Патент на изобретение №2159613
|
||||||||||||||||||||||||||
(54) ЗАМЕЩЕННЫЕ N-(ИНДОЛ-2-КАРБОНИЛ)-
(57) Реферат: Изобретение относится к производным индол-2-карбоксамидам, которые могут быть использованы в качестве ингибиторов гликогенфосфорилазы, и к способам лечения гликогенфосфорилазо-зависимых заболеваний или состояний с использованием указанных соединений и фармацевтических композиций, содержащих эти соединения. Изобретение обеспечивает возможность использования заявленных соединений для лечения диабета, гипергликемии, гиперхолестеринемии, гипертензии и других патологий. 3 с. и 33 з.п. ф-лы. Настоящее изобретение относится к ингибиторам гликогенфосфорилазы; к фармацевтическим композициям, содержащим такие ингибиторы; и к использованию указанных ингибиторов для лечения диабета, гипергликемии, гиперхолестеринемии, гипертензии, гиперинсулинемии, гиперлипемии, атеросклероза, и ишемии миокарда у млекопитающих. Несмотря на сделанное уже давно открытие инсулина и его последующее широкое применение для лечения диабета, а также более позднее открытие возможности использования сульфонилмочевин (например, ХлорпропамидаТМ (Pfiger), ТолбутамидаТМ (Upjohn), АцетогексамидаТМ (E.I.Zilly), ТолазамидаТМ (Upjohn)) и бигуанидов (например, фенформинаТМ (Ciba Jeigy), МетформинаТМ (G.Searle)) в качестве пероральных гипогликемических средств, лечение диабета этими средствами пока не дает удовлетворительных результатов. Использование инсулина, необходимое почти 10% больных диабетом, для которых синтетические гипогликемические средства являются неэффективными (диабет типа 1, инсулин-зависимый диабет), требует ежедневного введения многократных доз, обычно, путем инъекций, проводимых самим пациентом. Определение точной дозы инсулина требует частого проведения анализов мочи или крови на сахар. Введение избыточной дозы инсулина может вызвать гипогликемию с различной степенью тяжести, варьирующейся от легких изменений уровня глюкозы в крови до коматозного состояния или даже смерти. Лечение инсулин-независимого диабета (диабета типа 11, ИНСД) обычно предусматривает применение комплексных мер, а именно, диеты; упражнений; приема пероральных препаратов, например, сульфонилмочевины; и, в более тяжелых случаях, введения инсулина. Однако, имеющиеся клинические препараты гипогликемизирующего действия могут обладать другими побочными эффектами, ограничивающими их использование. В любом случае, там, где один из указанных препаратов не дает желаемого результата, может быть с успехом использован другой препарат. Поэтому постоянная потребность в получении новых гипогликемических средств, которые обладали бы меньшим побочным действием, и были бы более эффективными по сравнению с другими имеющимися средствами, является совершенно очевидным фактом. Как было уже признано специалистами, атеросклероз (системное поражение артерий) является одной из главных причин смертности в Соединенных штатах Америки и в Западной Европе. Цепь патологий, приводящая к атеросклерозу и окклюзивных заболеваний сердца, хорошо известна. На ранней стадии этой патологической цепи происходит образование “жировых пластов” в сонной, венечной и мозговой артериях, и в аорте. Эти поражения имеют желтый цвет благодаря присутствию липидных отложений, обнаруживаемых, главным образом, в клетках гладких мышц и в макрофагах внутренней оболочки (интимы) артерий и аорты. Кроме того, было высказано мнение, что большая часть холестерина, находящегося в указанных жировых пластах, в свою очередь, вызывает развитие “атеросклеротических бляшек”, которые состоят из аккумулированных клеток интимальных гладких мышц, нагруженных липидом и окруженных внеклеточным липидом, коллагеном, эластином и протеогликанами. Эти клетки вместе с матриксом образуют соединительнотканный слой, который покрывает более глубокие отложения клеточного дебриса и находящегося над ними клеточного липида. Этот липид представляет собой, в основном, свободный и этерифицированный холестерин. Такие бляшки из соединительной ткани образуются довольно медленно, и со временем подвергается кальцификации и некрозу, что приводит к развитию “осложнений”, являющихся причиной артериальной окклюзии, и способствующих возникновению пристеночного тромбоза и спазма артериальной мышцы, т.е., признаков, характерных для прогрессирующего атеросклероза. Эпидемиологические данные ясно свидетельствуют о том, что гиперлипидемия является главным фактором риска в развитии сердечно-сосудистых заболеваний (CVD), обусловленных атеросклерозом. В последнее время, ведущие специалисты с области медицины придают особое значение снижению уровня холестерина в крови, а особенно холестерина низкой плотности, как главной меры для предупреждения CVD. При этом, верхние пределы “нормального” уровня холестерина должны быть, как считают специалисты, значительно ниже тех уровней, которые были признаны до настоящего времени. Исходя из этих соображений, совершенно очевидно, что большая часть населения Западной Европы и Америки составляет группу повышенного риска. Такими независимыми факторами риска являются непереносимость глюкозы, гипертрофия левого желудочка, гипертензия, и мужской пол. Сердечно-сосудистые заболевания чаще всего встречаются у больных диабетом, что, по крайней мере, отчасти, можно объяснить наличием у этих индивидуумов множества независимых факторов риска. Поэтому, успешное лечение гиперлипидемии у основной части населения, а, в частности, у больных диабетом, имеет исключительно важное значение. Гипертензия (или повышенное кровяное давление) представляет собой состояние человека, которое имеет вторичный характер, и является лишь симптомом, связанным с различными другими нарушениями органов или систем, такими, как стеноз почечной артерии феохромоцитома, или эндокринные расстройства. Однако, гипертензия также наблюдается у многих пациентов, у которых отсутствуют какие-либо характерные для гипертензии этиологические факторы или расстройства. Хотя такая “первичная” гипертензия часто ассоциируется с различными расстройствами, такими, как ожирение, диабет и гипертриглицеридемия, однако, прямой связи гипертензии с указанными расстройствами не было установлено. Кроме того, симптомы высокого кровяного давления наблюдаются у пациентов, у которых полностью отсутствуют признаки каких-либо других заболеваний или расстройств. Известно, что гипертензия может непосредственно приводить к сердечной недостаточности, почечной недостаточности и к инсульту (кровоизлиянию в мозг). Эти состояния могут привести к скоропостижной смерти пациента. Гипертензия может также способствовать развитию атеросклероза и ишемической болезни сердца. Эти заболевания постепенно ослабляют организм пациента и могут, в конце концов, медленно привести к летальному исходу. Точная причина первичной гипертензии пока неизвестна, хотя имеются некоторые предположения о ряде факторов, способствующих возникновению этого заболевания. Такими факторами являются, например, стрессы; неконтролируемые эмоции; нерегулируемое высвобождение гормонов (ренина, ангиотензина, системы альдостерона); избыточное количество соли и воды в организме, обусловленное недостаточной функцией почек; утолщение стенок и гипертрофия сосудистой сети, приводящие у сужению кровеносных сосудов; и генетические факторы. Учитывая вышеуказанные факторы, были предприняты попытки лечения первичной гипертензии. В связи с этим, был разработан широкий ряд бета-блокаторов, сосудосуживающих средств, ингибиторов ангиотензин-конвертазы (т.е., фермента, конвертирующего ангиотензин), и т.п., которые были квалифицированы как гипотензивные средства. Лечение гипертензии с использованием указанных соединений было признано успешным для предупреждения скоропостижной смерти, например, от сердечной недостаточности, почечной недостаточности и кровоизлияния в мозг. Однако, применение указанных средств не решает проблем, связанных с развитием атеросклероза или заболеваний сердца, обусловленных гипертензией, протекающей в течение длительного периода времени. Это означает, что, хотя с помощью таких средств может быть снижено кровяное давление, однако, эти средства не могут устранить причину, лежащую в основе возникновения первичной гипертензии. Гипертензия также ассоциируется с повышенными уровнями инсулина в крови, т.е., состояния, известного как гиперинсулинемия. Инсулин, который представляет собой пептидный гормон, стимулирует утилизацию глюкозы, пептидный синтез, и образование и сохранение нейтральных липидов, а также, помимо всего прочего, он стимулирует рост сосудистых клеток и способствует удержанию натрия в почках. Эти две последние функции инсулина могут осуществляться независимо от уровней глюкозы в крови и являются известными причинами возникновения гипертензии. Так, например, рост периферической сосудистой сети может вызывать сужение периферических капилляров; а повышение концентрации натрия способствует увеличению объема крови. Поэтому, снижение уровня инсулина при гиперинсулинемии может способствовать предупреждению аномального роста сосудистой ткани, и повышения содержания натрия в почках, обусловленных высокими уровнями инсулина в крови, и следовательно, такое снижение уровня инсулина повлечет за собой ослабление симптомов гипертензии. Гипетрофия сердца является серьезным фактором риска, провоцирующим внезапную смерть, инфаркт миокарда и застойную сердечную недостаточность. Указанные явления, по крайней мере, частично, обусловлены повышенной восприимчивостью пациента к повреждению миокарда после ишемии и реперфузии, которое может происходить у амбулаторных больных, а также в процессе операции. При этом, не существует какого-либо подходящего лекарственного средства, которое позволило бы предотвратить или минимизировать неблагоприятный исход операции на сердце, а особенно операции по поводу инфаркта миокарда. Любая хирургическая операция, даже, если она проводится не на сердце, связана со значительным риском возникновения инфаркта миокарда, а иногда может приводить к летательному исходу. Примерно из 7 миллионов человек, подвергающихся операциям, не связанным с хирургией сердца, 20-25% пациентов либо умирают уже во время операции, либо получают серьезные осложнения на сердце. Кроме того, из 400000 пациентов, ежегодно подвергающихся операции на сердце с использованием аппаратов искусственного кровообращения, 1-2% пациентов умирают во время операции, а у 5% в ходе операции возникает инфаркт миокарда. В настоящее время, не существует какой-либо лекарственной терапии, которая способствовала бы снижению уровня повреждения сердечных тканей во время операций по поводу ишемии миокарда, при усилении резистентности сердечной ткани к приступам ишемии. Такая терапия, как ожидается, могла бы сократить жизнь пациентам, снизить число госпитализаций, повысить качество жизни и уменьшить затраты на медицинскую помощь пациентам группы высокого риска. Продуцирование глюкозы в печени является важным фактором лечения инсулин-независимого сахарного диабета (ИНСД). Перечень является главным регулятором уровней глюкозы в крови в постабсорбтивном (голодном) состоянии; при этом, у пациента с ИНСД, степень продуцирования глюкозы значительно выше, чем у нормальных индивидуумов. Аналогичным образом, после приема пищи, когда печень играет относительно меньшую роль в общей доставке глюкозы в плазму крови, продуцирование глюкозы в печени у пациентов с ИНСД является аномально высоким. Одним из основных способов предупреждения образования глюкозы в печени является прекращение процесса гликогенолиза. Печень продуцирует глюкозу благодаря процессам гликогенолиза (расщепление глюкозных остатков гликогенового полимера) и глюконеогенеза (синтез глюкозы из 2- и 3-углеродных предшественников). Имеется ряд данных, свидетельствующих о том, что гликогенолиз может вносить важный вклад в продуцирование глюкозы в печени у больных диабетом. Во-первых, по оценкам специалистов, до 75% продуцирования глюкозы в печени у нормальных индивидуумов в постабсорбтивном состоянии обусловлено гликогенолизом. И во-вторых, у пациентов, страдающих заболеванием, связанным с аномальным накоплением гликогена в печени, включая заболевание Герса (недостаточность гликогенфосфорилазы в печени), наблюдается эпизодическая гипогликемия. Эти наблюдения дают основание предположить, что гликогенолиз может играть важную роль в продуцировании глюкозы в печени. Гликогенолиз в печени, мышцах и головном мозге катализируется тканеспецифическими изоформами фермента гликогенфосфорилазы. Это фермент расщепляет макромолекулу гликогена с образованием глюкозо-1-фосфата и новой укороченной макромолекулы гликогена. В настоящее время известны два типа ингибиторов гликогенфосфорилазы: глюкоза и аналоги глюкозы (Martin. J.L. et al. Biochemistry, 1991, 30, 10101), и кофеин и другие аналоги пурина (Kasvinsky. P. J. et al., J.Biol. Chem., 1978, 253, 3343-3351 и 9102-9106). Было высказано предположение, что эти соединения и ингибиторы гликогенфосфорилазы, в целом, могут быть использованы для лечения инсулин-независимого сахарного диабета путем снижения продуцирования глюкозы в печени и уменьшении уровня глюкозы в крови (Blundell, T.B. et al., Diabеtologia 1992, 35, Suppl 2, 569-576; и Martin et al., Biochemistry 1991, 30, 10101). Механизм (или механизмы) восприимчивости организма к повреждению миокарда, наблюдаемому после ишемии и реперфузии, пока полностью не изучен. В связи с этим, сообщалось (M.F. Allaro et al., Am. J. Physiol, 267, H66-H74, 1994), что “у крыс с гипертрофией сердца, предишемическое восстановление гликогена… ассоциируется с улучшением постишемического восстановления функции левого желудочка”. Таким образом, хотя существует ряд способов лечения гипергликемии, гиперхолестеринемии, гипертензии, гиперлипидемии, атеросклероза и ишемии миокарда, однако, потребность в новых лекарственных средствах остается актуальной, а поэтому необходимо продолжать поиски альтернативных способов лечения. Настоящее изобретение относится к соединениям формулы I, ингибирующим гликогенфосфорилазу и предназначенным для использования в лечении диабета, гипергликемии, гиперхолестеринемии, гипертензии, гиперинсулинемии, гиперлипидемии, атеросклероза и ишемии миокарда. Соединениями настоящего изобретения являются соединения формулы I: 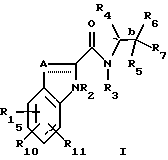 и их фармацевтически приемлемые соли и пролекарственные предшественники, где: пунктирная линия (—) означает необязательную связь; A представляет собой -C(H)=, -C((C1-C4)алкил)= или -C(галогено)=, если пунктирная линия означает связь; либо A представляет собой метилен или -CH(C1-C4)алкил, если пунктирная линия не является связью; R1, R10 или R11 независимо представляют собой H, галоген, 4-, 6- или 7-нитро, циано, (C1-C4)алкил, (C1-C4)алкокси, фторометил, дифторометил или трифторометил; R2 представляет собой H; R3 представляет собой H или (C1-C5)алкил; R4 представляет собой H, метил, этил, н-пропил, гидрокси(C1-C3)алкил, (C1-C3)алкокси(C1-C3)алкил, фенил(C1-C4)алкил, фенилгидрокси(C1-C4)алкил, фенил(C1-C4)алкокси(C1-C4)алкил, тиен-2- или -3-ил(C1-C4)алкил или фур-2- или -3-ил(C1-C4)алкил, где указанные кольца R4 являются независимо моно-, ди- или три-замещенными у атома углерода водородом, галогеном, (C1-C4)алкило(C1-C4)алкокси, трифторометилом, гидрокси, амино или циано; или R4 представляет собой пирид-2-, -3- или -4-ил(C1-C4)алкил, триазол-2-, -4- или -5-ил(C1-C4)алкил, имидазол-1-, -4- или -5-ил(C1-C4)алкил, пиррол-2- или -3-ил(C1-C4)алкил, оксазол-2-, -4- или -5-ил-(C1-C4)алкил, пиразол-3-, -4- или -5-ил(C1-C4)алкил, изоксазол-3-, -4- или -5-ил(C1-C4)алкил, изотиазол-3-, -4- или -5-ил(C1-C4)алкил, пиридазин-3- или -4-ил(C1-C4)алкил, пиримидин-2-, -4- -5- или -6-ил(C1-C4)алкил, пиразин-2- или -3-ил(C1-C4)алкил или 1,3,5-триазин-2-ил(C1-C4)алкил; причем предыдущие гетероциклы R4 являются независимо и необязательно моно- или ди-замещенными галогеном, трифторометилом, (C1-C4)алкилом, (C1-C4)алкокси, амино или гидрокси, а указанные моно- или ди-заместители связаны с углеродом; R5 представляет собой H, гидрокси, фтор, (C1-C5)-алкил, (C1-C5)алкокси, (C1-С5)алканоил, амино(C1-C4)алкокси, моно-N- или ди-N,N-(C1-C4)алкиламино(C1-C4)алкокси, карбокси(C1-C4)алкокси, (C1-C5)алкокси-карбонил (C1-C4)алкокси, бензилоксикарбонил, (C1-C4)алкокси) или карбонилокси, где указанная карбонилокси-группа является углерод-углерод-связанной с фенилом, триазолилом, имидазолилом, 1H-индолилом, фурилом, пирролилом, оксазолилом, изоксазолилом, изотиазолилом, пиридазинилом, пиримидинилом, пиразинилом или 1,3,5-триазинилом, и где указанные выше кольца R5 являются необязательно моно-замещенными галогеном, (C1-C4)алкилом, (С1-С4)алкокси, гидрокси, амино или трифторометилом, а указанные моно-заместители связаны с атомом углерода; R7 представляет собой H, фтор или (C1-C5)алкил; либо R5 и R7, взятые вместе, представляют собой оксо-группу; R6 представляет собой карбокси, (C1-C8)алкоксикарбонил, C(O)NR8R9 или (CO)R12, где: R8 представляет собой H, (C1-C3)алкил, гидрокси или (C1-C3)алкокси; и R9 представляет собой H, (C1-C8) алкил, гидрокси, (C1-C8)-алкокси, метилен-перфторированный (C1-C8)алкил, фенил, пиридил, тенил, фурил, пирролил, пирролидинил, оксазолил, тиазолил, имидазолил, пиразолил, пиразолидинил, изоксазолил, изотиазолил, пиранил, пиперидинил, морфолинил, пиридазинил, пиримидинил, пиразинил, пиперазинил или 1,3,5-тиазинил, где указанные выше кольца R9 присоединены через связь углерод-азот; или R9 представляет собой моно-, ди- или три-замещенный (C1-C5)алкил, где указанные заместители независимо представляют собой H, гидрокси, амино, моно-N- или ди- N,N-(C1-C5)алкиламино; или R9 является моно- или ди-замещенным (C1-C5)алкилом, где указанные заместители независимо представляют собой фенил, пиридил, фурил, пирролил, пирролидинил, оксазолил, тиазолил, имидазолил, пиразолил, пиразолинил, пиразолидинил, изоксазолил, изотиазолил, пиранил, пиридинил, пиперидинил, морфолинил, пиридазинил, пиримидинил, пиразинил, пиперазинил или 1,3,5-триазинил; где неароматические азот-содержащие кольца R9 являются необязательно замещенными у атома азота (C1-C6)алкилом, бензилом, бензоилом или (C1-C6)алкоксикарбонилом; и где кольца R9 являются необязательно монозамещенными у атома углерода галогеном, (C1-C4)алкилом, (C1-C4)алкокси, гидрокси, амино или моно- N- и ди- N,N-(C1-C5)алкиламино, при условии, что отсутствует кватернизированный азот, и отсутствуют связи азот-кислород, азот-азот или азот-галоген; R12 представляет собой пиперазин-1-ил, 4-(C1-C4)алкилпиперазин-1-ил, 4-формилпиперазин-1-ил, морфолино, тиоморфолино, 1-оксотиоморфолино, 1,1-диоксо-тиоморфолино, тиазилидин-3-ил, 1-оксо-тиазолидин-3-ил, 1,1-диоксо-тиазолидин-3-ил, 2-(C1-C5)алкоксикарбонилпирролидин-1-ил, оксазолидин-3-ил или 2(R)-гидроксиметилпирролидин-1-ил; или R12 представляет собой 3- и/или 4-моно- или ди-замещенный оксазетидин-2-ил, 2-, 4- и/или 5-моно- или ди-замещенный оксазолидин-3-ил, 2-, 4- и/или 5-моно или ди-замещенный тиазолидин-3-ил, 2-, 4- и/или 5-моно- или ди-замещенный 1-оксотиазолидин-3-ил, 2-, 4- и/или 5-моно- или ди-замещенный 1,1-диоксотиазолидин-3-ил, 3- и/или 4-, моно- или ди-замещенный пирролидин-1-ил, 3-, 4- и/или 5-, моно-, ди- или три-замещенный пиперидин-1-ил, 3-, 4-, и/или 5-моно, ди- или три-замещенный пиперазин-1-ил, 3-замещенный азетидин-1-ил, 4- и/или 5-, моно- или ди-замещенный 1,2-оксазинан-2-ил, 3- и/или 4-моно- или ди-замещенный пиразолидин-1-ил, 4- и/или 5-, моно- или ди-замещенный изоксазолидин-2-ил, 4- и/или 5-моно- и/или ди-замещенный изотиазолидин-2-ил, где указанные R12-заместители независимо представляют собой H, галоген, (C1-C5)алкил, гидрокси, амино, моно- N- или ди- N,N-(C1-C5)алкиламино, формил, оксо, гидроксиимино, (C1-C5)алкокси, карбокси, карбамоил, моно-N- или ди-N,N-(C1-C4)алкилкарбамоил, (C1-C4)алкоксиимино, (C1-C4)алкоксиметокси, (C1-C6)алкоксикарбонил, карбокси (C1-C5)алкил или гидрокси(C1-C5)алкил; при условии, что, если R4 является H, метилом, этилом или н-пропилом, что R5 является OH; при условии, что, если R5 и R7 являются H, то R4 не является H, метилом, этилом, н-пропилом, гидрокси(C1-C3)алкилом или (C1-C3)алкокси(C1-C3)алкилом, а R6 является (C(O)NR8R9, C(O)R12 или (C1-C4)алкоксикарбонилом. Первая группа предпочтительных соединений формулы I состоит из соединений, в которых: R1 представляет собой 5-H, 5-галоген, 5-метил или 5-циано; R10 и R11 независимо представляют собой H или галоген; A представляет собой -C(H)=; R2 и R3 представляют собой H; R4 представляет собой фенил(C1-C2)алкил, где фенильные группы независимо являются моно-, ди- или три-замещенными водородом или галогеном, либо они являются независимо моно- или ди-замещенными водородом, галогеном, (C1-C4)алкилом, (C1-C4)алкокси, трифторометилом, гидрокси, амино или циано; или R4 представляет собой тиен-2- или -3-ил(C1-C2)алкил, пирид-2-, -3- или -4-ил(C1-C2)алкил, тиазол-2-, -4- или -5-ил(C1-C2)алкил, имидазол, -1-, -2-, -4- или -5-ил(C1-C2)алкил, фур-2- или -3-ил(C1-C2)алкил, пиррол-2- или -3-ил(C1-C2)алкил, оксазол-2-, -4- или -5-ил(C1-C2)алкил, пиразол-3-, -4- или -5-ил(C1-C2)алкил, изоксазол-3-, -4- или -5-ил(C1-C2)алкил, где указанные выше гетероциклы R4 являются необязательно и независимо моно- или ди-замещенными галогеном, трифторометилом, (C1-C4)алкилом, (C1-C4)алкокси, амино или гидрокси; а указанные моно- или ди-заместители связаны с углеродом; R5 представляет собой гидрокси; R6 представляет собой C(O)NR8R9 или C(O)R12; и R7 представляет собой H. Из вышеуказанной первой группы предпочтительных соединений формулы I, первая группа особенно предпочтительных соединений состоит из соединений, в которых: атом углерода a имеет S-конфигурацию; атом углерода b имеет R-конфигурацию; R4 представляет собой фенил(C1-C2)алкил, тиен-2-ил-(C1-C2)алкил, тиен-3-ил-(C1-C2)алкил, фур-2-ил-(C1-C2)алкил или фур-3-ил(C1-C2)алкил, где указанные кольца являются независимо моно- или ди-замещенными водородом или фтором; R6 представляет собой C(O)NR8R9; R8 представляет собой (C1-C3)алкил, гидрокси или (C1-C3)алкокси; и R9 представляет собой H, (C1-C8)алкил, гидрокси-, гидрокси(C1-C6)алкил, (C1-C8)алкокси, пиридил, морфолинил, пиперазинил, пиперидинил, имидазолил или тиазолил, или (C1-C4)алкил, моно-замещенный пиридилом, морфолинилом, пиперазинилом, пирролидинилом, пиперидинилом, имидазолилом или тиазолилом. Из вышеуказанной первой группы особенно предпочтительных соединений, наиболее предпочтительными соединениями являются: 5-Хлоро-1H-индол-2-карбоновой кислоты [(1S)-((R)-гидрокси-диметилкарбамоил-метил)-2-фенил-этил]-амид; 5,6-Дихлоро-1H-индол-2-карбоновой кислоты { (1S)-[(R)-гидрокси-(метокси-метил-карбамоил)-метил]-2-фенил-этил}-амид; 5-Хлоро-1H-индол-2-карбоновой кислоты {(1S)-[(R)-гидрокси-(метокси-метил-карбамоил)-метил]-2-фенил-этил}-амино; 5-Хлоро-1H-индол-2-карбоновой кислоты ((1S)-{(R)-гидрокси[(2-гидрокси-этил)-метил-карбамоил]-метил}-2- фенил-этил)амид; 5-Хлоро-1H-индол-2-карбоновой кислоты {(1S)-[(R)-гидрокси-(метил-пиридин-2-ил-карбамоил)-метил]-2-фенил-этил}амид; или 5-Хлоро-1H-индол-2-карбоновой кислоты ((1S)-{(R)-гидрокси-[метил-(2-пиридин-2-ил-этил)карбамоил]-метил}-2- фенил-этил)-амид. В вышеуказанную первую группу особенно предпочтительных соединений входят соединения, в которых: a. R1 представляет собой 5-Хлоро; R10 и R11 представляют собой H; R4 представляет собой бензил; R8 представляет собой метил; и R9 представляет собой метил; b. R1 представляет собой 5-хлоро; R11 представляет собой H; R10 представляет собой 6-хлоро; R4 представляет собой бензил; R8 представляет собой метил; и R9 представляет собой метокси; c. R1 представляет собой 5-хлоро; R10 и R11 представляют собой H; R4 представляет собой бензил; R8 представляет собой метил; и R9 представляет собой метокси; d. R1 представляет собой 5-хлоро; R10 и R11 представляют собой H; R4 представляет собой бензил; R8 представляет собой метил; и R9 представляет собой 2-(гидрокси)этил; e. R1 представляет собой 5-хлоро; R10 и R11 представляют собой H; R4 представляет собой бензил; R8 представляет собой метил; и R9 представляет собой пиридин-2-ил; и f. R1 представляет собой 5-хлоро; R10 и R11 представляют собой H; R4 представляет собой бензил; R8 представляет собой метил; и R9 представляет собой 2-(пиридин-2-ил)этил. Из вышеуказанной первой группы предпочтительных соединений формулы I, вторая группа особенно предпочтительных соединений состоит из соединений, в которых: атом углерода a имеет (S)-конфигурацию; атом углерода b имеет (R)-конфигурацию; R4 представляет собой фенил(C1-C2)алкил, тиен-2-ил-(C1-C2)алкил; тиен-3-ил(C1-C2)алкил, фур-2-ил-(C1-C2)алкил или фур-3-ил-(C1-C2)алкил, где указанные кольца являются независимо моно- или ди-замещенными водородом или фтором; R6 представляет собой C(O)R12; и R12 представляет собой морфолино, 4-(C1-C4)алкилпиперазин-1-ил, 3-замещенный азетидин-1-ил, 3- и/или 4-, моно- или ди-замещенный пирролидин-1-ил, 4- и/или 5-, моно- или ди-замещенный изоксазолидин-2-ил, 4- и/или 5-моно- или ди-замещенный 1,2-оксазинан-2-ил, где указанные заместители независимо представляют собой H, галоген, гидрокси, амино, моно- N- или ди-N,N-(C1-C6)алкиламино, оксо, гидроксиимино или алкокси. Из вышеуказанной второй группы особенно предпочтительных соединений, наиболее предпочтительными являются следующие соединения: 5-Хлоро-1H-индол-2-карбоновой кислоты [(1S)-бензил-(2R)-гидрокси-3-(4-метил-пиперазин-1-ил)-3-оксо-пропил]амида гидрохлорид; 5-Хлоро-1H-индол-2-карбоновой кислоты [(1S)-бензил-(2R)-гидрокси-3-(3-гидрокси-азетидин-1-ил)-3-оксо-пропил]амид; 5-Хлоро-1H-индол-2-карбоновой кислоты (1S)-бензил-(2R)-гидрокси-3-изоксазолидин-2-ил-3-оксо-пропил)-амид; 5-Хлоро-1H-индол-2-карбоновой кислоты ((1S)-бензил-(2R)-гидрокси-3-[1,2] оксазинан-2-ил-3-оксо-пропил)-амид; 5-Хлоро-1H-индол-2-карбоновой кислоты [(1S)-бензил-(2R)-гидрокси-3-(3S)-гидрокси-пирролидин-1-ил)-3-оксо-пропил амид; 5-Хлоро-1H-индол-2-карбоновой кислоты [(1S)-бензил-3-((3S, 4S)-дигидрокси-пирролидин-1-ил)-(2R)-гидрокси-3- оксо-пропил]-амид; 5-Хлоро-1H-индол-2-карбоновой кислоты [(1S)-бензил-3-((3R, 4S)-дигидрокси-пирролидин-1-ил)-(2R)-гидрокси-3- оксо-пропил]амид; 5-Хлоро-1H-индол-2-карбоновой кислоты ((S)-бензил-(2R)-гидрокси-3-морфолин-4-ил-3-оксо-пропил)-амид. В вышеуказанную вторую группу особенно предпочтительных соединений входят соединения, в которых: a. R1 представляет собой 5-хлоро; R10 и R11 представляют собой H; R4 представляет собой бензил; и R12 представляет собой 4-метилпиперазин-1-ил; b. R1 представляет собой 5-хлоро; R10 и R11 представляют собой H; R4 представляет собой бензил; и R12 представляет собой 3-гидроксиазетидин-1-ил; c. R1 представляет собой 5-хлоро; R10 и R11 представляют собой H; R4 представляет собой бензил; и R12 представляет собой изоксазолидин-2-ил; d. R1 представляет собой 5-хлоро; R10 и R11 представляют собой H; R4 представляет собой бензил; и R12 представляет собой (1,2)-оксазинан-2-ил; e. R1 представляет собой 5-хлоро; R10 и R11 представляют собой H; R4 представляет собой бензил; и R12 представляет собой 3(S)-гидроксипирролидин-1-ил; f. R1 представляет собой 5-хлоро; R10 и R11 представляют собой H; R4 представляет собой бензил; и R12 представляет собой (3S,4S)-дигидроксипирролидин-1-ил; g. R1 представляет собой 5-хлоро; R10 и R11 представляют собой H; R4 представляет собой бензил; и R12 представляет собой (3R,4S)-дигидроксипирролидин-1-ил; и h. R1 представляет собой 5-хлоро; R10 и R11 представляют собой H; R4 представляет собой бензил; и R12 представляет собой морфолино. Вторая группа предпочтительных соединений формулы I состоит из соединений, в которых: R1 представляет собой H, галоген, метил или циано; R10 и R11 независимо представляют собой H или галоген; A представляет собой -C(H)=; R2 и R3 представляют собой H; R4 представляет собой фенил(C1-C2)алкил, где указанные фенильные группы являются независимо моно-, ди- или три-замещенными H или галогеном, либо они являются независимо моно- или ди-замещенными H, галогеном, (C1-C4)алкилом, (C1-C4)-алкокси, трифторометилом, гидрокси, амино или циано; или R4 представляет собой тиен-2- или -3-ил(C1-C2)алкил, пирид-2-, -3- или -4-ил(C1-C2)алкил, тиазол-2-, -4- или -5-ил(C1-C2)алкил, имидазол-1-, -2-, -4- или -5-ил(C1-C2)алкил, фур-2- или -3-ил(C1-C2)алкил, пиррол-2- или -3-ил(C1-C2)алкил, оксазол-2-, -4- или -5-ил(C1-C2)алкил, пиразил-3-, -4- или -5-ил(C1-C2)алкил, изоксазол-3-, -4- или -5-ил(C1-C2)алкил, где указанные выше гетероциклы R4 являются необязательно и независимо моно- или ди-замещенными галогеном, трифторметилом, (C1-C4)алкилом, (C1-C4)алкокси, амино или гидрокси; а указанные моно- или ди-заместители связаны с углеродом; R5 представляет собой карбокси; R6 представляет собой карбокси или (C1-C8)алкоксикарбонил; и R7 представляет собой H, фторо или (C1-C6)алкил. Из второй группы предпочтительных соединений формулы I, группу особенно предпочтительных соединений составляют соединения, в которых: атом углерода a имеет (S)-конфигурацию; атом углерода b имеет (R)-конфигурацию; R4 представляет собой фенил(C1-C2)алкил, тиен-2-ил(C1-C2)алкил, тиен-3-ил(C1-C2)алкил, фур-2-ил(C1-C2)алкил или фур-3-ил(C1-C2)алкил, где указанные кольца являются независимо моно- или ди-замещенными водородом или фтором; R10 и R11 представляют собой H; R6 представляет собой карбокси; и R7 представляет собой H. Из непосредственно предшествующей группы соединений, предпочтительными являются соединения, в которых: R1 представляет собой 5-хлоро; R10 и R11 представляют собой H; и R4 представляет собой бензил. Третья группа предпочтительных соединений формулы I состоит из соединений, в которых: R1 представляет собой H, галоген, метил или циано; R10 и R11 независимо представляют H или галоген; A представляет собой -C(H)=; R2 и R3 представляют собой H; R4 представляет собой фенил(C1-C2)алкил, где указанные фенильные группы являются независимо моно-, ди- или три-замещенными H или галогеном, либо они являются независимо моно- или ди-замещенными H, галогеном, (C1-C4)алкилом, (C1-C4)алкокси, трифторометилом, гидрокси, амино или циано; или R4 представляет собой тиен-2- или -3-ил(C1-C2)алкил, пирид-2-, -3- или -4-ил(C1-C2)алкил, тиазол-2-, -4- или -5-ил(C1-C2)алкил, имидазол-1-, -2-, -4- или -5-ил(C1-C2)алкил, фур-2- или -3-ил(C1-C2)алкил, пиррол-2- или -3-ил(C1-C2)алкил, оксазол-2-, -4- или -5-ил(C1-C2)алкил, пиразол-3-, -4- или -5-ил(C1-C2)алкил, изоксазол-3-, -4- или -5-ил(C1-C2)алкил, где указанные выше гетероциклы R4 являются независимо и необязательно моно- или ди-замещенными галогеном, трифторометилом, (C1-C4)алкилом, (C1-C4)алкокси, амино или гидрокси; а указанные моно- или ди-заместители связаны с углеродом; R5 представляет собой фтор, (C1-C4)алкил, (C1-C5)алкокси, амино(C1-C4)алкокси, моно-N- или ди-N,N-(C1-C4)алкиламино-(C1-C4)алкокси, карбокси(C1-C4)алкокси, (C1-C5)алкокси-карбонилл(C1-C4)алкокси, бензилоксикарбонил(C1-C4)алкокси; R6 представляет собой карбокси или (C1-C8)алкоксикарбонил; и R7 представляет собой H, фтор или (C1-C6)алкил. Четвертая группа предпочтительных соединений формулы I состоит из соединений, в которых: R1 представляет собой H, галоген, метил или циано; R10 и R11 независимо представляют собой H или галоген; A представляет собой -C(H)=; R2 и R3 представляют собой H; R4 представляет собой фенил(C1-C2)алкил, где указанные фенильные группы являются независимо моно-, ди- или три-замещенными H или галогеном, либо они являются независимо моно- или ди-замещенными H, галогеном, (C1-C4)алкилом, (C1-C4)алкокси, трифторометилом, гидрокси, амино или циано; или R4 представляет собой тиен-2- или -3-ил(C1-C2)алкил, тиазол-2-, -4- или -5-ил(C1-C2)алкил, имидазол-1-, -2-, -4- или -5-ил(C1-C2)алкил, фур-2- или -3-ил(C1-C2)алкил, пиррол-2- или -3-ил(C1-C2)алкил, оксазол-2-, -4- или -5-ил(C1-C2)алкил, пиразол-3-, -4- или -5-ил(C1-C2)алкил, изоксазол-3-, -4- или -5-ил(C1-C2)алкил, где указанные выше гетероциклы R4 являются необязательно и независимо моно- или ди-замещенными галогеном, трифторометилом, (C1-C4)алкилом, (C1-C4)алкокси, амино или гидрокси; а указанные моно- или ди-заместители связаны с углеродом; R5 представляет собой фтор, (C1-C4)алкил, (C1-C5)алкокси, амино(C1-C4)алкокси, моно-N- или ди-N,N-(C1-C4)алкиламино-(C1-C4)алкокси, карбокси(C1-C4)алкокси, (C1-C5)алкокси-карбонил(C1-C4)алкокси, бензилоксикарбонил(C1-C4)алкокси; R6 представляет собой C(O)NR8R9 или C(O)R12; R7 представляет собой H, фтор или (C1-C6)алкил. В другом своем аспекте, настоящее изобретение относится к способу лечения гликогенфосфорилазо-зависимых заболеваний или состояний у млекопитающего путем введения млекопитающему, страдающему указанным заболеванием или состоянием, эффективного для такого лечения количества соединения формулы I. В другом своем аспекте, настоящее изобретение относится к способу лечения гипергликемии у млекопитающего путем введения млекопитающему, страдающему гипергликемией, эффективного для такого лечения количества соединения формулы I. В другом своем аспекте, настоящее изобретение относится к способу лечения диабета у млекопитающего путем введения млекопитающему, страдающему диабетом, эффективного против диабета количества соединения формулы I. В другом своем аспекте, настоящее изобретение относится к способу лечения гиперхолестеринемии у млекопитающего путем введения млекопитающему, страдающему гиперхолестеринемией, эффективного для лечения гиперхолестеринемии количества соединения формулы I. Лечение диабета также предусматривает предупреждение или ослабление хронических осложнений, таких, как невропатия, нефропатия, ретинопатия или катаракта. В другом своем аспекте, настоящее изобретение относится к способу лечения атеросклероза у млекопитающего путем введения млекопитающему, страдающему атеросклерозом, эффективного для лечения атеросклероза количества соединения формулы I. В другом своем аспекте, настоящее изобретение относится к способу лечения гиперинсулинемии у млекопитающего путем введения млекопитающему, страдающему гиперинсулинемией, эффективного для лечения гиперинсулинемии количества соединения формулы I. В другом своем аспекте, настоящее изобретение относится к способу лечения гипертензии у млекопитающего путем введения млекопитающему, страдающему гипертензией, эффективного для лечения гипертензии количества соединения формулы I. В другом своем аспекте, настоящее изобретение относится к способу лечения гиперлипидемии у млекопитающего путем введения млекопитающему, страдающему гиперлипидемией, эффективного для лечения гиперлипидемии количества соединения формулы I. В другом своем аспекте, настоящее изобретение относится к способу предупреждения ишемических повреждений миокарда у млекопитающего путем введения млекопитающему, подвергающемуся риску ишемического повреждения миокарда при операции, эффективного для предупреждения указанного ишемического повреждения количества соединения формулы I. В другом своем аспекте, настоящее изобретение относится к способу предупреждения ишемических повреждений миокарда у млекопитающего путем введения млекопитающему, подвергающемуся риску ишемического повреждения миокарда при операции, эффективного для предупреждения указанного ишемического повреждения количества ингибитора гликогенфосфорилазы. Настоящее изобретение также относится к фармацевтическим композициям, содержащим терапевтически эффективное количество соединения формулы I и фармацевтически приемлемый носитель. Предпочтительными являются фармацевтические композиции для лечения гликогенфосфорилазо-зависимых заболеваний или состояний у млекопитающих, содержащие эффективное для лечения гликогенфосфорилазо-зависимых заболеваний или состояний количество соединения формулы I и фармацевтически приемлемый носитель. В другом своем аспекте, настоящее изобретение относится к фармацевтическим композициям для лечения диабета, содержащим терапевтически эффективное количество ингибитора гликогенфосфорилазы; одно или несколько противодиабетических средств, таких, как инсулин или его аналогов (например, LysPro-инсулин); GLP-1 (7-37) (инсулинотропин) и GLP-1 (7-36) – H2; сульфомочевины и их аналоги; хлоропропамид, глибенкламид, толбутамид, толазамид, ацетогексамид, глипизид, глимепирид, репаглинид, меглитинид; бигуаниды: метформин, фенформин, буформин;  2 -антагонисты и имидазолины: мидаглизол, изаглидол, дериглидол, идазоксан, эфароксан, флупароксан; другие средства, усиливающие секрецию инсулина; линоглирид, A-4166; глитазоны: циглитазон, пиоглитазон, энглитазон, троглитазон, даглитазон, BP 49653; ингибиторы окисления жирных кислот: кломоксир, этомоксир; ингибиторы -глюкозидазы: миглитол, акарбоза, эмиглитат, воглибоза, MDL-25637, камиглибоза, MDL-73945; 2 -антагонисты и имидазолины: мидаглизол, изаглидол, дериглидол, идазоксан, эфароксан, флупароксан; другие средства, усиливающие секрецию инсулина; линоглирид, A-4166; глитазоны: циглитазон, пиоглитазон, энглитазон, троглитазон, даглитазон, BP 49653; ингибиторы окисления жирных кислот: кломоксир, этомоксир; ингибиторы -глюкозидазы: миглитол, акарбоза, эмиглитат, воглибоза, MDL-25637, камиглибоза, MDL-73945;  -агонисты: BPL 35135, BPL 37344, Ro 16-8714, 1C1 D7114, CL 316243; ингибиторы фосфодиэстеразы; L-386398; средства, понижающие содержание липидов: бенфлуорекс; средства против ожирения: фенфлурамин; ванадатные и ванадиевые комплексы (например, нагливанR) и пероксованадиевые комплексы; антагонисты амилина; антагонисты глюкагона; ингибиторы глюконеогенеза; аналоги соматостатина; антилиполитические средства: никотиновая кислота, аципимокс, WAG994; и -агонисты: BPL 35135, BPL 37344, Ro 16-8714, 1C1 D7114, CL 316243; ингибиторы фосфодиэстеразы; L-386398; средства, понижающие содержание липидов: бенфлуорекс; средства против ожирения: фенфлурамин; ванадатные и ванадиевые комплексы (например, нагливанR) и пероксованадиевые комплексы; антагонисты амилина; антагонисты глюкагона; ингибиторы глюконеогенеза; аналоги соматостатина; антилиполитические средства: никотиновая кислота, аципимокс, WAG994; инеобязательно фармацевтически приемлемый носитель; Из описанной выше группы композиций, предпочтительными являются такие фармацевтические композиции, в которых ингибитором гликогенфосфорилазы является соединение формулы I. В другом своем аспекте, настоящее изобретение относится к способу лечения диабета у млекопитающих с использованием вышеописанных комплексных композиций. К гликогенфосфорилазо-зависимым заболеваниям или состояниям относятся такие состояния, которые частично или полностью опосредуются, инициируются или поддерживаются расщеплением макромолекулы гликогена гликогенфосфорилазами, в результате чего высвобождается глюкозо-1-фосфат и образуется новая более короткая молекула гликогена. Эти состояния характеризуются повышенной гликогенфосфорилазной активностью, и уменьшение интенсивности симптомов указанных состояний может быть достигнуто путем снижения такой активности. Примерами таких заболеваний или состояний являются диабет, гипергликемия, гиперхолестеринемия, гипертензия, гиперинсулинемия, гиперлипидемия, атеросклероз и ишемия миокарда. Термин “ингибитор гликогенфосфорилазы” относится к любому соединению или средству, или комбинации соединений и/или средств, которые способствуют уменьшению, замедлению или прекращению ферментативного действия гликогенфосфорилазы. Известное в настоящее время действие гликогенфосфорилазы заключается в расщеплении гликогена путем катализа обратимой реакции макромолекулы гликогена и неорганического фосфата с образованием глюкозо-1-фосфата и макромолекулы гликогена, которая становится на один глюкозильный остаток короче, чем исходная молекула гликогена (в направлении гликогенолиза). Под термином “лечение”, используемым в настоящем описании, подразумевается предупреждающее (например, профилактическое) и паллиативное лечение. Термин “галоген” означает хлор, бром, йод или фтор. Термин “алкил” означает насыщенный углеводород с прямой или разветвленной цепью. Примерами таких алкильных групп (длина которых определяется конкретным примером) являются метил, этил, пропил, изопропил, бутил, втор-бутил, трет-бутил, пентил, изопентил, гексил и изогексил. Термин “алкокси” означает насыщенный прямой или разветвленный алкил, связанный через окси-группу. Примерами таких алкокси-групп (длина которых определяется конкретным примером) являются метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, трет-бутокси, пентокси, изопентокси, гексокси и изогексокси. Выражение “фармацевтически приемлемая анионная соль” относится к нетоксичным анионным солям, содержащим анионы (но не ограничивается ими), таким, как хлорид, бромид, иодид, сульфат, бисульфат, фосфат, ацетат, малеат, фумарат, оксалат, лактат, тартрат, цитрат, глюконат, метансульфонат и 4-толуолсульфонат. Выражение “фармацевтически приемлемая катионная соль” относится к нетоксичным катионным солям (но не ограничивается ими), таким как соли натрия, калия, кальция, магния, аммония, или протонированный бензатин(N,N-дибензилэтилендиамин), холин, этаноламин, диэтаноламин, этилендиамин, мегламин (N-метилглюкамин), бенетамин (N-бензилфенэтиламин), пиперазин или трометамин (2-амино-2-гидроксиметил-1,3-пропандиол). Выражение “пролекарство” относится к соединениям, которые являются предшественниками лекарственного средства и которые, после их введения, способствуют высвобождению in vivo лекарственного средства посредством определенных химических или физиологических процессов (например, пролекарство, после его введения в среду с физиологическим pH, превращается в нужную лекарственную форму). Типичные пролекарства, после их расщепления, высвобождают соответствующую свободную кислоту, и такими гидролизуемыми эфирообразующими остатками соединений настоящего изобретения являются, но не ограничиваются ими, карбоново-кислотные заместители (например, R6 является карбокси, либо R8, R9 или R12 содержат карбокси), где свободный водород замещен (C1-C4)алкилом, (C2-C12)алканоилоксиметилом, 1-(алканоилокси)этилом, имеющим 4-9 атомов углерода, 1-метил-1-(алканоилокси)-этилом, имеющим 5-10 атомов углерода, алкоксикарбонилоксиметилом, имеющим 3-6 атомов углерода, 1-(алкоксикарбонилокси)этилом, имеющим 4-7 атомов углерода, 1-метил-1-(алкоксикарбонилокси)этилом, имеющим 5-8 атомов углерода, N-(алкоксикарбонил)аминометилом, имеющим 3-9 атомов углерода, 1-(N-(алкоксикарбонил)амино)этилом, имеющим 4-10 атомов углерода, 3-фталидилом, 4-кротонолактонилом, гамма-бутиролактон-4-илом, ди-N,N-(C1-C2)алкиламино(C2-C3)алкилом (таким, как -диметиламиноэтил), карбамоил-(C1-C2)алкилом, N, N-ди(C1-C2)-алкилкарбамоил-(C1-C2)алкилом и пиперидино-, пирролидино- или морфолино(C2-C3)алкилом.
Другие типичные пролекарства высвобождают спирт формулы I, где свободный водород гидрокси-заместителя (например, R5 является гидрокси-группой) замещен (C1-C6)алканоилоксиметилом, 1-((C1-C6)алканоилокси)этилом, 1-метил-1-((C1-C6)алканоилокси)-этилом, (C1-C6)алкоксикарбонилоксиметилом, N-(C1-C6)алкоксикарбониламинометилом, сукциноилом, (C1-C6)алканоилом,-амино-(C1-C4)алканоилом, арилактилом и -аминоацилом, либо -аминоацил--аминоацилом (где указанные -аминоацильные радикалы независимо представляют собой любую из натуральных L-аминокислот, присутствующих в белках), P(O)(OH)2, -P(O)(O)(C1-C6)-алкил)2, ом или гликозилом (радикал, образующийся в результате отщепления гидроксила полуацеталя углевода).
Другими характерными пролекарствами являются, но не ограничиваются ими, производные формулы I, где R2 является свободным водородом, который замещен R-карбонилом, RO-карбонилом, NRR’-карбонилом, где R и R’ независимо представляют собой ((C1-C10)алкил, (C3-C7)циклоалкил, бензил; или R-карбонил представляет собой натуральный -аминоацил-натуральный -аминоацил, -C(OH)C(O)OY, где Y представляет собой H, (C1-C6)алкил или бензил, -C(OY0)Y1, где Y0 представляет собой (C1-C4)алкил, а Y1 представляет собой (C1-C6)алкил, карбокси(C1-C6)алкил, амино(C1-C4)алкил, или моно-N- или ди-N, N-(C1-C6)алкиламиноалкил, -C(Y2)Y3, где Y2 представляет собой H или метил, а Y3 представляет собой моно-N- или ди-N,N-(C1-C6)алкиламино, морфолино, пиперидин-1-ил или пирролидин-1-ил.
Другими примерами пролекарств являются циклические структуры, такие, как соединения формулы I, где R2 и R3 имеют общий углерод, образуя, тем самым, 5-членное кольцо. Этот связывающий углерод может быть независимо моно- или ди-замещен H, (C1-C6)алкилом, (C3-C6)циклоалкилом или фенилом. Альтернативно, R3 и R5, взятые вместе, могут образовывать оксазолидиновое кольцо, а два углерода оксазолидинового кольца могут быть независимо моно- или ди-замещенными H, (C1-C6)алкилом, (C3-C6)циклоалкилом, или фенилом. Альтернативно, пролекарственными формами соединения формулы I являются соединения, в которых R5, взятый вместе с R8 или R9, может образовывать оксазолин-4-он’овое кольцо, а 2 атома углерода этого кольца могут быть независимо моно- или ди-замещенными H, (C1-C6)алкилом, (C3-C6)циклоалкилом, фенилом или оксо-группой.
Используемые в настоящем описании выражения “реакционно-инертный растворитель” и “инертный растворитель” относятся к растворителю, который не вступает в нежелательные реакции с исходными соединениями, реагентами, промежуточными соединениями или продуктами реакции, и не оказывает неблагоприятного влияния на выход целевого продукта.
Каждому среднему специалисту ясно, что определенные соединения настоящего изобретения содержат один или несколько атомов, которые могут находиться в конкретной стереохимической или геометрической конфигурации, образуя стереоизомеры и конфигурационные изомеры. Все такие изомеры и их смеси входят в объем настоящего изобретения. Кроме того, в объем настоящего изобретения входят также гидраты соединений настоящего изобретения.
Каждому среднему специалисту ясно, что определенные комбинации заместителей, содержащих гетероатом и входящих в объем настоящего изобретения, определяют соединения, которые являются менее стабильными в физиологических условиях (например, соединения, содержащие ацетальные или аминальные связи). Поэтому, такие соединения являются менее предпочтительными.
Термин “кольцо Rx“, где x является целым числом, например, “кольцо R9“, “кольцо R12” или “кольцо R4“, используемый в настоящем описании при указании на замещение на кольце, относится к тем частям молекулы, где кольцом является Rx, а также к тем частям молекулы, где кольцо содержится в Rx.
Используемый в настоящем описании термин “моно-N- или ди-N,N-(C1-Cx)алкил. .. относится к (C1-Cx)алкильному радикалу, взятому отдельно, в тех случаях, когда он является ди-N,N-(C1-Cx)алкилом… (где x – целое число).
Другие отличительные признаки и преимущества настоящего изобретения очевидны из нижеследующего подробного описания и формулы настоящего изобретения.
В основном, соединения формулы I могут быть получены способами, известными специалистам-химикам, особенно, в свете нижеприведенного подробного описания изобретения. Однако, некоторые способы получения соединений формулы I имеют отличительные признаки, присущие настоящему изобретению, а поэтому эти способы проиллюстрированы реакционными схемами (см. в конце описания).
В соответствии с реакционной схемой I, соединения формулы I, где R1, R10, R11, A1, R2, R3, R4, R5, R6 и R7 определены выше, могут быть получены любым из двух основных способов. В первом способе, желаемое соединение формулы I может быть получено реакцией взаимодействия соответствующей индол–2-карбоновой кислоты или индолин-2-карбоновой кислоты формулы I с соответствующим амином формулы III (т. е., реакции ацилирования амина). Во втором способе, желаемое соединение формулы I может быть получено реакцией взаимодействия соответствующего соединения формулы IV (т.е., соединения формулы I, в котором R6 представляет собой карбокси-группу) с соответствующим спиртом, или амином формулы R8R9NH или R12H, или спиртом, где R8, R9 и R12 определены выше (т.е., реакции ацилирования амина или спирта).
Обычно, соединение формулы II объединяют с соединением формулы III (или соединение формулы IV объединяют с соответствующим амином (например, R12H или R8R9NH)) или спиртом в присутствии соответствующего связующего агента. Подходящим связующим агентом является вещество, которое превращает карбоновую кислоту в соответствующее реакционноспособное соединение, которое образует амидную или сложноэфирную связь при взаимодействии с амином или спиртом.
Таким конденсирующим агентом может быть реагент, который после смешивания с карбоновой кислотой и амином или спиртом в одном реакционном сосуде осуществляет реакцию конденсации. Если проводят реакцию конденсации кислоты со спиртом, то спирт в качестве реакционного растворителя предпочтительно использовать в большом избытке без добавления или с добавлением 1,0-1,5 эквивалента диметиламинопиридина. Примерами конденсирующих реагентов являются 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид/гидроксибензотриазол (DEC/HBT), карбонилдиимидазол, дициклогексилкарбодиимид/гидроксибензотриазол (HBT), 2-этокси-1-этоксикарбонил-1,2-дигидрохинолин (EEDQ), карбонилдиимидазол/HBT, диэтилфосфорилцианнид. Указанная реакция взаимодействия протекает в инертном растворителе, предпочтительно в апротонном растворителе при температуре от около -20oC до около 50oC в течение периода времени от около 1 часа до около 48 часов. Примерами растворителей являются ацетонитрил, дихлорметан, диметилформамид и хлороформ. Примером подходящих методов проведения реакции конденсации является Методика A, описанная ниже (непосредственно перед примерами).
Конденсирующим агентом может быть также агент, превращающий карбоновую кислоту в активированное промежуточное соединение, которое выделяют, и/или которое, после его образования в первой стадии, оставляют для реакции с амином или спиртом во второй стадии. Примерами таких конденсирующих агентов и активированных промежуточных соединений являются тионилхлорид или оксалилхлорид, который образует хлорангидрид; фторангидрид циануровой кислоты, который образует фторангидрид; или алкилхлороформиат, такой, как изобутил- или изопропенилхлороформиат (с основанием, таким, как третичный амин), который образует смешанный ангидрид карбоновой кислоты. Если конденсирующим агентом является оксалилхлорид, то для катализа образования хлорангидрида предпочтительно использовать небольшое количество диметилформамида в качестве сорастворителя с другим растворителем (таким, как дихлорометан). Выбор конденсирующих агентов, соответствующих растворителей и температур может быть легко сделан самим специалистом, либо определен исходя из специальной литературы. Эти и другие условия реакции конденсации карбоновых кислот описаны в Houben-Weyl, vol. XV, part 11, E. Wunsch. bd. G. Thieme Verlad, 1974, Stuttgart, & M. Bodansky, Principles of Peptide Synthesis, Springer. Verlag Berlin 1984, and The Peptides Analysis, Synthesis and Biology (ed. E. Gross & J. Meienhofer), vols. 1-5 (Academic Press NY 1979-1983).
Соединения формулы IV, где R1, R10, R11, A, R2, R3, R4, R5 и R7 определены выше, могут быть получены из соответствующего сложного эфира формулы V (т.е., соединений формулы I, где R6 представляет собой (C1-C5)алкоксикарбонил или бензилоксикарбонил) реакцией гидролиза с водным раствором щелочи при температуре от около -20oC до около 100oC, как правило, при температуре около 20oC, в течение периода времени от около 30 минут до около 24 часов.
Альтернативно, соединения формулы IV могут быть получены активацией индолкарбоновой кислоты формулы II с использованием конденсирующего агента (описанного выше), в результате чего получают активированное промежуточное соединение (такое, как хлорангидрид, фторангидрид или смешанный ангидрид), которое затем подвергают реакции с соединением формулы III (где R3, R4, R5 и R7 определены выше, а R6 представляет собой карбокси-группу) в подходящем растворителе и в присутствии подходящего основания. Подходящими растворителями являются вода или метанол, или их смесь, взятые вместе с сорастворителями, такими, как дихлорометан, тетрагидрофуран, или диоксан. Подходящими основаниями являются гидроксиды натрия, калия или лития, бикарбонат натрия или калия, карбоната натрия или калия, или карбонат калия вместе с бромидом тетрабутиламмония (1 эквивалент), взятом в количестве, достаточном для потребления кислоты, высвобождающейся при реакции (обычно, это количество достаточно для поддержания pH реакции до значения более чем 8). Для получения нужного значения pH реакции, основание можно добавлять постепенно вместе с активированным промежуточным соединением. Обычно, реакцию проводят при температуре от -20oC до 50oC. Методика выделения для удаления примесей может быть разработана самим специалистом, но, обычно такая методика предусматривает удаление смешиваемых с водой сорастворителей путем выпаривания, экстракцию примесей при высоком pH с использованием органических растворителей, подкисление для понижения pH (1-2) и фильтрацию или экстракцию нужного продукта с использованием подходящего растворителя, такого, как этилацетат или дихлорметан.
Соединение формулы V может быть получено реакцией взаимодействия соединения формулы III, где R6 является алкоксикарбонилом, с соответствующим соединением формулы II в соответствии с методикой, аналогичной описанной выше (например, Методикой A).
Альтернативно, соединения формулы I, которые содержат атомы серы в состоянии сульфоксидного или сульфонового окисления, могут быть получены из соответствующих соединений формулы I, имеющих атом серы в неокисленном состоянии, обработкой соответствующим окислителем, таким, как м-хлоропероксибензойная кислота в дихлорметане, при температуре от около 0oC до около 25oC в течение периода времени от около 1 до около 48 часов, с использованием от около 1 до около 1,3 эквивалентов для превращения в состояние сульфоксидного окисления, и с использованием примерно более 2 эквивалентов для превращения в состояние сульфонового окисления.
Альтернативно, соединения формулы I, которые являются моно- или ди-алкилированными у аминоалкокси-группы R5, могут быть получены из соответствующего соединения формулы I, где R5 является аминоалкокси-группой моноалкилированием или диалкилированием по амину R5 с получением нужного соединения формулы I. Такое моно- или ди-алкилирование может быть осуществлено обработкой соединения, в котором R5 является аминоалкокси, 1 эквивалентом соответствующего карбонильного соединения (для моноалкилирования) или более, чем 2 эквивалентами соответствующего карбонильного соединения (для диалкилирования) и соответствующим восстановителем в подходящем растворителе. Подходящими условиями для проведения реакции восстановления являются использование цианоборогидрида натрия или борогидрида натрия в метаноле или этаноле, или катализатора гидрирования при гетерогенном катализе (такого, как палладий-на-угле) в полярном растворителе, таком, как вода, метанол или этанол при температуре примерно 0-60oC в течение 1 – 48 часов.
Альтернативно, соединения формулы I, в которых R5 является алканоилокси (RCOO-), могут быть получены O-ацилированием соответствующего соединения формулы I с соответствующим хлорангидридом или с другим активированным кислотным производным в присутствии, если это необходимо, соответствующего основания (например, третичного основания, такого, как триалкиламина или пиридин), предпочтительно в апротонном растворителе, таком, как тетрагидрофуран или дихлорметан, при температуре от около 0oC до около 50oC, в течение примерно 0,5-48 часов.
Альтернативно, соединения формулы I, где R5 и R7, взятые вместе, представляют собой оксогруппу, могут быть получены путем окисления соответствующего соединения формулы I, например, соединения, в котором R5 является гидроксигруппой, а R7 является водородом, с использованием подходящего окислителя. Примерами таких окислителей являются реагент Десс-Мартина в хлорометане, карбодиимиде, и диметилсульфоксиде, и кислотный катализатор (окисление по Пфицнеру-Моффету или его модификация, например, с использованием водорастворимого карбодиимида; или реакции типа Сверна, например, с использованием оксалилхлорида /ДМСО/триэтиламина). Соединения формулы I, имеющие другие чувствительные к окислению функциональные группы, могут быть получены в результате соответствующей защиты и деблокирования таких групп.
Некоторые способы получения соединений, описанные в настоящей заявке, могут потребовать защиты уходящих функциональных групп (например, первичный амин, вторичный амин, карбоксил в предшественниках соединений формулы I). Необходимость в такой защите может варьироваться в зависимости от природы уходящей функциональной группы и условий конкретных методов получения. Необходимость такой защиты может быть легко установлена самим специалистом. Использование указанных методов защиты/снятия защиты также хорошо известно специалистам. Общее описание защитных групп и их использование описано, например, T. W.Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991.
Например, в реакционной схеме I, некоторые соединения формулы I содержат первичную аминогруппу, вторичную аминогруппу или карбоновокислотную функциональную группу в части молекулы, определенной R5 или R6, которая может препятствовать нужной реакции взаимодействия, проводимой в соответствии с реакционной схемой I, в том случае, если промежуточное соединение формулы III, или амин R12H или R8R9NH остается незащищенным. Поэтому первичная или вторичная функциональная аминогруппа, если она присутствует в радикалах R5 или R6 промежуточного соединения формулы III или амина R8R9NH или R12H, может быть защищена с помощью соответствующей защитной группы в процессе реакции взаимодействия реакционной схемы I. Продуктом такой реакции взаимодействия является соединение формулы I, содержащей защитную группу. В последующей стадии, эту защитную группу удаляют с получением соединения формулы I. Подходящими защитными группами для защиты аминогруппы и карбоновокислотной группы являются защитные группы, которые обычно используются в пептидном синтезе (например, такие, как N-т-бутоксикарбонил, N-карбобензилокси, и 9-флуоренилметиленоксикарбонил для аминов, и низшие алкиловые или бензиловые сложные эфиры для карбоновокислотных групп), которые не вступают в химические реакции в условиях конденсации, описанных выше (а также в методике A, описанной непосредственно перед примерами), и которые могут быть удалены без какого-либо химического воздействия на другие функциональные группы соединения формулы I.
Исходные индол-2-карбоновые кислоты и индолин-2-карбоновые кислоты, используемые в реакционной схеме I, если они не являются коммерчески доступными или известными соединениями (такими, которые очень широко описаны в литературе), могут быть получены методами стандартного синтеза. Например, согласно реакционной схеме II, сложный эфир индола формулы VII может быть получен из соединения формулы VI (где Q выбирают так, чтобы получить нужное A, определенное выше) посредством синтеза индолов Фишера (см. The Fischer Indole Synthesis, Robinson, B. (Wiley, New York, 1982) и последующего омыления полученного сложного эфира индола формулы VII с получением соответствующей кислоты формулы VIII. Исходный арилгидразон может быть получен конденсацией легкодоступного гидразина с соответствующим карбониловым производным, или с помощью реакции Яппа-Клингемана (см. , Organic Reactions, Phillips, R.R. 1959, 10, 143).
Альтернативно, индол-2-карбоновая кислота формулы VIIIA может быть получена конденсацией орто-метилнитро-соединения формулы IX с оксалатом (сложным эфиром) с последующим восстановлением нитро-группы и последующим гидролизом.
Такой трехстадийный способ синтеза индолов известен как реакция Райссерта (Reissert, Chemische Berichte, 1987, 30, 1030). Условия для осуществления указанного способа и ссылки, относящиеся к этому синтезу, описаны в литературе (Kermack et al., J. Chem. Soc. 1921, 119, 1609; Cannon et al., J. Med. Chem. 1981, 24, 238; Julian et al., in Heterocyclic Compounds, vol. 3 (Wiley, New York, NY, 1962, R.C.Elderfield, ed.) p. 18). Примеры конкретного осуществления этого способа приведены в настоящем описании (примеры 10A-10C).
3-Галогено-5-хлоро-1H-индол-2-карбоновые кислоты могут быть также получены галогенированием 5-хлоро-1H-индол-2-карбоновых кислот.
Альтернативно (реакционной схеме II), замещенные индолы формулы XIV могут быть получены восстановлением соответствующих индолов формулы XV с использованием восстановителя, такого, как магний в метаноле, при температуре от около 25oC до около 65oC в течение времени от около 1 часа до около 48 часов (реакционная схема III).
Индолинкарбоновые кислоты формулы XVI получают омылением соответствующего сложного эфира формулы XVII (реакционная формула III). Соединение формулы XVII получают восстановлением соответствующего сложного эфира индола формулы VII с использованием восстановителя, такого, как магний в метаноле, как описано выше для превращения соединения формулы XV в соединение формулы XIV.
Ниже описано получение различных аминов, используемых в вышеуказанных реакционных схемах.
В соответствии с реакционной схемой IV, соединения формулы XXII (амины формулы III в реакционной схеме, где R5 является OH; R7 является H; а R6 является сложным эфиром), или соединения формулы XXVI (где R6 является C(O)NR8R9 или C(O)R12) получают исходя из N-защищенного (обозначенного PT) альдегида формулы XX. Альдегид формулы XX или натрийбисульфитный аддукт альдегида формулы XX обрабатывают цианидом калия или натрия в водном растворе с сорастворителем, таким, как диоксан или этилацетат, при температуре от около 0oC до около 50oC, в результате чего получают цианогидрин формулы XXI. Цианогидрин формулы XXI обрабатывают спиртом (например, (C1-C6)алканолом, таким, как метанол) в присутствии катализатора на основе сильной кислоты, такого, как хлороводород, при температуре от около 0oC до около 50oC с последующим добавлением воды, если это необходимо. Затем защитную группу (PT) удаляют, если она присутствует, методом соответствующего деблокирования, в результате чего получают соединение формулы XXII. Например, если N-защитной группой PT (формула XX) представляет собой трет-бутоксикарбонил (t-Boc), то соединение формулы XXIII может быть получено непосредственно из соединения формулы XXI, и добавления воды не требуется. Соединение формулы XXII может быть защищено у атома азота соответствующей защитной группой с образованием соединения формулы XXIII и последующим гидролизом сложного эфира водной щелочью при температуре от около 0oC до около 50oC в инертном растворителе, в результате чего образуются гидрокси-кислоты формулы XXIV. Соединение формулы XXIV подвергают взаимодействию (аналогичная реакция описана в реакционной схеме I) с соответствующим амином R8R9NH или HR12, и получают соединение формулы XXV, которое затем подвергают деблокированию, в результате чего получают соединение формулы XXVI (т.е., соединение формулы III, в котором R5 является OH, R7 является H, а R6 является C(O)R12 или C(O)NR8R9). Пример реакции превращения цианогидрина формулы XXI в соответствующий метиловый сложный эфир формулы XXII с удалением защитной группы t-Boc описан в PCT-публикации WO/9325574, в примере 1a. Другие примеры превращения цианогидрина в низшие алкиловые сложные эфиры формулы XXIII можно найти в патенте США N 4814342 и EPO-публикации 0438233.
Некоторые соединения формулы I являются стереоизомерами благодаря стереохимической конфигурации у атомов углерода, обозначенных a и b. В соответствии с реакционной схемой IV, любой специалист может получить промежуточные соединения формул XXII и XXVI с нужной стереохимией. Например, альдегид формулы XX в любой энантиомерной форме (стереохимия у a) может быть легко получен исходя из стандартной методики, описанной ниже (см. реакционную схему V). Цианогидрин формулы XXI может быть получен из соединения формулы XX обработкой цианидом натрия или калия, как описано выше, сохраняя, при этом, стереохимию у атома углерода a, в результате чего образуется смесь стереоизомеров у углерода b.
Для разделения изомеров или для выделения одного изомера на этой стадии можно использовать метод кристаллизации.
Например, получение соединения формулы XXI, где PT является Boc, R3 является H, R4 является бензилом, а атомы углерода a и b имеют конфигурацию (S) и (R), соответственно, с проведением очистки перекристаллизацией описано в Bio-Chemistry 1992, 31, 8125-8141.
Альтернативно, разделение изомеров может быть осуществлено с использованием методики хроматографии или перекристаллизации после превращения соединения формулы XXI (смесь изомеров) в соединение формул XXII, XXIII, XXIV, XXV, XXVI, X, IV или I в соответствии с методами и/или реакционными схемами, описанными в настоящей заявке. Промежуточные соединения формулы XXI, имеющие конкретную стереохимию у атомов углерода a и b, превращают в промежуточные соединения формулы XXII с сохранением этой стереохимии путем обработки спиртом в присутствии сильной кислоты в качестве катализатора, с последующим добавлением воды, если это необходимо, как описано выше.
Альтернативно, нужный изомер соединения формулы XXI может быть также получен путем дериватизации промежуточного соединения формулы XXI и хроматографического разделения диастереомерных производных (например, с использованием триметилсилилхлорида (TMS) или т-бутилдиметилсилилхлорида (TBDMS) с получением O-TMS- или O-TBDMS-производных). Например, в примере 24D (приведенном в настоящей заявке) описано разделение диастереомерных производных формулы XXI. Силиловое производное промежуточного соединения формулы XXI, имеющего единственную стереоизомерную форму у атомов углерода a и b, превращают с сохранением стереохимии, в промежуточное соединение формулы XXII (если в этой стадии силильную группу не удаляют, то ее удаляют в последующей стадии с использованием соответствующей методики, например, обработкой фторидом тетрабутиламмония в тетрагидрофуране) в соответствии с методикой, описанной выше для превращения соединения формулы XXI в соединение формулы XXII (см. пример 24C, приведенный ниже, где описано превращение силильного производного формулы XXI в один изомер формулы XXII с потерей силильной группы).
В соответствии с реакционной схемой V, альдегиды формулы (исходные соединения для реакционной схемы IV) получают из соответствующих аминокислот формулы XXX. Аминокислоты формулы XXX являются N-защищенными защитной группой (PT), такой, как Вос. Защищенное соединение подвергают этерификации спиртом и превращают в сложный эфир, предпочтительно в метиловый или этиловый сложный эфир соединения формулы XXXI. Эта реакция может быть осуществлена путем обработки соединения формулы XXX метил- или этилиодидом в присутствии подходящего основания (например, K2CO3) в полярном растворителе, таком как диметилформамид. Соединение формулы XXXI подвергают реакции восстановления, например, с гидридом диизобутиламмония в гексане или толуоле, или в их смеси при температуре от около -78oC до около -50oC с последующим гашением реакции метанолом при -78oC, как описано в работе J.Med. Chem. , 1985, 28, 1779-1790, в результате чего получают альдегид формулы XX. Альтернативно (на реакционной схеме V не показано), аналогичные N-метоксиметиламиды, соответствующие соединению формулы XXXI, в котором спиртовой заместитель сложного эфира заменен на N(OMe)Me, получают из соединения формулы XXX, N, O-диметилгидроксиламина и соответствующего конденсирующего агента (например, гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида (DEC)), как описано в Методике A. Полученное соединение восстанавливают с использованием алюмогидрида лития в инертном растворителе, таком, как простой эфир или тетрагидрофуран, при температуре от около 0oC до около 25oC, в результате чего получают альдегид формулы XX. Этот двухстадийный метод является общим методом превращения N-защищенных -аминокислот в альдегиды формулы XX (Fehenz & Castro, Synthesis 1983, 676-678).
Альтернативно, альдегиды формулы XX могут быть получены окислением защищенных аминоспиртов формулы XXXIII, например, с использованием пиридин-SO3 при температуре от около -10oC до около 40oC в инертном растворителе, предпочтительно в диметилсульфоксиде. Защищенные аминоспирты формулы XXXIII, если они не являются коммерческими продуктами, могут быть получены посредством защиты аминоспиртов формулы XXXII. Аминоспирты формулы XXXII получают путем восстановления аминокислот формулы XXX. Эту реакцию восстановления осуществляют обработкой соединений формулы XXX алюмогидридом лития в соответствии с методикой, описанной Dickman et al., Organic Synthesis; Wiley, New York, 1990; Collect vol.VII, p 530, либо сернокислым борогидридом натрия в соответствии с методикой, описанной Abiko & Masamune, Tеtrahedron Lett. 1992, 333, 5517-5518; либо иодоборогидридом натрия в соответствии с методикой, описанной McKennon & Meyers, J.Org.Chem. 1993, 58, 3568-3571, где дается также обзор других методов превращения аминокислот формулы XXX в аминоспирты формулы XXXII.
В соответствии с реакционной схемой VI формулы XXX, соединения, используемые в реакционной схеме V, могут быть получены, как описано ниже. Аминокислоты формулы XLI могут быть получены путем N-алкилирования защищенных (PT) аминокислот формулы XL путем обработки соответствующим основанием и алкилирующим агентом. Конкретная методика проведения такого алкилирования описана Benoiton, Can. J. Chem. 1977, 55, 906-910, и Hansen, J.Org.Chem.1985, 50, 945-950. Например, если R3 является метилом, то используют гидрид натрия и метилиодид в тетрагидрофуране. После деблокирования соединения формулы XLI получают нужное соединение формулы XXX.
Альтернативно, аминокислота формулы XLII может быть подвергнута N-алкилированию с использованием трехстадийного метода, предусматривающего восстановительное бензилирование (например, с использованием бензальдегида в присутствии катализатора гидрирования Pd/C) с получением моно-N-бензилового производного; и восстановительное аминирование с использованием соответствующего ацильного соединения (например, формальдегида и цианоборогидрида натрия для введения R3 в качестве метильной группы) с получением N-бензил-, N-R3-замещенной аминокислоты. N-бензильную защитную группу затем удаляют (например, путем гидрирования с использованием соответствующего катализатора), и получают соединение формулы XXX. Конкретные условия этого трехстадийного метода алкилирования описаны Reinhold et. al., J.Med.Chem. 1968, 11, 258-260.
Описанный выше метод может быть также использован для введения радикала R3 в промежуточное соединение формулы XLIV с образованием промежуточного соединения формулы XLV (которое представляет промежуточное соединение формулы III, где R7 является OH). Этот метод может быть также использован для введения радикала R3 в промежуточное соединение формулы IIIa (которое представляет собой промежуточное соединение формулы III, где R3 является H).
Аминокислоты, используемые в описываемых реакционных схемах (например, в схемах XL, XLII), если они не являются коммерчески доступными, могут быть получены различными методами, хорошо известными специалистам. Например, для этой цели может быть использован синтез Штреккера или его варианты. В соответствии с этим методом, альдегид (R4CHO), цианид натрия или калия, и хлорид аммония подвергают реакции с получением соответствующего аминонитрила. Этот аминонитрил затем гидролизуют с использованием минеральной кислоты и получают нужную аминокислоту R4C(NH2)COOH формулы XLII. Альтернативно, может быть использован метод Бухерера-Берга, где гидантоин может быть образован путем нагревания альдегида (R4CHO) с карбидом аммония и цианидом калия, и последующего кислотного или основного гидролиза (например, с использованием гидроксида бария в кипящем диоксане) с получением нужной аминокислоты формулы XLII (R4C(NH2)COOH).
В литературе также описаны и другие методы синтеза -аминокислоты, которые позволяют специалисту получить нужное промежуточное соединение R4C(NH2)COOH формулы XLII, необходимое для синтеза соединений формулы I.
Подходящие методы синтеза или выделения соединений формулы можно найти в обзорных работах Duthaler (Tetrahedron, 1994, 50, 1539-1650), или Williams (P.M.Williams Synthesis of optically active amino acids. Pergamon: Oxford.U. K.1989).
В частности, синтез промежуточного соединения формулы XLII в любой энантиомерной форме из соответствующего промежуточного соединения R4X (X=C1, Br или I) может быть осуществлен по методу Пиррунга и Кришнамурту (J.Org.Chem. 1993, 58, 957-958) или по методу O’Donnel et al. (J.Am.Chem.Soc. 1989, 111, 2353-2355). Необходимые промежуточные соединения R4X могут быть легко получены многими методами, известными специалистам. Например, соединения, где R4X является ArCH2X, могут быть получены радикальным галогенированием соединения ArCH3 или формилированием арена Ar-H и превращения спирта в бромид.
Другой конкретный метод синтеза промежуточных соединений формулы XLII в любой энантиомерной форме описан Corey и Link (J.Am.Chem.Soc.1992, 114, 1906-1908). Так, например, промежуточное соединение формулы R4CH(OH)CCl3 энантиоспецифически восстанавливают до промежуточного соединения R4CH(OH)CCl3, которое восстанавливают каталитическим гидрированием, в результате чего получают нужное соединение формулы XLII. Необходимый трихлорометилкетон R4COCCl3 получают с помощью реакции альдегида R4CHO с трихлорометил-анионом с последующим окислением (Gallina & Giordano, Synthesis 1989, 466-468).
Промежуточные амины формулы III (используемые в Реакционной схеме I), где R5 и R7 являются H, могут быть получены в соответствии с Реакционной схемой VII. Аминокислоту формулы L (соответствующим образом замещенной (PT)) активируют превращением в хлорангидрид, фторангидрид или смешанный ангидрид (например, с использованием изобутилхлороформата и триэтиламина в инертном растворителе, таком, как тетрагидрофуран или диоксан при температуре от около -0oC до около -40oC), и после обработки активированного промежуточного соединения диазометаном получают диазокетон формулы LI. Диазокетон формулы LI обрабатывают спиртом (ROH) (например, (C1-C6) алканолом, таким, как метанол) при нагревании или с использованием подходящего катализатора, такого, как оксид серебра или бензоат серебра, в результате чего получают сложный эфир формулы LII. Сложный эфир формулы LII подвергают деблокированию с образованием соединения формулы IIIA (перегруппировка Вольфа). Альтернативно, сложный эфир формулы LII гидролизуют, например, щелочью, а затем подвергают реакции с соответствующим амином R12H или HNR8R9, в результате чего получают соединение формулы IIIB, как описано ранее.
В соответствии с Реакционной схемой VIII, промежуточные амины формулы III, где R5 представляет собой связанный с кислородом заместитель (например, алкокси) (использованный в реакционной схеме I), могут быть получены следующим образом. Соединение формулы XI подвергают O-алкилированию обработкой соответствующим алкилирующим агентом (например, алкилиодидом, алкилбромидом, алкилхлоридом или алкилтозилатом) и достаточным основанием с образованием алкоксида (гидрида натрия или калия) в подходящем полярном апротонном растворителе (например, диметилформамиде или тетрагидрофуране) при температуре от около 0oC до около 150oC, в результате чего получают соединение формулы XII. Соединение формулы LXII деблокируют, и получают нужное аминовое промежуточное соединение.
Промежуточные амины формулы III, где R5 представляет собой (C1-C6) алкоксикарбонилокси-группу (используемую в Реакционной схеме I), могут быть получены следующим образом. Соединение формулы XI алкилируют с использованием галогено-алканоата (сложного эфира), и получают соединение формулы LXIII, которое затем деблокируют с получением нужного амина. Соответствующая кислота может быть получена гидролизом сложного эфира с использованием водной щелочи в соответствующем растворителе. Амины формулы III, где R6 содержит сложноэфирную группу, а R5 содержит карбоксигруппу, могут быть получены из амина формулы XIII (полученного как описано выше), где R5 содержит карбоновокислотную функциональную группу, такую, как т-бутиловый сложный эфир, путем обработки безводной кислотой с получением соответствующей кислоты без гидролиза сложного эфира в положении R6.
Соединения формулы LXVI (промежуточные амины формулы III, в которых R5 является защищенной аминоалкокси-группой) могут быть получены из соединения формулы LXI. Соединение формулы LXI алкилируют с использованием галогено-алкан-нитрила, и получают соединение формулы XIV. Соединение формулы LXIV подвергают восстановлению до первичного амина путем обработки водородом и соответствующим катализатором (например, родий-на-угле) в присутствии аммиака предпочтительно в полярном протонном растворителе, таком, как вода, метанол или этанол, в результате чего получают первичный амин формулы LXV. Соединение формулы LXV защищают у атома азота с использованием защитной группы PT, которая является ортогональной по отношению к другой защитной группе (PT), с последующим деблокированием защитной группы PT, в результате чего получают нужное соединение формулы III. Защищенное соединение формулы III подвергают взаимодействию с соответствующим соединением формулы II, в результате чего получают защищенное соединение формулы I, которое является незащищенным.
Соединения формул LXIII и LXIV, где n = 2, получают предпочтительно обработкой соединения формулы LXI избыточным количеством сложного эфира акриловой кислоты или акрилонитрилом, соответственно, в присутствии подходящего основания, такого, как гидроксид калия или натрия, в подходящем растворителе, предпочтительно в полярном протонном растворителе.
В соответствии с реакционной схемой I, соединения формулы LXVII и формулы LXIX (соединения формулы III, где R5 является F, либо R5 и R7 оба являются F) могут быть получены из соединения формулы LXI. Соединение формулы LXI обрабатывают подходящим фторирующим агентом, таким, как трифторид диэтиламиносеры в инертном растворителе, таком, как апротонный растворитель, предпочтительно дихлорметан, в результате чего получают соединение формулы LXVII. Соединение формулы LXVII обычно подвергают деблокированию.
Соединение формулы LXI окисляют до соединения формулы LXVIII в условиях, описанных выше для получения соединений формулы I, где R5 и R7, взятые вместе, образуют оксо-группу. Соединение формулы LXVIII подвергают дефторированию в подходящих условиях (например, с использованием трифторида диэтиламиносеры в дихлорометане).
В соответствии с реакционной схемой X, соединение формулы LXXIII или соединение формулы LXIV, где R7 является алкилом (т.е. соединение формулы III, где R7 является алкилом), получают из соединения формулы LXX (см. также реакционную схему V для получения аналогичных аминов). Соединение формулы LXX обрабатывают металлоорганическим реагентом R7M, и полученный вторичный спирт окисляют как описано непосредственно выше с образованием соединения формулы LXXI. Соединение формулы LXXIпревращают посредством цианогидрина формулы LXXII в соединение формулы LXXIII с использованием таких же самых условий, которые были использованы для превращения соединения формулы XXI в соединение формулы XXII в реакционной схеме IV. Альтернативно, соединение формулы LXXII превращают в соединение формулы LXIV, как описано выше для превращения промежуточного циано-соединения в амид в Реакционной схеме V. Соединение формулы R8NH2 или R9NH2 подвергают моноалкилированию с использованием карбонильного соединения, соответствующего R8 или R9, соответственно, в подходящих условиях восстановительного аминирования, в результате чего получают амин формулы R8R9NH. Во избежание диалкилирования, предпочтительно защитить амины R8NH2 или R9NH2 подходящей защитной группой PT с получением R8(PT)NH или R9(PT)NH, например, посредством реакции с бензальдегидом и восстановителем. Защищенные амины подвергают моноалкилированию с использованием карбонильного соединения в соответствии с R9 или R8 в подходящих условиях восстановительного аминирования, в результате чего получают R8R9N(PT). После удаления защитной группы (PT) (например, путем исчерпывающего каталитического гидрирования, если PT является бензилом), получают соединение формулы R8R9NH. Подходящие условия восстановительного аминирования любой специалист может найти в соответствующей литературе. Такие условия описаны Borch и др. (J.Am.Chem.Soc., 1971, 2897-2904), в обзорных статьях Emerson (Organic Reaction, Wiley. New York, 1948 (14), 174), Hufchins и др. (Org. Prep. Proced. Int. 1979 (11), 20; и Lans и др. (Synthesis 1975, 135). Условия восстановительного аминирования, благоприятствующие N-моноалкилированию, описаны Morales и др. (Systhetic Communications 1984, 1213-1200) и Verardo и др. (Synthesic 1992, 121-125). Амины R8NH2 или R9NH2 могут быть также подвергнуты моноалкилированию с использованием R9X или R8X, соответственно, где X является хлоридом, бромидом, тозилатом или мезилатом. Альтернативно, промежуточное соединение формулы R8(PT)NH или R9(PT)NH может быть подвергнуто алкилированию с использованием R9X или R8X с последующим удалением защитной группы и образованием соединения формулы R8R9NH. Для получения аминов формулы R8R9NH, где R8-NH или R9-NH имеют кислород-азотную связь, могут быть использованы другие методы. Например, легкодоступное соединение формулы (C1-C4)алкоксикарбонил-NHOH или NH2CONHOH подвергают диалкилированию по атому азота и по атому кислорода обработкой основанием и избыточным количеством подходящего алкилирующего агента (R-X), в результате чего получают соответствующий (C1-C4) алкоксикарбонил-N(R)OP, который затем гидролизуют, и получают нужное соединение, имеющее формулу R8R9NH (где R8=R9=R). Подходящие условия для этой реакции, основания и алкилирующие агенты описаны Jorl & Kroll (org. Prep.Procad. Int. 1987, 19, 75-78) и Major и Fleck (J.Am. Chem. Soc., 1928, 50, 1479). Альтернативно, амин формулы NH2CONH(OH) может быть последовательно алкилирован, сначала по кислороду с получением соединения NH2CONH(OR’), а затем по азоту с получением соединения NH2CON(R”) (OR’) путем последовательной обработки алкилирующими агентами R’X и R”X, соответственно, в присутствии подходящего основания. Подходящие основания и алкилирующие агенты описаны Kreutzkamp и Messinger (Chem.Ber. 100, 3463-3465 (1967) и Danen и др. (J.Am. Chem. Soc. 1973, 95, 5716-5724). После гидролиза этих алкилированных производных гидроксимочевины получают амины R’ONH2 и R’ONHR”, которые соответствуют некоторым аминам формулы R8R9NH. Любой специалист-химик может модифицировать описанный выше метод для других алкилирующих агентов R, R’ и R”-X в целях получения других аминов формулы R8R9NH, где R8-N или R9-N имеют кислородл-азотную связь. Uno и др. (Synlett, 1991, 559-560) описывают BF3-катализируемую реакцию присоединения металлоорганического реагента R-Li; с O-алкилоксимом формулы R’CH=N-OR”, в результате которой получают соединения формулы R’RCH-NH(OR”). Этот способ может быть также использован для получения соединения формулы R8R9NH, где один из R8NH или R9-NH имеет кислород-азотную связь. Пролекарственные формы соединений настоящего изобретения, где карбоксильная группа в карбоновой кислоте формулы I заменена на сложный эфир, могут быть получены путем реакции карбоновой кислоты с соответствующим алкилгалогенидом в присутствии основания, такого как карбонат калия в инертном растворителе, таком, как диметилформамид, при температуре от около 0 до 100oC в течение периода времени от около 1 до около 24 час. Альтернативно, указанную кислоту подвергают реакции с соответствующим спиртом, используемым в качестве растворителя в присутствии каталитического количества кислоты, такой, как концентрированная серная кислота при температуре от около 20oC до 120oC, предпочтительно при температуре кипения растворителя, в течение периода времени от около 1 часа до около 24 часов. Другой метод предусматривает проведение реакции указанной кислоты со стехиометрическим количеством спирта в присутствии каталитического количества кислоты в инертном растворителе, таком, как тетрагидрофуран, и с параллельным удалением выделяемой воды физическими (например, с помощью ловушки Дина-Старка) или химическими (например, с использованием молекулярных сит) средствами. Пролекарственные формы соединений настоящего изобретения, в которых спиртовая функциональная группа была преобразована в эфирную группу, могут быть получены реакцией спирта с соответствующим алкилбромидом или алкилиодидом в присутствии основания, такого, как карбонат калия в инертном растворителе, таком, как диметилформамид, при температуре от около 0oC до 100oC в течение периода времени от около 1 часа до около 24 часов. Алканоиламинометиловые простые эфиры могут быть получены реакцией спирта с бис-(алканоиламино)метаном в присутствии каталитического количества кислоты в инертном растворителе, таком, как тетрагидрофуран, в соответствии с методом, описанным в патенте США 4997984. Альтернативно, эти соединения могут быть получены методами, описанными Hoffman и др. в J.Org.Chem. 1994, 59, 3530. Сложные эфиры диалкилфосфорной кислоты (диалкилфосфаты) могут быть получены реакцией спирта с диалкилхлорофосфатом в присутствии основания в инертном растворителе, таком как тетрагидрофуран. Бифосфаты могут быть получены реакцией спирта с диарил- или дибензилхлорофосфатом, как описано выше, с последующим гидролизом или гидрированием в присутствии катализатора на основе благородного металла, соответственно. Гликозиды получают реакцией спирта с углеводом в инертном растворителе, таком, как толуол, и в присутствии кислоты. Воду, обычно образующуюся в процессе реакции, удаляют как описано выше. Альтернативный метод предусматривает проведение реакции с соответствующим образом защищенным гликозилгалогенидом в присутствии основания с последующим деблокированием. N-(1-гидроксиалкил)амиды, N-(1-гидрокси-1-(алкоксикарбонил)метил)амиды или основания, в которых R2 был заменен на C(OH)C(O)OY, могут быть получены реакцией исходного амида или индола с соответствующим альдегидом в нейтральных или основных условиях (например, с использованием этоксида натрия в этаноле) при температуре 25-70oC. N-алкоксиметилиндолы или N-1-(алкокси)алкилиндолы могут быть получены реакцией N-незамещенного индола с нужным алкилгалогенидом в присутствии основания и в инертном растворителе. 1-(N, N-диалкиламинометил)индол, 1-(1-(N, N-диалкиламино)этил)индол и N,N-диалкиламинометиламиды (например, R3=CH2(CH3)2) могут быть получены реакцией исходного соединения N-H с соответствующим альдегидом и амином в спиртовом растворителе при 25-70oC. Вышеуказанные пролекарственные формы (например, пролекарства настоящего изобретения, в которых R2 и R3 являются общим углеродом) могут быть получены реакцией исходного соединения (лекарственного средства) с альдегидом или кетоном или с его диметилацеталем в инертном растворителе в присутствии каталитического количества кислоты и с сопутствующим удалением воды или метанола. Альтернативно, эти соединения могут быть получены реакцией аминоспирта или гидроксиамида с гем-дибромоалканом в присутствии основания (например, карбоната калия) в инертном растворителе (например, диметилформамиде). Хотя получение большинства исходных соединений и реагентов, используемых в вышеописанных реакционных схемах (например, амины, замещенные индолкарбоновые кислоты, замещенные индолинкарбоновые кислоты, аминокислоты) описано в настоящей заявке, однако, все эти соединения могут быть легко получены самим специалистом в соответствии со стандартными методами органического синтеза. Например, многие промежуточные соединения, используемые для получения соединений формулы I, являются родственными природными аминокислотами или происходят от этих аминокислот, и представляют большой научный и коммерческий интерес, а поэтому, большинство таких промежуточных соединений производятся промышленностью, или широко описаны в литературе, либо, они могут быть легко получены из известных соединений методами, описанными в литературе. Такими промежуточными соединениями являются, например, соединения формулы XX, формулы XXX, формулы XXXI и формулы XXXII. Соединения формулы I имеют асимметрические атомы углерода, а поэтому могут образовывать энантиомеры или диастереомеры. Диастереомерные смеси могут быть разделены с получением отдельных диастереомеров методами, известными per se и основанными на физических и химических различиях этих диастереомеров, например, с помощью хроматографии и/или фракционированной кристаллизации. Энантиомеры (например, соединения формулы III, VIII или IX) могут быть разделены путем превращения энантиомерной смеси в диастереомерную смесь реакцией с соответствующим оптически активным соединением (например, спиртом), с последующим разделением этих диастереомеров и превращением (например, путем гидролиза) отдельных диастереомеров в соответствующие чистые энантиомеры. Все указанные изомеры, включая диастереомеры, энантиомеры и их смеси, входят в объем настоящего изобретения. Хотя многие соединения настоящего изобретения не ионизируются в физиологических условиях, однако, некоторые соединения настоящего изобретения являются ионизируемыми в физиологических условиях. Примерами таких ионизируемых соединений являются кислотные формы соединений настоящего изобретения, которые образуют соли с фармацевтически приемлемым катионом. Все указанные соли входят в объем настоящего изобретения и могут быть получены стандартными методами. Например, они могут быть получены взаимодействием кислотных и основных молекул, взятых, обычно, в стереохимическом соотношении, в водной, безводной или частично водной среде. Эти соли могут быть выделены фильтрацией, осаждением безводным растворителем с последующей фильтрацией, выпариванием растворителя, или, в случае использования водных растворов, лиофилизацией, если это целесообразно. Кроме того, некоторые из соединений настоящего изобретения являются основными и могут образовывать соли с фармацевтически приемлемым анионом. Все такие соли входят в объем настоящего изобретения и могут быть легко получены стандартными методами. Например, указанные соли могут быть получены посредством взаимодействия кислотных и основных молекул, взятых, обычно, в стехиометрическом соотношении, в водной, безводной или частично водной среде. Эти соли могут быть выделены фильтрацией, осаждением безводным растворителем с последующей фильтрацией, выпариванием растворителя или, в случае использования водных растворов, лиофилизацией, если это целесообразно. Кроме того, если соединения настоящего изобретения образуют гидраты или сольваты, то все они также входят в объем настоящего изобретения. Полезность соединений настоящего изобретения в качестве терапевтических средств для лечения заболеваний, связанных с нарушением метаболизма (подробно описанных в настоящей заявке), у млекопитающих (например, у человека), была подтверждена активностью этих соединений в стандартных анализах, а также в in vitro – in vivo – анализах, описанных ниже. Благодаря этим анализам можно также сравнить активности соединений настоящего изобретения с активностью других известных соединений. Результаты этих сравнений были использованы для определения дозы, необходимой для лечения млекопитающих, включая человека, от указанных заболеваний. Очищенная гликогенфосфорилаза печени человека (HLGP) была получена методом, описанным ниже. Экспрессия и ферментация HLGP-кДНК была экспрессирована из плазмиды рКК233-2 (Pharmacia Biotech. Inc. , Piscataway, New Jersey) в штамме XL-1 Blue E.coli (Stratagene Cloning Systems, LaJolla, CA). Этот штамм инокулировали в LB-среду (состоящую из 10 г триптона, 5 г дрожжевого экстракта, 5 г NaCl и 1 мл 1 н. NaOH на литр), в которую были добавлены 100 мг/л ампициллина, 100 мг/л пиридоксина и 600 мг/л MnCl2, и культивировали при 37oC до тех пор, пока ОП550 (оптическая плотность при 550 нм) не стала равной 1,0. На этой стадии, клетки индуцировали 1 мМ изопропил-1-тио- -D-галактозидом (IPTG). Через три часа после индуцирования, клетки собирали путем цетрифугирования, и клеточный осадок замораживали при -70oC и хранили до тех пор, пока они не понадобятся для очистки.
Очистка гликогенфосфорилазыКлеточный осадок, полученный как описано выше, ресуспендировали в 25 мМ -глицерофосфата (pH 7,0), содержащего 0,2 мМ ДТТ, 1 мМ MgCl2 и следующие ингибиторы протеазы:0,7 мкг/мл пепстатина A 0,5 мкг/мл лейпептина 0,2 мМ фенилметилcульфонилфторида (PMSF), и 0,5 мМ EDTA. и лизировали путем предварительной обработки 200 мкг/мл лизоцима и 3 мкг/мл ДНКазы с последующей обработкой ультразвуком в 250-миллиметровых партиях в течение 5 х 1,5 минут на льду с использованием ультразвукового дезинтегратора клеток (Bronson Sonic Power Co., Dcenbury CT). Клеточные лизаты очищали путем центрифугирования при 35 000 хг в течение 1 часа, а затем путем фильтрации через фильтр 0,45 мкм. HLGP в растворимой фракции лизатов (составляющей, как было оценено, менее 1% от всего белка) очищали путем перманентного контроля за ферментной активностью (как описано ниже, в главе Анализ на HLGP-активность) во фракциях, полученных по ходу хроматографирования (более подробно см. ниже). Хроматография, основанная на средстве иммобилизованного металла (Immobilized Metal Affinity Chromatography) (IMAC). Эта стадия основана на методе Zuong и др. (Zuong et al., Journal of Chromatography (1992), 584, 77-84). 500 мл отфильтрованной растворимой фракции клеточных лизатов (полученных приблизительно из 160 г исходного клеточного осадка) загружали в 130-миллилитровую колонку IMAC с хелатообразующей Сефарозой (Pharmacia LKB Biotechnology, Piscataway New Jersey), которая была загружена 50 мМ CuCl2 и 25 мМ -глицерофосфата, 250 мМ NaCl и 1 мМ имидазола с использованием уравновешивающего буфера при pH 7. Колонку промывали уравновешивающим буфером до тех пор, пока A280 не возвращалось к своему исходному значению (т.е. фоновому значению). Затем образец элюировали с колонки тем же самым буфером, содержащим 100 мМ имидазола для удаления связанной HLGP и других связанных белков. Фракции, содержащие HLGP-активность собирали и объединяли (примерно 600 мл), после чего добавляли этилендиаминтетрауксусную кислоту (EDTA), DL-дитиотреитол (DTT), фенилметилсульфонилфторид (PMSF), лейпептин и пестатин A до получения концентрацией указанных соединений: 0,3 мМ, 0,2 мМ, 0,2 мМ, 0,5 мкг/мл и 0,7 мкг/мл, соответственно. Полученную объединенную HLGP обессоливали на колонке с Сефадексом G-25 (Sigma Chemical Co. , St.Louis, Missoceri), уравновешивали буфером A (25 мМ Трис-HCl, pH 7,3, 3 мМ DTT) для удаления имидазола и хранили на льду до проведения второй хроматографической стадии.
Хроматография на 5′-АМР-СефарозеОбразец обессоленной объединенной HLGH (приблизительно 600 мл) смешивали с 70 мл 5′-АРМ-Сефарозы (Pharmacia, LKB, Biotechnology, Piscataway, New Jersey, Sepharose), а затем уравновешивали буфером A (см.выше). Полученную смесь слегка помешивали в течение 1 часа при 22oC, а затем загружали в колонку и промывали буфером A до тех пор, пока A280 не возвращалось к своему исходному значению. HLGP и другие белки элюировали с колонки Буфером B (25 мМ Трис-HCl, 0,2 мМ DTT и 10 мМ аденозин-5-монофосфата (АМР), pH 7,3). HLGP-содержащие фракции идентифицировали на гликогенфосфатазную активность (как описано ниже) и визуально наблюдали полосу HLGP-белка с Мг – 97 кДа путем электрофореза в полиакриламидном геле с додецилсульфатом натрия (ДСН-ПААГ) с последующим окрашиванием серебром (2D-Silver Stain 11 “Daiichi Kit”, Daiichi Pure Chemicals Co. , Ltd., Tokyo, Japan), а затем объединяли. Объединенные HLGP диализовали в Буфер C (25 мМ -глицерофосфата, 0,2 мМ DTT, 0,3 мМ EDTA, 200 мМ NaCl, pH 7,0) и хранили на льду до использования.
Определение HLGP-ферментативной активностиA) Активация HLGP: Превращение HLGP в HLGPa Перед определением HLGP-ферментативной активности, неактивную форму фермента HLGP, экспрессированную в штамме XL-1 Blue (E.coli, обозначенную HLGPb)(Stratagene Cloning Systems, La Jolla California), превращают в активную форму (обозначенную HLGPa) путем фосфорилирования HLGP с использованием киназы фосфорилазы методом, описанным ниже. Реакция HLGPb с иммобилизованной киназой фосфорилазы Киназу фосфорилазы (Sigma Chemical Company, St.Zouis, MO) иммобилизировали на гранулах Affi-Gel 10 (BioRad Corp., Melvile, NY) в соответствии с инструкциями производителя. Для этого фермент киназу фосфорилазы (10 мг) инкубировали с промытыми гранулами Affi-Jel (1 мл) в 2,5 мл 100 мМ HEPES и 80 мМ CaCl2 при pH 7,4 в течение 4 часов при 4oC. Затем гранулы промывали один раз тем же самым буфером, после чего подвергали блокированию с использованием 50 мМ HEPES и 1 M сложного эфира глицина при pH 8,0 в течение 1 часа при комнатной температуре. После этого блокирующий буфер удаляли и заменяли 50 мМ HEPES (pH 7,4), 1 мМ -меркаптоэтанола и 0,2% NaN3 для хранения. Перед проведением реакции превращения HLGPb в HLGPa, гранулы Affi-Gel с иммобилизованной киназой фосфорилазы уравновешивали путем промывания в буфере, используемом для осуществления киназной реакции и содержащем 25 мМ -глицерофосфата, 0,3 мМ DTT и 0,3 мМ EDTA при pH 7,8 (буфер для киназного анализа).
Частично очищенную неактивную HLGPb, полученную в результате хроматографии на 5′-АМР-Сефарозе, как описано выше, разводили в соотношении 1:10 буфером для киназного анализа, а затем смешивали с вышеупомянутой киназой фосфорилазы, иммобилизованной на гранулах Affi-Gel. Затем добавляли NaATP до 5 мМ и MgCl2 до 6 мМ. Полученную смесь слегка помешивали в течение 30-60 минут при 25oC. Из этих гранул брали образец и процент активации HLGPb посредством ее превращения в HLGPa оценивали путем определения HLGP-ферментативной активности в отсутствие или в присутствии 3,3 мМ АМР. Процент полной HLGP-ферментативной активности, обусловленной ферментной активностью HLGPa (АМР-независимой), вычисляли следующим образом: B. Анализ на HLGPa-активность Гипогликемическая активность (а также активность, способствующая лечению/предупреждению других заболеваний/состояний) соединений настоящего изобретения может быть непосредственно определена путем оценки воздействия соединений настоящего изобретения на активность активированной формы гликогенфосфорилазы (GPa) с использованием одного из двух методов: а именно, в одном методе, гликогенфосфорилазную активность измеряют в прямом направлении путем перманентного контроля за продуцированием гликозо-1-фосфата из гликогена; а в другом методе, гликогенфосфорилазную активность измеряют в обратном направлении путем оценки синтеза гликогена из глюкозо-1-фосфата с высвобождением неорганического фосфата. Все реакции проводили в тройном повторе в 96-луночных планшетах, а изменение оптической плотности, обусловленное образованием продукта реакции, измеряли на длине волны, конкретно указанной ниже, в ELISA – ридере MCC/340 МК11 (Lao Systems, Finland), соединенном с приемником Fitertech Microplate Stacker (ICN Biomedical Co., Huntsvillc, Alabama). Для измерения ферментативной активности HLGPa в прямом направлении, продуцирование глюкозо-1-фосфата из гликогена оценивали в соответствии с общим методом мультиферментного связывания, описанного Pesce и др. (Pesce, M. A., Bodocerian, S.H., Harris, P.C., & Nicholson, J.F.(1977) Clinical Chemistry 23, 1711-1717) и модифицированного следующим образом: 1-100 мкг фосфорилазы a 10 ед. фосфоглюкомутазы и 15 ед. глюкозо-6-фосфат-дегидрогеназы (Bochringer Mannheim Biochemicals, Indianapolis, IN) разводили до 1 мл в Буфере A (описанном выше). Буфер A (pH 7,2) содержит 50 мМ HEPES, 100 мМ KСl, 2,5 мМ этиленгликольтетрауксусной кислоты (ECTA), 2,5 мМ MgCl2, 3,5 мМ KH2PO4 и 0,5 мМ дитиотреитола. 20 мкл этого маточного раствора добавляли к 80 мкл Буфера A, содержащего 0,47 мг/мл гликогена, 9,4 мМ глюкозы, 0,63 мМ окисленной формы никотинамид-адениндинуклеотидфосфат (NADP+). Перед добавлением ферментов, добавляли испытуемые соединения в виде 5 мкл-раствора в 14% диметилсульфоксида (ДМСО). Начальный уровень ферментативной активности HLGPa при отсутствии ингибиторов определяли путем добавления 5 мкл 14% ДМСО, а полностью ингибированный уровень ферментативной активности HLGPa получали путем добавления 20 мкл 50 мМ кофеина (позитивный контроль). За ходом реакции, протекающей при комнатной температуре, следили путем оценки конверсии окисленного NADP+ в восстановленный NADPH при 340 нм. Для измерения ферментативной активности HLGPa в обратном направлении, превращение глюкозо-1-фосфата в гликоген с высвобождением неорганического фосфата оценивали в соответствии с общей методикой, описанной Engers и др. (Engers, H. D., Shechovsky S. & Madsen N.B. 91970) Can.J.Biochem, 48, 746-754), и модифицированный следующим образом: 1 – 100 мкг HLGPa разводили до 1 мл в Буфере B (описанном выше). Буфер B (pH 7,2) содержит 50 мМ HEPES, 100 мМ KCl, 2,5 мМ EGTA, 2,5 мМ MgCl2 и 0,5 мМ дитиотреитола. 20 мкл исходного раствора добавляли к 80 мкл Буфера B, содержащего 1,25 мг/мл гликогена, 9,4 мМ глюкозы и 0,63 мМ глюкозо-1-фосфата. Перед добавлением фермента, добавляли испытуемые соединения в виде 5 мкл-раствора в 14% ДМСО. Начальный уровень ферментативной активности HLGPa при отсутствии ингибиторов определяли путем добавления 5 мкл 14% ДМСО, а полностью ингибированный уровень ферментативной активности HLGPa получали путем добавления 20 мкл 50 мМ кофеина. Эту смесь инкубировали в течение 1 часа при комнатной температуре, и неорганический фосфат, высвобождаемый из глюкозо-1-фосфата, измеряли в соответствии с общей методикой, описанной Lanzetta и др. (Lanzetta P.A., Alvarez L. J., Reinach P.S. & Candia O.A. (1979) Anal. Biochem. 100, 95-97), и модифицированный следующим образом: 150 мкг 10 мг/мл молибдата аммония, 0,38 мг/мл малахитового зеленого в 1 н. HCl добавляли к 100 мкл ферментной смеси. После 20-минутного инкубирования при комнатной температуре измеряли оптическую плотность при 620 нм. Соединения настоящего изобретения могут быть легко адаптированы для клинического использования в качестве гипогликемических агентов. Гипогликемическая активность соединений настоящего изобретения может быть определена путем оценки количества испытуемого соединения, способствующего снижению уровней глюкозы по сравнению с уровнями глюкозы, полученными с использованием лишь наполнителя (без испытуемого соединения), вводимого ob/ob-мышам-самцам. Этот тест позволяет также определить приблизительную величину минимальной эффективной дозы (MED) для in vivo – снижения концентрации глюкозы в плазме крови указанных мышей с использованием испытуемых соединений. Поскольку концентрация глюкозы в крови тесно связана с развитием диабета, эти соединения, благодаря их гипогликемической активности, обладают способностью предупреждать, приостанавливать и/или способствовать обратному развитию диабета. Мышей-самцов C57BL/6J – ob/ob в возрасте от 5 до 8 недель (полученных из Jackson Laboratory, Bar Harbor, ME) помещали в клетки (по пять животных на клетку) в стандартные условия, обычно применяемые при содержании животных. После недельного периода акклиматизации, животных взвешивали, и перед каждой обработкой у мышей брали кровь (25 мкл) из ретроорбитальной полости. Пробу крови сразу разводили 1:5 физиологическим раствором, содержащим 0,025% натрий-гепарина, и выдерживали на льду для анализа метаболитов. Животных распределяли на группы обработки так, чтобы каждая группа имела одинаковую среднюю концентрацию глюкозы в плазме крови. После распределения на группы, животным каждый день в течение 4-х дней перорально вводили дозу наполнителя, содержащего, либо: 1) 0,25% масс. метилцеллюлозы в воде без коррекции pH; либо 2) 0,1% поверхностно-активное вещество, блок-сополимер Плуроник  P105 (BASF Corporation, Parsippany, NY) в 0,1% физиологическом растворе без коррекции pH. На 5-й день животных снова взвешивали, после чего вводили перорально дозу испытуемого соединения или наполнителя. Все лекарственные средства вводили в наполнителе, состоящем либо из: P105 (BASF Corporation, Parsippany, NY) в 0,1% физиологическом растворе без коррекции pH. На 5-й день животных снова взвешивали, после чего вводили перорально дозу испытуемого соединения или наполнителя. Все лекарственные средства вводили в наполнителе, состоящем либо из:1) 0,25% мас/об. метилцеллюлозы в воде без коррекции pH; либо из 2) 10% ДМСО/0,1% Плуроникa P105 (BASF Corporation, Parsippany, ny) в 0,1% физиологическом растворе без коррекции pH. Затем через три часа у животных брали кровь из ретроорбитальной пазухи для определения уровней метаболитов в крови. Свежесобранные образцы центрифугировали в течение двух минут при 10000 х г при комнатной температуре. Супернатант анализировали на глюкозу с помощью, например, Abbott VPTM (Abbott Laboratiries, Diagnostics Division, Irving, TX) и автоматического анализатора (VP Super System Autoanalyzer, Abbott Laboratories, Irving, TX), используя при этом тест-систему реагентов A-GentTM Glucose-UV (Abbott Laboratories, Irving TX) (модификация метода Richterich & Dauwalder, Schweizerische Medizinische Wochenschrift, 101, 860 (1971)) (гексокиназный метод), и используя 100 мг/дл-стандарт. Уровень глюкозы в плазме вычисляли по следующему уравнению: Глюкоза в плазме (мг/мл) = Измеренная величина для данной пробы х 5 х 1,784 = 8,92 х измеренная величинаx) где 5 – кратность разведения, а 1,784 – поправка на гематократное число плазмы (которое принимают равным 44%). У животных, которым вводили дозы лишь одного наполнителя, наблюдались, в основном, неизмененные гипергликемические уровни глюкозы в крови (например, равные или превышающие 250 мг/дл), а у животных, которым вводили испытуемые соединения в соответствующих дозах, наблюдалось значительное снижение уровней глюкозы в крови. Гипогликемическую активность испытуемых соединений определяли путем статистического анализа (непарного т-критерия Стьюдента) средней концентрации глюкозы в плазме путем сравнения группы, обработанной испытуемым соединением, и группы, которой был введен наполнитель на 5-й день. Описанный выше анализ проводили с использованием ряда доз испытуемых соединений, что позволило определить приблизительное значение минимальной эффективной дозы (МЭД) для in vivo – снижения концентрации глюкозы в плазме. Соединения настоящего изобретения могут быть легко адаптированы для клинического использования в качестве средств, способствующих снижению уровня инсулина в крови до нормы; в качестве средств, снижающих концентрацию триглицеридов в крови, и в качестве гипохолестеринемических средств. Такая активность может быть определена путем оценки количества испытуемого соединения, способствующего снижению уровней инсулина, триглицеридов, или холестерина в крови, по сравнению с контролем (т.е., контрольным ob/ob/мышам-самцам вводили наполнитель без испытуемого соединения). Поскольку концентрация холестерина в крови тесно связана с развитием сердечно-сосудистых заболеваний, а также нарушений кровообращения головного мозга и периферической системы кровообращения, соединения настоящего изобретения, благодаря их гипохолестеринемической активности, способны предупреждать, приостанавливать и/или вызывать обратное развитие атеросклероза. Поскольку концентрация инсулина в крови тесно связана со стимуляцией роста сосудистых клеток и с увеличением накопления натрия в почках (помимо всех прочих функций, например, стимулирования утилизации глюкозы), а эти функции, как известно, приводят к развитию гипертензии, соединения настоящего изобретения, благодаря их гипоинсулинемическому действию, способны предупреждать, приостанавливать и/или вызывать обратное развитие гипертензии. Поскольку концентрация триглицеридов в крови вносит свой вклад в общий уровень липидов в крови, соединения настоящего изобретения, благодаря их действию, приводящему к снижению уровней триглицеридов, способны предупреждать, приостанавливать и/или вызывать обратное развитие гиперлипидемии. Мышей-самцов C57BL/6J-ob/ob в возрасте от 5 до 8 недель (полученных из Jackson Laboratory, Bar Harbor, ME) помещали в клетки (по пять животных на клетку) в стандартные условия, при которых обычно содержатся животные, и кормили ad libitum стандартным рационом для грызунов. После недельного периода акклиматизации, животных взвешивали, и перед каждой обработкой, из ретроорбитальной пазухи брали 25 микролитров крови. Пробы крови сразу разводили 1: 5 физиологическим раствором, содержащим 0,025% натрий-гепарин, и выдерживали на льду для анализа на содержание глюкозы в плазме. Животных распределяли на группы обработки так, чтобы каждая группа имела одинаковую среднюю концентрацию глюкозы в плазме крови. Испытуемые соединения вводили с помощью желудочного зонда в виде примерно 0.02%/ – 2,0%-ного раствора (масс/объем(масс. /об. )) либо в: 1) 10% ДМСО/0,1% Плуроник P105 (ПАВ, блок-сополимер) (BASF Corporation, Parsippany, NY) в 0.1% -ном физиологическим растворе, без коррекции pH; либо в: 2) 0,25% (масс./об.) метилцеллюлоза в воде, без коррекции pH. Эти соединения вводили ежедневно в течение 1 – 15 дней в виде разовой дозы (s.i.d.), либо в виде разделенной дозы два раза в день (b. i. d. ). Контрольные мыши получали 10% ДМСО/0,1% Плуроник P105 в 0,1%-ном физиологическом растворе без коррекции pH, либо 0,25% (масс./об.) метилцеллюлозу в воде без коррекции pH.
Через три часа после введения последней дозы, мышей умерщвляли путем декапитации (обезглавливания), и кровь собирали в 0,5-миллилитровые пробирки сепаратора для отделения сыворотки, содержащие 3,6 мг смеси фторида натрия и оксалата калия (1:1 масс./масс.). Свежесобранные образцы центрифугировали в течение двух минут при 10000 x г при комнатной температуре, и сывороточный супернатант собирали и разводили (1:1 об./об.) 1 TME/мл (TME – международная туберкулиновая единица) раствора апротинина в 0,1%-ном физиологическим растворе, без коррекции pH.
Разведенные образцы плазмы хранили при -80oC до использования в анализах. После оттаивания, разведенные образцы сыворотки анализировали на уровни инсулина, триглицеридов и холестерина. Концентрацию инсулина в сыворотке определяли с использованием наборов Equate P1A INSULIN (в соответствии с методом двойного антитела, как описано производителем), которые были закуплены у фирмы Binax, South Portland, ME. Коэффициент вариации для серии анализов составлял  10%. Уровень триглицеридов в сыворотке определяли с использованием автоматического анализатора Abbott VPTM и VP Super System Autoanalyzer (Abbott Laboratories, Irving, TX) и тест-системы реагентов A-GentTM Triglycerides Test reagent System (Abbott Laboratories, Diagnostics Division, Irving, TX) (иммуноферментный метод с использованием липазы: модификация метода Сэмпсона (Sampson et al., Clinical Chemistry 21. 1983 (1975). Уровень общего холестерина в сыворотке определяли с помощью автоматического анализатора Abbott VPTM and VP Super SystemR Autoanalyzer (Abbott Laboratories, Irving TX) и тест-системы реагентов A-JentTM Cholesterol Test reagent System (Иммуноферментный метод с использованием холестеринэстеразы; модификация метода Аллена (Allain, et al., Clinical Chemistry 20, 470 (1974)), с использованием 100- и 300 мг/дл-стандартов. Уровни инсулина, триглицеридов, а также общего холестерина в сыворотке вычисляли по следующему уравнению: 10%. Уровень триглицеридов в сыворотке определяли с использованием автоматического анализатора Abbott VPTM и VP Super System Autoanalyzer (Abbott Laboratories, Irving, TX) и тест-системы реагентов A-GentTM Triglycerides Test reagent System (Abbott Laboratories, Diagnostics Division, Irving, TX) (иммуноферментный метод с использованием липазы: модификация метода Сэмпсона (Sampson et al., Clinical Chemistry 21. 1983 (1975). Уровень общего холестерина в сыворотке определяли с помощью автоматического анализатора Abbott VPTM and VP Super SystemR Autoanalyzer (Abbott Laboratories, Irving TX) и тест-системы реагентов A-JentTM Cholesterol Test reagent System (Иммуноферментный метод с использованием холестеринэстеразы; модификация метода Аллена (Allain, et al., Clinical Chemistry 20, 470 (1974)), с использованием 100- и 300 мг/дл-стандартов. Уровни инсулина, триглицеридов, а также общего холестерина в сыворотке вычисляли по следующему уравнению:Уровень инсулина в сыворотке (мкЕД./мл)=Измеренная величина для данной пробы х 2 Уровень триглицеридов в сыворотке (мг/дл) = измеренная величина для данной пробы х 2 Уровень общего холестерина в сыворотке = измеренная величина для данной пробы х 2 где 2 – кратность разведения. У животных, которым вводили дозы одного наполнителя, наблюдались, в основном, неизменные повышенные уровни инсулина (например, 225 мкЕд), уровни триглицеридов (например, 225 мг/дл) и уровни общего холестерина (например, 160 мг/дл) в сыворотке, тогда, как у животных, обработанных соединениями настоящего изобретения, наблюдалось снижение сывороточных уровней инсулина, триглицеридов и общего холестерина. Активность соединений настоящего изобретения, направленную на снижение сывороточных уровней инсулина, триглицеридов и общего холестерина, определяли с помощью статистического анализа (непарного т-критерия Стьюдента) средних концентраций инсулина, триглицеридов, или общего холестерина в сыворотке путем сравнения результатов, полученных для группы, обработанной испытуемыми соединениями, и для контрольной группы, обработанной лишь одним наполнителем. Активность соединений настоящего изобретения, направленная на защиту сердечной ткани от повреждения, может быть продемонстрирована in vitro с помощью методики, предложенной в Butwell и др. (Am.J.Physiol., 264, H1884-H1889, 1993) и Allard и др. (Am.J.Physiol., 1994, 267, H66-H74). Эксперименты проводились с использованием изохорных препаратов сердца крыс, полученных как описано в вышеуказанной статье. Нормальных самцов крыс Sprague-Dawley; самцов крыс Sprague-Dawley с сердечной гипертрофией, индуцированной путем операции с перевязыванием аорты; самцов крыс BB/W с быстро прогрессирующим диабетом; или контрольных крыс соответствующего возраста (BB/W), не имеющих диабета, предварительно обрабатывали гепарином (1000 ед., i.p.), а затем пентобарбиталом (65 мг/кг, i.p.). После достижения крысами состояния глубокой анестезии (определяют по рефлексу конечности), сердце быстро иссекали, и помещали в охлажденный льдом физиологический раствор. В течение 2 минут сердце подвергали ретроградной перфузии через аорту. Частоту сердечных сокращений и внутрижелудочковое давление определяли с использованием в левом желудочке латексного баллона с высоконапорным шлангом, соединенным с датчиком давления. Перфузию сердца проводили перфузирующим раствором, содержащим 118 мМ NaCl, 4,7 мМ KCl, 1,2 мМ MgCl2, 1,2 мМ CaCl2, 25 мМ NaHCO3, 11 мМ глюкозы. Перфузионный аппарат был снабжен нагреваемой баней для непосредственного регулирования температуры перфузата и воды, циркулирующей вокруг перфузионного шланга для поддержания температуры сердца 37oC. Насыщение кислородом перфузата осуществляли с помощью педиатрического волоконного оксигенатора (Capiax, Terumo Corp., Tokyo, Japan) в непосредственной близости к сердцу. Сначала осуществляли перфузию крысиных сердец перфузатом  испытуемым соединением в течение 10 минут или более, затем, в течение 20 минут сердца подвергали глобальной ишемии, а после этого, в течение 60 минут осуществляли реперфузию в отсутствие испытуемого соединения. Частоту сердечных сокращений для контроля и для сердец, обработанных испытуемым соединением, сравнивали за период времени после ишемии. Кроме того, за период времени после ишемии сравнивали давление внутри левого желудочка для контрольных сердец и для сердец, обработанных испытуемым соединением. В конце эксперимента, сердца также подвергали перфузии и окрашиванию для определения уровня инфарктных зон по отношению к зонам риска (% 1A/AAP), как описано ниже.
Терапевтическое действие соединений настоящего изобретения, способствующее предупреждению повреждения сердечной ткани, являющегося результатом ишемического нарушения кровоснабжения, может быть также продемонстрировано in vivo в соответствии с методикой, предложенной Liu и др. (Circulation Vol. 84, N 1 (July 1991)) и описанной ниже. В этом in vivo – анализе проводили испытания соединений настоящего изобретения на их способность к защите тканей сердца по сравнению с контрольной группой, которой вводили наполнитель (физиологический раствор). В качестве основной информации следует отметить, что короткие периоды ишемии миокарда с последующей коронарной артериальной реперфузией способствуют защите сердца от последующей тяжелой ишемии миокарда (Murry et al., Circulation 74:1124-1136, 1986). Защита сердечной ткани, определяемая как уменьшение участков ткани миокарда, подверженных некрозу (инфаркту), может быть индуцирована фармакологически путем внутривенного введения агонистов рецептора аденозина интактным анестезированным кроликам, исследованным как in situ – модель для предварительной подготовки к экспериментам по изучению ишемии миокарда (Liu et al. Circulation 84: 350-356, 1991). В этом in vivo – эксперименте проводили исследования на способность соединений обеспечивать фармакологическую защиту сердечной ткани, т.е. , на способность этих соединений уменьшить участки инфаркта миокарда при парентеральном введении интактным анестезированным кроликам. Действие этих соединений можно затем сравнить с результатами, полученными для кроликов с предварительной ишемической подготовкой с использованием агониста аденозина Al, N6-1-(фенил-2P-изопропил)аденозин (P1A), который, как было показано, обеспечивает фармакологическую защиту сердечной ткани у интактных анестезированных кроликов, исследованных in situ (Liu et al., Circulation 84: 350-356, 1991). Точная методика этого эксперимента описана ниже.
Хирургия: Самцов новозеландских белых кроликов (3-4 кг) анестезировали натрий-пентобарбиталом (30 мг/кг, i. v. ). Трахеотомию осуществляли путем вентрально-цервикального срединного разреза, и проводили искусственную вентиляцию легких 100%-ным кислородом с использованием аппарата искусственной вентиляции при положительном давлении. Для введения лекарственного средства, в левую яремную вену вводили катетер, а в левую сонную артерию вводили катетер для измерения кровяного давления. Затем сердца обнажали посредством левой торакотомии, и на выступающую ветвь левой венечной артерии надевали петлю (из шелка ОО). Ишемию индуцировали путем тугого затягивания петли и ее закрепления зажимом. Для реперфузии пораженной зоны петлю освобождали. Ишемия миокарда характеризуется региональным цианозом; а реперфузия характеризуется восстановленной гиперемией.
Протокол: Эксперимент начинали после стабилизации артериального давления и частоты сердечных сокращений в течение, по крайней мере, 30 минут. Предварительное ишемическое состояние индуцировали посредством двухкратной окклюзии (пережимания) венечной артерии в течение 5 минут с последующей реперфузией в течение 10 минут. Предварительную фармакологическую обработку осуществляли путем двухкратного влияния испытуемого соединения, например, в течение 5 минут с перерывом на 10 минут перед вторым вливанием, либо путем вливания агониста аденозина P1A (0,25 мг/кг). После индуцирования ишемического состояния, предварительной фармакологической обработки или при отсутствии указанной подготовки (необработанный контроль, введения наполнителя) артерию пережимали на 30 минут, а затем подвергали реперфузии в течение двух часов для индуцирования инфаркта миокарда. Испытуемое соединение и P1A растворяли в физиологическом растворе или в другом подходящем наполнителе и вводили в дозе 1 – 5 мл/кг, соответственно.
Окрашивание: (Liu et al., Circulation 84:350-356, 1991): По истечении 2-часового периода реперфузии, сердца быстро удаляли, подвешивали на аппарате Лангендорфа, и в течение 1 мин промывали сильной струей нормального физиологического раствора, нагретого до температуры тела (38oC). Затем петлю (из щелка, применяемого для хирургических швов) туго затягивали для повторной окклюзии артерии, и с перфузатом вливали 0,5%-ную суспензию из флуоресцентных частиц (1 – 10 мкм) для окрашивания всего миокарда за исключением зон риска (нефлуоресцентный желудочек). После этого сердца быстро замораживали и хранили в течение ночи при -20oC. На следующий день, сердца разрезали на 2-миллилитровые среды, и эти среды окрашивали 1% хлористым трифенилтетразолием (ТТС). Поскольку ТТС реагирует с “живой” тканью, то эта окраска позволяет дифференцировать “живые” ткани (окрашенные красным цветом) от омертвелой ткани (неокрашенная ткань, подвергнутая инфаркту). Инфарктные зоны (неокрашенные) и зоны риска (в которых отсутствуют флуоресцентные частицы) подсчитывали для каждого среза левого желудочка с использованием предварительно прокалиброванного визуального анализатора. Для нормализации данных ишемического повреждения вследствие различия зон риска у разных сердец, эти данные выражали как отношение зоны, поврежденные инфарктом, к зоне риска (% 1A/AAP). Все данные представляют собой средние значения ср.кв. отклонение; при этом, сравнение проводили с использованием однофакторного дисперсионного анализа ANOVA или непарного т-критерия Стьюдента. Статистическая значимость составляла p < 0,05.
Введение соединений настоящего изобретения может быть осуществлено любым способом, обеспечивающим доставку этих соединений предпочтительно в ткани печени и/или сердца. Такими способами являются пероральное, парентеральное, интрадуоденальное введение, и т.п. Обычно, соединения настоящего изобретения вводят в виде одноразовой (например, один раз в день) дозы или в виде разделенных доз.
Однако, при этом очевидно, что количество и режим введения соединений настоящего изобретения зависит от конкретного заболевания/состояния, от конкретного индивидуума, подвергающегося лечению, от тяжести заболевания, от способа введения и от назначения лечащего врача. Таким образом, в зависимости от индивидуальности каждого конкретного пациента, лечащий врач может варьировать дозы лекарственного средства для достижения нужной активности (например, активности, снижающей уровень глюкозы и крови), используя при этом рекомендованные ниже диапазоны приемлемых доз. Исходя из желаемой степени активности, лечащий врач должен учитывать такие факторы, как начальный уровень, другие факторы риска (сердечно-сосудистые факторы), наличие предшествующих заболеваний, возраст и жалобы пациента.
В основном, эффективная доза для достижения нужной активности соединений настоящего изобретения, направленной, например, на снижение уровней глюкозы, триглицеридов, холестерина и инсулина в крови, составляет в пределах от 0,005 до 50 мг/кг/день, предпочтительно от 0,01 до 25 мг/кг/день, а наиболее предпочтительно от 0,1 до 15 мг/кг/день.
Соединения настоящего изобретения, в основном, предназначены для перорального введения, но в некоторых случаях, может быть также использовано парентеральное введение (например, внутривенное, внутримышечное, подкожное или интрамедуллярно (в костный мозг)), например, в тех случаях, когда пероральное введение неприемлемо для данной конкретной цели, либо, когда пациент не способен проглотить лекарственное средство. Соединение настоящего изобретения может быть введено путем местного применения, например, в тех случаях, когда пациент страдает желудочно-кишечными расстройствами, либо в тех случаях, когда по мнению врача, лучший эффект может быть достигнут путем нанесения лекарственного средства на поверхность ткани или органа.
Соединения настоящего изобретения обычно вводят в виде фармацевтической композиции, содержащей, по крайней мере, одно из соединений настоящего изобретения в сочетании с фармацевтически приемлемым наполнителем или разбавителем. Таким образом, соединения настоящего изобретения могут быть введены отдельно или в виде смеси в качестве стандартной пероральной или парентеральной лекарственной формы, либо в качестве лекарственной формы для чрезкожного введения.
Фармацевтическая композиция для перорального введения может быть изготовлена в виде растворов, суспензий, таблеток, драже, капсул, порошков, и т. п. Таблетки, помимо различных наполнителей, таких, как цитрат натрия, карбонат кальция и фосфат кальция, могут содержать различные дезинтегрирующие агенты, такие, как крахмал, предпочтительно, картофельный или маниоковый крахмал; некоторые силикатные комплексы; связующие агенты, такие, как поливинилпирролидон, сахара, желатин и аравийская камедь. Кроме того, при изготовлении таблеток часто используются смазывающие агенты, такие, как стеарат магния, испытуемым соединением в течение 10 минут или более, затем, в течение 20 минут сердца подвергали глобальной ишемии, а после этого, в течение 60 минут осуществляли реперфузию в отсутствие испытуемого соединения. Частоту сердечных сокращений для контроля и для сердец, обработанных испытуемым соединением, сравнивали за период времени после ишемии. Кроме того, за период времени после ишемии сравнивали давление внутри левого желудочка для контрольных сердец и для сердец, обработанных испытуемым соединением. В конце эксперимента, сердца также подвергали перфузии и окрашиванию для определения уровня инфарктных зон по отношению к зонам риска (% 1A/AAP), как описано ниже.
Терапевтическое действие соединений настоящего изобретения, способствующее предупреждению повреждения сердечной ткани, являющегося результатом ишемического нарушения кровоснабжения, может быть также продемонстрировано in vivo в соответствии с методикой, предложенной Liu и др. (Circulation Vol. 84, N 1 (July 1991)) и описанной ниже. В этом in vivo – анализе проводили испытания соединений настоящего изобретения на их способность к защите тканей сердца по сравнению с контрольной группой, которой вводили наполнитель (физиологический раствор). В качестве основной информации следует отметить, что короткие периоды ишемии миокарда с последующей коронарной артериальной реперфузией способствуют защите сердца от последующей тяжелой ишемии миокарда (Murry et al., Circulation 74:1124-1136, 1986). Защита сердечной ткани, определяемая как уменьшение участков ткани миокарда, подверженных некрозу (инфаркту), может быть индуцирована фармакологически путем внутривенного введения агонистов рецептора аденозина интактным анестезированным кроликам, исследованным как in situ – модель для предварительной подготовки к экспериментам по изучению ишемии миокарда (Liu et al. Circulation 84: 350-356, 1991). В этом in vivo – эксперименте проводили исследования на способность соединений обеспечивать фармакологическую защиту сердечной ткани, т.е. , на способность этих соединений уменьшить участки инфаркта миокарда при парентеральном введении интактным анестезированным кроликам. Действие этих соединений можно затем сравнить с результатами, полученными для кроликов с предварительной ишемической подготовкой с использованием агониста аденозина Al, N6-1-(фенил-2P-изопропил)аденозин (P1A), который, как было показано, обеспечивает фармакологическую защиту сердечной ткани у интактных анестезированных кроликов, исследованных in situ (Liu et al., Circulation 84: 350-356, 1991). Точная методика этого эксперимента описана ниже.
Хирургия: Самцов новозеландских белых кроликов (3-4 кг) анестезировали натрий-пентобарбиталом (30 мг/кг, i. v. ). Трахеотомию осуществляли путем вентрально-цервикального срединного разреза, и проводили искусственную вентиляцию легких 100%-ным кислородом с использованием аппарата искусственной вентиляции при положительном давлении. Для введения лекарственного средства, в левую яремную вену вводили катетер, а в левую сонную артерию вводили катетер для измерения кровяного давления. Затем сердца обнажали посредством левой торакотомии, и на выступающую ветвь левой венечной артерии надевали петлю (из шелка ОО). Ишемию индуцировали путем тугого затягивания петли и ее закрепления зажимом. Для реперфузии пораженной зоны петлю освобождали. Ишемия миокарда характеризуется региональным цианозом; а реперфузия характеризуется восстановленной гиперемией.
Протокол: Эксперимент начинали после стабилизации артериального давления и частоты сердечных сокращений в течение, по крайней мере, 30 минут. Предварительное ишемическое состояние индуцировали посредством двухкратной окклюзии (пережимания) венечной артерии в течение 5 минут с последующей реперфузией в течение 10 минут. Предварительную фармакологическую обработку осуществляли путем двухкратного влияния испытуемого соединения, например, в течение 5 минут с перерывом на 10 минут перед вторым вливанием, либо путем вливания агониста аденозина P1A (0,25 мг/кг). После индуцирования ишемического состояния, предварительной фармакологической обработки или при отсутствии указанной подготовки (необработанный контроль, введения наполнителя) артерию пережимали на 30 минут, а затем подвергали реперфузии в течение двух часов для индуцирования инфаркта миокарда. Испытуемое соединение и P1A растворяли в физиологическом растворе или в другом подходящем наполнителе и вводили в дозе 1 – 5 мл/кг, соответственно.
Окрашивание: (Liu et al., Circulation 84:350-356, 1991): По истечении 2-часового периода реперфузии, сердца быстро удаляли, подвешивали на аппарате Лангендорфа, и в течение 1 мин промывали сильной струей нормального физиологического раствора, нагретого до температуры тела (38oC). Затем петлю (из щелка, применяемого для хирургических швов) туго затягивали для повторной окклюзии артерии, и с перфузатом вливали 0,5%-ную суспензию из флуоресцентных частиц (1 – 10 мкм) для окрашивания всего миокарда за исключением зон риска (нефлуоресцентный желудочек). После этого сердца быстро замораживали и хранили в течение ночи при -20oC. На следующий день, сердца разрезали на 2-миллилитровые среды, и эти среды окрашивали 1% хлористым трифенилтетразолием (ТТС). Поскольку ТТС реагирует с “живой” тканью, то эта окраска позволяет дифференцировать “живые” ткани (окрашенные красным цветом) от омертвелой ткани (неокрашенная ткань, подвергнутая инфаркту). Инфарктные зоны (неокрашенные) и зоны риска (в которых отсутствуют флуоресцентные частицы) подсчитывали для каждого среза левого желудочка с использованием предварительно прокалиброванного визуального анализатора. Для нормализации данных ишемического повреждения вследствие различия зон риска у разных сердец, эти данные выражали как отношение зоны, поврежденные инфарктом, к зоне риска (% 1A/AAP). Все данные представляют собой средние значения ср.кв. отклонение; при этом, сравнение проводили с использованием однофакторного дисперсионного анализа ANOVA или непарного т-критерия Стьюдента. Статистическая значимость составляла p < 0,05.
Введение соединений настоящего изобретения может быть осуществлено любым способом, обеспечивающим доставку этих соединений предпочтительно в ткани печени и/или сердца. Такими способами являются пероральное, парентеральное, интрадуоденальное введение, и т.п. Обычно, соединения настоящего изобретения вводят в виде одноразовой (например, один раз в день) дозы или в виде разделенных доз.
Однако, при этом очевидно, что количество и режим введения соединений настоящего изобретения зависит от конкретного заболевания/состояния, от конкретного индивидуума, подвергающегося лечению, от тяжести заболевания, от способа введения и от назначения лечащего врача. Таким образом, в зависимости от индивидуальности каждого конкретного пациента, лечащий врач может варьировать дозы лекарственного средства для достижения нужной активности (например, активности, снижающей уровень глюкозы и крови), используя при этом рекомендованные ниже диапазоны приемлемых доз. Исходя из желаемой степени активности, лечащий врач должен учитывать такие факторы, как начальный уровень, другие факторы риска (сердечно-сосудистые факторы), наличие предшествующих заболеваний, возраст и жалобы пациента.
В основном, эффективная доза для достижения нужной активности соединений настоящего изобретения, направленной, например, на снижение уровней глюкозы, триглицеридов, холестерина и инсулина в крови, составляет в пределах от 0,005 до 50 мг/кг/день, предпочтительно от 0,01 до 25 мг/кг/день, а наиболее предпочтительно от 0,1 до 15 мг/кг/день.
Соединения настоящего изобретения, в основном, предназначены для перорального введения, но в некоторых случаях, может быть также использовано парентеральное введение (например, внутривенное, внутримышечное, подкожное или интрамедуллярно (в костный мозг)), например, в тех случаях, когда пероральное введение неприемлемо для данной конкретной цели, либо, когда пациент не способен проглотить лекарственное средство. Соединение настоящего изобретения может быть введено путем местного применения, например, в тех случаях, когда пациент страдает желудочно-кишечными расстройствами, либо в тех случаях, когда по мнению врача, лучший эффект может быть достигнут путем нанесения лекарственного средства на поверхность ткани или органа.
Соединения настоящего изобретения обычно вводят в виде фармацевтической композиции, содержащей, по крайней мере, одно из соединений настоящего изобретения в сочетании с фармацевтически приемлемым наполнителем или разбавителем. Таким образом, соединения настоящего изобретения могут быть введены отдельно или в виде смеси в качестве стандартной пероральной или парентеральной лекарственной формы, либо в качестве лекарственной формы для чрезкожного введения.
Фармацевтическая композиция для перорального введения может быть изготовлена в виде растворов, суспензий, таблеток, драже, капсул, порошков, и т. п. Таблетки, помимо различных наполнителей, таких, как цитрат натрия, карбонат кальция и фосфат кальция, могут содержать различные дезинтегрирующие агенты, такие, как крахмал, предпочтительно, картофельный или маниоковый крахмал; некоторые силикатные комплексы; связующие агенты, такие, как поливинилпирролидон, сахара, желатин и аравийская камедь. Кроме того, при изготовлении таблеток часто используются смазывающие агенты, такие, как стеарат магния,лаурилсульфат натрия, и тальк. Твердые композиции аналогичного типа могут быть также использованы для выполнения мягких и жестких желатиновых капсул; при этом, предпочтительными материалами, в этом случае, являются лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли. Если для перорального введения используются водные суспензии и/или эликсиры, то в этом случае, соединения настоящего изобретения могут быть объединены с различными подслащивающими агентами, ароматизаторами, красителями, эмульгирующими агентами и/или суспендирующими агентами, а также с такими разбавителями, как вода, этанол, пропиленгликоль, глицерин, и их различные комбинации. Для парентерального введения, могут быть использованы растворы в кунжутном или арахисовом масле, или в водном пропиленгликоле; а также водные стерильные растворы соответствующих водорастворимых солей. Если необходимо, то указанные водные растворы могут быть забуферены, причем, жидкому разбавителю придают изотоничность с помощью достаточного количества физиологического раствора или глюкозы. Такие водные растворы являются особенно подходящими для внутривенных, внутримышечных, подкожных и внутрибрюшинных инъекций. В связи с этим следует отметить, что стерильные водные растворы могут быть легко получены в соответствии со стандартной методикой, хорошо известной специалистам. Для чрезкожного введения (например, для наружного нанесения) могут быть использованы разведенные стерильные водные или частично водные растворы (обычно в концентрации примерно 0,1 – 0,5%), либо растворы, изготовленные аналогично парентеральным растворам. Методы получения фармацевтических композиций с определенным количеством активного ингредиента хорошо известны специалистам и могут быть легко осуществлены, исходя из вышеприведенного описания. См., например, Remington’s Pharmaceutical science, Mack Publishing Company, baster. PA. 15th Edition (1975). Фармацевтические композиции настоящего изобретения могут содержать 0,1 – 95% соединения (или соединений) настоящего изобретения, а предпочтительно 1 – 70% указанного соединения. В любом случае, вводимая композиция или препарат должны содержать такое количество соединения (соединений) настоящего изобретения, которое является эффективным для лечения соответствующего заболевания/состояния, в частности, гликогенфосфорилазо-зависимого заболевания/состояния. Методы ЯМР-спектры получены на спектрометре Varian X1 – 300 (Varian Co., Palo Alto, Калифорния) или Boucker AM-330 (Boucker Cо. Billerics Массачузетс) при около 23oC и при 300 МГц для протона и 75,4 МГц для ядер углерода. Химические сдвиги измеряли по отношению к триметилсилану и выражали в миллионных долях. Резонансы, определяемые как обмениваемые, не наблюдались в отдельных ЯМР-экспериментах, где образец встряхивали вместе с несколькими каплями D2О в том же самом растворителе. Масс-спектроскопию путем бомбардировки быстрыми атомами (FAB-MC) проводили на спектрометре VG70-2505 (V4 Analytical D., Wythanshaw, Манчестер, Великобритания) с использованием жидкой матрицы, состоящей из дитиотреитола/дитиоэритритола. Масс-спектроскопия путем ионизации тепловым электронным ударом (TSPMS) проводили на спектрометре Fisons Trio – 1000 (Fisons Co., Valencia Калифорния) с использованием ионизации аммиаком. Масс-спектры при химической ионизации получали на приборе Hewlett-Packard 5989 (Hewlett-Packard Co. , Palo Alto, California) (ионизация аммонием, PBMS). Там, где описана интенсивность хлор- или бром-содержащих ионов, наблюдался ожидаемый уровень интенсивности (приблизительно 3:1 для 35Cl/37Cl – содержащих ионов и 1:1 для 79Br/81Br – содержащих ионов, при этом, данные интенсивности приводятся лишь для ионов с меньшей массой. ЖХВР осуществляли с детекцией при 214 нМ на C-18-колонке (250 х 4,6 мм) Rainin Microsord (Rainin Cо., Woburn, Massachusetts) с использованием системы с двумя насосами/смесителями для изократического элюирования смесью ацетонитрила и водного 0,1М KH2PO4 (pH 2,1, H3PO4) при скорости потока 1,5 мл/мин. Образцы вводили в смесь (1:1) ацетонитрила и фосфатного буфера с pH 7,0 (0,025 М в каждом из Na2HPO4 и KH2PO4). Процент чистоты относится к проценту полной суммарной площади в течение 10-15-минутного прогона. Точки плавления не корректировали и определяли на приборе Buchi 510 (Buchi Laboratorums-Technic AO., Jlawi, Швейцария), где были получены точки плавления 120 – 122oC для бензойной кислоты и 237,5o – 240,5oC для п-хлоробензойной кислоты (сорта aldrich 99+%). Колоночную хроматографию осуществляли на силикагеле Amicon (30 мкМ, 60А – размер пор) (Amicon D Vision, W.P.Grace & Co, Beverly, Mass. ) в стеклянных колонках при низком давлении азота. Если это не указано особо, то были использованы реагенты, полученные из коммерческих источников. Диметилформамид, 2-пропанол, тетрагидрофуран и дихлорметан, используемые в качестве реакционных растворителей, были безводными и поставлялись фирмой Aldrich, Chemical Company (Milwaukee, Wisconson). Микроанализы осуществлялись лабораторией Schwartzkopf Microanalytical Laboratory, Woodside, NY. Термины “концентрировали” и “совместно выпаривали” относятся к удалению растворителя с использованием выпарного вакуумного роторного испарителя при температуре бани менее чем 45oC. Методика A (Пептидный синтез с использованием DEC) 0,1-0,7М-раствор первичного амина (1,0 экв. или гидрохлорида первичного амина, и 1,0-1,3 экв. триэтиламина на один экв. HCl) в дихлорометане (если это не указано особо) последовательно обрабатывали при 25oC 0,95-1,2 эквивалентами конкретно указанной карбоновой кислоты, 1,2-1,8 эквивалентами гидрата гидроксибензотриазола (обычно 1,5 экв. по отношению к карбоновой кислоте) и 0,95-1,2 эквивалентами (в молярном отношении к карбоновой кислоте) 1-(3-диметиламинопропил)-3- этилкарбодиимида гидрохлорида (DEC), и полученную смесь размешивали в течение 14-20 часов (см. ниже, прим. 1). Смесь разводили этилацетатом, промывали 2-3 раза в 1 или 2 н. NaOH, 2-3 раза 1 или 2 н. HCl (прим. 2), после чего органический слой сушили над MgSO4, концентрировали, а затем очищали путем хроматографии на силикагеле, растирания или перекристаллизации с использованием конкретно указанных растворителей. Очищенные продукты анализировали с помощью ОФ-ЖХВР, которая подтверждала чистоту продукта более 95%, если это не указано особо. Изменения в методике A, если они имеются, указаны отдельно ниже. Реакции проводили при 0-25oC с первоначальным охлаждением сосуда в изолированной ледяной бане, что позволяло оставлять смесь на несколько часов до ее нагревания до комнатной температуры. Примечание 1: При крупномасштабных реакциях (> 50 мл растворителя) смесь концентрировали на этой стадии, а остаток растворяли в этилацетате. Примечание 2: Если продукт содержал ионизируемую функциональную амино группу, то промывку кислотой не проводили. Пример 1 (3S)-[(5-хлоро-1H-индол-2-карбонил)амино]-(2R)-гидрокси-4- фенилмасляной кислоты изопропиловый сложный эфир. К раствору, содержащему (3S)-амино-4-фенил-(2R)- гидроксимасляной кислоты изопропиловый сложный эфир (1,35 г, 4,93 мМ) 5-хлоро-1H-индол-2-карбоновую кислоту (1,06 г, 5,4 мМ), и гидрат 1-гидроксибензотриазола (1,15 г, 7,5 мМ) в дихлорметане (15 мл) при температуре 25oC, одной порцией добавляли 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорид (DEC, 1,03 г, 5,38 мМ). Полученную смесь перемешивали в течение 18 часов при температуре 25oC, а затем разводили этилацетатом, после чего, полученный раствор дважды промывали 2 н. гидроксидом натрия и дважды промывали 2 н. соляной кислотой. Затем раствор осушали сульфатом магния и концентрировали. Остаток хроматографировали на 112 г двуокиси кремния, используя в качестве элюента сначала этилацетат/гексан (1: 4, 1,5 л), а затем этилацетат/гексан (1:3), в результате чего получали целевое соединение. Выход: 91%; ВРЖХ (70/30): 5,69 минут (78%) и 21,5 минут (19%). TSPMS 415/417 (MH+, 100%). 1H-ЯМР (CDCl3)  : 9,7 (с, 1H), 7,57 (д, 1H, J=2 Гц), 7,38-7,18 (м, 7-8H), 6,73 (д, 1H, J = прибл. 2 Гц), 6,57 (д, 1H, J=9,7 Гц) 5,04 (септет, 1H, J= 6,3 Гц), 4,83 (м, 1H), 4,19 (дд, 1H, J=2Гц), 3,51 (д, 1H, J=3,6 Гц), 3,05 (м, 2H), 1,17 (д, 3H, J=6,3 Гц), 1,11 (д, 3H, J=6,3 Гц). Присутствовало также приблизительно 15% другого вещества, предпочтительно являющегося N, O-бис(5-хлоро-1H-индол-2-карбониловым производным): д. (частотный): 9,80 (с, 1H), 5,28 (дд, 1H, индол-CO2CH).
Пример 1A : 9,7 (с, 1H), 7,57 (д, 1H, J=2 Гц), 7,38-7,18 (м, 7-8H), 6,73 (д, 1H, J = прибл. 2 Гц), 6,57 (д, 1H, J=9,7 Гц) 5,04 (септет, 1H, J= 6,3 Гц), 4,83 (м, 1H), 4,19 (дд, 1H, J=2Гц), 3,51 (д, 1H, J=3,6 Гц), 3,05 (м, 2H), 1,17 (д, 3H, J=6,3 Гц), 1,11 (д, 3H, J=6,3 Гц). Присутствовало также приблизительно 15% другого вещества, предпочтительно являющегося N, O-бис(5-хлоро-1H-индол-2-карбониловым производным): д. (частотный): 9,80 (с, 1H), 5,28 (дд, 1H, индол-CO2CH).
Пример 1A3(S), 2(R)-3-Амино-2-гидрокси-4-фенилмасляной кислоты изопропиловый сложный эфир Раствор 3(S), 2(R)-N-[(1,1-диметилэтокси)карбонил] -3-амино-2- гидрокси-4-фенилбутиронитрила (Parris et al. , Biochemistry 1992, 31, 8125-8141 (252 г, 0,912 М)) в безводном 2-пропаноле (6 л) обрабатывали при температуре 5-17oC безводным хлористым водородом (374 г), и этот раствор перемешивали в течение 20 часов при температуре 25oC (защищенный от атмосферного воздействия трубкой с осушителем Drierit). После добавления еще 348 г безводного хлористого водорода при температуре менее чем 10oC, смесь перемешивали в течение 72 часов при температуре 25oC. Полученную смесь концентрировали, а образовавшийся остаток растворяли в 0,1 н. соляной кислоте. После выдерживания в течение одного часа при температуре 25oC, этот раствор экстрагировали эфиром (3 х 1 л), а водный слой доводили до pH 12 добавлением 6 н. гидроксида натрия (приблизительно 450 мл). Полученную суспензию экстрагировали этилацетатом (4 х 1 л), после чего, экстракты промывали водой (500 мл) и солевым раствором (500 мл), а затем сушили и концентрировали, в результате чего получали 177 г желтого твердого вещества. Это твердое вещество растворяли в кипящем изопропиловом эфире (2 литра), фильтровали в нагретом состоянии и концентрировали путем кипячения до объема 1,4 литра. Образовавшееся после охлаждения твердое вещество собирали фильтрацией, промывали холодным изопропиловым эфиром и сушили (107 г). Второй сбор (12,2 г) получали из маточного раствора. Твердый сбор получали хроматографированием концентрированных маточных растворов на силикагеле (градиент 2-пропанолоа в дихлорметане, 1-4%) и перекристаллизацией очищенного продукта из изопропилового эфира (4,4 г, полный выход = 123,6 г, 57%), т.пл. 106-109oC; 1H-ЯМР (CDCl3) : 7,35 – 7,2 (м, 5H), 5,11 (септет, 1H, J=6,2 Гц), 4,01 (д, 1H, J=2,2 Гц), 3,30 (ддд, 1H), 2,91 (A от AB, 1H, J=6,3 и 13,3 Гц), 2,71 (B от AB, 1H, J=8,5 и 13,3 Гц), 1,8 (шир., 2-3H), 1,25 (д, 6H, J=6,2 Гц), TSP-MC: 238 (MH+).
Пример 25-Хлоро-1H-индол-2-карбоновой кислоты [(1S)-бензил-(2R)- гидрокси-3-(4-метил-пиперазин-1-ил)-3-оксо-пропил]-амида гидрохлорид Дигидрохлорид (3S)-амино(2R)-гидрокси-1-(4-метилпиперазин- 1-ил)-4-фенил-бутан-1-она (0,25 мМ) и 5-хлоро-1H-индол-2-карбоновую кислоту (0,30 мМ) подвергали реакции взаимодействия в соответствии с методикой A, после чего, продукт очищали с помощью хроматографии на силикагеле, используя в качестве элюента 0,5-8% этанол в дихлорметане, в результате чего было получено целевое соединение (42%-ный выход). Это соединение вместе с менее полярным соединением (13%) анализировали с помощью ЯМР, который указывал на сходство полученного соединения с N,O-бис(5-хлоро-1H-индол-2-карбониловым производным). Целевое более полярное соединение (48 мг) растворяли в смеси метанола и 0,25 мл 1 н. соляной кислоты, а затем, полученный раствор концентрировали, и образовавшееся твердое вещество растирали с эфиром, в результате чего было получено 42 мг нужного продукта; ВРЖХ (70/30), 2,53 мин (80%), 4,04 мин (13%), причем последнее время удерживания соответствовало времени удерживания N,O-бис-O-ацилированного производного, выделенного, как описано выше. 1H-ЯМР (D2O) : 7,70 (с, 1H), 7,5-7,2 (м, 7H), 7,05 (с, 1H), 4,57 (м, 1H), 4,47 (м, 1H), 4,04 (м, 1H), 3,58 (м, 4H), 3,34 (м, 4-5H), 2,97 (с, 1,5H), 2,97 (с, 1,5H), 2,91 (с, 1,5H).
PBMC: 455/457 (MH+, 100%).
Пример 2A(3S)-Амино-(2R)-гидрокси-1-(4-метил-пиперазин-1-ил)-4- фенилбутан-1-она дигидрохлорид [(1S)-Бензил-(2R)-гидрокси-3-(4-метил-пиперазин-1-ил)- 3-оксо-пропил]-карбаминовой кислоты трет-бутиловый сложный эфир (0,190 г, 0,5 мМ) растворяли в 4 М смеси соляной кислоты – диоксана при температуре 25oC в течение 0,5 часа. Полученную смесь концентрировали, и остаток растирали с эфиром, а затем сушили. Выход: 212 мг, ВРЖХ (15/85) 2,85 мин; PB-MC 278 (MH+, 100%). Пример 2B [(1S)-Бензил-(2R)-гидрокси-3-(4-метил-пиперазин-1-ил)-3- оксопропил] -карбаминовой кислоты трет-бутиловый сложный эфир N-Метилпиперазин (75 мг, 0,75 мМ) и (2R, 3S)-3-трет-бутоксикарбониламино-2-гидрокси-4-фенилмасляную кислоту (0,200 грамм, 0,68 мМ) подвергали реакции взаимодействия в соответствии с методикой A, в результате чего получали бесцветную пену, которую использовали без очистки. Выход: 225 мг (88%); PB-MC: 378 (MH+, 100%). Пример 3 5-Хлоро-1H-индол-2-карбоновой кислоты [(1S)-((R)- гидроксиметилкарбамоил-метил)-2-фенил-этил-амид Гидрохлорид метиламина (0,38 мМ) и (3S)-[(5-хлоро-1H-индол-2-карбонил)амино] -(2R)-гидрокси-4-фенилмасляную кислоту (0,35 мМ) подвергали реакции взаимодействия в соответствии с методикой A (с использованием ДМФ, при 0-25oC), и полученный неочищенный продукт очищали с помощью хроматографии на силикагеле (элюент: 1-8% этанол в дихлорметане и 0,5 % гидроксид аммония), в результате чего получали целевое соединение. Выход: 82%; ВРЖХ (70/30), 3,09 мин (98%); PB-MC 386/388 (MH+, 100%). Анализ для C20H20ClN3O3 + 0,25 H2O; Вычислено: C 61,54; H 5,29; N 10,76; Найдено: C 61,17; H 5,63; N 10,83. Пример 4 (3S)-[(5-Фторо-1H-индол-2-карбонил)-амино] -(2R)-гидрокси- 4-фенилмасляной кислоты метиловый сложный эфир (3S)-Амино-(2R)-гидрокси-4-фенилмасляной кислоты метиловый сложный эфир (0,8 мМ, WO 9325574 Пример 1A) и 5-фторо-1H-индол-2-карбоновую кислоту (0,8 мМ) подвергали реакции взаимодействия в соответствии с методикой A (при 0-25oC и экстракции кислотой, а затем основанием), и полученный неочищенный продукт очищали путем растирания с эфиром; Выход = 71%, ВРЖХ (60/40), 4,51 мин (98%), т.пл. 219,5-210oC, PBMC: 371 (MH+ 100%). Анализ для C20H19FN2O4 + 0,25 H2O; Вычислено: C 64,08; H 5,27; N 7,44; Найдено: C 64,14; H 5,30; N 7,48. Пример 5 (3S) [(5-Бромо-1H-индол-2-карбонил)-амино]-(2R)-гидрокси-4- фенилмасляной кислоты метиловый сложный эфир (3S)-Амино-(2R)-гидрокси-4-фенилмасляной кислоты метиловый сложный эфир (WO 93/25574, Пример 1A) (0,7 мМ) и 5-бромо-1H-индол-2-карбоновую кислоту (0,7 мМ) подвергали реакции взаимодействия в соответствии с Методикой A (за исключением того, что температура составляла 0-25oC). Неочищенный продукт, содержащий 25% N,O-бис-ацилированное вещество (ВРЖХ-анализ), использовали в последующих реакциях без дополнительной очистки; Выход = 97%, ВРЖХ (70/30), 4,03 мин (73%), 11,8 мин (25%). Образец растирали с эфиром/гексаном для идентификации и биологического тестирования: ВРЖХ (70/30) 4,03 мин (94%), 11,7 мин (4%), FAB-MC 431/433 (MH+, 35%), 307 (100%). 1H-ЯМР (CDCl3) : 9,31 (шир., 1H), 7,75 (д, 1H, J=прибл. 2 Гц), 7,35-7,20 (м, 7H), 6,73 (д, 1H, J=1,6 Гц), 6,47 (д, 1H, J=9,6 Гц), 4,80 (м, 1H), 4,21 (дд, 1H, J=2,5 Гц), 3,72 (с, 3H), 3,33 (д, 1H, J=4 Гц), 3,06 (м, 2H).
Анализ для C20H19BrN2O4:Вычислено: C 55,70; H 4,44; N 6,50; Найдено: C 56,12; H 4,62; N 6,16. Пример 6 5-Фторо-1H-индол-2-карбоновой кислоты [(1S)-((R)- диметилкарбамоил-гидрокси-метил)-2-фенил-этил]-амид Гидрохлорид диметиламина (0,52 мМ) и (3R)-[(5-фторо-1H-индол-2-карбонил)-амино] -(2S)-гидрокси-4-фенилмасляную кислоту (0,43 мМ) подвергали реакции взаимодействия в соответствии с Методикой A (за исключением того, что температура составляла 0-25oC). Неочищенный продукт растворяли в дихлорметане, после чего, полученный раствор перемешивали в течение одного часа приблизительно с 200 мг смеси диметиламинопиридина/полистироловой смолы (Aldrich Chemical Co. , Milwaukee, WI), а затем фильтровали и концентрировали, в результате чего получали желаемый продукт в виде бесцветного твердого вещества: Выход = 62%, ВРЖХ (60/40), 4,15 мин (97%), т.пл. 213-214oC; TSP-MC: 384 (MH+, 100%). Анализ для C21H22FN3O3: Вычислено: C 65,78; H 5,78; N 10,96; Найдено: C 65,89; H 6,16; N 11,00. Пример 7. 5-Бромо-1H-индол-2-карбоновой кислоты {(1S)-[(R)-гидрокси-(метокси-метил-карбамоил)-метил]-2-фенил-этил}-амид (3S)-Амино-(2R)-гидрокси-N-метокси-N-метил-4-фенил-бутирамида гидрохлорид (0,36 мМ) и 3-[(5-бромо-1H-индол-2-карбонил)-амино]-2-гидрокси-4-фенилмасляную кислоту (0,36 мМ) подвергали реакции взаимодействия в соответствии с Методикой A, а затем, неочищенный продукт очищали с помощью хроматографии на силикагеле (элюент: 30-40% смесь этилацетата/гексана) и с помощью растирания со смесью эфира/гексана (1:1). Выход: 65%, ВРЖХ (60/40), 5,77 мин (100%); РВ-МС 460/462 (МН+, 90%). Анализ для C21H22BrN3O4: Вычислено: C 54,79; H 4,82; N 9,13; Найдено: C 54,88; H 5,22; N 8,83. Пример 8 5-Хлоро-3-метил-1H-индол-2-карбоновой кислоты [(1S)-[(R)-гидрокси-(метокси-метил-карбамоил)-метил]-2-фенил-этил]-амид (3S)-Амино-(2R)-гидрокси-N-метокси-N-метил-4-фенилбутирамида гидрохлорид (0,3 мМ) и 5-хлоро-3-метил-1H-индол-2-карбоновую кислоту (0,3 мМ) подвергали реакции взаимодействия в соответствии с методикой A, и полученный неочищенный продукт очищали растиранием с эфиром: Выход=59%, ВРЖХ: (60/40), 7,45 мин (100%), РВ-МС 430/432 (MH+, 100/40%). 1H-ЯМР (CDCl3) : 8,98 (шир., 1H), 7,56 (д, 1H, J=2Гц), 7,4-7,15 (м, 7H), 6,35 (д, 1H, J = 9 Гц), 4,95 (м, 1H), 4,32 (д, 1H, J=5,1 Гц), 3,81 (д, 1H, J= 5 Гц), 3,36 (с, 3H), 3,15 (с, 3H), 3,15 (дд, 1H), 3,03 (дд, 1H, J= 13,16 Гц), 2,51 (с, 3H).
Анализ для C22H24ClN3O4:Вычислено: C 61,46; H 5,63; N 9,77; Найдено: C 61,13; H 5,53; N 9,74. Пример 8A 5-Хлоро-3-метил-1H-индол-2-карбоновая кислота К суспензии, состоящей из сложного этилового эфира 5-хлоро-3-метил-1H-индол-2-карбоновой кислоты (7,0 г, 29,4 мМ) и метанола (50 мл), добавляли 20 мл 2 н. гидроксида натрия, и полученную смесь перемешивали в течение 18 часов при температуре 25oC. После добавления 100 мл тетрагидрофурана, полученный раствор нагревали с обратным холодильником в течение 30 минут и концентрировали. Остаток растворяли в воде и полученный раствор дважды экстрагировали этилацетатом. Водный слой подкисляли, а затем, осадок собирали путем фильтрации и промывали водой (5,24 г). Пример 8B 5-Хлоро-3-метил-1H-индол-2-карбоновой кислоты этиловый сложный эфир п-Хлорофенилгидразон этил-2-оксобутаноата получали путем модификации реакции Яппа-Клингеманна, описанной Lions & Hughes (J.Proc. Roy. Soc. N.S. Wales 1939.71: 445) с получением п-хлороанилина и этил-2-этилацетоацетата. Фенилгидразон обрабатывали смесью соляной кислоты и этанола в соответствии с методикой, описанной Lions & Heghes (J.Proc. Roy. Soc. N.S.Wales 1939, 71: 445 с получением соответствующего бромофенилгидразона. После суспендирования концентрированного остатка в воде, целевое соединение в виде оранжевого твердого вещества собирали фильтрованием. Пример 8C (3S)-Амино-(2R)-гидрокси-N-метокси-N-метил-4-фенил-бутирамида гидрохлорид 31055-274-2.31055-85-1 (1S)-[(R)-Гидрокси-(метокси-метил-карбамоил)-метил]-2-метил-фенил-этил]- карбаминовой кислоты трет-бутиловый сложный эфир (791 мг, 2,3 мМ) растворяли в 4 М HCl/диоксане в течение 45 минут при температуре 25oC, а затем, раствор концентрировали. Остаток выпаривали вместе с эфиром, суспендировали в эфире и фильтровали, в результате чего получали 583 мг (91%) целевого продукта. Пример 8 { (1S)-[(R)-Гидрокси-(метокси-метил-карбамоил)-метил]-2-фенил-этил] -карбаминовой кислоты сложный трет-бутиловый эфир (3S)-трет-Бутоксикарбониламино-(2R)-гидрокси-4-фенилмасяную кислоту (10,06 г, 34,1 мМ; Schweizerhalls, Inc., S,Plainfield, N.J.) и N,O-диметилгидроксиламина гидрохлорид (3,49 г, 35,7 мМ) подвергали реакции взаимодействия в соответствии с Методикой A, и полученный неочищенный продукт (10,7 г) очищали с помощью хроматографии на силикагеле (элюент: 25-50% этилацетат/гексан), в результате чего получали целевое соединение в виде пены (9,5 г, 83%); МС: 339 (МН+, 100%). Пример 9. (3S)-[(5,6-Дихлоро-1H-индол-2-карбонил)-амино]-(2R)-гидрокси-4- фенилмасляной кислоты сложный метиловый эфир (3S)-Амино-(2R)-гидрокси-4-фенилмасляной кислоты сложный метиловый эфир (1,2 мМ) и 5,6-дихлоро-1H-индол-2-карбоновую кислоту (1,2 мМ) подвергали реакции взаимодействия в соответствии с методикой A (время реакции = 72 часа), и полученный продукт очищали с помощью хроматографии на двуокиси кремния (элюент: 20-40% этилацетата/гексана). Выход 52%; т.пл. 198-202oC; TSP-MC 421/423 (МН+, 100%). Анализ для C20H18Cl N2O4 + 0,25 H2O: Вычислено: C 56,42; H 4,38; N 6,58; Найдено: C 56,25; H 4,17; N 6,58. Пример 9A 5,6-Дихлоро-1H-индол-2-карбоновая кислота К нагретому раствору 3,4-дихлоро-5-нитрофенилпировиноградной кислоты (1,5 г, 5,4 мМ) в уксусной кислоте (15 мл) медленно добавляли цинковую пыль (3,52 г, 54 мМ). Через несколько минут наблюдалась сильная экзотермическая реакция. После нагревания раствора до 80oC, реакция была полностью завершена, о чем свидетельствовал ТСХ-анализ. Полученную смесь фильтровали, а затем, отфильтрованные твердые вещества промывали уксусной кислотой, и фильтрат концентрировали. Остаток растворяли в 2N NaOH, а полученный раствор три раза промывали эфиром, два раза дихлорметаном, и подкисляли до pH 1 путем добавления 6N соляной кислоты. После экстрагирования этилацетатом, экстракты сушили и концентрировали, в результате чего получали 458 мг (34%) целевого соединения в виде светло-коричневого твердого вещества. ВРЖХ (60/40), 5,31 мин (93%). Пример 9B 3,4-Дихлоро-5-нитрофенилпировиноградной кислоты калиевая соль К перемешанной смеси, состоящей из металлического калия (2,67 г, 68 мМ) и эфира (100 мл), добавляли 25 мл абсолютного этанола при температуре 3-15oC. Полученный раствор обрабатывали при температуре 3oC раствором диэтилоксалата (10,0 г, 62 мМ) в 2-метил-3,4-дихлоро-1-нитробензоле (10,0 г, 62 мМ) в течение 5-10 минут, а затем, этот раствор перемешивали в течение 30 минут при 3oC и в течение 18 часов при 25oC. Полученную смесь фильтровали, и образовавшееся твердое вещество промывали эфиром и осушали (13,7 г). 12,7 г полученного вещества растворяли в 400 мл горячей воды, а затем, раствор охлаждали и экстрагировали эфиром. После этого, водный слой подкисляли до pH 2 добавлением концентрированной соляной кислоты, а эфирный слой отделяли, сушили и концентрировали, с получением 7,5 г твердого вещества, которое растирали с гексаном, и получали целевое соединение в виде желтого твердого вещества (7,01 г, 41%). Пример 10 5-Циано-1H-индол-2-карбоновой кислоты {(1S)-[(R)-гидрокси-(метокси-метил-карбамоил)-метил]-2-фенил-этил}-амид (3S)-Амино-(2R)-гидрокси-N-метокси-N-метил-4-фенил-бутирамида гидрохлорид (0,3 мМ) и 5-циано-1H-индол-2-карбоновую кислоту (0,3 мМ) подвергали реакции взаимодействия в соответствии с методикой A (время реакции = 5 дней). Неочищенный продукт растворяли в метаноле, содержащем 1,0 эквивалента 1N NaOH при 25oC в течение 45 минут, а затем концентрировали. Остаток растворяли в этилацетате, после чего, раствор промывали дважды 2N соляной кислотой и дважды 2N гидроксидом натрия, а затем сушили и концентрировали. Полученный остаток хроматографировали на силикагеле, используя в качестве элюента смесь гексана/этилацетата (20-25%). Очищенный продукт растирали со смесью эфира/гексана (1: 1) и получали целевое соединение. Выход: 66%, ВРЖХ (60/40); 3,9 минут (100%), т.пл. 210-211oC; РВМС: 407 (МН+ 100%). 1Н-ЯМР (CDCl3) : 9,83 (шир., 1H), 7,97 (с,1H), 7,46 (м, 2H), 7,36 (м, 4H), 6,88 (д, 1H, J=2 Гц), 6,56 (д, 1H, J=10 Гц), 4,95 (м, 1H), 4,32 (д,1H, J=5,5 Гц), 3,83 (д, 1H, J=5,4 Гц), 3,36 (с, 3H), 3,13 (с, 3H), 3,10 (м, 2H).
Анализ для C22H22N4O4:Вычислено: C 65,01; H 5,46; N 13,78; Найдено: C 64,92; H 5,60; N 13,78. Пример 10A 5-Циано-1H-индол-2-карбоновая кислота К раствору, содержащему этанол (10 мл) и гидроксид калия (2 г), добавляли сложный этиловый эфир 5-циано-1H-индол-2-карбоновой кислоты (1,71 г, 8,0 мМ), и полученную смесь нагревали с обратным холодильником в течение одного часа. Для растворения осадка, к смеси добавляли воду, а pH доводили до 1 добавлением 6N соляной кислоты. После добавления образовывался осадок. Затем, смесь охлаждали на ледяной бане, фильтровали, и полученное бесцветное твердое вещество промывали холодной водой и сушили (1,51 г). Часть (1,4 г) этого вещества суспендировали в горячей уксусной кислоте (40 мл) и охлаждали, в результате чего получали твердое вещество, которое фильтровали, промывали холодным этилацетатом и сушили. Выход: 980 мг (70%); ВРЖХ (60/40) 3,09 минут (97%). Пример 10B 5-Циано-1H-индол-2-карбоновой кислоты сложный этиловый эфир К горячей суспензии, содержащей сложный этиловый эфир 3-циано-5-нитрофенилпировиноградной кислоты (23,2 г, 88 мМ) в уксусной кислоте (225 мл) и воде (225 мл), добавляли цинковую пыль (57,8 г, 887 мМ) (Внимание! возникает бурная экзотермическая реакция) так, чтобы поддерживалось нагревание с обратным холодильником, а затем, реакционную смесь нагревали с обратным холодильником в течение получаса. После фильтрования смеси, отфильтрованные соли промывали 150 л горячей уксусной кислоты, а фильтрат охлаждали в течение ночи с получением кристаллов, которые фильтровали, промывали холодной смесью уксусной кислоты и воды (1:1), а затем водой, и наконец, сушили (10,11 г, 53%). После концентрирования фильтрата, остаток растворяли в этилацетате и полученный раствор промывали насыщенным водным раствором бикарбоната натрия, а затем солевым раствором, и наконец, сушили и концентрировали, в результате чего получали вторую партию вещества (5,05 грамм). Основную партию вещества использовали в последующих реакциях. Пример 10C 3-Циано-5-нитрофенилпировиноградной кислоты этиловый эфир К смеси, состоящей из дистиллированного диэтилоксалата (120 г, 821 мМ) и 3-метил-4-нитробензонитрила (32 г, 197 мМ) при 0oC добавляли раствор этоксида натрия в этаноле (из 2,2 г 400 мМ металлического натрия в 400 мл этанола). Полученный раствор красного цвета нагревали в течение 18 часов при температуре 40oC. Охлажденную смесь разводили 600 л воды и подкисляли до pH 2,1 добавлением концентрированной соляной кислоты. Образовавшийся осадок собирали фильтрацией смеси (13oC), а затем сушили и очищали хроматографией на двуокиси кремния, элюируя 15%, 30% и 50% ацетоном/гексаном, в результате чего получали целевое соединение в виде оранжевого твердого продукта, который использовали без очистки (23,6 г, 31%). Пример 11 5-Метил-1H-индол-2-карбоновой кислоты {(1S)-[(R)-гидрокси-(метокси-метил-карбамоил)-метил]-2-фенил-этил}-амид (3S)-Амино-(2R)-гидрокси-N-метокси-N-метил-4-фенилбутирамида гидрохлорид (0,5 мМ) и 5-метил-1H-индол-2-карбоновую кислоту (0,5 мМ) подвергали реакции взаимодействия в соответствии с методикой A (за исключением того, что температура составляла 0-25oC, а экстрагирование осуществляли сначала кислотой, а затем основанием), и полученный продукт хроматографировали на двуокиси кремния (элюент: смесь 20-50% этил-ацетата/гексана). Выход: 75%; ВРЖХ (60/40%), 5,06 минут (99%), РВ-МС 396 (МН+, 100%). 1H-ЯМР (CDCl3) : 9,14 (шир., 1H), 7,4-7,2 (м, 6H), 7,07 (дд, 1H, J=2, прибл. 8 Гц), 6,76 (д, 1H, J=2 Гц), 6,45 (д, 1H, J=9,7 Гц), 4,90 (м, 1H), 4,29 (д, 1H, J=5,5 Гц), 3,83 (д, 1H, J=5,5 Гц), 3,35 (с, 3H), 3,13 (c, 3H), 3,09 (дд, 1H, J=6,13 Гц), 3,00 (дд, 1H, J=9,13 Гц), 2,42 (c, 3H).
Анализ для C22H25N3O4:Вычислено: C 66,82; H 6,37; N 10,18; Найдено: C 66,97; H 6,48; N 10,33. Пример 12 5-Фторо-1H-индол-2-карбоновой кислоты [(1S)-[(R)-гидрокси-(метокси-метил-карбамоил)-метил]-2-фенил-этил]-амид (3S)-Амино-(2R)-гидрокси-N-метокси-N-метил-4-фенилбутирамида гидрохлорид (0,5 мМ) и 5-фторо-1H-индол-2-карбоновую кислоту (0,5 мМ) подвергали реакции взаимодействия в соответствии с методикой A (за исключением того, что промывание осуществляли кислотой, а затем основанием), и полученный продукт очищали с помощью хроматографии на силикагеле (элюент: смесь 20-25% этилацетата и гексана). Выход: 69%, ВРЖХ (60/40), 4,55 минут (95%); РB-MC: 400 (MH+, 100%). 1H-ЯМР (CDCl3) : 9,34 (шир., 1H), 7,4 – 7,2 (м, 7H), 7,00 (дт, 1H, J = 2,5 и 9,1 Гц), 6,80 (д, 1H, J = 1,6 Гц), 6,48 (д, 1H, J = 9, Гц), 4,93 (м, 1H), 4,30 (д, 1H, J = 5,3 Гц), 3,83 (д, 1H, J = 5,3 Гц), 3,35 (с, 3H), 3,14 (с, 3H), 3,08 (дд, 1H, A от AB), 3,02 (дд, 1H, J = 5,11 Гц, B от AB).
Анализ для C11H22FN3O4:Вычислено: C 63,15; H 5,55; N 10,52. Найдено: C 64,19; H 6,07; N 10,91. Пример 13 1H-Индол-2-карбоновой кислоты { (1S)-[(R)-гидрокси-(метокси-метил-карбамоил)-метил]-2-фенил-этил}-амид (3S)-Амино-(2R)-гидрокси-N-метокси-N-метил-4-фенил-бутирамида гидрохлорид (0,26 мМ) и 1H-индол-2-карбоновую кислоту (0,28 мМ) подвергали реакции взаимодействия в соответствии с методикой A (за исключением того, чтоо реакционная температура составляла 0-25oC), и полученный продукт очищали с помощью хроматографии на силикагеле (элюент: смесь этилацетата/гексана, 20-50%). Выход: 87%, ВРЖХ (60/40), 4,26 минут (96%); PB-MC 382 (MH+, 100%); 1H-ЯМР (CDCl3) : 9,24 (шир., 1H), 7,63 (д, 1H, J = 8,0 Гц), 7,4 – 7,15 (м, 8H), 7,11 (дт, 1H, J = 8,0 1,5 Гц), 6,85 (д, 1H, J = 1,5 Гц), 6,48 (д, 1H, J = 9,8 Гц), 4,94 (м, 1H), 4,30 (д, 1H, J = 5,5 Гц), 3,84 (д, 1H, J = 1,5 Гц), 3,36 (с, 3H), 3,14 (с, 3H), 3,09 (дд, 1H, J = 6,13 Гц, A от AB), 3,03 (дд, 1H, J = 10,13 Гц, B от AB).
Анализ для C21H23N3O4:Вычислено: C 66,13; H 6,08; N 11,02. Найдено: C 66,19; H 6,08; N 11,02. Пример 14 5-Хлоро-1H-индол-2-карбоновой кислоты { [(1S)-[(метокси-метил-карбамоил-метил]-2-фенил-этил}-амид (3S)-[(5-Хлор-1H-индол-2-карбонил)-амино] -4-фенилмасляную кислоту (357 мг, 1,0 мМ) и 98% N,O-диметилгидроксиламина гидрохлорид (98 мг, 1,0 мМ) подвергали реакции взаимодействия в соответствии с методикой A (за исключением того, что в качестве растворителя использовали диметилформамид). Полученное вещество в виде пены растирали с эфиром, а затем, образовавшееся вязкое твердое вещество растворяли в дихлорметане, концентрировали и растирали с гексаном. Выход: 215 мг, 54%; ВРЖХ (60/40), 6,38 минут (98%) PB-MC 400/402 (MH+, 100%). Анализ для C21H22ClN3O3: Вычислено: C 63,08; H 5,55; N 10,51. Найдено: C 62,91; H 5,79; N 10,21. Пример 14A (3S)-[(5-Хлоро-1H-индол-2-карбонил)амино]-4-фенилмасляная кислота К суспензии, содержащей сложный метиловый эфир (3S)-[(5- хлоро-1H-индол-2-карбонил)амино-4-фенил-масляной кислоты (1,28 г, 3,45 мМ) в метаноле (10 мл) при 25oC добавляли 3,0 мл 2N NaOH. После выдерживания в течение 18 часов, реакционную смесь разводили тетрагидрофураном (10 мл), а затем, раствор нагревали с обратным холодильником в течение 10 минут и концентрировали. Полученное твердое вещество перемешивали с 6N соляной кислотой в течение 15 минут, а затем, суспензию фильтровали и полученное твердое вещество промывали 2N соляной кислотой и осушали. Выход: 1,15 г (93%); ВРЖХ (60/40), 5,18 минут (100%). Пример 15 (3S)-[(5-Хлоро-1H-индол-2-карбонил)-амино]-(2R)-гидрокси-4- фенилмасляной кислоты метиловый эфир К смеси, содержащей сложный метиловый эфир (3S)-амино-(2R)-гидрокси-4-фенилмасляной кислоты (WO 93/25574, пример 1A, 77,5 г, 370 мМ), 5-хлоро-1H-индол-2-карбоновую кислоту (72,45 грамм, 370 мМ) и 1-гидроксибензотриазола гидрат в дихлорметане (640 мл) при 25oC, добавляли 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (DEC, 71 г, 370 мМ). Полученную смесь перемешивали в течение 18 часов, концентрировали, а остаток растворяли в этилацетате, и полученный раствор промывали дважды 2N гидроксидом натрия и дважды 1N соляной кислотой, а затем солевым раствором. После сушки и концентрирования получали целевое соединение в виде желтой пены (140,7 г, 98%), которое использовали в последующем гидролизе без дополнительной очистки (ВРЖХ (70/30), 3,61 минуты (82%), 9,57 минут (13%). Чистый образец получали с помощью хроматографии на двуокиси кремния (элюент: смесь этилацетата/гексана), т.пл. 180-183oC. 1H-ЯМР (CDCl3) : 9,52 (шир., 1H), 7,55 (д, 1H, J = 2 Гц), 7,35 – 7,15 (м, 7H), 6,70 (д, 1H, J = 2 Гц), 6,50 (д, 1H, J = 10 Гц), 4,82 (м, 1H), 4,22 (с, 1H), 3,72 (с, 3H), 3,4 (шир., 1H), 3,05 (м, 2H).
13C-ЯМР (CDCl3, 75,5 мГц) : 174,2, 164,4, 137,1, 135,0, 131,1, 129,8, 128,8, 128,3, 127,0, 127,0, 126,2, 125,0, 121,0, 113,2, 102,3, 70,4, 43,3, 43,1, 38,1.
TSР-МС 387/389 (MH+, 100/30%).
Анализ для C20H19ClN2O4 + 0,5H2O:Вычислено: C 60,69; H 5,09; N 7,08. Найдено: C 60,30; H 4,98; N 6,86. Пример 16 3-[(5-Хлоро-1H-индол-2-карбонил)-амино] -(2RS)-гидрокси-пропионовая кислота Гидрохлорид сложного метилового эфира (RS)-3-амино-2-гидроксипропионовой кислоты (6,6 мМ) и 5-хлоро-1H-индол-2-карбоновую кислоту (6,6 мМ) подвергали реакции взаимодействия в соответствии с методикой A (за исключением того, что экстрагирование осуществляли сначала кислотой, а затем основанием, а в течение промывки кислотой образовывался осадок, после чего, смесь фильтровали и фильтрат использовали так, как описано в Методике A). Неочищенный продукт (920 мг) растворяли в метаноле и обрабатывали 6,6 мл 1N гидроксида натрия в течение 2 часов при 25oC. После добавления 6,6 мл 1N NaOH, смесь концентрировали, а полученный остаток растворяли в этилацетате. Полученный раствор промывали 2N соляной кислотой и солевым раствором, а затем осушали и концентрировали. Образовавшееся бесцветное твердое вещество перемешивали с хлороформом и фильтровали, в результате чего получали целевой продукт: Выход: 763 мг (40%); ВРЖХ (60/40), 2,86 минут (89%), т.пл. 214-215oC, PB-MC 283/285 (MH+, 100%). 1H-ЯМР (ДМСО-d6) : 11,78 (с, 1H), 8,62 (т, 1H), 7,70 (д, 1H, J = 2 Гц), 7,42 (д, 1H, J = 8,7 Гц), 7,17 (дд, 1H, J = 2, 8,7 Гц), 7,14 (д, 1H, J = 2 Гц), 4,18 (дд, 1H, J = 5,8 Гц), 3,58 (м, 2H).
Анализ для C12H11ClN2O4 + 0,1 H2O:Вычислено: C 50,66; H 3,97; N 9,85. Найдено: C 50,80; H 4,06; N 9,48. Пример 16A (RS)-3-Амино-2-гидроксипропионовой кислоты метилового эфира гидрохлорид Смесь D, L-изосерина (2,06 г, 19,6 мМ), метанола (20 мл) и хлоротриметилсилана (9,5 г, 88 мМ) нагревали с обратным холодильником в течение 5 часов, а затем охлаждали и концентрировали, в результате чего получали целевое соединение (3,20 г). Пример 17 5-Хлоро-1H-индол-2-карбоновой кислоты [(1S)-((R)-метокси-метилкарбамоил-метил)-2-фенил-этил]-амид (3S)-Амино-(2R)-метокси-N,N-диметил-4-фенил-бутирамида гидрохлорид (0,84 мМ) и 5-хлоро-1H-индол-2-карбоновую кислоту (0,80 мМ) подвергали реакции взаимодействия в соответствии с методикой A (за исключением того, что реакционная температура составляла 0-25oC, а в качестве растворителя использовали смесь (2:1) дихлорметана/диметилформамида), и полученный продукт очищали хроматографией на двуокиси кремния (элюент: смесь этилацетата/гексана (1: 1). Выход: 81%; ВРЖХ (60/40), 5,44 минуты (100%), TSP-MC 414/416 (MH+, 100/30%). 1H-ЯМР (CDCl3) : 9,38 (шир., 1H), 7,60 (д, 1H, J = 2 Гц), 7,4 – 7,2 (м, 6H), 7,20 (дд, 1H, J = 2,9 Гц), 7,03 (д, 1H, J = 8 Гц), 6,92 (д, 1H, J = 2 Гц), 4,50 (м, 1H), 4,00 (д, 1H, J = 2 Гц), 3,40 (, 3H), 3,22 (дд, A от AB, 1H, J = 5,13 Гц), 3,00 (дд, B от AB, 1H, J = 10,13 Гц), 2,86 (с, 3H), 2,65 (с, 3H).
Анализ для C22H24ClN3O3:Вычислено: C 63,48; H 5,84; N 10,15. Найдено: C 63,48; H 5,97; N 9,97. Пример 17A (3S)-Амино-(2R)- метокси-N,N-диметил-4-фенил-бутирамида гидрохлорид (1S, 2R)-(1-Бензил-2-диметилкарбамоил-2-метокси-этилкарбаминовой кислоты трет-бутиловый эфир (283 мг, 0,84 мМ) растворяли в смеси 4N HCl и диоксида (1 мл) в течение 1,5 часа при температуре 25oC и концентрировали. Остаток выпаривали вместе с эфиром и сушили. Пример 17B (1S, 2R)-(1-Бензил-2-диметилкарбамоил-2-метокси-этил)-карбаминовой кислоты трет-бутиловый эфир К раствору, содержащему трет-бутиловый эфир (1S,2R)-(1-бензил-2-диметилкарбамоил-2-гидрокси-этил)-карбаминовой кислоты (322 мг, 1,0 мМ) в тетрагидрофуране (4 мл) при 0oC добавляли гидрид натрия (53 мг 50% дисперсии в масле). После того, как бурное выделение пузырьков газа было завершено (несколько минут), к смеси добавляли метилиодид (155 мг), и через 15 минут к этой смеси добавляли еще 11 мг дисперсии гидрида натрия в масле и 23 мг метилиодида. Еще через 15 минут к смеси добавляли водный раствор хлорида аммония и этилацетат, после чего, органический слой отделяли, промывали водой и 2N гидроксидом натрия, а затем сушили и концентрировали, в результате чего получали вязкое маслообразное вещество, которое использовали без дополнительной очистки. Выход: 283 мг, 84%. Пример 17C (1S, 2R)-1-Бензил-2-диметилкарбамоил-2-гидрокси-этил)-карбаминовой кислоты трет-бутиловый эфир (3S)-Трет-бутоксикарбониламино-(2R)-гидрокси-4-фенилмасляную кислоту (Schweizerhall, Ins., S. Plainfield, nI, 1,02 г, 3,4 мМ) и диметиламина гидрохлорид (338 г, 4,1 мМ) подвергали реакции взаимодействия в соответствии с методикой A (за исключением того, что реакционная температура составляла 0-25oC, в качестве растворителя использовали смесь диметилформамида и дихлорметана, а экстрагирование осуществляли кислотой, а затем основанием), и получали неочищенный продукт, который хроматографировали на двуокиси кремния (элюент: смесь этанола в дихлорметане (1-8%), в результате чего получали вещество в виде пены. Выход: 995 мг, 91%. Пример 18 5-Хлоро-1H-индол-2-карбоновой кислоты (3-азетидин-1-ил-(1S)-бензил-(2R)-гидрокси-3-оксо-пропил)-амид Азетидин (0,44 мМ) и (3S)-[(5-хлоро-1H-индол-2-карбонил)-амино]-(2R)-гидрокси-4-фенилмасляную кислоту (0,4 мМ) подвергали реакции взаимодействия в соответствии с методикой A (за исключением того, что в качестве растворителя использовали смесь (1: 1) диметилформамида и дихлорметана), и получали желаемое вещество. Выход: 94%, ВРЖХ (60/40), 4,55 минут (> 98%); PB-MC 412/414 (MH+, 100%). Анализ для C22H22ClN3O3 + 0,25 H2O: Вычислено: C 63,46; H 5,45; N 10,09. Найдено: C 63,61; H 5,66; N 10,27. Пример 19 5-Хлоро-1H-индол-2-карбоновой кислоты [(1S)-бензил-(2R)-метокси-2-(метокси-метил-карбамоил)-этил]-амид (3S, 2R)-3-Амино-(2R)N-диметокси-N-метил-4-фенилбутирамид (0,31 мМ) и 5-хлоро-1H-индол-2-карбоновую кислоту (0,31 мМ) подвергали реакцию взаимодействия в соответствии с Методикой A, и полученный продукт очищали хроматографией на силикагеле (элюент: смесь (20-40%) этилацетата-гексана). Выход: 81%, ВРЖХ (60/40), 7,39 минут (98%), PB-MC 430/432 (MH+, 100%). 1H-ЯМР (CDCl3) : 9,44 (с, 1H), 7,58 (д, 1H, J = прибл. 2 Гц), 7,4 – 7,22 (м, 6H), 7,19 (дд, 1H, J = 2,0 и 8,8 Гц), 6,89 (д, 1H, J = прибл. 2 Гц), 6,80 (д, 1H, J = 8 Гц), 4,72 (м, 1H), 3,93 (с, 1H), 3,93 (с, 3H), 3,24 (с, 3H), 3,19 (дд, 1H, J = 5,1, 13 Гц, A от AB), 3,06 (с, 3H), 2,95 (дд, 1H, J = 10,9 Гц и 13 Гц, и B от AB).
Анализ для C22H24ClN3O4 + 0,33 C6H14:Вычислено: C 62,85; H 6,30; N 9,16. Найдено: C 62,91; H 6,29; N 8,95. Пример 19A (3S,2R)-3-Амино-(2R), N-диметокси-N-метил-4-фенил-бутирамид Трет-бутиловый эфир (1S, 2R)-(1-бензил-2-метокси-метилкарбамоил-2-метокси-этил)-карбаминовой кислоты (113 мг, 0,32 мМ) растворяли в смеси 4N соляной кислоты и диоксана (4 мл) при температуре 25oC в течение 1 часа, а затем концентрировали, после чего остаток растирали с эфиром, и получали целевой продукт (93 мг, 100%). Пример 19B (1S, 2R)-(1-Бензил-2-метокси-метил-карбамоил-2-метокси- этилкарбаминовой кислоты трет-бутиловый эфир. К раствору трет-бутилового эфира (1S,2R)-(1-бензил-2-метокси-метил-карбамоил-2-гидрокси-этил)-карбаминовой кислоты в тетрагидрофуране (2 мл) при 0oC добавляли дисперсию гидрида натрия (30 мг 50% дисперсии в масле). После выдерживания в течение 5 минут, к смеси добавляли метилиодид (175 мг), и эту смесь оставляли на 18 часов при температуре 25oC. После добавления этилацетата и насыщенного водного раствора хлорида аммония, органический слой отделяли, промывали водой, сушили, концентрировали и хроматографировали на двуокиси кремния (элюент: смесь (10-20%) этилацетата/гексана). Выход: 113 мг (52%); ВРЖХ (60/40); 6,45 минут (>96%). Пример 20 { (2S)-[(5-Хлоро-1H-индол-2-карбонил)-амино] -(1R)-(метокси-метил-карбамоил) -1-фенил-пропокси}-уксусной кислоты бензиловый эфир. Гидрохлорид бензилового эфира (1R,2S)-[2-амино-1-(метокси-метил-карбамоил)-3-фенил-пропокси] -уксусной кислоты (162 мг, 0,38 мМ) и 5-хлоро-1H-индол-2-карбоновую кислоту (77 мг, 0,36 мМ) подвергали реакции взаимодействия в соответствии с методикой A (за исключением того, что реакционная температура составляла 0 – 25oC), и полученный неочищенный продукт очищали с помощью хроматографии на силикагеле (элюент: смесь 20-75% этилацетата в гексане), в результате чего получали целевое соединение в виде стеклообразного твердого вещества. Выход: 61%, TSP-MC 564/566 (MH+, 90/60%), 581/583 (МН+ +NH3, 100/50%). Пример 20A (1R,2S)-[2-Амино-1-(метокси-метил-карбамоил)-3-фенил-пропокси]- уксусной кислоты бензилового эфира гидрохлорид Бензиловый эфир (1R, 2S)-[2-трет-бутоксикарбониламино-1- (метокси-метил-карбамоил)-3-фенил-пропокси] -уксусной кислоты (170 мг, 0,35 мМ) растворяли в смеси 4N соляной кислоты и диоксана (2 мл) при 25oC в течение полутора часов, а затем концентрировали, и образовавшийся остаток выпаривали вместе с эфиром, и сушили с получением маслообразного вещества (163 мг), MC:387 (MH+, 100%). Пример 20B (1R, 2S)-[2-трет-бутоксикарбониламино-1-(метокси-метил-карбамоил)- 3-фенил-пропокси]-уксусной кислоты бензиловый эфир К раствору, содержащему трет-бутиловый эфир (1S,2R)-(1-бензил-2- метокси-метил-карбамоил)-2-гидрокси-этил-карбаминовой кислоты (858 мг, 2,5 мМ) в тетрагидрофуране (8 мл) при 0oC добавляли дисперсию гидрида натрия (120 мг 50% дисперсии в масле, 2,8 мМ). После прекращения бурного выделения пузырьков газа, к смеси добавляли бензилбромоацетат (0,56 г, 2,5 мМ) и эту смесь нагревали до 25oC. Через два часа, к смеси добавляли еще 12 мг дисперсии гидрида натрия, и полученную смесь перемешивали в течение одного часа, разводили этилацетатом и насыщенным хлоридом аммония, а органический слой отделяли, промывали водой, сушили и концентрировали с получением маслообразного вещества, которое хроматографировали на силикагеле (элюент: смесь (20 – 75%) этилацетата/гексана). Наиболее чистые фракции объединяли и получали маслообразное вещество (175 мг, 15%), MC: 487 (МН+), 387 (100%). Пример 21 { (2S)-[(5-Хлоро-1H-индол-2-карбонил)-амино]-(1R)-(метокси- метил-карбамоил)-3-фенил-пропокси}-уксусная кислота Смесь, содержащую бензиловый эфир {(2S)-[(5-хлоро-1H-индол- 2-карбонил)амино]-(1R)-(метокси-метил-карбамоил)-3-фенил-пропокси}- уксусной кислоты (120 мг, 0,2 мМ) и 50% влажный гидроксид палладия-на-угле в метаноле (50 мг), перемешивали путем встряхивания в течение одного часа при температуре 25oC и под давлением водорода 40 фунт/кв.дюйм (2,812 кг/см2). Полученную смесь оставляли на 30 минут, а затем фильтровали, и этот фильтрат концентрировали с получением 121 мг твердого вещества, которое хроматографировали на двуокиси кремния (элюент: смесь этилацетата/гексана, 25-100%), в результате чего получали 84 мг твердого вещества: ВРЖХ (60/40), 40,81 мин (37%) и 6,24 мин (63%). 1H-ЯМР-анализ и MC-анализ указывали на присутствие сложных метиловых эфиров 5-дез-Cl и целевого продукта, соответственно. Полученное твердое вещество растворяли в тетрагидрофуране и обрабатывали 170 мкл 1N гидроксида натрия при 25oC в течение 30 минут, после чего раствор концентрировали и образовавшийся остаток распределяли между этилацетатом и 1N соляной кислотой. Органический слой отделяли, промывали водой, сушили и концентрировали, в результате чего получали смесь целевого соединения и аналога дез-5-Cl: Выход: 85 мг (71%); ВРЖХ (60/40), 3,49 минут (37%), 4,23 минут (61%), МС: 338 (МН+, 100%); TSP-MC: 474/476 (МН+ для целевого соединения, 40%), 440 (МН+ для аналога дез-5-Cl, 95%). Пример 22 (3S)-[(1-Индол-2-карбонил)амино]-(2R)-гидрокси-4-фенил- бутирамид (2R,3S)-3-амино-2-гидрокси-4-фенилбутирамид (0,59 мМ, WS 4599198, пример 1D) и индол-2-карбоновую кислоту (0,71 мМ) подвергали реакции взаимодействия в соответствии с методикой A (за исключением того, что промывали сначала кислотой, а затем основанием), и полученный продукт очищали с помощью хроматографии на силикагеле (элюент: смесь 66 – 100% этилацетата-гексана). Выход: 89% ВРЖХ (60/40, колонка C-8 Zorbax Dupont) 99%; MC: 338 (MH+, 100%), 1H-ЯМР (ДМСО-d6) : 11,53 (с, 1H), 7,95 (д, 1H, J=9 Гц), 7,63 (д, 1H, J=8 Гц), 7,5 – 7,15 (м,7-8 Гц),7,12 (д, 1H, J=прибл. 7 Гц), 7,09 (д, 1H, J=прибл. 8 Гц), 5,95 (д, 1H, J=6 Гц), 4,55 (м, 1H), 3,93 (м, 1H), 2,98 (дд, 1H, A от AB, J=6,13 Гц), 2,88 (дд, 1H, B от AB, J=8,13 Гц).
Пример 23{ (3S)-[(5-Хлоро-1H-индол-2-карбонил)амино] -(2S)-гидрокси-4- фенилбутирамид К раствору, содержащему гидрохлорид (2S,3S)-3-амино-2-гидрокси- 4-фенилбутирамида (0,319 г, 1,61 мМ) и триэтиламин (145 мг, 1,42 мМ) в дихлорметане (2 мл) при 25oC, добавляли 5-хлоро-1H-индол-2-карбонилфторид (0,30 г, 1,29 мМ). Через 18 часов, смесь разводили этилацетатом, и полученный раствор дважды промывали 1N соляной кислотой, дважды насыщенным водным раствором бикарбоната натрия и один раз солевым раствором, а затем сушили и концентрировали. Остаток хроматографировали на двуокиси кремния (элюент: смесь 50 – 100% этилацетата/гексана) и получали 0,31 г целевого продукта в виде твердого вещества, которое перекристаллизовывали из изопропилового спирта. Выход: 0,020 г; FAB-MC 372/374 (MH+, 21%), 217 (100%), 1H-ЯМР (ДМСО-d6, частичный) : 8,5 (д, 1H, J=9 Гц), 7,48 (д, 1H, J=2 Гц), 7,4 – 7,1 (м, 9H), 5,95 (д, 1H, J=7 Гц), 4,56 (м, 1H), 4,08 (м, 1H), 2,92 (дд, 1H, J=11,13 Гц), 2,68 (дд, J=3,13 Гц).
Пример 23A5-Хлоро-1H-индол-2-карбонилфторид Раствор, содержащий 5-хлоро-1H-индол-карбоновую кислоту (10,0 г, 51,1 мМ) и пиридин (33,1 мМ) в ацетонитриле, добавляли к раствору, содержащему фторид циануровой кислоты (2,76 г, 20,4 мМ) в ацетонитриле (всего 340 мл) при температуре 25oC. Реакцию анализировали с помощью TCX на аликвотах, а затем гасили путем добавления бутиламина, после чего, реакция завершалась приблизительно через 1 час. Полученную смесь выливали на лед, экстрагировали эфиром, сушили над сульфатом натрия и концентрировали с получением твердого вещества, которое использовали без дополнительной очистки (10,0 г, 99%). TCX-анализ аликвот, погашенных путем добавления бутиламина, указывал на присутствие некоторого количества 5-хлороиндрол-2-карбоновой кислоты и менее полярного N-бутиламина. Образец очищали с помощью хроматографии на силикагеле, элюируя смесью этилацетата/гексана (50-100%), для последующей идентификации (16368-130-1). Пример 23B (2S,3S)-3-Амино-2-гидрокси-4-фенил-бутирамида гидрохлорид (1S)-[(S)-Карбамоил-гидрокси-метил)-2-фенил-этил] -карбаминовой кислоты трет-бутиловый эфир (0,50 г, 1,7 мМ) растворяли в смеси 4 М HCl/диоксана в течение одного часа при 25oC. После концентрирования смеси, остаток растирали с эфиром, и получали бесцветное твердое вещество (430 мг). ВРЖХ (60/40), 2,68 мин (100%). Пример 23C { (1S)-((S)-Карбамоил-гидрокси-метил]-2-фенил-этил}-карбаминовой кислоты трет-бутиловый эфир. К раствору, содержащему трет-бутиловый эфир {(1S)-[(S)-(трет- бутил-диметил-силанилокси)-карбамоил-метил]-2-фенил-этил]- карбаминовой кислоты в тетрагидрофуране (6 мл) при 0oC добавляли фторид тетрабутиламмония (23 мл 1 М раствора в тетрагидрофуране). Через 30 минут, смесь разводили этилацетатом и водой, а органический слой отделяли, промывали водой, а затем, два раза 1N соляной кислотой, два раза 1N бикарбонатом натрия и один раз солевым раствором. Полученную эмульсию фильтровали, а фильтрат осушали, и концентрировали с получением бесцветного твердого вещества (0,5 г, 20%). Часть (3,1 г) отфильтрованного твердого вещества (3,3 г) перекристаллизовывали из горячего этилацетата, и получали 1,33 г нужного продукта в виде бесцветного твердого вещества. Пример 23D {(1S)-(S)-(Трет-бутил-диметил-силанилокси)карбамоил-метил]-2-фенил-этил] -карбаминовой кислоты трет-бутиловый эфир К раствору, содержащему трет-бутиловый эфир {1(S)-бензил-(2S)-(трет-бутил-диметил-силанилокси)-2-цианоэтил} – карбаминовой кислоты (пример 24D, 5,0 г, 12,8 мМ) и 1N NaOH (22 мл) в этаноле (110 мл) при температуре 0oC, в течение 15 минут добавляли 30% пероксид водорода (7,2 мл 64 мМ). Полученную смесь перемешивали в течение полутора часов, обрабатывали 175 мл водного раствора 10% тиосульфата натрия, концентрировали и экстрагировали этилацетатом. Экстракты осушали сульфатом натрия и концентрировали. Остаток хроматографировали на двуокиси кремния (элюент, смесь 20 – 33% этилацетата/гексана) и получали целевое соединение в виде бесцветного твердого вещества (3,17 г, 61%). Пример 24 5-Хлоро-1H-индол-2-карбоновой кислоты { (1S)-[(S)-гидрокси(метокси-метил-карбамоил)-метил]-2-фенил-этил}-амид N, O-Диметилгидроксиламина гидрохлорид (0,4 мМ) и (3S)-[(5-хлоро-1H-индол-2-карбонил)амино] -(2S)-гидрокси-4-фенил- масляную кислоту (0,38 мМ) подвергали реакции взаимодействия в соответствии с методикой A, и полученный продукт очищали хроматографией на двуокиси кремния (элюент: смесь 20 – 50% этил-ацетата/гексана), выход=72%; ВРЖХ (60/40), 5,05 минут (98%); PB-MC 416/418 (MH+, 100%). 1H-ЯМР (CDCl3) : 9,30 (шир. 1H), 7,60 (д, 1H, J=2 Гц), 7,33 (д, 1H, J=8 Гц), 7,3-7,15 (м, 6-7H), 6,75 (м, 2H), 5,00 (м, 1H), 4,65 (д, 1H, J=4 Гц), 3,71 (с, 3H), 3,06 (с, 3H), 2,87 (м, 2H), 1,6 (шир.).
Анализ для C21H22ClN3O4+0,35 H2O:Вычислено: C 59,74; H 5,42; N 9,95. Найдено: C 60,14; H 5,65; N 9,55. Пример 24A (3S)-[(5-Хлоро-1H-индол-2-карбонил)амино] -(2S)гидрокси-4- фенилмасляная кислота К раствору, содержащему метиловый эфир (3S)-[(5-хлоро-1H-индол-2-карбонил)амино] -(2S)-гидрокси-4- фенилмасляной кислоты (500 мг, 1,29 мМ) в метаноле, при 25oC добавляли 2,6 мл водного раствора 1N NaOH. Через 18 часов, смесь концентрировали, а остаток растворяли в этилацетате и воде, после чего полученный раствор подкисляли до pH 1 добавлением 6 н. соляной кислоты. Водный слой отделяли, а затем три раза экстрагировали этилацетатом. Органический слой объединяли, сушили и концентрировали, в результате чего получали твердое вещество (417 мг, 87%); ВРЖХ (60/40), 4,23 мин (>98%). Пример 24B { ((3S)-[(5-Хлоро-1H-индол-2-карбонил)-амино] -(2S)-гидрокси-4- фенилмасляной кислоты метиловый эфир (3S)-Амино-(2S)-гидрокси-4-фенилмасляной кислоты метиловый эфир (1,4 мМ) и 5-хлоро-1H-индол-2-карбоновую кислоту (1,37 мМ) подвергали реакции взаимодействия в соответствии с методикой A (реакционная температура = 0-25oC, время протекания реакции = 40 часов; растворитель: смесь дихлорметана и диметилформамида), в результате чего получали целевой продукт, выход: 94%; ВРЖХ (60/40), 5,38 минут (97%), т.пл. 214-221oC; РВ-МС 387/389 (MH+, 100%). Анализ для C20H19ClN2O4: Вычислено: C 62,10; H 4,95; N 7,24. Найдено: C 62,12; H 5,07; N 7,11. Пример 24C (3S)-Амино-(2S)-гидрокси-4-фенилмасляной кислоты метиловый эфир К раствору безводной соляной кислоты (3,2 г) и метанола (20 мл) добавляли [(1(S)-бензил-(2S)-(трет-бутил-диметилсиланилокси)-2-циано- этил]-карбаминовой кислоты трет-бутиловый эфир (417 мг), и полученный раствор закрывали пробкой и выдерживали в течение 5 дней при температуре 25oC. Смесь концентрировали с получением 308 мг бесцветного твердого вещества, которое являлось гомогенным (1H-ЯМР (D2O)-анализ). Полученное вещество объединяли со спектрально эквивалентным веществом, полученным тем же самым способом из 400 мг аналогичного исходного соединения, и всю эту смесь растворяли в насыщенном водном растворе гидрокарбоната натрия, и десять раз экстрагировали хлороформом. Объединенные экстракты сушили и концентрировали, в результате чего получали целевое соединение (328 мг, 75%). Пример 24 [1(S)-Бензил-(2S)-(трет-бутил-диметил-силанилокси)-2-цианоэтил] – карбаминовой кислоты трет-бутиловый эфир N-т-Бутоксикарбонил-(3S)-амино-(2RS)-гидрокси-4-фенилбутиронитрил превращали в соответствующее O-трет-бутилдиметилсилиловые эфиры с помощью методики, описанной в примере 1B патента США N 4599198, а изомеры разделяли хроматографией на силикагеле (элюент: 7-8% эфир/гексан). Целевое соединение отделяли от его наименее полярного изомера 2R (последний пример 1B в патенте США N 4599198). Пример 24E N-т-Бутоксикарбонил-(3S)-амино-(2RS)-гидрокси-4-фенилбутиронитрил К раствору, содержащему N-т-бутоксикарбонил-N-фенилаланин (J. Med. Chem. 1985, Vol. 28, 1799-1790, 10,0 г, 40,1 мМ) в диметоксиэтане (100 мл) при температуре 0-5oC добавляли раствор (5oC) бисульфита натрия (4,38 г) в воде (100 мл). Полученную смесь перемешивали в течение 2 часов при 0oC, а затем в течение ночи при 25oC. Эту смесь концентрировали до объема 80 мл, разводили 250 мл этилацетата, и полученный раствор обрабатывали цианидом калия (2,61 г, 40,1 мМ). После выдерживания в течение 4 часов при температуре 25oC, органический слой отделяли, дважды промывали водой и один раз солевым раствором, а затем сушили и концентрировали. Полученное маслообразное вещество кристаллизовали из смеси эфира/гексана, и получали бесцветное твердое вещество (3,53 г, т.пл. 95-98oC). Второй сбор получали путем перекристаллизации маточного раствора (5,0 г) из смеси эфира/гексана (бесцветное твердое вещество 2,44 г) (т.пл. 88-92oC). Второе вещество, имеющее низкую температуру плавления, использовали в последующей реакции силилирования. Пример 25 (3S)-[(5-Хлоро-1H-индол-2-карбонил)амино] -(2R)-гидрокси-4-фенилмасляная кислота К раствору, содержащему неочищенный (3S)-[(5-хлоро-1H-индол-2-карбонил)-амино]-(2R)-гидрокси-4-фенилмасляной кислоты метиловый эфир (содержащий в виде примеси 13% N,O-бис-5-хлоро-1H-индол-2-карбонил) (140,7 г, 363 мМ) и метанол (1900 мл), при температуре 10-22oC добавляли 375 мл водного раствора 2N NaOH, и полученную смесь перемешивали при температуре 25oC. Через два часа, раствор концентрировали, а остаток растворяли в 2 л этилацетата и 500 мл 2N соляной кислоты. Водный слой отделяли и дважды промывали 2N соляной кислотой, а органические слои объединяли, промывали солевым раствором, сушили над сульфатом натрия и концентрировали. Остаток помещали в 100 мл горячего этилацетата (с получением суспензии), добавляли 1300 мл хлороформа, и эту суспензию нагревали с обратным холодильником в течение 5 минут при механическом перемешивании, а затем фильтровали в нагретом состоянии, и отфильтрованные твердые вещества промывали смесью хлороформа и этилацетата (3: 1, 400 мл) при температуре, близкой к температуре кипения. Полученное твердое вещество сушили в вакууме до получения постоянной массы (101 г, 75%). Фильтрат концентрировали и перекристаллизовывали растворением в горячем тетрагидрофуране (70 мл), добавлением горячего гексана (200 мл), охлаждением в течение ночи, фильтрованием и промывкой полученного твердого вещества смесью тетрагидрофурана/гексана (1:5) с получением 7,03 г (5%) желаемого продукта. Маточные растворы с последней стадии концентрировали и перекристаллизовывали тем же самым способом, в результате чего получали 11,07 грамм (8%) желаемого вещества. Все три продукта анализировали с помощью ВРЖХ (60/40), 4,2 мин (>98%). Анализ на присутствие 5-хлоро-1H-индол-2-карбоновой кислоты осуществляли ВРЖХ (C8, колонка Zorbax 15 см (600:400:2:1) вода/ацетонитрил/триэтиламина/уксусная кислота, 600:400:2:1), которая указывала на ее присутствие в трех продуктах в 0,4%, 0,7% и 21%, соответственно, как описано выше. Данные для главного продукта: т.пл. 209-212oC, TSP-МС (MH+, 100%). 1H-ЯМР (ДМСО-d6) : 12,6 (шир., 1H), 11,7 (с, 1H), 8,17 (д, 1H, J = 9,1 Гц), 7,71 (д, 1H, J = 2 Гц), 7,39 (д, 1H, J = 8,7 Гц), 7,28 (м, 4H), 7,17 (м, 3H), 5,55 (шир., 1H), 4,57 (м, 1H), 4,05 (д, 1H, J = 3,6 Гц), 2,97 (дд, 1H, A от AB, J = 6,5 и 13,5 Гц), 2,87 (дд, 1H, B от AB, J = 8,5 и 13,5 Гц).
Анализ для C19H17ClN2O4:Вычислено: C 61,21; H 4,60; N 7,51. Найдено: C 61,09; H 4,63; N 7,59. Пример 26 (3R)-[(5-фторо-1H-индол-2-карбонил)-амино] -(2R)-гидрокси-4- фенил-масляная кислота Раствор, содержащий метиловый эфир (3S)-[(5-фторо-1H-индол-2-карбонил)-амино]-(2R)-гидрокси-4-фенил-масляной кислоты (190 мг, 0,5 мМ), 1N NaOH (1 мл) и метанол (5 мл), перемешивали в течение 18 часов при температуре 25oC, pH доводили до 1-2 путем добавления 1N соляной кислоты, а затем раствор концентрировали, и образовавшиеся твердые вещества помещали в воду при 25oC и фильтровали. Полученное твердое вещество промывали эфиром и сушили, в результате чего получали бесцветное стеклообразное вещество (160 мг, 87%); ВРЖХ (60/40), 3,49 минут (99%); 1H-ЯМР (частичный, ДМСО-d6) : 8,15 (д, 1H, J = 8 Гц), 7,42 (м, 2H), 7,3 (м, 4H), 7,15 (м, 2H), 7,03 (дт, 1H), 4,60 (м, 1H), 4,03 (д, 1H), 3,00 (дд, 1H, J = 8,13 Гц), 2.90 (дд, 1H, J = 8,13 Гц).
Пример 27(3S)-[(5-Бромо-1H-индол-2-карбонил)-амино]-(2R)-гидрокси-4-фенилмасляная кислота К раствору, содержащему метиловый эфир (3S)-[(5-бромо-1H-индол-2-карбонил)-амино] -(2R)-гидрокси-4-фенилмасляной кислоты (2,45 г, 5,7 мМ) в метаноле (60 мл) при температуре 25oC, добавляли водный раствор 1N NaOH (60 мл). Через 2 часа смесь концентрировали и распределяли между этилацетатом и 2N соляной кислотой. Водный слой отделяли и экстрагировали этилацетатом, а объединенные органические слои промывали 1N соляной кислотой и солевым раствором, а затем сушили и концентрировали. Полученное твердое вещество растирали с хлороформом при 25oC. Выход: 85%; ВРЖХ (60/40), 4,24 минуты (100%); т.пл. 213-216oC; TSP-МС 417/419 (MH+, 98%). 1H-ЯМР (частичный, ДМСО-d6) : 11,72 (шир., 1H), 8,20 (д, 1H, J = 10 Гц), 7,86 (д, 1H, J = 2 Гц), 7,4-7,1 (м, 8H), 4,60 (м, 1H), 4,04 (д, 1H, J = 3,5 Гц), 3,00 (дд, A от AB, J = 7,13 Гц), 2,88 (дд, 1H, B от AB, J = 8,5 и 13 Гц).
Анализ для C19H17BrN2O4 + 0,25H2O:Вычислено: C 54,11; H 4,18; N 6,64. Найдено: C 54,15; H 4,15; N 6,64. Пример 28 (3S)-[(5,6-Дихлоро-1H-индол-2-карбонил)-амино] -(2R)-гидрокси-4- фенилмасляная кислота К суспензии, содержащей метиловый эфир (3S)-[(5,6-дихлоро-1H-индол-2-карбонил)-амино] -(2R)-гидрокси-4-фенил-масляной кислоты (249 мг, 0,6 мМ) и метанол (5 мл) при температуре 25oC, добавляли водный раствор 1N NaOH (1,18 мл). После отстаивания в течение 18 часов, смесь концентрировали, и полученный остаток распределяли между избыточным количеством 2N соляной кислоты и этилацетатом. Водный слой отделяли и промывали этилацетатом, а объединенные органические слои промывали солевым раствором, сушили и концентрировали, в результате чего получали желтое твердое вещество, выход, 259 мг; ВРЖХ (60/40), 4,96 минут (100%); TSP-МС 407/409 (MH+, 100/40%); 1H-ЯМР (частичный, ДМСО-d6) : 11,8 (шир., 1H), 8,28 (д, 1H, J = 9 Гц), 7,98 (с, 1H), 7,58 (с, 1H), 7,3-7,15 (м, 6H), 4,60 (м, 1H), 4,07 (д, 1H, J = 3-4 Гц), 2,98 (дд, 1H, A от AB, J = 6,13 Гц), 2,88 (дд, 1H, J = 9,13 Гц).
Анализ для C19H16Cl2N2O4 + 0,5H2O:Вычислено: C 54,82; H 4,12; N 6,73. Найдено: C 54,86; H 4,08; N 6,76. Пример 29 (3R)-[(5-Хлоро-1H-индол-2-карбонил)-амино]-(2R)-гидрокси-4-фенилмасляная кислота К суспензии, содержащей метиловый эфир (3R)-[(5-хлоро-1H-индол-2-карбонил)-амино] -(2R)-гидрокси-4-фенилмасляной кислоты (326 мг, 0,8 мМ) и метанол при 25oC, добавляли водный раствор 1 н. NaOH (1,69 мл). Через 2,5 часа, смесь концентрировали (обнаруживался исходный продукт) и растворяли в метаноле и водном растворе 1 н. NaOH (0,5 мл). Через один час, смесь концентрировали, а остаток распределяли между избытком 2N соляной кислоты и этилацетатом, после чего, органический слой отделяли, сушили и концентрировали. Выход: 288 мг (92%); ВРЖХ (60/40), 3,89 минут (93%); т.пл. 215-223oC; TSP-МС 375/373 (MH+, 100%). 1H-ЯМР (ДМСО-d6) : 12,7 (шир., 1H), 11,65 (с, 1H), 8,50 (д, 1H, J = 8,8 Гц), 7,70 (д, 1H, J = 2 Гц), 7,37 (д, H1, J = 8,7 Гц), 7,4-7,1 (м, 7H), 5,7 (шир., 1H), 4,50 (м, 1H), 4,17 (д, 1H, J = 4,8 Гц), 2,94 (дд, 1H, A от AB, J = 10,14 Гц), 2,78 (дд, 1H, B от AB, J = 3,14 Гц).
Анализ для C19H17ClN2O4 + 0,1H2O:Вычислено: C 60,92; H 4,63; N 7,48. Найдено: C 60,72; H 4,78; N 7,53. Пример 29A (3R)-[(5-Хлоро-1H-индол-2-карбонил)-амино] -(2R)-гидрокси-4- фенил-масляной кислоты метиловый эфир Гидрохлорид метилового эфира (2R,3R)-3-амино-2-гидрокси-4-фенилмасляной кислоты (239 мг, 1,0 мМ) и 5-хлоро-1H-индол-2-карбоновую кислоту (200 мг, 1,05 мМ) подвергали реакции взаимодействия в соответствии с методикой A (за исключением того, что реакционная температура составляла 0-25oC, а промывку осуществляли сначала кислотой, а затем основанием), в результате чего получали неочищенный продукт, который использовали без дополнительной очистки. Выход: 328 мг, 87%. Пример 29B (2R, 3R)-Амино-2-гидрокси-4-фенилмасляной кислоты метилового эфира гидрохлорид Смесь (2R, 3R)-3-амино-2-гидрокси-4-фенилмасляной кислоты (200 мг, 1,0 мМ, Sigma Chemical Co. (St. Louis, MO), хлортриметилсилана (500 мг, 4,6 мМ) и метанола (2 мл) нагревали с обратным холодильником в течение 5,5 часа и концентрировали, в результате чего получали целевое соединение в виде пены. Выход: 244 мг (100%). Пример 30 5-Хлоро-1H-индол-2-карбоновой кислоты [(2R)-гидрокси-2-(метокси-метил-карбамоил)-этил]-амид N, O-Диметилгидроксиламина гидрохлорид (1,0 мМ) и 3-[(5-хлоро-1H-индол-2-карбонил)-амино] -(2RS)-гидрокси-пропионовую кислоту (0,95 мМ) подвергали реакции взаимодействия в соответствии с методикой A (за исключением того, что реакционная температура составляла 0-25oC, а промывку осуществляли сначала кислотой, а затем основанием), и полученный неочищенный продукт растирали с эфиром, в результате чего получали твердое бесцветное вещество. Выход: 69%; ВРЖХ (60/40), 3,18 минут (96%); т.пл. 192-192,5oC; РВМС 326/328 (MH+, 100%). 1H-ЯМР (ДМСО-d6) : 11,80 (с, 1H), 8,62 (т, 1H), 7,70 (д, 1H, J = 2 Гц), 7,41 (д, 1H, J = 8,8 Гц), 7,17 (дд, 1H, J = 2 и 8,7 Гц), 7,13 (с, 1H), 5,35 (м, 1H), 4,65 (м, 1H), 3,69 (с, 3H), 3,47 (м, 2H), 3,34 (с, 3H).
Анализ для C14H16ClN3O4:Вычислено: C 51,62; H 4,95; N 12,90. Найдено: C 51,78; H 5,07; N 12,75. Пример 31 (3S)-[(5-Хлоро-1H-индол-2-карбонил)амио]-(2R)-гидрокси-4-фенилбутирамид Избыток безводного аммиака вводили в раствор, содержащий (3S)-[(5-хлоро-1H-индол-2-карбонил)-амино]-(2R)-гидрокси-4-фенилмасляной кислоты сложный метиловый эфир (100 мг, 0,27 мМ) в метаноле (10 мл), и полученную смесь нагревали в реакторе Парра из нержавеющей стали под давлением 50 фунт/кв. дюйм (3,515 кг/см2) при 70oC в течение 48 часов. Эту смесь охлаждали, концентрировали, и полученное твердое вещество растирали с эфиром. Выход: прибл. 60%, ВРЖХ= 3,52 минуты (95%), PB-MC 372/374 (MH+, 100%). 1H-ЯМР (частичный, ДМСО-d6) : 11,75 (с, 1H), 8,04 (д, 1H), 7,70 (д, 1H, J = 2 Гц), 7,5 – 7,1 (м, 9H), 9,90 (шир., 1H), 4,52 (шир., 1H), 3,93 (шир., 1H), 2,95 (дд, 1H), 2,88 (дд, 1H).
Анализ для C19H18ClN3O3 + 0,5 H2O:Вычислено: C 59,92; H 5,03; N 11,03. Найдено: C 59,66; H 5,10; N 11,40. Пример 32 5,6-Дихлоро-1H-индол-2-карбоновой кислоты [(1S)-[(R)-гидрокси-(метокси-метил-карбамоил)-метил]-2-фенил-этил]-амид N, O-Диметилгидроксиламина гидрохлорид (0,24 мМ) и (3S)-[(5,6-дихлоро-1H-индол-2-карбонил)-амино] -(2R)-гидрокси-4-фенилмасляную кислоту (0,22 мМ) подвергали реакции взаимодействия в соответствии с методикой A (за исключением того, что время проведения реакции составляло 96 часов, а промывку осуществляли сначала кислотой, а затем основанием), и полученный продукт очищали с помощью хроматографии на двуокиси кремния (элюент: смесь 20-40% этилацетата/гексана). Выход: 72%; ВРЖХ (60/40), 7,2 мин (99%); т.пл. 210-211,5oC, PB-MC: 450/452 (M+, 100%). 1H-ЯМР (CDCl3) : 10,41 (шир., 1H), 7,73 (с, 1H), 7,68 (с, 1H), 7,4 – 7,2 (м, 6H), 6,78 (д, 1H, J = прибл. 1 Гц), 6,58 (д, 1H, J = 10 Гц), 5,03 (м, 1H), 4,34 (д, 1H, J = 5 Гц), 3,85 (д, 1H, J = 5 Гц), 3,37 (с, 3H), 3,2 – 3,0 (м, 2H), 3,10 (с, 3H).
Анализ для C21H21Cl2N3O4:Вычислено: C 56,01; H 4,70; N 9,33. Найдено: C 55,61; H 4,68; N 9,22. Пример 33 5-Хлоро-1H-индол-2-карбоновой кислоты [(1S)-((R)-гидрокси-диметилкарбамоил-метил)-2-фенил-этил]-амид Диметиламина гидрохлорид (262 мг, 3,22 мМ) и (3S)-[(5-хлоро-1H-индол-2-карбонил)-амино] -(2R)-гидрокси-4-фенилмасляную кислоту (1,0 г, 2,68 мМ) подвергали реакции взаимодействия в ДМФ (4 мл) с использованием триэтиламина (530 мг, 3,22 мМ), гидрата 2-гидроксибензотриазола (612 мг, 4 мМ) и гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида при температуре 25oC в течение 18 часов. Полученную смесь разводили хлороформом (80 мл) и этилацетатом (10 мл), а затем промывали 2N гидроксидом натрия и 2N соляной кислотой, осушали и концентрировали с получением 1,2 г вещества в виде бесцветной пены. Это вещество растворяли в этилацетате, и полученный раствор дважды промывали 2N гидроксидом натрия, а затем сушили и концентрировали с получением 1,02 г бесцветного твердого вещества. Полученное вещество вводили в 10 мл холодного эфира и фильтровали путем промывания 5 мл холодного эфира, а после сушки получали желаемый продукт в виде бесцветного твердого вещества. Выход: 715 мг, 67%; т.пл. 190-192oC; ВРЖХ (60/40), 4,53 мин (100%); FАВ-МС 400/402 (MH+, 80%), 178 (100%). 1H-ЯМР (CDCl3) : 9,40 (с, 1H), 7,55 (с, 1H), 7,4 – 7,1 (м, 7H), 6,86 (д, 1H, J = 2 Гц), 6,62 (д, 1H, J = 9,6 Гц), 4,65 (м, 1H), 4,40 (м, 2H), 3,10 (м, 2H), 2,88 (с, 3H), 2,72 (с, 3H).
Анализ для C21H22ClN3O3:Вычислено: C 63,08; H 5,55; N 10,51. Найдено: C 63,03; H 5,68; N 10,25. Пример 34 5-Хлоро-1H-индол-2-карбоновой кислоты [(1S)-(R)-гидрокси-(гидрокси-метил-карбамоил)-метил]-2-фенил-этил]-амид N-Метилгидроксиламина гидрохлороид (167 мг, 2,0 мМ) и (3S)-[(5-хлоро-1H-индол-2-карбонил)-амино] -(2R)-гидрокси-4-фенилмасляную кислоту (373 мг, 10 мМ) подвергали реакции взаимодействия в соответствии с методикой A (за исключением того, что в качестве растворителя использовали диметилформамид, а промывкой основанием пренебрегали), и неочищенный продукт очищали хроматографией на двуокиси кремния (элюент: смесь 0,5-4% этанола в дихлорметане, содержащем 0,5%-ную уксусную кислоту). Очищенный продукт растирали со смесью гексана/эфира. Выход: 13%; ВРЖХ (60/40), 4,26 мин (97%); т.пл. 182-184,5oC; TSP-MC 402/404 (MH+, 100%). 1H-ЯМР (ДМСО-d6, частичный) : 11,67 (шир., 1H), 9,89 (шир., 1H), 8,08 (д, 1H, J = 10 Гц), 7,71 (д, 1H, J = 1,9 Гц), 7,39 (д, 1H, J = 8,8 Гц), 7,35 – 7,1 (м, 7H), 4,73 (м, 2H), 4,51 (м, 1H), 3,05 (м, 1H), 3,05 (с, 3H), 2,93 (м, 2H).
Пример 355-Хлоро-1H-индол-2-карбоновой кислоты [(1S)-((R)-гидроксиметоксикарбамоил-метил)-2-фенил-этил]-амид N-Метоксиламина гидрохлорид (0,77 мМ) и (3S)-[(5-хлоро-1H-индол-2-карбонил)-амино] -(2R)-гидрокси-4-фенил-масляную кислоту (0,70 мМ) подвергали реакции взаимодействия в соответствии с Методикой A (с использованием в качестве растворителя диметилформамида), и полученный продукт очищали с помощью хроматографии на двуокиси кремния (элюент: смесь 1 – 10% этанола в дихлорметане), а затем растирали с эфиром/гексаном. Выход: 72%; ВРЖХ (60/40), 3,35 мин (> 99%); т.пл. 215-216,5oC (с разлож. ); FAB-MC 402/404 (MH+, 100%). Анализ для C20H20ClN3O4 + 0,7 H2O: Вычислено: C 57,96; H 5,20; N 10,14. Найдено: C 57,90; H 5,15; N 10,10. Пример 36 5-Хлоро-1H-индол-2-карбоновой кислоты [(1S)-{(R)-гидрокси-(метокси-метил-карбамоил-метил]-2-фенил-этил}-амид N, O-Диметилгидроксиламина гидрохлорид (7,4 мМ) и (3S)-[(5-хлоро-1H-индол-2-карбонил)-амино] -(2R)-гидрокси-4-фенил-масляную кислоту (6,7 мМ) подвергали реакции взаимодействия в соответствии с Методикой A (с использованием диметилформамида в качестве растворителя), и получали неочищенный продукт, который хроматографировали на двуокиси кремния (элюент: 40-50% этилацетат/гексан) с получением неочищенного продукта, который перемешивали в течение ночи со смесью эфира/гексана (1:1), и получали твердое вещество, которое собирали фильтрацией и сушили. Выход: 70%, ВРЖХ (60/40), 5,36 минут (99%); т.пл. 189-190oC; 1H-ЯМР (CDCl3) : 9,52 (шир., 1H), 7,56 (д, 1H, J = 2,0 Гц), 7,4 – 7,3 (м, 5H), 7,38 (м, 1H), 7,18 (дд, 1H, J = 2,0 и 8,8 Гц), 6,76 (д, 1H, J = 1,4 Гц), 6,53 (д, 1H, J = 9 Гц), 4,94 (м, 1H), 4,31 (д, 1H, J = 5,2 Гц, коллапсируют в синглет с D2O), 3,86 (д, 1H, J = 5,6 Гц, обмен с D2O), 3,35 (с, 3H), 3,13 (с, 3H), 3,13 – 2,98 (м, 2H); PB-MC 593/595 (MH+, 65%), 200 (100%).
Ниже приводится анализ вещества, перекристаллизованного из этилацетата/гексана (1:3) (усадка при 150oC, т.пл. 189-190oC); Анализ для C21H22ClN3O4: Вычислено: C 60,65; H 5,33; N 10,10; Анализ: C 60,52; H 5,34; N 10,32.
Пример 375-Хлоро-1H-индол-2-карбоновой кислоты (2S)-[(5-хлоро-1H-индол-2-карбонил)-амино]-(1R)-(метокси-метил-карбамоил)- 3-фенил-пропиловый эфир (3S)-Амино-(2R)-гидркоси-N-метокси-N-метил-4-фенил-бутирамида гидрохлорид (4,2 мМ) и 5-хлоро-1H-индол-2-карбоновую кислоту (4,2 мМ) подвергали реакции взаимодействия в соответствии с Методикой A. Полученную смесь очищали с помощью хроматографии на двуокиси кремния (элюент: 33-50% этилацетат/гексан, и получали целевое соединение (100 мг) и более полярное главное соединение, а именно, 5-хлоро-1H-индол-2-карбоновой кислоты {(1S)-[(R)-гидрокси-(метокоси-метил-карбамоил)-метил] -2-фенил-этил} -амид (970 мг) и смесь двух соединений (159 мг наиболее полярного продукта). Данные для целевого соединения: РВ-МС 593/595 (MH+, 60%), 400 (100%). 1H-ЯМР (CDCl3) : 9,62 (шир., 2H), 7,69 (д, 1H, J = 2 Гц), 7,56 (д, 1H, J = 2 Гц), 7,4 – 7,2 (м, 10H), 7,04 (д, 1H, J = 8,8 Гц), 6,91 (д, 1H, J = 1-2 Гц), 5,50 (д, 1H, J = 2 Гц), 5,09 (м, 1H), 3,47 (с, 3H), 3,26 (дд, 1H, J = 6,13 Гц), 3,14 (с, 3H), 2,99 (дд, 1H, J = 10,13 Гц).
Пример 385-Хлоро-1H-индол-2-карбоновой кислоты (1S)-бензил-(2R)-гидрокси-3-оксо-3-пирролидин-1-ил-пропил)-амид Пирролидин (0,5 мМ) и (3S)-[(5-хлоро-1H-индол-2-карбонил)-амино]-(2R)-гидрокси-4-фенил-масляную кислоту (0,5 мМ) подвергали реакции взаимодействия в соответствии с Методикой A (с использованием диметилформамида в качестве растворителя), и получали неочищенный продукт, который растирали с эфиром. Выход: 65%; ВРЖХ (60/40), 6,3 мин (98%); PB-MC 426/428 (MH+, 100%); Анализ для C23H24ClN3O3 + 0,25 H2O: Вычислено: C 64,18; H 5,74; N 9,76. Найдено: C 64,02; H 5,71; N 9,61. Пример 39 5-Хлоро-1H-индол-2-карбоновой кислоты [(1S)-бензил-(2R)-гидрокси-3-(3-гидрокси-азетидин-1-ил)-3-оксо-пропил]-амид 3-Гидроксиазетидина гидрохлорид (0,56 мМ) и (3S)-[(5- хлоро-1H-индол-2-карбонил)-амино]-(2R)-гидрокси-4-фенилмасляную кислоту (0,5 мМ) подвергали реакции взаимодействия в соответствии с Методикой A (за исключением того, что реакционная температура составляла 0-25oC, а в качестве растворителя использовали смесь дихлорметана/диметилформамида (1:1)), и полученный неочищенный продукт очищали с помощью хроматографии на двуокиси кремния, с использованием в качестве элюента 2-10% этанола/дихлорметана. Выход: 69%, ВРЖХ (60/40), 3,38 минут (96%); РВ-МС 428/430 (MH+, 100%). Анализ для C22H22ClN3O4 + 0,125 H2O: Вычислено: C 61,43; H 5,21; N 9,77. Найдено: C 61,09; H 5,57; N 9,68. Пример 40 5-Хлоро-1H-индол-2-карбоновой кислоты ((1S)-бензил-(2R)-гидрокси-3-изоксазолидин-2-ил-3-оксо-пропил)-амид Изоксазолидина гидрохлорид (Cupps T.L, et al., J. Org. Chem, 1985, 50, 3972-3979, 0,83 мМ) и (3S)-[(5-хлоро-1H-индол-2-карбонил)-амино]-(2R)-гидрокси-4-фенилмасляную кислоту (0,79 мМ) подвергали реакции взаимодействия в соответствии с Методикой A, и продукт очищали с помощью хроматографии на силикагеле, с использованием в качестве элюента 50% и 75% этилацетата/гексана. Выход = 75%; ВРЖХ (60/40), 4,94 минуты (95%); TSP-MC 428/430 (MH+, 100%). 1H-ЯМР (ДМСО-d6) : 11,70 (с, 1H), 8,17 (д, 1H, J = 9,3 Гц), 7,71 (с, 1H, J = 2 Гц), 7,38 (д, 1H, J = 8,7 Гц), 7,27 (м, 4H), 7,15 (м, 3H), 5,02 (д, 1H), 4,61 (м, 1H), 4,42 (дд, 1H), 4,10 (м, 1H), 3,93 (м, 1H), 3,55 (м, 1H), 2,95 (м, 2H), 2,26 (м, 2H).
Анализ для C22H22ClN3O4:Вычислено: C 61,75; H 5,18; N 9,82; Найдено: C 61,59; H 5,35; N 9,44. Пример 41 5-Хлоро-1H-индол-2-карбоновой кислоты [(1S)-((R)-диэтилкарбамоил-гидрокси-метил)-2-фенил-этил]-амид Диэтиламин (0,45 мМ) и (3S)-[(5-хлоро-1H-индол-2-карбонил)-амино] – (2R)-гидрокси-4-фенилмасляную кислоту (0,4 мМ) подвергали реакции взаимодействия в соответствии с методикой A, и полученный продукт очищали с помощью хроматографии на двуокиси кремния (элюент: 10-25% этилацетата/гексана). Выход = 35%; ВРЖХ (60/40), 7,06 минут (96%); т.пл. 218-222oC, PB-МС 428/430 (MH+, 100%). 1H-ЯМР (CDCl3) : 9,14 (с, 1H), 7,61 (с, 1H), 7,44-7,15 (м, 7H), 6,81 (д, 1,3H), 6,55 (д, 1H, J = 10 Гц), 4,55 (м, 1H), 4,37 (д, 1H, J = 5,2 Гц), 4,29 (д, 1H, J = 5,3 Гц), 3,43 (м, 1H), 3,2-3,0 (м, 3H), 2,88 (кв., 2H, J = 7 Гц), 1,05 (т, 3H, J = 7,1 Гц), 0,98 (т, 3H, J = 7,1 Гц).
Пример 425-Хлоро-1H-индол-2-карбоновой кислоты { (1S)-[(R)-гидрокси-[(2-гидрокси-этил)-метил-карбамоил]-метил-2- фенил-этил)-амид N-(2-Гидроксиэтил)метиламина гидрохлорид (0,77 мМ) и (3S)-[(5-хлоро-1H-индол-2-карбонил)-амино]-(2R)-гидрокси-4-фенил-масляную кислоту (0,70 мМ) подвергали реакции взаимодействия в соответствии с методикой A (за исключением того, что в качестве растворителя использовали диметилформамид, а экстрагирование осуществляли кислотой, а затем основанием), и полученный продукт очищали хроматографией на двуокиси кремния (элюент: 0,5-8% этанола/дихлорметана), и растирали с эфиром/гексаном. Выход: 65%; ВРЖХ (60/40), 3,67 мин (93%); т.пл. 192,5-195oC; TSP-МС 430/432 (MH+, 100%). 1H-ЯМР (CDCl3) : 9,18 (шир., 1H), 7,60 (д, 1H, J = 2 Гц), 7,4-7,25 (м, 6H), 7,24 (дд, 1H, J = 2,9 Гц), 6,85 (д, 1H, J = 2 Гц), 6,63 (д, 1H, J = 9 Гц), 4,85 (м, 1H), 4,47 (м, 1H), 4,06 (м, 1H), 3,63 (м, 2H), 3,12 (м, 2H), 2,95 (с, 3H), 2,85 (м, 1H), 2,5 (шир., 2H).
Анализ для C22H24ClN3O4:Вычислено: C 61,46; H 5,63; N 9,77. Найдено: C 61,45; H 5,95; N 9,85. Пример 43 5-Хлоро-1H-индол-2-карбоновой кислоты ((1S)-бензил-(2R)-гидрокси-3-оксо-3-пиперидин-1-ил-пропил)-амид Пиперидина гидрохлорид (0,42 мМ) и (3S)-[(5-хлоро-1H-индол-2- карбонил)-амино] -(2R)-гидрокси-4-фенил-масляную кислоту (0,4 мМ) подвергали реакции взаимодействия в соответствии с методикой A (с использованием в качестве растворителя дихлорметана/диметилформамида, 1:1), и полученный продукт очищали с помощью хроматографии на двуокиси кремния (элюент: 20-25% этилацетата/гексана). Выход – 97%; ВРЖХ (60/40), 6,92 минуты (100%); PB-МС 440/442 (MH+, 100%). Анализ для C24H26ClN3O3: Вычислено: C 65,52; H 5,96; N 9,55. Найдено: C 65,27; H 6,12; N 9,29. Пример 44 5-Хлоро-1H-индол-2-карбоновой кислоты ((1S)-бензил-2(R)-гидрокси-3-морфолин-4-ил-3-оксо-пропил)-амид Морфолин (0,55 мМ) и (3S)-[(5-хлоро-1H-индол-2-карбонил)-амино]- (2R)-гидрокси-4-фенил-масляную кислоту (0,5 мМ) подвергали реакции взаимодействия в соответствии с методикой A (растворитель: диметилформамид), и полученный продукт очищали растиранием с эфиром. Выход: 50%; ВРЖХ (60/40), 5,37 минут (>98%); TSP-МС 442/444 (MH+, 100%); 1H-ЯМР (CDCl3) : 9,13 (шир., 1H), 7,59 (д, 1H, J = 2 Гц), 7,35-7,1 (м, 7H), 6,79 (д, 1H, J = 2 Гц), 6,51 (д, 1H, J = 9 Гц), 4,55 (м, 1H), 4,30 (м, 1H), 4,27 (м, 1H), 3,77 (м, 1H), 3,62 (м, 2H), 3,50 (м, 3H), 3,05 (м, 3H), 2,94 (м, 1H).
Анализ для C23H24ClN3O4:Вычислено: C 62,51; H 5,47; N 9,51. Найдено: C 62,11; H 5,39; N 9,19. Пример 45 5-Хлоро-1H-индол-2-карбоновой кислоты ((1S)-бензил-(2R)-гидрокси-3-[1,2] оксазинан-2-ил-3-оксо-пропил)-амид [1,2] Оксазинана гидрохлорид (0,42 мМ) и (3S)-[(5-хлоро-1H-индол-2-карбонил)-амино] -(2R)-гидрокси-4-фенил-масляную кислоту (0,4 мМ) подвергали реакции взаимодействия в соответствии с методикой A (растворитель: смесь дихлорметана/диметилформамида, 1:1), и полученный продукт очищали с помощью хроматографии на двуокиси кремния (элюент: 25% этилацетат/гексан). Выход: 76%; ВРЖХ (60/40), 6,07 минут (99%); PB-МС: 442/444 (MH+, 100%). 1H-ЯМР (CDCl3) : 9,41 (шир. с, 1H), 7,58 (д, 1H, J = 2 Гц), 7,38-7,18 (м, 7H), 6,78 (д, 1H, J = 2 Гц), 6,55 (д, 1H, J = 9 Гц), 4,89 (м, 1H), 4,58 (с, 1H), 4,00 (м, 1H), 3,67 (м, 3H), 3,10 (м, 2H), 1,9 (шир.), 1,7 (м, 4H).
Анализ для C23H24ClN3O4:Вычислено: C 62,51; H 5,47; N 9,51. Найдено: C 62,18; H 5,59; N 9,29. Пример 46 5-Хлоро-1H-индол-2-карбоновой кислоты [(1S)-бензил-(2R)-гидрокси- 3-((3S)-гидрокси-пирролидин-1-ил)-3-оксо-пропил]-амид (R)-3-Гидроксипирролидин (0,58 мМ) и (3S)-[(5-хлоро-1H-индол-2-карбонил)-амино] -(2R)-гидрокси-4-фенилмасляную кислоту (0,56 мМ) подвергали реакции взаимодействия в соответствии с методикой A, и полученный продукт дважды очищали с помощью хроматографии на двуокиси кремния (элюент: 25-100% этилацетата/гексана). Выход = 9%; ВРЖХ (60/40), 3,87 минут (96%); PB-МС 442/444 (MH+, 100%). Пример 47 5-Хлоро-1H-индол-2-карбоновой кислоты [(1S)-((R)-трет-бутоксикарбамоил-гидрокси-метил)-2-фенил-этил]-амид 5-Хлоро-O-трет-бутилгидроксиламина гидрохлорид (2,0 мМ) и (3S)-[(5-хлоро-1H-индол-2-карбонил)-амино] -(2R)-гидрокси-4-фенил-масляную кислоту (1,0 мМ) подвергали реакции взаимодействия в соответствии с методикой A (за исключением того, что в качестве растворителя использовали диметилформамид, а промыванием кислотой пренебрегали), и полученный продукт очищали хроматографией на двуокиси кремния (элюент: 30-50% этилацетата/гексана). Выход = 77%; ВРЖХ (60/40) 4,96 минут (98%); FAB-МС 444 (MH+, 90%), 511 (100%). 1H-ЯМР (CDCl3) : 9,38 (шир., 1H), 9,18 (шир., 1H), 7,85 (шир, H), 7,53 (с, 1H), 7,3-7,0 (м, 7H), 6,87 (с, 1H), 4,40 (д, 1H, J = 4 Гц), 4,30 (м, 1H), 3,20 (м, 2H), 1,12 (с, 9H).
Пример 485-Хлоро-1H-индол-2-карбоновой кислоты ((1S)-бензил-(2R)-гидрокси-3-оксо-3-тиазолидин-3-ил-пропил)-амид Тиазолидин (0,70 мМ) и (3S)-[(5-хлоро-1H-индол-2-карбонил)-амино] -(2R)-гидрокси-4-фенил-масляную кислоту (0,67 мМ) подвергали реакции взаимодействия в соответствии с методикой A (за исключением того, что в качестве растворителя использовали смесь дихлорметана/диметилформамида, 1:1), и получали продукт, который использовали без дополнительной очистки. Выход: 93%; ВРЖХ (60/40) 5,78 минут (96%); PB-МС 444/446 (MH+, 100%). Анализ для C22H22ClN3O3: Вычислено: C 59,52; H 5,00; N 9,47. Найдено: C 59,29; H 5,22; N 9,22. Пример 49 5-Бромо-1H-индол-2-карбоновой кислоты [(1S)-((R)-диметилкарбамоил-гидрокси-метил)-2-фенил-этил]-амид Диметиламина гидрохлорид (0,39 мМ) и (3S)-[(5-бромо-1H-индол-2-карбонил)-амино] -(2R)-гидрокси-4-фенил-масляную кислоту (0,32 мМ) подвергали реакции взаимодействия в соответствии с методикой A (реакционная температура = 0-25oC). Неочищенный продукт (159 мг) перемешивали с 200 мг полистирола-DMAP (Aldrich Chemical Co., Milwaukee, WI) в дихлорметане при 25oC в течение одного часа, а затем фильтровали, и фильтрат концентрировали. Выход = 68%; ВРЖХ (60/40) 5,4 минут (>98%); т.пл. 171-176oC; TSP-МС 444/446 (MH+, 85%); Анализ для C21H22N3O3Br: Вычислено: C 56,77; H 4,99; N 9,46. Найдено: C 56,42; H 5,33; N 9,08. Пример 50 5-Хлоро-1H-индол-2-карбоновой кислоты [(1S)-[(R)-гидрокси-(пиридин-3-илкарбамоил)-метил]-2-фенил-этил]-амид 3-Аминопиридин (0,7 мМ) и (3S)-[(5-хлоро-1H-индол-2-карбонилл)-амино]-(2R)-гидрокси-4-фенил-масляную кислоту (0,70 мМ) подвергали реакции взаимодействия в соответствии с методикой A (с использованием в качестве растворителя диметилформамида), и полученный продукт очищали с помощью хроматографии на двуокиси кремния, элюируя 0,5-8% этанола в дихлорметане, содержащем 0,5% гидроксид аммония, а затем растирали с эфиром. Выход = 45%; ВРЖХ (60/40) 3,08 минут (>99%); TSP-МС 449/451 (MH+, 100%); Анализ для C24H21ClN4O3 + 0,3H2O: Вычислено: C 63,45; H 4,79; N 12,33. Найдено: C 63,35; H 5,03; N 12,37. Пример 51 5-Хлоро-1H-индол-2-карбоновой кислоты {(1S)-[(R)-гидрокси-(2,2,2-трифторо-этилкарбамоил)-метил]-2-фенил}-этил]-амид 2,2,2-Трифтороэтиламин (0,28 мМ) и (3S)-[(5-хлоро-1H-индол-2-карбонил)-амино] -(2R)-гидрокси-4-фенил-масляную кислоту (0,28 мМ) подвергали реакции взаимодействия в соответствии с методикой A (с использованием диметилформамида в качестве растворителя), и полученный продукт очищали растиранием с эфиром; т. пл. 228-229, 5oC; выход = 81%; PB-МС 454/456 (100%, MH+), 471/473 (MH+, NH3, 80%). Анализ для C21H19ClF3N3O3: Вычислено: C 55,58; H 4,22; N 9,26. Найдено: C 55,29; H 4,25; N 9,04. Пример 52 (S)-5-Хлоро-1H-индол-2-карбоновой кислоты [1-(метокси-метил-карбамоанкарбонил)-2-фенил-этил]-амид К раствору безводного диметилсульфоксида (4 мл) и безводного толуола (4 мл) при 0oC последовательно добавляли 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (DEC, 790 мг, 4,12 мМ), дихлоруксусную кислоту (136 мг, 1,06 мМ) и 5-хлоро-1H-индол-2-карбоновой кислоты [(1S)-[(R)-гидрокси-(метокси-метил-карбамоил)-метил]-2-фенил-этил]-амид (287 мг, 0,69 мМ). После отстаивания в течение 18 часов при температуре 25oC, реакционную смесь разводили этилацетатом, и полученный раствор промывали 2N соляной кислотой и насыщенным водным раствором гидрокарбоната натрия. Органический слой сушили и концентрировали. Полученное вещество в виде пены перекристаллизовывали из эфира. Выход = 100 мг, 35%; ВРЖХ (60/40) 10,72 минуты (87%); исходный продукт элюировали в течение 6,68 минут, выход = >0,5%; РВ-МС 414/416 (МН+, 70%), 384/386 (100%). 1H-ЯМР (CDCl3, содержащий 10-20% ДМСО-d6) : 9,90 (шир., 1H), 7,54 (д, 1H, J=1,7 Гц), 7,3-7,1 (м, прибл. 7H), 7,04 (м, 1H), 6,77 (с, 1H), 5,40 (м, 1H), 3,58 (с, 3H), 3,2 (м, 2H), 3,08 (с, 3H).
Пример 535-Хлоро-1H-индол-2-карбоновой кислоты [(1S)-бензил-(2R)-гидрокси-3-(4-гидрокси-пиперидин-1-ил)-3-оксо-пропил]-амид 4-Гидроксипиперидина гидрохлорид (0,51 мМ) и (3S)-[(5-хлоро-1H-индол-2-карбонил)-амино] -(2R)-гидрокси-4- фенилмасляную кислоту (0,48 мМ) подвергали реакции взаимодействия в соответствии с Методикой A (за исключением того, что реакционная температура составляла 0-25oC), и полученный продукт очищали растиранием с эфиром, а затем растиранием в кипящем этилацетате, после чего, продукт хроматографировали на двуокиси кремния (элюент: 50-100% этилацетат/гексан). Выход: 57%; ВРЖХ (60/40) 3,92 мин (96%); т.пл. 230-232oC; TSP-MC 456/458 (МН+,100%). 1H-ЯМР (ДМСО-d6) : 11,65 (шир., 0,5H), 11,60 (шир., 0,5H), 8,24 (м, 1H), 7,70 (д, 1H, J=2Гц), 7,38 (д, 0,5H, J=9 Гц), 7,37 (д, 0,5 H, J=9 Гц), 7,3-7,1 (м, 7H), 4,0-4,7 (м, 2H), 4,5 (м, 2H), 3,8-3,65 (м, 3H), 3,2 (м, 1H), 3,1 (дд, 1H), 3,0 (дд, 1H), 1,95 (м, 0,5H), 1,7-1,65 (м, 2H), 1,4-1,25 (м, 1, 5H).
Пример 545-Хлоро-1H-индол-2-карбоновой кислоты [(1S)-бензил(2R)-гидрокси- 3-((3R, S)-гидрокси-пиперидин-1-ил)-3-оксо-пропил]-амид 3-Гидроксипиперидин (0,56 мМ) и (3S)-[(5-хлоро-1H-индол-2-карбонил)-амино] -(2R)-гидрокси-4-фенил-масляную кислоту (0,54 мМ) подвергали реакции взаимодействия в соответствии с методикой A, и полученный продукт очищали с помощью хроматографии на двуокиси кремния (элюент: 20-40% этилацетат/гексан), а затем, растиранием со смесью эфира/гексана (1:1). Выход= 47%; ВРЖХ (60/40) 4,44 минуты (92%); РВ-МС 456/458 (МН+, 100%). Анализ для C24H26ClN3O4: Вычислено: C 63,22; H 5,75; N 9,22. Найдено: C 62,93; H 5,90; N 8,92. Пример 55 5-Хлоро-1H-индол-2-карбоновой кислоты [(1S)-бензил-(2R)-гидрокси-3-((2R)-гидроксиметил-пирролидин-1-ил)-3- оксо-пропил]-амид P-2-Пирролидинметанол (1,1 Мм) и (3S)-[(5-хлоро-1H-индол-2-карбонил)-амино] -(2R)-гидрокси-4-фенил-масляную кислоту (1,1 мМ) подвергали реакции взаимодействия в соответствии с Методикой A, и полученный продукт очищали с помощью хроматографии на двуокиси кремния, используя в качестве элюента 1-8% этанол/дихлорметан, а затем, с помощью хроматографии на двуокиси кремния, используя в качестве элюента 50% этилацетат/гексан. Выход = 9%; ВРЖХ (60/40) 5,17 минут (84%); т.пл. 236-239oC; TSP-MC 456/458 (МН+, 100%). Анализ для C24H26ClN3O4: Вычислено: C 63,22; H 5,75; N 9,22. Найдено C 63,23; H 6,11; N 8,52. Пример 56 5-Хлоро-1H-индол-2-карбоновой кислоты ((1S)-{ (R)-[(2-диметиламиноэтил)-метил-карбамоил]-гидрокси-метил}-2-фенил- этил)-амид N-(2-Диметиламиноэтил)метиламин (0,77 мМ) и (3S)-[(5-хлоро-1H- индол-2-карбонил)-амино] -(2R)-гидрокси-4-фенил-масляную кислоту (0,70 мМ) подвергали реакции взаимодействия в соответствии с методикой A (растворитель: диметилформамид), и полученный продукт очищали с помощью хроматографии на двуокиси кремния ( элюент: 1-8% этанол в дихлорметане, содержащем 0,5% NH4OH), а затем, путем растирания с эфиром/гексаном. Выход =87%; ВРЖХ (60/40) 2,89 мин (96%); ТSР-МС 457/549 (МН+, 100%). Анализ для C24H29ClN4O3 + 0,2H2O: Вычислено: C 62,59; H 6,43; N 12,16. Найдено: C 62,85; H 6,82; N 12,06. Пример 57 5-Хлоро-1H-индол-2-карбоновой кислоты [(1S)-бензил-3-((3R, 4R)-дигидрокси-пирролидин-1-ил)-2-гидрокси-3-оксо-пропил)-амид (3R,4R) -3,4-Дигидроксипирролидин (полученный на 2S,3S -(-)-винной кислоты (синтетического изомера) с помощью методики, описанной в Патенте США N 4634775 (1,0 мМ), и (3S)-[(5-хлоро-1H-индол-2-карбонил)-амино]-(2R)-гидрокси-4-фенил- масляную кислоту (1,0 мМ) подвергали реакции взаимодействия в соответствии с методикой A (растворитель: диметилформамид), и продукт очищали с помощью хроматографии на двуокиси кремния (элюент: этилацетат), а затем, растиранием с эфиром. Выход=72%; ВРЖХ (60/40) 3,21 мин (97%); TSP-MC 458/460 (МН+, 100%). Анализ для C23H24ClN3O5: Вычислено: C 60,33; H 5,28; N 9,18. Найдено: C 60,09; H 5,21; N 9,08. Пример 58 5-Хлоро-1H-индол-2-карбоновой кислоты [(1S)-бензил-3-((3S, 4S)-дигидрокси-пирролидин-1-ил)-(2R)-гидрокси-3-оксо-пропил]-амид (3S, 4S)-Дигидроксипирролидин, полученный из 2R,3R -(+)-винной кислоты (Патент США N 4634775; 1,0 мМ), и (3S)-[(5-хлоро-1H-индол-2-карбонил)амино] -(2R)-гидрокси-фенил-масляную кислоту (1,0 мМ) подвергали реакции взаимодействия в соответствии с Методикой A ( с использованием в качестве растворителя диметилформамида), и полученный продукт очищали хроматографией на двуокиси кремния (элюент: этилацетат), а затем, растиранием с эфиром. Выход-60%; ВРЖХ (60/40) 3,02 мин (98%); ТSР-МС 458/460 (МН+, 100%). Анализ для C23H24ClN3O5: Вычислено: C 59,16; H 5,40; N 9,00. Найдено: C 59,44; H 5,29; N 8,95. 1H-ЯМР (ДМСО-d6) : 11,7 (шир.,1H), 8,18 (д, 1H, J=9 Гц), 7,70 (д, 1H, J= 2Гц), 7,38 (д,1H, J=8,6 Гц), 7,26 (м,4H), 7,15 (м,3H), 5,18 (д,1H, J=4,0 Гц, обмен), 5,11 (д,1H), 5,08 (д, 1H), 4,47 (м, 1H), 4,27 (дд, 1H, J=5,9 Гц, коллапсируют в дублет с D2O), 3,95 (м, 1H), 3,89 (м, 1H), 3,64 (дд, 1H, J= 4,9 Гц), 3,34 (м, 3H), 2,92 (м, 2H).
Пример 595-Хлоро-1H-индол-2-карбоновой кислоты [(1S)-бензол-3-((3R, 4S)-дигидрокси-пирролидин-1-ил)-(2P)-гидрокси-3-оксо-пропил]-амид (3R, 4S)-Дигидроксипирролидина гидрохлорид (цис- или мезо-изомер, 0,86 мМ) и (3S)-[(5-хлоро-1H-индол-2-карбонил)-амино] -(2R)-гидрокси-4-фенил-масляную кислоту (0,82 мМ) подвергали реакции взаимодействия в соответствии с методикой A (с использованием в качестве растворителя диметилформамида), и полученный продукт очищали с помощью хроматографии на двуокиси кремния (элюент: 1-10% этанол в дихлорметане). Выход: 39%; ВРЖХ (60/40) 2,92 мин (96%); РВ-МС 458/460 (МН+, 100%). Анализ для C23H24ClN3O5 + 0,75H2O: Вычислено: C 58,60; H 5,45; N 8,91. Найдено: C 59,22; H 5,52; N 8,59. Пример 59A Цис-3,4-дигидроксипирролидина гидрохлорид (цис- или мезо-изомер) Цис-3,4-дигидроксипирролидин-2,5-дигидро-пиррол-1-карбоновой кислоты трет-бутиловый эфир (1,99 г, 9,8 мМ) растворяли в смеси 4 М соляной кислоте и диоксана при температуре 5oC, и полученную суспензию перемешивали в течение 1 часа при температуре 25oC. Эту смесь концентрировали, а остаток растирали с эфиром, в результате чего получали целевой продукт в виде светло-пурпурного порошка (1,30 г, 95%). Пример 59B Цис-3,4-дигидрокси-пирролидин-1-карбоновой кислоты трет-бутиловый сложный эфир Раствор неочищенного трет-бутилового эфира 2,5-диидропиррол-1-карбоновой кислоты последовательно обрабатывали тетраоксидом осмия (2,5% в т-бутаноле, 6 мл) и N-метилморфолин -N-оксидом при температуре 25oC. Через 48 часов к смеси добавляли водный раствор 10% тиосульфата натрия, и эту смесь перемешивали в течение 30 минут, частично концентрировали для удаления тетрагидрофурана, а затем, полученную водную смесь дважды экстрагировали эфиром. Эфирные экстракты промывали 10% тиосульфатом натрия и 0,1 М соляной кислотой, а затем осушали и концентрировали. Полученное темно-оранжевое маслообразное вещество хроматографировали на двуокиси кремния (элюент: 1%, 2%, 4%, 8% и 10% этанол-дихлорметан), в результате чего получали целевое соединение в виде сиропа янтарного цвета (4,09 г). Пример 59C 2,5-Дигидро-пиррол-1-карбоновой кислоты трет-бутиловый эфир Ди-т-бутилдикарбонат (83 г, 380 мМ) добавляли к раствору 3-пирролидина (содержащего 35%-ный пирроллидин; 25 г, 362 мМ) в тетрагидрофуране (500 мл) при температуре 0oC. Полученную смесь перемешивали в течение 1 часа при температуре 25oC, а затем концентрировали, в результате чего получали 76,2 г желтого маслообразного вещества, которое использовали без дополнительной очистки. Пример 60 5-Хлоро-1H-индол-2-карбоновой кислоты ((1S)-бензил-(2R)-гидрокси-3-оксо-3-тиоморфолин-4-ил-пропил)-амид Тиоморфолин (0,52 мМ) и (3S)-[3S)-[(5-хлоро-1H-индол-2-карбонил)-амино] -(2R)-гидрокси-4-фенил- масляную кислоту (0,49 мМ) подвергали реакции взаимодействия в соответствии с методикой A (за исключением того, что реакционная температура составляла 0-25oC), и полученный продукт очищали с помощью хроматографии на двуокиси кремния (элюент: смесь этилацетата/гексана). Выход = 75%, ВРЖХ (60/40) 7,12 минут (97%); PB-MC 458/460 (MH+, 100%). 1H-ЯМР (CDCl3, частичный) : 9,15 (шир., 1H), 7,60 (д, 1H, J = 2 Гц), 7,4 – 7,2 (м, 7H), 6,80 (д, 1H, J = 2 Гц), 6,52 (д, 1H, J = 9 Гц), 4,55 (м, 1H), 4,29 (с, 1H), 4,10 (м, 1H), 3,48 (м, 1H), 3,30 (м, 1H), 3,2 – 2,85 (м, 4H), 2,62 (м, 1H), 2,5 (м, 1H), 2,4 (м, 1H).
Пример 615-Хлоро-1H-индол-2-карбоновой кислоты {(1S)-[(R)-гидрокси-(метил-пиридин-1-ил-карбамоил)метил]-2-фенил-этил}-амид 2-Метиламинопиридин (3,4 мМ) и (3S)-[(5-хлоро-1H-индол-2-карбонил)-амино] -(2R)-гидрокси-4-фенил-масляную кислоту (4,4 мМ) подвергали реакции взаимодействия в соответствии с методикой A (за исключением того, что в качестве растворителя использовали диметилформамид, вместо 1-гидрокси-7-азабензотриазола использовали 1-гидроксибензотриазол, кроме протекания реакции составляло 18 часов, а промывку кислотой не проводили), и продукт очищали с помощью хроматографии на двуокиси кремния (элюент: 0,5-4% этанол в дихлорметане), а затем, путем четырехкратного растирания с эфиром. Выход = 5%; ВРЖХ (60/40) 5,57 мин (95%): TSP-MC 463/464 (MH+, 100%). 1H-ЯМР (ДМСО-d6) : 11,73 (шир., 1H), 8,24 (м, 1H), 8,18 (д, 1H, J = 9 Гц), 7,78 (дт., 1H, J = 2,9 Гц), 7,72 (д, 1H, J = 2 Гц), 7,43 (с, 1H), 7,41 (с, 1H), 7,28 (м, 1H), 7,25 – 7,1 (м, 5H), 7,02 (м, 2H), 5,05 (д, 1H, J = 9 Гц), 4,60 (м, 1H), 4,35 (м, 1H), 3,22 (с, 3H), 2,70 (м, 2H).
Анализ для C25H23ClN4O3 + 1,3H2O;Вычислено: C 61,74; H 5,31; N 11,52. Найдено: C 61,84; H 5,00; N 11,52. Пример 62 5-Хлоро-1H-индол-2-карбоновой кислоты [(1S)-бензил-3-(4-формилпиперазин-1-ил)-(2R)-гидрокси-3-оксо-пропил]-амид 3-Формилпиперазин (0,77 мМ) и (3S)-[(5-хлоро-1H-индол-2-карбонил)-амино] -(2R)-гидрокси-4-фенил-масляную кислоту (0,70 мМ) подвергали реакции взаимодействия в соответствии с методикой A (за исключением того, что в качестве растворителя использовали диметилформамид, а промывку осуществляли кислотой, а затем основанием), и полученный продукт очищали с помощью хроматографии на двуокиси кремния (элюент: 0,5-4% этанол в дихлорметане), а затем растирали с эфиром/гексаном. Выход = 78%; ВРЖХ (60/40) 3,45 минут (96%), PB-MC 469/471 (MH+, 100%). Анализ для C24H25ClN4O4 + 0,3 H2O: Вычислено: C 60,77; H 5,44; N 11,81. Найдено: C 60,65; H 5,70; N 11,85. Пример 63 5-Хлоро-1H-индол-2-карбоновой кислоты [(1S)-бензил-(2R)-гидрокси-3-(4-гидроксиметил-пиперидин-1-ил)-3-оксо-пропил] -амид 4-(Гидроксиметил)пиперидин (1,5 мМ) (J. Med. Chem, 1991, 34, 1073) и (3S)-[(5-хлоро-1H-индол-2-карбонил)-амино] -(2R)-гидрокси-4-фенил-масляную кислоту (1,4 мМ) подвергали реакции взаимодействия в соответствии с Методикой A, и полученный продукт очищали с помощью хроматографии на двуокиси кремния (элюент: 50-100% этилацетат/гексан). Выход = 70%; ВРЖХ (60/40) 4,09 мин (97%); TSP-MC 470/472 (MH+, 100%). Анализ для C25H28ClN3O4 + 0,25H2O: Вычислено: C 63,29; H 6,05; N 8,86. Найдено: C 63,39; H 6,00; N 8,63. Пример 64 5-Хлоро-1H-индол-2-карбоновой кислоты ((1S)-[(R)-гидрокси-[метил(2-пиридин-2-ил-этил)-карбамоил]-метил]-2-фенил- этил)-амид Метил-(2-пиридин-2-ил-этил)-амин (0,77 мМ) и (3S)-[(5-хлоро-1H-индол-2-карбонил)-амино] -(2R)-гидрокси-4-фенил-масляную кислоту (0,70 мМ) подвергали реакции взаимодействия в соответствии с Методикой A (с использованием в качестве растворителя диметилформамида), и полученный продукт очищали с помощью хроматографии (элюент: 0,5-8% этанол/дихлорметан) на двуокиси кремния, выход = 82%, ВРЖХ (60/40) 3,33 минуты (97%); TSP-MC 491/493 (MH+, 100%). 1H-ЯМР (CDCl3) : 9,84 (шир., 0,67H), 9,35 (шир., 0,3H), 8,49 (м, 1H), 7,7 – 7,5 (м, 2H), 7,4 – 7,1 (м, 9H), 6,92 (д, 0,3H, J = 8 Гц), 6,8 (м, 14H), 6,65 (д, 0,3H, J = 9 Гц), 4,62 (м, 1,5H), 4,5 (м, 0,5H), 4,34 (с, 0,7H), 4,29 (с, 0,3H), 3,82 (м, 1H), 3,48 (м, 2H), 3,05 (м, 3H), 2,86 (с, 1H), 2,70 (с, 2H).
Анализ для C27H27ClN4O3 + 0,2H2O:Вычислено: C 65,57; H 5,58; N 11,33. Найдено: C 65,56; N 5,84; N 11,36. Пример 65 1-[(3S)-[(5-Хлоро-1H-индол-2-карбонил)-амино] -(2R)-гидрокси-4-фенил- бутилил]-пиперидин-4-карбоновой кислоты этиловый эфир. Этилизонипекотат и (3S)-[(5-хлоро-1H-индол-2-карбонил)-амино]-(2R)-гидрокси-4-фенил-масляную кислоту (0,75 мМ) подвергали реакции взаимодействия в соответствии с методикой A, и полученный продукт очищали хроматографией на двуокиси кремния, используя в качестве элюента 20-40% этилацетата/гексана. Выход = 95%; ВРЖХ (60/40) 7,96 минут (95%): PM-MC 512/514 (MH+, 100%). Пример 66 1-[(3S)-[(5-Хлоро-1H-индол-2-карбонил)-амино] -(2R)-гидрокси-4-фенил- бутирил-пирролидин-2(S)-карбоновой кислоты трет-бутиловый эфир (S)-Пирролидин-2-карбоновой кислоты трет-бутиловый эфир и (3S)-[(5-хлоро-1H-индол-2-карбонил)-амино] -(2R)-гидрокси-4-фенил-масляную кислоту (2,1 мМ) подвергали реакции взаимодействия в соответствии с методикой A (время протекания реакции = 60 часов), и продукт очищали с помощью хроматографии на двуокиси кремния (элюент: 25-50% этилацетат/гексан). Выход: 74%, ВРЖХ (60/40) 8,27 минуты (99%), TSP-MC (MH+, 100%). Анализ для C28H32ClN3O5: Вычислено: C 63,93; H 6,13; N 7,99. Найдено: C 64,05; H 6,32; N 7,79. Пример 67 5-Хлоро-1H-индол-2-карбоновой кислоты [(1R)-[(S)-гидрокси-(метокси-метил-карбамоил)-метил]-2-фенил-этил]-амид 5-Хлоро-1H-индол-2-карбоновую кислоту (0,25 мМ) и (2S,3R)-3-амино-2-гидрокси-N-метокси-N-метил-4-фенилбутирамида гидрохлорид (0,25 мМ) подвергали реакции взаимодействия в соответствии с методикой A (за исключением того, что реакционная температура составляла 0-25oC, а промывку осуществляли сначала кислотой, а затем основанием). Неочищенный продукт растворяли в метаноле, содержащем 0,25 эквивалента 1 н. NaOH при 25oC в течение 2 часов, и еще один час растворяли в метаноле, содержащем 0,25 эквивалента 1N NaOH (для гидролиза менее полярного N,O-бис-5-хлоро-1H-индолкарбонилового производного), а затем, раствор концентрировали, остаток растворяли в этилацетате, и полученный раствор промывали 2N соляной кислотой и солевым раствором, после чего сушили и концентрировали. Остаток очищали с помощью хроматографии на двуокиси кремния, элюируя 30-50% этилацетатом/гексаном. Хроматографированное вещество (содержащее полярную примесь) растворяли в этилацетате, и полученный раствор дважды промывали 2N гидроксидом натрия, сушили и концентрировали. Выход = 57%; ВРЖХ (60/40) 5,36 мин (98%); т.пл. 165-167oC; PB-MC 416/418 (MH+, 100%). 1H-ЯМР (CDCl3) : 9,4 (шир., 1H), 7,58 (д, 1H, J = 2 Гц), 7,4 – 7,1 (м, 7H), 6,77 (д, 1H, J = 2 Гц), 6,51 (д, 1H, J = 10 Гц), 4,91 (м, 1H), 4,30 (д, 1H, J = 5 Гц), 3,83 (д, 1H, J = 5 Гц), 3,35 (с, 3H), 3,13 (с, 3H), 3,09 (м, 2H).
Анализ для C21H22ClN3O4 + 1,0H2O:Вычислено: C 58,13; H 5,58; N 9,68. Найдено: C 58,05; H 5,24; N 9,54. Пример 67A (2S, 3R)-3-Амино-2-гидрокси-N-метокси-N-метил-4-фенил-бутирамида гидрохлорид { 1(R)-[Гидрокси-((S)-метокси-метил-карбамоил)-метил]-2-фенил-этил} -карбаминовую кислоту (285 мг, 0,8 мМ) растворяли в холодной смеси 4N HCl и диоксана, и полученный раствор перемешивали в течение одного часа при температуре 0oC. Затем, смесь концентрировали, а остаток растирали с эфиром и сушили, в результате чего получали 207 мг (90%) твердого вещества. Пример 67B {(1S)-[Гидрокси-((R)-метокси-метил-карбамоил)-метил]-2-фенил-этил}- карбаминовая кислота (2S, 3R)-3-(т-Бутоксикарбониламино)-2-гидрокси-4-фенил-масляную кислоту (300 мг, 1,0 мМ, Sigma Chemical Co., St. Louis, MO) и N,O-диметилгидроксиламина гидрохлорид (104 мг, 1,1 мМ) подвергали реакции взаимодействия в соответствии с методикой A (за исключением того, что реакционная температура составляла 0-25oC). Выход = 88%; ВРЖХ (60/40); 4,90 мин (95%). Пример 68 5-Хлоро-1H-индол-2-карбоновой кислоты [(1R)-[гидрокси-((R)-метокси-метил-карбамоил)-метил]-2-фенил-этил]-амид N, O-Деметилгидроксиламина дигидрохлорид (0,32 мМ) и (3R)-[(5-хлоро-1H-индол-2-карбонил)-амино] -(2R)-гидрокси-4-фенил-масляную кислоту (0,3 мМ) подвергали реакции взаимодействия в соответствии с методикой A (за исключением того, что реакционная температура составляла 0-25oC, а промывку осуществляли сначала кислотой, а затем основанием), и полученный продукт очищали с помощью хроматографии на двуокиси кремния (элюент: 20-50% этилацетат/гексан). Выход: 73%; ВРЖХ (60/40) 4,86 мин (95%); PB-MC 416/418 (MH+, 100%). 1H-ЯМР (CDCl3) : 9,47 (шир., 1H), 7,58 (д, 1H, J = 1,7 Гц), 7,31 (д, 1H, J = 8,7 Гц), 7,30 – 7,10 (м, 6H), 6,78 (д, 1H, J = 10 Гц), 6,74 (с, 1H), 5,00 (м, 1H), 4,63 (м, 1H), 3,80 (шир., прибл. 1H), 3,70 (с, 3H), 3,04 (с, 3H), 2,87 (м, 2H).
Анализ для C21H22ClN3O4 + 0,1H2O:Вычислено: C 60,39; H 5,36; N 10,06. Найдено: C 60,76; H 5,74; N 9,78. Пример 69 5-Хлоро-1H-индол-2-карбоновой кислоты [(1S)-бензил-(2R)-гидрокси-3-оксо-3-(1-оксо-1-тиазолидин-3-ил)-пропил]-амид К раствору, содержащему 5-хлоро-1H-индол-2-карбоновой кислоты ((1S)-бензил-(2R)-гидрокси-3-оксо-3-тиазолидин-3-ил-пропил)-амид (80 мг, 0,18 мМ) в дихлорметане (2 мл) при температуре 25oC добавляли м-хлоропероксибензойную кислоту (62 мг 50% раствора, 0,18 мМ). После выдерживания в течение одного часа, смесь выливали в смесь насыщенного водного раствора бикарбоната натрия (12 мл), водного раствора 10% тиосульфата натрия (12 мл) и этилацетата. Водный слой отделяли и дважды экстрагировали этилацетатом. Органические слои объединяли, промывали насыщенным водным раствором бикарбоната натрия, сушили и концентрировали, в результате чего получали желтое маслообразное вещество (80 мг, 96%), ВРЖХ (60/40) 3,37 мин (97%), PB-MC: 460/462 (MH+, 100%). Примеры 70 и 71 5-Хлоро-1H-индол-2-карбоновой кислоты [(1S)-бензил-(2R)-гидрокси-3-оксо-3-(1-оксо-1-тиомофролин-4-ил)-пропил]- амид (Пример 70) и 5-Хлоро-1H-индол-2-карбоновой кислоты [(1S)-бензил-3-(1,1-диоксо-1-тиоморфолин-4-ил)-(2R)-гидрокси-3-оксо- пропил]-амид (Пример 71) К раствору, содержащему 5-хлоро-1H-индол-2-карбоновой кислоты ((1S)-бензил-(2R)-гидрокси-3-оксо-3-тиоморфолин-4-ил-пропил)-амид (60 мг, 0,13 мМ) в дихлорметане (1,5 мл) при температуре 25oC добавляли м-хлоропероксибензойную кислоту (45 мг 50% раствора, 0,13 мМ). Через один час, смесь выливали в смесь насыщенного водного раствора бикарбоната натрия (12 мл), водного раствора 10% тиосульфата натрия (12 мл) и этилацетата. Водный слой отделяли и дважды экстрагировали этилацетатом. Органические слои объединяли, промывали насыщенным водным раствором бикарбоната натрия, сушили и концентрировали с получением целевого сульфоксида (пример 70) в виде желтого твердого вещества, который хроматографировали на силикагеле, используя в качестве элюента 1% этанол/дихлорметан. Выход = 44 мг, 72%, ВРЖХ (60/40) 6,14 мин (98%); PB-MC 474/476 (MH+, 100%). Был выделен также менее полярный продукт (8 мг), который является идентичным целевому сульфону (пример 71): ВРЖХ (60/40) 6,44 мин (96%); PB-MC 490/492 (MH+, 100%). Пример 72 1-[(3S)-{ (5-Хлоро-1H-индол-2-карбонил)-амино} -(2R)-гидрокси-4- фенил-бутирил]-пиперидин-4-карбоновая кислота К раствору, содержащему этиловый эфир 1-{(3S)-[(5-хлоро-1H-индол-2-карбонил)-амино] -(2R)-гидрокси-4-фенил-бутирил} – пиперидин-4-карбоновой кислоты (111 мг, 0,22 мМ) в тетрагидрофуране (2 мл) при температуре 25oC добавляли раствор гидроксида лития (0,2 мл 1 н. раствора в воде). После отстаивания в течение 18 часов, смесь концентрировали, а остаток растирали с эфиром. Полученное твердое вещество распределяли между водой и этилацетатом, а затем доводили до pH 1 путем добавления 6N соляной кислоты. Органический слой отделяли, сушили и концентрировали, в результате чего получали 109 мг (100%) соединения в виде твердого вещества. ВРЖХ (60/40) 3,79 мин (99%); TSP-MC 484/486 (MH+, 100%). 1H-ЯМР (ДМСО-d6) : 12,25 (шир., 1H), 11,65 (шир., 1H), 8,17 (д, 0,5H, J= 9 Гц), 8,14 (д, 0,5H, J=9 Гц), 7,70 (д, 1H, J=2 Гц), 7,38 (д, 1H, J=8,8 Гц), 7,35-7,1 (м, 7H), 4,78 (м, 1H, обмен с D2O), 4,5 (м, 2H), 4,1 (м, 1H), 3,8 (м, 0,5H), 3,7 (м, 0,5H), 3,15 (м, 0,5H), 3,0 (м, 2-2,5H), 2,75 (м, 1H), 1,5 (возможный м., 1H), 1,8 (м, 2-2,5H), 1,5 (м., прибл. 1,5H).
Анализ для C25H26ClN3O5 + 0,55 H2O:Вычислено: C 60,80; H 5,53; N 8,51. Найдено: C 61,15; H 5,68; N 8,11. Пример 73 5-Хлоро-1H-индол-2-карбоновой кислоты [(1S)-((R)-гидрокси- гидроксикарбамоил-метил)-2-фенил-этил]-амид К раствору, содержащему 5-хлоро-1H-индол-4-карбоновой кислоты [(1S)-((R)-трет-бутоксикарбамоил-гидрокси-метил-2-фенил-этил-амид (256 мг, 0,58 мМ) в дихлорметане (2 мл) добавляли трифторуксусную кислоту (2 мл), и полученный раствор перемешивали в течение 18 часов при температуре 25oC. После добавления еще 2 мл трифторуксусной кислоты, смесь оставляли на 72 часа, а затем концентрировали, и остаток хроматографировали на силикагеле (элюент: 2,5%, 5% и 10% этанол/дихлорметан, содержащий 1% уксусную кислоту). Очищенный продукт растирали со смесью эфира/гексана и сушили. Выход: 70 мг, 31%, ВРЖХ (60/40) 3,11 мин (96%). Анализ для C19H18ClN3O + 1,0 OH2O: Вычислено: C 56,23; H 4,97; N 10,35. Найдено: C 56,63; H 4,94; N 9,95. Пример 74 5-Хлоро-1H-индол-2-карбоновой кислоты ((1S)-{ [(бензил- пиперидин-4-ил)-метил-карбамоил]-(R)-гидрокси-метил]-2-фенил-этил)- амид (3S)-[(5-Хлоро-1H-индол-2-карбонил)амино]-(2R)-гидрокси-4- фенилмасляную кислоту (310 мг, 0,8 мМ) и (1-бензил-пиперидин-4-ил)- метил-амина гидрохлорид (Публикация EPO N 0457686, Пример 1A; 200 мг, 0,8 мМ) подвергали реакции взаимодействия в соответствии с Методикой A (растворитель: диметилформамид). Неочищенный продукт очищали с помощью хроматографии на силикагеле (элюент: 0,5-4% этанол в дихлорметане, содержащем 0,5% гидроксид аммония), и получали целевое соединение в виде бесцветной пены. Выход = 140 мг, 30%, ВРЖХ (60/40); 4,15 мин (95%); TSP-MC 559/562 (MH+, 100%). Анализ для C32H35ClN4O3 + HCl + 1,5 H2O: Вычислено: C 61,73; H 6,31; N 9,00. Найдено: C 61,61; H 6,29; N 8,71. Пример 75 4-({ 3S)-[(5-Хлоро-1H-индол-2-карбонил)-амино] – (2R)-гидрокси-4-фенил-бутирил] -метил-амино)-пиперидин-1-карбоновой кислоты трет-бутиловый сложный эфир (3S)-[(5-Хлоро-1H-индол-2-карбонил)-амино]-(2R)-гидрокси-4- фенил-масляную кислоту (1,0 г, 2,6 мМ) и 4-метиламино- пиперидин-1-карбоновой кислоты трет-бутиловый эфир (575 мг, 2,6 мМ) подвергали реакции взаимодействия в соответствии с Методикой A (растворитель : диметилформамид). Неочищенный продукт очищали с помощью хроматографии на силикагеле, элюируя 20%, 30%, 40%, 50% и 75% этилацетатом/гексаном. Выход = 319 мг, 21%, ВРЖХ (60/40) 10,31 мин (94%); 569/571 (MH+, 100%). Пример 75A 4-Метиламино-пиперидин-1-карбоновой кислоты трет-бутиловый эфир К раствору трет-бутилового эфира 4-оксопиперидин-1-карбоновой кислоты в метаноле (400 мл) при температуре 0oC последовательно добавляли измельченные молекулярные сита 3A (5,2 г), гидрохлорид метиламина (16,96 г, 251 мМ), безводный ацетат натрия (41,21 г, 502 мМ) и 95%-ный цианоборогидрид натрия (3,99 г, 60 мМ), после чего, смесь нагревали до 25oC в течение нескольких часов. После выдерживания в течение 18 часов при температуре 25oC, реакционную смесь фильтровали через Целит , твердые остатки промывали метанолом и этилацетатом, и фильтрат концентрировали. Остаток растворяли в этилацетате, а затем, полученный раствор дважды промывали 2N гидроксидом натрия и один раз солевым раствором. После осушки и концентрирования получали целевое маслообразное вещество (12,79 г, 119%).
Пример 765-Хлоро-1H-индол-2-карбоновой кислоты {[(1-S)-(R)-гидрокси- (метил-пиперидин-4-ил-карбамоил)-метил]-2-фенил-этил}-амида гидрохлорид 4-({ (3S)-[(5-Хлоро-1H-индол-2-карбонил)-амино]-(2R)- гидрокси-4-фенил-бутирил-метил-амино)-пиперидин-1-карбоновой кислоты трет-бутиловый эфир (292 мг, 0,5 мМ) растворяли в 4 М HCl/диоксане при температуре 0oC, и полученный раствор перемешивали в течение одного часа при комнатной температуре. Полученную смесь концентрировали, а затем, остаток растирали с эфиром и сушили. Выход = 249 мг, 96%; ВРЖХ (60/40) 2,59 минут (96%); PB-MC 469/471 (MH+, 100%). 1H-ЯМР (DMCO-d6) : 11,7 (с, 0,3H), 11,6 (с, 0,7H), 8,75 (шир., 2H, обмен с D2O), 7,70 (д, 1H, J=2 Гц), 7,4-7,1 (м, 8H), 4,94 (д, 0,3H, J=7,8 Гц, обмен с D2O), 4,77 (д, 0,7H, J=7,7 Гц, обмен с D2O), 4,6 (м, 1H), 4,47 (дд, J= 3,8 Гц), 4,4 (м, 0,7H), 3,9 (м, 0,3H), 3,4-3,2 (м, прибл. 1,5H), 2,95 (м, 2H), 2,15-1,8 (м, прибл. 2,5H), 1,75-1,50 (м, 2H).
Анализ для C25H29ClN4O3 +HCl + 0,7 H2O:Вычислено: C 57,96; H 6,11; N 10,82. Найдено: C 58,22; H 6,23; N 10,46. Пример 77 5-Хлоро-1H-индол-2-карбоновой кислоты { ((1S)-(R)-гидрокси-[метил-(1-метил-пиперидин-4-ил)-карбамоил]-метил}- 2-фенил-этил)амида гидрохлорид К раствору, содержащему гидрохлорид (1S)-[(R)-гидрокси -(метил-пиперидин-4-ил-карбамоил)-метил] -2-фенил-этил] -амида 5-хлоро-1H-индол-2-карбоновой кислоты (100 мг, 0,2 мМ) в метаноле (2 мл) при температуре 25oC последовательно добавляли 100 мг измельченных в порошок молекулярных сит 3A, 22 мг триэтиламина (0,2 мМ), ледяную уксусную кислоту (64 мг, 1,1 мМ), цианоборогидрид натрия (95%, 18 мг, 0,3 мМ) и водный раствор формальдегида (37 мас.% в воде, 22 мг, 0,3 мМ). После выдерживания в течение 18 часов, реакционную смесь фильтровали через Целит , а твердые вещества промывали метанолом и концентрировали. Остаток растворяли в этилацетате, и полученный раствор дважды промывали 2N гидроксидом натрия в один раз солевым раствором, а затем сушили и концентрировали. Бесцветный твердый остаток очищали с помощью хроматографии на силикагеле (элюент: 1-8% этанол в дихлорметане) и получали бесцветное твердое вещество (93 мг, 91%). Полученное вещество растворяли в метаноле при температуре 0oC, и полученный раствор обрабатывали 0,21 литрами 1,01 н. соляной кислоты, а затем сразу же концентрировали. Остаток растирали с эфиром и сушили. Выход = 87 мг, 79%; ВРЖХ (60/40) 2,86 мин (95%); TSP-MC 483/485 (MH+, 100%).
Пример 78(3S)-[(5-Хлоро-1H-индол-2-карбонил)-амино] -4-фенил-масляной кислоты метиловый эфир (3S)-3-Амино-4-фенил-масляной кислоты метилового сложного эфира гидрохлорид (1,15 г, 5 мМ) и 5-хлоро-1H-индол-2-карбоновую кислоту подвергали реакции взаимодействия в соответствии с методикой A. Полученный продукт очищали путем растирания с эфиром. Выход: 1,46 г (79%), ВРЖХ (60/40) 8,85 мин (100%); PB-MC 371/373 (MH+, 100%/35%). Анализ для C20H19ClN2O3: Вычислено: C 64,78; H 5,16; N 7,55. Найдено: C 64,81: H 5,34; N 7,46. Пример 78A (3S)-Амино-4-фенил-масляной кислоты метилового сложного эфира гидрохлорид (3S)-Трет-бутоксикарбониламино-4-фенил-масляной кислоты метиловый эфир (см. Heferocyсles, стр. 1835 (1989) и J. Med, Chem. 1975, стр. 761, 3,49 г, 12,1 мМ) растворяли в смеси 4 М HCl/диоксана при 0oC, а затем, раствор перемешивали в течение получаса при 2-25oC. Полученную смесь концентрировали, и остаток растирали с эфиром и сушили. Выход = 2,56 г (92%). При этом следует иметь в виду, что настоящее изобретение не ограничивается приведенными выше примерами его осуществления, и в него могут быть внесены различные изменения и модификации, не выходящие, однако, за рамки существа и объема изобретения, сформулированные в нижеследующей формуле изобретения. Формула изобретения
-аланинамидов общей формулы I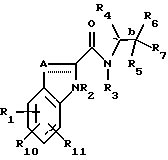 и их фармацевтически приемлемые соли и сложные эфиры, где пунктирная линия (—) обозначает связь; А представляет собой -С(Н)=, -С((С1 – С4)алкил)= или -С(галогено); R1, R10, R11 независимо представляют водород, галоген, циано, (С1 – С4) алкил, (С1 – С4) алкокси; R2 представляет водород; R3 представляет водород; R4 представляет водород, метил, этил, н-пропил, гидрокси(С1 – С3) алкил, (С1 – С3) алкокси (С1 – С3) алкил, фенил (С1 – С4) алкил, тиен-2- или -3-ил(С1 – С4) алкил, где указанные кольца R4 являются независимо моно- или дизамещенными y атома углерода галогеном; R5 представляет водород, гидрокси, (С1 – С5) алкил, (С1 – С5)алкокси, бензилоксикарбонил(С1-С4)алкокси, карбокси(С1-С4)алкокси, 5-хлор-1Н-индол-2-карбонилокси; R7 представляет водород, (С1-С5) алкил или R5 и R7 вместе представляют оксогруппу; R6 представляет карбокси, (С1-С8) алкоксикарбонил, С(О)NR8R9 или С(О)R12, где R8 представляет водород, (С1-С3) алкил, гидрокси, (С1-С3) алкокси и R9 представляет водород, (С1-С8)алкил, гидрокси, гидрокси (С1-С5) алкил, (С1-С8) алкокси, метиленперфторированный (С1-С8) алкил, пиридил, пиперидинил, где указанные выше кольца R9 присоединены через связь углерод – азот, где неароматические азотсодержащие кольца R9 являются необязательно замещенными y атома азота (С1-С4)алкилом, бензилом, (С1-С6) алкоксикарбонилом; R12 представляет собой пиперазин-1-ил, 4-(С1-С4) алкилпиперазин-1-ил, 4-формилпиперазин-1-ил, морфолино, тиоморфолино, 1-оксотиоморфолино, 1,1-диоксотиоморфолино, тиазолидин-3-ил, 1-оксотиазолидин-3-ил или R12 представляет собой 2-, 4- и/или 5-моно- или дизамещенный тиазолидин-3-ил, 3- и/или 4-моно- или дизамещенный пирролидин-1-ил, 3-, 4- и/или 5-моно-, ди- или тризамещенный пиперидин-1-ил, 3-, 4- и/или 5-моно-, ди- или тризамещенный пиперазин-1-ил, 3-замещенный азетидин-1-ил, 4- и/или 5-моно- или дизамещенный 1,2-оксазинан-2-ил, 4- и/или 5-моно или дизамещенный изоксазолидин-2-ил, где указанные R12 заместители независимо представляют водород, гидрокси, формил, оксо, карбокси, (С1-С6) алкоксикарбонил или гидрокси (С1-С5) алкил; при условии, что если R4 является водородом, метилом, этилом или н-пропилом, то R5 является ОН; при условии, что если R5 и R7 являются водородом, то R4 не является водородом, метилом, этилом, н-пропилом, гидрокси (С1-С3) алкилом, а R6 является С(О)NR8R9, С(О)R12 или (С1-С4) алкоксикарбонилом. 2. Соединение по п.1, где R1 представляет собой 5-Н, 5-галоген, 5-метил или 5-циано; R10 и R11 независимо представляют собой Н или галоген; А представляет собой -С(Н)=; R2 и R3 представляют собой Н; R4 представляет собой фенил (С1-С2) алкил, где указанные фенильные группы независимо являются моно-, дизамещенными галогеном, или R4 представляет собой тиен-2- или -3-ил (С1-С2) алкил; R5 представляет собой гидрокси; R6 представляет собой С(О)NR8R9 или С(О)R12 и R7 представляет собой Н. 3. Соединение по п. 2, где атом углерода а имеет S-конфигурацию; атом углерода b имеет R-кофигурацию; R4 представляет собой фенил (С1-С2) алкил, тиен-2-ил (С1-С2) алкил, тиен-3-ил-(С1-С2) алкил, где указанные кольца являются независимо моно- или дизамещенными фтором; R6 представляет собой С(О)NR8R9; R8 представляет собой (С1-С3) алкил, гидрокси или (С1-С3) алкокси, R9 представляет собой Н, (С1-С8) алкил, гидрокси (С1-С6) алкил, (С1-С8) алкокси, пиридил, пиперидинил. 4. Соединение по п.3, выбранное из группы, включающей в себя 5-хлоро-1Н-индол-2-карбоновой кислоты [(1S)-{(R)-гидрокси-диметилкарбамоилметил}-2-фенил-этил] -амид; 5,6-дихлоро-1Н-индол-2-карбоновой кислоты [(1S)-{(R)-гидрокси-(метокси-метилкарбамоил)-метил} -2-фенил-этил] -амид; 5-хлоро-1Н-индол-2-карбоновой кислоты { (1S)-[(R)-гидрокси-(метокси-метилкарбамоил)-метил] -2-фенил-этил} -амид; 5-хлоро-1Н-индол-2-карбоновой кислоты ((1S)-[(R)-гидрокси-[(2-гидрокси-этилметил-карбамоил)-метил] -2-фенил-этил)-амид; 5-хлоро-1Н-индол-2-карбоновой кислоты {(1S)-[(R)-гидрокси-(метил-пиридин-2-ил-карбамоил)-метил] -2-фенил-этил}-амид или 5-хлоро-1Н-индол-2-карбоновой кислоты {(1S)-[(R)-гидрокси-метил-(2-пиридин-2-ил-этил)-карбамоил]-метил)-2-фенил-этил}-амид. 5. Соединение по п.3, где R1 представляет собой 5-хлоро; R10 и R11 представляют собой Н; R4 представляет собой бензил; R8 представляет собой метил и R9 представляет собой метил. 6. Соединение по п.3, где R1 представляет собой 5-хлоро; R11 представляет собой Н; R10 представляет собой 6-хлоро; R4 представляет собой бензил; R8 представляет собой метил и R9 представляет собой метокси. 7. Соединение по п. 3, где R1 представляет собой 5-хлоро; R10 и R11 представляет собой Н; R4 представляет собой бензил; R8 представляет собой метил и R9 представляет собой метокси. 8. Соединение по п. 3, где R1 представляет собой 5-хлоро; R10 и R11 представляет собой Н; R4 представляет собой бензил; R8 представляет собой метил и R9 представляет собой 2-(гидрокси) этил. 9. Соединение по п. 3, где R1 представляет собой 5-хлоро; R10 и R11 представляют собой Н; R4 представляет собой бензил; R8 представляет собой метил и R9 представляет собой пиридин-2-ил. 10. Соединение по п. 3, где R1 представляет собой 5-хлоро; R10 и R11 представляют собой Н; R4 представляет собой бензил; R8 представляет собой метил и R9 представляет собой 2-(пиридин-2-ил) этил. 11. Соединение по п.2, где атом углерода а имеет (S)-конфигурацию; атом углерода b имеет (R-)-конфигурацию; R4 представляет собой фенил (С1 – С2) алкил, тиен-2-ил-(С1 – С2) алкил, тиен-3-ил-(С1-С2) алкил, где указанные кольца являются независимо моно- или дизамещенными фтором; R6 представляет собой С(О)R12 и R12 представляет собой морфолино, 4-(С1-С4) алкилпиперазин-1-ил, 3-замещенный азетидин-1-ил, 3- и/или 4-, моно- или дизамещенный пирролидин-1-ил, 4- и/или 5-моно или дизамещенный изоксазолидин-2-ил, 4- и/или 5-, моно- или дизамещенный 1,2-оксазинан-2-ил, где указанные заместители независимо представляют собой водород, гидрокси, оксо. 12. Соединение по п.1, выбранное из группы, включающей в себя 5-хлоро-1Н-индол-2-карбоновой кислоты [(1S)-бензил-(2R)-гидрокси-3-(4-метилпиперазин-1-ил)-3-оксо-пропил] -амида гидрохлорид; 5-хлоро-1Н-индол-2-карбоновой кислоты [(1S)-бензил-(2R)-гидрокси-3-(3-гидроксиазетидин-1-ил)-3-оксо-пропил] -амид; 5-хлоро-1Н-индол-2-карбоновой кислоты [(1S)-бензил-(2R)-гидрокси-3-изоксазолидин-2-ил-3-оксо-пропил] -амид; 5-хлоро-1Н-индол-2-карбоновой кислоты ((1S)-бензил-(2R)-гидрокси-3-[1,2] -оксазинан-2-ил-3-оксо-пропил)-амид; 5-хлоро-1Н-индол-2-карбоновой кислоты [(1S)-бензил-(2R)-гидрокси-3-((3S)-гидрокси-пирролидин-1-ил)-3-оксо-пропил] -амид; 5-хлоро-1Н-индол-2-карбоновой кислоты [(1S)-бензил-3-((3S), 4S)-дигидроксипирролидин-1-ил)-(2R)-гидрокси-3-оксо-пропил] -амид; 5-хлоро-1Н-индол-2-карбоновой кислоты [(1S)-бензил-3-((3R, 4S)-дигидроксипирролидин-1-ил)-(2R)-гидрокси-3-оксо-пропил] -амид; или 5-хлоро-1Н-индол-2-карбоновой кислоты [(1S)-бензил-(2R)-гидрокси-3-морфолин-4-ил-3-оксо-пропил]-амид. 13. Соединение по п. 11, где R1 представляет собой 5-хлоро; R10 и R11 представляют собой Н; R4 представляет собой бензил и R12 представляет собой 4-метилпиперазин-1-ил. 14. Соединение по п. 11, где R1 представляет собой 5-хлоро; R10 и R11 представляют собой Н; R4 представляет собой бензил и R12 представляет собой 3-гидроксиазетидин-1-ил. 15. Соединение по п. 11, где R1 представляет собой 5-хлоро; R10 и R11 представляют собой Н; R4 представляет собой бензил и R12 представляет собой изоксазолидин-2-ил. 16. Соединение по п. 11, где R1 представляет собой 5-хлоро; R10 и R11 представляют собой Н; R4 представляет собой бензил и R12 представляет собой (1,2)-оксазинан-2-ил. 17. Соединение по п. 11, где R1 представляет собой 5-хлоро; R10 и R11 представляют собой Н; R4 представляет собой бензил и R12 представляет собой 3(S)-гидроксипирролидин-1-ил. 18. Соединение по п. 11, где R1 представляет собой 5-хлоро; R10 и R11 представляют собой Н; R4 представляет собой бензил и R12 представляет собой (3S, 4S)-дигидроксипирролидин-1-ил. 19. Соединение по п. 11, где R1 представляет собой 5-хлоро; R10 и R11 представляют собой Н; R4 представляет собой бензил и R12 представляет собой (3S, 4S)-дигидроксипирролидин-1-ил. 20. Соединение по п. 11, где R1 представляет собой 5-хлоро; R10 и R11 представляют собой Н; R4 представляет собой бензил и R12 представляет собой морфолино. 21. Соединение по п.1, где R1 представляет собой водород, галоген, метил или циано; R10 и R11 независимо представляют собой Н или галоген; А представляет собой -С(Н)= ; R2 и R3 представляют собой Н; R4 представляет собой фенил (С1-С2) алкил, где указанные фенильные группы независимо являются моно-, дизамещенными галогеном, или R4 представляет собой тиен-2- или -3-ил(С1-С2) алкил, R5 представляет собой гидрокси; R6 представляет собой карбокси или (С1-С8) алкоксикарбонил и R7 представляет собой водород, (С1-С6) алкил. 22. Соединение по п.21, где атом углерода а имеет (S)-конфигурацию; атом углерода b имеет (R)-конфигурацию; R4 представляет собой фенил (С1-С2) алкил, тиен-2-ил-(С1-С2) алкил, тиен-3-ил-(С1-С2) алкил, где указанные кольца являются независимо моно- или дизамещенными фтором; R10 и R11 представляют собой Н; R6 представляет собой карбокси и R7 представляет собой Н. 23. Соединение по п. 22, где R1 представляет собой 5-хлоро; R10 и R11 представляют собой Н и R4 представляет собой бензил. 24. Соединение по п.1, где R1 представляет собой Н, галоген, метил или циано; R10 и R11 независимо представляют собой водород или галоген; А представляет собой -С(Н)= ; R2 и R3 представляют собой Н; R4 представляет собой фенил (С1-С2) алкил, где указанные фенильные группы являются независимо моно-, дизамещенными галогеном, или R4 представляет собой тиен-2- или 3-ил(С1-С2) алкил; R5 представляет собой (С1-С4) алкил, (С1-С5)-алкокси, карбокси (С1 – С4) алкокси, бензилоксикарбонил (С1-С4) алкокси; R6 представляет собой карбокси или (С1-С8) алкоксикарбонил и R7 представляет собой Н, (С1-С6) алкил. 25. Соединение по п.1, где R1 представляет собой Н, галоген, метил или циано; R10 и R11 независимо представляют собой Н или галоген; А представляет собой -С(Н)= ; R2 и R3 представляют собой Н; R4 представляет собой фенил (С1-С2) алкил, где указанные фенильные группы являются независимо моно-, дизамещенными галогеном, или R4 представляет собой тиен-2- или 3-(С1-С2) алкил; R5 представляет собой (С1-С4) алкил, (С1-С5)-алкокси, карбокси (С2-С4) алкокси, бензилоксикарбонил (С1-С4) алкокси; R6 представляет собой С(О)NR8R9 или С(О)R12 и R7 представляет собой водород, (С1-С6) алкил. 26. Способ лечения гликогенфосфорилазозависимого заболевания или состояния у млекопитающего, предусматривающий введение млекопитающему, страдающему гликогенфосфорилазозависимым заболеванием или состоянием, терапевтически эффективного для лечения указанного заболевания или состояния количества соединения по п.1. 27. Способ по п.26 для лечения гипергликемии у млекопитающего, предусматривающий введение млекопитающему, страдающему гипергликемией, эффективного для лечения гипергликемии количества соединения по п.1. 28. Способ по п.26 для лечения диабета у млекопитающего, предусматривающий введение млекопитающему, страдающему диабетом, эффективного для лечения диабета количества соединения по п.1. 29. Способ по п. 26 для лечения гиперхолестеринемии у млекопитающего, предусматривающий введение млекопитающему, страдающему гиперхолестеринемией, эффективного для лечения гиперхолестеринемии количества соединения по п.1. 30. Способ по п.26 для лечения атеросклероза у млекопитающего, предусматривающий введение млекопитающему, страдающему атеросклерозом, эффективного для лечения атеросклероза количества соединения по п.1. 31. Способ по п.26 для лечения гиперинсулинемии у млекопитающего, предусматривающий введение млекопитающему, страдающему гиперинсулинемией, эффективного для лечения гиперинсулинемии количества соединения по п.1. 32. Способ по п.26 для лечения гипертензии у млекопитающего, предусматривающий введение млекопитающему, страдающему гипертензией, эффективного для лечения гипертензии количества соединения по п.1. 33. Способ по п.26 для лечения гиперлипидемии у млекопитающего, предусматривающий введение млекопитающему, страдающему гиперлипидемией, эффективного для лечения гиперлипидемии количества соединения по п.1. 34. Способ по п.26 для предупреждения ишемического повреждения миокарда у млекопитающего, предусматривающий введение млекопитающему, подверженному риску ишемического повреждения миокарда в ходе операции, эффективного для предупреждения указанного повреждения количества соединения по п.1. 35. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении гликогенфосфорилазы, содержащая терапевтически эффективное количество соединения по п.1 и фармацевтически приемлемый носитель. 36. Фармацевтическая композиция по п.35, предназначенная для лечения гликогенфосфорилазозависимых заболеваний или состояний у млекопитающих и включающая в себя терапевтически эффективное для лечения указанного заболевания или состояний количество соединения по п.1 и фармацевтически приемлемый носитель. РИСУНКИ
MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 06.06.2004
Извещение опубликовано: 20.09.2007 БИ: 26/2007
|
||||||||||||||||||||||||||