Патент на изобретение №2160277
|
||||||||||||||||||||||||||
(54) АНСА-ЦИРКОНОЦЕНЫ, ФУНКЦИОНАЛИЗИРОВАННЫЕ ПО ЦИКЛОСИЛАНОВОМУ МОСТИКУ, И СПОСОБ ИХ ПОЛУЧЕНИЯ
(57) Реферат: Изобретение относится к новым мостичным цирконоценам (ЦЦ), а именно к анса-цирконоценам с циклосилановым мостиком, функционализированным непосредственно по мостику, которые могут быть использованы как катализаторы в химической промышленности при производстве полиолефинов (ПО). Предложены новые анса-цирконоцены с циклосилановым мостиком общей формулы IV, где R1=H, CH3, C2H5; R2=H, алкил от C1 до C4, нормальный или разветвленный; арил; А= BC8H14; MR3, M – олово или кремний, R – алкил от C1 до C4, нормальный или разветвленный; арил; и способ их получения, включающий синтез замещенного индена, получение литиевой соли индена и его взаимодействие с 1,1-дихлор-2,5-дигидросилом в диэтиловом эфире с последующим взаимодействием полученной дилитиевой соли соответствующего бисинденильного лиганда с кремниевым мостиком с тетрахлоридом циркония для получения соответствующего цирконоцена и нагреванием его в тетрагидрофуране с алкил(арил)производным моногидрида бора, олова или кремния. Данный способ отличается высоким выходом промежуточных и целевого продукта и позволяет вдвое увеличить содержание активной рацемической формы в металлокомплексе. Полученные соединения обладают высокой каталитической активностью и стереоселективностью. 2 с. и 1 з. п. ф-лы, 2 табл. 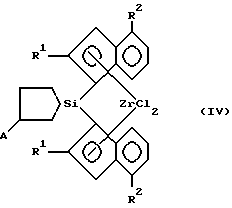
Изобретение относится к новым мостичным цирконоценам (ЦЦ), а именно к анса-цирконоценам с циклосилановым мостиком, функционализированным непосредственно по мостику, которые могут быть использованы как катализаторы в химической промышленности при производстве полиолефинов (ПО). Мостичные (анса-) ЦЦ, особенно ЦЦ с кремниевым мостиком, относятся к наиболее активным гомогенным металлоценовым катализаторам (МЦК) полимеризации олефинов, открытие которых произвело революцию в синтезе ПО благодаря их чрезвычайно высокой каталитической активности и стереоселективности. МЦК применяются совместно с полиметилалюмоксаном (МАО) в качестве сокатализатора (Sinclair K., Wilson R. Chemistry and Jndustry, 1994, N 7, p. 857). В настоящее время известно большое количество анса-металлоценов и цирконоценов. Наиболее близкими к предлагаемым анса-цирконоценам и способу их получения являются стерически затрудненные ЦЦ с кремниевым мостиком и способ их получения, описанные в работе Spaleck W. et al. Organometallics, 1994, 13, p. 954 – прототип, которые представляют собой бис-инденильные производные анса-цирконоценов C2 – симметрии с кремниевым мостиком формулы I, II или III. 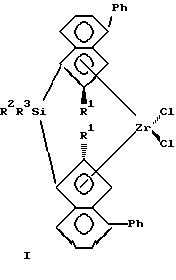 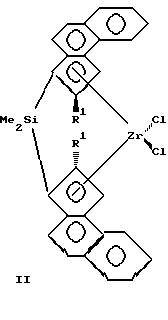 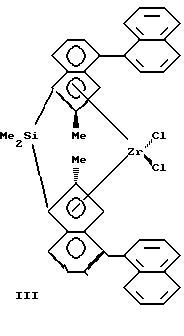 Ia R1=R2=R3=Me; Ib R1=R2=Me, R3=Ph; Ic R1=H, R2=R3=Me; IIa R=Me; IIb R=H. Получение известных соединений I-III представлено на примере 1 a-c схемой 1. Первой стадией синтеза является получение инденильных лигандов (4). Исходным соединением является 2-фенилбензилбромид (1), который конденсируют с малоновым или метилмалоновым эфиром в присутствии этилата натрия. Последующие реакции омыления эфира (в присутствии водной щелочи) и декарбоксилирования путем нагревания при 130oC дают соответствующую кислоту (2) с хорошим выходом (на неочищенную (2) 85% от теор.). Взаимодействием кислоты (2) с тионилхлоридом получают ее хлорангидрид, который подвергают циклизации в инданон (3) в присутствии AlCl3 в качестве катализатора Фриделя-Крафтса. Выход инданонов был удовлетворительный кроме нафтилинданона – промежуточного продукта при синтезе соединения III, так как более высокая реакционная способность нафтильного производного привела к побочным реакциям, и выход нафтилинданона был только 13%. Инданон (3) затем восстанавливают боргидридом натрия в смеси тетрагидрофурана (ТГФ) и метанола при 0oC, кипятят в присутствии p-толуолсульфокислоты и выделяют инден (4) методом препаративной ТСХ (тонкослойной хроматографии) на силикагеле с хорошим выходом. Следующей стадией является получение бисинденильных лигандов с кремниевым мостиком (5). Инден (4) растворяют в смеси сухого толуола и ТГФ в атмосфере аргона и прибавляют при комнатной температуре раствор н-бутиллития в гексане. Нагревают до 80oC, выдерживают при 80oC 1 час и охлаждают до 0oC, прибавляют дихлорид диметилсилана (а, с) или метилфенилсилана (b). Греют 1 час при 80oC и обрабатывают водой. Из органического слоя методом ТСХ выделяют соединение (5). Выход (5a) 70%, (5b) 44%, (5c) 62%. Заключительной стадией является получение цирконоценов 1 а-с. К раствору (5) в сухом толуоле в атмосфере аргона прибавляют при комнатной температуре раствор н-BuLi в гексане. Кипятят с обратным холодильником 3 часа, охлаждают до -25oC и прибавляют тетрахлорид циркония. Оставляют на 2 часа. Отфильтровывают циркониевый комплекс, представляющий собой смесь рацемической (активной) и мезо (неактивной) – форм (рац:мезо = 1:1). Перекристаллизовывают из метиленхлорида. Выход 1 а 33%, I b 10%, 1 с 15% В работе, выбранной за прототип, проанализированы более ранние результаты по исследованию влияния структуры металлоценов на их поведение в реакции полимеризации олефинов, и с целью изучения влияния ароматических заместителей в инденильном ядре мостичных ЦЦ были синтезированы соединения I-III, которые были испытаны с МАО в качестве сокатализатора (Al : Zr = 15000 : 1) в полимеризации пропилена и этилена. Полимеризацию пропилена в работе-прототипе проводили в среде жидкого мономера при 70oC в термостатируемом стальном реакторе. МАО применяли в виде 10% -ного раствора в толуоле, ЦЦ растворяли в таком же растворе МАО. Полученные результаты показали (см. таблицу 1), что введение объемного ароматического заместителя в 4-положение инденильного ядра существенно улучшает все показатели процесса полимеризации по сравнению с незамещенным в этом положении (2a*) или 4-алкилзамещенным (4a*) ЦЦ, описанными ранее в других работах. При проведении полимеризации этилена (в растворе) наивысшую каталитическую активность, как и в реакции с пропиленом, показали соединения Ia и III, они же показали и наивысший MW [2]. Следует подчеркнуть, что беспрецедентно высокие показатели процесса полимеризации, полученные с использованием соединений Ia и III, по сравнению с другими мостичными цирконоценами позволяют считать соединения Ia и III наилучшими из известных на сегодня в качестве катализаторов полимеризации олефинов совместно с МАО, а сам факт открытия такого влияния ароматического заместителя в 4-ом положении инденильного кольца крупным вкладом в химию МЦК. Однако ЦЦ I-III, как и другие известные МЦК, являются синтетически труднодоступными. Так, соединение III, проявляющее наивысшую каталитическую активность, является наиболее труднодоступным из ЦЦ I-III из-за низких выходов промежуточных продуктов при синтезе. Кроме того, известные ЦЦ I-III отличаются высоким содержанием неактивной в реакции полимеризации мезо-формы (соотношение рац- и мезо-изомеров 1:1), от которой желательно освобождать металлокомплекс, что весьма затруднительно и часто невозможно. Необходимо отметить еще один недостаток известных МЦК – все известные модификации структуры металлоценов осуществляются на самых ранних этапах их синтеза, что сильно тормозит исследования каталитической активности, так как нельзя заранее предвидеть ни выход на промежуточных стадиях, ни саму возможность получения как мостичного лиганда, так и конечного целевого продукта – металлокомплекса, а также будут ли полученные металлоцены растворимы в органических растворителях (толуоле), чтобы быть перспективными для получения гомогенных катализаторов. Задачей предлагаемого изобретения является создание новых анса-ЦЦ, обладающих высокой активностью и стереоселективностью в качестве катализатора полимеризации олефинов и имеющих принципиально новую структуру, а именно функционализированных непосредственно по кремниевому мостику, что позволит использовать их в качестве объекта исследований при изучении влияния функциональных заместителей в мостике, то есть в непосредственной близости от каталитического центра, а также разработка способа их получения с достаточно высоким выходом промежуточных и конечного продуктов с улучшенными методиками выделения промежуточных продуктов и с увеличенным содержанием активной рацемической формы в циркониевом комплексе. Решение поставленной задачи достигается: – предлагаемыми новыми анса-цирконоценами, функционализированными по циклосилановому мостику формулы IV: 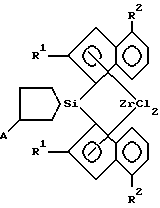 R1 = H, CH3, С2H5; R2 = H, алкил от С1 до С4, нормальный или разветвленный; арил; А = BC8H14; MR3, где М – олово или кремний, R = алкил от C1 до C4, нормальный или разветвленный; арил. – и способом их получения, в котором литиевую соль индена подвергают взаимодействию с 1,1 – дихлор-2,5- дигидросилолом в диэтиловом эфире с последующим взаимодействием полученной дилитиевой соли соответствующего бисинденильного лиганда с кремниевым мостиком с тетрахлоридом циркония для получения соответствующего цирконоцена и нагреванием его в тетрагидрофуране с алкил(арил) производным моногидрида бора, олова или кремния. Цирконоцен нагревают в тетрагидрофуране с алкил(арил) производными (моно)гидрида бора, олова или кремния. Получение дилитиевой соли бис-инденильного продукта с кремниевым мостиком можно осуществлять в виде кристаллического аддукта с диэтиловым эфиром, который затем подвергают взаимодействию с тетрахлоридом циркония. При разработке предлагаемого изобретения первоначально были созданы новые ЦЦ с непредельным циклосилановым мостиком (описанные в отдельной заявке, подаваемой одновременно с данной), что и позволило получить анса-ЦЦ, функционализированные по мостику. Введение функциональных заместителей, особенно объемных, в мостик ЦЦ, то есть в непосредственной близости от каталитического центра, представляет большой интерес для химии МЦК, так как создает совершенно новые пространственные эффекты в металлокомплексе, которые неизбежно должны влиять на стереоселективные свойства катализатора. Синтез предлагаемых ЦЦ IV описан в примерах и представлен на схеме 2. Полученные соединения IV (и промежуточные продукты) охарактеризованы данными элементного анализа и ЯМР – спектрами (1H, 13C). Соединения IV совместно с МАО в качестве сокатализатора были испытаны в реакции полимеризации пропилена. Процесс полимеризации проводили по методике, аналогичной описанной в работе-прототипе. Полученные результаты представлены в таблице 2. Примеры. I. Синтез замещенного индена (42). Синтез диэтилового эфира пропандикарбоновой кислоты CH3CH2CH(COOEt)2 (этилмалоновый эфир). К раствору этилата натрия, приготовленному из 16.2 г (0.705 г-атом) натрия и 350 мл абс. этанола добавили при перемешивании 53 мл (55.92 г, 0.349 моль) диэтилового эфира малоновой кислоты. К приготовленному таким образом раствору медленно при охлаждении на ледяной бане добавили 53 мл (77.38 г, 0.710 моль) этилбромида, дали смеси нагреться до комнатной температуры и перемешивали еще 30 минут. Этиловый спирт отогнали, добавили воду до полного растворения осадка, полученную смесь экстрагировали эфиром, объединенные эфирные вытяжки сушили сульфатом натрия, растворитель отогнали и оставшееся масло подвергли перегонке, что дало 52.33 г (80%) целевого продукта. Аналогично получали метилмалоновый эфир (вместо этилата натрия используют метилат натрия). Синтез 2-(гидроксиметил)дифенила 2-PhC6H4CH2OH (2-фенилбензиловый спирт). К суспензии 5.7 г (150.0 ммоль) литийалюминийгидрида в 50 мл кипящего тетрагидрофурана в течение 4 часов добавили при перемешивании раствор 12.5 г (63,1 ммоль) 2-фенилбензойной кислоты в 100 мл абсолютного тетрагидрофурана, кипятили реакционную смесь еще 3 часа и оставили на ночь. Реакционную смесь осторожно при интенсивном охлаждении обработали ледяной водой до окончания выделения водорода и далее серной кислотой (1:1) до полного растворения осадка. Полученный таким образом раствор трижды экстрагировали эфиром, объединенные эфирные вытяжки сушили сульфатом натрия, растворитель отогнали и оставшееся масло охладили до начала кристаллизации. Хроматографически чистый продукт. Т.пл. 29oC. Выход 9.54 г (82%). Аналогично получают 2-алкил и 2-нафтилбензиловый спирт (вместо-2-фенилбензойной кислоты используют 2-алкил или 2-нафтилбензойную кислоту). Синтез 2-(бромометил)дифенила 2-PhC6H4CH2Br (2-фенилбензилбромид) (12). Смесь 3.20 г (17.37 ммоль) 2-(гидроксиметил)дифенила, 5 мл конц. HBг и 1.2 мл конц. H2SO4 кипятили с обратным холодильником в течение 6 часов, охладили и вылили в 20 мл ледяной воды. Органическую фазу (нижний слой) отделили, промыли насыщенным раствором карбоната натрия; водную фазу нейтрализовали карбонатом натрия и трижды экстрагировали эфиром. Объединенную органическую фазу сушили сульфатом натрия, растворитель отогнали и оставшееся масло перегнали при уменьшенном давлении, собирая фракцию 160-165oC/7 мм рт. ст., что дало 3.64 г чистого продукта. Выход 85%. Осторожно! Сильный лакриматор! Аналогично получали 2-алкил- и 2-нафтилбензилбромид. Синтез 2-(2-дифенилметил)бутановой кислоты (Et)(2- PhC6H4CH2)CHCOOH (22). К раствору этилата натрия, приготовленному из 1.40 г (60.9 мг-атом) натрия и 47 мл абс. этанола добавили при комнатной температуре и перемешивании раствор 11.70 г (62.2 ммоль) диэтилового эфира этилмалоновой кислоты в 15 мл абс. этанола. К приготовленному таким образом раствору при слабом кипении добавили по каплям раствор 15.60 г (63.1 ммоль) 2-(бромометил)дифенила в 20 мл абс. этанола, реакционную смесь кипятили еще 3 часа. Реакционную смесь охладили, обработали при охлаждении раствором 9 г КОН в 25 мл воды, снова кипятили в течение 4 часов и охладили. Растворители отогнали в вакууме водоструйного насоса и остаток подкислили концентрированной соляной кислотой до pH 1. Выпавший белый осадок отфильтровали и нагревали на бане из сплава Вуда в течение 1 часа при 130oC. Бледно-желтое масло использовали в дальнейшем без дополнительной очистки. Синтез хлорангидрида 2-(2-дифенилметил)бутановой кислоты (Et)(2-PhC6H4CH2)CHC(O)Cl. К 7.20 г (28.2 ммоль) 2-(2- дифенилметил)бутановой кислоты добавили 20 мл свежеперегнанного тионилхлорида и полученный раствор кипятили с обратным холодильником в течение 1 часа. Избыток тионилхлорида отогнали; для полноты удаления тионилхлорида в смесь трижды вводили порции абс. толуола по 30 мл и отгоняли его в вакууме. Остаток растворили в 50 мл абс. толуола и использовали в дальнейшем без дополнительной обработки. Синтез 4-фенил-2-этилинданона-1 2-Et-4-Ph-C9H6O(32). Приготовленный как указано выше раствор хлорангидрида 2-(2- дифенилметил)бутановой кислоты в 50 мл абс. толуола в течение 1 часа добавили при перемешивании и охлаждении до 10oC к суспензии 5.70 г (42.7 ммоль) безводного хлорида алюминия в 50 мл абс.толуола. Смесь перемешивали при 80oC в течение 1 часа, вылили на 300 г колотого льда, подкислили конц. HCl до pH 1, органическую фазу отделили, а водную трижды экстрагировали эфиром. Объединенные органические фазы промыли водой, раствором соды и высушили над сульфатом натрия и удалили растворители в вакууме. Сырой продукт подвергли очистке методом препаративной колоночной хроматографии (силикагель, петролейный эфир – эфир 2:1). Светло-желтые кристаллы. Выход 4.65 г (70% в расчете на 2-(2- дифенилметил)бутановую кислоту). Аналогично получали 4-фенил-2- метилинданон, 4-фенилинданон, 4-алкил-2-алкилинданон и 4-нафтил-2- R1-инданон (R1 = H, CH3, C2H5). Синтез 4-фенил-2-этилиндена 2-Et-4-PhlndH (42). К раствору 7.10 г (30.0 ммоль) 4-фенил-2-этилинданона-1 в смеси 30 мл абс. тетрагидрофурана и 15 мл абс. метанола медленно при перемешивании и охлаждении до 0oC добавляли небольшими порциями 1.70 г (44.9 ммоль) измельченного борогидрида натрия. Смесь перемешивали еще 16 часов, вылили на 100 г колотого льда, подкислили конц. HCl до pH 1, трижды экстрагировали эфиром. Органические вытяжки промыли насыщенным раствором поваренной соли и сушили сульфатом магния. Растворители отогнали в вакууме и оставшееся масло растворили в 100 мл сухого толуола, содержащего 0.3 г моногидрата n-толуолсульфокислоты, и кипятили в течение 2 часов. Охлажденный раствор промыли насыщенным водным раствором бикарбоната натрия, высушили сульфатом натрия, растворитель удалили в вакууме и остаток очистили препаративной колоночной хроматографией на силикагеле, используя в качестве элюента смесь петролейный эфир – дихлорометан 9:1. Бесцветное масло. 3.70 г (56% в расчете на 4-фенил-2-этилинданон-1). ЯМР 1H (400 МГц, CDCl3, 25oC)  м.д.: 7.63-7.17 (м., 8H, ароматические протоны индена и фенильных групп); 6.59 (т., 4J = 1.6 Гц, 1H, олефиновый протон индена); 3.43 (с., 2H, аллильные протоны индена); 2.52 (уш.кв., 3J = 7.6 Гц, 2H, CH2 – протоны этильной группы); 1.24 (т., 3J = 7.6 Гц, 3H, CH3-протоны этильной группы); м.д.: 7.63-7.17 (м., 8H, ароматические протоны индена и фенильных групп); 6.59 (т., 4J = 1.6 Гц, 1H, олефиновый протон индена); 3.43 (с., 2H, аллильные протоны индена); 2.52 (уш.кв., 3J = 7.6 Гц, 2H, CH2 – протоны этильной группы); 1.24 (т., 3J = 7.6 Гц, 3H, CH3-протоны этильной группы);13С{ 1H} (100 МГц, CDCl3, 25) м.д.: 152.72, 146.24, 141.37, 140.56, 137.46 (четвертичные ароматические и олефиновый углероды индена и фенильной группы); 128.44, 128.35, 126.98, 125.22, 125.19, 124.28, 119.09 (третичные ароматические и олефиновый углероды индена и фенильной группы); 41.00 (аллильный углерод индена); 24.26 (метиленовый углерод этильной группы); 13.28 (метильный углерод этильной группы).
Аналогично получали другие замещенные индены (42) с R1 и R2, указанные на схеме 2.
II. Синтез ЦЦ с непредельным циклосилановым мостиком.
Синтез 4-фенил-2-этилиндениллития 2-Et-4-PhlndLi.
Синтез проводили в вакууммированной цельнопаянной аппаратуре типа сосудов Шленка.
К раствору 11.00 г (49.93 ммоль) 4-фенил-2-этилиндена (42) в 70 мл диэтилового эфира добавили при охлаждении до -20oC и интенсивном перемешивании 23.8 мл (49.98 ммоль) 2.1 М раствора н-бутиллития в легком петролейном эфире, дали смеси нагреться до комнатной температуры и оставили на ночь. Примерно половину общего объема растворителей отогнали, желтый объемный осадок отделили фильтрованием от интенсивно-красного маточного раствора, трижды промыли на фильтре пентаном (порции по 70 мл), высушили в высоком вакууме и полученное снежно-белое твердое вещество растворили в 100 мл диэтилового эфира. Прозрачный желтый раствор отделили от тонкого осадка двукратной декантацией, оттитровали и использовали в виде 0.35 М раствора. Получено 105 мл 0.35 М раствора, выход 73.5% от теории. Аналогично получали литиевые соли с другими замещенными инденами.
Синтез 1,1′-(бутен-2-диил-1,4)силилиденбис(4-фенил-2- этилиндена) (1,4-CH2CH=CHCH2)Si(2-Et-4-PhlndH)2(52).
К раствору 1.14 г (7.44 ммоль) 1,1-дихлоро-2,5-дигидросилола в 30 мл диэтилового эфира при охлаждении до -30 – -40oC и интенсивном перемешивании в течение 30 минут добавили 42.5 мл 0.35 М эфирного раствора (4-фенил-2-этилиндениллития) (14.86 ммоль), дали смеси постепенно нагреться до комнатной температуры и оставили на ночь. Раствор отфильтровали от выпавшего хлорида лития, растворитель отогнали, ввели 50 мл пентана и после повторного отфильтровывания и отгонки растворителя оставшееся слегка желтоватое стеклующееся масло высушили в высоком вакууме. Выход практически количественный. Полную очистку бидентатного лиганда (52) проводили в форме его дилитиевого производного.
Аналогично осуществляли взаимодействие 1,1-дихлоро-2,5- дигидросилола с Li-солями других замещенных инденов.
1,1′-(бутен-2-диил-1,4)силилиденбис(4-фенил-2- этилинденил)дилития аддукт с диэтиловым эфиром (1,4- CH2CH=CHCH2)Si(2-Et-4-Phlnd)2Li2 Et2O (add). К раствору полученного на предыдущей стадии лиганда (52) в смеси 10 мл диэтилового эфира и 40 мл пентана при охлаждении до -20oC и интенсивном перемешивании добавили 6.54 мл 2.16 М раствора н-бутиллития в гексане (14.12 ммоль) и дали смеси нагреться до комнатной температуры. Отделившееся первоначально желтое масло постепенно закристаллизовалось. Осадок отфильтровали, трижды промыли на фильтре той же смесью растворителей и полученное снежно-белое порошкообразное вещество высушили в высоком вакууме. Получено 3.19 г (5.26 ммоль) дилитиевой соли (add). Выход 70.7% в расчете на исходно взятый 4-фенил-2-этилиндениллитий.
NMR (THF-d8, 30oC) d м.д. 1H: 1.31 (т, CH2CH3); 2.18 (с, CH2Si); 3.04 (кв, CH2CH3); 6.03 (с, CH= CH); 6.32 (с, H3); 6.52 (м, H5, H6); 7.12 (т, пара-H); 7.30 (т, мета-H); 7.69 (м, H7), 7.79 (д, орто-H).
13С: 17.58 (CH2CH3); 22.63 (CH2Si); 25.89 (CH2CH3); 95.97 (С3); 96.54 (С1); 113.82, 114.14, 119.86 (С5, C6, C7); 125.02 (пара-C); 128.22 (мета-C); 129.24 (орто-C); 132.60 (CH= CH); 130.34, 131.16, 137.77, 145.22, 147.36 (С3a, С7a, 2, С4, ипсо-C).
Аналогично получали аддукты с диэтиловым эфиром дилитиевых солей других бидентатных лигандов с непредельным циклосилановым мостиком.
Синтез { Et2O (add). К раствору полученного на предыдущей стадии лиганда (52) в смеси 10 мл диэтилового эфира и 40 мл пентана при охлаждении до -20oC и интенсивном перемешивании добавили 6.54 мл 2.16 М раствора н-бутиллития в гексане (14.12 ммоль) и дали смеси нагреться до комнатной температуры. Отделившееся первоначально желтое масло постепенно закристаллизовалось. Осадок отфильтровали, трижды промыли на фильтре той же смесью растворителей и полученное снежно-белое порошкообразное вещество высушили в высоком вакууме. Получено 3.19 г (5.26 ммоль) дилитиевой соли (add). Выход 70.7% в расчете на исходно взятый 4-фенил-2-этилиндениллитий.
NMR (THF-d8, 30oC) d м.д. 1H: 1.31 (т, CH2CH3); 2.18 (с, CH2Si); 3.04 (кв, CH2CH3); 6.03 (с, CH= CH); 6.32 (с, H3); 6.52 (м, H5, H6); 7.12 (т, пара-H); 7.30 (т, мета-H); 7.69 (м, H7), 7.79 (д, орто-H).
13С: 17.58 (CH2CH3); 22.63 (CH2Si); 25.89 (CH2CH3); 95.97 (С3); 96.54 (С1); 113.82, 114.14, 119.86 (С5, C6, C7); 125.02 (пара-C); 128.22 (мета-C); 129.24 (орто-C); 132.60 (CH= CH); 130.34, 131.16, 137.77, 145.22, 147.36 (С3a, С7a, 2, С4, ипсо-C).
Аналогично получали аддукты с диэтиловым эфиром дилитиевых солей других бидентатных лигандов с непредельным циклосилановым мостиком.
Синтез {  5,5 5,5 -[1,1′-(Бутeн-2-диил-1,4)cилилиденбиc(4-фeнил-2- этилинденил)]}дихлороциркония (1,4-CH2CH=CHCH2)Si(2-Et-4-Phlnd)2ZrCl2, смесь рац- и мезо-форм (2:1).
Суспензию 3.19 г (5.26 ммоль) эфирата дилитиевой соли (add) и 1.24 г (5.26 ммоль) ZrCl4 в 100 мл толуола перемешивали в течение суток при комнатной температуре, ввели еще 500 мл толуола, оранжевый маточный раствор отделили от очень мелкодисперсного осадка декантациями (3 недели), толуол отогнали и остаток перекристаллизовали из минимального объема хлористого метилена, что дало 2.40 г (3.53 ммоль) целевого комплекса, представляющего собой смесь рац- и мезо-форм в соотношении 2:1. Выход 67,1%.
NMR 1H (CD2Cl2, 30oC) d м.д.: 1.11 (т, CH2CH3, рац); 1.21 (т, CH2CH3, мезо); 2.41 (м), 2.74 (м) (CH2CH3, рац); 2.76 (м, CH2CH3, мезо); 2.55 (ушир. с), 2.89 (ушир. с) (CH2Si, мезо); 2.61 (AB-система), 2.78 (AB-система, 2J(AB) = 18 Гц) (CH2Si, рац); 6.33 (с, HC=CH, рац); 6.33 (м), 6.36 (м) (HC= CH, мезо); 6.86 (с, H3, мезо); 6.97 (с, H3, рац); 6.93 (м), 7.10 – 7.65 (м) (H5, H6, H7, Ph).
C38H34Cl2Si1Zr1, М = 680,96. Найдено: Cl = 9,80%,. Вычислено: Cl = 10,43%.
Аналогично получали ЦЦ с непредельным циклосилановым мостиком с другими замещенными бисинденильными лигандами.
Синтез { 5,5-[1,1′-(Бутен-2-диил-1,4)силилиденбисинденил]} дихлороциркония (1,4-CH2CH=CHCH2)Silnd2ZrCl2, смесь рац- и мезо-форм (1:1).
Синтез индениллития IndLi.
К раствору 11.62 г (100.00 ммоль) индена производства фирмы “Aldrich” в 50 мл диэтилового эфира добавили при охлаждении до -20oC и интенсивном перемешивании 53.2 мл (100.01 ммоль) 1.88 М раствора н-бутиллития в легком петролейном эфире, дали смеси нагреться до комнатной температуры и оставили на ночь. Примерно две трети общего объема растворителей отогнали, выпавший белый кристаллический осадок отделили фильтрованием от интенсивно-красного маточного раствора, трижды промыли на фильтре пентаном (порции по 70 мл), высушили в высоком вакууме. Снежно-белое кристаллическое вещество. Выход 10.90 г (89.27 ммоль, 89.3% от теории).
Синтез 1,1′-(бутен-2-диил-1,4)силилиденбисиндена (1,4- CH2CH= CHCH2)Si(IndH)2 (52), R1=R2=H.
К раствору 2.86 г (37.51 ммоль) 1,1-дихлоро-2,5-дигидросилола в 30 мл диэтилового эфира при охлаждении до -30 – -40oC и интенсивном перемешивании в течение 30 минут добавили раствор 4.58 г (37.51 ммоль) индениллития в диэтиловом эфире, дали смеси постепенно нагреться до комнатной температуры и оставили на ночь. Раствор отфильтровали от выпавшего хлорида лития, растворитель отогнали, ввели 50 мл пентана и после повторного отфильтровывания и отгонки растворителя оставшееся слегка желтоватое масло высушили в высоком вакууме. Выход практически количественный. Полную очистку лиганда (52) (R1 = R2 = Н) проводили в форме его дилитиевого производного.
Синтез [1,1′-(бутен-2-диил- 1,4)силилиденбисинденил] дилития (аддукт с диэтиловым эфиром) (1,4- CH2CH=CHCH2)Silnd2Li2 Et2O (add, R1=R2=H).
К раствору 5.14 г (16.45 ммоль) полученного на предыдущей стадии лиганда (52) в 50 мл диэтилового эфира при охлаждении до -20oC и интенсивном перемешивании добавили 15.2 мл 2.16 М раствора н-бутиллития в гексане (16.45 ммоль), дали смеси нагреться до комнатной температуры и оставили на ночь. Выпавший белый осадок отфильтровали, трижды промыли на фильтре той же смесью растворителей и полученное снежно-белое порошкообразное вещество высушили в высоком вакууме. Получено 4.91 г (12.32 ммоль) дилитиевой соли (add). Выход 74.9% в расчете на исходно взятый индениллитий.
NMR (THF-d8, 30oC) d м.д. 1H: 1.91 (с, CH2Si); 6.01 (с, CH=CH); 6.04 (д, 3J(H2-H3) = 3.2 Гц, H2); 6.47 (м, H5, H6); 6.88 (д, H3); 7.32 (м, H7); 7.64 (м, H4).
13С: 20.50 (CH2Si); 95.14 (С3); 98.26 (С1); 114.33, 114.63, 119.40, 121.67, 125.99 (C2, C4, С5, С6, C7); 132.78 (CH=CH); 133.89, 136.28 (C3a, C7a).
Синтез { 5,5 -[1,1′-(Бутен-2-диил- 1,4)силилиденбисинденил]}дихлороциркония (1,4-CH2CH=CHCH2)SiInd2ZrCl2, смесь рац- и мезо-форм (1:1).
Суспензию 1.34 г (3.36 ммоль) эфирата дилитиевой соли (add) и 0.78 г (3.36 ммоль) ZrCl4 в 70 мл толуола перемешивали в течение суток, сухой остаток многократно экстрагировали толуолом, толуольный экстракт сконцентрировали, кристаллический желтый осадок отфильтровали от маточника, промыли толуолом, пентаном и высушили в высоком вакууме, что дало 1.02 г (2.17 ммоль) металлокомплекса, представляющего собой смесь рац- и мезо-форм в соотношении 1:1. Выход 64.5%.
NMR (CD2Cl2, 30oC) d м.д.:1H (CD2Cl2, 30oC) 2.29 (ушир. с), 2.64 (ушир. с) (CH2Si, мезо) 2.37 (AB-система), 2.56 (AB-система, 2J(AB) = 17 Гц) (CH2Si, рац); 6.11 (д, 3J(H2-H3) = 3.5 Гц, H2, рац); 6.17 (д, 3J(H2-H3) = 3.5 Гц, H2, мезо); 6.29 (с, CH=CH, рац); 6.29 (м), 6.30 (м) (CH=CH, мезо); 6.90 (д, H3, рац); 6.95 (д, H3, мезо); 6.93 (м), 7.22 (м) (H5, H6, мезо); 7.10 (м), 7.39 (м) (H5, H6 рац); 7.51 (м), 7.60 (м) (H4, H7, рац, мезо).
13C: 16.45, 16.86 (CH2Si, мезо); 16.99 (CH2Si, рац); 87.94 (С1, рац); 89.08 (С1, мезо); 117.95, 118.14, 124.38, 126.34, 127.18, 128.00 (C2, C3, C4, C5, C6, С7, рац); 119.62, 119.85, 125.75, 126.06, 126.34, 127.54 (C2, C3, C4, C5, C6, С7, мезо); 130.65, 130.88 (CH=CH, мезо); 130.70 (CH=CH, рац); 125.89, 133.79 (С3a, С7a, рац); 128.13, 134.93 (С3a, С7a, мезо). C22H18Cl2Si1Zr1, М = 472,67. Найдено: Cl = 14,38%. Вычислено: Cl = 15,02%.
III. Синтез функционализированных по кремниевому мостику ЦЦ формулы IV.
Синтез борированного анса-цирконоцена с незамещенными бисинденильными лигандами IVa ( 5:5-1,1′ (3-(9- борабициклононанил-9) силоландиил-1,1)бисинденил]дихлороциркония).
Раствор 125 мг (264,5 ммоль) анса-цирконоцена -1,1′ [(Бут-2-диил-1,4)силилиден] -бисинденил-дихлороциркония (смесь рац- и мезо-формы) и 9-борабицикло [3, 3, 1] нонана (32 мг, 132,3 ммоль) в ТГФ смешали при комнатной температуре, нагрели при 70 – 80oC в запаянном сосуде, охладили, растворитель удалили, а остаток высушили в вакууме. Получено кристаллическое вещество желто-оранжевого цвета. Выход количественный. C30H33B1Cl2Si1Zr1. М = 594,69.
Найдено: Zr = 15,88; Cl = 11,38. Вычислено: Zr = 15,34; Cl = 11,92.
Синтез станнилированного анса-цирконоцена с незамещенными бисинденильными лигандами IV b ([ 5:5-1,1′(3-(триметилстанил) силоландиил-1,1) бисинденил] дихлороциркония).
Смесь 125 мг (264,5 ммоль) анса-цирконоцена 7 (смесь рац- и мезо-форм) и 43 мг (264,5 ммоль) триметилстаннана в 30 мл ТГФ нагревали при 70 – 80oC в запаянном сосуде в течение 5 часов. Смесь охладили и растворитель удалили в вакууме. Полученное желто-оранжевое масло промыли пентаном (2 раза по 30 мл). Полученный сухой остаток растворили в эфире, профильтровали раствор и удалили эфир. Получено 155 мг (92% от теор.) комплекса IVb. C25H28Cl2Si1Sn1Zr1, M = 637,49.
Найдено: Zr = 14,90; Cl = 10,85. Вычислено: Zr = 14,31; Cl = 11,12.
Синтез борированного анса-цирконоцена с замещенными бисинденильными лигандами IVc ([ 5:5-1,1′ (3-(9-борабициклононанил-9) силоландиил-1,1) бис-4-фенил-2-этилинденил]дихлороциркония).
Синтез комплекса IVс проведен по методике синтеза цирконоцена IVа. Основное отличие и основная трудность заключаются в том, что продукт образуется в виде плохо кристаллизующегося масла. Твердый продукт получается в результате многократного промывания продуктов пентаном при -20 (-30)oC. Выход IVc 85% (от теоретического). C46H49B1Cl2Si1Zr1, M = 802,98.
Найдено: Zr = 11,80; Cl = 8,25. Вычислено: Zr = 11,36; Cl = 8,83.
IV. Полимеризация пропилена.
Методика.
Предварительно откачанный реактор объемом 0,25 л заполняют жидким пропиленом, добавляют 2-3 мл 10%-ого толуольного раствора МАО и предварительно растворенный в 10%-ом толуольном растворе МАО цирконоцен в количестве 0,1-110-6 моль. Нагревают до 50-70oC и ведут полимеризацию в течение 20-30 минут.
Выход ИПП определяют взвешиванием. Молекулярно-массовые характеристики полипропилена получены методом гель-проникающей хроматографии на высокотемпературном гель-хроматографе “Waters 150-С”. Параметры стереорегулярности определены из соотношения полос поглощения в ИК-спектрах D998/D973. Температуры плавления (Тпл) полученных полимеров определены методом дифференциальной сканирующей калориметрии на калориметре ДСК-910 (“DuPont Instrument”). Результаты опытов и свойства полученных полимеров приведены в таблице 2.
Приведенные данные показывают, что предлагаемые ЦЦ с функционализированным циклосилановым мостиком обладают высокой каталитической активностью и стереоселективностью.
Способ их получения отличается высокими выходами промежуточных и целевого продуктов, улучшенной методикой выделения дилитиевых производных мостичных лигандов и позволяет вдвое увеличить содержание активной рацемической формы в металлокомплексе. -[1,1′-(Бутeн-2-диил-1,4)cилилиденбиc(4-фeнил-2- этилинденил)]}дихлороциркония (1,4-CH2CH=CHCH2)Si(2-Et-4-Phlnd)2ZrCl2, смесь рац- и мезо-форм (2:1).
Суспензию 3.19 г (5.26 ммоль) эфирата дилитиевой соли (add) и 1.24 г (5.26 ммоль) ZrCl4 в 100 мл толуола перемешивали в течение суток при комнатной температуре, ввели еще 500 мл толуола, оранжевый маточный раствор отделили от очень мелкодисперсного осадка декантациями (3 недели), толуол отогнали и остаток перекристаллизовали из минимального объема хлористого метилена, что дало 2.40 г (3.53 ммоль) целевого комплекса, представляющего собой смесь рац- и мезо-форм в соотношении 2:1. Выход 67,1%.
NMR 1H (CD2Cl2, 30oC) d м.д.: 1.11 (т, CH2CH3, рац); 1.21 (т, CH2CH3, мезо); 2.41 (м), 2.74 (м) (CH2CH3, рац); 2.76 (м, CH2CH3, мезо); 2.55 (ушир. с), 2.89 (ушир. с) (CH2Si, мезо); 2.61 (AB-система), 2.78 (AB-система, 2J(AB) = 18 Гц) (CH2Si, рац); 6.33 (с, HC=CH, рац); 6.33 (м), 6.36 (м) (HC= CH, мезо); 6.86 (с, H3, мезо); 6.97 (с, H3, рац); 6.93 (м), 7.10 – 7.65 (м) (H5, H6, H7, Ph).
C38H34Cl2Si1Zr1, М = 680,96. Найдено: Cl = 9,80%,. Вычислено: Cl = 10,43%.
Аналогично получали ЦЦ с непредельным циклосилановым мостиком с другими замещенными бисинденильными лигандами.
Синтез { 5,5-[1,1′-(Бутен-2-диил-1,4)силилиденбисинденил]} дихлороциркония (1,4-CH2CH=CHCH2)Silnd2ZrCl2, смесь рац- и мезо-форм (1:1).
Синтез индениллития IndLi.
К раствору 11.62 г (100.00 ммоль) индена производства фирмы “Aldrich” в 50 мл диэтилового эфира добавили при охлаждении до -20oC и интенсивном перемешивании 53.2 мл (100.01 ммоль) 1.88 М раствора н-бутиллития в легком петролейном эфире, дали смеси нагреться до комнатной температуры и оставили на ночь. Примерно две трети общего объема растворителей отогнали, выпавший белый кристаллический осадок отделили фильтрованием от интенсивно-красного маточного раствора, трижды промыли на фильтре пентаном (порции по 70 мл), высушили в высоком вакууме. Снежно-белое кристаллическое вещество. Выход 10.90 г (89.27 ммоль, 89.3% от теории).
Синтез 1,1′-(бутен-2-диил-1,4)силилиденбисиндена (1,4- CH2CH= CHCH2)Si(IndH)2 (52), R1=R2=H.
К раствору 2.86 г (37.51 ммоль) 1,1-дихлоро-2,5-дигидросилола в 30 мл диэтилового эфира при охлаждении до -30 – -40oC и интенсивном перемешивании в течение 30 минут добавили раствор 4.58 г (37.51 ммоль) индениллития в диэтиловом эфире, дали смеси постепенно нагреться до комнатной температуры и оставили на ночь. Раствор отфильтровали от выпавшего хлорида лития, растворитель отогнали, ввели 50 мл пентана и после повторного отфильтровывания и отгонки растворителя оставшееся слегка желтоватое масло высушили в высоком вакууме. Выход практически количественный. Полную очистку лиганда (52) (R1 = R2 = Н) проводили в форме его дилитиевого производного.
Синтез [1,1′-(бутен-2-диил- 1,4)силилиденбисинденил] дилития (аддукт с диэтиловым эфиром) (1,4- CH2CH=CHCH2)Silnd2Li2 Et2O (add, R1=R2=H).
К раствору 5.14 г (16.45 ммоль) полученного на предыдущей стадии лиганда (52) в 50 мл диэтилового эфира при охлаждении до -20oC и интенсивном перемешивании добавили 15.2 мл 2.16 М раствора н-бутиллития в гексане (16.45 ммоль), дали смеси нагреться до комнатной температуры и оставили на ночь. Выпавший белый осадок отфильтровали, трижды промыли на фильтре той же смесью растворителей и полученное снежно-белое порошкообразное вещество высушили в высоком вакууме. Получено 4.91 г (12.32 ммоль) дилитиевой соли (add). Выход 74.9% в расчете на исходно взятый индениллитий.
NMR (THF-d8, 30oC) d м.д. 1H: 1.91 (с, CH2Si); 6.01 (с, CH=CH); 6.04 (д, 3J(H2-H3) = 3.2 Гц, H2); 6.47 (м, H5, H6); 6.88 (д, H3); 7.32 (м, H7); 7.64 (м, H4).
13С: 20.50 (CH2Si); 95.14 (С3); 98.26 (С1); 114.33, 114.63, 119.40, 121.67, 125.99 (C2, C4, С5, С6, C7); 132.78 (CH=CH); 133.89, 136.28 (C3a, C7a).
Синтез { 5,5 -[1,1′-(Бутен-2-диил- 1,4)силилиденбисинденил]}дихлороциркония (1,4-CH2CH=CHCH2)SiInd2ZrCl2, смесь рац- и мезо-форм (1:1).
Суспензию 1.34 г (3.36 ммоль) эфирата дилитиевой соли (add) и 0.78 г (3.36 ммоль) ZrCl4 в 70 мл толуола перемешивали в течение суток, сухой остаток многократно экстрагировали толуолом, толуольный экстракт сконцентрировали, кристаллический желтый осадок отфильтровали от маточника, промыли толуолом, пентаном и высушили в высоком вакууме, что дало 1.02 г (2.17 ммоль) металлокомплекса, представляющего собой смесь рац- и мезо-форм в соотношении 1:1. Выход 64.5%.
NMR (CD2Cl2, 30oC) d м.д.:1H (CD2Cl2, 30oC) 2.29 (ушир. с), 2.64 (ушир. с) (CH2Si, мезо) 2.37 (AB-система), 2.56 (AB-система, 2J(AB) = 17 Гц) (CH2Si, рац); 6.11 (д, 3J(H2-H3) = 3.5 Гц, H2, рац); 6.17 (д, 3J(H2-H3) = 3.5 Гц, H2, мезо); 6.29 (с, CH=CH, рац); 6.29 (м), 6.30 (м) (CH=CH, мезо); 6.90 (д, H3, рац); 6.95 (д, H3, мезо); 6.93 (м), 7.22 (м) (H5, H6, мезо); 7.10 (м), 7.39 (м) (H5, H6 рац); 7.51 (м), 7.60 (м) (H4, H7, рац, мезо).
13C: 16.45, 16.86 (CH2Si, мезо); 16.99 (CH2Si, рац); 87.94 (С1, рац); 89.08 (С1, мезо); 117.95, 118.14, 124.38, 126.34, 127.18, 128.00 (C2, C3, C4, C5, C6, С7, рац); 119.62, 119.85, 125.75, 126.06, 126.34, 127.54 (C2, C3, C4, C5, C6, С7, мезо); 130.65, 130.88 (CH=CH, мезо); 130.70 (CH=CH, рац); 125.89, 133.79 (С3a, С7a, рац); 128.13, 134.93 (С3a, С7a, мезо). C22H18Cl2Si1Zr1, М = 472,67. Найдено: Cl = 14,38%. Вычислено: Cl = 15,02%.
III. Синтез функционализированных по кремниевому мостику ЦЦ формулы IV.
Синтез борированного анса-цирконоцена с незамещенными бисинденильными лигандами IVa ( 5:5-1,1′ (3-(9- борабициклононанил-9) силоландиил-1,1)бисинденил]дихлороциркония).
Раствор 125 мг (264,5 ммоль) анса-цирконоцена -1,1′ [(Бут-2-диил-1,4)силилиден] -бисинденил-дихлороциркония (смесь рац- и мезо-формы) и 9-борабицикло [3, 3, 1] нонана (32 мг, 132,3 ммоль) в ТГФ смешали при комнатной температуре, нагрели при 70 – 80oC в запаянном сосуде, охладили, растворитель удалили, а остаток высушили в вакууме. Получено кристаллическое вещество желто-оранжевого цвета. Выход количественный. C30H33B1Cl2Si1Zr1. М = 594,69.
Найдено: Zr = 15,88; Cl = 11,38. Вычислено: Zr = 15,34; Cl = 11,92.
Синтез станнилированного анса-цирконоцена с незамещенными бисинденильными лигандами IV b ([ 5:5-1,1′(3-(триметилстанил) силоландиил-1,1) бисинденил] дихлороциркония).
Смесь 125 мг (264,5 ммоль) анса-цирконоцена 7 (смесь рац- и мезо-форм) и 43 мг (264,5 ммоль) триметилстаннана в 30 мл ТГФ нагревали при 70 – 80oC в запаянном сосуде в течение 5 часов. Смесь охладили и растворитель удалили в вакууме. Полученное желто-оранжевое масло промыли пентаном (2 раза по 30 мл). Полученный сухой остаток растворили в эфире, профильтровали раствор и удалили эфир. Получено 155 мг (92% от теор.) комплекса IVb. C25H28Cl2Si1Sn1Zr1, M = 637,49.
Найдено: Zr = 14,90; Cl = 10,85. Вычислено: Zr = 14,31; Cl = 11,12.
Синтез борированного анса-цирконоцена с замещенными бисинденильными лигандами IVc ([ 5:5-1,1′ (3-(9-борабициклононанил-9) силоландиил-1,1) бис-4-фенил-2-этилинденил]дихлороциркония).
Синтез комплекса IVс проведен по методике синтеза цирконоцена IVа. Основное отличие и основная трудность заключаются в том, что продукт образуется в виде плохо кристаллизующегося масла. Твердый продукт получается в результате многократного промывания продуктов пентаном при -20 (-30)oC. Выход IVc 85% (от теоретического). C46H49B1Cl2Si1Zr1, M = 802,98.
Найдено: Zr = 11,80; Cl = 8,25. Вычислено: Zr = 11,36; Cl = 8,83.
IV. Полимеризация пропилена.
Методика.
Предварительно откачанный реактор объемом 0,25 л заполняют жидким пропиленом, добавляют 2-3 мл 10%-ого толуольного раствора МАО и предварительно растворенный в 10%-ом толуольном растворе МАО цирконоцен в количестве 0,1-110-6 моль. Нагревают до 50-70oC и ведут полимеризацию в течение 20-30 минут.
Выход ИПП определяют взвешиванием. Молекулярно-массовые характеристики полипропилена получены методом гель-проникающей хроматографии на высокотемпературном гель-хроматографе “Waters 150-С”. Параметры стереорегулярности определены из соотношения полос поглощения в ИК-спектрах D998/D973. Температуры плавления (Тпл) полученных полимеров определены методом дифференциальной сканирующей калориметрии на калориметре ДСК-910 (“DuPont Instrument”). Результаты опытов и свойства полученных полимеров приведены в таблице 2.
Приведенные данные показывают, что предлагаемые ЦЦ с функционализированным циклосилановым мостиком обладают высокой каталитической активностью и стереоселективностью.
Способ их получения отличается высокими выходами промежуточных и целевого продуктов, улучшенной методикой выделения дилитиевых производных мостичных лигандов и позволяет вдвое увеличить содержание активной рацемической формы в металлокомплексе.
Формула изобретения
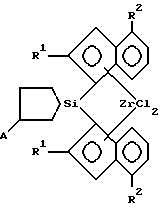 R1 = H, CH3, C2H5; R2 = H, алкил от C1 до C4, нормальный или разветвленный, арил; A = BC8H14, MR3, где M – олово или кремний, R – алкил от C1 до C4, нормальный или разветвленный, арил. 2. Способ получения анса-цирконоценов, функционализированных по циклосилановому мостику формулы IV по п.1, отличающийся тем, что литиевую соль индена подвергают взаимодействию с 1,1-дихлор-2,5-дигидросилолом в диэтиловом эфире с последующим взаимодействием полученной дилитиевой соли соответствующего бисинденильного лиганда с кремниевым мостиком с тетрахлоридом циркония для получения соответствующего цирконоцена и нагреванием его в тетрагидрофуране с алкил(арил)производным моногидрида бора, олова или кремния. 3. Способ по п.2, отличающийся тем, что получение дилитиевой соли бисинденильного лиганда с кремниевым мостиком осуществляют в виде кристаллического аддукта с диэтиловым эфиром, который затем подвергают взаимодействию с тетрахлоридом циркония. РИСУНКИ
MM4A Досрочное прекращение действия патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 22.06.2003
Извещение опубликовано: 20.12.2004 БИ: 35/2004
|
||||||||||||||||||||||||||