Патент на изобретение №2325658
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) СПОСОБ ОПРЕДЕЛЕНИЯ ЙОДИД-ИОНА В МОЧЕ
(57) Реферат:
Изобретение относится к электрохимическим, в частности, к ионометрическим методам анализа, и предназначено для определения йодид-иона в моче. Сущность способа: аликвоту пробы мочи смешивают с буферным раствором с рН 6,8-7,5, содержащим от 4,2 до 8 мас.% пероксида водорода, в соотношении 1:1 или 1:2, после чего выдерживают в темноте при комнатной температуре в течение не менее одних суток. Затем проводят измерение потенциала йодидселективного электрода непосредственно в растворе законсервированной пробы без отделения мешающих компонентов. Использование способа позволяет повысить точность определения йодид-ионов в моче, при высокопроизводительности и мобильности способа. Способ может быть использован в диагностике, лечении и профилактике заболеваний человека, связанных с дефицитом и метаболизмом йода для проведения широкомасштабных скрининговых и мониторинговых обследований населения. 2 ил., 2 табл.
(56) (продолжение): Изобретение относится к электрохимическим, в частности к ионометрическим методам анализа, и предназначено для определения йодид-иона в моче с помощью ионосслективных электродов. Изобретение может быть использовано в биохимических, клинических и санитарно-гигиенических исследованиях, в мероприятиях по диагностике, лечению и профилактике заболеваний человека, связанных с дефицитом и метаболизмом йода. Известны способы определения йода в моче. Кинетические способы [1, 2] основаны на каталитическом действии йода на реакцию восстановления церия (IV) мышьяком (III). Скорость уменьшения окраски раствора церия (IV) зависит от концентрации йода и измеряется спектрофотометрически. Подготовка к измерению заключается либо в обработке пробы окислительной смесью с NaClO3 или с (NH4)2S2O8 при повышенной температуре [1], либо в стабилизации пробы путем выдерживания се в течение 2 недель в смеси с раствором тимола в изопропаноле и последующего центрифугирования [2]. Процедуры определения в способах [1, 2] сложны и трудоемки, требуют применения токсичных и малодоступных реактивов. Многостадийность пробоподготовки снижает надежность определения. В ряде случаев кинетические способы вообще не применимы, например, к анализу мочи, содержащей глюкозу (у больных сахарным диабетом) или другие восстановители. Инверсионно-вольтамперометрический способ [3] основан на разбавлении пробы мочи в 100 раз, ультрафиолетовом облучении ее в присутствии пероксида водорода и нитрата калия и последующем измерении концентрации йода методом инверсионной вольтамперометрии на ртутно-пленочном электроде с серебряной подложкой. На электроде вначале осаждают йодид-ион в виде Hg2I2, затем проводят восстановление осадка при линейном изменении потенциала. Для реализации способа [3] необходима малодоступная дорогостоящая аппаратура – вольтамперометрический анализатор с встроенной УФ-лампой. Измерение аналитического сигнала отличается сложностью и методическими тонкостями, требует высокой квалификации оператора. Наиболее простыми и доступными для практического применения являются ионометрические методы, основанные на измерении мембранно-концентрационного потенциала йодидселективного электрода. Нижний предел ионометрического определения йода выше, чем с помощью кинетических и вольтамперометрических способов. Однако это не следует считать недостатком, так как даже тяжелый дефицит йода характеризуется содержанием его в моче на уровне 20 мкг/л, надежно определяемым ионометрически. В известных ионометрических способах [4, 5] концентрацию йодид-иона измеряют и рассчитывают по методу двойной стандартной добавки. Практика показывает, что результаты определения с использованием этого приема недостаточно надежны. Правильность определения в значительной степени зависит от выбора концентрации и объема добавок и от связанного с ним изменения величины потенциала электрода ( По технической сущности предлагаемый способ наиболее близок к ионометрическому способу [6], в котором пробу предварительно консервируют добавлением азида натрия, затем отделяют сопутствующие галогенид-ионы с помощью ионообменной хроматографии в вакууме (40-50 мм рт.ст.) и в относительно «чистом» элюате выполняют прямые потенциометрические измерения с йодидселективным электродом. Способ [6] отличается трудоемкостью и многостадийностью не только этапа подготовки пробы, но и построения градировочного графика. Для реализации способа необходимо оборудование для хроматографирования в вакууме, а также специально обработанная анионообменная смола. Целью изобретения является упрощение процедуры анализа, повышение точности определения, а также создание высокопроизводительного и мобильного способа определения йодид-иона в моче. Предлагаемый способ включает следующие операции. Аликвоту пробы мочи смешивают в соотношении 1:1 или 1:2 с буферным раствором, содержащим пероксид водорода, выдерживают в темноте при комнатной температуре (18-25°С) в течение не менее одних суток и измеряют эдс раствора относительно хлоридсеребряного электрода сравнения. Концентрацию йодид-иона находят по градуировочному графику. Градуировочные растворы готовят смешиванием стандартных растворов йодида калия с буферным раствором в соотношении 1:1 или 1:2. Необходимость выдерживания проб мочи в присутствии буферного раствора до проведения измерений в течение не менее одних суток установлена экспериментально (фиг.1). Измерения выполнены через 3 мин после погружения электродной пары в раствор. При этом в растворах, выдержанных сутки и более, потенциал устанавливается равновесным, а в остальных – дрейфует в сторону увеличения. Существенно важно то, что равновесный потенциал и соответствующая ему концентрация йодид-иона сохраняют постоянное значение в течение длительного времени, не менее 20 суток. Через сутки выдерживания изменяется внешний вид раствора пробы: он становится практически бесцветным, прозрачным и лишенным характерного для мочи запаха. Эти показатели сохраняются и через 20 суток. Таким образом, в предлагаемом способе измеряется устойчивый равновесный потенциал йодидселективного электрода и автоматически решается задача консервации пробы, сохранения ее бактериальной стабильности в течение длительного времени. Предлагаемый способ основан на том, что ионометрическое определение йодид-иона в моче проводят в экспериментально установленных условиях, при которых устраняется мешающее влияние как органических, так и неорганических компонентов пробы на мембрану электрода и стабилизируется определяемая ионная форма йода. К рядовой пробе мочи добавляли буферный раствор с разной концентрацией пероксида водорода, таким путем одновременно варьируя величину рН в измеряемом растворе, и через сутки проводили измерения. Результаты представлены на фиг.2. Как видно, на фиг.2 имеется плато, где совпадают обе зависимости и потенциал сохраняется постоянным. Плато ограничено интервалом значений рН 6,8÷7,5 и интервалом концентраций пероксида водорода – 4,2÷8 мас.%. Подобные зависимости получены и при анализе других проб мочи с использованием различных образцов йодидселективных электродов. За границами плато при уменьшении рН (достигается увеличением концентрации Н2O2) возможно частичное окисление йодида вследствие возрастания окислительного потенциала буфера, что приводит к увеличению потенциала электрода, а при увеличении рН (достигается уменьшением концентрации Н2O2) возможно неполное устранение мешающих компонентов, например, ионов серы, в результате чего потенциал убывает. На функцию йодидселективного электрода оказывают существенное мешающее влияние (в сторону уменьшения потенциала) ионы двух- и четырехвалентной серы, а также значительные количества бромид- и хлорид-ионов. Установлено, что в буферном растворе с рН 6,8-7,5, содержащем от 4,2 до 8 мас.% Н2O2, окисляются и переходят в электродно-неактивную форму до 1 г/л сульфид-, 15 г/л сульфит- и 13 г/л тиосульфат-ионов без удаления их из анализируемого раствора пробы. Мешающее влияние хлорида натрия – основного неорганического макрокомпонента мочи (его содержание в среднем составляет 7-10 г/л) – изучено экспериментально. На фоне выбранного буферного раствора по методу изопотенциальной точки определен коэффициент селективности йодидного электрода к хлорид-иону. Найдено, что КI-Cl=3,5·10-7. Следовательно, избыток хлорид-иона по отношению к йодиду не должен превышать величины 2,9·106. При нижнем пределе определения йодида 20 мкг/л реальный избыток хлорида составляет не более 1·106, то есть в три раза ниже допустимого значения, что указывает на селективность отклика в присутствии избытка хлорид-иона. Бромид-ион присутствует в моче в незначительном количестве по сравнению с Cl–-ионом и заведомо не влияет на мембрану йодидного электрода. Установлено, что другой макрокомпонент мочи – мочевина на фоне буферного раствора с пероксидом водорода также не мешает определению йодида. Градуировочные графики в отсутствие и в присутствии 25 г/л мочевины (ее среднее содержание в моче составляет 15-25 г/л) совпадают. В предлагаемом способе устраняется мешающее влияние не только мочевины, но и всех других органических компонентов, включая и бактериальные. Для доказательства этого брали две аликвоты пробы: одну анализировали предлагаемым способом, а вторую – озоляли, как рекомендовано в [7], в щелочной среде при 480°С до получения зольного остатка белого цвета, выщелачивали и затем определяли концентрацию йодид-иона с помощью йодидселективного электрода. К части проб перед введением щелочи добавляли точное количество стандартного раствора КI. Результаты представлены в табл.1.
Расчеты, произведенные по данным табл.1, показывают, что при сжигании (удалении) органической матрицы, содержащей также и глюкозу, относительное среднеквадратичное отклонение концентрации добавки от среднего значения (Сср.=31 мкг/л) составляет 18,2%, а систематическая составляющая погрешности (dr=-4%отн) с вероятностью Р=0,95 не значима по критерию Стьюдента (tэкс=0,76 Следовательно, при озолении йод не теряется, результаты определения правильны, и поэтому сравнивать между собой результаты параллельных определений в законсервированных и озоленных пробах вполне правомочно. Рассчитано, что среднеквадратическое отклонение средней концентрации йодид-иона, установленной предлагаемым способом (Сср.=115 мкг/л), от средней концентрации в этих же, но озоленных пробах (Сср.=116 мкг/л) составляет 8,9% отн. При этом систематическая составляющая погрешности (dr=0,95) не значима по критерию Стьюдента (tэкс.=0,95 Результаты статистической обработки данных свидетельствуют о правильности определения йодида предлагаемым способом и после озоления проб. Кроме того, полученная величина межметодической погрешности (8,9%) оказалась ниже, чем межсерийная ошибка, приведенная в прототипе (10,5-18%). Таким образом, в буферном растворе с рН 6,8-7,5, содержащем 4,2-8 мас.% Н2O2, происходит консервирование пробы мочи на длительное время и устраняется мешающее влияние неорганических и всех органических компонентов без удаления их из анализируемого раствора пробы. Пример конкретной реализации способа. В пластмассовый сосуд для измерений вместимостью 30-50 мл помещают 10 мл мочи, добавляют 10 мл буферного раствора с рН 7,1, содержащего 7,5 мас.% Н2O2. Сосуд закрывают крышкой и выдерживают в темноте при комнатной температуре в течение одних суток. Затем в сосуд помещают измерительный и вспомогательный электроды и при перемешивании регистрируют установившееся значение потенциала. Концентрацию йодид-иона находят по градуировочному графику. Оценка воспроизводимости и правильности определения йодид-иона предлагаемым способом представлена в табл.2. Воспроизводимость оценивали по результатам 4 параллельных измерений в 12 пробах. Измерения выполнены в каждый из четырех последующих после консервации дней. Правильность оценивали по методу «введено-найдено», для чего к законсервированной пробе добавляли точное количество стандартного раствора КI.
Как видно из табл.2, относительное среднеквадратическое отклонение (sr) результатов определения предлагаемым способом от среднего значения (Сср.=167 мкг/л) не превышает 6,5%, что в 1,6-2,8 раза ниже аналогично полученной межсерийной погрешности (10,5-18%) в прототипе. Если рассчитать стандартное отклонение концентрации йодид-иона, найденной в добавке, от введенной концентрации, оно составит 14,5% отн., что примерно вдвое выше, чем для самой пробы, однако оно также находится в пределах межсерийной погрешности прототипа. Важно, что систематическая составляющая стандартного отклонения для добавки (dr=-0,3%) не значима по критерию Стьюдента (tэкс.=0,07 Следовательно, результаты определения концентрации йодид-иона предлагаемым способом являются правильными и более точными, чем в прототипе. Таким образом, предлагаемый способ определения йодид-иона в моче отличается высокой селективностью и точностью, позволяет проводить анализ в присутствии органических и неорганических компонентов матрицы. Отсутствие необходимости отделения мешающих компонентов существенно упрощает процедуру анализа и увеличивает его производительность. Время, необходимое для консервации проб, является свободным и может быть использовано для подготовительной и любой другой работы. С использованием одного комплекта оборудования возможно выполнение анализа в 100-120 законсервированных пробах в течение 6 часов. Низкая стоимость и долговечность, удобство и простота в эксплуатации оборудования, необходимого для анализа, делают предлагаемый способ мобильным, позволяют использовать его как в стационарных, так и в полевых условиях, а возможность длительного хранения законсервированных проб – удобным для проведения широкомасштабных скрининговых и мониторинговых обследований населения. Источники информации Формула изобретения
Способ определения йодид-иона в моче, включающий предварительное консервирование пробы мочи в темноте при комнатной температуре и прямое измерение концентрации с помощью йодидселективного электрода, отличающийся тем, что аликвоту пробы мочи смешивают с буферным раствором с рН 6,8-7,5, содержащим от 4,2 до 8 мас.% пероксида водорода, в соотношении 1:1 или 1:2, выдерживают в течение не менее одних суток, затем проводят измерение электродного потенциала непосредственно в растворе законсервированной пробы без отделения мешающих компонентов.
РИСУНКИ
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
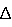 Е, мВ). Эти показатели в свою очередь существенно зависят от концентрации определяемого иона и состава пробы, однако они в способах [4, 5] не регламентированы.
Е, мВ). Эти показатели в свою очередь существенно зависят от концентрации определяемого иона и состава пробы, однако они в способах [4, 5] не регламентированы.
