Патент на изобретение №2315994
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) СПОСОБ ОПРЕДЕЛЕНИЯ ДИГИДРОКСИБЕНЗОЛОВ В ВОДНЫХ РАСТВОРАХ
(57) Реферат:
Способ позволяет контролировать содержание дигидроксибензолов в очищенных сточных водах предприятий лакокрасочной и фотографической промышленности. Способ включает определение дигидроксибензолов в водных растворах методом обращенно-фазовой микроколоночной высокоэффективной жидкостной хроматографии с предварительной стадией экстракционного концентрирования при рН 2-3. При этом экстракционное концентрирование дигидроксибензолов осуществляют ацетонитрилом при соотношении равновесных объемов водной и органической фаз 100:2 в присутствии высаливателя сульфата аммония в количестве 36,5-40,0 мас.% по отношению к массе пробы. Способ обеспечивает повышение степени концентрирования, снижение пределов обнаружения, снижение расхода экстракционных реагентов и себестоимости анализа. 2 табл.
Изобретение относится к аналитической химии органических соединений и может быть рекомендовано для аналитического контроля содержания химических соединений в очищенных сточных водах предприятий лакокрасочной и фотографической промышленности. Известен способ определения гидрохинона и метола в водных растворах (RU патент 2172952, БИ №24 от 27.08.2001). Способ предполагает предварительное концентрирование гидрохинона из анализируемой водной пробы диоксаном при соотношении равновесных объемов водной и органической фаз 10:1 в присутствии сульфата аммония и последующее его определение вольтамперометрическим методом. Недостатки способа – применение нестандартного оборудования (трехэлектродная ячейка для вольтамперометрического детектирования), неселективность, невысокая степень концентрирования. Наиболее близким по технической сущности и достигаемому эффекту является способ определения гидрохинона и пирокатехина в водных растворах (RU патент 2205398, БИ №15 от 27.05.2003), который включает концентрирование гидрохинона и пирокатехина гексановым раствором триоктилфосфиноксида (ТОФО) и определение их в выделившейся органической фазе методом обращенно-фазовой микроколоночной жидкостной хроматографии. Недостатки способа: применение дорогостоящего и дефицитного реактива ТОФО; сложность пробоподготовки (высушивание органического слоя в токе азота); невысокая степень концентрирования (коэффициент концентрирования равен 10). Технической задачей изобретения является повышение степени концентрировании, снижение пределов обнаружения, упрощение способа определения дигидроксибензолов в водных растворах (за счет исключения стадии пробоподготовки органической фазы), снижение расхода экстракционных реагентов и себестоимости анализа, а также расширение области его применения. Поставленная техническая задача достигается тем, что в способе определения дигидроксибензолов в водных растворах, включающем определение дигидроксибензолов в водных растворах методом обращенно-фазовой микроколоночной высокоэффективной жидкостной хроматографии с предварительной стадией экстракционного концентрирования при рН 2-3, новым является то, что экстракционное концентрирование дигидроксибензолов осуществляют ацетонитрилом при соотношении равновесных объемов водной и органической фаз 100:2 в присутствии высаливателя сульфата аммония в количестве 36,5-40,0 мас.% по отношению к массе пробы. Технический результат изобретения заключается в упрощении способа за счет исключения стадии пробоподготовки органической фазы (исключение из анализа дефицитного реактива ТОФО и стадии высушивания органического слоя в токе азота), повышении степени концентрирования (коэффициент концентрирования равен 50), снижении пределов обнаружения и расхода экстракционных реагентов. Способ определения дигидроксибензолов в водных растворах осуществляется следующим образом. К 100 см3 водной пробы, содержащей гидрохинон, пирокатехин и резорцин, подкисляют до рН 2,5, добавляют высаливатель сульфата аммония в количестве 36,5-40,0 мас.% по отношению к массе пробы и 2,5 см3 ацетонитрила (Коренман Я.И., Ермолаева Т.Н., Мишина А.В. и др. Тройные диаграммы вода – сульфат аммония – гидрофильный растворитель. Липецк, 1995. – Деп. В ВИНИТИ №295-В-95. – 10 с.), экстрагируют на на вибросмесителе 15 мин. Через 15 мин после расслаивания фаз (соотношение равновесных объемов водной и органической фаз 100:2) отделяют органический слой и анализируют на хроматографе «Милихром-4» с УФ-детектором. Определение дигидроксибензолов проводят методом обращенно-фазовой микроколоночной высокоэффективной жидкостной хроматографии: колонка 80×2 мм; сорбент – силасорб С 18; размер частиц сорбента 7 мкм; подвижная фаза – смесь ацетонитрил: вода в объемном соотношении 20:80; аналитическая длина волны 272 нм; расход подвижной фазы 10-4 дм3/мин; объем вводимой пробы 5×10-6 дм3; время анализа 15 мин. Концентрацию каждого из дигидроксибензолов в водном растворе вычисляют по формуле: С=(С0·100)/(R·r), где С0 – концентрация дигидроксибензола (пирокатехина, гидрохинона, резорцина) в экстракте, которую находят методом абсолютной градуировки, мг/дм3; С – концентрация дигидроксибензола в исходной пробе, мг/дм3; r – соотношение равновесных объемов водной и органической фаз; R – степень извлечения дигидроксибензола в системе ацетонитрил – водно-солевой раствор, %. Степень извлечения дигидроксибензолов (R, %) рассчитывают по формуле: R=Д·100/Д+r, где Д – коэффициент распределения дигидроксибензолов в системе ацетонитрил – водно-солевой раствор; r – соотношение равновесных объемов водной и органической фаз. Способ осуществим. Степень извлечения дигидроксибензолов предложенным способом составляет 94-96%. Способ поясняется следующими примерами. Пример 1. Растворы дигидроксибензолов готовят из препаратов квалификации «ч», очищенных возгонкой. Степень чистоты препаратов контролируют по температурам плавления. Исходные водные растворы с точно известной концентрацией готовят методом разбавления стандартных растворов. К 100 см3 водной пробы, содержащей гидрохинон, пирокатехин и резорцин, подкисляют до рН 2,5, добавляют высаливатель сульфата аммония в количестве 38,5 мас.% по отношению к массе пробы и 2,5 см3 ацетонитрила, экстрагируют на на вибросмесителе 15 мин. Через 15 мин после расслаивания фаз (соотношение равновесных объемов водной и органической фаз 100:2) отделяют органический слой и анализируют на хроматографе «Милихром-4» с УФ-детектором. Определение дигидроксибензолов проводят методом обращенно-фазовой микроколоночной высокоэффективной жидкостной хроматографии: колонка 80×2 мм; сорбент – силасорб С 18; размер частиц сорбента 7 мкм; подвижная фаза – смесь ацетонитрил: вода в объемном соотношении 20:80; аналитическая длина волны 272 нм; расход подвижной фазы 10-4 дм3/мин; объем вводимой пробы 5×10-6 дм3; время анализа 15 мин. Концентрацию каждого из дигидроксибензолов в водном растворе вычисляют по формуле: С=(С0·100)/(R·r), где С0 – концентрация дигидроксибензола (пирокатехина, гидрохинона, резорцина) в экстракте, которую находят методом абсолютной градуировки, мг/дм3; С – концентрация дигидроксибензола в исходной пробе, мг/дм3; r – соотношение равновесных объемов водной и органической фаз; R – степень извлечения дигидроксибензола в системе ацетонитрил – водно-солевой раствор, %. Степень извлечения дигидроксибензолов (R, %) рассчитывают по формуле: R=Д·100/Д+r, где Д – коэффициент распределения дигидроксибензолов в системе ацетонитрил – водно-солевой раствор; r – соотношение равновесных объемов водной и органической фаз. Коэффициенты распределения дигидроксибензолов определяют фотометрически на фотоэлектроколориметре КФК-3 по реакции с диазотированной сульфаниловой кислотой. Способ осуществим. Степень извлечения дигидроксибензолов 94-96%. Данные анализа представлены в табл.1, 2. Как видно из таблиц, коэффициент концентрирования равен 50, что позволяет снизить предел обнаружения в 5 раз, ошибка определения дигидроксибензолов не превышает 10%. Пример 2. К 100 см3 водной пробы, содержащей гидрохинон, пирокатехин и резорцин, подкисляют до рН 2,5, добавляют высаливатель сульфата аммония в количестве 36,5 мас.% по отношению к массе пробы и 2,5 см3 ацетонитрила, экстрагируют на на вибросмесителе 15 мин. Далее анализируют аналогично примеру 1. Способ осуществим. Данные анализа представлены в таблице 1, 2. Как видно из таблиц, коэффициент концентрирования равен 50, ошибка определения не превышает 10%. Пример 3. К 100 см3 водной пробы, содержащей гидрохинон, пирокатехин и резорцин, подкисляют до рН 2,5, добавляют высаливатель сульфата аммония в количестве 40,0 мас.% по отношению к массе пробы и 2,5 см3 ацетонитрила, экстрагируют на вибросмесителе 15 мин. Далее анализируют аналогично примеру 1. Способ осуществим. Данные анализа представлены в таблице 1, 2. Как видно из таблиц, коэффициент концентрирования равен 50, ошибка определения не превышает 10%. При уменьшении количества высаливателя (меньше 36,5 мас.%) выделившийся экстракт содержит значительное количество воды, что мешает дальнейшему определению; увеличение содержания высаливателя (более 40,0 мас.%) приводит к выделению кристаллов соли, которые адсорбируют дигидроксибензолы. Изменение соотношения равновесных объемов в сторону увеличения органической фазы приводит к уменьшению коэффициента концентрирования, уменьшение объема органической фазы снижает степень извлечения. Правильность способа проверена на модельных растворах дигидроксибензолов методом «введено – найдено». Результаты определения представлены в таблице 1. Относительная ошибка определения пирокатехина не превышает 10%.
В таблице 2 приведена сравнительная характеристика предлагаемого способа и прототипа.
Предлагаемый способ позволяет проводить определение дигидроксибензолов в водных растворах без стадии пробоподготовки органической фазы, снизить расход экстракционных реагентов, повысить степень концентрирования, снизить предел обнаружения в 5 раз, удешевить способ, а также расширить области применения для контроля содержания дигидроксибензолов.
Формула изобретения
Способ определения дигидроксибензолов в водных растворах методом обращенно-фазовой микроколоночной высокоэффективной жидкостной хроматографии с предварительной стадией экстракционного концентрирования при рН 2-3, отличающийся тем, что экстракционное концентрирование дигидроксибензолов осуществляют ацетонитрилом при соотношении равновесных объемов водной и органической фаз 100:2 в присутствии высаливателя сульфата аммония в количестве 36,5-40,0 мас.% по отношению к массе пробы.
MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 17.10.2008
Извещение опубликовано: 27.06.2010 БИ: 18/2010
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||
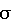 , мг/дм3
, мг/дм3