Патент на изобретение №2159770
|
||||||||||||||||||||||||||
(54) ЗАМЕЩЕННЫЕ БИФЕНИЛСУЛЬФОНАМИДЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, СПОСОБ АНТАГОНИСТИЧЕСКОГО ВОЗДЕЙСТВИЯ НА ЭНДОТЕЛИН
(57) Реферат: Описываются новые замещенные бифенилсульфонамиды общей формулы I, где один из X и Y – кислород, другой – азот; R1 и R2 – каждый непосредственно связан с атомом углерода кольца и каждый независимо друг от друга обозначает водород или низший алкоксил, R3 и R4 каждый связаны с атомом углерода кольца и каждый независимо друг от друга обозначает низший алкил, R12, R12 и R14 каждый обозначает водород, R11 – водород или низший алкил, который может быть замещен Z1, Z2, Z3, где Z1, Z2, Z3 каждый обозначает независимо друг от друга водород, или Z1 – группа Z4-NZ7Z8 или Z4-N(Z11) Z5-Z6, где Z4 – одинарная связь, Z5 – одинарная связь или группа C=O, Z6 – низший алкил, алкил, замещенный 1-3 атомами галогена, циклоалкилалкил, Z7 – водород, Z8 – низший алкил, или Z1 – 2-оксопирролидинил или замещенный пиразол, J, K, L, T и U – каждый независимо друг от друга обозначает N или C, при условии, что, по крайней мере, один обозначает N и самое большее два обозначают N и, если только один из J, K, L, T, U обозначает N, то N может быть замещен O–, так что образуется N-оксид, p – ноль. Соединения 1 ингибируют активность эндотелина. Описываются фармацевтические композиции на их основе и способ антагонического воздействия на эндотелин. 3 с. и 6 з.п. ф-лы, 1 табл. 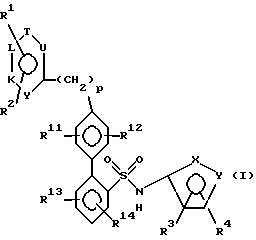
Настоящее изобретение относится к антагонистам эндотелина, пригодным также для лечения гипертонии. Соединения формулы (I): 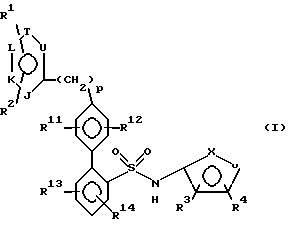 их энантиомеры и диастереомеры, и их фармацевтически приемлемые соли являются антагонистами рецептора эндотелина, пригодными, между прочим, в качестве антигипертонических агентов. На всем протяжении настоящего описания вышеуказанные символы имеют следующие значения: один из X и Y обозначает азот, а другой – кислород; R1 и R2, каждый, прямо связаны с атомом углерода кольца и, каждый независимо друг от друга, обозначают: водород или низший; алкоксил; R3 и R4, каждый прямо связаны с атомом углерода кольца и, каждый, независимо друг от друга, обозначает низший алкил; R12, R13 и R14, каждый обозначает атом водорода; R11 обозначает водород, низший алкил, или формил, который может быть замещен Z1, Z2 и Z3, где Z1 Z2 и Z3, каждый, независимо друг от друга, обозначает водород или Z1 обозначает замещенный 2-оксопирролидинил, или замещенный пиразол, или группу Z4-NZ7Z8 или Z4-N(Z11)-Z5-Z6, где Z4 – одинарная связь, Z5 обозначает одинарную связь или группу C=O; Z6 обозначает низший алкил; алкил, замещенный одним, двумя или тремя атомами галогенов, циклоалкилалкил; Z7 обозначает водород, Z8 обозначает низший алкил; J, K, L, T и U, каждый, независимо друг от друга, обозначают N или C, при условии, что, по крайней мере, один обозначает N и самое большее два обозначают N; и если только один из J, K, L, T и U обозначает N, то N может быть замещен -O–, так что образуется N-оксид; “p” обозначает 0. Ниже приводятся значения терминов, используемых в настоящем описании. Эти значения относятся к терминам, используемым на протяжении настоящего описания, индивидуально или в качестве части другой группы, если не указано ничего другого в особых случаях. Выражение “низший алкил” относится к алкильным группам с 1-4 атомами углерода. Выражение “низший алкоксил” относится к (низший алкил) – O-группе. Термины “галоген” и “halo” относятся к фтору, хлору, брому и иоду. На всем протяжении описания группы и их заместители выбирают так, чтобы обеспечить стабильные составляющие (части) и соединения. Соединения формулы (1) образуют соли, которые также входят в рамки настоящего изобретения. Предпочтительны фармацевтически приемлемые (т.е. нетоксичные, физиологически приемлемые) соли, хотя также пригодны другие соли, например, при выделении или очистке соединений настоящего изобретения. Соединения формулы (1) могут образовывать соли со щелочными металлами, такими, как натрий, калий и литий; со щелочноземельными металлами, такими, как кальций и магний; с органическими основаниями, такими, как дициклогексиламин, трет. – бутиламин, бензатин, N-метил-D-глюкамид и гидрабамин; и с аминокислотами, такими, как аргинин, лизин и т.п. Такие соли могут быть получены путем взаимодействия соединения формулы (1) с желательным ионом в среде, в которой соль осаждается, или в водной среде с последующей лиофилизацией. Некоторые из заместителей R1-R4 и R11-R14 соединения формулы (I) могут содержать асимметрические атомы углерода. Такие соединения формулы (I) могут существовать, следовательно, в энантиомерной и диастереомерной формах и в виде их рацемических смесей. Все они входят в рамки настоящего изобретения. Дополнительно, соединения формулы (1) могут существовать в виде энантиомеров даже в отсутствие асимметрических атомов углерода. Все такие энантиомеры также входят в рамки настоящего изобретения. Соединения формулы (1) являются антагонистами ET-1, ET-2 и/или ET-3 и пригодны для лечения состояний, связанных с увеличивающимися уровнями ET (эндотелина) (например, диализ, травмы и хирургия), и всех зависимых от эндотелина нарушений. Таким образом они пригодны в качестве антигипертонических агентов. Путем введения композиции, содержащей одно (или в комбинации) из соединений настоящего изобретения, понижают кровяное давление у больных гипертонией млекопитающих (например, людей). Они также пригодны при индуцированной беременностью гипертонии и коме (предэклампсия и эклампсия), острой воротной гипертонии и вторичной гипертонии при лечении с помощью эритропоетина. Соединения настоящего изобретения также пригодны для лечения нарушений, связанных с почечной, гломерулярной и мезангиальной клеточной дисфункцией, включая острую и хроническую почечную недостаточность, гломерулярное повреждение, вторичное почечное повреждение в старческом возрасте или связанное с диализом, нефросклероз (главным образом, гипертонический нефросклероз), нефротоксичность (в том числе нефротоксичность, связанную с агентами изображения и контраста [imaging and contrast agents] и с циклоспоринами), почечную ишемию, первичный везикоуретральный рефлюкс, гломерулосклероз и т.п. Соединения настоящего изобретения также могут быть пригодны для лечения нарушений, связанных с паракринной и эндокринной функциями. Соединения настоящего изобретения также пригодны для лечения эндотоксемии или эндоксинового шока, также, как геморрагического шока. Соединения настоящего изобретения также пригодны при гипоксической и ишемической болезни, и в качестве антиишемических агентов для лечения, например, ишемии сердца, почек и головного мозга, и повторной перфузии (такой, как повторная перфузия, происходящая вследствие сердечно-легочной обходной хирургии), коронарного и церебрального ангиоспазма и т.п. Кроме того, соединения настоящего изобретения также могут быть пригодны в качестве антиаритмических агентов, агентов против ангины, антифибриллярных агентов, антиастматических агентов, агентов против атеросклероза и артериосклероза, добавок к кардиоплегическим растворам для сердечно-легочных обходов, дополнений к тромболитической терапии, и агентов против диареи. Соединения настоящего изобретения могут быть пригодны для терапии инфаркта миокарда, для терапии периферических сосудистых заболеваний (например, болезнь Рейно и синдром Такаясу), для лечения гипертрофии сердца (например, гипертрофическая кардиомиопатия), для лечения первичной легочной гипертонии (например, плексогенической, эмболической) у взрослых и у новорожденных, и вторичной легочной гипертонии при сердечной недостаточности, радиационном и химиотерапевтическом повреждении или других травмах; для лечения сосудистых нарушений центральной нервной системы, таких, как паралич, мигрень и кровотечение в подпаутинном пространстве, для лечения бихивеоральных расстройств центральной нервной системы, для лечения желудочно-кишечных заболеваний, таких, как язвенный колит, болезнь Крона, повреждение слизистой оболочки желудка, язвенная и ишемическая болезнь кишечника, для лечения болезней желчного пузыря или желчных протоков, таких, как холангит, для лечения панкреатита; для регуляции роста клеток, для лечения не дающей рецидивов (излечимой) гипертрофии предстательной железы, для лечения рецидива стеноза вследствие ангиопластики или после любых процедур, включая трансплантацию, для терапии застойной сердечной недостаточности, в том числе для ингибирования фиброза, для ингибирования расширения, реконструкции и дисфункции левого желудочка сердца, и для лечения гепатотоксичности и предупреждения скоропостижной смерти. Соединения настоящего изобретения могут быть пригодны для лечения заболевания серповидными эритроцитами, включая инициирование и/или развитие болевых кризов этого заболевания, для лечения вредных (разрушительных) последствий продуцирующих ET опухолей, таких, как образующиеся при гипертонии из гемангиоперицитомы; для лечения раннего и прогрессирующего заболевания и поражения печени, включая сопровождающие осложнения (например, гепатотоксичность, фиброз и цирроз), для лечения спастических заболеваний мочевого тракта и/или мочевого пузыря, для лечения гепаторенальных синдромов, для лечения иммунологических заболеваний, включая васкулит (воспаление сосудов), такой, как волчанка, системный склероз, смешанная криоглобулинемия, и для лечения фиброза, ассоциированного с почечной дисфункцией и гепатотоксичностью. Соединения настоящего изобретения могут быть пригодны для терапии метаболических и неврологических нарушений; для терапии рака; для лечения инсулинзависимого и инсулиннезависимого сахарного диабета, невропатии, ретинопатии, синдрома одышки у беременных, дисменореи, эпилепсии, гемморагических и ишемических приступов; для реконструирования костей; для лечения псориаза и хронических воспалительных заболеваний, таких, как ревматоидный артрит, остеоартрит, саркоидозный и экзематозный дерматит (все типы дерматита). Соединения настоящего изобретения также могут быть сформулированы в комбинации с ингибиторами преобразующих эндотелии ферментов (ECE), такими, как фосфорамидон; с антагонистами тромбоксановых рецепторов; с агентами открытия калиевых каналов; с ингибиторами тромбина (например, гирудин и т.п.); с ингибиторами факторов роста, такими, как модуляторы PDCF-активности; с антагонистами фактора активации тромбоцитов (PAF); с антагонистами рецепторов ангиотензина-II (AII); с ингибиторами ренина; с ингибиторами ферментов преобразования ангиотензина (ACE), такими, как каптоприл, зофеноприл, фозиноприл, церанаприл, алацеприл, эналаприл, делаприл, пентоприл, хинаприл, рамиприл, лизиноприл и соли этих соединений; с ингибиторами нейтральной эндопептидазы (NEP); с ингибиторами пары NEP-ACE; с ингибиторами HMG-CoA-редуктазы, такими, как правастатин и мевакор; с ингибиторами скваленсинтетазы; с секвестрантами желчной кислоты, такими, как квестран; с блокирующими кальциевые каналы средствами; с активаторами калиевых каналов; с бета-адренергическими агентами; с антиаритмическими агентами; с диуретиками, такими, как хлортиацид, гидрохлортиацид, флуметацид, гидрофлуметацид, бендрофлуметацид, метилхлортиацид, трихлорметиацид, политиацид или бензотиацид, также, как этакриновая кислота, трикринафен, хлорталидон, фуросемид, музолимин, буметанид, триамтерен, амилорид и спиронолактон, и соли таких соединений; и с тромболитическими агентами, такими, как тканевый плазминогенный активатор (tPA), рекомбинант tPA, стрептокиназа, урокиназа, проурокиназа и анизоилированный плазминогенный стрептокиназный комплекс – активатор (APSAC). Если формулируют в определенной дозе, то для комбинированных продуктов используют соединения настоящего изобретения в нижеуказанных дозировочных пределах, а другие фармацевтически активные агенты – в указанных для них дозировочных пределах. Соединения настоящего изобретения также могут быть сформулированы с, или пригодны в сочетании с, противогрибковыми и иммуносупрессивными агентами, такими, как амфотерицин B, циклоспорины и т.п., для противодействия гломурулярному сжатию и вторичной нефротоксичности за счет таких соединений. Соединения настоящего изобретения также могут быть использованы в сочетании с гемодиализом. Соединения настоящего изобретения можно вводить орально или парентерально различным известным видам млекопитающих, которые больны, например, людям, в эффективном количестве в пределах доз около 0,1-100 мг/кг, предпочтительно примерно 0,2-50 мг/кг и наиболее предпочтительно примерно 0,5-25 мг/кг (или примерно от 1 до примерно 2500 мг, предпочтительно примерно от 5 до примерно 2000 мг), в виде разовых или в виде разделенных на 2-4 части суточных доз. Активные вещества могут быть использованы в композиции, такой, как таблетка, капсула, раствор или суспензия, содержащей примерно 5-500 мг на дозировочную единицу соединения или смеси соединений формулы (1), или в топической форме для заживления ран (0,01-5 мас.% соединения формулы (1), 1-5 обработок в день). Они могут быть смешаны обычным образом с физиологически приемлемой основой для приготовления лекарственного средства или носителем, эксципиентом, связующим, консервантом, стабилизатором, вкусовым веществом и т. д. , или с топическим носителем, таким, как Plastibase (минеральное масло, желатинированное с помощью полиэтилена), как требует общепринятая фармацевтическая практика. Соединения изобретения также можно вводить топически для лечения периферических сосудистых заболеваний и они также могут быть сформулированы в виде крема или мази. Соединения формулы (1) также могут быть сформулированы в виде композиций, таких, как стерильные растворы или суспензии для парентерального введения. Примерно 0,1-500 мг соединения формулы (1) смешивают с физиологически приемлемой основой для приготовления лекарственного средства, носителем, эксципиентом, связующим, консервантом, стабилизатором и т.д. в виде дозировочной единицы, приготовляемой согласно требованиям общепринятой фармацевтической практики. Количество активного вещества в этих композициях или препаратах такое, что получается желательная дозировка в указанных пределах. Что касается данных, затребованных в запросе, то, заявитель обращает внимание на табл. 1, в которой активность представителей рассматриваемых соединений как антагонистов эндотелина (“ETA Ki” представляет собой константу связывания EPA-рецепторов). Представлена в табл. 1 (см. в конце описания). Соединения настоящего изобретения могут быть получены методами, аналогичными описанным в заявке США с порядковым номером 08/493331, зарегистрированной 24 июля 1995 г., Murugesan и др., (Attorney Docket N HA662c), включенной здесь в виде ссылки, или такими способами, как описанные ниже. В любом из нижеприведенных способов R11, R12, R13 и/или R14 в любой пригодный момент могут быть превращены в другую R11, R12, R13 и/или R14-группу, так как описано в вышеуказанной заявке США с порядковым номером 08/493331. Например, R11-группа, которая представляет собой алкил, может быть превращена в R11-группу, которая является замещенным амино или амидной группой алкилом, с помощью известных в технике способов. A. Как представлено в схеме 1, целевые соединения формулы 4 могут быть получены путем катализируемой с помощью Pd(O) реакции сочетания соответствующим образом защищенной фенилсульфонамид-2-борной кислоты в качестве промежуточного соединения формулы 2 с 4-гетероцикло-арилгалогенидом формулы 1. Реакцию сочетания осуществляют в присутствии пригодного основания, такого, как водный раствор карбоната калия, и растворителя, такого, как смесь толуола с этанолом. Результирующее соединение формулы 3 превращают в целевое соединение формулы 4 путем удаления защитной группы. Если группу борной кислоты в промежуточном соединении формулы 2 заменяют триалкилолово-составляющей, то промежуточное соединение формулы 2 также можно вводить в реакцию сочетания с соединением формулы 1, где OSO2CF3 заменена на галоидную группу. Промежуточная борная кислота формулы 2, указанная в схеме 1, может быть получена из 2-бромфенилсульфонамида формулы 5 (получение которого описано в Европейской патентной заявке N 0569193 (1993)), как указано в схеме II, путем получения литийорганического соединения с помощью пригодного алкиллития (такого, как н-бутиллитий), последующей обработки с помощью триалкилбората (например, триизопропилбората) и, в заключение, добавления водной кислоты, такой, как водная соляная кислота: (В приведенных схемах I и II, “Prot” обозначает соответствующую защитную группу для сульфонамидной функции. Это также описано в Европейской патентной заявке N 0569193 (1993)). B. Целевые соединения также могут быть получены альтернативными путями, указанными в схемах III и IV: Как представлено в схеме III, 4′-гетероцикло-арилгалогенид формулы 1 может быть превращен в промежуточную борную кислоту формулы 6 через указанную последовательность реакций. Это соединение формулы 6, путем катализируемой с помощью Pd(O) реакции сочетания с соединением формулы 5, дает биарильный аналог формулы 3. Путем удаления защитной группы из биарильного аналога 3 получают целевое соединение формулы 4. Если группу борной кислоты в соединении формулы 6 заменяют триалкилолово-составляющей, то соединение формулы 6 также можно вводить в реакцию сочетания с соединением формулы 5, где OSO2C3F3 заменена галоидной группой. Как представлено в схеме IV, целевое соединение формулы 4′ может быть получено путем катализируемой с помощью Pd(O) реакции сочетания производного борной кислоты формулы 2 с 1,4-дибромбензолом формулы 7 с получением 4′-бромбифенильного производного формулы 8. Катализируемая с помощью Pd(O) реакция сочетания соединения формулы 8 с любым соединением – гетероциклической борной кислотой или гетероциклическим производным олова (где R15 обозначает низший алкил), затем приводит после удаления защитной группы к целевому соединению формулы 4′. (В отношении общих приемов в биарильном синтезе см., например, Bringmann и др. , Angew.Chem. Int., Ed. Engl. 29 (1990), 977-991. В отношении гетероциклов см., например, В.Н.Калинин, Synth., 413-432 (1992) и Т.R.Bailey, Tet. Lett., 27 (1986), 4407-4410). C. 4′-Гетероцикло-арил-галогенид формулы 1 может быть получен путем использования ряда способов, описанных в литературе. Несколько характерных примеров приводится ниже: 1). Как показано в схеме V, катализируемая с помощью Pd(O) перекрестная реакция сочетания 4-бром-фенилборной кислоты 10 с гетероциклическим галогенидом формулы 9 в присутствии основания и пригодных растворителей приводит к 4′-гетероцикло-арил-галогениду формулы 1′. Множество 4′-гетероциклов, таких, как пиридины и пиримидины, может быть получено при использовании этого метода. См., например, Mitchell и Wallbank, Tet Lett., 32, 2273 (1991). Как видно, гетероциклическая борная кислота или родственное производное олова формулы 9 может быть введена в реакцию сочетания с 1,4-дибромбензолом формулы 7 в присутствии Pd(O) с получением 4′-гетероцикло-арил-галогенида формулы 1′ (где p=0). Соединения формулы 9 каждое, имеются в продаже или могут быть получены известными в уровне техники способами. См. “The Chemistry of Heterocyclic Compounds”, John Wiley and Sons. В некоторых отдельных случаях борная кислота может быть заменена производным олова для осуществления реакции сочетания. (Снова, по поводу общих приемов в биарильном синтезе см. : Bringmann и др., Angew.Chem. Int. Ed. Engl. 29 (1990), 977-991). 4′-Гетероцикло-арил-галогенид формулы 1 также может быть получен из соответствующего соединения, где в положении 4′ вместо гетероцикла находится реактивная группа, такая, как цианогруппа. Такое получение можно осуществлять путем превращения реактивной группы в желательный 4′-гетероцикл известными в технике способами. Например, 4′-циано-арил-галогенид может быть превращен в соответствующий 4′-амидино-арил-галогенид:  и последний может быть превращен в 4′-пиримидин-арил-галогенид формулы 1 способами, такими, как способы, описанные Wagner и др. Chem.Ber. 104, 2975-2983 (1971)), включенные здесь в виде ссылки. D. 4′-алкилпиридиновые или пиримидиновые целевые соединения также могут быть получены альтернативным путем, указанным в схеме VII. В этих соединениях K, L и T обозначают углерод, по крайней мере, один из J и U обозначает азот, и “p” = 1 или 2. Как видно из схемы VII, обработка 2-галоген-пиридина или 2-галогенпиримидина формулы 11, с помощью илида Виттига формулы 12, получаемого известными в технике способами, с последующим гидролизом промежуточного соединения, осуществляемым при использовании водного основания, такого, как карбонат натрия, приводит к соответствующему 4-бромфенил-алкилпиридину/пиримидину формулы 1”. Катализируемая с помощью Pd(O) перекрестная реакция сочетания соединения формулы 1” с соединением формулы 2 с последующим удалением защитной группы дает целевое соединение формулы 4” (см. например, E.C.Taylor и S.F.Martin, J.Am. Chem. Soc., 94 (1972), 2874). E. Типичный пример образования N-оксида представлен в схеме VIII: R=CH3, м-хлорфенил, и т.д. Соответствующие пиридиновые аналоги могут быть окислены до соответствующих N-оксидных производных при применении ряда известных окислителей, таких, как м-хлорнадбензойная кислота или надуксусная кислота. Схема VIII также применима для других изомерных пиридиновых аналогов. Теперь изобретение описывается далее с помощью нижеследующих примеров осуществления, которые представляют собой предпочтительные варианты изобретения. Эти примеры приводятся с целью иллюстрации и не ограничивают объема охраны изобретения. ПРИМЕР 1 N-(3,4-Диметил-5-изоксазолил)-4′-(2-пиримидинил)[1,1′-бифенил] – 2-сульфонамид 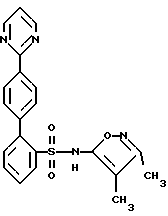 A. 2-(4-Бромфенил)пиримидин К раствору 0,8 г (4,0 ммоль) п-бром-фенилборной кислоты и 0,23 г (0,2 ммоль) тетракис(трифенилфосфин) палладия (O) в 20 мл толуола, в атмосфере аргона, добавляют 10 мл 2 М водного раствора карбоната натрия, затем 1,0 г (6,23 ммоль) 2-бром-пиримидина в 10 мл 95%-ного этанола. Смесь кипятят с обратным холодильником в течение 1,5 часов, разбавляют с помощью 100 мл воды и экстрагируют 3 раза по 50 мл этилацетатом. Объединенные органические экстракты промывают один раз с помощью 100 мл рассола, сушат и выпаривают. Остаток хроматографируют на 100 г силикагеля, используя смесь гексана с этилацетатом в соотношении 3: 1, с получением 0,82 г соединения A в виде белого твердого вещества. В. N-(3,4-Диметил-5-изоксазолил)-N-[(2-метоксиэтокси)-метил]-4′-(2-пиридимидинил)[1,1′-бифенил]-2-сульфонамид К раствору 0,35 г (0,91 ммоль) 2-бор-N-(3,4-диметил-5-изоксазолил)-N-[(2-метоксиэтокси)метил] бензолсульфонамида и 0,052 г (0,045 ммоль) тетракис(трифенилфосфин) палладия (O) в 20 мл толуола, в атмосфере аргона, добавляют 10 мл 2 М водного раствора карбоната натрия, затем 0,278 г (1,18 ммоль) соединения A в 10 мл 95%-ного этанола. Смесь кипятят с обратным холодильником в течение 1,5 часов, разбавляют с помощью 100 мл воды и экстрагируют 3 раза по 50 мл этилацетатом. Объединенные органические экстракты промывают один раз с помощью 100 мл рассола, сушат и выпаривают. Остаток хроматографируют на 75 г силикагеля, используя смесь гексана с этилацетатом в соотношении 2:1, с получением 0,14 г (39%) соединения В в виде бесцветной смолы. C. N-(3,4-Диметил-5-изоксазолил)-4′-(2-пиридимидинил)-[1,1′-бифенил]-2-сульфонамид К раствору 0,13 г (0,25 ммоль) соединения В в 6 мл 95%-ного этанола добавляют 6 мл 6 н. водного раствора соляной кислоты и кипятят с обратным холодильником в течение 1,5 часов. Смесь затем концентрируют и разбавляют с помощью 10 мл воды, и раствор нейтрализуют до установления pH 7, используя водный раствор бикарбоната натрия. Смесь затем снова подкисляют до pH 4, используя ледяную уксусную кислоту, и раствор экстрагируют 3 раза по 50 мл этилацетатом. Объединенные органические экстракты промывают один раз водой, сушат и выпаривают (0,11 г). Этот материал очищают путем препаративной ВЭЖХ (высокоэффективной жидкостной хроматографии) с обращенной фазой на колонке размером 30х500 мм, заполненной ODS S10, используя 70% растворителя В (90% метанола, 10%, 0,1% трифторуксусной кислоты) и 30% растворителя А (10% метанола, 90% воды, 0,1% трифторуксусной кислоты). Соответствующие фракции собирают и нейтрализуют с помощью водного раствора бикарбоната натрия до установления pH 7 и концентрируют до объема 10 мл. Раствор затем подкисляют до pH 4, используя ледяную уксусную кислоту, и твердое вещество белого цвета отфильтровывают и высушивают, получая 0,09 г (87%) целевого соединения. Т. пл. >210oC. 1H-ЯМР (CDCl3):  1,70 (с, 3H), 2,00 (с, 3H), 6,49 (ш.с, 1H), 7,09-8,70 (м, 10H).
13C-ЯМР (CDCl3): 6,6, 10,8, 108,5, 119,4, 127,3, 127,8, 128,0, 129,0, 130,3, 132,5, 133,1, 137,4, 138,0, 140,7, 141,0, 154,1, 157,3, 161,8, 164,1.
ПРИМЕР 2 1,70 (с, 3H), 2,00 (с, 3H), 6,49 (ш.с, 1H), 7,09-8,70 (м, 10H).
13C-ЯМР (CDCl3): 6,6, 10,8, 108,5, 119,4, 127,3, 127,8, 128,0, 129,0, 130,3, 132,5, 133,1, 137,4, 138,0, 140,7, 141,0, 154,1, 157,3, 161,8, 164,1.
ПРИМЕР 2N-(3,4-Диметил-5-изоксазолил)-4′-(2-пиридинил)[1,1′-бифенил] -2-сульфонамид 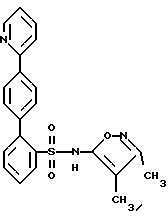 А. 2-(4-Бромфенил)пиридин К раствору 2,0 г (9,96 ммоль) п-бром-фенилборной кислоты и 0,57 г (0,5 ммоль) тетракис(трифенилфосфин) палладия (O) в 20 мл толуола, в атмосфере аргона, добавляют 15 мл 2 М водного раствора карбоната натрия, после чего добавляют 3,16 г (20 ммоль) 2-бромпиридина в 15 мл 95%-ного этанола. Смесь кипятят с обратным холодильником в течение 2 часов, разбавляют с помощью 100 мл воды и экстрагируют 3 раза по 50 мл этилацетатом. Объединенные органические экстракты промывают один раз с помощью 100 мл рассола, сушат и выпаривают. Остаток хроматографируют на 100 г силикагеля, используя смесь гексана с этилацетатом в соотношении 4:1, с получением 2,3 г соединения A в виде слегка желтоватого твердого вещества, которое при стоянии затвердевает. B. N-(3,4-Диметил-5-изоксазолил)-N-[(2-метоксиэтокси)метил] -4′-(2-пиридинил)[1,1′-бифенил]-2-сульфонамид К раствору 1,47 г (3,84 ммоль) 2-бор-N-(3,4-диметил-5-изоксазолил)-N-[(2-метоксиэтокси)метил] бензолсульфонамида и 0,173 г (0,15 ммоль) тетракис(трифенилфосфин) палладия (O) в 40 мл толуола, в атмосфере аргона, добавляют 20 мл 2 М водного раствора карбоната натрия, после чего добавляют 1,0 г (4,27 ммоль) соединения A в 20 мл 95%-ного этанола. Смесь кипятят с обратным холодильником в течение 2 часов, разбавляют с помощью 100 мл воды и экстрагируют 3 раза по 50 мл этилацетатом. Объединенные органические экстракты промывают один раз с помощью 100 мл рассола, сушат и выпаривают. Остаток хроматографируют на 50 г силикагеля, используя смесь гексана с этилацетатом в соотношении 2:1, с получением 0,91 г (47%) соединения B в виде бесцветной смолы. C. N-(3,4-Диметил-5-изоксазолил)-4′-(2-пиридинил)[1,1′-бифенил] -2-сульфонамид К раствору 0,9 г (1,78 ммоль) соединения B в 12 мл 95%-ного этанола добавляют 12 мл 6 н. водного раствора соляной кислоты и смесь кипятят с обратным холодильником в течение 2 часов. Смесь затем концентрируют и разбавляют с помощью 25 мл воды и раствор нейтрализуют до pH 7, используя водный раствор бикарбоната натрия. Смесь подкисляют до pH 4, используя ледяную уксусную кислоту, и раствор затем экстрагируют 3 раза по 50 мл этилацетатом. Объединенные органические экстракты промывают один раз водой, сушат и выпаривают (0,75 г). Остаток хроматографируют на 25 г силикагеля, используя смесь гексана с этилацетатом в соотношении 3:2, с получением 0,53 г (73%) целевого соединения. Т.пл. = 85-90oC. Анализ, рассчитанный для C22H19N3O3S  1,17H2O 1,17H2OРассчитано,%: C 61,94; H 5,04; N 9,85; S 7,52. Найдено,%: C 62,10; H 4,66; N 9,69; S 7,81. ПРИМЕР 3 N-(3,4-Диметил-5-изоксазолил)-4′-(3-пиридинил)[1,1′-бифенил] -2-сульфонамид 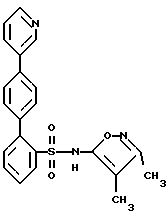 A. 3-(4-Бромфенил)пиридин К раствору 4-бромфенилборной кислоты (1,41 г, 7 ммоль), 3-бромпиридина (6,64 г, 42 ммоль) в 50 мл толуола и 40 мл 95%-ного этанола, в атмосфере аргона, добавляют тетракис(трифенилфосфин) палладий (О) (809 мг, 0,7 ммоль), после чего добавляют 30 мл 2 М водного раствора бикарбоната натрия. Реакционную смесь нагревают при 85oC в течение 1,5 часов, охлаждают и разбавляют с помощью 150 мл этилацетата. Органическую жидкость отделяют и промывают с помощью 20 мл воды и 20 мл рассола, сушат и концентрируют. Остаток хроматографируют на силикагеле, используя смесь 4:1 гексана с этилацетатом, с получением соединения A (1,5 г, 92%) в виде бесцветного масла. B. N-(3,4-Диметил-5-изоксазолил)-N-[(2-метоксиэтокси)метил] -4′-(3-пиридинил)[1,1′-бифенил]-2-сульфонамид К раствору 2-бор-N-(3,4-диметил-5-изоксазолил)-N-[(2-метоксиэтокси)метил] бензолсульфонамида (384 мг, 1,0 ммоль) соединения A (351 мг, 1,5 ммоль) в 9 мл толуола и 7,2 мл 95%-ного этанола в атмосфере аргона, добавляют тетракис(трифенилфосфин)палладия (О) (116 мг, 0,1 ммоль) с последующим добавлением 5,5 мл 2 М водного раствора карбоната натрия. Реакционную смесь нагревают при 75oC в течение 3 часов, охлаждают и разбавляют с помощью 50 мл этилацетата. Органическую жидкость отделяют и промывают с помощью 10 мл воды и 10 мл рассола, сушат и концентрируют. Остаток хроматографируют на силикагеле, используя смесь 1:2 гексана с этилацетатом, с получением соединения B (346 мг, 70%) в виде бесцветной смолы. C. N-(3,4-Диметил-5-изоксазолил)-4′-(3-пиридинил)1,1′-бифенил]-2-сульфонамид К раствору соединения B (346 мг, 0,70 ммоль) в 10 мл 95%-ного этанола, добавляют 10 мл 6 н. водного раствора соляной кислоты и кипятят с обратным холодильником в течение 1 часа. Реакционную смесь концентрируют и pH-значение раствора доводят до 8, используя раствор бикарбоната натрия. Его затем снова подкисляют до pH 5 с помощью ледяной уксусной кислоты. Смесь экстрагируют 3 раза по 40 мл этилацетатом. Органическую жидкость промывают с помощью 10 мл воды и 10 мл рассола, сушат и концентрируют. Остаток хроматографируют на силикагеле, используя смесь в соотношении 100:2,5 дихлорметана с метанолом, с получением целевого соединения (199 мг, 70%) в виде белого твердого вещества. Т.пл. 96-106oC (аморфное). Анализ, рассчитанный для C22H19N3O3S 0,44H2O:Рассчитано, %: C 63,92; H 4,83; N 10,17; S 7,76. Найдено, %: C 63,95; H 4,64; N 10,14; S 7,55. ПРИМЕР 4 4′-(4,6-Диметокси-2-пиримидинил)-N-(3,4-диметил-5-изоксазолил) [1,1′-бифенил]-2-сульфонамид 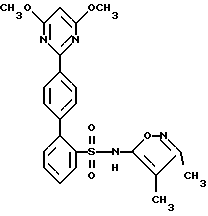 A. 2-Хлор-4,6-диметоксипиримидин К раствору 2-амино-4,6-диметоксипиримидина (12,41 г, 80 ммоль) в 66 мл концентрированной соляной кислоты, при температуре от -8oC до -12oC, в течение 30 минут добавляют раствор нитрита натрия (11,04 г, 160 ммоль) в 22 мл воды. Смесь перемешивают при комнатной температуре в течение ночи. Осадок отделяют путем отфильтровывания и промывают небольшим количеством воды. Твердое вещество растворяют в 200 мл дихлорметана, сушат (сульфат магния) и отфильтровывают, фильтрат концентрируют с получением соединения A (3,74 г, 27%) в виде белого твердого вещества. B. 2-(4-Бромфенил)-4,6-диметоксипиримидин К раствору 4-бромфенилборной кислоты (602 мг, 3 ммоль), соединения A (524 мг, 3 ммоль) в 22,5 мл толуола и 18 мл 95%-ного этанола, в атмосфере аргона, добавляют тетракис(трифенилфосфин)палладий (О) (208 мг, 0,18 ммоль), после чего добавляют 13,5 мл 2 М водного раствора карбоната натрия. Реакционную смесь нагревают при 80oC в течение 1,25 часа, охлаждают и разбавляют с помощью 60 мл этилацетата. Органическую жидкость отделяют и промывают с помощью 15 мл воды и 15 мл рассола, сушат и концентрируют. Остаток хроматографируют на силикагеле, используя смесь 4:1 гексана с дихлорметаном, с получением соединения B (170 мг, 19%) в виде белого твердого вещества. C. 4′-(4,6-Диметокси-2-пиримидинил)-N-(3,4-диметил-5-изоксазолил)-N-[(2-метоксиэтокси)метил][1,1′-бифенил]-2-сульфонамид К раствору 2-бop-N-(3,4-диметил-5-изоксазолил)-N-[(2-метоксиэтокси)метил] бензолсульфонамида (384 мг, 1,0 ммоль), соединения B (339 мг, 1,15 ммоль) в 9 мл толуола и 7,2 мл 95%-ного этанола, в атмосфере аргона, добавляют тетракис(трифенилфосфин)палладий (О) (116 мг, 0,1 ммоль), после чего добавляют 5,43 мл 2 М водного раствора карбоната натрия. Реакционную смесь нагревают при 75oC в течение 3 часов, охлаждают и разбавляют с помощью 50 мл этилацетата. Органическую жидкость отделяют, промывают с помощью 10 мл воды и 10 мл рассола, сушат и концентрируют. Остаток хроматографируют на силикагеле, используя смесь 3:1 гексана с этилацетатом, с получением соединения C (370 мг, 67%) в виде бесцветной смолы. D. 4′-(4,6-Диметокси-2-пиримидинил)-N-(3,4-диметил-5-изоксазолил)-[1,1′-бифенил]-2-сульфонамид К раствору соединения C (370 мг, 0,67 ммоль) в 15 мл 95%-ного этанола добавляют 15 мл 6 н. водного раствора соляной кислоты и кипятят с обратным холодильником в течение 45 минут. Реакционную смесь концентрируют и pH-значение раствора доводят до величины 8, используя раствор бикарбоната натрия. Смесь затем подкисляют до pH 5 с помощью ледяной уксусной кислоты. Смесь экстрагируют 3 раза по 50 мл этилацетатом. Органическую жидкость промывают с помощью 15 мл воды и 15 мл рассола, сушат и концентрируют. Остаток хроматографируют на силикагеле, используя смесь в соотношении 10:1 и затем в соотношении 5:1 дихлорметана с этилацетатом, с получением целевого соединения (185 мг, 60%) в виде белого твердого вещества. Т.пл. 83-93oC (аморфное). Анализ, рассчитанный для C23H22N4O5S: Рассчитано, %: C 59,22; H 4,75; N 12,01; S 6,87. Найдено, %: C 59,15; H 4,76; N 11,73; S 6,56. ПРИМЕР 5 N-(3,4-Диметил-5-изоксазолил-4′-(4-пиримидинил)[1,1′-бифенил]-2-сульфонамид 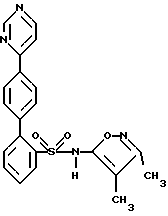 A. 4-(4-Бромфенил)пиримидин К находящемуся в высушенной в сушильной печи колбе и перемешиваемому при 0oC и в атмосфере аргона раствору 1,4- дибромбензола (3,6 г, 15 ммоль) в течение 8 минут прикапывают н-бутиллитий (1,6 М, в гексане; 8,0 мл; 13 ммоль). После перемешивания при 0oC в течение 30 минут, реакционный раствор охлаждают до -35oC и в течение 10 минут добавляют раствор пиримидина (1,2 г, 1,2 мл, 15 ммоль) в эфире (15 мл). После перемешивания при -35oC в течение 20 минут, реакцию прекращают с помощью воды (4,5 мл) и переносят в делительную воронку. Экстракция с помощью эфира (2 раза по 30 мл) и дихлорметана (30 мл) и высушивание объединенных органических слоев над сульфатом магния дают, после выпаривания растворителя, 4,2 г вещества. Остаток растворяют в ацетоне, и насыщенный ацетоновый раствор перманганата калия (примерно 30 мл) добавляют до тех пор, пока не будет оставаться фиолетовое окрашивание. Этот раствор кипятят с обратным холодильником в течение 10 минут. Реакционную смесь фильтруют через Целит  , который промывают ацетоном и этанолом, и фильтрат выпаривают с получением 2,3 г сырого продукта. Перекристаллизация из гептана дает 0,78 г (24%) соединения A. Т.пл. 81,0-83,5oC.
B. N-(3,4-Диметил-5-изоксазолил)-N-[(2-метоксиэтокси)метил] -4′-(4-пиримидинил) [1,1′-бифенил]-2-сульфонамид , который промывают ацетоном и этанолом, и фильтрат выпаривают с получением 2,3 г сырого продукта. Перекристаллизация из гептана дает 0,78 г (24%) соединения A. Т.пл. 81,0-83,5oC.
B. N-(3,4-Диметил-5-изоксазолил)-N-[(2-метоксиэтокси)метил] -4′-(4-пиримидинил) [1,1′-бифенил]-2-сульфонамидРаствор 2-бop-N-(3,4-диметил-5-изоксазолил)-N-[(2-метоксиэтокси)метил] бензолсульфонамида (1,0 г, 1,6 ммоль) в этаноле (через который пропускают путем барботирования в течение 20 минут аргон; 7,2 мл) добавляют к раствору соединения A (0,5 г, 2,0 ммоль) в толуоле (через который в течение 20 минут пропускают путем барботирования аргон; 14 мл). К этому раствору добавляют раствор карбоната натрия (0,45 г) в воде (через которую пропускают путем барботирования аргон в течение 20 минут; 7,2 мл), после чего добавляют тетракис(трифенилфосфин)палладий (О) (0,13 г, 0,11 ммоль). После кипячения с обратным холодильником в атмосфере аргона в течение 4,5 часов, раствор охлаждают и выливают в рассол (20 мл). Экстракция с помощью этилацетата (2 раза по 70 мл) и высушивание объединенных органических слоев над сульфатом магния, после выпаривания растворителя, дает 1,6 г сырого продукта. Флэш-хроматография (диоксид кремния; диаметр колонки 50 мм; 50% этилацетат/дихлорметан) дает 0,20 г (25%) соединения B. C. N-(3,4-Диметил-5-изоксазолил)-4′-(4-пиримидинил)[1,1′-бифенил]-2-сульфонамид Раствор соединения B (0,20 г, 0,40 ммоль) в этаноле (6,0 мл) и 6 н. соляной кислоты (6,0 мл) перемешивают при 80oC. Спустя 7 часов, этанол выпаривают в вакууме и остаток переносят в делительную воронку со смесью дихлорметана с водой. Водный слой доводят до pH 1,5 с помощью насыщенного раствора бикарбоната натрия. Экстракция с помощью дихлорметана (4 раза по 20 мл) и высушивание над сульфатом магния, после выпаривания растворителя, приводят к 0,24 г сырого продукта. Флэш-хроматография (диоксид кремния; диаметр колонки 15 мм; 8% метанол/дихлорметан) с последующей перекристаллизацией из горячего метанола дают 42 мг (26%) целевого соединения. Т. пл. = 211,0-214,0oC. Анализ, рассчитанный для C21H18N4O3S 0,16H2O:Рассчитано, %: C 61,61; H 4,51; N 13,68; S 7,83. Найдено, %: C 61,66; H 4,42; N 13,63; S 7,60. ПРИМЕР 6 N-(3,4-Диметил-5-изоксазолил)-4′-(2-пиразинил)[1,1′-бифенил]-2-сульфонамид 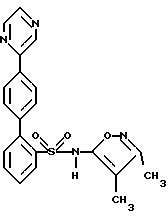 A. 2-(4-Бромфенил)пиразин К находящемуся в высушенной в сушильной печи колбе и перемешиваемому при 0oC и в атмосфере аргона раствору 1,4- дибромбензола (3,6 г, 15 ммоль) в эфире (22 мл) в течение 8 минут прикапывают н-бутиллитий (1,6 М в гексане; 8,0 мл; 13 ммоль). После перемешивания при 0oC в течение 30 минут, этот раствор в течение 13 минут добавляют к раствору пиразина (0,86 г; 11 ммоль; 0,86 мл) в тетрагидрофуране (34 мл), перемешиваемому при -78oC. Когда добавление закончено, через раствор барботируют сухой воздух (пропускаемый через слой сульфата кальция). Спустя 30 минут охлаждающую баню удаляют и затем, спустя следующие 30 минут, реакцию прекращают путем добавления воды (4,5 мл) и реакционную смесь переносят в делительную воронку. Экстракция дихлорметаном (2 раза по 50 мл) и высушивание объединенных органических слоев над сульфатом магния дают, после выпаривания растворителя, 3,9 г продукта. Флэш-хроматография (диоксид кремния; диаметр колонки 75 мм; 5% этилацетат/дихлорметан) дает 0,93 г (36%) соединения A. Т.пл. = 105,0-107,5oC. B. N-(3,4-Диметил-5-изоксазолил)-N-[(2-метоксиэтокси)метил] -4′-(2-пиразинил)[1,1′-бифенил]-2-судьфонамид Раствор 2-бop-N-(3,4-диметил-5-изоксазолил)-N-[(2-метоксиэтокси)метил] бензолсульфонамида (1,0 г, 1,6 ммоль) в этаноле (через который продувают путем барботирования аргон в течение 20 минут; 7,2 мл) добавляют к раствору соединения A (0,50 г, 2,0 ммоль) в толуоле (через который продувают путем барботирования аргон в течение 20 минут; 14 мл). К этому раствору добавляют раствор карбоната натрия (0,45 г) в воде (через которую в течение 20 минут продувают путем барботирования аргон; 7,2 мл), после чего добавляют тетракис(трифенилфосфин)палладий(O) (0,13 г; 0,11 ммоль). После кипячения с обратным холодильником в атмосфере аргона в течение 4 часов, раствор охлаждают и выливают в рассол (20 мл). Экстракция с помощью этилацетата (2 раза по 70 мл) и высушивание объединенных органических слоев над сульфатом магния дают, после выпаривания растворителя, 1,6 г сырого продукта. Флэш-хроматография (диоксид кремния; диаметр колонки 50 мм; 50% этилацетат/дихлорметан) дает 0,15 г (19%) соединения B. C. N-(3,4-Диметил-5-изоксазолил)-4′-(2-пиразинил)[1,1′-бифенил] -2-сульфонамид Раствор соединения B (0,15 г, 0,30 ммоль) в этаноле (4,6 мл) и 6 н. соляной кислоты (4,6 мл) перемешивают при 90oC. Спустя 3 часа, этанол выпаривают в вакууме и остаток переносят в делительную воронку со смесью дихлорметана с водой. Водный слой доводят до pH 1,5 с помощью насыщенного раствора бикарбоната натрия. Экстракция дихлорметаном (4 раза по 20 мл) и высушивание над сульфатом магния дают, после выпаривания растворителя, 0,18 г сырого продукта. Флэш-хроматография (диоксид кремния; диаметр колонки 15 мм; 7% метанол/дихлорметан) дает 94 мг почти чистого продукта. Препаративная ВЭЖХ (YMC S-10; 30х500 мм (размер колонки)); 68,4% метанола в воде с 1% трифторуксусной кислоты; скорость 25 мл/мин; фракции 17-19,5 минут) дает 64 мг (52%) целевого соединения. Т.пл. = 131,0-134,0oC. Анализ, рассчитанный для C21H18N4O3S 0,50H2O:Рассчитано, %: C 60,71; H 4,61; N 13,49; S 7,72. Найдено, %: C 60,49; H 4,37; N 13,17; S 8,09. ПРИМЕР 7 N-(3,4-Диметил-5-изоксазолил)-4′-(5-пиримидинил)[1,1′-бифенил] -2-сульфонамид 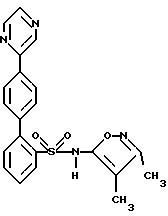 A. 5-(4-Бромфенил)пиримидин Раствор 4-бромфенилборной кислоты (2,0 г, 10 ммоль) в этаноле (через который продувают путем барботирования аргон в течение 30 минут; 44 мл) добавляют к раствору 5-бромпиримидина (1,9 г, 12 ммоль) в толуоле (через который продувают путем барботирования аргон в течение 30 минут, 87 мл). К этому раствору добавляют раствор карбоната натрия (2,7 г) в воде (через которую продувают путем барботирования аргон в течение 30 минут, 44 мл), после чего добавляют тетракис(трифенилфосфин)палладий (О) (0,76 г, 0,66 ммоль). После перемешивания в атмосфере аргона при 75oC в течение 1 часа, раствор охлаждают и выливают в рассол (50 мл). Экстракция этилацетатом (2 раза по 250 мл) и высушивание объединенных органических слоев над сульфатом магния дает после выпаривания растворителя 3,4 г сырого продукта. Флэш-хроматография (диоксид кремния, диаметр колонки 50 мм, 5-10% этилацетат/дихлорметан) с последующей перекристаллизацией из горячего эфира дает 1,0 г (42%) соединения A. Т.пл. = 144,0-146,5oC. B. N-(3,4-Диметил-5-изоксазолил)-N-[(2-метоксиэтокси)метил]-4′-(5-пиримидинил) [1,1′-бифенил]-2-сульфонамид Раствор 2-бор-N-(3,4-диметил-5-изоксазолил)-N-[(2-метоксиэтокси)метил] бензосульфонамида (1,0 г, 1,6 ммоль) в этаноле (через который в течение 20 минут продувают путем барботирования аргон; 7,2 мл) добавляют к раствору соединения A (0,50 г, 2,0 ммоль) в толуоле (через который путем барботирования продувают аргон в течение 20 минут, 14 мл). К этому раствору добавляют раствор карбоната натрия (0,45 г) в воде (через которую путем барботирования продувают в течение 20 минут аргон, 7,2 мл), после чего добавляют тетракис(трифенилфосфин)палладий (О) (0,13 г, 0,11 ммоль). После перемешивания при 75oC в атмосфере аргона в течение 4 часов, раствор охлаждают и выливают в рассол (20 мл). Экстракция этилацетатом (2 раза по 70 мл) и высушивание объединенных органических слоев над сульфатом магния дают, после выпаривания растворителя, 1,8 г сырого продукта. Флэш-хроматография (диоксид кремния, диаметр колонки 50 мм, 50% этилацетат/гексан, затем 10% метанол/этилацетат) дает 0,15 г (19%) соединения B. C. N-(3,4-Диметил-5-изоксазолил)-4′-(5-пиримидинил)[1,1′-бифенил]-2-сульфонамид Раствор соединения B (0,15 г, 0,30 ммоль) в этаноле (4,6 мл) и 6 н. соляной кислоты (4,6 мл) перемешивают при 90oC. Спустя 2 часа, этанол выпаривают в вакууме и остаток переносят в делительную воронку со смесью дихлорметана с водой. Водный слой доводят до pH 2,0 с помощью насыщенного раствора бикарбоната натрия. Экстракция дихлорметаном (4 раза по 20 мл) и высушивание над сульфатом магния дает, после выпаривания растворителя 0,13 г сырого продукта. Флэш-хроматография (диоксид кремния; диаметр колонки 15 мм, 7% метанол/дихлорметан) дает 54 мг почти чистого продукта. Препаративная ВЭЖХ (YMC S-10, размеры колонки 30х500 мм, 66,8% метанола в воде с 1% трифторуксусной кислоты, скорость 25 мл/мин, фракции 16-18,7 минут) дает 25 мг (20%) целевого соединения. Т.пл. = 105,0-109,0oC. Анализ, рассчитанный для C21H18N4O3S 0,40C2HF3O20,90H2O:Рассчитано, %: C 55,91; H 4,35; N 11,96; S 6,85. Найдено, %: C 55,92; H 3,97; N 11,61; S 7,29. ПРИМЕР 8 N-(3,4-Диметил-5-изоксазолил)-4′-(2-пиридинил)[1,1′-бифенил] -2-сульфонамид, N4  -оксид -оксид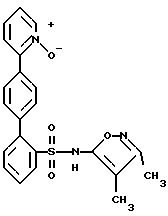 A. N-(3,4-Диметил-5-изоксазолил)-4′-(2-пиридинил)[1,1′-бифенил] -2-сульфонамид, N4 -оксидК раствору 0,15 г (0,37 ммоль) целевого соединения вышеприведенного примера 2 в 5 мл дихлорметана добавляют 0,088 г м-хлорнадбензойной кислоты и смесь перемешивают при комнатной температуре в течение 3 часов. Смесь затем выпаривают и остаток очищают путем препаративной ВЭЖХ с обращенной фазой на колонке размером 30х500 мм, заполненной ODS-10, используя 58% растворителя B (90% метанола, 10% воды, 0,1% трифторуксусной кислоты) и 42% растворителя A (10% метанола, 90% воды, 0,1% трифторуксусной кислоты). Соответствующие фракции собирают, нейтрализуют с помощью водного раствора бикарбоната натрия до pH 7 и концентрируют до объема 10 мл. Раствор затем подкисляют до pH 4 при использовании ледяной уксусной кислоты, и твердое белое вещество отфильтровывают и высушивают с получением 0,088 г (56%) целевого соединения. Т.пл. = 110-117oC. Анализ, рассчитанный для C22H19N3O4S 1,70 H2O:Рассчитано, %: C 58,44; H 4,99; N 9,29; S 7,09. Найдено, %: C 58,82; H 4,36; N 9,91; S 6,75. ПРИМЕР 9 N-(3,4-Диметил-5-изоксазолил)-4′-(6-метокси-2-пиридинил)-[1,1′-бифенил] -2-сульфонамид 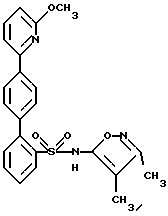 A. 2-Бром-6-метоксипиридин Раствор 5,0 г (21,21 ммоль) 2,6-дибромпиридина и 8,2 мл (35,875 ммоль) 25%-ного метоксида натрия в метаноле в 15 мл метанола перемешивают при кипячении с обратным холодильником в течение примерно 26 часов до тех пор, пока тонкослойная хроматография (этилацетат:гексан в соотношении 1:1) не будет показывать непрореагировавшего 2,6-дибромпиридина. Реакционную смесь охлаждают до комнатной температуры, выливают в холодный 5%-ный раствор бикарбоната натрия и экстрагируют диэтиловым эфиром. После выпаривания, остаток растворяют в диэтиловом эфире, раствор промывают рассолом, сушат (сульфат магния) и выпаривают досуха при получении 3,6 г (90%) бесцветного масла. B. 2-(4-Бромфенил)-6-метоксипиридин К 1,88 г (10 ммоль) соединения A и 0,5 г (2,5 ммоль) 4-бромфенилборной кислоты в смеси из 20 мл дегазированного толуола и 16 мл дегазированного этанола добавляют 0,175 г (0,15 ммоль) тетракис(трифенилфосфин)палладий (О) и 12 мл дегазированного 2 н. водного раствора карбоната натрия. Смесь перемешивают при 75oC в течение 3 часов. Реакционную смесь охлаждают до комнатной температуры, разбавляют с помощью 200 мл этилацетата, промывают водой и затем рассолом, сушат (сульфат магния) и выпаривают. Флэш-хроматография (50 ммх6”, 25-50% этилацетат/гексан) дает 1,86 г смеси исходного материала с продуктом, которую хроматографируют дополнительно 2 раза (50 ммх6”, 1% этилацетата/гексан, затем 50 ммх7”, 0,75% этилацетат/гексан), получая 230 мг (35%) бесцветного масла из всех вместе смешанных фракций. C. N-(3,4-Диметил-5-изоксазолил)-N-[(2-метоксиэтокси)метил1-4′-(6-метокси-2-пиридинил) [1,1′-бифенил]-2-сульфонамид К 230 мг (0,87 ммоль) соединения B и 280 мг (0,73 ммоль) 2-бop-N-(3,4-диметил-5-изоксазолил)-N-[(2-метоксиэтокси)метил] бензолсульфонамида в смеси из 5,5 мл дегазированного толуола и 3,7 мл дегазированного этанола добавляют 84 мг (0,072 ммоль) тетракис(трифенилфосфин)палладия (О) и 3,2 мл дегазированного 2 н. водного раствора карбоната натрия. Смесь перемешивают при 85oC в течение 3 часов. Реакционную смесь охлаждают до комнатной температуры, разбавляют этилацетатом, промывают водой и затем рассолом, сушат (сульфат магния) и выпаривают. Флэш-хроматография (50 ммх6”; 30%-40%-50% этилацетат/гексан) дает 172 мг маслянисто-твердого остатка (45%). D. N-(3,4-Диметил-5-изоксазолил)-4′-(6-метокси-2-пиридинил) [1,1′-бифенил]-2-сульфонамид Смесь из 170 мг (0,325 ммоль) соединения C и 7,5 мл 6 н. соляной кислоты в 7,5 мл этанола перемешивают при 75oC в течение 30 минут. Добавляют дополнительную воду. pH-значение доводят примерно до величины 8 с помощью насыщенного раствора бикарбоната натрия и затем снова доводят примерно до 4,5 с помощью уксусной кислоты. Смесь экстрагируют 3 раза по 50 мл этилацетатом и органические экстракты промывают с помощью 25 мл воды и 25 мл рассола, сушат (сульфатом магния) и выпаривают. Флэш-хроматография (25 ммх6”; 40%-50% этилацетат/гексан) дает 50 мг полутвердого продукта, который частично очищают путем порошкования с гексаном, получая 15 мг целевого соединения в виде белого твердого вещества. 120 мг регенерированного соединения C (полученного в п. C) также выделяют и снова подвергают вышеуказанным реакционным условиям (4 мл 6 н. соляной кислоты и 4 мл этанола). После осуществления таких же операций, как указанные выше, сырой продукт объединяют с маточными жидкостями из вышеуказанного порошкования и очищают путем флэш-хроматографии (2х25 ммх8”; 40%-50% этилацетат/гексан) получая дополнительные 50 мг целевого соединения в виде белого твердого вещества (65 мг в целом; выход = 46%). Т. пл. 132-134oC. Анализ, рассчитанный для C23H21N3O4S 0,39H2O:Рассчитано, %: C 62,42; H 4,96; N 9,49; S 7,24. Найдено, %: C 62,70; H 4,87; N 9,21; S 6,93. ПРИМЕР 10 Следующие соединения с (1) по (7) также получают описанными здесь способами, например, из 4′-амидинарилгалогенида, как описано выше: (1) N-[[2′-[[3,4-диметил-5-изоксазолил)амино] сульфонил]-4-(2-пиримидинил) [1,1′-бифенил] -2-ил] метил] -N-метилциклогексанпропанамид следующей формулы: 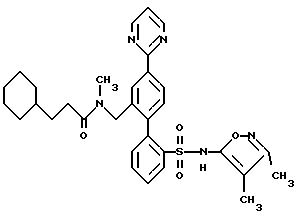 (2) N-[[2′-[[(3,4-диметил-5-изоксазолил) амино]сульфонил]-4-(2-пиримидинил) [1,1′-бифенил] -2-ил] метил] -N-метилциклогексанацетамид следующей формулы: 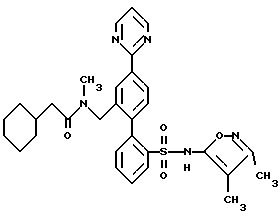 (3) N-[[2′-[[(3,4-диметил-5-изоксазолил)амино] сульфонил] -4-(2-пиримидинил) [1,1′-бифенил]-2-ил]метил]-N-метилбензолпропанамид следующей формулы:  (4) N-(3,4-диметил-5-изоксазолил)-2′-[(3,3-диметил-2-оксо-1-пирролидинил)метил]-4′-(2-пиримидинил)[1,1′-бифенил]-2-сульфонамид следующей формулы: 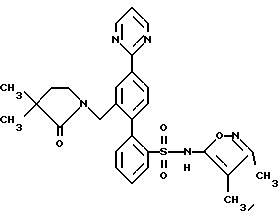 (5) N-(3,4-диметил-5-изоксазолил)-2′-[[метил(2,2,2-трифторэтил)амино] метил]-4′-(2-пиримидинил)[1,1′-бифенил]-2-сульфонамид следующей формулы: 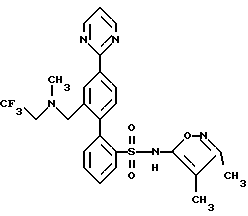 (6) N-[[2′-[[(3,4-диметил-5-изоксазолил)амино] сульфонил] -4-(2-пиримидинил)[1,1′-бифенил] -2-ил] метил] -N-метил-2-тиофенбутанамид следующей формулы: 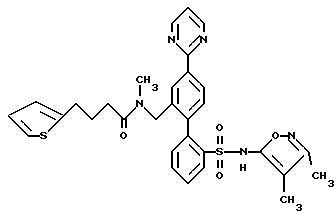 (7) N-(3,4-диметил-5-изоксазолил)-2′-[[(4-метоксифенил)метиламино]метил] -4′-(2-пиримидинил)[1,1′-бифенил]-2-сульфонамид следующей формулы: 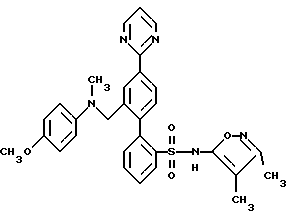 ПРИМЕР 11 N-(3,4-диметил-5-изоксазолил)-2′-формил-4′-(2-пиримидинил)[1,1-бифенил] -2-сульфонамид 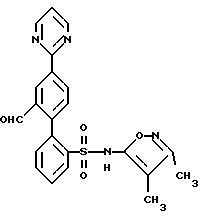 A. 4-бром-3-метилбензолкарбоксамидамид В суспензию 2,73 г (51,0 ммоль) хлорида аммония в 40 мл толуола при 0oC добавили 2,0 М раствор триметилалюминия (25,5 мл), добавку производили в течение 5 минут. Затем смесь медленно прогревали до комнатной температуры и перемешивали еще в течение 2 часов. Эту смесь затем добавили в раствор 5,0 г (25,5 ммоль) 4-бром-3-метилбензонитрила в 15 мл толуола в атмосфере аргона, и полученную смесь нагревали до 80oC в течение 6 дней. Затем смесь охлаждали до комнатной температуры и добавили в суспензию силикагеля (100 г) в 200 мл CH2Cl2 и фильтровали. Силикагель промывали 400 мл метанола и объединенный фильтрат выпаривали до получения 6,8 г (100%) искомого соединения на этой стадии, в виде белого твердого вещества. C. N-[3-(диметиламино)-2-пропенилиден]-N-метилметанаммония перхлорат В раствор диметиламина в метаноле (раствор 2,0 М, 25 мл) при 0oC добавили перхлорную кислоту (7,17 г, 50 ммоль), добавку производили в течение 10 минут, затем смесь концентрировали до около 5 мл. В эту смесь добавили раствор 3-диметиламиноакролеина (4,96 г, 50 ммоль) в 20 мл абсолютного раствора этанола (EtOH), затем смесь нагревали с обратным холодильником в течение 4 часов. После этого смесь охлаждали до 5oC и белое твердое вещество отфильтровывали и промывали 25 мл EtOH до получения искомого на этой стадии соединения (11,5 г, 100%). B. 2-(4-бром-3-метилфенил)пиримидин В раствор 5,0 г (23,46 ммоль) соединения, полученного на этапе A, в 15 мл метанола (MeOH) добавили 7,94 г (35,2 ммоль) соединения, полученного на этапе C, и 1,9 г (35,2 ммоль) метилата натрия и смесь перемешивали при комнатной температуре в атмосфере аргона в течение 1 часа. Затем раствор перегоняли в течение 4 часов и концентрировали до около 10 мл. Остаток добавили в 200 мл воды и экстрагировали 4х150 мл этилацетатом (EtOAc). Объединенные органические экстракты промывали один раз водой, высушивали и выпаривали. Остаток подвергали хроматографии на 100 г силикагеля, используя смесь гексан/EtOAc 4: 1 до получения 1,65 г (28%) искомого на данной стадии соединения в виде белого твердого вещества. D. 2-[4-бром-3-(бромметил)фенил]пиримидин Смесь соединения, полученного на стадии C (1,0 г, 4,01 ммоль), N-бромсукцинимида (2,69 г, 15,14 ммоль) и бензоилпероксида (100 мг) в 150 мл тетрахлорметана нагревали с обратным холодильником в течение 6 часов, освещая раствор солнечной лампой. После этого смесь охлаждали и фильтровали. Фильтрат концентрировали до получения 1,75 г светло-желтого твердого вещества, содержащего искомое на данной стадии соединение, которое использовали на последующей стадии без дополнительной очистки. Е. 2-бром-4-(2-пиримидинил)бензальдегид В раствор 1,75 г неочищенного соединения, полученного на стадии D, в 6 мл безводного диметилсульфоксида (ДМСО) в атмосфере аргона добавили 3,0 г безводного триметиламин N-оксида (полученного как описано в работе Soderquist и др. , Tet. Letters. , 27.3961 (1986); это соединение выпускается промышленностью), и смесь перемешивали при 55oC в течение 12 часов. Затем смесь охлаждали и добавляли в 100 мл смеси воды со льдом и экстрагировали 3х50 мл EtOAc. Объединенные органические экстракты промывали один раз 100 мл соляного раствора, высушивали и выпаривали. Остаток очищали хроматографией на 100 мл силикагеля, используя смесь гексан/EtOAc 6:1 до получения 0,86 г (81,5% для двух стадий) искомого на этой стадии соединения в виде белого твердого вещества. F. N-(3,4-диметил-5-изоксазолил)-2′-формил-N-[(2-метоксиэтокси)метил]-4′-(2-пиримидинил)[1,1′-бифенил]-2-сульфонамид В раствор 0,554 г (1,188 ммоль) 2-бром-N-(3,4-диметил-5-изоксазолил)-N-[(2-метоксиэтокси)метил]бензолсульфонамида (полученного как описано в заявке на патент США N 08/603975, поданной 20 февраля 1996 г. от имени Murugesan и др. (регистрационный N в журнале поверенного HA662d) и 0,0578 г (0,05 ммоль) тетракис(трифенилфосфин)палладий (О) в 20 мл толуола в атмосфере аргона добавили 10 мл 2 М водного раствора карбоната натрия, затем 0,25 г (0,95 ммоль) соединения, полученного на стадии E, в 10 мл 95% EtOH. Смесь нагревали с обратным холодильником в течение 2 часов, а затем разбавляли 100 мл воды и экстрагировали 3х50 мл EtOAc. Объединенные органические экстракты промывали один раз 100 мл соляного раствора, высушивали и выпаривали. Остаток подвергали хроматографии на 100 мл силикагеля, используя смесь гексан/EtOAc 1:1 до получения 0,48 г (80%) искомого на данной стадии соединения в виде бесцветной смолы. G. N-(3,4-диметил-5-изоксазолил)-2′-формил-4′-(2-пиримидинил)[1,1′-бифенил]-2-сульфонамид В раствор 0,26 г (0,497 ммоль) соединения, полученного на стадии F, в 10 мл 95% EtOH, добавили 10 мл 6 н. водного раствора HCl и нагревали с обратным холодильником в течение 1 часа. Затем смесь концентрировали и разбавляли 100 мл воды и экстрагировали 3х50 мл EtOAc. Объединенные органические экстракты промывали один раз водой, высушивали и выпаривали до получения 0,21 г (97%) искомого соединения по настоящему примеру в виде бесцветной смолы. 1H-ЯМР (CDCl3): 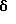 1,79 (с, 3H), 2,12 (с, 3H), 7,24-8,87 (м, 10H), 9,76 (с, 1H).
ПРИМЕР 12 1,79 (с, 3H), 2,12 (с, 3H), 7,24-8,87 (м, 10H), 9,76 (с, 1H).
ПРИМЕР 12N-(3,4-диметил-5-изоксазолил)-2′-[(метиламино)метил] -4′-(2-пиримидинил)[1,1′-бифенил]-2-сульфонамид  В раствор 0,21 г (0,483 ммоль) соединения по примеру 11 в 15 мл CH2Cl2 добавили 1 мл 3 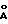 молекулярных сит, 0,18 мл (33% раствор в EtOH, 1,45 ммоль) метиламина и 0,087 г (1,45 ммоль) уксусной кислоты, смесь перемешивали в атмосфере аргона в течение 10 минут. Затем добавили триацетоксиборгидрид натрия (0,307 г, 1,45 ммоль) и смесь перемешивали при комнатной температуре в течение ночи. После этого раствор фильтровали через броунмиллерит, броунмиллерит промывали 25 мл CH2Cl2, и объединенный фильтрат промывали 2х50 мл воды, высушивали и выпаривали до получения 0,16 г (74%) искомого соединения по данному примеру в виде светло-желтой смолы.
Rf = 0,23 (5% метанол в CH2Cl2).
ПРИМЕР 13 молекулярных сит, 0,18 мл (33% раствор в EtOH, 1,45 ммоль) метиламина и 0,087 г (1,45 ммоль) уксусной кислоты, смесь перемешивали в атмосфере аргона в течение 10 минут. Затем добавили триацетоксиборгидрид натрия (0,307 г, 1,45 ммоль) и смесь перемешивали при комнатной температуре в течение ночи. После этого раствор фильтровали через броунмиллерит, броунмиллерит промывали 25 мл CH2Cl2, и объединенный фильтрат промывали 2х50 мл воды, высушивали и выпаривали до получения 0,16 г (74%) искомого соединения по данному примеру в виде светло-желтой смолы.
Rf = 0,23 (5% метанол в CH2Cl2).
ПРИМЕР 13N-[2′-[[3,4-диметил-5-изоксазолил)амино] сульфонил] -4-(2-пиримидинил)[1,1′-бифенил]-2-ил]метил]-N-3,3-триметилбутанамид 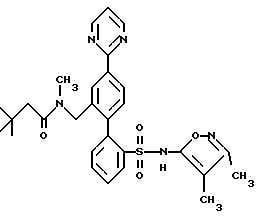 В раствор 0,14 г (0,31 ммоль) соединения по примеру 12 в 10 мл CH2Cl2 добавили 0,046 г (0,342 ммоль) трет-бутил-ацетилхлорида и 0,063 г (0,624 ммоль) триэтиламина. После этого смесь перемешивали при комнатной температуре в течение 4 часов и выпаривали. Остаток очищали препаративной ВЭЖХ с обращенной фазой на колонке ODS S10 30х500 мм, используя 78% растворителя B (90% MeOH, 10% H2O, 0,1% (трифторуксусной кислоты (ТФК) и 22% растворителя A (10% MeOH, 90% H2O, 0,1% ТФК). Нужные фракции собирали и нейтрализовывали водным раствором бикарбоната натрия до pH 7 и концентрировали до 10 мл. После этого раствор подкисляли до pH 4 при помощи водного раствора бисульфата натрия и белое твердое вещество отфильтровывали и высушивали, получая 0,031 г (18%) искомого соединения по данному примеру в виде белого твердого вещества, т.пл. 105-115oC. ПРИМЕР 14 N-(4,5-диметил-3-изоксазолил)-2′-[(3,3-диметил-2-оксо-1-пирролидинил)метил]-4′-(2-пиримидинил)[1,1′-бифенил]-2-сульфонамид 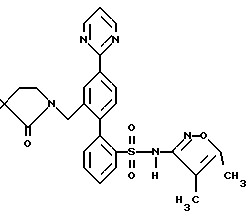 A. 2′-(бромметил)-N-(4,5-диметил-3-изоксазолил)-N-[(2-метоксиэтокси)метил]-4′-(2-пиримидинил)[1,1′-бифенил]-2-сульфонамид В N-(4, 5-диметил-3-изоксазолил)-2′-формил-N-[(2-метоксиэтокси)метил]-4′-(2-пиримидинил)[1,1′-бифенил] -2-сульфонамид (460 мг, 0,88 ммоль, полученный как описано на стадии C примера 16) в 10 мл MeOH, добавили NaBH4 (40 мг, 1,06 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов и концентрировали. В полученный остаток добавили 10 мл H2O и 50 мл EtOAc. Органический слой отделяли, промывали H2O, соляным раствором, высушивали и концентрировали до получения N-(4,5-диметил-3-изоксазолил)-2′-(гидроксиметил)-N-[(2-метоксиэтокси)метил] -4′-(2-пиримидинил)[1,1′-бифенил]-2-сульфонамида. В N-(4,5-диметил-3-изоксазолил)-2′-(гидроксиметил)-N-[(2-метоксиэтокси)метил] -4′-(2-пиримидинил)[1,1′-бифенил] -2-сульфонамид в 4,4 мл ДМФ при 0oC добавили тетрабромметан (438 мг, 1,32 ммоль), а затем трифенилфосфин (346 мг, 1,32 ммоль). Смесь перемешивали при 0oC в течение 4 часов, разбавляли 100 мл смесью 1:2 гексан/EtOAc. Органический раствор промывали 3х20 мл H2O, соляным раствором и концентрировали. Остаток подвергали хроматографии на силикагеле, используя смесь 1,2:1 гексан/EtOAc до получения искомого на данной стадии соединения (370 мг, 72% за две стадии) в виде смолы. B. N-(4,5-диметил-3-изоксазолил)-2′-[(3,3-диметил-2-оксо-1-пирролидинил)метил]-4′-(2-пиримидинил)[1,1′-бифенил]-2-сульфонамид В 3,3-диметил-2-пирролидинон (58 мг, 0,51 ммоль, полученный как описано в примере 15) в 0,5 мл ДМФ при 0oC добавили гидрид натрия (60% в минеральном масле, 26 мг, 0,64 ммоль) и смесь перемешивали при комнатной температуре в течение 20 минут. В полученную смесь добавили соединение по стадии A (150 мл, 0,26 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов. Затем смесь разбавили 50 мл EtOAc, промыли H2O, соляным раствором и концентрировали до получения N-(4,5-диметил-3-изоксазолил)-2′-[(3,3-диметил-2-оксо-1-пирролидинил)метил] -N-[(2-метоксиэтокси)метил] -4′-(2-пиримидинил)[1,1′-бифенил]-2-сульфонамида в виде смолы. В раствор N-(4,5-диметил-3-изоксазолил)-2′-[(3,3-диметил-2-оксо-1-пирролидинил)метил]-N-[(2-метоксиэтокси)метил]-4′-(2-пиримидинил)[1,1′-бифенил] -2-сульфонамида в 5,1 мл CH3CN добавили триметилсилилхлорид (Me3SiCl, 166 мг, 1,53 ммоль), затем Nal (229 мг, 1,53 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа. Затем добавили дополнительное количество Mе3SiCl (166 мг, 1,53 ммоль) и Nal (229 мг, 1,53 ммоль), добавку производили тремя порциями и реакционную смесь перемешивали еще в течение 1 часа и 20 минут. Затем реакционную смесь добавили в 5 мл H2O и 30 мл EtOAc. Органический слой отделили и промыли 2 мл насыщенного водного раствора тиосульфата натрия, соляным раствором, высушивали и концентрировали. Остаток очищали препаративной ВЭЖХ на колонке ODS S10, используя 32% растворителя A (10% MeOH, 90% H2O, 0,1% ТФК) и 68% растворителя B (90% MeOH, 10% H2O, 0,1% ТФК) до получения искомого соединения по данному примеру (29 мг, 2,1% за две стадии) в виде белого твердого вещества, темп. плавления 125-132oC (аморфное). ПРИМЕР 15 N-(3,4-диметил-5-изоксазолил)-2′-[(3,3-диметил-2-оксо-1-пирролидинил)метил]-4′-(2-пиримидинил)[1,1′-бифенил]-2-сульфонамид 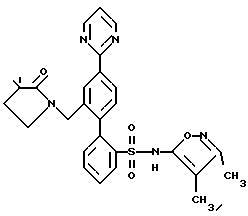 A. N-(3,4-диметил-5-изoкcaзoлил)-2′-[(3,3-диметил-2-оксо-1-пирролидинил)метил] -N-[(2-метоксиэтокси)метил] -4′-(2-пиримидинил)[1,1′-бифенил]-2-сульфонамид В колбу, содержащую 3,3-диметил-2-оксо-1-пирролидин-карбоновую кислоту, 1,1-диметилэтилового сложного эфира гидрохлорид (0,5 г, 2,34 ммоль, полученный как описано в работе J.Chem. Res. (Synopsis), 414-415 (1993)), добавили 1 н. HCl в простом эфире (1,5 мл) и смесь перемешивали в течение ночи. Затем раствор выпаривали и остаток высушивали под вакуумом до получения 0,26 г (98%) 3,3-диметил-2-пирролидинона в виде светло-желтой смолы, которая отверждалась при отстаивании. В раствор 3,3-диметил-2-пирролидинона, полученный таким образом (0,033 г, 0,29 ммоль), в 1 мл диметилформамида (ДМФ) добавляли NaH (60% суспензию в минеральном масле (0,015 г, 0,36 ммоль) и смесь перемешивали при комнатной температуре в атмосфере аргона в течение 10 минут. Затем добавили 2′-(бромметил)-N-(3,4-диметил-5-изоксазолил)-N-[(2-метоксиэтокси)метил] -4′-(2-пиримидинил)[1,1′-бифенил] -2-сульфонамид (полученный как описано в примере 14, стадия A, за исключением того, что вместо N-(4,5-диметил-3-изоксазолил)-2′-формил-N-[(2-метоксиэтокси)метил] -4′-(2-пиримидинил)[1,1′-бифенил] -2-сульфонамида использовали соединение, полученное на стадии F по примеру 11, 0,085 г, 0,145 ммоль), и смесь перемешивали в течение 3 часов. Затем смесь добавили в 50 мл воды и раствор экстрагировали 3х25 мл EtOAc. Объединенные органические экстракты промывали водой, высушивали и выпаривали до получения 0,086 г (95%) искомого соединения по данной стадии в виде бесцветной смолы. B. N-(3,4-диметил-5-изоксазолил)-2′-[(3,3-диметил-2-оксо-1-пирролидинил)метил]-4′-(2-пиримидинил)[1,1′-бифенил]-2-сульфонамид В раствор соединения, полученного на этапе A (0,081 г, 0,13 ммоль) в ацетонитриле добавили хлортриметилсилан (0,085 г, 0,78 ммоль) и иодид натрия (0,117 г, 0,78 ммоль) и смесь перемешивали при комнатной температуре в течение 30 минут. Затем добавили дополнительные количества хлортриметилсилана (0,085 г, 0,78 ммоль) и иодида натрия (0,117 г, 0,78 ммоль) и смесь перемешивали еще в течение 1 часа. Смесь разбавляли 25 мл 2% водного раствора тиосульфата натрия и экстрагировали 3х25 мл EtOAc. Объединенные органические экстракты затем промывали один раз водой, высушивали и выпаривали до получения 0,071 г бесцветной смолы. Смолу соединяли еще с 0,071 г того же материала, полученного в другой порции, и смесь очищали препаративной ВЭЖХ с обращенной фазой на 30х500 мм колонке ODS S10, используя 71% растворителя B (90% MeOH, 10% H2O, 0,1% ТФК) и 29% растворителя A (10% MeOH, 90% H2O, 0,1% ТФК). Соответствующие фракции собирали и нейтрализовывали водным раствором бикарбоната натрия до pH 7 и концентрировали до 5 мл. Затем раствор подкисляли до pH 2, используя водный раствор бисульфата натрия, и белое твердое вещество отфильтровывали и высушивали до получения 0,039 г (28%) искомого соединения по данному примеру в виде белого твердого вещества, темп.плавл. 105-115oC (аморфного). ПРИМЕР 16 N-[[2′-[[3,5-диметил-3-изоксазолил)амино] сульфонил] -4′-(2-пиримидинил)[1,1′-бифенил]-2-ил]метил]-N-3,3-триметилбутанамид 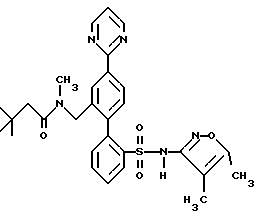 A. 2-бром-N-(4,5-диметил-3-изоксазолил)бензолсульфонамид В раствор 4,5-диметил-3-изоксазоламина (1,62 г, 13,00 ммоль, получен как описано в работе T. Konoike и др. Tetrahedron Letters, 37, 3339 (1996)) и 4-диметиламинопиридина (159 мг, 1,3 ммоль) в 6,5 мл пиридина при 0oC добавили 2-бромбензосульфонилхлорид (3,65 г, 14,3 ммоль), добавку производили порциями в течение 10 минут. После перемешивания при комнатной температуре в течение ночи смесь по капле добавили в 40 мл 6 н. HCl при 0oC. Смесь экстрагировали 3х50 мл EtOAc. Объединенные органические экстракты промывали 30 мл каждого из 1н. HCl и соляным раствором, высушивали и концентрировали. Остаток растворяли в 160 мл MeOH и добавляли 160 мл 3% водного раствора NaHCO3, после чего смесь концентрировали под вакуумом для удаления большей части MeOH. Твердое вещество отфильтровывали и водный фильтрат подкисляли до pH 2 с помощью NaHSO4 и экстрагировали 3х100 мл EtOAc. Экстракты промывали соляным раствором, высушивали и концентрировали до получения искомого соединения по данной стадии (3,0 г, 70%). Rf = 0,57, силикагель, 1:1 гексан/EtOAc. B. 2-N-(4,5-диметил-3-изоксазолил)-N-[(2-метоксиэтокси)метил] бензолсульфонамид В соединение, полученное на стадии A (3,19 г, 10,05 ммоль), в 10 мл ДМФ порциями добавили NaH (60% в минеральном масле, 442 мг, 11,05 ммоль). После перемешивания при комнатной температуре в течение 10 минут по капле добавили 2-метоксиэтоксиметилхлорид (1,57 г, 12,56 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Затем в смесь добавили 20 мл H2O и 100 мл смеси 1:1 гексан/EtOAc. Органический слой отделяли и промывали 2х50 мл воды, 30 мл соляного раствора, высушивали и концентрировали. Остаток подвергли хроматографии на силикагеле, используя смесь 3:1 гексан/EtOAc до получения искомого соединения по данной стадии (2,99 г, 71%) в виде масла. C. N-(4,5-диметил-3-изоксазолил)-2′-формил-N-[(2-метоксиэтокси)метил]-4′-(2-пиримидинил) [1,1′-бифенил]-2-сульфонамид В раствор соединения, полученного на стадии B (1,28 г, 3,04 ммоль), в 15 мл тетрагидрофурана (ТГФ) при -95oC добавили п-бутиллитий (“n-BuLi”, 2 М в пентане, 1,75 мл, 3,5 ммоль). Смесь перемешивали при -95oC в течение 10 минут и добавили триметилборат (411 мг, 3,95 ммоль). Холодную баню удаляли и смесь перемешивали при комнатной температуре в течение 1 часа и концентрировали. В остаток добавили 2-бром-5-(2-пиримидинил)бензальдегид (250 мг, 0,95 ммоль, получен как описано в примере 11, стадия E), 4,75 мл толуола, 1,43 мл 95% EtOAc, 1,43 мл 2 М водного раствора карбоната натрия и тетракис(трифенилфосфин)палладий (О) (110 мг, 0,095 ммоль) и реакционную смесь нагревали в атмосфере аргона до 85oC в течение 4 часов, охлаждали и разбавляли 40 мл EtOAc. Органический слой отделяли и промывали 10 мл H2O и 10 мл соляного раствора, высушивали и концентрировали. Остаток подвергли хроматографии на силикагеле, используя смесь 1:1 гексан/EtOAc, до получения искомого соединения по данной стадии (250 мг, 50%) в виде бесцветной смолы. Rf = 0,40, силикагель 1:2 гексан/EtOAc. D. N-[[2′-[[(4,5-диметил-3-изоксазолил)[(2-метоксиэтокси)метил]амино]сульфонил] -4′-(2-пиримидинил)[1,1′-бифенил1-2-ил] метил] -N-3,3-триметилбутанамид В соединение, полученное на стадии C (130 мг, 0,25 ммоль) добавили метиламин (MeNH2, 8,03 М в EtOH, 0,16 мл, 1,24 ммоль) и 3  молекулярные сита в 2,5 мл CH2Cl2 уксусной кислоты (AcOH, 45 мг, 0,75 ммоль), после чего добавили триацетоксиборгидрид натрия (NаВ(AcO)3H, 158 мг, 0,75 ммоль). После перемешивания при комнатной температуре в течение 16 часов добавили MeNH2, (8,03 М в EtOAc, 0,155 мл, 1,24 ммоль), AcOH (45 мг, 0,75 ммоль) и NaB (AcO)3H (263 мг, 1,25 ммоль) и реакционную смесь перемешивали еще в течение 24 часов. Смесь разбавляли EtOAc и фильтровали через броунмиллерит. Фильтрат промывали водным раствором NaHCO3, H2O, соляным раствором, высушивали и концентрировали до получения N-(4,5-диметил-3-изоксазолил)-N-[(2-[(метоксиэтокси)метил] -2′-[(метиламино)метил] -4′-(2-пиримидинил) [1,1′-бифенил]-2-сульфонамида.
В N-(4,5-диметил-3-изоксазолил)-N-[(2-метоксиэтокси)метил] -2′-[(метиламино)метил] -4′-(2-пиримидинил)[1,1′-бифенил]-2-сульфонамид в 2,5 мл CH2Cl2 при 0oC добавили трет-бутилацетилхлорид (35 мг, 0,26 ммоль), а затем триэтиламин (50 мг, 0,49 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали. Остаток подвергли хроматографии на силикагеле, используя смесь 1:1,2 гексан/EtOAc до получения искомого соединения по данной стадии (73 мг, 39% за две стадии) в виде бесцветной смолы. Rf = 0,71, силикагель 20:1 CH2Cl2/MeOH.
E. N-[[2′-[[4,5-диметил-3-изоксазолил)амино] сульфонил] -4′- (2-пиримидинил)[1,1′-бифенил]-2-ил]метил]-N-3,3-триметилбутанамид молекулярные сита в 2,5 мл CH2Cl2 уксусной кислоты (AcOH, 45 мг, 0,75 ммоль), после чего добавили триацетоксиборгидрид натрия (NаВ(AcO)3H, 158 мг, 0,75 ммоль). После перемешивания при комнатной температуре в течение 16 часов добавили MeNH2, (8,03 М в EtOAc, 0,155 мл, 1,24 ммоль), AcOH (45 мг, 0,75 ммоль) и NaB (AcO)3H (263 мг, 1,25 ммоль) и реакционную смесь перемешивали еще в течение 24 часов. Смесь разбавляли EtOAc и фильтровали через броунмиллерит. Фильтрат промывали водным раствором NaHCO3, H2O, соляным раствором, высушивали и концентрировали до получения N-(4,5-диметил-3-изоксазолил)-N-[(2-[(метоксиэтокси)метил] -2′-[(метиламино)метил] -4′-(2-пиримидинил) [1,1′-бифенил]-2-сульфонамида.
В N-(4,5-диметил-3-изоксазолил)-N-[(2-метоксиэтокси)метил] -2′-[(метиламино)метил] -4′-(2-пиримидинил)[1,1′-бифенил]-2-сульфонамид в 2,5 мл CH2Cl2 при 0oC добавили трет-бутилацетилхлорид (35 мг, 0,26 ммоль), а затем триэтиламин (50 мг, 0,49 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали. Остаток подвергли хроматографии на силикагеле, используя смесь 1:1,2 гексан/EtOAc до получения искомого соединения по данной стадии (73 мг, 39% за две стадии) в виде бесцветной смолы. Rf = 0,71, силикагель 20:1 CH2Cl2/MeOH.
E. N-[[2′-[[4,5-диметил-3-изоксазолил)амино] сульфонил] -4′- (2-пиримидинил)[1,1′-бифенил]-2-ил]метил]-N-3,3-триметилбутанамидВ раствор соединения, полученного на стадии D (71 мг, 0,11 ммоль) в 2,3 мл CH3CN, добавили триметилсилилхлорид (Me3SiCl, 73 мг, 0,67 ммоль), а затем Nal (100 мг, 0,67 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут. Затем добавили дополнительные количества Me3SiCl (24 мг, 0,22 ммоль) и Nal (33 мг, 0,22 ммоль) и реакционную смесь перемешивали еще в течение 1 часа. Затем реакционную смесь добавили в 1 мл H2O и 30 мл EtOAc. Органический слой отделяли и промывали 2 мл насыщенного водного раствора тиосульфата натрия, соляным раствором, высушивали и концентрировали. Остаток очищали препаративной ВЭЖХ на колонке ODS S10, используя 25% растворителя A (10% MeOH, 90% H2O, 0,1% ТФК) и 75% растворителя B (90% MeOH, 10% H2O, 0,1% ТФК) до получения искомого соединения по данному примеру (43 мг, 70%) в виде белого твердого вещества, темп. плавления 120-128oC (аморфный). Rf = 0,44 (силикагель, 20:1 CH2Cl2/MeOH). ПРИМЕР 17 N-(4,5-диметил-3-изоксазолил)-4′-(2-пиримидинил)-2′-[[3-(трифторметил)-1Н-пиразол-1-ил]метил][1,1′-бифенил]-2-сульфонамид 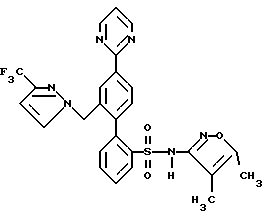 В раствор 3-(трифторметил)пиразола (56 мг, 0,41 ммоль) в 0,4 мл ДМФ при 0oC добавили гидрид натрия (60% в минеральном масле, 16,3 мг, 0,41 ммоль) и смесь перемешивали при комнатной температуре в течение 10 минут. В смесь добавили соединение, полученное на стадии A примера 14 (120 мг, 0,20 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, разбавляли 30 мл EtOAc и 10 мл гексана, затем промывали 10 мл H2O и соляным раствором, высушивали и концентрировали до получения N-(4,5-диметил-3-изоксазолил)-N-[(2-метоксиэтокси)метил]-4′-(2-пиримидинил)-2′-[[(трифторметил)-1H-пиразол-1-ил] метил][1,1′-бифенил]-2-сульфонамида в виде смолы. В раствор N-(4,5-диметил-3-изоксазолил)-N- [(2-метоксиэтокси)метил]-4′-(2-пиримидинил)-2′-[[3-(трифторметил)-1H-пиразол-1-ил] метил] [1,1′-бифенил] -2-сульфонамида в 3 мл 95% EtOH, добавили 3 мл 6н. HCl. Смесь нагревали с обратным холодильником в течение 1 часа и концентрировали. Затем добавили 10 мл H2O и смесь экстрагировали 3х30 мл EtOAc. Объединенные органические экстракты промывали соляным раствором, высушивали и концентрировали. Остаток очищали препаративной ВЭЖХ на колонке ODS S10, используя 24% растворителя A (10% MeOH, 90% H2O, 0,1% ТФК) и 76% растворителя B (90% MeOH, 10% H2O, 0,1% ТФК) до получения искомого соединения по данному примеру (87 мг 77% за 2 стадии) в виде белого твердого вещества с температурой плавления 108-114oC (аморфного). ПРИМЕР 18 N-(3,4-Диметил-5-изоксазолил)-4′-(2-пиримидинил)-2′-[[3-(трифторметил)-1H-пиpaзoл-1-ил]метил][1,1′-бифенил]-2-сульфонамид 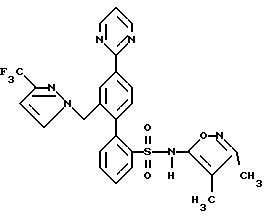 Искомое соединение по данному примеру получали способом, аналогичным способу по примеру 17, за исключением того, что 2′- (бромметил)-N-(3,4-диметил-5-изоксазолил)-N-[[2-метоксиэтокси)метил] – 4′-(2-пиримидинил)[1,1-бифенил] -2-сульфонамид (полученный на стадии A примера 15) использовали вместо соединения стадии A примера 14. Формула изобретения
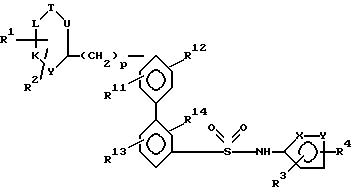 где один из X и Y – кислород, другой – азот; R1 и R2 каждый непосредственно связан с атомом углерода кольца и каждый независимо друг от друга обозначают водород или низший алкоксил; R3 и R4 каждый связаны с атомом углерода кольца и каждый независимо друг от друга обозначают низший алкил; R12, R13 и R14 каждый обозначают водород; R11 – водород, формил или низший алкил, который может быть замещен Z1, Z2, Z3, где Z1, Z2, Z3 каждый обозначают независимо друг от друга водород, или Z1 – группа Z4 – NZ7Z8 или Z4 – N(Z11)Z5 – Z6, где Z4 – одинарная связь, Z5 – одинарная связь или группа C = O, Z6 – низший алкил, алкил, замещенный 1 – 3 атомами галогена, циклоалкилалкил, Z7 – водород, Z8 – низший алкил, или Z1 – замещенный 2-оксопирролидинил или замещенный пиразол; J, K, L, T и U каждый независимо друг от друга, обозначают N или C при условии, что, по крайней мере, один обозначает N и самое большее два обозначают N, и если только один из J, K, L, T, U обозначает N, то N может быть замещен O–, так что образуется N-оксид; p = 0. 2. Соединение по п.1, где R3 и R4 каждый независимо друг от друга обозначают алкил с 1 – 4 атомами углерода. 3. Соединение по п.1, где R3 и R4 каждый обозначают метил. 4. Соединение по п.1, где R1 и R2 каждый независимо друг от друга обозначают водород или низший алкоксил, R3 и R4 каждый обозначают метил, R11, R12, R13, R14 каждый обозначают водород. 5. Соединение по п.4, где R12, R13, R14 каждый обозначают водород, а R11 – замещенный алкил. 6. Соединение по п. 5, где R11 обозначает алкил, замещенный Z4 – N(Z11)Z5Z6. 7. Соединение по п.1, выбираемое из группы, состоящей из N-(3,4-диметил-5-изоксазолил)-4′-(2-пиримидинил)[1,11-бифенил] -2-сульфонамида; N-(3,4-диметил-5-изоксазолил)-4′-(2-пиридинил)[1,11-бифенил] -2-сульфонамида; N-(3,4-диметил-5-изоксазолил)-4′-(3-пиридинил)[1,11-бифенил]-2-сульфонамида; 4′-(4,6-диметокси-2-пиримидинил)-N-(3,4-диметил-5-изоксазолил)[1,1′-бифенил] -2-сульфонамида; N-(3,4-диметил-5-изоксазолил)-4′-(4-пиримидинил)[1,11-бифенил]-2-сульфонамида; N-(3,4-диметил-5-изоксазолил)-4′-(2-пиразинил)[1,11-бифенил] -2-сульфонамида; N-(3,4-диметил-5-изоксазолил)-4′-(5-пиримидинил)[1,11-бифенил] -2-сульфонамида; N-(3,4-диметил-5-изоксазолил)-4′-(2-пиридинил)[1,11-бифенил] -2-сульфонамид-N4-оксида; N-(3,4-диметил-5-изоксазолил)-4′-(6-метокси-2-пиридинил)[1,11-бифенил] -2-сульфонамида; N-[[2′-[[(3,4-диметил-5-изоксазолил)амино]сульфонил]-4-(2-пиримидинил)[1,11-бифенил] -2-ил] метил]-N’-метилбензолпропанамида; N-(3,4-диметил-5-изоксазолил)-2′-[(3,3-диметил-2-оксо-1-пирролидинил)метил] -4′-(2-пиримидинил)[1,11-бифенил] -2-сульфонамида; N-(3,4-диметил-5-изоксазолил)-2′-формил-4′-(2-пиримидинил)[1,11-бифенил] -2-сульфонамида; N-(3,4-диметил-5-изоксазолил)-2′-[(метиламино)метил] -4′-(2-пиримидинил)[1,1′-бифенил]-2-сульфонамида; N-[[2-[[(3,4-диметил-5-изоксазолил)амино]сульфонил]-4-(2-пиримидинил)[1,1′-бифенил] -2-ил]метил]-N,3,3-триметилбутанамида; N-(4,5-диметил-3-изоксазолил)-2′-[(3,3-диметил-2-оксо-1-пирролидинил)метил] -4′-(2-пиримидинил)[1,1′-бифенил] -2-сульфонамида; N-[[2′-[[(4,5-диметил-5-изоксазолил)амино] сульфонил] -4′-(2-пиримидинил)[1,1′-бифенил] -2-ил]метил]-N,3,3-триметилбутанамида. 8. Способ антагонистического воздействия на эндотелин, отличающийся тем, что вводят эффективное для этого количество соединения по п.1. 9. Фармацевтическая композиция, обладающая антагонистической активностью против эндотелина, включающая активное вещество и фармацевтически приемлемый носитель, отличающаяся тем, что в качестве активного вещества содержит соединение по п.1 в эффективном количестве. РИСУНКИ
MM4A Досрочное прекращение действия патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 06.02.2003
Извещение опубликовано: 20.11.2004 БИ: 32/2004
|
||||||||||||||||||||||||||