Патент на изобретение №2323208
|
||||||||||||||||||||||||||||||||||||
(54) 4-ЦИАНФЕНИЛОВЫЙ ЭФИР 4[4
(57) Реферат:
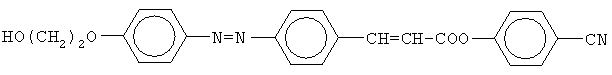
Изобретение относится к новому соединению – 4-цианфениловому эфиру 4-[4′-(2-гидроксиэтилокси)фенилазо]коричной кислоты, применяемому в качестве жидкокристаллической стационарной фазы для газовой хроматографии. Данное соединение обладает высокой структурной селективностью по отношению к позиционным изомерам ксилола. 1 табл.
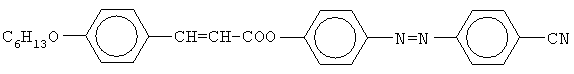
Изобретение относится к химической промышленности, а именно к получению жидких кристаллов, в частности к 4-цианфениловому эфиру 4-[4′-(2-гидроксиэтилокси)фенилазо]-коричной кислоты, который может быть использован в качестве жидкокристаллической стационарной фазы для газовой хроматографии. Уровень техники
Это вещество обладает широким температурным интервалом жидкокристаллического состояния (от 134 до 300°С), способностью к разделению позиционных изомеров и может быть использовано в качестве жидкокристаллической стационарной фазы в газовой хроматографии для анализа смесей органических соединений. Основным недостатком 4-гексилоксициннамоилокси-4′-цианазобензола является его низкая структурная селективность (коэффициент селективности по отношению к изомерам ксилола Сущность изобретения Изобретательской задачей является поиск нового химического соединения класса цианфениловых эфиров азокоричных кислот, которое обладало бы жидкокристаллическими свойствами и более высокой структурной селективностью по отношению к позиционным изомерам ксилола. Поставленная задача решена соединением 4-цианфениловый эфир 4-[4′-(2-гидроксиэтилокси)фенилазо]коричной кислоты формулы:
Структура заявленного соединения доказана методами элементного анализа, спектроскопии ИК и ЯМР 1Н. В ИК спектре 4-цианфенилового эфира 4-[4′-(2-гидроксиэтилокси)фенилазо]коричной кислоты в таблетке с KBr наблюдаются полосы поглощения (приведены частоты в см-1) нитрильной группы – 2223, гидроксильной группы – 3431, связей С-Н алифатической цепи – 2853; 2923; связей С-Н ароматических колец 1127-1734. В спектре ЯМР 1H 4-цианфенилового эфира 4-[4′-(2-гидроксиэтилокси)фенилазо]коричной кислоты в хлороформе-D наблюдаются резонансные сигналы ароматических протонов (приведены химические сдвиги в м.д. относительно ГМДС) 7.03д (2Н); 7,31д (2Н); 7,70д (2Н); 7,74д (2Н); 7,82д (2Н); 7,94д (2Н); сигналы других групп 3,84т (2Н, O-СН2-), 4,31т (2Н, -СН2-O-), 5,1с (1Н, Н-O-), 6,75с и 6,55д (1Н, =СН-СОО-), 7,93д (1Н, -СН=). Заявленное соединение обладает более высокой структурной селективностью по отношению к структурным изомерам ксилола. Сведения, подтверждающие возможность воспроизведения изобретения Для синтеза 4-цианфенилового эфира 4-[4′-(2-гидроксиэтилокси)фенилазо]коричной кислоты используют следующие вещества: 1. 4-аминобензальдегид получен по методике [Синтезы органических препаратов, М.: Ин. лит-ра, 1953, сб.4, с.30]. 2. Нитрит натрия ГОСТ 19906-74. 3. Соляная кислота ГОСТ 857-95. 4. Гидроокись натрия ГОСТ 11078-78. 5. Фенол ГОСТ 23519-93. 6. Хлорид натрия ГОСТ 13830. 7. Этиленхлоргидрин ТУ 6-01-05757587-58-94. 8. Поташ ГОСТ 4221-76. 9. Диметилформамид ТУ 6-09-3720-79. 10. Хлороформ ТУ 2631-020-11291058-96. 11. Тионилхлорид ТУ 6-01-4689387-41-90. 12. Пиридин ГОСТ 13647. 13. Малоновая кислота ТУ 6-09-2608-77. 14. Серный эфир ГОСТ 6255-52. 15. Муравьиная кислота ГОСТ 5848-73. 16. 4-Оксибензальдегид ТУ 18-16-144-82. 17. 4-гидроксибензонитрил получен по методике [Авторское свид. НРБ №26591, заявл. 31.03.78 №39249, опубл. 26.05.79]. Заявленное соединение получают следующим образом. Стадия 1. 18,3 г (0,2 моль) 4-аминобензальдегида суспендируют при температуре 0°C в 100 мл воды. Отдельно растворяют при той же температуре 9,9 г (0,2 моль) нитрита натрия в 50 мл воды. При интенсивном перемешивании к суспензии 4-аминобензальдегида приливают одновременно раствор нитрита натрия и 50 мл 24%-ного раствора (0,33 моль) соляной кислоты, соблюдая равенство расходов последних (рН смеси 7). Конец реакции диазотирования контролируют по йодкрахмальной бумаге. Раствор диазосоставляющей доводят до рН=7 добавлением 4%-ного раствора NaOH. К нейтральному раствору диазосоставляющей при перемешивании и охлаждении приливают охлажденный до 0°C раствор 14,2 г (0,15 моль) фенола в 50 мл 6%-ного (0,15 моль) раствора щелочи. Конец азосочетания контролируют капельной реакцией с фенолятом натрия. Продукт для полноты выделения высаждают добавлением 60 г NaCl в реакционную смесь, отделяют фильтрованием и перекристаллизовывают из 80%-ной уксусной кислоты. Получают ярко-оранжевые кристаллы 4-гидрокси-4′-формилазобензола, т.пл. 185°C. Выход 29,9 г (87%). Стадия 2. 2,3 г (0,01 моль) 4-гидрокси-4′-формилазобензола, 0,81 г (0,01 моль) этиленхлоргидрина и 1,66 г (0,012 моль) поташа в 150 мл диметилформамида кипятят при интенсивном перемешивании в течение 4 ч. Горячую реакционную смесь выливают в 400 мл ледяной воды, отфильтровывают и высушивают осадок. Затем пропускают через колонку с Al2О3, перекристаллизовывают из этанола. Получают оранжевые кристаллы 4-(2-гидроксиэтилокси)-4′-формилазобензола, т.пл. 131,7°C, т.пр. 154,0°C. Выход 2,5 г (92%). Стадия 3. 2,7 г (0,01 моль) 4-(2-гидроксиэтилокси)-4′-формилазобензола и 1,74 г (0,015 моль) малоновой кислоты кипятят в 50 мл сухого пиридина в течение 4 ч. Реакционную смесь выливают в ледяную воду, осадок отфильтровывают, промывают водой, перекристаллизовывают из 80%-й уксусной кислоты. Получают ярко-оранжевые кристаллы 4-[4′-(2-гидроксиэтилокси)фенилазо]коричной кислоты, т.пл.=240°С. Выход 2,12 г (68%). Стадия 4. 3,12 г (0,01 моль) 4-[4′-(2-гидроксиэтилокси)фенилазо]коричной кислоты, 2 мл тионилхлорида и 3 капли пиридина нагревают при 100°С в течение 2,5 ч. Затем из реакционной массы под вакуумом отгоняют непрореагировавший тионилхлорид. Этот свежеприготовленный хлорангидрид 4-[(4′-2-гидроксиэтилокси)фенилазо]коричной кислоты растворяют в 50 мл серного эфира и добавляют раствор 1,19 г (0,01 моль) 4-гидроксибензонитрила в 70 мл пиридина и кипятят 3,5 ч. Раствор выливают в ледяную воду, подкисленную HCl. Осадок экстрагируют хлороформом, хроматографируют на оксиде алюминия и перекристаллизовывают из этилацетата и этанола. Получают целевой продукт 4-цианфениловый эфир 4-[4′-(2-гидроксиэтилоксифенилазо]коричной кислоты. Выход 2,14 г (52%). Т.пл.=, т.пр.=. Найдено (%): С 65,74; Н 4,49; N 8,93. Вычислено (%): С 69,73; Н 4,6; N10,17. Анализ полученного соединения методом поляризационной микроскопии (микроскоп «Полам 211» с термостоликом) свидетельствует об образовании нематической фазы в интервале температур 138-306°С. Использование заявленного соединения в качестве стационарной фазы в газовой хроматографии для разделения позиционных изомеров ксилола иллюстрируется следующими примерами. Пример 1. Навеску жидкого кристалла (4-цианфенилового эфира 4-[4′-(2-гидроксиэтилокси)фенилазо]коричной кислоты или 4-гексилоксициннамоилокси-4′-цианазобензола в качестве прототипа) в количестве 0,36 г растворяют в 30 мл этилового эфира марки ХЧ. Полученный раствор добавляют к 3,6 г твердого носителя марки Chromaton N-AW (0.40-0.63 Chemapol, Чехия) и нагревают на водяной бане при перемешивании до полного испарения растворителя. Для удаления следов этилового эфира проводят сушку в течение 12 часов в вакууме при 100°С и остаточном давлении 2 мм рт.ст. Далее насадку (твердый носитель с нанесенной на него стационарной фазой) помещают в колонку из нержавеющей стали (1000×3 мм) и кондиционируют 6 ч в потоке гелия при 100°С. Количество неподвижной фазы составляет 10% от массы насадки. Неизменность состава неподвижной жидкой фазы в колонке контролируют взвешиванием колонки перед каждой серией опытов. Времена удерживания сорбатов измеряют на газовом хроматографе Chrom-5 (Чехия) с пламенно-ионизационным детектором при чувствительности, обеспечивающей регистрацию ионизационного тока 3.2·10-10 А. Измерения проводят в изотермическом режиме в интервале температур 120-200°С. Точность термостатирования 0.1°C. Температуры испарителя и детектора устанавливают на 20°C выше температуры колонки. В качестве газа-носителя используют гелий с содержанием основного вещества 99.99%. Расход гелия поддерживают в пределах 30-35 мл/мин, измеряя его пенным расходомером. Замеры расхода выполняют при каждой температуре опыта по окончании определения времени удерживания сорбата. Давление на выходе, равное атмосферному, определяют барометром БР-52 с ценой деления 0.5 мм рт.ст. Для того чтобы условия эксперимента соответствовали предельному разбавлению, а концентрация сорбата – линейному участку изотермы растворения, в колонку вводят малые – не более 0.1 мкл – объемы сорбатов. Применяют шприц объемом 1 мкл (Hamilton, Швейцария). «Мертвое» время удерживания определяют по метану. Времена удерживания регистрируют интегратором ИТ-2 с погрешностью не более 0.01 с. Это позволяет измерять времена удерживания соединений в пяти параллельных опытах с отклонением от среднестатистического значения не более 0.5%. Коэффициент селективности по Херингтону определяют как частное от деления времени удерживания пара-ксилола на время удерживания мета-ксилола с учетом мертвого времени удерживания. Рассчитывают средний коэффициен селективности из пяти измерений. В таблице приведены результаты испытаний жидкокристаллических стационарных фаз на основе 4-цианфенилового эфира 4-[4′-(2-гидроксиэтилокси)фенилазо]коричной кислоты и 4-гексилоксициннамоилокси-4′-цианазобензола. Данные таблицы с очевидностью подтверждают, что заявленное соединение проявляет более высокую структурную селективность по отношению к позиционным изомерам ксилола, что делает возможным его использование в качестве стационарной фазы для газовой хроматографии в процессах количественного анализа смесей органических соединений.
Формула изобретения
4-цианфениловый эфир4-[4′-(2-гидроксиэтилокси)фенилазо]коричной кислоты, проявляющий свойства жидкокристаллической стационарной фазы для газовой хроматографии.
|
||||||||||||||||||||||||||||||||||||
 (2-ГИДРОКСИЭТИЛОКСИ)ФЕНИЛАЗО]КОРИЧНОЙ КИСЛОТЫ, ПРОЯВЛЯЮЩИЙ СВОЙСТВА ЖИДКОКРИСТАЛЛИЧЕСКОЙ СТАЦИОНАРНОЙ ФАЗЫ ДЛЯ ГАЗОВОЙ ХРОМАТОГРАФИИ
(2-ГИДРОКСИЭТИЛОКСИ)ФЕНИЛАЗО]КОРИЧНОЙ КИСЛОТЫ, ПРОЯВЛЯЮЩИЙ СВОЙСТВА ЖИДКОКРИСТАЛЛИЧЕСКОЙ СТАЦИОНАРНОЙ ФАЗЫ ДЛЯ ГАЗОВОЙ ХРОМАТОГРАФИИ м/п=1,13, позволяющий фиксировать отдельные пики изомеров на хроматограмме смеси. Однако недостатком 4-метокси-4′-этоксиазоксибензола является узкий температурный диапазон жидкокристаллического состояния (от 91,5 до 150°С), что ограничивает его применение в хроматографическом анализе.
м/п=1,13, позволяющий фиксировать отдельные пики изомеров на хроматограмме смеси. Однако недостатком 4-метокси-4′-этоксиазоксибензола является узкий температурный диапазон жидкокристаллического состояния (от 91,5 до 150°С), что ограничивает его применение в хроматографическом анализе.