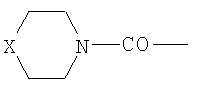Патент на изобретение №2322450
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) МОДИФИЦИРОВАННЫЕ 5′-ФОСФОНАТЫ АЗТ В КАЧЕСТВЕ АКТИВНЫХ КОМПОНЕНТОВ ДЛЯ ПОТЕНЦИАЛЬНЫХ ПРОТИВОВИРУСНЫХ ПРЕПАРАТОВ
(57) Реферат:
Изобретение относится к новым 5′-фосфонатам АЗТ общей формулы (I), обладающим анти-ВИЧ активностью и применению 5′-фосфонатов АЗТ в качестве активного компонента для приготовления лекарственных средств, обладающих анти-ВИЧ активностью. В общей формуле (I):
при n=0-2, R1=
(56) (продолжение): CLASS=”b560m”десорбции масс-спектрометрии производных нуклеозидов и нуклеотидов. – Молекулярная биология. – М., 1994, т.28, №3, с.708-13. О.А.МАМАЕВА И ДР. Изучение анти-HIV активности 5′-фосфонатов активности азидотимидина и фтортимидина. – Молекулярная биология. – М., 1994, т.28, №1, с.137-142. М.К.КУХАНОВА И ДР. Гидролиз 5′-фосфонатов и фосфатов нуклеозидов фосфатазами различного происхождения и сыворотками крови человека и теленка. – Молекулярная биология. – М., 1992, т.26, №5, с.1148-59. А.КРАЕВСКИЙ И ДР. Новые 5′-фосфонаты, модифицированные по сахарному остатку нуклеозидов как ингибиторы продукции ВИЧ. – Молекулярная биология. – М., 1992, т.28, №3, с.624-633. Н.В.ТАРУСОВА И ДР. Ингибирование ВИЧ репродукции в клеточных культурах 5′-фосфонатами 3′-азидо-2′,3′-дидеоксинуклеозидов. – Молекулярная биология. – М., 1989, т.23, №6, с.1716-24. ЯНВАРЕВ Д.В. И ДР. Аминокарбонилфосфонильные производные 3′-азидо-3′-дезокситимидина как потенциальные анти-ВИЧ-препараты. Материалы Международной конференции студентов и аспирантов по фундаментальным наукам “Ломоносов-2003”. Секция Химия. – М., Изд-во Хим. фак. МГУ, 15-18 апр., 2003, т.1, с.151. SOMOGYI GABOR ET AL. Targeted drug delivery to the brain via phosphonate derivatives II/Anionic chemical delivery system for zidovudine(AZT)”EInternational Journal of Pharmaceutics, 1998, 166(1)27-35. CASARA PATRICK J. ET AL. “Synthesis of acid stable5′-О-fluoromhetyl phosphonates of nucleosides. Evalution as inhibitors of reverse transcriptase” Bioorganic & Medicinal Chemistry Letters, 1992, 2(2), 145-148. SOMOGYI G. ET AL. “Metabolic properies of phosphonate esters” PHARMAZIE, 2004, 59(5), 378-381. KRAEVSKII A.A. ET AL. “Nucleosides.156.5′-Hydrogenphosphonates and 5′-methylhosphonates of sugar-modified pyrimidine nucleosides as potential anti-HIV-1 agents”. Nucleosides & Nucleotides, 1992, 11(2), 177-96.
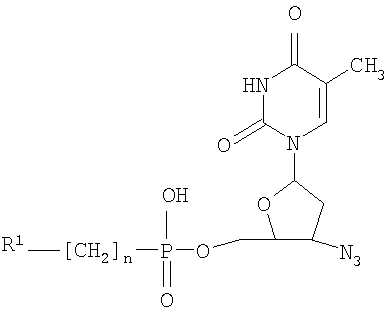
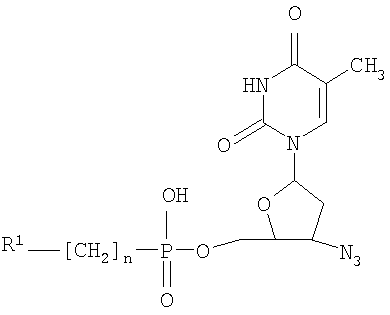
Изобретение относится к области молекулярной биологии, вирусологии и медицины, а именно к новым производным нуклеозидов, а именно, к замещенным 5′-фосфонатам АЗТ. Эти соединения обладают противовирусным действием и могут быть применены для подавления репродукции вируса иммунодефицита человека. В настоящее время в медицинской практике используется целый ряд соединений, обладающих противовирусной активностью в отношении ВИЧ. Среди них различают нуклеозидные и ненуклеозидные ингибиторы. Среди производных нуклеозидов наиболее часто применяются 3′-азидо-3′-дезокситимидин (АЗТ, Зидовудин®), 2′,3′-дидезоксицитидин (ddC, Зальцитабин®), 2′,3′-дидезоксиинозин (ddl, Диданозин®), 2′,3′-дидезокси-2′,3′-дидегидротимидин (d4T, Ставудин®) и 2′,3′-дидезокси-3′-тиацитидин (ЗТС, Ламивудин®) [De Clercq, E., 2002. New development in anti-HIV chemotherapy. Biochim. Biophys. Acta, 1587, 258-275]. Механизм действия указанных соединений состоит в том, что после проникновения в инфицированные клетки они подвергаются трифосфорилированию и специфично блокируют синтез ДНК, катализируемый обратной транскриптазой ВИЧ. Высокая изменчивость ВИЧ приводит к быстрому возникновению резистентных штаммов вируса [Groschel, В., Cinatl, J.H., and CinatI J. Jr., 1997. Viral and cellular factors for resistance against antiretroviral agents. Intervirology, 40, 400-407; Antonelli, G, Turriziani, О., Verri, A., Narciso, P., Ferri, F., D’Offizi, G., and Dianzini, F., 1996. Long-term exposure to zidovudine affects in vitro and in vivo the efficiency of thymidine kinase. AIDS Res Hum Retrovir., 12, 223-228], и, следовательно, к необходимости смены препарата. К тому же из-за низкой эффективности внутриклеточных превращений используемые препараты требуют высоких доз применения, что вызывает появление выраженных токсических эффектов. Следствиями токсичности АЗТ являются подавление деятельности клеток спинного мозга, нарушения функции печени и миопатия [Chariot, P., Drogou, I., De Lacroix-Szmania, I., Eliezer-Vanerot, M.C., Chazaud, В., Lombes, A., Schaeffer, A., and Zafrani, E.S., 1999. Zidovudine-induced mitochondrial disorder with massive liver steanosis, myopathy, lactic acidosis, and mitochondial DNA depletion. J. Hepatol. 30, 156-160; Kellam, P., Boucher, C.A., and Larder, B.A., 1992. Fifth mutations in HIV reverse transcriptase contributes to the development of high level resistance to zidovudine. Proc. Natl. Acad. Sci. U.S.A. 89, 1934-1938; Ren, J., Esnouf, R.M., Hopkins, A.L., Jones, E.Y., Kirby, I., Keeling, J., Ross, C.K., Larder, B.A., Stuart, D.I., and Stammers, D.K., 1998. 3′-Azido-3′-deoxythymidine drug resistance mutations in HIV-1 reverse transcriptase can induce long range conformational changes. Proc. Natl. Acad. Sci. U.S.A, 95, 9518-9523]. Быстрое выведение АЗТ из организма требует частого приема препарата. Кроме того, при длительном применении АЗТ достаточно быстро вырабатываются резистентные штаммы вируса и лечение теряет эффективность. Несмотря на все вышеперечисленные недостатки, АЗТ по-прежнему остается наиболее широко применяемым анти-ВИЧ препаратом. Известен Н-фосфонат АЗТ (Никавир®), одобренный в России для терапии СПИД, менее токсичен, чем АЗТ [Intracellular metabolism and pharmacokinetics of 5′-hydrohenphosphonate of 3′-azido-2′,3′-dideoxythymidine, a prodrug of 3′-azido-2′,3′-dideoxythymidine. Antiviral Reserch 63 (2004), 107-113]. Действие Никавира основано на способности высвобождать АЗТ, который после внутриклеточного превращения в АЗТ-5′-трифосфат ингибирует репликацию ВИЧ. По данным фармакокинетических исследований клинические преимущества Никавира объясняются более медленным и плавным нарастанием концентрации АЗТ в крови, чем при приеме собственно АЗТ; при этом Смакс АЗТ из Никавира <Смакс АЗТ из Зидовудина, a T1/2 АЗТ из Никавира >T1/2 АЗТ из Зидовудина [Y.Skoblov et al./Antiviral Research 63 (2004) 107-113]. Тем не менее, токсичность Никавира остается достаточно высокой. Другим недостатком является возникновение резистентности к Никавиру. Данным изобретением решена задача создания низкотоксичных производных АЗТ, обладающих способностью проникать внутрь клетки и постепенно высвобождать активный нуклеозид (АЗТ). Это позволит поддерживать внутриклеточную концентрацию препарата, достаточную для проявления терапевтического действия в течение длительного времени, и, таким образом, снизить разовую дозу препарата и/или частоту приема и уменьшить побочные эффекты. Задача решена созданием соединений 5′-фосфонил-3′-азидо-3′-дезокситимидин общей формулы:
при n=0-2, при n=0, R1=Cl3С-, R1 означает Cl-, Br-, n=1-6 и R1 означает R2C(O)O- (где R2-C1-С6алкил), при n=2-6. Новые соединения подавляют репродукцию вируса иммунодефицита человека 1-го типа в культуре перевиваемых лимфоцитов МТ-4, обеспечивают защиту клеток от цитопатогенного действия вируса и не проявляют токсичности в отношении хозяйских клеток вплоть до крайне высоких концентраций (табл.1). Из полученных экспериментальных данных видно, что исследуемые соединения, не оказывая токсического действия на клетки в эффективных концентрациях (50% токсические дозы на 2-4 порядка превышают 50% ингибирующие дозы) в высокой степени подавляют репродукцию вируса иммунодефицита 1 типа в культуре клеток МТ-4. Терапевтические индексы исследуемых соединений (IS), определяемые как отношение токсической дозы препарата к его эффективной дозе, сравнимы с таковыми для Н-фосфоната АЗТ. Вирусологические тесты проведены в соответствии с описанными ранее протоколами. Показано, что заявляемые фосфонаты АЗТ в организме собак медленно высвобождают АЗТ, таким образом, представляя собой латентные формы АЗТ (Пример 10, табл.2). Исследования показывают, что для всех заявляемых фосфонатов единственным продуктом метаболизма, детектируемым в крови животных, является АЗТ. Фармакокинетические параметры, приведенные в табл.2, были определены по генерированному АЗТ и зависели от структуры фосфоната. Целевые фосфонаты получали по следующей схеме в одну или несколько стадий:
Ниже приведены конкретные примеры, раскрывающие сущность изобретения. Пример 1. 5′-Хлорметилфосфонил-3′-азидо-3′-дезокситимидин (Ia). К раствору 3′-азидо-3′-дезокситимидина (0,8 г, 3 ммоль) в триэтилфосфате (10 мл), охлажденному до 0°С, добавляли хлорметилфосфонилдихлорид (0,92 мл, 9 ммоль). Смесь перемешивали 18 часов при 18°С, разбавляли охлажденной смесью пиридина (10 мл) и воды (10 мл), перемешивали 30 минут и выливали в воду (700 мл). Раствор наносили на колонку с DEAE целлюлозой, элюировали в линейном градиенте NH4HCO3 (0 Пример 2. 5′-(Диметиламиноэтиламино)карбонилфосфонил-3′-азидо-3′-дезокситимидина (Ib) К раствору 5′-этоксикарбонилфосфонил-3′-азидо-3′-дезокситимидина (300 мг, 0,75 ммоль) в смеси диоксан-вода (1:1, 5 мл), добавляли N,N-диметиламиноэтиламин (300 мг, 3,4 ммоль). Смесь перемешивали 18 часов при 37°С, упаривали, переупаривали с водой (10 мл), остаток наносили на колонку с LiChroprep RP-8, элюировали в линейном градиенте метанола (0 5′-(Триметиламмониоэтиламино)карбонилфосфонил-3′-азидо-3′-дезокситимидин (Ic). К суспензии 5′-(диметиламиноэтиламино)карбонилфосфонил-3′-азидо-3′-дезокситимидина ((1b), 90 мг, 0,2 ммоль) в триметилфосфате (1 мл) прибавили трибутиламин (0,1 мл) и перемешивали 3 суток при 37°С. Затем упаривали, переупаривали с водой (3×10 мл), остаток наносили на колонку с LiChroprep RP-8, элюировали в линейном градиенте метанола (0 Пример 3. 5′-Метоксиметилфосфонил-3′-азидо-3′-дезокситимидин (Id) Раствор пиридиниевой соли метоксиметилфосфоновой кислоты (1,2 ммоль) в пиридине (3 мл) прибавляли к раствору 3′-азидо-3′-дезокситимидина (267 мг, 1 ммоль) в пиридине (2 мл), прибавляли дициклогексилкарбодиимид (520 мг, 2,5 ммоль), реакционную массу перемешивали при комнатной температуре 10 часов и разбавляли водой (5 мл). После 30 минут дополнительного перемешивания отделяли осадок, раствор упаривали, остаток растворяли в воде (100 мл). Раствор наносили на колонку с DEAE целлюлозой в НСО3 –-форме, элюировали в линейном градиенте NH4HCO3 (0 Пример 4. 5′-(Циклогексиламино)карбонилфосфонил-3′-азидо-3′-дезокситимидин (Ie). Раствор 5′-этоксикарбонилфосфонил-3′-азидо-3′-дезокситимидина (80 мг, 0,2 ммоль) в циклогексиламине (1 мл) перемешивали 18 ч при комнатной температуре, а затем сконцентрировали в вакууме. Остаток растворили в воде и хроматографировали на DEAE-Toyopearl, элюировали в линейном градиенте NH4HCO3 (0 Пример 5. 5′-(Аминоэтиламино)карбонилфосфонил-3′-азидо-3′-дезокситимидин (If). К раствору 5′-этоксикарбонилфосфонил-3′-азидо-3′-дезокситимидина (80 мг, 0,2 ммоль) в этаноле (1 мл) прибавляли этилендиамин (1 мл). Через 18 ч при 37°С растворители упаривали в вакууме. Остаток растворили в воде и хроматографировали на DEAE-Toyopearl (в ОН–-форме), элюировали в линейном градиенте NH4HCO3 (0 Пример 6 5′-(Гидроксиэтиламино)карбонилфосфонил-3′-азидо-3′-дезокситимидин (Ig). К раствору 5′-этоксикарбонилфосфонил-3′-азидо-3′-дезокситимидина (80 мг, 0,2 ммоль) в этаноле (1 мл) прибавляли аминоэтанол (1 мл). Через 18 ч при 37°С растворители упаривали в вакууме. Остаток растворили в воде и хроматографировали на DEAE-Toyopearl, элюировали в линейном градиенте NH4НСО3 (0 Пример 7 5′-(Гексиламино)карбонилфосфонил-3′-азидо-3′-дезокситимидин (lh). К раствору 5′-этоксикарбонилфосфонил-3′-азидо-3′-дезокситимидина (80 мг, 0,2 ммоль) в этаноле (2 мл) прибавляли н-гексиламин (1 мл). Через 18 ч при 37°С растворители упаривали, остаток растворили в воде и хроматографировали на DEAE-Toyopearl, элюировали в линейном градиенте NH4НСО3 (0 Пример 8 5′-Трихлорметилфосфонил-3′-азидо-3′-дезокситимидин (Ii) Раствор пиридиниевой соли трихлорметилфосфоновой кислоты (1,2 ммоль) в пиридине (3 мл) прибавляли к раствору 3′-азидо-3′-дезокситимидина (267 мг, 1 ммоль) в пиридине (2 мл), прибавляли при перемешивании дициклогексилкарбодиимид (520 мг, 2,5 ммоль), реакционную массу перемешивали при комнатной температуре 10 часов и разбавляли водой (5 мл). После 30 минут дополнительного перемешивания отделяли осадок. Раствор наносили на колонку с DEAE целлюлозой в НСО3 –-форме, элюировали в линейном градиенте NH4НСО3 (0 Пример 9 Исследование ингибирования репродукции ВИЧ включает культивирование первично инфицированных лимфоидных клеток линии МТ-4 в присутствии исследуемых соединений, конечные концентрации которых в культуральной среде составляют 0,001-100 мкг/мл, на протяжении одного пассажа – в течение 4 суток. Ингибирование репродукции ВИЧ в культуре чувствительных клеток определяют по снижению накопления вирусспецифического белка р24 (по данным иммуноферментного анализа), а также по увеличению жизнеспособности клеток в присутствии препарата по сравнению с контролем, определяемому на 4-е сутки культивирования при окрашивании бромидом 3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолия (МТТ). Оценка цитотоксичности соединений. Цитотоксичность препарата оценивают путем добавления его разведении в бессывороточной среде RPMI-1640 к клеточной суспензии МТ-4, помещенной в лунки 96-луночного планшета (“Cel-Cult”, England), до конечных концентраций 0,001-100 мкг/мл (по три лунки на каждую дозу) с последующим культивированием при 37°С в течение 4 суток. Посевная концентрация составляет 0,5×106 клеточных частиц в миллилитре. Контролем служат клетки без добавления препарата, вместо которого вносят такое же количество бессывороточной среды. Жизнеспособность клеток подсчитывают на 4 сутки культивирования, пользуясь формазановым методом (прижизненным окрашиванием клеток МТТ). Токсичность различных доз препарата определяют по жизнеспособности клеток относительно контроля, по полученным результатам строят дозозависимую кривую и определяют концентрацию, на 50% снижающую жизнеспособность клеток (CD50). Исследуемые соединения не оказывают токсического действия на клетки МТ-4 в эффективных концентрациях. Следует также отметить, что 50% токсичные дозы на 2-4 порядков превышают эффективные в отношении ВИЧ-1 дозы (таблица 1). Влияние исследуемых соединений на репродукцию ВИЧ-1 в культуре клеток МТ-4 исследовано по известной методике. Терапевтический индекс, или индекс селективности (IS) считают как отношение 50%-ной токсической концентрации соединения к его 50%-ной эффективной дозе (результаты представлены в таблице 1). На основании этих количественных показателей ингибирования можно судить об эффективности противовирусного действия заявляемых соединений, заключающейся в высокой степени подавления репликации ВИЧ-1 в культуре клеток МТ-4, сравнимой с эффективностью Никавира. Пример 10. Собаке весом 12 кг вводили 250 мг исследуемого вещества орально (в смеси с творогом). Через определенные промежутки времени отбирали пробы крови (1 мл) из бедренной вены. Пробы центрифугировали (10 минут, 2000 об/мин), супернатант отделяли. Из супернатанта отбирали аликвоты (0,25 мл), добавляли оксетан (0,25 мкг, как внутренний стандарт) и метанол (0,75 мл). Полученную смесь центрифугировали 3 минуты при 5000 об/мин. Супернатант отделяли и упаривали в токе воздуха при 40°С, к остатку добавляли воду (1 мл). Аликвоты (20 мкл) анализировали методом ВЭЖХ на жидкостном хроматографе Gynkotec, Германия; аналитическая колонка Ultrasphere ODC “Beckman”, USA. Элюент: 6% ацетонитрил в 0,1% Н3PO4 (рН 2,1) в присутствии 0,15% три-этиламина. Детекция при
Таким образом, показано, что заявленные соединения обладают низкой токсичностью, способны эффективно ингибировать репродукцию вируса иммунодефицита человека 1 типа в культуре клеток МТ-4 и генерировать АЗТ в организме млекопитающих, обеспечивая плавное нарастание его концентрации в крови.
Формула изобретения
1. 5′-Фосфонаты АЗТ, имеющие общую формулу
при n=0-2, R1= (где Х=СН2, NH); R2-NH-C(O)- (где R2-C2-С6алкил, С5-С7циклоалкил), HO(CH2)k– (где k=2-4); при n=0, R1-Cl3С- R1 означает Cl-, Br-, n=1-6 и R1 означает R3C(О)О- (где R3-C1-С6алкил), при n=2-6. 2. Применение 5′-фосфонатов АЗТ, общей формулы
где n=0-2, R1= (где Х=СН2, NH, О); R2-NH-C(O)- (где R2=Н, C1-С6алкил, С5-С7циклоалкил), HO(CH2)k– (где k=2-4); при n=0, R1=Cl3С; R1=Cl-, Br-, I-, и при n=1-6 и R3C(О)О- (где R3-C1-С6алкил), при n=2-6, обладающих анти-ВИЧ активностью, в качестве активного компонента для приготовления лекарственных средств.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
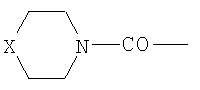 (где Х=СН2, NH, О); R2-NH-C(O)- (где R2=Н, C1-С6алкил, С5-С7циклоалкил), HO(CH2)k– (где k=2-4); при n=0, R1=Cl3С; при n=1-6 R1=Cl-, Br-, I-, и при n=2-6: R3C(O)O- (где R3-C1-С6алкил), при n=2-6. 2 н.п. ф-лы, 2 табл.
(где Х=СН2, NH, О); R2-NH-C(O)- (где R2=Н, C1-С6алкил, С5-С7циклоалкил), HO(CH2)k– (где k=2-4); при n=0, R1=Cl3С; при n=1-6 R1=Cl-, Br-, I-, и при n=2-6: R3C(O)O- (где R3-C1-С6алкил), при n=2-6. 2 н.п. ф-лы, 2 табл.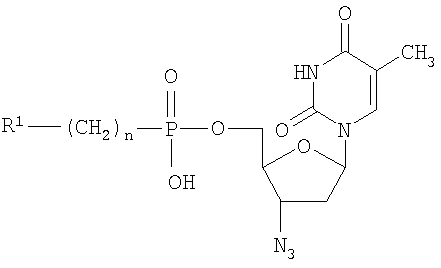
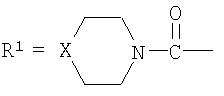 (где Х=СН2, NH); R2-NH-C(O)- (где R2=C2-С6алкил, С5-С7циклоалкил), HO(CH2)k– (где k=2-4),
(где Х=СН2, NH); R2-NH-C(O)- (где R2=C2-С6алкил, С5-С7циклоалкил), HO(CH2)k– (где k=2-4),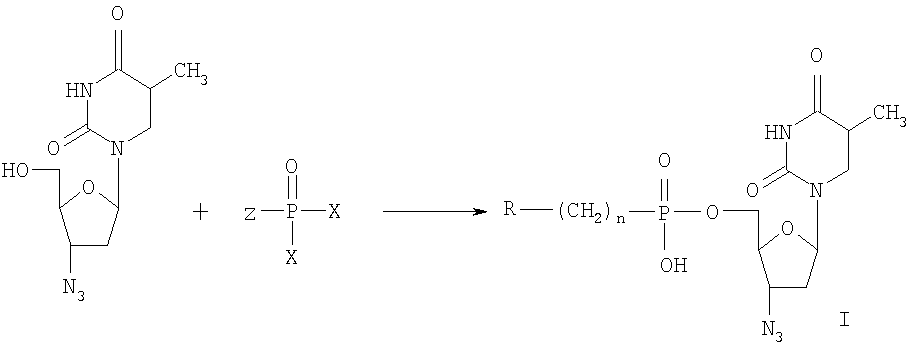
 0,15 М). Целевые фракции упаривали, остаток разбавляли водой (3 мл) и дополнительно очищали на колонке с LiChroprep RP-18, элюируя водой. Целевую фракцию лиофилизовали, получали 1 г (84%) фосфоната (Ia). 1H ЯМР (D2O): 7,72 kb (1H, J=0,5 Гц, H-6), 6,27 т (1H, J=6 Гц, Н-1′), 4,55 м (1Н, Н-3′), 4,21 м (3Н, Н-4′ и Н-5′), 3,58 д (2Н, J=8,5 Гц, СН2-Р), 2,54 м (2Н, Н-2′), 1,95 д (3Н, J=0,5 Гц, 5-Ме). 31Р ЯМР (D2O): 16,03 с.
0,15 М). Целевые фракции упаривали, остаток разбавляли водой (3 мл) и дополнительно очищали на колонке с LiChroprep RP-18, элюируя водой. Целевую фракцию лиофилизовали, получали 1 г (84%) фосфоната (Ia). 1H ЯМР (D2O): 7,72 kb (1H, J=0,5 Гц, H-6), 6,27 т (1H, J=6 Гц, Н-1′), 4,55 м (1Н, Н-3′), 4,21 м (3Н, Н-4′ и Н-5′), 3,58 д (2Н, J=8,5 Гц, СН2-Р), 2,54 м (2Н, Н-2′), 1,95 д (3Н, J=0,5 Гц, 5-Ме). 31Р ЯМР (D2O): 16,03 с. 3,31 т (2Н,
3,31 т (2Н, 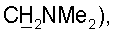 2,90 с (6Н, NMe2), 2,47 т (J=6,4 Гц, 2Н, Н-2′), 1,82 с (3Н, 5-Ме). 31Р-ЯМР (D2O): -1,97 с.
2,90 с (6Н, NMe2), 2,47 т (J=6,4 Гц, 2Н, Н-2′), 1,82 с (3Н, 5-Ме). 31Р-ЯМР (D2O): -1,97 с.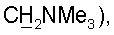 3,17 с (9Н, NMe3), 2,48 м (2Н, Н-2′), 1,82 с (3Н, 5-Ме). 31Р-ЯМР (D2O): -2,30 с.
3,17 с (9Н, NMe3), 2,48 м (2Н, Н-2′), 1,82 с (3Н, 5-Ме). 31Р-ЯМР (D2O): -2,30 с.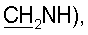 3,12 т (2Н, J=5,9 Гц,
3,12 т (2Н, J=5,9 Гц, 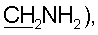 2,42 т (2Н, J=6,2 Гц, Н-2′), 1,82 с (3Н, 5-Ме). 31P ЯМР (D2O): -1,90 с.
2,42 т (2Н, J=6,2 Гц, Н-2′), 1,82 с (3Н, 5-Ме). 31P ЯМР (D2O): -1,90 с.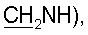 3,28 дд (2Н, J=5,3 и 5,6 Гц,
3,28 дд (2Н, J=5,3 и 5,6 Гц, 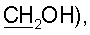 2,36 т (2Н, J=6,2 Гц, Н-2′), 1,77 с (3Н, 5-Ме). 31Р ЯМР (D2O): -1,41 с.
2,36 т (2Н, J=6,2 Гц, Н-2′), 1,77 с (3Н, 5-Ме). 31Р ЯМР (D2O): -1,41 с. max 265 нм, температура 30°С. Фармакокинетические параметры, полученные в результате анализа данных, приведены в таблице 2.
max 265 нм, температура 30°С. Фармакокинетические параметры, полученные в результате анализа данных, приведены в таблице 2. М
М