Патент на изобретение №2320655
|
||||||||||||||||||||||||||
(54) УЛУЧШЕННЫЙ СПОСОБ ПОЛУЧЕНИЯ
(57) Реферат:
Изобретение относится к способу получения
Настоящее изобретение относится к улучшенному способу получения Элетриптан, 3-{[1-метилпирролидин-2(R)-ил]метил}-5-(2-фенилсульфонилэтил)-1Н-индол и способ его получения описаны в US-B-5607951. Дополнительные способы получения элетриптана описаны в ЕР-В-1088817 и WO-A-02/50063. Бромгидрат элетриптана имеет структуру формулы (I) ниже.
WO-A-96/06842 описывает бромгидрат элетриптана, две из его кристаллических форм и способы их получения. Одна из кристаллических форм, описанная в настоящем описании, обозначенная как WO-A-00/32589 описывает кристаллический моногидрат бромгидрата элетриптана и способы его получения. Два способа для превращения свободного основания элетриптана в В широких масштабах выход бромгидрата элетриптана с использованием таких способов предшествующей области техники находится в области 73%. Необходимо принимать во внимание, что в получении достаточных количеств бромгидрата элетриптана для удовлетворения общего рынка Relpax В настоящее время было неожиданно обнаружено, что надежный способ с высоким выходом получения Выход, получаемый при использовании этого способа в крупномасштабном производстве, составляет от 93 до 96%. Получаемый продукт является исключительно Считают, что продукт стадии (а) является гидратом, вероятно, моногидратом, описанным в WO-A-00/32589. Этот гидрат затем превращается на стадии (b) с образованием желаемого продукта. Исходное вещество элетриптана является предпочтительно сухим (менее чем 0,3% воды по анализу Карла Фишера) и свободным от загрязнения частицами (раствор в 2-бутаноне может быть отфильтрован, если необходимо). Бромистоводородная кислота является предпочтительно 48% водным раствором и преимущественно добавляется в реакционный сосуд в виде раствора в 2-бутаноне в течение, по меньшей мере, часа и при комнатной температуре. При таком добавлении рН реакционной смеси не падает ниже 5 и образуется более чистая реакционная смесь с более высоким выходом. Применение от 0,95 до 1,05 молярных эквивалентов бромистоводородной кислоты является предпочтительным, применение 0,98 молярных эквивалентов является оптимальным. Всего около 21 литра 2-бутанона на килограмм исходного вещества элетриптана должно предпочтительно расходоваться. После добавления бромистоводородной кислоты реакционную смесь перемешивают предпочтительно в течение, по меньшей мере, 3 часов. Во время азеотропной перегонки практически вся вода должна быть удалена из реакционной смеси. Остаточное содержание воды менее чем 0,5% мас./мас. является предпочтительным. Когда около 21 литра 2-бутанона на килограмм исходного вещества элетриптана расходуется в сочетании с 0,98 эквивалентами 48% бромистоводородной кислоты, окончательный объем около 11 литров на килограмм элетриптана является идеальным. Продукт легко выделяют фильтрацией. Обычно реакционной смеси давали медленно остыть до комнатной температуры, необязательно гранулировали, фильтровали, далее промывали 2-бутаноном и сушили.
Исходный объем 15 литров толуола на килограмм бромгидрата элетриптана является предпочтительным. Для оптимальных результатов перегонка толуола должна повторяться дважды, и необходимо поддерживать нагревание реакционной смеси при субтемпературе флегмы в течение минимум двух часов между перегонками. Субтемпература флегмы около 106°C является идеальной. Конечный продукт обычно выделяют фильтрацией. Обычно реакционную смесь охлаждали до комнатной температуры, гранулировали, фильтровали, промывали дополнительным количеством толуола и сушили. Стадию (с) преимущественно проводили в атмосфере азота для предотвращения обесцвечивания продукта. Следующий вариант воплощения изобретения обеспечивает способ образования стабильного Этот процесс превращения включает стадии (а) кристаллизации раствора исходного вещества бромгидрата элетриптана в смеси 2-бутанона и воды и (b) отгонки азеотропа 2-бутанона/воды до завершения образования безводного Этот способ является особенно преимущественным, так как ранее только осуществимый крупномасштабный способ превращения смешанных полиморфных и/или сольватированных/гидратированных форм бромгидрата элетриптана в чистый Элетриптан предпочтительно получают в соответствии со схемой 1 ниже. Схема 1
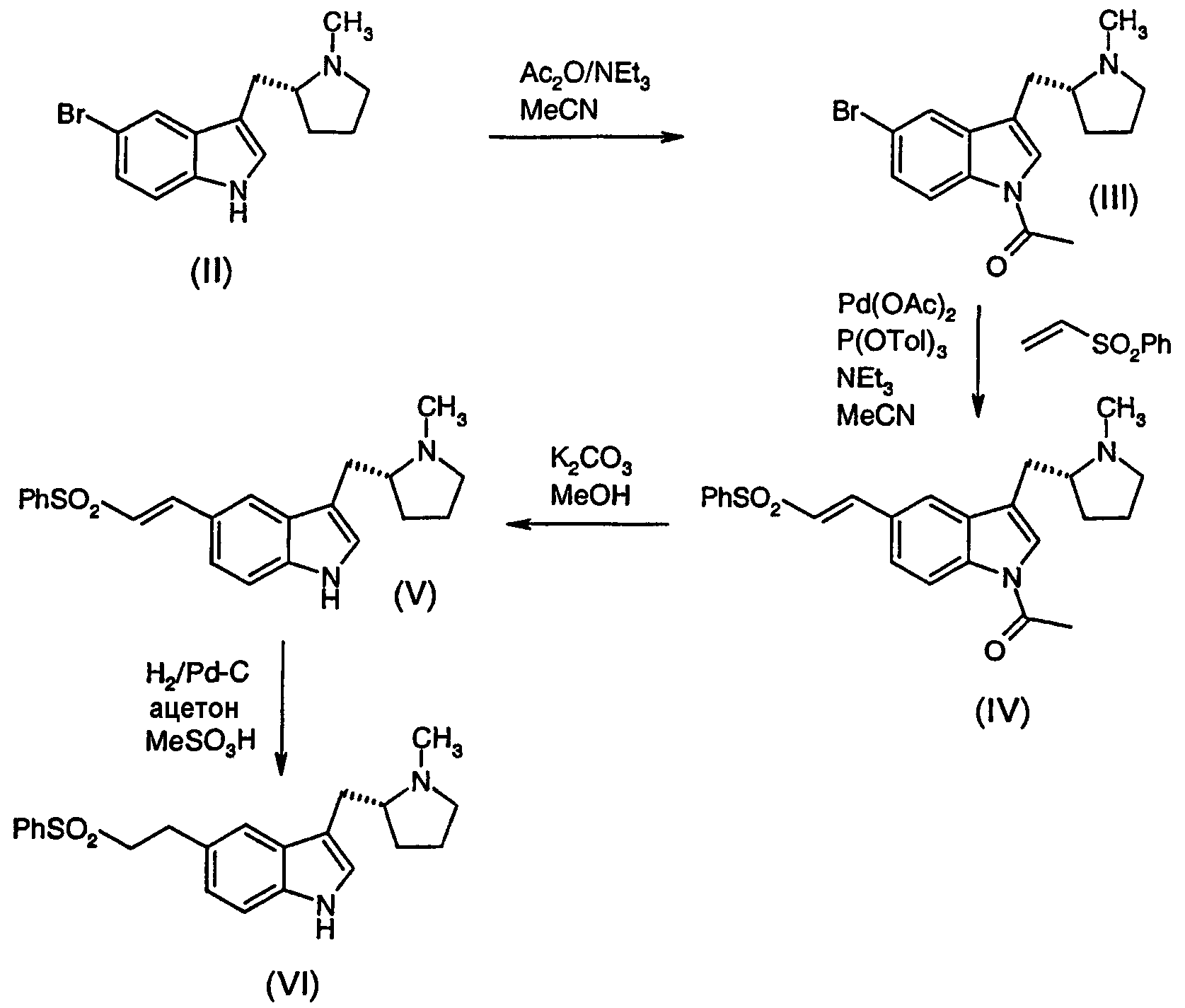
Соединение (II) ((R)-5-бром-3-(N-метилпирролидин-2-илметил)-1Н-индол) может быть получено способами, описанными в US-B-5607951 или EP-B-1088817. Соединение (III) ((R)-1-ацетил-5-бром-3-(N-метилпирролидин-2-илметил)-1Н-индол) может быть получено обработкой раствора соединения (II) в ацетонитриле ангидридом уксусной кислоты и триэтиламином. Реакцию предпочтительно проводят при нагревании с обратным холодильником. Соединение (IV) ((R)-1-ацетил-5-(2-фенилсульфонилэтенил)-3-(N-метилпирролидин-2-илметил)-1Н-индол) может быть получено обработкой раствора соединения (III) в ацетонитриле триэтиламином, три-о-толилфосфином, ацетатом палладия и фенилвинилсульфоном. Реакцию предпочтительно проводят при нагревании с обратным холодильником. Соединение (II) может быть легко преобразовано в соединение (IV) без выделения соединения (III). Соединение (V) ((R)-5-(2-фенилсульфонилэтенил)-3-(N-метилпирролидин-2-илметил)-1Н-индол) может быть получено обработкой раствора соединения (IV) в метаноле карбонатом калия. Реакцию предпочтительно проводят при комнатной температуре. Подобный способ, описанный в предшествующей области техники (см. пример 58 US-B-5607951), использует перекристаллизацию для очистки и выделения соединения (V). Перекристаллизация является самым эффективным средством очистки, особенно из смеси ацетонитрила и воды или смеси ацетона и воды, но выход полученного продукта ниже оптимального. Неожиданно было обнаружено, что колоночная хроматография является более эффективным способом очистки сырого соединения (V), даже в большом многокилограммовом объеме, и значительно больший выход продукта может быть достигнут этим способом. Когда используется хроматография для очистки соединения (V), метанольный раствор, получающийся в реакции, фильтровали и нейтрализовали водной кислотой, предпочтительно фосфорной кислотой. Затем смесь загружали в колонку, упакованную с подходящей стационарной фазой (предпочтительно полимером стирола, таким как смола CG-161, доступная от Tosoh Bioscience). Колонку элюировали смесью ацетона и водной кислоты (предпочтительно уксусной или фосфорной кислоты) и фракции, содержащие продукт, объединяли и концентрировали. Ацетон добавляли к концентрированному раствору и рН доводили до от 10 до 11 с использованием подходящего основания, такого как карбонат калия, для того чтобы осадить продукт. Продукт собирали, промывали водой и сушили. Соединение (VI) (элетриптан) может быть получено обработкой раствора соединения (V) в смеси ацетона и воды палладием на угле и метансульфоновой кислотой в атмосфере водорода. Катализатор, включающий 5% палладия на угле, является предпочтительным. Особенно преимущественным катализатором для использования в этом способе является РМС 2020С (поставляемый Precious Metals Corporation), требующий такую же низкую загрузку катализатора, как 7% палладия на угле. Следующие примеры иллюстрируют определенные способы осуществления изобретения. Дифференциальную сканирующую калориметрию (ДСК) проводили с использованием прибора DSC-7 Perkin Elmer. Приблизительно 10 мг каждого образца тщательно отвешивали в 50-микролитровый алюминиевый лоток. Образцы нагревали при 20°С/минуту в диапазоне от 40°С до 220°С с продувкой газообразным азотом. Пример 1 (а) (R)-5-Бром-3-(N-метилпирролидин-2-илметил)-1Н-индол (256 кг), ацетонитрил (380 кг), триэтиламин (115 кг) и ангидрид уксусной кислоты (115 кг) загружали в сухой остеклованный сосуд. Реакционную смесь нагревали до образования обратного тока и поддерживали при такой температуре в течение 4,5 часов. (b) Смесь ацетонитрила (375 кг), ацетата палладия (12,5 кг) и три-о-толилфосфина (60 кг) перемешивали в течение 1 часа. Добавляли фенилвинилсульфон (160 кг), триэтиламин (92 кг) и, наконец, раствор, полученный в части (а), и смесь нагревали до образования обратного тока в течение 7,5 часов. Реакционную смесь охлаждали и добавляли раствор 190 кг концентрированной соляной кислоты в 1200 кг воды в течение 4 часов. Полученную смесь фильтровали для удаления отработанного катализатора и добавляли еще 3000 кг воды и 300 кг 50% масс./масс. водного раствора гидроксида натрия к фильтрату для осаждения продукта. Полученную суспензию фильтровали и промывали водой (500 кг) для получения сырого (R)-1-ацетил-5-(2-фенилсульфонилэтенил)-3-(N-метилпирролидин-2-илметил)-1Н-индола в виде темно-коричневого, влажного, кристаллического твердого вещества (535 кг влажного, эквивалентного 338 кг сухого). Такой сырой продукт добавляли к 530 кг ацетона и смесь нагревали до 60°С. При достижении этой температуры добавляли 814 кг воды в течение 2 часов, при этом одновременно охлаждая смесь обратно до температуры окружающей среды. Затем партию гранулировали в течение 2 часов и фильтровали для получения очищенного продукта (350 кг влажного, эквивалентные 280 кг сухого, 83%). Пример 2 Метанол (660 кг) и (R)-1-ацетил-5-(2-фенилсульфонилэтенил)-3-(N-метилпирролидин-2-илметил)-1Н-индол (77 кг сухого эквивалента перекристаллизованного продукта примера 1) загружали в сосуд и полученную смесь перемешивали в течение 5 минут. Добавляли карбонат калия (8,9 кг) и смесь перемешивали при комнатной температуре в течение 30 минут. Затем реакционную смесь нагревали до 35°С и добавляли фолиевый углерод (11,6 кг) и воду (235 кг). Полученную смесь фильтровали и фильтрат разводили добавлением воды (1300 кг, добавленной в течение двух часов) и гранулировали в течение 2 часов при комнатной температуре. Фильтрация давала сырой (R)-5-(2-фенилсульфонилэтенил)-3-(N-метилпирролидин-2-илметил)-1Н-индол (75 кг влажного, эквивалентные 60,5 кг сухого, 88%). Пример 3 Смесь ацетонитрила (940 кг) и сырого (R)-5-(2-фенилсульфонилэтенил)-3-(N-метилпирролидин-2-илметил)-1Н-индола (478 кг сухого эквивалента, продукт способа примера 2) нагревали до 55°С. Добавляли воду (720 кг) и смесь охлаждали до 20°С и гранулировали в течение 2 часов при этой температуре. Чистый (R)-5-(2-фенилсульфонилэтенил)-3-(N-метилпирролидин-2-илметил)-1Н-индол (482 кг влажного, эквивалентные 393 кг сухого, 82%) извлекали фильтрацией. Пример 4 Ацетон (1140 кг) и (R)-5-(2-фенилсульфонилэтенил)-3-(N-метилпирролидин-2-илметил)-1Н-индол (482 кг сухого эквивалента, перекристаллизованный продукт примера 3) загружали в сосуд и смесь нагревали до 55°С. Добавляли воду (1520 кг) и смесь охлаждали до 20°С и гранулировали в течение 2 часов. Перекристаллизованный (R)-5-(2-фенилсульфонилэтенил)-3-(N-метилпирролидин-2-илметил)-1Н-индол (492 кг влажного, 343 кг сухого эквивалента, 87%) выделяли фильтрацией. Пример 5 (R)-5-(2-фенилсульфонилэтенил)-3-(N-метилпирролидин-2-илметил)-1Н-индол (200 кг сухого эквивалента) и ацетон (1186,5 кг) загружали в сухой, остеклованный сосуд. Добавляли деионизированную воду (300 кг), дополнительный ацетон (237 кг), метансульфоновую кислоту (55 кг) и суспензию палладия на угле (22,4 кг сухого эквивалента) в деионизированной воде (200 кг) и смесь гидрировали в атмосфере газообразного водорода. Реакционную взвесь фильтровали для удаления катализатора. Добавляли деионизированную воду (1300 кг) и 48% водный раствор гидроксида натрия (60 кг) для осаждения продукта, который выделяли фильтрацией и промывали смесью деионизированной воды (210 кг), ацетона (83 кг) и дополнительной деионизированной воды (710 кг) для получения 3-{[1-метилпирролидин-2(R)-ил]метил}-5-(2-фенилсульфонилэтил)-1Н-индола (220 кг влажного, 157,65 кг сухого, 78,41%). Пример 6 3-{[1-Метилпирролидин-2(R)-ил]метил}-5-(2-фенилсульфонилэтил)-1Н-индол (15 кг) и 2-бутанон (204 кг) загружали в сухой остеклованный сосуд. Добавляли раствор 48% водной бромистоводородной кислоты (6,45 кг) в 2-бутаноне (63 кг) и полученную взвесь подвергали азеотропной перегонке до остаточного объема 150 литров. Реакционную смесь охлаждали до 17,5°C и продукт выделяли фильтрацией. Продукт промывали 2-бутаноном (16 кг) для получения ДКС: Наблюдали один основной эндотерм с максимумом пика в диапазоне 173°С-179°С, показательный для Пример 7
ДКС: Наблюдали один основной эндотерм с максимумом пика в диапазоне 173°С-179°С, показательный для Пример 8 Бромгидрат 3-{[1-метилпирролидин-2(R)-ил]метил}-5-(2-фенилсульфонилэтил)-1Н-индола (10 кг), 2-бутанон (63 кг) и деионизированную воду (0,65 кг) загружали в сухой остеклованный сосуд и нагревали до 67,5°С для образования раствора. Раствор затем охлаждали до 60°С и добавляли еще 2-бутанон (42 кг) для осаждения продукта. Полученную взвесь подвергали азеотропной перегонке до остаточного объема реакционной смеси 50 литров и затем охлаждали до 22,5°С. Продукт выделяли фильтрацией и промывали 2-бутаноном (10,5 кг) для получения ДКС: Наблюдали один основной эндотерм с максимумом пика в диапазоне 173°С-179°С, показательный для Сравнительный пример 3-{[1-Метилпирролидин-2(R)-ил]метил}-5-(2-фенилсульфонилэтил)-1Н-индол (274 кг) и ацетон (3419 кг) загружали в сухой остеклованный сосуд. Добавляли раствор 48% водной бромистоводородной кислоты (114,7 кг) в ацетоне (1383 кг) при температуре от 50 до 55°С в течение 1 часа и полученную суспензию перемешивали в течение 4 часов. Реакционную смесь охлаждали до от 30 до 35°С и продукт выделяли фильтрацией. Продукт промывали ацетоном (861 кг) для получения ДКС: Наблюдали один основной эндотерм с максимумом пика в диапазоне 173°С-179°С, показательный для
Формула изобретения
1. Способ получения 2. Способ по п.1, где содержание воды в реакционной смеси уменьшено до менее чем 0,5 мас.% /мас. на стадии (b). 3. Способ по п.1 или 2, включающий дополнительную стадию суспендирования продукта стадии (b) в орошающем толуоле и удаления части толуола перегонкой. 4. Способ по п.3, где, по меньшей мере, 12% толуола удаляют. 5. Способ по любому из предшествующих пунктов, где исходное вещество элетриптана получают обработкой раствора (R)-5-(2-фенилсульфонилэтенил)-3-(N-метилпирролидин-2-илметил)-1Н-индола в ацетоне палладием на угле и метансульфоновой кислотой в атмосфере водорода. 6. Способ по п.5, где (R)-5-(2-фенилсульфонилэтенил)-3-(N-метилпирролидин-2-илметил)-1Н-индол получают обработкой раствора (R)-1-ацетил-5-(2-фенилсульфонилэтенил)-3-(N-метилпирролидин-2-илметил)-1H-индола в метаноле карбонатом калия. 7. Способ по п.6, где (R)-5-(2-фенилсульфонилэтенил)-3-(N-метилпирролидин-2-илметил)-1Н-индола очищают колоночной хроматографией. 8. Способ по п.6 или 7, где (R)-1-ацетил-5-(2-фенилсульфонилэтенил)-3-(N-метилпирролидин-2-илметил)-1Н-индол получают обработкой раствора (R)-1-ацетил-5-бром-3-(N-метилпирролидин-2-илметил)-1Н-индола в ацетонитриле триэтиламином, три-о-толилфосфином, ацетатом палладия и фенилвинилсульфоном. 9. Способ по п.8, где (R)-1-ацетил-5-бром-3-(N-метилпирролидин-2-илметил)-1Н-индол получают обработкой раствора (R)-5-бром-3-(N-метилпирролидин-2-илметил)-1Н-индола в ацетонитриле ангидридом уксусной кислоты и триэтиламином. 10. Способ получения 11. Способ по п.10, включающий дополнительную стадию суспендирования продукта стадии (b) в орошающем толуоле и удаления части толуола перегонкой.
MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 13.07.2009
Извещение опубликовано: 27.07.2010 БИ: 21/2010
|
||||||||||||||||||||||||||
 -ПОЛИМОРФНОГО БРОМГИДРАТА ЭЛЕТРИПТАНА
-ПОЛИМОРФНОГО БРОМГИДРАТА ЭЛЕТРИПТАНА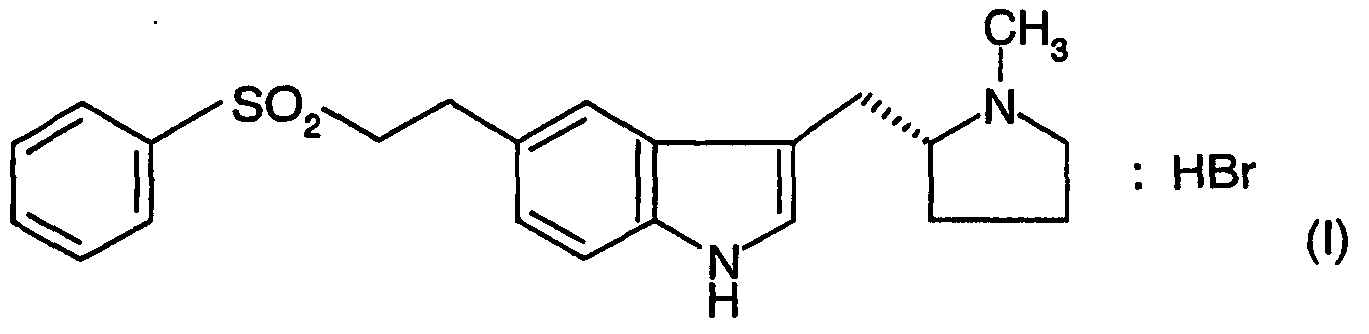
 .
.