Неочищенный продукт использовали без дополнительной очистки для синтеза 11b. Масс-спектр: m/z (%) 253 (90, M+), 217 (100, M+-Cl), 189 (35), 153 (97, M+-SO2Cl), 125 (94), 119 (65, фенилпропилкарбений+), 91(90), 77 (29, фенил+). Пример 11 1-(3-Хлорэтил)-4-метилсульфанилбензол (11а) 11а получали из 10а (12,0 г; 0,05 моль) с использованием способа, описанного для синтеза 11b. 1Н-ЯМР (CDCl3): 1-(3-Хлорпропил)-4-метилсульфанилбензол (11b) При комнатной температуре раствор 10b (12,7 г; 5,0 ммоль) в диэтиловом эфире (75 мл) добавляли по каплям в течение 2,5 часов к суспензии LiAlH4 (2,9 г; 7,6 ммоль) в диэтиловом эфире (50 мл). По окончании прибавления реакционную смесь перемешивали при комнатной температуре время от времени добавляя LiAlH4 до тех пор, пока исходное вещество не перестанет обнаруживаться с помощью тонкослойной хроматографии (2,5 ч). При охлаждении льдом в реакционную смесь вводили лед и водную фазу подкисляли 10% соляной кислотой (рН 1). Органическую фазу удаляли и водную фазу экстрагировали диэтиловым эфиром (3×). Объединенный органический экстракт промывали водным раствором гидроксида натрия 10%-ной концентрации (4×50 мл) до тех пор, пока он не станет визуально бесцветным. К объединенному раствору гидроксида натрия добавляли диметилсульфат (9,0 г; 7,0 ммоль) и смесь перемешивали при комнатной температуре в течение 16,5 часов. Маслянистый осадок помещали в диэтиловый эфир. Органическую фазу отделяли и водную фазу опять экстрагировали диэтиловым эфиром (2×). Объединенный органический экстракт сушили над Na2SO4 и концентрировали. Коричневый маслянистый остаток подвергали перегонке c использованием трубки с шаровым расширением (0,2 мбар. 250°С). 1Н-ЯМР (CDCl3): Пример 12 1-(2-Хлорэтил)-4-метансульфинилибензол (12а) При охлаждении к раствору 11а (1,5 г; 8,0 ммоль) в ледяной уксусной кислоте (20 мл) добавляли раствор Н2О2 35%-ной концентрации (0,9 г; 9,3 ммоль). По окончании прибавления реакционную смесь перемешивали при комнатной температуре в течение 2,5 часов, разбавляли при охлаждении ледяной водой и доводили до рН 8 с использованием аммиачной воды 25%-ной концентрации. Маслянистый белый осадок помещали в диэтиловый эфир и водную фазу экстрагировали диэтиловым эфиром (3×). Объединенные органические экстракты сушили над Na2SO4 и концентрировали. 1Н-ЯМР (CDCl3): 1-(3-Хлорпропил)-4-метансульфинилибензол (12b) 12b получали из 11b (2,0 г; 10,0 ммоль) с использованием способа, описанного при синтезе 12а. Т.пл. 46°С Общие способы получения соединений формулы I. Получение 2-арилалкил- или алкилсульфанилимидазолов (общий способ А) Суспензию соответствующего имидазол-2-тиона (1 эквивалент), соответствующего основания (1,2 эквивалента) и соответствующего арилалкил- или алкилгалогенида (1 эквивалент) в смеси этанол/ТГФ (8+2) перемешивали при кипении с обратным холодильником до тех пор, пока имидазол-2-тион не перестанет обнаруживаться по данным тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры и фильтровали. Фильтрат, который в большинстве случаев имел красную/оранжевую окраску, концентрировали и остаток очищали колоночной хроматографией, перекристаллизацией или растиранием. Таким образом получали соединения 13a-с, 14a-с и 17a-m. Получение 2-бензилсульфанилимидазолов, содержащих фенольную функциональную группу в радикале R2 (общий способ В). Добавляя соляную кислоту 10%-ной концентрации (10-15 капель), имидазол-2-тион 1а (1 эквивалент) растворяли в ледяной уксусной кислоте (5 мл). К первоначальной смеси, имеющей светло-желтую окраску, добавляли соответствующий бензиловый спирт (1 эквивалент) и реакционную смесь перемешивали при подходящей температуре (температура/время) до тех пор, пока 1а не перестанет обнаруживаться по данным тонкослойной хроматографии. В случае сульфоксидов 18g-i добавляли раствор Н2О2 35%-ной концентрации и реакционную смесь перемешивали при комнатной температуре дополнительно в течение 4 часов. Реакционную смесь разбавляли Н2О (5 мл) и доводили до рН 8 с использованием аммиачной воды 25%-ной концентрации. Осадок отфильтровывали и промывали водой. Неочищенный продукт очищали колоночной хроматографией, перекристаллизацией или растиранием. Таким образом получали имидазол-2-илсульфанилметилфенолы 18а-i. Получение N-замещенных 2-аминопиридинов (общий способ С) В атмосфере аргона соответствующий 5-(2-галогенпиридин-4-ил)имидазол (1 эквивалент) суспендировали в соответствующем амине (примерно 10 эквивалентов). Реакционную смесь перемешивали при соответствующей температуре до тех пор, пока исходное вещество больше не обнаруживалось тонкослойной хроматографией. Реакционную смесь охлаждали до комнатной температуры и помещали в 10%-ную лимонную кислоту, которую предварительно доводили до рН 5 с использованием NaOH 20%-ной концентрации. Водную эмульсию экстрагировали этилацетатом (3×). Объединенный органический экстракт промывали 10%-ной лимонной кислотой/рН 5 (1×), раствором Na2CO3 10%-ной концентрации (2×) и насыщенным раствором NaCl (1×), сушили над Na2SO4 и концентрировали. Маслянистый остаток разделяли колоночной хроматографией. Аминопиридины 25f-p, 26c-e и 27c-d получали таким образом. Пример 13 3-[5-(4-Фторфенил)-2-(4-метилсульфанилбензилсульфанил)-3Н-имидазол-4-ил]пиридин (13а) С использованием общего способа А указанное в заголовке соединение получали из 1b (0,42 г; 1,5 ммоль) и 2 (0,25 г; 1,4 ммоль) после проведения реакции в течение 4,5 часов и разделения колоночной хроматографией (SiO2 60, CH2Cl2/EtOH 9+1). Т.пл. 163°С. ИК (ATR) (затухающее суммарное отражение): 1505, 1493, 1222 (C-F), 837, 806 см-1. 1Н-ЯМР (ДМСО-d6): 3-[5-(4-Фторфенил)-2-(4-метансульфинилбензилсульфанил)-3Н-имидазол-4-ил]пиридин (13b) С использованием общего способа А указанное в заголовке соединение получали из 1b (0,42 г; 1,5 ммоль) и 3 (0,27 г; 1,5 ммоль) после проведения реакции в течение 8 часов и разделения колоночной хроматографией (SiO2 60, CH2Cl2/EtOH 9+1)/ Т.пл. 127°С. ИК (ATR): 1506, 1222 (C-F), 1027 (S=O), 1013, 838, 811 см-1. 1Н-ЯМР (ДМСО-d6): 3-[5-(4-Фторфенил)-2-(4-метансульфонилбензилсульфанил)-3Н-имидазол-4-ил]пиридин (13c) С использованием общего способа А указанное в заголовке соединение получали из 1b (0,42 г; 1,5 ммоль) и 4 (0,29 г; 1,43 ммоль) с добавлением Na2CO3 (0,43 г; 4,1 ммоль) после проведения реакции в течение 6,5 часов и растирания с горячим этилацетатом. Т.пл. 129°С. ИК (ATR): 1506, 1296 (SO2), 1222 (C-F), 1145 (SO2), 1089, 839, 812 см-1. 1Н-ЯМР (ДМСО-d6): Пример 14 4-[5-(4-Хлорфенил)-2-(4-метилсульфанилбензилсульфанил)-3Н-имидазол-4-ил]пиридин (14а) С использованием общего способа А указанное в заголовке соединение получали из 1с (0,26 г; 0,9 ммоль) и 6 (0,15 г; 0,87 ммоль) с добавлением Na2CO3 (две порции на кончике шпателя) после проведения реакции в течение 6,5 часов и разделения колоночной хроматографией (SiO2 60, CH2Cl2/EtOH 9+1). Т.пл. 236°С. ИК (ATR): 1600, 1492, 1094, 1005, 968, 829, 684 (C-Cl), 561 см-1. 1Н-ЯМР (ДМСО-d6): 4-[5-(4-Хлорфенил)-2-(4-метансульфинилбензилсульфанил)-3Н-имидазол-4-ил]пиридин (14b) С использованием общего способа А указанное в заголовке соединение получали из 1с (0,26 г; 0,9 ммоль) и 3 (0,16 г; 0,85 ммоль) с добавлением Na2CO3 после проведения реакции в течение 6,5 часов и разделения колоночной хроматографией (SiO2 60, CH2Cl2/EtOH 9+1). Т.пл. 224°С. ИК (ATR): 1600, 1510, 1490, 1033 (S=O), 1001, 967, 829, 677 (C-Cl) см-1. 1Н-ЯМР (ДМСО-d6): 4-[5-(4-Хлорфенил)-2-(4-метансульфонилбензилсульфанил)-3Н-имидазол-4-ил]пиридин (14с) С использованием общего способа А указанное в заголовке соединение получали из 1с (0,26 г; 0,9 ммоль) и 3 (0,16 г; 0,85 ммоль) с добавлением Na2CO3 (две порции на кончике шпателя) после проведения реакции в течение 6,5 часов и разделения колоночной хроматографией (SiO2 60, CH2Cl2/EtOH 9+1). Т.пл. 232°С. ИК (ATR): 1603, 1490, 1300 (SO2), 1141 (SO2), 1086, 1002, 952, 829, 681 (C-Cl), 550 см-1. 1Н-ЯМР (ДМСО-d6): Пример 15 4-[5-(4-Бромфенил)-2-(4-метилсульфанилбензилсульфанил)-3Н-имидазол-4-ил]пиридин (15а) С использованием общего способа А указанное в заголовке соединение получали из 1d (0,25 г; 0,75 ммоль) и 2 (0,13 г; 0,72 ммоль) после проведения реакции в течение 5 часов и разделения колоночной хроматографией (SiO2 60, CH2Cl2/EtOH 9+1). ИК (ATR): 1600, 1517, 1490, 1089, 1069, 1003, 968, 826 см-1. 1Н-ЯМР (ДМСО-d6): 4-[5-(4-Бромфенил)-2-(4-метансульфинилбензилсульфанил)-3Н-имидазол-4-ил]пиридин (15b) С использованием общего способа А указанное в заголовке соединение получали из 1d (0,25 г; 0,75 ммоль) и 3 (0,14 г; 0,72 ммоль) после проведения реакции в течение 10 часов и разделения колоночной хроматографией (SiO2 60, CH2Cl2/EtOH 9+1). Т.пл. 222°С. ИК (ATR): 1604, 1487, 1035 (S=O), 1010, 1000, 966, 822 см-1. 1Н-ЯМР (ДМСО-d6): 4-[5-(4-Бромфенил)-2-(4-метансульфонилбензилсульфанил)-3Н-имидазол-4-ил]пиридин (15с) С использованием общего способа А указанное в заголовке соединение получали из 1d (0,25 г; 0,75 ммоль) и 4 (0,15 г; 0,72 ммоль) после проведения реакции в течение 5 часов и разделения колоночной хроматографией (SiO2 60, CH2Cl2/EtOH 9+1). Т.пл. 226°С. ИК (ATR): 1605, 1318, 1303 (SO2), 1145 (SO2), 1003, 967, 957, 827, 822 см-1. 1Н-ЯМР (ДМСО-d6): Пример 16 4-[2-(4-Метилсульфанилбензилсульфанил)-5-фенил-3Н-имидазол-4-ил]пиридин (16а) С использованием общего способа А указанное в заголовке соединение получали из 1е (0,38 г; 1,5 ммоль) и 2 (0,25 г; 1,4 ммоль) после проведения реакции в течение 5,75 часов и разделения колоночной хроматографией (SiO2 60, CH2Cl2/EtOH 9+1). Т.пл. 213°С. ИК (ATR): 1601, 1491, 1417, 1094, 1004, 967, 828, 771, 700 см-1. 1Н-ЯМР (ДМСО-d6): 4-[2-(4-Метансульфинилбензилсульфанил)-5-фенил-3Н-имидазол-4-ил]пиридин (16b) С использованием общего способа А указанное в заголовке соединение получали из 1е (0,38 г; 1,5 ммоль) и 3 (0,27 г; 1,43 ммоль) после проведения реакции в течение 5,5 часов и разделения колоночной хроматографией (SiO2 60, CH2Cl2/EtOH 9+1). Т.пл. 189°С. ИК (ATR): 1603, 1494, 1051 (S=O), 1003, 833, 701 см-1. 1Н-ЯМР (ДМСО-d6): 4-[2-(4-Метансульфонилбензилсульфанил)-5-фенил-3Н-имидазол-4-ил]пиридин (16с) С использованием общего способа А указанное в заголовке соединение получали из 1е (0,38 г; 1,5 ммоль) и 4 (0,29 г; 1,43 ммоль) после проведения реакции в течение 4,25 часов и разделения колоночной хроматографией (SiO2 60, CH2Cl2/EtOH 9+1). Т.пл. 247°С. ИК (ATR): 1602, 1298 (SO2), 1145 (SO2), 1006, 953, 827, 775, 701 см-1. 1Н-ЯМР (ДМСО-d6): Пример 17 4-{5-(4-Фторфенил)-2-[2-(4-метансульфинилфенил)этилсульфанил]-1Н-имидазол-4-ил}пиридин (17а) С использованием общего способа А указанное в заголовке соединение получали из 1а (0,25 г; 0,9 ммоль) и 12а (0,22 г; 1,1 ммоль) с добавлением Na2CO3 (1 порция на кончике шпателя) и каталитического количества NaI, после проведения реакции в течение 50 часов и разделения колоночной хроматографией (SiO2 60, CH2Cl2/EtOH 9+1). Т.пл. 177°С. ИК (ATR): 1221 (C-F), 1032 (S=O) см-1. 1Н-ЯМР (ДМСО-d6): 4-{5-(4-Фторфенил)-2-[2-(4-метансульфинилфенил)пропилсульфанил]-1Н-имидазол-4-ил}пиридин (17b) С использованием общего способа А указанное в заголовке соединение получали из 1а (0,25 г; 0,9 ммоль) и 12b (0,22 г; 1,0 ммоль) с добавлением Na2CO3 (1 порция на кончике шпателя) и каталитического количества NaI, после проведения реакции в течение 40 часов и разделения колоночной хроматографией (SiO2 60, CH2Cl2/EtOH 9+1). Т.пл. 142°С. ИК (ATR): 1222 (C-F), 1043 (S=O) см-1. 1Н-ЯМР (ДМСО-d6): 4-[2-Бензилсульфанил-5-(4-фторфенил)-1Н-имидазол-4-ил]пиридин (17с) С использованием общего способа А указанное в заголовке соединение получали из 1а (0,28 г; 1,0 ммоль) и 1-хлорметилбензола (0,13 г; 1,0 ммоль) после проведения реакции в течение 6 часов и растирания с МеОН. Т.пл. 223°С. ИК (ATR): 1233 см-1 (C-F). 1Н-ЯМР (ДМСО-d6): 4-[5-(4-Фторфенил)-2-фенэтилсульфанил-1Н-имидазол-4-ил]пиридин (17d) С использованием общего способа А указанное в заголовке соединение получали из 1а (0,5 г; 1,9 ммоль) и 2-хлорэтилбензола (0,28 г; 2,0 ммоль) с добавлением Na2CO3 (1 порция на кончике шпателя) и каталитического количества NaI после проведения реакции в течение 70 часов и растирания с EtOH. Т.пл. 257°С. ИК (ATR): 1223 см-1 (C-F). 1Н-ЯМР (ДМСО-d6): 4-[5-(4-Фторфенил)-2-(3-фенилпропилсульфанил)-1Н-имидазол-4-ил]пиридин (17е) С использованием общего способа А указанное в заголовке соединение получали из 1а (0,5 г; 1,9 ммоль) и 3-хлорпропилбензола (0,28 г; 2,0 ммоль) с добавлением Na2CO3 (1 порция на кончике шпателя) и каталитического количества NaI после проведения реакции в течение 70 часов и растирания с EtOH. Т.пл. 183°С. ИК (ATR): 1226 см-1 (C-F). 1Н-ЯМР (ДМСО-d6): 4-[5-(4-Фторфенил)-4-пиридин-4-ил-1Н-имидазол-2-илсулдьфанил]ацетонитрил (17f) С использованием общего способа А указанное в заголовке соединение получали из 1а (1,1 г; 4,0 ммоль) и хлорацетонитрила (0,30 г; 4,0 ммоль) после проведения реакции в течение 18 часов и очистки колоночной хроматографией (SiO2 60, этилацетат). Т.пл. 219°С. ИК (ATR): 2243 (CN), 1226 см-1 (C-F). 1Н-ЯМР (ДМСО-d6): 4-[5-(4-Фторфенил)-2-(нафталин-1-илметилсульфанил)-1Н-имидазол-4-ил]пиридин (17g) С использованием общего способа А указанное в заголовке соединение получали из 1а (0,28 г; 1,0 ммоль) и 1-хлорметилнафтола (0,18 г; 1,0 ммоль) после проведения реакции в течение 6,5 часов и очистки колоночной хроматографией (SiO2 60, этилацетат). Т.пл. 364°С. ИК (ATR): 1225 см-1 (C-F). 1Н-ЯМР (ДМСО-d6): 4-[2-2-Циклогексилметилсульфанил-5-(4-фторфенил)-1Н-имидазол-4-ил]пиридин (17h) С использованием общего способа А указанное в заголовке соединение получали из 1а (0,25 г; 0,9 ммоль) и 1-хлорметилциклогексана (0,18 г; 1,0 ммоль) с добавлением Na2CO3 (1 порция на кончике шпателя) и каталитического количества NaI после проведения реакции в течение 47 часов и растирания с EtOH. Т.пл. 235°С. ИК (ATR): 2922, 2852 (с-Нех), 1222 см-1 (C-F). 1Н-ЯМР (ДМСО-d6): 4-[5-(4-Фторфенил)-2-метилсульфанил-1Н-имидазол-4-ил]пиридин (17i) С использованием общего способа А указанное в заголовке соединение получали из 1а (0,41 г; 1,5 ммоль) и иодистого метила (0,27 г; 1,9 ммоль) после проведения реакции в течение 8 часов и растирания с EtOH. Т.пл. 263°С. ИК (ATR): 1226 см-1 (C-F). 1Н-ЯМР (ДМСО-d6): 4-[5-(4-Фторфенил)-2-(2-метилсульфанилбензилсульфанил)-1Н-имидазол-4-ил]пиридин (17j) С использованием общего способа А указанное в заголовке соединение получали из 1а (0,28 г; 1,0 ммоль) и 1-хлорметил-2-метилсульфанилбензола (0,17 г; 1,0 ммоль) после проведения реакции в течение 5,5 часов и очистки колоночной хроматографией (SiO2 60, этилацетат). Т.пл. 223°С. ИК (ATR): 1228 см-1 (C-F). 1Н-ЯМР (CD3OD): 4-[5-(4-Фторфенил)-2-(2-метансульфинилбензилсульфанил)-1Н-имидазол-4-ил]пиридин (17k) С использованием общего способа А указанное в заголовке соединение получали из 1а (0,28 г; 1,0 ммоль) и 1-хлорметил-2-метансульфинилбензола (0,18 г; 1,0 ммоль) после проведения реакции в течение 4 часов и перекристаллизации из смеси метанол/этилацетат (1+1). Т.пл. 205°С. ИК (KBr): 1213 (C-F), 1033 см-1 (S=O). 1Н-ЯМР (CD3OD): 4-[5-(4-Фторфенил)-2-(3-метилсульфанилбензилсульфанил)-1Н-имидазол-4-ил]пиридин (17l) С использованием общего способа А указанное в заголовке соединение получали из 1а (1,1 г; 4,1 ммоль) и 1-хлорметил-3-метилсульфанилбензола (0,7 г; 4,1 ммоль) после проведения реакции в течение 11 часов и перекристаллизации из EtOH. Т.пл. 218°С. ИК (KBr): 1225 см-1 (C-F). 1Н-ЯМР (ДМСО-d6): 4-[5-(4-Фторфенил)-2-(3-метансульфинилбензилсульфанил)-1Н-имидазол-4-ил]пиридин (17m) Раствор Н2О2 (0,13 мл; 1,3 ммоль) 35%-ной концентрации добавляли по каплям к суспензии 17l (0,50 г; 1,2 ммоль) в ледяной уксусной кислоте (7 мл). Реакционную смесь перемешивали при комнатной температуре в течение 20,5 часов, разбавляли Н2О (5 мл), доводили до рН 9 с использованием аммиачной воды 25%-ной концентрации и экстрагировали этилацетатом (3×). Объединенный органический экстракт промывали насыщенным раствором NaCl (3×) и сушили над Na2SO4. Маслянистый неочищенный продукт, полученный после удаления растворителя, растирали со смесью диэтиловый эфир/этилацетат (1+1) и полутвердый остаток очищали колоночной хроматографией (RP-18, MeOH). Т.пл. 171°С. ИК (KBr): 1228 (C-F), 1019 см-1 (S=O). 1Н-ЯМР (CD3OD): Пример 18 2-[5-(4-Фторфенил)-4-пиридин-4-ил-1Н-имидазол-2-илсульфанилметил]фенол (18а) С использованием общего способа В (23 часа, комнатная температура) указанное в заголовке соединение получали из 1а (0,20 г; 0,7 ммоль) и 2-гидроксиметилфенола (0,10 г; 0,8 ммоль) после растирания с EtOH. Т.пл. 200°С (разложение). ИК (ATR): 1266 (ОН изгиб), 1222 (C-F), 1005 (С-О) 1Н-ЯМР (ДМСО-d6): 3-[5-(4-Фторфенил)-4-пиридин-4-ил-1Н-имидазол-2-илсульфанилметил]фенол (18b) С использованием общего способа В (9 часов, нагревание при кипении с обратным холодильником) указанное в заголовке соединение получали из 1а (0,20 г; 0,7 ммоль) и 3-гидроксиметилфенола (0,10 г; 0,8 ммоль) после очистки колоночной хроматографией (SiO2 60, CH2Cl2/EtOH 9+1) Т.пл. 230°С ИК (ATR): 1287 (ОН изгиб), 1241 (C-F), 1007 см-1 (С-О) 1Н-ЯМР (ДМСО-d6): 4-[5-(4-Фторфенил)-4-пиридин-4-ил-1Н-имидазол-2-илсульфанилметил]фенол (18c) С использованием общего способа В (14 часов, комнатная температура) указанное в заголовке соединение получали из 1а (0,20 г; 0,7 ммоль) и 3-гидроксиметилфенола (0,10 г; 0,8 ммоль) после очистки колоночной хроматографией (SiO2 60, CH2Cl2/EtOH 9+1) Т.пл. 250°С (разложение) ИК (ATR): 1271 (ОН изгиб), 1232 (C-F), 1004 см-1 (С-О) 1Н-ЯМР (ДМСО-d6): 2-[5-(4-Фторфенил)-4-пиридин-4-ил-1Н-имидазол-2-илсульфанилметил]-4-метилсульфанилфенол (18d) С использованием общего способа В (1 час, комнатная температура) указанное в заголовке соединение получали из 1а (0,50 г; 2,9 ммоль) и 8а (0,50 г; 2,9 ммоль) после растирания с МеОН. Т.пл. 243°С ИК (KBr): 1275 (ОН изгиб), 1230 (C-F), 1005 см-1 (С-О) 1Н-ЯМР (ДМФА-d7): 4-Хлор-2-[5-(4-фторфенил)-4-пиридин-4-ил-1Н-имидазол-2-илсульфанилметил]-6-метилсульфанилфенол (18е) С использованием общего способа В (1,5 часа, 75°С) указанное в заголовке соединение получали из 1а (0,80 г; 3,0 ммоль) и 8b (0,60 г; 3,0 ммоль) после растирания с МеОН. Т.пл. 220°С (разложение) ИК (KBr): 1259 (ОН изгиб), 1225 (C-F), 1007 см-1 (С-О) 1Н-ЯМР (ДМСО-d6): 4-[5-(4-Фторфенил)-4-пиридин-4-ил-1Н-имидазол-2-илсульфанилметил]-4-метилсульфанилфенол (18f) С использованием общего способа В (2 часа, комнатная температура) указанное в заголовке соединение получали из 1а (0,20 г; 0,7 ммоль) и 8с (0,14 г; 0,8 ммоль) после растирания с МеОН. Т.пл. 230°С (разложение) ИК (KBr): 1227 (C-F), 1019 см-1 (С-О) 1Н-ЯМР (CD3OD): 2-[5-(4-Фторфенил)-4-пиридин-4-ил-1Н-имидазол-2-илсульфанилметил]-4-метансульфинилфенол (18g) С использованием общего способа В (1 час, комнатная температура) указанное в заголовке соединение получали из 1а (0,27 г; 1,0 ммоль) и 8а (0,17 г; 1,0 ммоль) с добавлением раствора Н2О2 35%-ной концентрации после перекристаллизации из смеси толуол/ТГФ (1+1). Т.пл. 216°С ИК (KBr): 1278 (ОН изгиб), 1232 (C-F), 1031 (S=O), 1003 см-1 (С-О) 1Н-ЯМР (CD3OD): 4-Хлор-2-[5-(4-фторфенил)-4-пиридин-4-ил-1Н-имидазол-2-илсульфанилметил]-6-метансульфинилфенол (18h) С использованием общего способа В (1,5 часа, 75°С) указанное в заголовке соединение получали из 1а (0,27 г; 1,0 ммоль) и 8b (0,21 г; 1,0 ммоль) с добавлением раствора Н2О2 35%-ной концентрации после очистки колоночной хроматографией (SiO2 60, ацетон). Т.пл. 175°С (разложение) ИК (KBr): 1265 (ОН изгиб), 1236 (C-F), 1051 (S=O), 1005 см-1 (С-О) 1Н-ЯМР (CD3OD): Пример 19 4-[5-(4-Фторфенил)-4-пиридин-4-ил-1Н-имидазол-2-илсульфанилметил]-2-метансульфинилфенол (19) С использованием общего способа В (2,5 часа, комнатная температура) указанное в заголовке соединение получали из 1а (0,27 г; 1,0 ммоль) и 8с (0,14 г; 0,8 ммоль) с добавлением раствора Н2О2 35%-ной концентрации после растирания с ацетоном. Т.пл. 185°С (разложение) ИК (KBr): 1296 (ОН изгиб), 1230 (C-F), 1062 (S=O), 1013 см-1 (С-О) 1Н-ЯМР (CD3OD): Пример 20 4-Фтор-N-метокси-N-метилбензамид (20) Суспензию 4-фторбензойной кислоты (20 г, 143 ммоль) в хлористом тиониле (130 г; 1,1 моль) перемешивали при кипении с обратным холодильником в течение 6 часов; интенсивное выделение газа, прозрачный раствор примерно через 10 минут, углубление окраски с желтой до оранжевой. Избыток хлористого тионила удаляли перегонкой (сначала атмосферное давление/40°С, затем мембранный вакуумный насос/40°С). Перегонкой остатка 4-фторбензоилхлорид отгоняли с использованием мембранного вакуумного насоса при 90°С над короткой колонной. Продукт реакции кристаллизовался при хранении в холодильнике (n20 D 1,5315; т.пл. 9°С; выход 20г/89%). Свежеперегнанный триэтиламин (29 мл) добавляли к суспензии гидрохлорида N,O-диметилгидроксиламина (9,0 г; 92 ммоль) в CH2Cl2 (75 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем охлаждали до -10°С. При охлаждении к первоначальной загрузке в течение 6 минут добавляли по каплям 4-фторбензоилхлорид (13,5 г; 85 ммоль). По окончании прибавления охлаждение удаляли и реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов. Светло-коричневую суспензию выливали на Н2О (100 мл). Органическую фазу удаляли и водную фазу экстрагировали диэтиловым эфиром (2×). Объединенный экстракт промывали насыщенным раствором NaCl, сушили над Na2SO4 и концентрировали. Маслянистый коричневый остаток кристаллизовался при охлаждении и потирании. Неочищенный продукт сушили с использованием масляного насоса (остаточный триэтиламин) и вводили в реакции без дополнительной очистки. 1Н-ЯМР (CDCl3): Пример 21 2-(2-Хлорпиридин-4-ил)-1-(4-фторфенил)этанон (21а) В двухгорлую колбу, высушенную нагреванием и продувкой аргоном, добавляли по каплям n-BuLi (раствор 15%-ной концентрации в н-гексане, 45 мл, 104 ммоль) к охлажденному до -85°С раствору диизопропиламина (15 мл, 106 ммоль) в абс.ТГФ (150 мл), при этом температура повышалась до -50°С. По окончании добавления светло-желтый раствор перемешивали при -85°С в течение 55 мин. При -85°С к первоначальной смеси добавляли раствор 2-хлор-4-метилпиридина (2-хлор- 1Н-ЯМР (CDCl3): 1-(4-Фторфенил)-2-(2-фторпиридин-4-ил)этанон (21b) 21b получали из 2-фтор-4-метилпиридина (13,9 г; 125 ммоль) с использованием способа, описанного для синтеза 21а. 1Н-ЯМР (CDCl3): 2-(2-Бромпиридин-4-ил)-1-(4-фторфенил)этанон (21c) 21с получали из 2-бром-4-метилпиридина (9,6 г; 56 ммоль) с использованием способа, описанного для синтеза 21а. 1Н-ЯМР (CDCl3): Пример 22 1-(2-Хлорпиридин-4-ил)-2-(4-фторфенил)этан-1,2-дион-1-оксим (22а) При перемешивании и охлаждении на водяной бане (примерно 10°С) раствор NaNO2 (0,85 г; 12,3 ммоль) в Н2О (10 мл) добавляли по каплям в течение 2,5 минут к раствору 21а (3,0 г; 12 ммоль) в ледяной уксусной кислоте. По окончании прибавления реакционную смесь перемешивали при комнатной температуре в течение 0,5 часа, добавляли Н2О (60 мл) и перемешивание при комнатной температуре продолжали в течение 3 часов. Светло-бежевый осадок отфильтровывали, промывали водой и сушили при пониженном давлении над CaCl2. 1Н-ЯМР (ДМСО-d6): 1-(2-Фторпиридин-4-ил)-2-(4-фторфенил)этан-1,2-дион-1-оксим (22b) 22b получали из 21b (10,0 г; 43 ммоль) с использованием способа, описанного для синтеза 22а. 1Н-ЯМР (ДМСО-d6): 1-(4-Фторфенил)-2-(2-изопропоксипиридин-4-ил)-2)этан-1,2-дион-1-оксим (22с) Раствор 22b (200 мг; 0,76 ммоль) в насыщенном HCl изопропаноле (15 мл) перемешивали при кипении с обратным холодильником в течение 2,5 часов. Раствор концентрировали и желтовато-белый остаток растирали с небольшим количеством этанола, отфильтровывали и сушили. 1Н-ЯМР (ДМСО-d6): 1-(2-Бромпиридин-4-ил)-2-(4-фторфенил)этан-1,2-дион-1-оксим (22d) 22d получали из 21с (5,0 г; 17 ммоль) с использованием способа, описанного для синтеза 22а. 1Н-ЯМР (ДМСО-d6): Пример 23 Гидрохлорид 2-амино-2-(2-хлорпиридин-4-ил)-1-(4-фторфенил)этанона (23а) При осторожном нагревании 22а (1,5 г; 45,4 ммоль) растворяли в метаноле (15 мл). Раствор охлаждали до комнатной температуры, добавляли HCl-содержащий метанол и смесь переносили в двухгорлую колбу. Вводили в колбу с первоначальной смесью 10% Pd-C (15 мг). Реакционную колбу вакуумировали с использованием масляного насоса и затем вводили Н2 через капилляр для ввода газа (4×). При комнатной температуре суспензию встряхивали в закрытой трехгорлой колбе в атмосфере Н2 (240 ударов в минуту) до тех пор, пока исходное вещество не переставало обнаруживаться по данным тонкослойной хроматографии (6 часов). Суспензию фильтровали и катализатор промывали большим количеством метанола. Объединенный фильтрат концентрировали и твердый аморфный остаток цвета горчицы сушили с использованием масляного насоса. Неочищенный продукт использовали без дополнительной очистки на следующей реакционной стадии. 1Н-ЯМР (ДМСО-d6): Гидрохлорид 2-амино-2-(2-фторпиридин-4-ил)-1-(4-фторфенил)этанона (23b) При осторожном нагревании 22b (5,0 г; 19 ммоль) растворяли в HCl-содержащем изопропаноле (IsOH/IsOH, насыщенный HCl, 1+1, 60 мл). Желтоватый раствор охлаждали до комнатной температуры и переносили в двухгорлую колбу (100 мл). Реакционную колбу вакуумировали с использованием масляного насоса и затем вводили Н2 через капилляр для ввода газа (4×). При комнатной температуре суспензию встряхивали в закрытой трехгорлой колбе в атмосфере Н2 (240 ударов в минуту) до тех пор, пока исходное вещество не переставало обнаруживаться по данным тонкослойной хроматографии (6,5 часов). Катализатор отфильтровывали. Остаток после фильтрования промывали большим количеством метанола (примерно 800 мл). Объединенные фильтраты концентрировали и твердый аморфный остаток сушили на масляном насосе. Неочищенный продукт использовали без дополнительной очистки на следующей реакционной стадии. 1Н-ЯМР (ДМСО-d6): Гидрохлорид 2-амино-1-(4-фторфенил)-2-(2-изопропоксипиридин-4-ил)этанона (23с) 23с получали из 22с (2,0 г; 7,6 ммоль) с использованием способа, описанного для синтеза 23а. 1Н-ЯМР (ДМСО-d6): Гидрохлорид 2-амино-1-(4-фторфенил)-2-(2-метоксипиридин-4-ил)этанона (23d) 23d получен при обработке 22b (7,5 г; 29 ммоль) в условиях, описанных для синтеза 23а. 1Н-ЯМР (ДМСО-d6): Гидрохлорид 2-амино-1-(4-фторфенил)-2-пиридин-4-илэтанона (23е) 23е получен при обработке 22с (4,0 г; 12,4 ммоль) в условиях, описанных для синтеза 23b. 1Н-ЯМР (ДМСО-d6): Гидрохлорид 2-амино-2-(2-бромпиридин-4-ил)-1-(4-фторфенил)этанона (23f) Раствор 22d (1,8 г; 5,6 ммоль) в абсолютном этаноле (30 мл) охлаждали до -10°С и добавляли концентрированную серную кислоту (1,3 мл). При охлаждении к первоначальной смеси добавляли постепенно цинковую пыль (1,1 г). Реакционную смесь перемешивали при -10°С в течение 30 минут и затем нагревали до комнатной температуры. Серо-зеленую суспензию фильтровали и белый остаток (ZnSO4) промывали большим количеством этанола. Объединенный желтый фильтрат концентрировали и твердый желтоватый остаток сушили с использованием масляного насоса. 1Н-ЯМР (ДМСО-d6): Пример 24 4-(2-Хлорпиридин-4-ил)-5-(4-фторфенил)-1,3-дигидроимидазол-2-тион (24а) При осторожном нагревании 23а (2,9 г; примерно 9,6 ммоль) растворяли в абсолютном ДМФ (75 мл). В прозрачный оранжево-красный раствор вводили тиоцианат калия (1,9 г; 19,6 ммоль), при этом сразу же наблюдалась опалесценция и окраска становилась более светлой. Реакционную смесь перемешивали при кипении с обратным холодильником в течение 1,5 часов. Суспензию охлаждали до комнатной температуры и при охлаждении Н2О разбавляли по каплям Н2О (примерно 140 мл). Желтый осадок отфильтровывали, промывали Н2О и сушили при пониженном давлении над CaCl2. 1Н-ЯМР (ДМСО-d6): 4-(4-Фторфенил)-5-(2-фторпиридин-4-ил)-1,3-дигидроимидазол-2-тион (24b) 24b получали из 23b (6,1 г; 20 ммоль) с использованием способа, описанного для синтеза 24а. 1Н-ЯМР (ДМСО-d6): 4-(4-Фторфенил)-5-(2-изопропоксипиридин-4-ил)-1,3-дигидроимидазол-2-тион (24с) 24с получали из 23с (2,5 г; 7,6 ммоль) с использованием способа, описанного для синтеза 24а. 1Н-ЯМР (ДМСО-d6): 4-(4-Фторфенил)-5-(2-метоксипиридин-4-ил)-1,3-дигидроимидазол-2-тион (24d) Тиоцианат калия (2 г, 20,6 ммоль) вводили в раствор 23d (3,2 г; 10,8 ммоль) в соляной кислоте 10%-ной концентрации (50 мл). Реакционную смесь перемешивали при кипении с обратным холодильником в течение 30 минут. Оранжевый раствор охлаждали и нейтрализовали с использованием раствора NaHCO3 10%-ной концентрации. Осадок отфильтровывали, промывали Н2О и сушили при пониженном давлении над CaCl2. Неочищенный продукт растирали с этанолом и нерастворимые компоненты отфильтровывали. При хранении 24d выпадал в осадок из этанольного фильтрата. 1Н-ЯМР (ДМСО-d6): Пример 25 2-Хлор-4-[5-(4-фторфенил)-2-метилсульфанил-3Н-имидазол-4-ил]пиридин (25а) С использованием общего способа А указанное в заголовке соединение получали из 24а (0,5 г; 1,6 ммоль) и иодистого метила (0,35 г; 2,5 ммоль) после проведения реакции в течение 12 часов и очистки колоночной хроматографией (Al2O3, CH2Cl2/этилацетат 1+1). Т.пл. 236°С. ИК (ATR): 3126, 3057, 2929, 1591, 1529, 1499, 1389, 1231 (C-F), 1159, 996, 976, 844, 780 см-1. 1Н-ЯМР (ДМСО-d6): 2-Фтор-4-[5-(4-фторфенил)-2-метилсульфанил-3Н-имидазол-4-ил]пиридин (25b) С использованием общего способа А указанное в заголовке соединение получали из 24b (1,4 г; 9,9 ммоль) и иодистого метила (1,4 г; 9,9 ммоль) после проведения реакции в течение 40 часов. Неочищенный продукт кипятили со смесью CH2Cl2/этилацетат (1+1). Объединенный органический экстракт обесцвечивали с использованием Al2O3 и полученный после концентрирования фильтрата остаток растирали с небольшим количеством EtOH. Т.пл. 224°С. ИК (ATR): 3073, 1609, 1497, 1421, 1234 (C-F), 1159, 1002, 883, 851, 833, 815 см-1. 1Н-ЯМР (ДМСО-d6): 4-[5-(4-Фторфенил)-2-метилсульфанил-3Н-имидазол-4-ил]-2-изопропоксипиридин (25с) NaH (55-65%; 1,0 г; примерно 23 ммоль) вводили в раствор 24с (4,0 г; 13,8 ммоль) в абсолютном ТГФ (60 мл). Эту первоначальную смесь перемешивали при комнатной температуре в течение 5 минут и добавляли по каплям раствор иодистого метила (2,2 г; 17,3 ммоль) в абсолютном ТГФ (5 мл) при охлаждении Н2О. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Прозрачный коричневый раствор концентрировали и остаток помещали в Н2О. Водный раствор нейтрализовали с использованием соляной кислоты 10%-ной концентрации и экстрагировали этилацетатом (2×). Объединенный органический экстракт промывали насыщенным раствором NaCl, сушили над Na2SO4 и концентрировали. Полутвердый остаток экстрагировали кипящим трет-бутилметиловым эфиром (2×) и фильтровали. Прозрачный эфирный фильтрат концентрировали и твердый остаток растирали с небольшим количеством трет-бутилметилового эфира, фильтровали и сушили. Дополнительное количество реакционного продукта получали разделением маточного раствора колоночной хроматографией (SiO2 60, CH2Cl2/этилацетат, 1+1). Т.пл. 141°С. ИК (ATR): 2928, 1610, 1544, 1509, 1412, 1314, 1222 (C-F), 1104, 1005, 954, 865, 843, 816 см-1. 1Н-ЯМР (ДМСО-d6): 4-[5-(4-Фторфенил)-2-метилсульфанил-3Н-имидазол-4-ил]-2-метоксипиридин (25d) Раствор 24d (1,0 г; 3,3 ммоль) и иодистого метила (5,6 г; 39 ммоль) в метаноле (50 мл) перемешивали при кипении с обратным холодильником в течение 3 часов. Реакционную смесь охлаждали и фильтровали. Фильтрат концентрировали и остаток помещали в этанол. Нерастворимые компоненты отфильтровывали и фильтрат концентрировали. Остаток помещали в CH2Cl2/EtOH (9+1). Нерастворимые компоненты отфильтровывали и фильтрат разделяли колоночной хроматографией (SiO2 60, CH2Cl2/EtOH, 9+1). Т.пл. 158°С. ИК (ATR): 1618, 1608, 1497, 1391, 1222 (C-F), 1212, 1036, 835, 825 см-1. 1Н-ЯМР (ДМСО-d6): 4-[5-(4-Фторфенил)-2-метилсульфанил-3Н-имидазол-4-ил]-1Н-пиридин-2-он (25е) При обработке 23d (8,8 г; 31 ммоль) тиоцианатом калия в кипящем ДМФ аналогично способу, описанному для 24а, 25е, был получен в качестве единственного продукта реакции. Т.пл. 314°С (разложение). После циклизации, приводящей к 1,3-дигидроимидазолтиону, метильная группа из метоксильного заместителя переходит к нуклеофильному атому серы тиона с образованием, во-первых, 2-метилсульфанил-3Н-имидазола и, во-вторых, 2-гидроксипиридин/1Н-пиридин-2-она. ИК (ATR): 1634 (пиридон I), 1610, 1557 (пиридон II), 1493, 1220 (C-F), 968, 837, 800 см-1. 1Н-ЯМР (ДМСО-d6): Бензил-{4-[5-(4-фторфенил)-2-метилсульфанил-3Н-имидазол-4-ил]пиридин-2-ил}амин (25f) С использованием общего способа С указанное в заголовке соединение получали из 25b (0,2 г; 0,7 ммоль) и бензиламина (0,8 г; 7,5 ммоль) после проведения реакции в течение 5 часов при температуре 160°С и разделения колоночной хроматографией (Al2O3, CH2Cl2/этилацетат 1+1). Т.пл. 152°С (разложение). ИК (ATR): 3234 (NH), 3006, 2916, 1601, 1583, 1501, 1451, 1432, 1353, 1225 (C-F), 1074, 844, 813, 729, 695 см-1. Н-ЯМР (CD3OD): {4-[5-(4-Фторфенил)-2-метилсульфанил-3Н-имидазол-4-ил]пиридин-2-ил}-(4-метоксибензил)амин (25g) С использованием общего способа С указанное в заголовке соединение получали из 25b (0,44 г; 1,5 ммоль) и 4-метоксибензиламина (2,0 г; 14.6 ммоль) после проведения реакции в течение 7 часов при температуре 160°С и разделения колоночной хроматографией (SiO2, CH2Cl2/EtOH 9+1). Т.пл. 207°С. ИК (ATR): 1598, 1558, 1510, 1244, 1217 (C-F), 846, 812 см-1. 1Н-ЯМР (CD3OD): {4-[5-(4-Фторфенил)-2-метилсульфанил-3Н-имидазол-4-ил]пиридин-2-ил}-(4-метилбензил)амин (25h) С использованием общего способа С указанное в заголовке соединение получали из 25b (0,2 г; 0,7 ммоль) и 4-метилбензиламина (0,85 г; 7,0 ммоль) после проведения реакции в течение 6 часов при 160°С и разделения колоночной хроматографией (SiO2, CH2Cl2/EtOH 9+1). Т.пл. 185°С. ИК (ATR): 1600, 1559, 1502, 1427, 1218 (C-F), 844, 809 см-1. 1Н-ЯМР (CD3OD): (4-Хлорбензил)-{4-[5-(4-фторфенил)-2-метилсульфанил-3Н-имидазол-4-ил]пиридин-2-ил}амин (25i) С использованием общего способа С указанное в заголовке соединение получали из 25b (0,2 г; 0,7 ммоль) и 4-хлорбензиламина (1,0 г; 7,0 ммоль) после проведения реакции в течение 5,5 часов при кипении с обратным холодильником и разделения колоночной хроматографией (SiO2, CH2Cl2/EtOH 9+1). Т.пл. 195°С. ИК (ATR): 3409, 1597, 1549, 1502, 1489, 1422, 1218 (C-F), 843, 814, 793 см-1. Н-ЯМР (CD3OD): (3,4-Дихлорбензил)-{4-[5-(4-фторфенил)-2-метилсульфанил-3Н-имидазол-4-ил]пиридин-2-ил}амин (25j) С использованием общего способа С указанное в заголовке соединение получали из 25b (0,2 г; 0,7 ммоль) и 3,4-дихлорбензиламина (1,2 г; 6,8 ммоль) после проведения реакции в течение 7,5 часов при 160°С и разделения колоночной хроматографией (SiO2, CH2Cl2/EtOH 9+1). Т.пл. 212°С. ИК (ATR): 3409, 1600, 1552, 1509, 1490, 1424, 1225 (C-F), 842, 827, 813 см-1. 1Н-ЯМР (CD3OD): {4-[5-(4-Фторфенил)-2-метилсульфанил-3Н-имидазол-4-ил]пиридин-2-ил}фениламин (25k) С использованием общего способа С указанное в заголовке соединение получали из 25b (0,2 г; 0,7 ммоль) и анилина (0,65 г; 7,0 ммоль) после проведения реакции в течение 6 часов при кипении с обратным холодильником и разделения колоночной хроматографией (SiO2, CH2Cl2/EtOH 9+1). Т.пл. 228°С. ИК (ATR): 3031, 1610, 1590, 1561, 1504, 1433, 1265, 1225 (C-F), 839, 827, 749, 695 см-1. 1Н-ЯМР (ДМСО-d6): {4-[5-(4-Фторфенил)-2-метилсульфанил-3Н-имидазол-4-ил]пиридин-2-ил}фенэтиламин (25l) С использованием общего способа С указанное в заголовке соединение получали из 25b (0,2 г; 0,7 ммоль) и 2-фенилэтиламина (0,85 г; 7,0 ммоль) после проведения реакции в течение 5,5 часов при 160°С и разделения колоночной хроматографией (SiO2, CH2Cl2/EtOH 9+1). Т.пл. 99°С. ИК (ATR): 3409, 1604, 1546, 1504, 1220 (C-F), 838, 813, 698 см-1. 1Н-ЯМР (CD3OD): (RS)-{4-[5-(4-Фторфенил)-2-метилсульфанил-3Н-имидазол-4-ил]пиридин-2-ил}-(1-фенилэтил)амин (25m) С использованием общего способа С указанное в заголовке соединение получали из 25b (0,2 г; 0,7 ммоль) и (RS)-1-фенилэтиламина (0,80 г; 6,6 ммоль) после проведения реакции в течение 7 часов при 160°С и разделения колоночной хроматографией (SiO2, CH2Cl2/EtOH 9+1). Т.пл. 117-119°С. ИК (ATR): 2926, 1607, 1547, 1502, 1434, 1221 (C-F), 1157, 838, 814, 699 см-1. 1Н-ЯМР (ДМСО-d6): (R)-{4-[5-(4-Фторфенил)-2-метилсульфанил-3Н-имидазол-4-ил]пиридин-2-ил}-(1-фенилэтил)амин (25n) С использованием общего способа С указанное в заголовке соединение получали из 25b (0,2 г; 0,7 ммоль) и (R)-1-фенилэтиламина (0,80 г; 6,6 ммоль) после проведения реакции в течение 7 часов при 170°С и разделения колоночной хроматографией (SiO2, CH2Cl2/этилацетат 1+1). Т.пл. 117-119°С. ИК (ATR): 2926, 1607, 1547, 1502, 1434, 1221 (C-F), 1157, 838, 814, 699 см-1. 1Н-ЯМР (CD3OD): (S)-{4-[5-(4-Фторфенил)-2-метилсульфанил-3Н-имидазол-4-ил]пиридин-2-ил}-(1-фенилэтил)амин (25о) С использованием общего способа С указанное в заголовке соединение получали из 25b (0,2 г; 0,7 ммоль) и (S)-1-фенилэтиламина (0,80 г; 6,6 ммоль) после проведения реакции в течение 13 часов при 170°С и разделения колоночной хроматографией (SiO2, CH2Cl2/этилацетат 1+1). Т.пл. 117-119°С. ИК (ATR): 2926, 1607, 1547, 1502, 1434, 1221 (C-F), 1157, 838, 814, 699 см-1. 1Н-ЯМР (CD3OD): Бензил-{4-[5-(4-фторфенил)-2-метилсульфанил-3Н-имидазол-4-ил]пиридин-2-ил}метиламин (25р) С использованием общего способа С указанное в заголовке соединение получали из 25b (0,2 г; 0,7 ммоль) и N-метилбензиламина (0,85 г; 7,0 ммоль) после проведения реакции в течение 7 часов при 180°С и двух разделений колоночной хроматографией (SiO2, CH2Cl2/этилацетат 1+1). Т.пл. 79°С. ИК (ATR): 2924, 1601, 1494, 1407, 1219 (C-F), 837, 810, 730, 696 см-1. 1Н-ЯМР (CD3OD): Соединения, перечисленные ниже в таблице 2, были получены с использованием вышеуказанного способа Таблица 2
Пример 26 4-[2-Бензилсульфанил-5-(4-фторфенил)-3Н-имидазол-4-ил]-2-хлорпиридин (26а) С использованием общего способа А указанное в заголовке соединение получали из 24а (0,3 г; 1,0 ммоль) и хлористого бензила (0,12 г; 1,0 ммоль) после проведения реакции в течение 6 часов и разделения колоночной хроматографией (SiO2 60, CH2Cl2/EtOH 9+1). Т.пл. 223°С. ИК (ATR): 2939, 1591, 1530, 1505, 1233 (C-F), 997, 838, 782, 700 см-1. 1Н-ЯМР (ДМСО-d6): 4-[2-Бензилсульфанил-5-(4-фторфенил)-3Н-имидазол-4-ил]-2-фторпиридин (26b) С использованием общего способа А указанное в заголовке соединение получали из 24b (5,1 г; 17,6 ммоль) и бромистого бензила (9,2 г; 54 ммоль) после проведения реакции в течение 1,5 часов и разделения колоночной хроматографией (Al2O3, CH2Cl2/этилацетат 1+1). Т.пл. 174°С. ИК (ATR): 3028, 2948, 1611, 1496, 1413, 1228 (C-F), 1203, 1003, 879, 838, 698 см-1. 1Н-ЯМР (ДМСО-d6): Бензил-{4-[2-бензилсульфанил-5-(4-фторфенил)-3Н-имидазол-4-ил]пиридин-2-ил}амин (26с) С использованием общего способа С указанное в заголовке соединение получали из 26b (0,2 г; 0,53 ммоль) и бензиламина (0,60 г; 5,6 ммоль) после проведения реакции в течение 6 часов при 180°С и разделения колоночной хроматографией (SiO2 60, CH2Cl2/этилацетат 1+1). Т.пл. 185°С. ИК (ATR): 3407 (NH), 3025, 2855, 2713, 1599, 1550, 1489, 1356, 1220 (C-F), 1155, 840, 814, 693 см-1. 1Н-ЯМР (CD3OD): (RS)-{4-[2-Бензилсульфанил-5-(4-фторфенил)-3Н-имидазол-4-ил]пиридин-2-ил}-(1-фенилэтил)амин (26d) С использованием общего способа С указанное в заголовке соединение получали из 26b (0,2 г; 0,53 ммоль) и (RS-)-1-фенилэтиламина (0,65 г; 5,4 ммоль) после проведения реакции в течение 15 часов при 150°С и разделения колоночной хроматографией (SiO2 60, CH2Cl2/этилацетат 1+1). Т.пл. 145°С. ИК (ATR): 3028, 1606, 1546, 1494, 1450, 1221 (C-F), 1157, 837, 813, 697 см-1. 1Н-ЯМР (CD3OD): {4-[2-Бензилсульфанил-5-(4-фторфенил)-3Н-имидазол-4-ил]пиридин-2-ил}-(4-метоксибензил)амин (26е) С использованием общего способа С указанное в заголовке соединение получали из 26а (0,2 г; 0,5 ммоль) и 4-метоксибензиламина (2,0 г; 14,6 ммоль) после проведения реакции в течение 22 часов при кипении с обратным холодильником и разделения колоночной хроматографией (Al2O3, CH2Cl2/этилацетат 1+1). Т.пл. 196-200°С. ИК (ATR): 1605, 1574, 1507, 1245, 1225 (C-F), 843, 814, 698 см-1. 1Н-ЯМР (ДМСО-d6): 4-[2-Бензилсульфанил-5-(4-фторфенил)-3Н-имидазол-4-ил]-2-метоксипиридин (26f) Суспензию 25а (0,1 г; 0,25 ммоль) в метанольном растворе NaOCH3 (30%, 2 мл) разбавляли метанолом (5 мл) и перемешивали при кипении с обратным холодильником в течение 13 часов. Реакционную смесь разбавляли Н2О и водный раствор экстрагировали (CH2Cl2) (3×). Объединенный органический экстракт промывали насыщенным раствором NaCl, сушили над Na2SO4 и концентрировали. Маслянистый остаток очищали колоночной хроматографией (SiO2 60, CH2Cl2/этилацетат 1+1). 1Н-ЯМР (CDCl3): Пример 27 2-Хлор-4-[5-(4-фторфенил)-2-(4-метансульфинилбензилсульфанил)-3Н-имидазол-4-ил]пиридин (27а) NaH (55-65%; 0,1 г; примерно 2 ммоль) вводили в раствор 24а (0,31 г; 1,0 ммоль) в абсолютном ТГФ (15 мл). Первоначальную смесь перемешивали при комнатной температуре в течение 5 минут и добавляли 4-метилсульфинилбензилхлорид (3, 0,19 г; 1,0 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Желто-коричневый раствор разбавляли Н2О и нейтрализовали лимонной кислотой 10%-ной концентрации. ТГФ удаляли и водный раствор экстрагировали этилацетатом (2×). Объединенный органический экстракт промывали насыщенным раствором NaCl (2×), сушили над Na2SO4 и концентрировали. Твердый остаток очищали колоночной хроматографией (SiO2 60, CH2Cl2/EtOH 9,5+0,5). Т.пл. 179°С. ИК (ATR): 3049, 1592, 1505, 1374, 1224 (C-F), 1086, 1030 (S=O), 1014, 989, 839, 816,781 см-1 (C-Cl). 1Н-ЯМР (ДМСО-d6): 2-Фтор-4-[5-(4-фторфенил)-2-(4-метансульфинилбензилсульфанил)-3Н-имидазол-4-ил]пиридин (27b) С использованием общего способа А указанное в заголовке соединение получали из 24b (4,2 г; 14,5 ммоль) и 3 (4,1 г; 22 ммоль) после проведения реакции в течение 2 часов и разделения колоночной хроматографией (1. Al2O3, CH2Cl2/этилацетат 1+1; 2. SiO2 60, CH2Cl2/EtOH 9+1). Т.пл. 150°С. ИК (ATR): 3061, 1610, 1506, 1408, 1227 (C-F), 1030 (S=O), 1016, 995, 978, 882, 839, 815 см-1. 1Н-ЯМР (ДМСО-d6): Бензил-{4-[5-(4-фторфенил)-2-(4-метансульфинилбензилсульфанил)-3Н-имидазол-4-ил]пиридин-2-ил}амин (27с) С использованием общего способа С указанное в заголовке соединение получали из 27b (0,3 г; 0,68 ммоль) и бензиламина (0,75 г; 7,0 ммоль) после проведения реакции в течение 7 часов при 170°С и разделения колоночной хроматографией (SiO2 60, CH2Cl2/EtOH 19+1). Т.пл. 149°С. ИК (ATR): 3238, 3064, 1600, 1558, 1514, 1495, 1227 (C-F), 1034 (S=O), 1006, 982, 839, 814 см-1. 1Н-ЯМР (CD3OD): (RS)-{4-[5-(4-фторфенил)-2-(4-метансульфинилбензилсульфанил)-3Н-имидазол-4-ил]пиридин-2-ил}-(1-фенилэтил)амин (27d) С использованием общего способа С указанное в заголовке соединение получали из 27b (0,3 г; 0,68 ммоль) и (RS)-1-фенилэтиламина (0,85 г; 7,0 ммоль) после проведения реакции в течение 10 часов при 170°С и разделения колоночной хроматографией (SiO2 60, CH2Cl2/EtOH 9+1). Т.пл. 193°С. ИК (ATR): 2967, 1606, 1547, 1502, 1221 (C-F), 1085, 1031 (S=O), 1014, 838, 814, 670 см-1. 1Н-ЯМР (CD3OD): Соединения, полученные способом С, представлены ниже в таблице 3 (однако соединение №31 было получено способом D)
Пример 37а Циклогексил-{4-[5-(4-фторфенил)-2-метилсульфанил-1Н-имидазол-4-ил]пиридин-2-ил}амин (37а) 2-Фтор-4-[5-(4-фторфенил)-2-метилсульфанил-1Н-имидазол-4-ил]пиридин (1,2 г, 4 ммоль), который отвешивали в одногорлую колбу емкостью 25 мл, предварительно продутую аргоном, суспендировали в циклогексиламине (3,97 г, 0,04 моль), вводили защитную атмосферу аргона и затем нагревали при кипении амина с обратным холодильником на масляной бане при температуре 160°С в течение 48 часов. Коричневую суспензию оставляли охлаждаться до комнатной температуры. Затем добавляли 30 мл раствора цитрата Na (10%-ный раствор лимонной кислоты, рН 5, с концентрированным NaOH) и смесь перемешивали в течение 10 минут и экстрагировали дважды 30 мл этилацетата. Объединенные органические фазы опять экстрагировали дважды в каждом случае 30 мл раствора цитрата Na (10%-ный раствор лимонной кислоты, рН 5, с концентрированным NaOH), затем 30 мл раствора NaHCO3 и экстрагировали один раз насыщенным раствором NaCl. Органические фазы сушили над Na2SO4 и концентрировали с использованием роторного испарителя, остаток кристаллизовали из 10 мл. Перекристаллизацию проводили из смеси изопропанол/воды (примерно 5 мл iPrOH нагревают и оставляют охлаждаться и медленно добавляют примерно 5 мл дистиллированной воды). Выход: 0,76 г (49,7%), чистота (по ВЭЖХ) 96,6%. Таким способом получают соединения формулы I, представленные ниже в таблице 4.
Соединения, представленные ниже в таблице 5, были получены с использованием вышеуказанного способа.
Формула изобретения
1. 2-Тиозамещенное производное имидазола формулы I
где R1 представляет собой арил, который является замещенным атомом галогена или галоген-С1-С6-алкилом; R2 выбран из группы, включающей a) арил-С1-С4-алкил, и b) C1-С6-алкил; R3 выбран из группы, включающей а) NR4R10; b) NR7COR10; c) OR10; d) NH2; R4 представляет собой Н, R5 и R6, которые могут быть одинаковыми или различными, представляют собой Н, галоген, ОН, C1-С6-алкокси, C1-С6-алкил или галоген-С1-С6-алкил; R7 представляет собой R4; R10 имеет одно из следующих значений: а) А-В b) c) d) e) f) C1-С6-алкил, который замещен 2 фенильными группами; А представляет собой линейный или разветвленный C1-С6-алкилен; В выбран из группы, включающей a) Н b) c) d) e) f) ОС1-С6-алкил; g) OH; Ну представляет собой 3-10-членный неароматический, моно-, би- или трициклический карбоцикл, который может быть или может не быть конденсирован с бензольным кольцом; Ar представляет собой 5- или 6-членный ароматический гетероцикл, который имеет 1 гетероатом, выбранный из группы, состоящей из О, S и N, и который может не быть конденсирован с бензольным кольцом; Het представляет собой 5- или 6-членный неароматический гетероцикл, который имеет 1 гетероатом, который представляет собой О, который может не быть конденсирован с бензольным кольцом; m равно 0, 1 или 2; или его оптические изомеры или физиологически приемлемые соли. 2. Соединение по п.1 формулы I, где R1 представляет собой арил, который замещен атомом галогена; R2 выбран из группы, включающей a) арил-С1-С4-алкил, и b) C1-С6-алкил; R3 выбран из группы, включающей a)NR4Rl0; b) NR7COR10; и с) C1-С6-алкокси; R4 представляет собой H; R10 представляет собой
R5 и R6, которые могут быть одинаковыми или различными, представляют собой Н, галоген, C1-С6-алкокси или C1-С6-алкил; R7 представляет собой Н; А представляет собой линейный или разветвленный C1-С6-алкилен, и m равно 0, 1 или 2, или его оптический изомер или физиологически приемлемая соль. 3. Соединение по п.1 или 2 формулы Ia
где R1, R2, R3 и m являются такими, как определено в п.1. 4. Соединение по п.1 или 2, где R3 представляет собой
где А, R5 и R6 являются такими, как определено в п.1. 5. Соединение по п.1 формулы I, где R10 представляет собой один из представленных ниже радикалов а) где Ar представляет собой 5- или 6-членный ароматический гетероцикл, который содержит гетероатом, выбранный из группы, включающей N, О и S; А представляет собой C1-С3-алкилен, и R5 и R6 представляют собой Н; b) где Het представляет собой 5- или 6-членный неароматический гетероцикл, который содержит О гетероатом; А представляет собой C1-С3-алкилен, и R5 и R6 представляют собой Н; с) где А представляет собой C1-С3-алкилен, и R5 и R6 представляют собой H, и Ну представляет собой циклопентил или циклогексил; d) циклопентил или циклогексил; e) фенил-С1-С6-алкил, причем алкильный радикал может иметь дополнительный фенильный заместитель; и f) С2-С6-алкил, который замещен фенилом. 6. Соединение по п.1 формулы I, где R3 представляет собой NR7COR10; где R10 выбран из группы, включающей фенил и С2-С6-алкил, который замещен фенилом. 7. Соединение по любому из предшествующих пунктов, где А представляет собой С1-С2-алкилен. 8. Соединение по любому из предшествующих пунктов, где А представляет собой этилен. 9. Соединение по любому из предшествующих пунктов, где R5 и R6 представляют собой Н. 10. Соединение по любому из предшествующих пунктов, где R1 представляет собой галогензамещенный фенил или фенил, замещенный CF3. 11. Соединение по любому из предшествующих пунктов, где R2 представляет собой бензил или C1-C6-алкил. 12. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении высвобождения цитокинов, включающая эффективное количество, по крайней мере, одного соединения по любому из пп.1-11, вместе с одним или несколькими фармацевтически приемлемыми носителями и/или вспомогательными добавками. 13. Применение по крайней мере одного соединения по любому из пп.1-11 при получении фармацевтической композиции, обладающей ингибирующей активностью в отношении высвобождения цитокинов.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
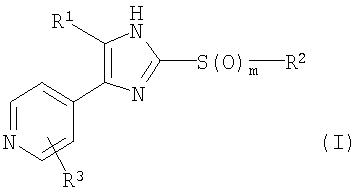
 ,
,  ,
,  ,
, 




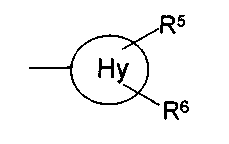
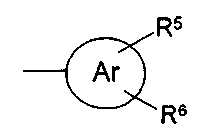


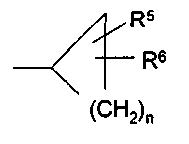


 ,
,
 ,
, ,
,
 ,
,
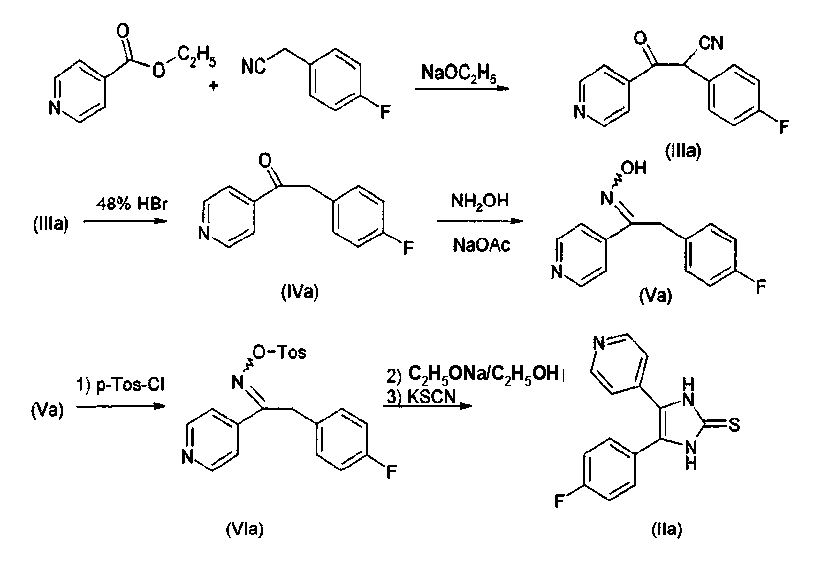
 -пиколин кетонов.
-пиколин кетонов.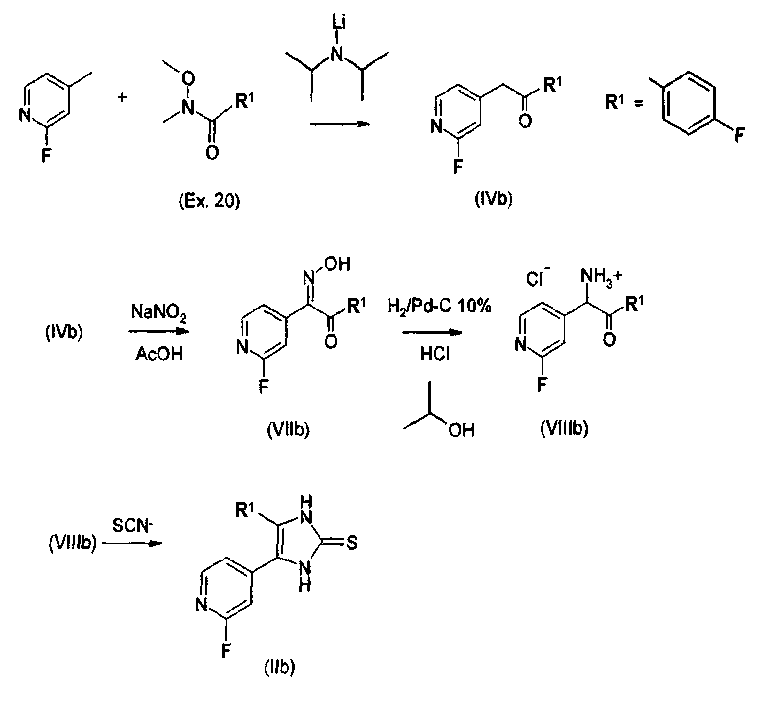


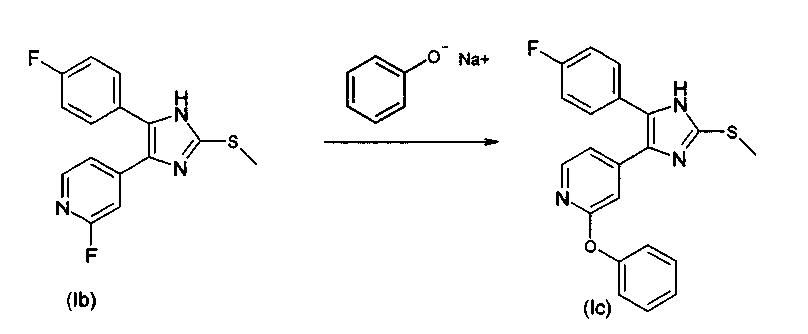








 и IL-
и IL- , которые играют важную роль в многочисленных воспалительных заболеваниях. В свете их ингибирующего высвобождение цитокина действия соединения согласно изобретению являются подходящими для лечения нарушений, которые связаны с расстройством иммунной системы. Они являются подходящими, например, для лечения аутоиммунных заболеваний, рака, ревматоидного артрита, подагры, септического шока, остеопороза, невропатической боли, распространения ВИЧ, слабоумия при ВИЧ, вирусного миокардита, инсулинзависимого диабета, перидонтальных нарушений, рестеноза, алопеции, уменьшения количества Т-клеток, связанного с ВИЧ-инфекцией или СПИДом, псориаза, острого панкреатита, реакций отторжения аллогенных трансплантатов, аллергической пневмонии, артериосклероза, рассеянного склероза, кахексии, болезни Альцгеймера, инсульта, приступов язвенного колита, болезни Крона, воспалительного заболевания кишечника (ВЗК), ишемии, застойной сердечной недостаточности, легочного фиброза, гепатита, глиобластомы, синдрома Гуйлайна-Барра, системной красной волчанки, респираторного дистресс-синдрома у взрослых (ARDS) и респираторного дистресс-синдрома.
, которые играют важную роль в многочисленных воспалительных заболеваниях. В свете их ингибирующего высвобождение цитокина действия соединения согласно изобретению являются подходящими для лечения нарушений, которые связаны с расстройством иммунной системы. Они являются подходящими, например, для лечения аутоиммунных заболеваний, рака, ревматоидного артрита, подагры, септического шока, остеопороза, невропатической боли, распространения ВИЧ, слабоумия при ВИЧ, вирусного миокардита, инсулинзависимого диабета, перидонтальных нарушений, рестеноза, алопеции, уменьшения количества Т-клеток, связанного с ВИЧ-инфекцией или СПИДом, псориаза, острого панкреатита, реакций отторжения аллогенных трансплантатов, аллергической пневмонии, артериосклероза, рассеянного склероза, кахексии, болезни Альцгеймера, инсульта, приступов язвенного колита, болезни Крона, воспалительного заболевания кишечника (ВЗК), ишемии, застойной сердечной недостаточности, легочного фиброза, гепатита, глиобластомы, синдрома Гуйлайна-Барра, системной красной волчанки, респираторного дистресс-синдрома у взрослых (ARDS) и респираторного дистресс-синдрома. (м.д.) 7,1 (м, 2Н, 4-F-Ph), 7,3 (м, 2Н, 4-Pyr), 7,5 (м, 2Н, 4-F-Ph), 8,5 (м, 2Н, 4-Pyr), 12,7 (д, 2Н, обмениваемый, NH).
(м.д.) 7,1 (м, 2Н, 4-F-Ph), 7,3 (м, 2Н, 4-Pyr), 7,5 (м, 2Н, 4-F-Ph), 8,5 (м, 2Н, 4-Pyr), 12,7 (д, 2Н, обмениваемый, NH).